
Thermally activated processes in an organic long-persistent luminescence system†
Kazuya
Jinnai
 ab,
Naohiro
Nishimura
ab,
Chihaya
Adachi
*abc and
Ryota
Kabe
*bd
ab,
Naohiro
Nishimura
ab,
Chihaya
Adachi
*abc and
Ryota
Kabe
*bd
aCenter for Organic Photonics and Electronics Research (OPERA), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: ryota.kabe@oist.jp
bJST, ERATO, Adachi Molecular Exciton Engineering Project, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: adachi@cstf.kyushu-u.ac.jp
cInternational Institute for Carbon Neutral Energy Research (I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
dOrganic Optoelectronics Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, Kunigami-gun, Okinawa 904-0495, Japan
First published on 28th April 2021
Abstract
Glow-in-the-dark materials can store absorbed photon energy and emit light for long periods of time. While inorganic long-persistent luminescence (LPL) materials are crystalline and often require rare metals, organic LPL (OLPL) materials are flexible and require no rare metals. The emission process of OLPL systems consists of photoinduced charge separation, charge accumulation, and emission from charge recombination. Although emission processes of OLPL systems have been investigated, the charge separation and accumulation processes remain enigmatic. In this study, we investigated the charge carrier dynamics of a binary OLPL system comprising of electron donors and acceptors. We confirmed the presence of thermal activation processes, thermally activated delayed fluorescence and thermoluminescence in the OLPL system.
1. Introduction
Glow-in-the-dark materials, which can store absorbed photon energy and emit light for long periods of time, are currently made entirely of inorganic materials.1,2 Inorganic long-persistent luminescence (LPL) materials are used as light sources that do not require electrical power, such as in emergency signs and watch dials. Inorganic LPL materials have good luminescence properties and durability, but require various fabrication processes like powdering and dispersing into polymeric media because of their insoluble crystalline properties. High-performance LPL materials also require rare metal dopants.1,2In contrast, organic LPL (OLPL) systems,3 consisting of organic electron donors and acceptors, do not require rare metals and can form transparent and flexible films by solution processes.4,5 Unlike long-lived phosphorescence,6,7 which is a radiative transition from a triplet excited state to a singlet ground state, OLPL systems accumulate energy in charge-separated states, similar to inorganic LPL systems.8,9 While long-lived phosphorescence shows simple exponential emission decay, the emission from charge recombination is a higher-order reaction and frequently shows emission decay according to the power law.10 Thus, the LPL decay is empirically fitted with the following power-law equation:11
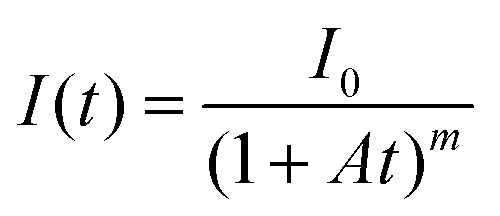 | (1) |
Here, A (s−1) is the rate constant of the entire emission process and m is a parameter that depends on the materials (0.5 < m < 2). However, each parameter depends on the number of accumulated carriers that are affected by the emission intensity and irradiation time.12
The emission process of OLPL systems consists of photoinduced charge separation, charge accumulation, and emission from charge recombination. The mixture of electron donors and acceptors forms the charge-transfer (CT) excited state between them, which is called an exciplex, after photoexcitation (Fig. 1).13 Then, some of the CT excited states become radical ion pairs of donor radical cations and acceptor radical anions.14 After successive charge recombination of these radical ion pairs, CT excited states are regenerated and light is emitted from the CT excited state (Fig. 1(b)).
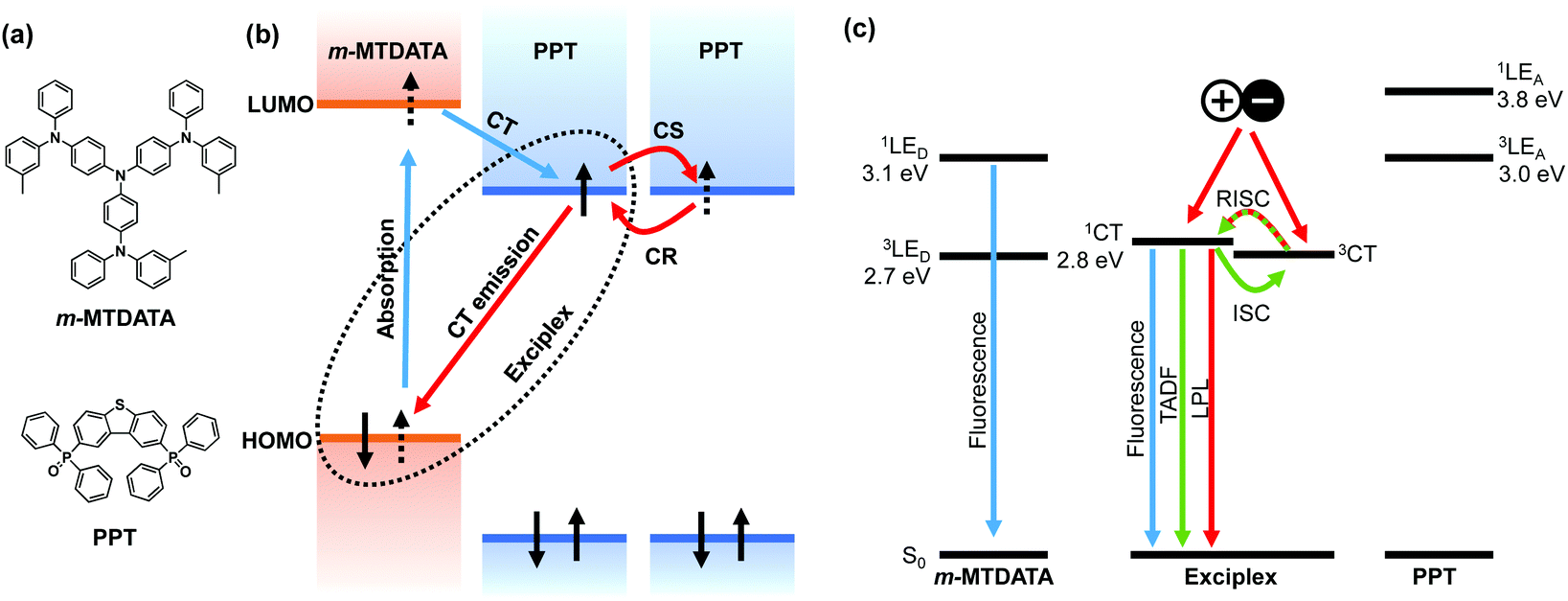 | ||
| Fig. 1 (a) Chemical structures of the electron donor, m-MTDATA, and the electron acceptor, PPT. (b) HOMO and LUMO energy diagrams and the emission mechanism of an OLPL system. (c) Singlet and triplet energy diagram of an m-MTDATA/PPT system. The OLPL system exhibits fluorescence, TADF, and LPL from the 1CT state. | ||
The charge recombination process generates both singlet and triplet CT excited states of exciplexes (1CT and 3CT, respectively), but most of the emission is expected to occur from the radiative transition of 1CT since exciplexes exhibit thermally activated delayed fluorescence (TADF) through reverse intersystem crossing (RISC) due to the small energy gap (ΔEST) between 1CT and 3CT.15,16 Local triplet excited states of donors and acceptors (3LE) also influence the emission process. If 1CT is the lowest excited state, LPL emission originates from 1CT, whereas if 3LE is much lower than 1CT, LPL emission occurs from both 1CT and 3LE.8,9
Although emission processes of OLPL systems have been investigated, the charge separation and accumulation processes remain unclarified. In this study, the effects of excitation power intensity, excitation time, and sample temperature on photoluminescence (PL) and LPL intensities during photoexcitation were observed in order to understand the charge separation process. The initial PL process was also analyzed by time-resolved spectroscopy to understand the contribution of the charge separation process. The thermal activation process of OLPL was demonstrated by thermoluminescence (TL) measurements.
2. Experimental section
m-MTDATA was obtained from Sigma-Aldrich (St Louis, MA, USA). PPT was synthesized according to the literature.17 All compounds were purified by sublimation and stored in a nitrogen-filled glovebox. A ∼0.5 mm thick mixed film was fabricated by the melt cast method. Mixtures of m-MTDATA (1 mol%) and PPT (99 mol%) were placed on a glass substrate with a 1 cm2 area recessed to a depth of 0.5 mm and heated to 250 °C for 30 s in a nitrogen-filled glovebox. After melting, the substrate was rapidly cooled to room temperature.For time-resolved spectroscopy studies, a pulsed Nd:YAG laser (PL2250, EKSPLA) was used as an excitation source (excitation wavelength 355 nm, pulse width 20 ps). Sample emission was detected using a gated streak camera (C10910-04, Hamamatsu Photonics). The sample was placed in a cryostat (PS-HT-200, Nagase Techno-Engineering) connected to a turbo molecular pump (HiPace 80, Pfeiffer Vacuum) and the temperature was controlled from 10 K to 500 K. The sample was excited with a 355 nm pulsed laser (PL2210, EKSPLA) at 10 Hz.
Temperature-dependence measurements and TL measurements were conducted in a cryostat (PS-HT-200, Nagase Techno-Engineering) connected to a turbo molecular pump (HiPace 80, Pfeiffer Vacuum). Emission spectra during (steady-state photoluminescence) and after (LPL) excitation were recorded using a multichannel spectrometer (QE-Pro, Ocean Photonics). Emission decay profiles of LPL were obtained using a silicon photomultiplier (C13366-1350GA, Hamamatsu Photonics) connected to a multimeter (34461A, Keysight).
3. Results and discussion
3.1 Photoluminescence with and without the charge separation process
A mixed film of m-MTDATA as an electron donor and PPT as an electron acceptor was fabricated using a melt cast process.18 The sample was placed in a cryostat and the temperature was controlled from 10 K to 500 K under vacuum. The sample was photoexcited with a 355 nm pulsed laser (pulse width = 20 ps), and the transient emission intensity and decay were recorded with a gated streak camera. The temperature dependence of emission decay profiles and time-resolved emission spectra are shown in Fig. 2. Two exponential decays at nanosecond and microsecond timescales and a long emission tail at the millisecond timescale were observed at room temperature (Fig. 2(a)). Emission spectra were slightly redshifted and then blueshifted over time (Fig. 2(d)), but most emission spectra are attributed to CT emission because they were broader than those of m-MTDATA and PPT (Fig. S1†). Within 10 ns after photoexcitation, the emission spectrum was slightly bluer than a steady-state PL spectrum due to m-MTDATA fluorescence, indicating insufficient CT formation.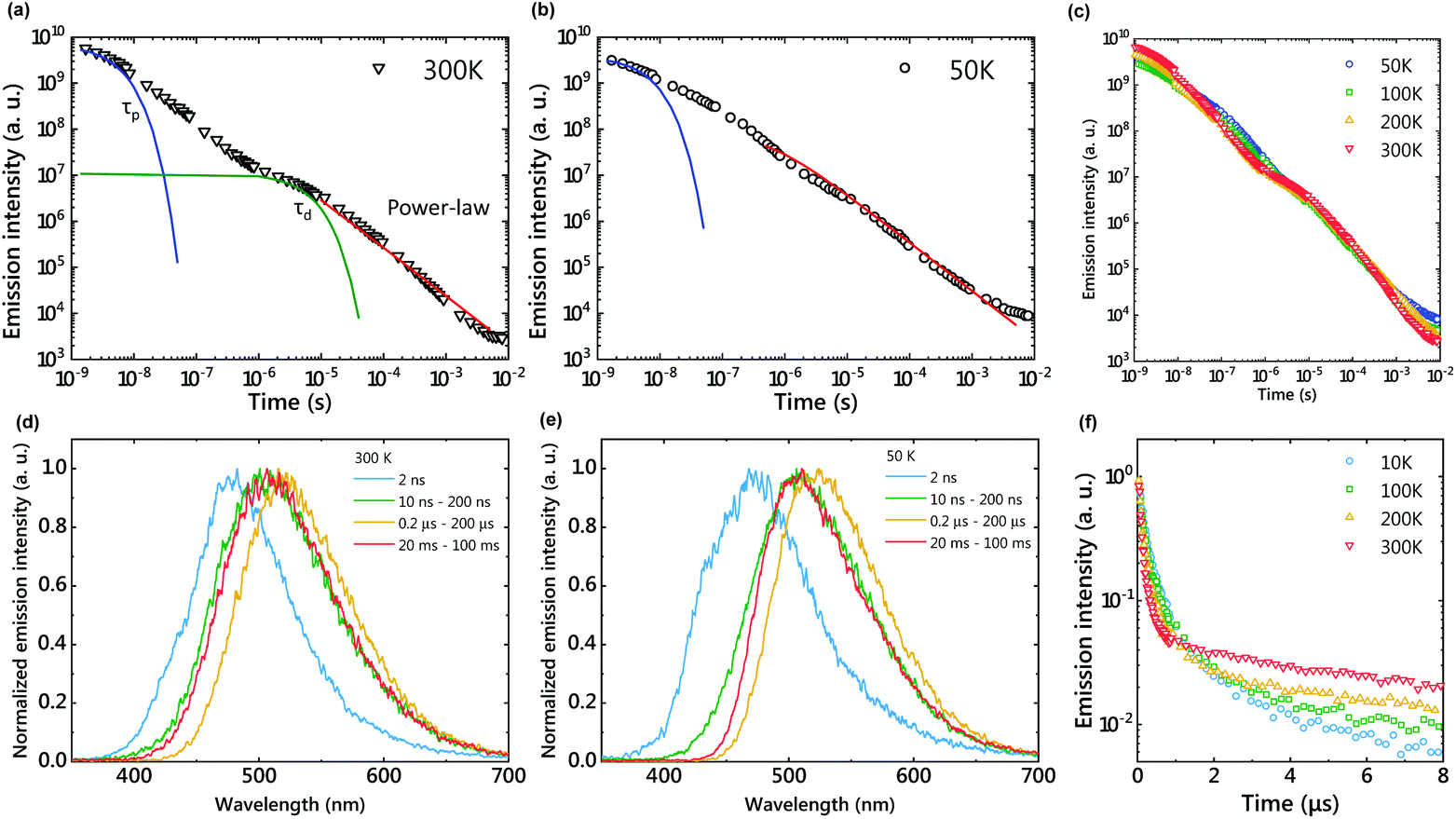 | ||
| Fig. 2 Emission decay profiles in a log–log plot of the m-MTDATA/PPT film at 300 K (a), 50 K (b), and various temperatures (c). Solid lines show fitting of τP, τd, and power-law decay. Time-dependent emission spectra at 300 K (d) and 50 K (e). Emission decay profiles in a semi-log plot (f). | ||
The first exponential decay, at the nanosecond timescale, corresponds to the fluorescence of the exciplex, because the fluorescence lifetime (τp) of 350 ns is longer than that of m-MTDATA. τp becomes longer at a lower temperature due to suppression of the nonradiative decay. The second exponential decay (τd = 21.5 μs) corresponds to TADF, since the emission intensity increased and τd decreased by increasing the sample temperature (Fig. 2(f) and Table S1†). Due to structural relaxation, the CT emission was slightly redshifted during the TADF process.19
The delayed emission after the TADF process corresponds to LPL emission, since the emission decay follows a power-law decay (Fig. 2(a)). Since LPL is not an exponential decay phenomenon, we cannot use the lifetime to discuss this long emission tail. The LPL emission spectra are identical to the fluorescence spectra of the exciplex, because the charge recombination process generates both 1CT and 3CT states and the 3CT excitons are upconverted to 1CT. At 50 K, a power-law emission decay was observed at the microsecond timescale since the RISC process was suppressed (Fig. 2(b)). Although LPL by charge recombination is present in the whole timescale, LPL is difficult to observe at a short timescale, because the emission intensities of fluorescence and TADF are much stronger than that of LPL.
3.2 Charge accumulation process
Unlike long-lived phosphorescence, the charge separation process of the OLPL system requires greatly prolonged photoexcitation. The LPL duration was observed by changing the photoexcitation time (Fig. 3). The LPL duration became longer and approached saturation as the excitation time increased (Fig. 3(b)). However, a slight increase was observed even with 1 hour photoexcitation, suggesting that more than 20 minutes of photoexcitation is required for sufficient charge accumulation. In the plot of excitation power intensity versus PL intensity (during photoexcitation) and integrated LPL intensity, the slope of PL is 1, but that of LPL is close to 0.5 (Fig. 3(c)). A slope of 1 indicates a single-photon process, since fluorescence without charge separation is dominant in PL. In contrast, a slope of 0.5 indicates that the LPL intensity is proportional to the square root of the excitation intensity.20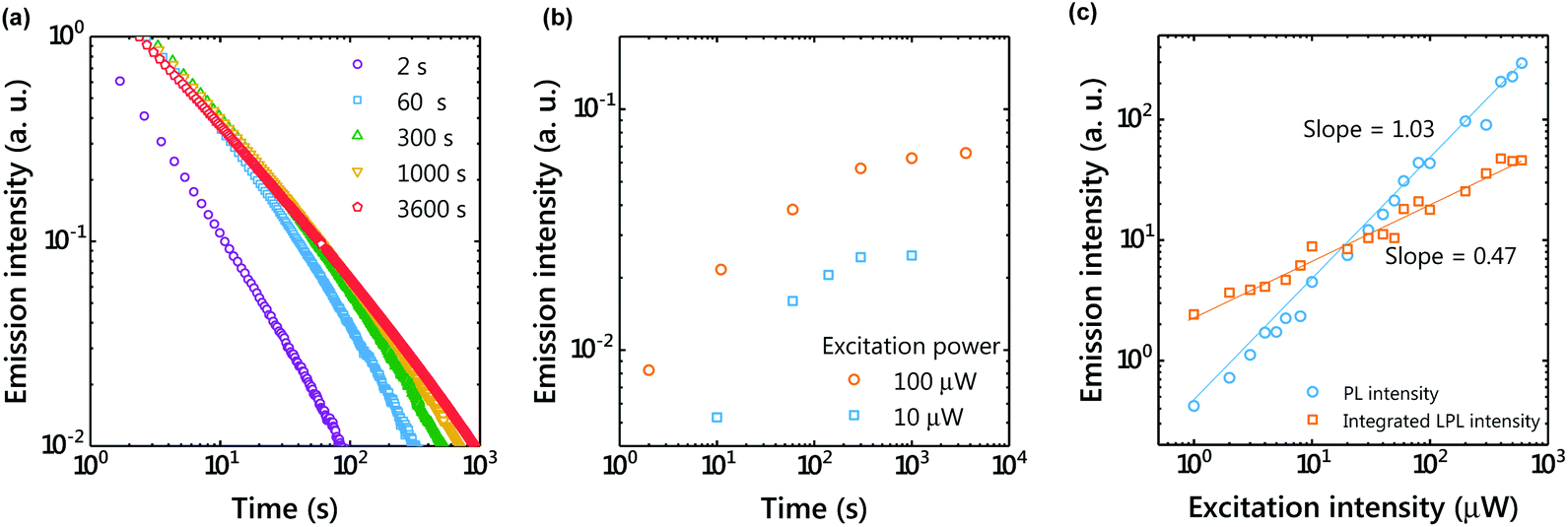 | ||
Fig. 3 (a) Emission decay profiles showing the dependence of LPL on the excitation time (1 molar ratio of the donor; excitation power, 100 μW; temperature, 300 K). (b) Excitation-time dependence of the emission intensity 100 s after photoexcitation. (c) Excitation-power dependence of the emission intensity under photoexcitation (PL) and integrated LPL intensity. The LPL intensity was integrated from 100 s to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s after photoexcitation. The solid lines show linear fitting. 000 s after photoexcitation. The solid lines show linear fitting. | ||
The charge recombination process can be considered as either geminate ion recombination,21,22 which means the recombination between the original donor–acceptor pair, or as bulk recombination,23 which means the recombination between a different donor–acceptor pair. In the steady state, the generation rate of ion pairs (g) and the charge recombination rate (kCRn2) are at equilibrium (kCR: charge recombination rate constant, n: the concentration of the radical cation or radical anion). Because the generation rate of ion pairs is proportional to the excitation intensity, the carrier concentration is proportional to the square root of the excitation intensity. A slope of 0.5 indicates that bulk recombination is the dominant process in this LPL emission. Previously, we reported that the LPL slope was close to 1 in the 3,3′,5,5′-tetramethylbenzidine (TMB)/PPT system,4 but the TMB/PPT system exhibits strong room temperature phosphorescence since 3LED is lower than 1CT. Because we used the intensity at 2 s after excitation in the previous report, the contribution of room temperature phosphorescence of TMB without charge separation (one photon process, slope = 1) was significant in that time range.
To understand the charge accumulation process, time-dependent PL intensities during photoexcitation were recorded (Fig. 4 and S2†). When the excitation power was constant, the PL intensity gradually increased with time due to the increased number of molecules excited by weak photoexcitation of 1 μW (Fig. 4(a and d)). When the excitation power was 10 μW, the PL intensity increased for about 100 s and then became almost constant. With the excitation power of 100 μW, the emission intensity reached its maximum in about 10 s and then decreased. Since charge accumulation is rapid at 100 μW, exciton quenching by accumulated excitons and polaron absorption by the accumulated charge are considered.24
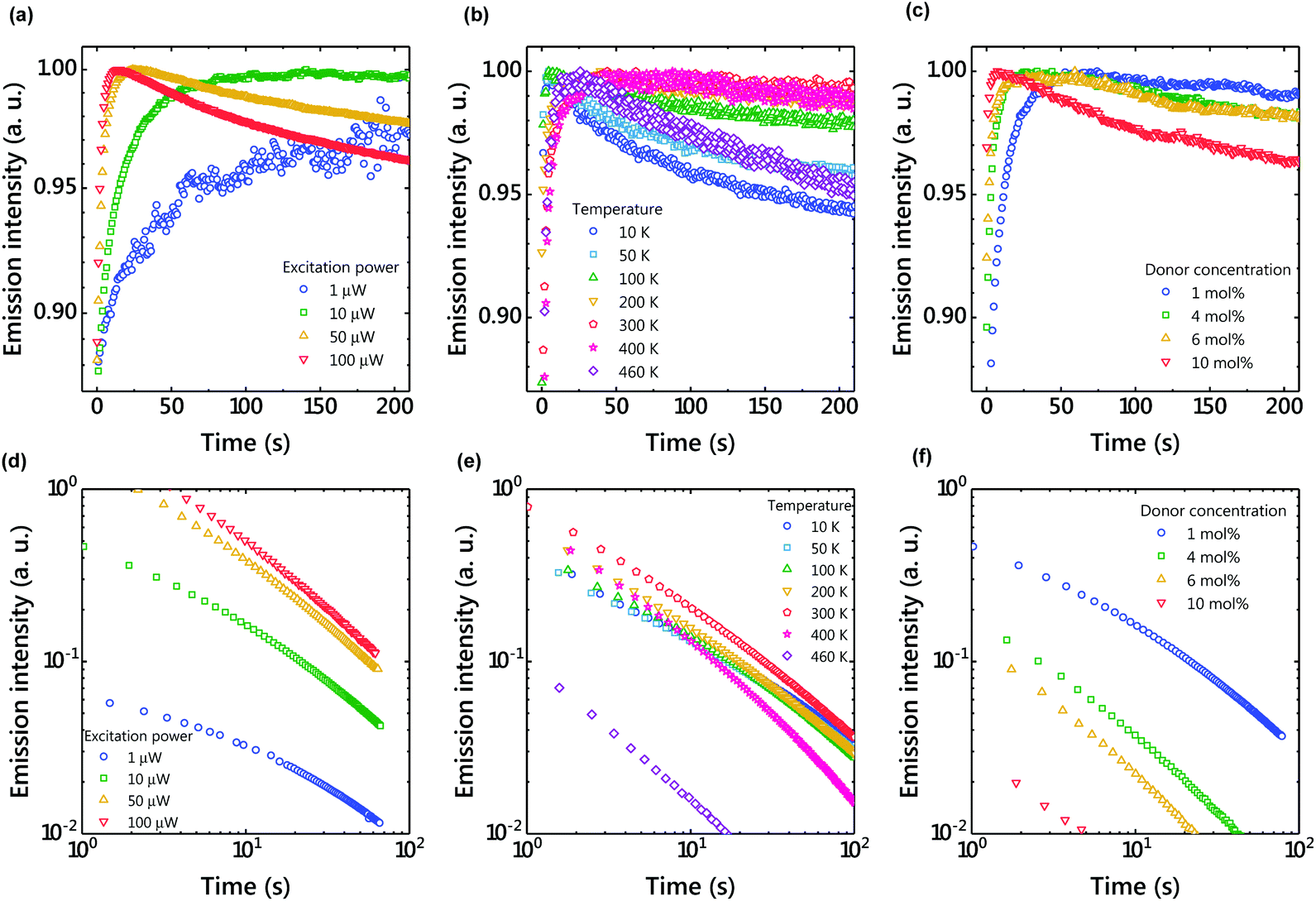 | ||
| Fig. 4 (a) Excitation power dependence of the time-dependent emission intensity under photoexcitation (1 molar ratio of the donor; temperature, 300 K). (b) Temperature dependence of the time-dependent emission intensity under photoexcitation (1 molar ratio of the donor; excitation power, 10 μW). (c) Donor concentration dependence of the time-dependent emission intensity under photoexcitation (excitation power, 10 μW; temperature, 300 K). (d) Excitation power dependence of the time-dependent emission intensity after photoexcitation (1 molar ratio of the donor; temperature, 300 K). (e) Temperature dependence of the time-dependent emission intensity after photoexcitation (1 molar ratio of the donor; excitation power, 10 μW). (f) Donor concentration dependence of the time-dependent emission intensity after photoexcitation (excitation power, 10 μW; temperature, 300 K). | ||
When the excitation intensity was kept constant and the sample temperature was changed, the PL intensity reached its maximum in about 30 seconds and then became constant at 300 K (Fig. 4(b)). In contrast, the PL intensity quickly reached a maximum and then decreased at low temperatures. Since charge diffusion is limited at low temperature, the PL intensity saturates very quickly (Fig. 5). At high temperatures such as 460 K, charge diffusion increases, but the effect of nonradiative deactivation also increases. Therefore, the PL intensity decreased over time. Since LPL is observed even at 10 K (Fig. 4(e)), charge recombination is thought to proceed via electron tunneling without thermal activation at low temperatures. The slope of LPL emission is nearly m = 1 at room temperature, although it varies slightly with temperature and excitation intensity. When charge recombination proceeds by a random walk after de-trapping, the slope is m = 1.5, suggesting the contribution of electron tunneling as well as thermal de-trapping.25
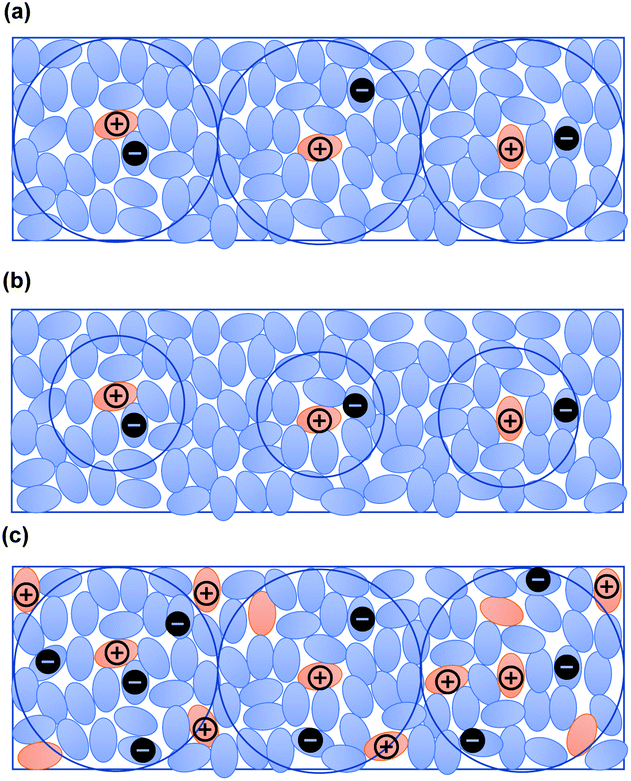 | ||
| Fig. 5 (a) Schematic diagram of a donor/acceptor mixed film. Charge separation occurs at the interface between electron donors and acceptors. Due to the low donor concentration, holes are localized to the donors and electrons diffuse to neighboring acceptor molecules. (b) Schematic diagram of charge separation at low temperature. Electrons can diffuse shorter distances at low temperature. (c) Schematic diagram of charge separation at a higher doping concentration. Generated charges quickly recombine with neighboring donors. | ||
The donor/acceptor ratio is important for long-term charge accumulation, and the current optimum donor concentration is 1% for OLPL systems (Fig. 4(f)). Although equal concentrations of the donor and acceptor can form more exciplexes and are suitable for PL, charge carriers generated by charge separation quickly recombine with neighboring donors when the donor concentration is high (Fig. 4(c) and 5(c)).
3.3 Thermoluminescence
TL is often used to analyze inorganic LPL materials since the TL curve represents the trap depth of LPL systems.26 The OLPL sample was kept at a constant temperature (10, 100, 200, and 300 K) in a cryostat and photoexcited with a 340 nm LED for 300 s. After photoexcitation, the initial LPL was observed. Then, the sample temperature was increased at 5 K min−1 and the TL intensity was obtained. The luminescence intensity was plotted against temperature to evaluate TL behavior.When the TL measurement of the m-MTDATA/PPT film was started from 10 K, a clear TL curve peaked at around 120 K was observed. This TL curve indicates the presence of a thermal activation process in the OLPL system (Fig. 6).27 Note that the onset of the TL curve is located at around 50 K, which is much lower than that of inorganic LPL systems. When the TL measurement was started from 300 K, a very weak TL curve peaked at around 360 K was observed. These results indicate that most of the stored charge carriers of the m-MTDATA/PPT film can be detrapped and recombined until it reaches room temperature. Thus, additional trap mechanisms like a ternary OLPL system4 are required for efficient charge trapping.
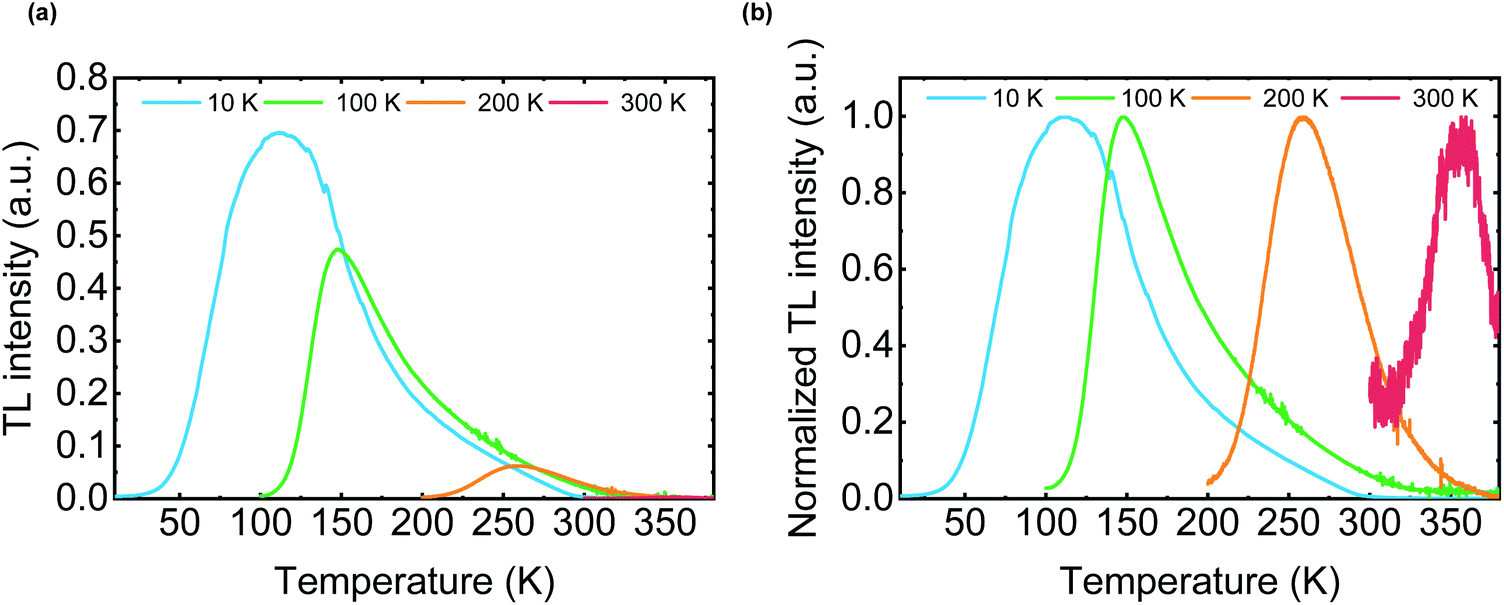 | ||
| Fig. 6 (a) TL curves of an m-MTDATA/PPT film. The sample was photoexcited at the starting temperature (10 K, 100 K, 200 K, and 300 K) for 300 s. After 1 h, the sample temperature was increased at 5 K min−1. (b) Normalized TL curves. | ||
3.4 Oxygen quenching of OLPL
Because the triplet excitons of TADF systems are often quenched by molecular oxygen, optical properties were also examined under oxygen gas (Fig. 7). As a result, the LPL emission was quenched by oxygen. Since charges accumulate in the radical cation of m-MTDATA and the radical anion of PPT in the LPL system, the chemical reaction of the radical species with oxygen should quench LPL emission. In contrast, interestingly the TADF emission was not completely quenched by oxygen due to a very thick film.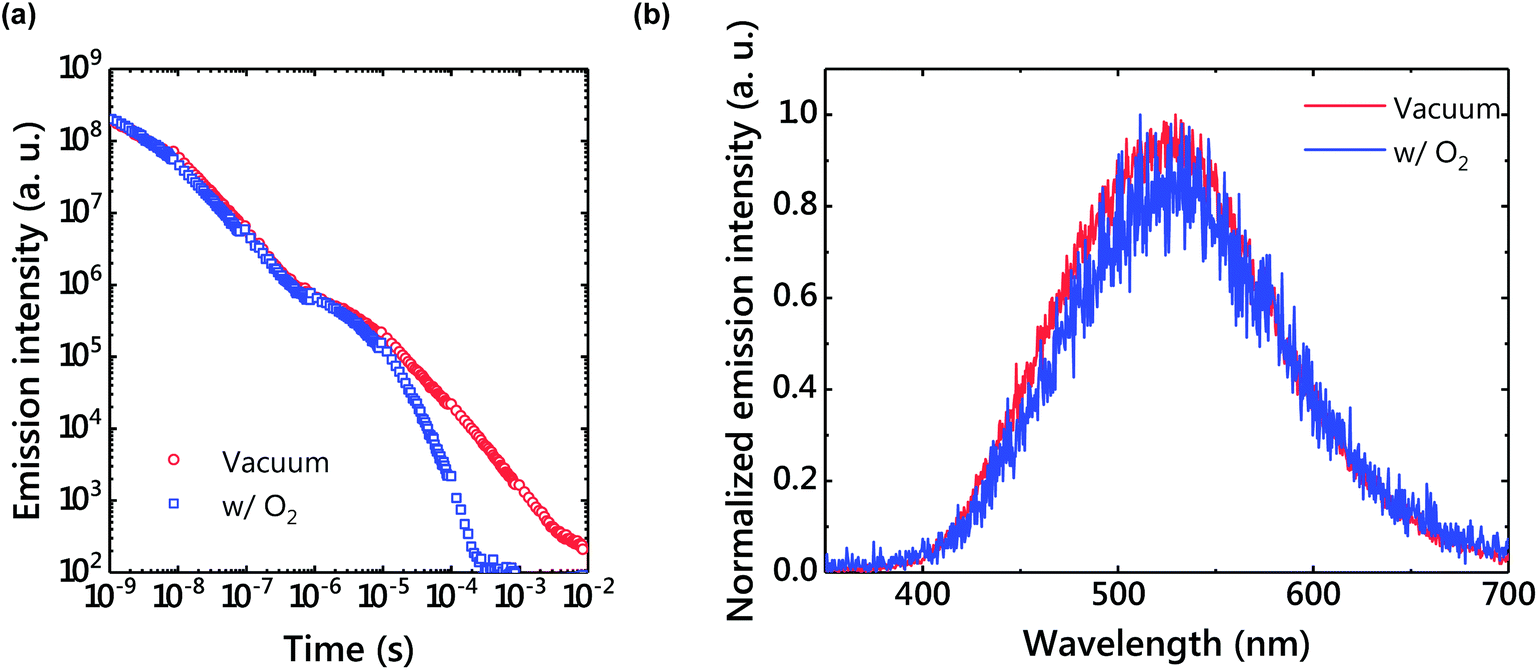 | ||
| Fig. 7 (a) An emission decay profile under vacuum and in air. The power-law decay disappeared in air. (b) Delayed emission spectra under vacuum and in air. | ||
4. Conclusions
We demonstrated the presence of thermal activation and TL in an m-MTDATA/PPT film. The TADF process represents not only the normal TADF process without charge separation, but also LPL from charge recombination. Therefore, triplet exciton management is important for highly efficient OLPL systems, similar to TADF OLEDs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Science and Technology Agency (JST), ERATO, Adachi Molecular Exciton Engineering Project, under JST ERATO Grant Number JPMJER1305, Japan, JSPS Core-to-Core Program, and JSPS KAKENHI Grant Numbers JP18H02049 and JP18H04522. We thank K. Kusuhara and N. Nakamura for their assistance with the preparation of PPT.References
- J. Xu and S. Tanabe, J. Lumin., 2019, 205, 581 CrossRef CAS.
- S. Wu, Z. Pan, R. Chen and X. Liu, Long Afterglow Phosphorescent Materials, Springer International Publishing, Cham, 2017 Search PubMed.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384 CrossRef CAS PubMed.
- K. Jinnai, R. Kabe and C. Adachi, Adv. Mater., 2018, 30, 1800365 CrossRef PubMed.
- Z. Lin, R. Kabe, N. Nishimura, K. Jinnai and C. Adachi, Adv. Mater., 2018, 30, 1803713 CrossRef PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- N. Nishimura, Z. Lin, K. Jinnai, R. Kabe and C. Adachi, Adv. Funct. Mater., 2020, 30, 2000795 CrossRef CAS.
- Z. Lin, R. Kabe, K. Wang and C. Adachi, Nat. Commun., 2020, 11, 191 CrossRef CAS PubMed.
- D. Hertel, H. Bässler, R. Guentner and U. Scherf, J. Chem. Phys., 2001, 115, 10007 CrossRef CAS.
- H. Ohkita, W. Sakai, A. Tsuchida and M. Yamamoto, Bull. Chem. Soc. Jpn., 1997, 70, 2665 CrossRef CAS.
- K. Gouda and Y. Hama, Radiat. Phys. Chem., 1985, 26, 285 CrossRef CAS.
- M. Sarma and K.-T. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 19279 CrossRef CAS PubMed.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736 CrossRef CAS PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253 CrossRef CAS.
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084 CrossRef CAS PubMed.
- C. Fan, C. Duan, Y. Wei, D. Ding, H. Xu and W. Huang, Chem. Mater., 2015, 27, 5131 CrossRef CAS.
- K. Jinnai, N. Nishimura, R. Kabe and C. Adachi, Chem. Lett., 2019, 48, 270 CrossRef CAS.
- T.-C. Lin, M. Sarma, Y.-T. Chen, S.-H. Liu, K.-T. Lin, P.-Y. Chiang, W.-T. Chuang, Y.-C. Liu, H.-F. Hsu, W.-Y. Hung, W.-C. Tang, K.-T. Wong and P.-T. Chou, Nat. Commun., 2018, 9, 3111 CrossRef PubMed.
- K. Seki, K. Marumoto and M. Tachiya, Appl. Phys. Express, 2013, 6, 051603 CrossRef.
- M. Wojcik, A. Nowak and K. Seki, J. Chem. Phys., 2017, 146, 054101 CrossRef PubMed.
- K. Seki, K. Murayama and M. Tachiya, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 235212 CrossRef.
- J. S. Manser and P. V. Kamat, Nat. Photonics, 2014, 8, 737 CrossRef CAS.
- D. Jia, Opt. Mater., 2003, 22, 65 CrossRef CAS.
- M. Tachiya and K. Seki, Appl. Phys. Lett., 2009, 94, 081104 CrossRef.
- K. Van den Eeckhout, P. F. Smet and D. Poelman, Materials, 2010, 3, 2536 CrossRef CAS.
- J. Ueda, K. Kuroishi and S. Tanabe, Appl. Phys. Lett., 2014, 104, 101904 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr09227d |
| This journal is © The Royal Society of Chemistry 2021 |