
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural evolution of iron forming iron oxide in a deep eutectic-solvothermal reaction†
Oliver S.
Hammond
 *a,
Ria S.
Atri
ab,
Daniel T.
Bowron
c,
Liliana
de Campo
d,
Sofia
Diaz-Moreno
e,
Luke L.
Keenan
e,
James
Doutch
c,
Salvador
Eslava
f and
Karen J.
Edler
*a
*a,
Ria S.
Atri
ab,
Daniel T.
Bowron
c,
Liliana
de Campo
d,
Sofia
Diaz-Moreno
e,
Luke L.
Keenan
e,
James
Doutch
c,
Salvador
Eslava
f and
Karen J.
Edler
*a
aDepartment of Chemistry and Centre for Doctoral Training in Sustainable Chemical Technologies, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: k.edler@bath.ac.uk
bDepartment of Chemical Engineering, University of Bath, Claverton Down, Bath BA2 7AY, UK
cISIS Neutron and Muon Source, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Didcot OX11 0QX, UK
dAustralian Centre for Neutron Scattering, ANSTO, Lucas Heights, New South Wales 2234, Australia
eDiamond Light Source, Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, UK
fDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, UK
First published on 2nd January 2021
Abstract
Deep eutectic solvents (DES) and their hydrated mixtures are used for solvothermal routes towards greener functional nanomaterials. Here we present the first static structural and in situ studies of the formation of iron oxide (hematite) nanoparticles in a DES of choline chloride![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea where xurea = 0.67 (aka. reline) as an exemplar solvothermal reaction, and observe the effects of water on the reaction. The initial speciation of Fe3+ in DES solutions was measured using extended X-ray absorption fine structure (EXAFS), while the atomistic structure of the mixture was resolved from neutron and X-ray diffraction and empirical potential structure refinement (EPSR) modelling. The reaction was monitored using in situ small-angle neutron scattering (SANS), to determine mesoscale changes, and EXAFS, to determine local rearrangements of order around iron ions. It is shown that iron salts form an octahedral [Fe(L)3(Cl)3] complex where (L) represents various O-containing ligands. Solubilised Fe3+ induced subtle structural rearrangements in the DES due to abstraction of chloride into complexes and distortion of H-bonding around complexes. EXAFS suggests the complex forms [–O–Fe–O–] oligomers by reaction with the products of thermal hydrolysis of urea, and is thus pseudo-zero-order in iron. In the hydrated DES, the reaction, nucleation and growth proceeds rapidly, whereas in the pure DES, the reaction initially proceeds quickly, but suddenly slows after 5000 s. In situ SANS and static small-angle X-ray scattering (SAXS) experiments reveal that nanoparticles spontaneously nucleate after 5000 s of reaction time in the pure DES before slow growth. Contrast effects observed in SANS measurements suggest that hydrated DES preferentially form 1D particle morphologies because of choline selectively capping surface crystal facets to direct growth along certain axes, whereas capping is restricted by the solvent structure in the pure DES.
:urea where xurea = 0.67 (aka. reline) as an exemplar solvothermal reaction, and observe the effects of water on the reaction. The initial speciation of Fe3+ in DES solutions was measured using extended X-ray absorption fine structure (EXAFS), while the atomistic structure of the mixture was resolved from neutron and X-ray diffraction and empirical potential structure refinement (EPSR) modelling. The reaction was monitored using in situ small-angle neutron scattering (SANS), to determine mesoscale changes, and EXAFS, to determine local rearrangements of order around iron ions. It is shown that iron salts form an octahedral [Fe(L)3(Cl)3] complex where (L) represents various O-containing ligands. Solubilised Fe3+ induced subtle structural rearrangements in the DES due to abstraction of chloride into complexes and distortion of H-bonding around complexes. EXAFS suggests the complex forms [–O–Fe–O–] oligomers by reaction with the products of thermal hydrolysis of urea, and is thus pseudo-zero-order in iron. In the hydrated DES, the reaction, nucleation and growth proceeds rapidly, whereas in the pure DES, the reaction initially proceeds quickly, but suddenly slows after 5000 s. In situ SANS and static small-angle X-ray scattering (SAXS) experiments reveal that nanoparticles spontaneously nucleate after 5000 s of reaction time in the pure DES before slow growth. Contrast effects observed in SANS measurements suggest that hydrated DES preferentially form 1D particle morphologies because of choline selectively capping surface crystal facets to direct growth along certain axes, whereas capping is restricted by the solvent structure in the pure DES.
1 Introduction
Neoteric solvents could offer new opportunities for the preparation of nanostructured functional materials, able to address the emergent challenges faced by society.1,2 In particular, solvent design has been used as an extension of the hydrothermal method.3–6 In solvothermal or ionothermal processes, informed selection of an organic solvent, ionic liquid (IL),7 or eutectic mixture allows control over the morphology of materials, offering scalable routes to new materials with greater performance.8–15 The successful application of these synthetic methodologies can be limited by insufficient understanding of intermolecular interactions and reaction mechanisms. Such ‘designer’ solvents often have a more intricate solvent structure than single molecule liquids, both in the bulk,16,17 and at the solvent-solid interface,18–20 which is further convoluted in mixtures and eutectics.21–29 In this context, it is important to fundamentally understand the processes occurring during these synthesis methods across multiple length scales, hence making their systematic assets and drawbacks clear. This will facilitate the development of scalable, environmentally-mindful and designer routes towards a broad library of potentially useful materials.30 Hematite (α-Fe2O3), with its band gap of 2.2 eV, has gained substantial current interest as a stable and active photoanode material for the photoelectrochemical oxidation of water to O2. This reaction represents the bottleneck in photoelectrochemical cells for water splitting and carbon dioxide solar conversion.30 Iron compounds such as hematite are attractive due to low toxicity,31 and iron accounts for 32.1% of Earth's mass.32 However, the performance of hematite in photoelectrochemical applications is reduced by poor photon penetration, picosecond-scale recombination of charge carriers, and the often long distance between these carriers and the active junction at the solid/liquid interface.30,33–35 Ornately nanostructured hematite thin films can mitigate these drawbacks; many of these preparations require intensive methods such as CVD.33,34 There is therefore a need to develop and understand new synthetic methodologies for nanostructured hematite. The desirable properties of DES are already leading to real-world applications36 and the production of nanomaterials;37 for example, Morris’ group have pioneered DES in the templated synthesis of porous framework materials.11,13–15,38 Our recent work has demonstrated the solvothermal preparation of nanostructured metal oxides including CeO2 and α-Fe2O3 from metal nitrate salts in DES, both under normal heating and under microwave irradiation.39,40 Interestingly, mixing DES with a cosolvent such as water offers an effective route for morphological control, driving the formation of 1D nanostructures, and corresponding with unusual interactions and solvation behavior in the bulk structure of hydrated DES.41 Overall, DES show broad promise in materials synthesis, unifying an inherently mild process with apparently endless opportunities for the design and optimisation of products, provided that an understanding of such syntheses is developed.Whereas the starting configuration for organic transformations is generally well-defined, the same understanding comes less easily for an inorganic reaction in a complex solvent. Ion speciation depends on variables such as the solvent structure and composition, pH and presence of co-solutes.42 The multicomponent nature of DES leads to an array of interactions and thus complicated and disordered bulk structuring, which is not readily comparable to either ionic liquids (ILs) or molecular solvents. Structuring in DES is heavily composition-dependent43 and further convoluted by solutes.22 Moreover, studying such processes requires techniques which are sensitive to the variable length scales and chemical evolution as the reaction progresses, but unaffected by the properties of the reacting mixtures such as high viscosity, gas evolution, and magnetism.
Here, we explore the molecular and mesoscopic mechanisms of nanoparticulate iron oxide synthesis (nominally α-Fe2O3 or hematite) from iron(III) nitrate solutions in a pure choline chloride:urea DES (1:2) and its hydrated counterpart(1:2:10w; ca. 42 wt% H2O). The self-assembly, structure and speciation of iron oxide nanoparticles (NPs) in pure and hydrated DES are assessed using SANS and SAXS, EXAFS, and neutron diffraction and modelling studies, to allow resolution of the full reaction. This work therefore aims to understand the atomistic configurations found within the system as the reaction begins, and how these evolve as the reaction proceeds, ultimately forming nanoparticles which change in size and shape over time depending on conditions. The only other in situ measurements of material production in DES were of metal film electrodeposition using reflectometry techniques;44,45 this is the first time-resolved study of a solvothermal reaction occurring in a DES.
2 Experimental
2.1 Preparation of DES
The choline chloride–urea DES was prepared according to the typical literature procedure of mechanically stirring vacuum-dried choline chloride (Acros, 99.5%) and urea (Acros, 99.5%) in a sealed vessel and with gentle heating until a clear monophasic liquid was obtained.46 We have found this methodology to consistently yield intrinsic water contents of ca. 2000 ppm by KF titration.21 DES–water mixtures were obtained by mixing pure DES stock solution with water (Elga, 18.2 MΩ cm) until a homogenous solution of 1:2:10 choline chloride:urea:water (a hydration level described as 10w) was made. Iron(III) nitrate nonahydrate (Acros, 99.9%) was mixed at room temperature with the pure or hydrated DES at constant molalities of 0.15–0.35 mol kg−1, and the resulting bright yellow solution was used as the precursor material for all subsequent techniques. The iron nitrate nonahydrate salt has a fairly high water content (slightly lower by mass fraction than that of the DES-hydrate) and is dissolved at relatively high concentrations. Varying the concentration of this species therefore has a non-negligible effect on the water content of the mixture in the absence of other added water. The water contents of the various DES mixtures are reported in Table 1.
| [Fe(NO3)3·9H2O] (mol kg−1) | Pure DES water content (wt%) | Hydrated DES water content (wt%) | Pure DES molar ratio choline:Fe3+ |
Hydrated DES molar ratio choline:Fe3+ |
|---|---|---|---|---|
| 0.15 | 2.29 | 40.91 | 25.67 | 15.16 |
| 0.20 | 3.00 | 40.89 | 19.25 | 11.37 |
| 0.25 | 3.68 | 40.88 | 15.40 | 9.09 |
| 0.35 | 4.97 | 40.85 | 11.00 | 6.50 |
Water content is calculated neglecting water in the pure solvent upon preparation from freshly vacuum-dried choline chloride and urea. Mole fractions of the components can be calculated from the eutectic stoichiometry of 1:2:(10) choline chloride:urea:(water) and the iron complex stoichiometry and molal concentration. The variable choline:Fe3+ stoichiometry arises from the preparation of the hydrated solvent with 40.9 wt% water.
2.2 Synthesis of iron oxide nanoparticles
Precursor solutions (10 g) were placed into a Teflon-lined stainless-steel autoclave and reacted in an oven at 90 °C for respectively 8 h (dry DES) or 3 h (hydrated DES), which were determined to be past the reaction end point from analysis of SANS and EXAFS data. The products were decanted directly into cellulose dialysis tubing (MWCO = 12.4 kDa) and dialysed several times against deionised water to remove ionic impurities. They were then dried in air (60 °C) from ethanol, yielding dry aggregated nanoparticulates. TEM images showing the fine nanoparticles formed in the dry reaction, and the 1D nanostructures formed in the hydrated reaction, are shown in the ESI (Fig. S11 to S14†).The prepared samples of iron oxide NPs produced off-line were characterised using a Bruker D8-ADVANCE X-ray diffractometer operating in Bragg–Brentano geometry with Cu Kα radiation (λ = 1.5418 Å) over 20–90° 2ϑ, giving a momentum transfer range from 1.42–5.76 Å−1 and therefore a structural resolution (π/Qmax) of 0.55 Å. Transmission electron micrographs and electron diffraction measurements were made using a JEOL JEM-2100Plus TEM with access provided by the Bath Microscopy and Analysis Suite.
2.3 Neutron and X-ray diffraction
Liquid-phase neutron diffraction measurements were made upon a series of 0.25 mol kg−1 solutions of Fe(NO3)3·9H2O, in isotope-substituted pure and hydrated (10w) DES using the NIMROD total scattering instrument,47 at TS2 of the ISIS Neutron & Muon Source, Rutherford Appleton Laboratory, UK, under beamtime allocation RB1620292. NIMROD covers a wide q-range of 0.01 ≤ Q ≤ 50 Å−1, using a wide angular detector coverage of 0.6–37.5° and time-of-flight neutrons of wavelength 0.05 ≤ λ ≤ 11 Å. Isotope-substituted DES were prepared in the same way as the hydrogenous samples, using the aforementioned reagents and, where necessary, substituting the hydrogenous components with d9-choline chloride (CK Isotopes, 99% pure, 98 atom% D), d4-urea (QMX Laboratories, 99.6% pure, 99.8 atom% D), and D2O (Sigma-Aldrich, 99.9% pure, 99.9 atom% D). The deuteration of the water of crystallisation of the iron nitrate nonahydrate was not controlled. The resultant isotopic contrast sets of choline chloride:urea in the pure DES were H:H, H:D, D:H, and D:D, whereas for the hydrated DES the choline chloride:urea:water compositions were H:H:H, H:D:D, D:H:D, D:D:H and D:D:D. Samples were transferred into null-scattering Ti0.32Zr0.68 cells with a wall thickness and path length of 1 mm, which were sealed against vacuum and leak-tested before transferring to the NIMROD sample changer environment, which was regulated to ca. 0.1 mbar and 30 ± 0.1 °C for the duration of the measurement. Diffraction measurements were made for a median of two hours and scans were averaged for improved counting statistics. GudrunN was used to process the raw neutron scattering data. Scans of the empty sample changer, a vanadium plate (3 mm), and each individual sample cell were used for background reduction and normalisation, before corrections for attenuation and multiple scattering, iterative subtraction of the inelastic scattering contribution of hydrogen, and normalisation to absolute units. The final set of corrected and normalised data were then simulated using empirical potential structure refinement (EPSR).48 Additionally, the samples were reacted in cells on the beamline at 90 °C following static measurements to observe any changes in the state of the samples; these data are shown in S15 and S16 of the ESI.† Minimal qualitative difference was observed for either DES in the high-Q (solvent structure) region, and a small-angle scattering signal evolved commensurate with the dedicated SANS measurements.
X-ray diffraction measurements were made of 0.25 mol kg−1 solutions of Fe(NO3)3·9H2O in pure and hydrated (10w) choline chloride–urea DES using a PANalytical Empyrean instrument, with Ag Kα radiation (λ = 0.5594 Å) and a solid-state GaliPIX3D area detector with a CdTe sensor. Samples containing hydrogenous DES were sealed in 2 mm quartz glass X-ray capillaries with a wall thickness of 0.01 mm and measured on a rotating goniometer sample stage for one hour at room temperature (21 °C). Due to the inorganic salt content, this temperature is above the transition temperature where liquid forms for the pure solution, whereas it would not be for pure ChCl:urea.49 Data were processed using GudrunX,50 which sequentially reduces and normalises the data by accounting for Compton and multiple scattering events, sample absorption, fluorescence, composition and geometry, and the background contribution from air and the empty capillary. The data were finally normalised to the single-atom scattering. The X-ray diffraction data were used alongside the neutron diffraction data as an additional experimental constraint upon the EPSR model. Complementary information is provided by the two techniques. Broadly, neutrons allow the determination of hydrogen site correlations as they have approximately equal sensitivity to the nuclear positions of all the atoms in the system. Conversely, X-rays are sensitive to electron density and hence reinforce the structural information for heavy atoms, namely the Cl− and Fe3+ based bonding modes, which are important in this system.51
2.4 Empirical potential structure refinement (EPSR) modelling of diffraction data
EPSR models were set up from both the reduced neutron and X-ray data, using the same potentials and geometries for urea, choline, chloride, water, and nitrate that have been described previously.39 The potentials for Fe3+ were initially derived from the parameters provided by Li et al. in their 12-6-4 Lennard-Jones type nonbonded model for highly charged metal ions.52 However, it was found that these values yielded poor fits, particularly for the close-range ‘nearest-neighbour’ interaction and especially when the X-ray contrast was introduced, which is most sensitive to the Fe and Cl parameterisation due to the heightened electron density of these atom types. Therefore, the Fe σ value (the Lennard-Jones well distance) was iteratively refined from the literature value of 2.886 Å to 1.9 Å to achieve a better quality-of-fit. While the σ of Cl could also be varied to improve the fit, Fe was chosen as the Cl parameters already appeared to give a good fit for the Cl-rich pure solvent, and keeping this constant reduces the number of fitted parameters. Following this, fitting proceeded routinely within the EPSR framework; the models were first equilibrated to stability under the reference potential, then the empirical potential was introduced, and the model equilibrated for ≥10000 iterations. Each iteration of the structure refinement involves the move or attempted move of each atom, molecule and molecular functional group in the model. Over this time, structural statistics were accumulated for parameters such as the radial distribution functions and hence coordination numbers and probabilistic spatial distribution. Further modelling details are provided in the ESI (Fig. S1–S3 and Table S1†).
2.5 Small-angle neutron scattering
Small-angle neutron scattering was used as an ideal non-destructive, non-interacting probe to evaluate the self-assembly of nanoparticles in the reacting DESs in situ, with high sensitivity to light elements (i.e. CHNO) and iron (bFe = 9.45 fm). Data were taken for reacting mixtures of pure and hydrated (10w) DES, held at a calibrated 90 °C at the sample position, with iron nitrate concentrations ranging from 0.15–0.35 mol kg−1, with H/D isotopic contrasts of choline chloride:urea:(water) of D:D:(D) and H:D:(D), using d9-choline chloride, d4-urea and D2O. Event-mode SANS measurements of nanoparticle growth were performed using the BILBY instrument,53 located at the OPAL reactor neutron source of the Australian Nuclear Science and Technology Organisation, Sydney, Australia, under beamtime award P6216. Using four serially phased mechanical choppers, BILBY was operated using time-of-flight neutrons to provide an effective q-range of 0.00229 ≤ q ≤ 0.29031 Å−1, with the rear detector placed in the 12 m position and the curtain detectors at 4.8 and 5.8 m. High levels of deuteration were favoured to reduce the incoherent scattering background and hence improve the signal-to-noise for the rapid kinetic measurements, resulting in the two choline chloride:urea:water substitutions of H:D:D and D:D:D. Samples (ca. 300 μL) were placed into ‘banjo’ Hellma cells of pathlength 1 mm and sealed with a PTFE plug. The quartz cells were placed into individual metal holders to ensure uniform heat distribution. They were stored at room temperature so that no reaction occurred prior to placing into the sample changer. The calculated neutron scattering length density values (SLDs) used for fitting are included in the ESI (Table S2†).
2.6 Extended X-ray absorption fine structure (EXAFS)
High-quality EXAFS measurements of the samples reacted off-line were made using the I20-Scanning instrument at Diamond Light Source, UK.54 Samples were measured under static conditions, with optimal spectrometer settings for energy resolution, signal sensitivity and k-range. The scanning branch of the I20 beamline has high flux (>1012 photons per s) and spectral purity, using an insertion device and four-bounce monochromator.55 Samples were mounted into flat-plate cells with 1.5 mm pathlength and Kapton thin film windows. The pure and hydrated DES were prepared with 0.15, 0.20 and 0.25 mol kg−1 Fe(NO3)3·9H2O and measured both in their unreacted state, and following reaction for 3 h (hydrated) and 8 h (pure), after which they are anticipated to have reacted to completion. This provides measurements of both the pre-reaction (‘before’) and post-reaction (‘after’) states to be fitted. Due to the high incident photon flux that results in photoreduction of the iron(III) ions, measurements on I20-Scanning were taken while Diamond Light Source was operating in low-α mode with a reduced storage ring beam current. Data were then processed and fitted using the Demeter software package, giving a high quality measure of the reaction starting and finishing points.56 EXAFS measurements were made of the reacting system using the parallel I20-EDE branch of the I20 beamline at Diamond Light Source, UK. I20-EDE was optimised for kinetic data collection, with energy resolution and k-range sacrificed relative to I20-Scanning to facilitate data collection with shorter acquisition times. I20-EDE is an energy-dispersive XAS beamline, operating with a fan-shaped convergent polychromatic incident beam. To prevent beam damage caused by extensive exposure of pink-beam synchrotron radiation, the beamline was operated in the ‘TurboXAS’ configuration rather than energy-dispersive mode, with radiation tuned to 10% bandwidth across the Fe absorption edge at 7112 eV.57 As before, samples of the pure and hydrated DES were prepared with 0.15, 0.20 and 0.25 mol kg−1 Fe(NO3)3·9H2O and syringed into aluminium flat-plate cells with 1.5 mm pathlength, Kapton windows and lined with lead tape to prevent spurious scattering. Filled cells were sealed using epoxy resin to prevent loss of sample during the reaction. The sample environment was then heated to a calibrated 90 °C at the sample position, before placing samples in the instrument and measuring continuously over the course of the reaction. Kinetic measurements were made for 120 s each until no further apparent change was observed. Data were then processed in batch using a routine which removed a flat background from the pre-edge slope and subtracted a low-order Chebyshev polynomial fitted to the post-edge region. Kinetic data were then interpreted using a 2-state component analysis of the data,58 having fitted the ‘before’ and ‘after’ data from the high-resolution I20-Scanning measurements using Artemis software.562.7 Small-angle X-ray scattering
When using a high-intensity synchrotron source, beam damage was observed, so SAXS measurements were made using a Xenocs Nano-inXider instrument provided by the Materials Characterisation Laboratory of the STFC ISIS Neutron and Muon source. The Xenocs instrument uses a vertical pinhole geometry with a Dectris Pilatus detector and Cu-Kα radiation, giving an accessible Q-range of 0.0045 ≤ Q ≤ 0.37 Å−1. Samples were prepared by sealing 0.5 g of ChCl:urea:iron nitrate:(water) stock solutions into small vials. Samples were placed into an air-circulating temperature-regulated oven at 90 °C and removed after hourly intervals and at 90 min. At the time of removal, samples were placed directly into a freezer until use. Prior to X-ray measurement, the samples were placed into quartz glass X-ray capillaries of 1.5 mm diameter and 10 μm wall thickness and sealed using beeswax. Samples were measured using a multi-capillary sample changer, under vacuum at room temperature (21 °C), for the measurement time of several hours. The data were radially averaged and background subtracted using an unreacted stock solution of iron nitrate in either pure or hydrated ChCl:urea. Data were analysed using the ATSAS software package;59 seven cycles of a simulated annealing routine in DAMMIF were averaged and filtered, before passing the filtered system through a final refinement in DAMMIN.
3 Results and discussion
3.1 Initial speciation of Fe3+
EXAFS measurements were made of precursor solutions of pure and hydrated (10w) choline chloride:urea DES containing 0.15 mol kg−1 of the precursor Fe(NO3)3·9H2O, held at room temperature. Data (markers), processed using Demeter, are shown for both pure and hydrated DES in Fig. 1 alongside fits performed in r-space using Artemis (lines), which are shown in both k- and r-space.56 Multiple ligand identities, configurations and geometries were trialled for the fitting, and the best quality-of-fit was obtained for an octahedral configuration of [Fe(L)3(Cl)3], as shown by close agreement between data and fit in Fig. 1, despite plotting with k2 weighting to exaggerate the high-k region. This suggested 6-fold coordination is consistent with Fe3+ in basic aqueous solution,60 which follows because of the slightly basic nature of the choline chloride:urea DES,61 and presence of water of crystallisation from Fe(NO3)3·9H2O.62 Ligands were either Cl, or a non-specified oxygen-containing ligand ‘L’ which could be any of the O-containing donor species present in the mixture, such as nitrate, water, urea, or choline. EXAFS provides a reliable determination of the metal–ligand complex geometry, especially coordination number and bond lengths, but is not sensitive to the nature of the oxygen-containing ligand ‘L’ in these multicomponent solvents. Both Cl and ‘L’ (O-donor) ligands were fitted to a single environment, and the urea –NH2 group was not considered an effective donor ligand because its lone pair is in resonance with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.26
O bond.26
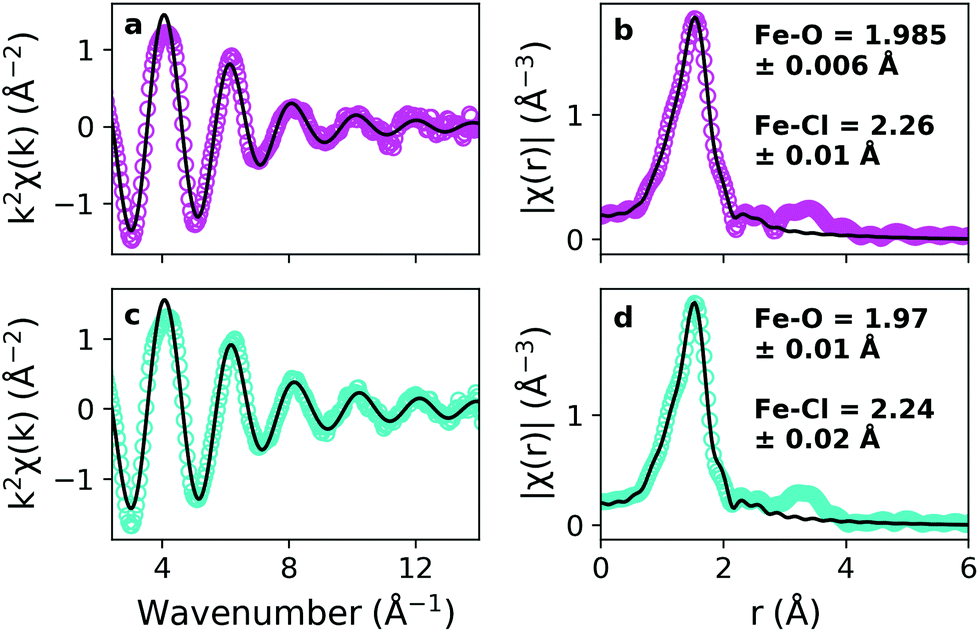 | ||
| Fig. 1 EXAFS data (coloured markers) and fits to the data (black solid lines) for the pure DES (magenta; a and b) and hydrated DES (teal; c and d). Data and fits are shown in k-space (left; a and c) and r-space (right; b and d). Fits were made in Artemis, in r-space, to a 6-coordinate octahedral complex with 3 O and 3 Cl ligands. Fitting ranges were 1–2.21 Å (pure DES; corresponding k-range 2.40–13.24 Å−1) and 1–2.13 Å (hydrated DES; corresponding k-range 2.50–13.23 Å−1). Calculated bond lengths for the Fe–O and Fe–Cl binding are shown in insets to b and c. The geometry of the complex was fitted using a hexacoordinate Fe(O)3(Cl)3 complex as a starting point by floating the bond lengths and altering the ligand degeneracy and composition to obtain the best chemically-sensible quality of fit. The exact identity of the ligands corresponding with Fe–O bonds cannot be resolved from this technique. | ||
The same complex structure fitted well to both the pure and hydrated systems, despite the high fraction of water in the latter by mass (ca. 42 wt%) or by mole (ca. 77 mol%). It is also noteworthy that the initial states were measured at Fe(NO3)3·9H2O concentrations of 0.20 and 0.25 mol kg−1, and no qualitative differences were observed in the data, likely due to the large excess of Cl- and O-containing ligands in the DES even at high Fe loadings. Our fitted structures differ slightly from the anionic metal perchloro complexes observed by Hartley et al., who measured the speciation of Cr, Mn, Fe, Co, Ni, Cu, Zn, Pt, Au, Pd, Ag, and Sn chlorides in ChCl:urea, ChCl:ethylene glycol, and ChCl:glycerol DES using EXAFS.63 In the polyol-based DES, some chelation of the metal centres was also observed. Although EXAFS is not acutely sensitive to these minor differences, the systems are different; except for Cr3+, all of the previous examples contained anhydrous M2+ chloride salts which may speciate differently from the Fe3+ nitrate salts measured here. Moreover, the earlier study was done under the assumption that DES were complex-ionic liquids,46,64 whereas an extended H-bond network (aka. “alphabet soup”) model has become the preferred descriptor for the solution structure of DES.22,24,26,39,65–67 The static EXAFS measurements therefore demonstrate the speciation of iron prior to the onset of reaction.
3.2 Initial solvent nanostructure and solvation of Fe3+
Values are compared with data in the pure solvent containing 10w (41 wt%) for the hydrated system and 1w (6.5 wt%) for the ‘pure’ system, as published previously.41 This is because the pure 1w solvent is closer in hydration to the pure solvent with 0.25 mol kg−1 of iron nitrate nonahydrate, which contains 3.7 wt% water, whereas the analysis of the pure iron-free DES was assumed to be anhydrous because the water content was confirmed to be low under representative conditions using Karl Fischer titration.21 Non-integer coordination numbers represent some complex fluxionality, the degree of which can be inferred from the stated deviation.Whereas EXAFS provides detail on the coordination number and bond lengths of the precursor complex, elastic scattering can reveal the nature of these ligands and the bulk solvent environment. Understanding all of these elements is key to elucidating the reaction mechanism. The structures of both pure and hydrated DES containing 0.25 mol kg−1 of Fe(NO3)3·9H2O were therefore measured at 303 K using neutron diffraction with isotopic labelling. An atomistic model was fitted to the neutron data using empirical potential structure refinement (EPSR) and co-refined with X-ray pair distribution function (XPDF) measurements. X-ray data improves the fits by imposing the interactions of more electron-dense species, i.e. Fe, as a further structural constraint. Data (markers) are shown alongside fits (lines) in Q-space in Fig. 2 and in R-space in the ESI (Fig. S1†). The fits converge well upon the data, except the typical low-Q divergence (≤1 Å−1) due to residual inelasticity,68 and only minor discrepancies are seen with the intensity of the X-ray data. Ensemble structural information was then obtained from equilibrated EPSR models (≥10000 refinement cycles).
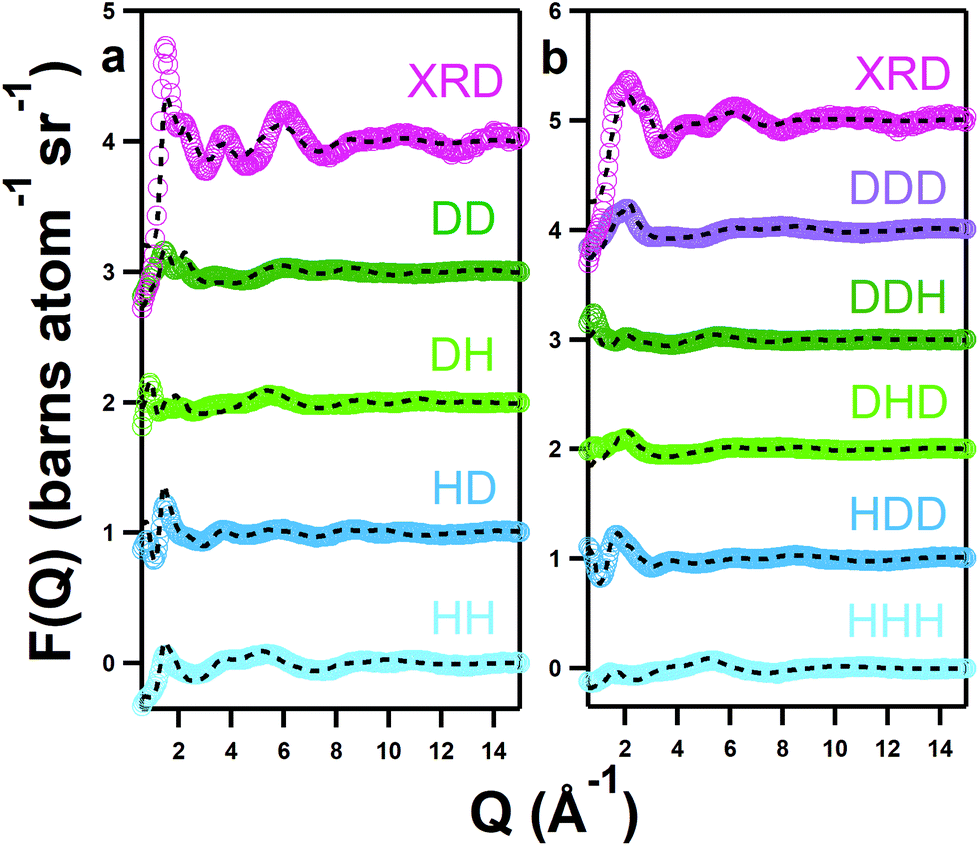 | ||
| Fig. 2 Neutron and X-ray diffraction data (coloured markers) and EPSR fits (black dashed lines) for the pure DES (a) and hydrated DES (b). Isotopic contrast labels refer to choline:urea:water deuteration state. For XRD, hydrogenous samples were measured. | ||
Calculated Fe-centred intermolecular coordination numbers (Ncoord) are shown in Table 2. Except choline, all O-containing species play some role in iron binding; choline is likely excluded from the primary solvation shell by like-charge repulsion. Similarly, Fe–Fe interactions in both systems are essentially zero. 2.77 ± 0.97 chlorides are bound to iron centres on average in the pure DES, close to the 3 suggested by EXAFS, though in the hydrated solvent only 1.65 ± 1.15 chlorides are bound. The number of coordinating urea molecules similarly falls from 0.89 ± 1.04 in the pure DES to 0.65 ± 0.79 in the hydrated system, whereas one nitrate ion is present in both pure (1.04 ± 0.96) and hydrous solvents (1.05 ± 0.80). Most notably, the coordination of water is negligible in the pure DES, but 2.41 ± 1.13 water molecules are coordinated to iron on average in the hydrated DES, making it the dominant species. The spatial orientations of Fe3+ cations around the various components of the mixtures are shown in Fig. 3, which highlight the Fe–O bonding modes, and also demonstrate the ability of Fe3+ to form intercalated complexes with species such as nitrate and water, as in the iron/nitrate @ urea 0w SDF (mid-right), and the iron/nitrate @ choline SDFs (mid-left). This suggests strong association of ionic species with Fe3+, especially nitrate, in addition to chloride. The scattering data generally agrees with the spectroscopic picture of a [Fe(L)3(Cl)3] octahedral complex with a mixed and fluxional coordination sphere, with Ncoord summing respectively to 5.02 and 5.76 for the pure and hydrated DES, rather than the anionic perchloro complexes shown by Hartley et al.63 A 5-fold Fe coordination for the pure DES as suggested by EPSR is possible, and would be consistent with the pentacoordinate iron-chloride complexes seen in concentrated aqueous chloride solution.60 However, the EPSR model is not refined against the EXAFS data, and EXAFS is a more accurate probe for the direct environment of the Fe ion. Therefore, the EPSR data for the complex should be considered as a proxy to understand how the mix of ligands changes with water content, and the discussion will continue to focus on a [Fe(L)3(Cl)3] complex.
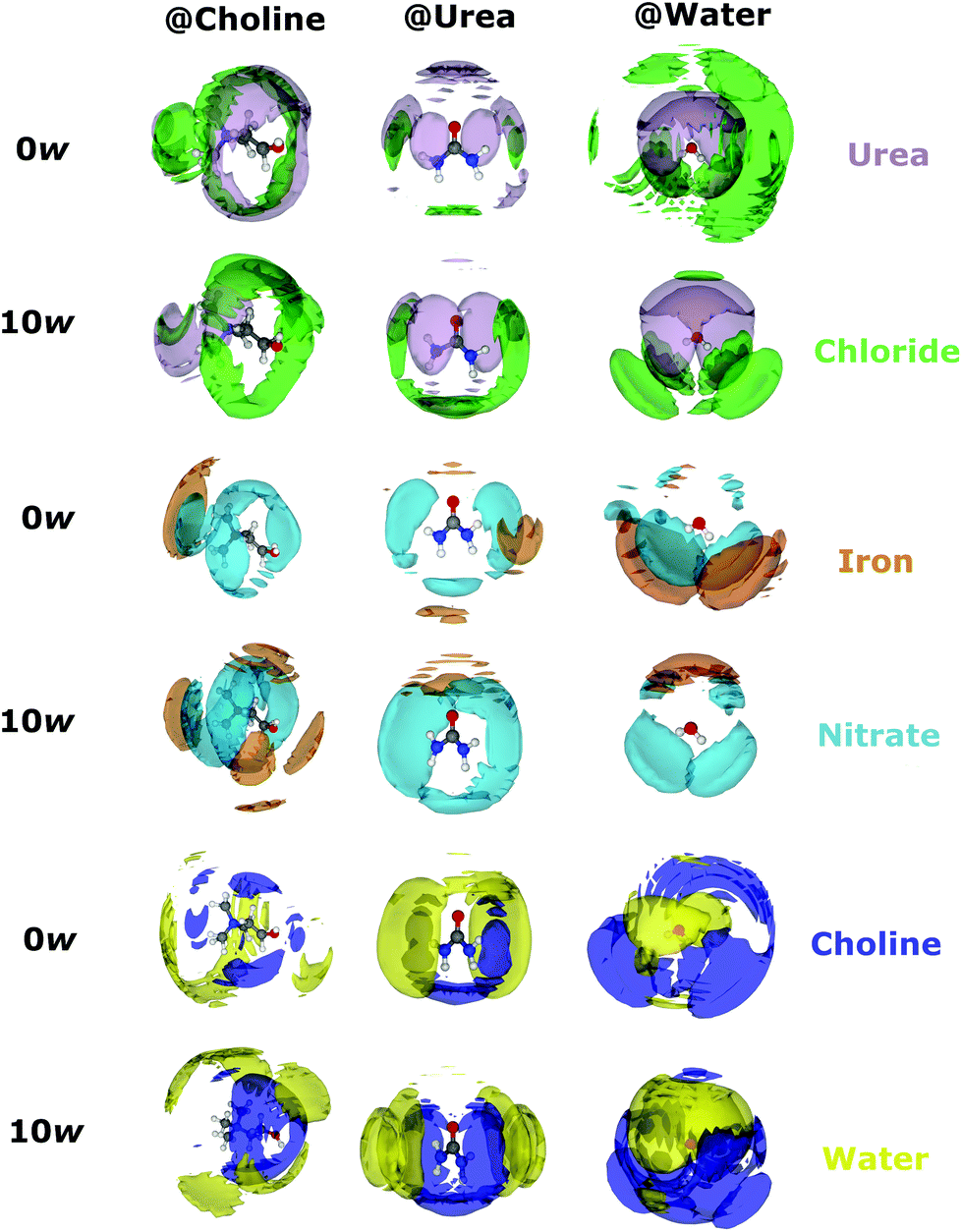 | ||
| Fig. 3 SDF plots showing most likely 3D configurations of chloride, iron, nitrate, choline, and water, around central choline, urea, and water molecules, in both the pure and in the hydrated DES in the presence of iron nitrate nonahydrate. Isosurfaces are universally plotted at the 7.5% probability level to facilitate comparison of the degree of order. | ||
:urea DES from EPSR modelling. Non-integer coordination number values and variances (which represent one standard deviation in coordination number) arise from the accumulation of statistics over ≥10000 iterations of refinement against the experimental potential. Coordination numbers are calculated by integrating each pRDF to their first minima; Rmax describes this maximum radius of integration. Molecular centres for polyatomic species were: urea CN, nitrate NO, choline C2N, water O1
| ‘A’ | ‘B’ | R max (0w) (Å) | R max (10w) (Å) | N coord (1w) | N coord (1w), with [Fe] | N coord (10w) | N coord (10w), with [Fe] |
|---|---|---|---|---|---|---|---|
| Fe3+ | H2O | 2.3 | 2.3 | — | 0.32 ± 0.52 | — | 2.41 ± 1.13 |
| Fe3+ | Cl− | 3.0 | 3.0 | — | 2.77 ± 0.97 | — | 1.65 ± 1.15 |
| Fe3+ | Urea | 4.0 | 4.0 | — | 0.89 ± 1.04 | — | 0.65 ± 0.79 |
| Fe3+ | NO3− | 3.5 | 3.5 | — | 1.04 ± 0.96 | — | 1.05 ± 0.80 |
| Fe3+ | Fe3+ | 6.7 | 5.5 | — | 0.12 ± 0.32 | — | 0.10 ± 0.30 |
| Cho+ | H2O | 6.2 | 6.2 | 2.50 ± 1.56 | 2.54 ± 1.66 | 15.37 ± 3.08 | 15.45 ± 2.82 |
| Urea | H2O | 4.9 | 4.9 | 1.48 ± 1.20 | 1.39 ± 1.15 | 8.09 ± 2.12 | 8.30 ± 2.23 |
| Cl− | H2O | 4.5 | 4.4 | 1.18 ± 1.08 | 1.20 ± 1.15 | 5.78 ± 1.79 | 6.82 ± 1.82 |
| H2O | H2O | 4.0 | 3.1 | 0.91 ± 0.97 | 0.77 ± 0.77 | 2.21 ± 1.05 | 1.98 ± 1.04 |
| Cho+ | Cho+ | 7.8 | 7.8 | 4.58 ± 1.64 | 4.42 ± 1.42 | 2.48 ± 1.45 | 1.84 ± 1.14 |
| Cho+ | Cl− | 4.7 | 4.7 | 1.04 ± 0.78 | 0.87 ± 0.82 | 0.58 ± 0.65 | 0.32 ± 0.53 |
| Cho+ | Urea | 6.8 | 6.8 | 6.69 ± 2.18 | 6.47 ± 1.91 | 3.25 ± 1.63 | 3.40 ± 1.49 |
| Urea | Cl− | 5.2 | 5.2 | 1.76 ± 0.94 | 1.66 ± 1.15 | 0.90 ± 0.78 | 0.67 ± 0.78 |
| Urea | Urea | 5.7 | 5.7 | 4.01 ± 1.71 | 3.79 ± 1.70 | 2.39 ± 1.36 | 2.05 ± 1.33 |
| Cl− | Cl− | 6.0 | 6.0 | 1.23 ± 0.91 | 1.93 ± 1.50 | 0.58 ± 0.69 | 1.22 ± 1.15 |
Coordination numbers (Ncoord) were also calculated for non-Fe intermolecular interactions, allowing observation of the change in bulk solvent structure when iron nitrate is added to the DES. Using previous work as a reference, the percentage change in Ncoord is shown in the ESI (Fig. S2 and S3†), alongside partial (site–site) coordination numbers.21,41 Compared to the iron-free examples, little change is generally seen, particularly for the choline–water and urea–water interactions. However, some of the intermolecular coordination numbers vary markedly, especially the interactions involving chloride or water, and several of these interactions increase on addition of iron. The chloride-water coordination rises from 5.78 ± 1.79 to 6.82 ± 1.82 with the addition of the iron salt to the water rich system. This can be contrasted with the pure, low-water system where this coordination measure only changes from 1.18 ± 1.08 to1.20 ± 1.15. Moreover, like-charge ordering (i.e. choline–choline interactions), already subtle in DES and their mixtures when compared to pure ILs,1 is disrupted further in the high-water system after iron addition with up to 20% changes in the N–N coordination, but less so in the pure DES. These structural differences most likely arise because chloride is abstracted from the bulk by Fe3+ due to strong Fe–Cl complexation, with cascading effects on the other interactions. For example, the chloride–chloride Ncoord increases most significantly on addition of Fe3+, but the choline–chloride interactions universally decrease. Interestingly, the choline HOH–Cl H-bond mode is clear in the pure DES at room temperature,21 but is sensitive to disruption, with high temperatures sufficient to diminish this ordering,69 as well as additional salt.39 This could be another manifestation of the structural fluxionality between the α- and β- (low- and high-temperature) polymorphs of crystalline choline chloride.70,71 The small molecular volume of introduced water allows it to more efficiently solvate the various DES components than the DES itself can. At high water contents, water hydrates choline better (Fig. 3; bottom-left SDF) and chloride becomes more well-ordered around water (top-right SDFs). Pronounced urea–urea association is observed both when dry and hydrated, relatable to the strong urea H-bonding networks formed in concentrated aqueous solutions,72 but significant interaction between urea and water is seen too (bottom–middle SDFs).
Combined, the EXAFS data and neutron and X-ray scattering measurements provide a picture of the solvation environment in the DES prior to reaction. The bulk structure is similar to the systems without solvated Fe3+, where there is an extensive disordered network of H-bonds. Stoichiometric cages of DES components interact with stable, solvated [Fe(L)3(Cl)3] complexes. The presence of these complexes slightly depletes the bulk Cl concentration, which subtly modifies the intermolecular interactions.
3.3 Evolution of mesoscale iron structure
Example in situ SANS data for the 0.35 mol kg−1 samples of pure DES are shown as a function of reaction time in Fig. 4, alongside extracted radius of gyration (Rg) and scale factor for the various contrasts and concentrations from Guinier analysis. The complete set of time-resolved data is shown in the ESI (Fig. S4 and S5†). In the pure DES, zero scattering is observed after removal of the DES background until approximately 2 h of reaction, which is repeatable across all measured contrasts and concentrations. Visually, at this point the reaction mixture changes from bright yellow to blood red. After this point, the nanoparticle volume fraction (scale factor, Fig. 4c and f) increases sigmoidally over the course of the measurement. Growth is particularly slow in the D:D contrasts at low concentrations of iron nitrate, which may be due to a subtle kinetic isotope effect, or slight differences between the H/D choline purity. Guinier fits were adequate despite the lack of modelled polydispersity or particle–particle interactions (S(Q); structure factor), with scattering patterns showing Porod exponents of (or near to) 4, signifying particles with a smooth surface. The lack of interparticle interaction is interesting as it implies effective interparticle screening by the DES, whereas a remarkable structure factor is observed between charged surfactant micelles in DES at far lower concentrations.73–75 The colloidal suspensions of NPs in pure DES formed after these reactions were found to be stable for long timescales until sufficient water was added or absorbed.
 | ||
| Fig. 4 Example background-subtracted time-resolved SANS data measured using BILBY for 0.35 mol kg−1 of Fe(NO3)3·9H2O in pure DES; DES were prepared with choline:urea deuteration of H:D (a) and D:D (d). Data for the concentration series were fitted using batch Guinier modelling and the extracted radius of gyration (Rg) and Guinier scale factors are shown in (b) and (c) for the H:D contrast, and (e) and (f) for the D:D contrast respectively. Note that the magnitude of the ordinate in (b) and (e) is identical, but that of (c) is approximately double that of (f). | ||
The calculated Rg in Fig. 4b and e shows nanoparticles of 5 to 9 nm at first observation, depending on the concentration and contrast. Nucleation therefore occurs after some time has elapsed, and is a rapid process which exceeds the time resolution of the 300 s SANS measurements. Following first observation the Rg grows slowly, suggesting slow individual growth of nanoparticles, rather than stepwise fusion and aggregation of individual particles. Although there are subtle differences between contrasts and concentrations, iron nitrate concentration and nanoparticle size correlate positively. As the iron precursor concentration is increased from 0.15 to 0.25 and 0.35 mol kg−1 for the H:D contrast, the final Rg (averaged from the final 5 datapoints, at 35–40 ks of elapsed reaction time) measures 6.92 ± 0.05, 8.27 ± 0.04, and 9.92 ± 0.13 nm respectively, whereas for the D:D contrast the same values are calculated as 7.03 ± 0.23, 8.92 ± 0.22 and 13.29 ± 0.08 nm respectively. Intuitively, higher concentrations of precursor lead to larger particles, because of higher growth rates following nucleation. The sizes measured for the D:D contrast data are universally larger than the H:D contrast. This suggests that under D:D contrast conditions, SANS images the nanoparticle ‘superstructure’ inclusive of a solvation shell, whereas this corona is somewhat hidden in the H:D contrast. Indeed, one feature which Guinier modelling does not completely account for is a diffuse structural feature observed in the mid-Q region at around 0.07 Å−1 at long reaction times, as shown in the ESI (Fig. S6†). This feature is more prominent in the H:D (hydrogenous choline) contrast, and may therefore suggest a choline-enriched near-surface layer at the DES/solid interface. This observation corresponds both with accounts of interfacial DES structuring from AFM and calculations,23,76,77 and the hypothesis that cholinium acts as a nanoparticle surface capping agent, inhibiting growth along specific planes. Therefore, cholinium may function like alkylammonium halide surfactants, which drive 1D nanoparticle growth.39,78 Surface enrichment also offers an explanation for the observed screening of interparticle interactions, and is consistent with long-range ordering observed at DES/nanoparticle interfaces by Hammons et al.20,79,80
Model-based fitting was used to extract more detailed structural information from the SAS data, and calculated polar and equatorial radii (Rpo and Req) are shown in Fig. 5.81 The SLDs used for these fits are shown in the ESI (Table S2†) alongside exemplar fits using an ellipsoid model with oblate spheroid platelet geometry (i.e. Req > Rpo, in Fig. S7†). This provided better fits than Guinier analysis, particularly at mid-Q values ca. 0.1 Å−1. At first observation the nanoparticles which appear in the pure DES are relatively large, as seen in Fig. 5a; after 10000 s the NPs have respective Rpo and Req of 2.2 and 8.4 nm. As the particles grow, following the same trend as highlighted by Guinier analysis, there is a subtle contraction in particle aspect ratio from 3.5 at initial appearance to approximately 2.5. After 37000 s, the particles from the 0.35 mol kg−1 sample have respective Rpo and Req of 6.7 and 15.5 nm, meaning the poles and equators of each nanoparticle do not grow equally, with a polar:equatorial growth rate ratio of 3:2. Model-based analysis therefore confirms the initial rapid nucleation step, which occurs at a critical point faster than the instrumental timescale, and followed by slow classical growth. For the 0.35 mol kg−1 sample, where this process is the most rapid, the particles undergo 0.67 nm h−1 of growth in Req and 0.38 nm−1 growth in Rpo by a linear approximation.
 | ||
| Fig. 5 (a) Fitted equatorial (Req) and polar (Rpo) radii as a function of time for the pure DES and (b) for the hydrated DES, in the H:D contrast. The tie line for (b) is drawn at 65.5 nm, the approximate length scale maximum. Above this length the model fit is no longer sensitive to changes in this parameter, when using model-based (oblate-ellipsoid) fitting to the data within SasView. | ||
Complementary structural data was provided by laboratory SAXS measurements of the reacting systems at timepoints of 1 h (3600 s), 90 min, (5400 s), 2 h (7200 s), 3 h (9600 s) and 4 h (14400 s). The strong theoretical X-ray scattering length contrast between the iron oxide nanoparticles and the organic solvent makes SAXS ideal for determining the presence of low volume fraction structures before the proposed nucleation step. The corrected, background-reduced data are shown in Fig. 6 alongside fits to the data using ATSAS software.59Table 3 shows extracted Rg values from the X-ray fits alongside analogous values calculated from Guinier analysis of SANS data. Size distributions obtained by P(R) inversion of the SAXS data are shown in the ESI (Fig. S8†).
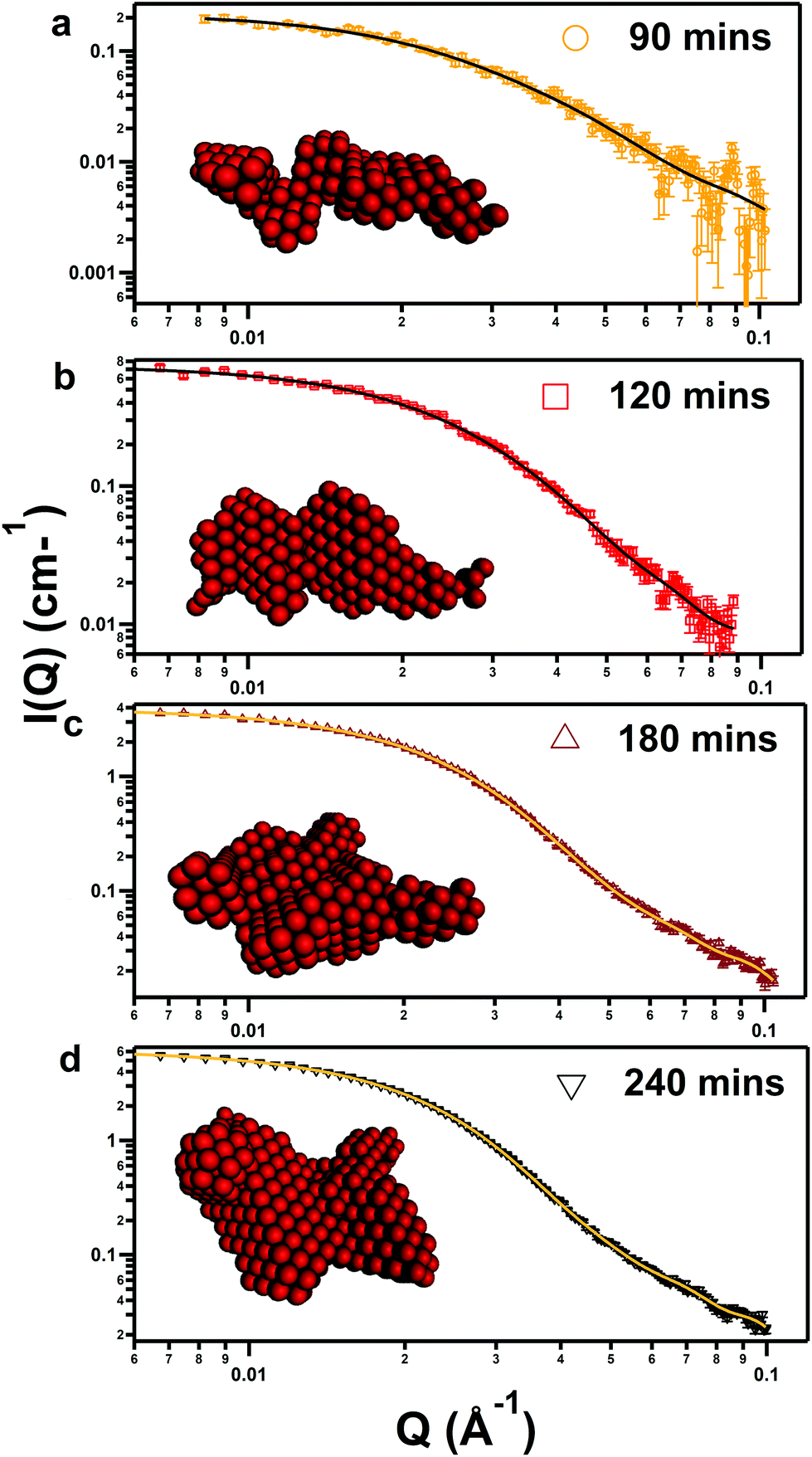 | ||
| Fig. 6 SAXS data of iron oxide (markers) corresponding with pure DES samples containing 0.25 mol kg−1 of iron, reacted in an oven at 90 °C for (a) 90 min (5400 s), (b) 120 min (7200 s), (c) 180 min (10800 s), (d) 240 min (14400 s). Fits to the data (lines) use software from the ATSAS package,59 which determines the overall nanoparticle shapes (inset) by simulated monophase annealing of dummy atoms. | ||
| Time (s) |
R
g (H:D) (nm) |
R
g (D:D) (nm) |
R g (SAXS) (nm) |
|---|---|---|---|
| 3600 | — | — | — |
| 5400 | — | — | 7.12 |
| 7200 | — | — | 7.21 |
| 10800 |
6.11 | — | 7.71 |
| 14400 |
6.79 | 6.78 | 8.15 |
After background subtraction, only a flat SAXS pattern was observed in the 60 min sample (3600 s), thus the mixture remains monophasic at this point. However, after 90 min (5400 s) nanoparticles were observed with a Rg of 7.12 nm, with a low volume fraction as shown by the measurement statistics. After this point, slow growth of nanoparticles over time follows, as shown in Table 3 and the evolution in P(R), with the Rg increasing to 7.21, 7.71, and 8.15 nm after 2, 3, and 4 h respectively. This also corresponds with a slight increase in polydispersity, inferred from the greater peak width at FWHM in the real-space size distributions shown in the ESI (Fig. S8†). Analysis using the DAMMIN subroutine of ATSAS64 fitted the data to an array of dummy atoms to provide an interpretation of the true nanoparticle shape, shown in the insets in Fig. 5. Visually, the nanoparticles become more globular over time, in agreement with analysis of the in situ SANS measurements, and just after nucleation, the particle is narrow, disordered and wormlike. The spontaneous nucleation of these particles from the reacting pure DES solution at a critical timepoint is therefore caused by two simultaneous effects: the concentration of reacted iron complexes increases, while the solubility of these complexes decreases over time due to the active degradation of the solvent. The solubility threshold is then reached, and nanoparticles nucleate and undergo slow growth. The lack of structure factor suggests that particles are non-interacting and do not fuse due to a solvation corona which efficiently screens charge in the DES environment. Additionally, the SAXS experiment shows that it is feasible to ‘freeze’ the reaction at selected timepoints to obtain fairly monodisperse nanoparticles of known size and morphology, which is facilitated by the slow (<1 nm h−1) growth rates after the initial step. TEM confirms the formation of globular particles in the pure DES (see ESI Fig. S11 and S13†). This may be useful to target preparation of particles for use in biomedical applications applying superparamagnetic iron oxide nanoparticles, such as MRI.9
Conversely, in the hydrated DES nanoparticles nucleate immediately and grow rapidly, as shown in Fig. 5b. The reacted pure and hydrated systems differ visually and in terms of stability. Whereas the pure solvent formed dark-red stable colloidal suspensions of iron oxide, the hydrated system formed opaque yellow/orange suspensions which separated over time to a floc and a colourless liquid phase, all of which are characteristics of large-scale aggregates. Nanoparticles are observed within the first 5 min SANS measurement and at this point are already larger than the nanoparticles ever grow in the pure DES, at around 80 × 25 nm for the 0.15 mol kg−1 sample when fitted with an oblate spheroid model. After this, growth occurs predominantly along the equatorial axis, in line with previous reports which have highlighted the 1D morphology of nanoparticles formed from reactions in hydrated DES.39,40 After a period of time, which varies by concentration, there is then a rapid stepwise fusion of nanoparticles to form highly elongated 1D nanostructures which exceed the available instrumental resolution (major axis >65 nm; Qmin = 0.00229 Å−1). During this stage, the crystallites almost double in size every 10 min. The ellipsoid model was used throughout for continuity, but these much longer particles fit more adequately to cylinders, which corresponds with the structures observed in the TEM of the products (see ESI Fig. S12 and S14†). PXRD on these larger particles demonstrates formation of the hematite structure (Fig. S10†). Overall, the hydrated DES shows iron oxide formation and growth behavior more relatable to known aqueous solution chemistry.9,82
3.4 Evolution of local iron structure
In situ time-resolved EXAFS measurements were made of the reacting pure and hydrated DES systems alongside static snapshots of the local structural environments at ‘before’, ‘after’ and ‘intermediate’ states. Examples for the systems containing 0.15 mol kg−1 of Fe(NO3)3·9H2O are shown in Fig. 7. Qualitatively, the variable EXAFS signal between 1–4 Å shows the progress of the reaction. In the low-water pure DES system, a structural feature at 1–2 Å (with a shoulder at 2–2.5 Å) linearly diminishes until 12000 s, and a second structural feature begins to grow after 5000 s of reaction at between 3–4 Å. The appearance of this second structural feature aligns with the 5000 s nucleation point from SANS and SAXS. Increasing iron concentration extends the reaction time, with no further change after 10000 s and 12000 s for 0.15 and 0.20 mol kg−1 samples respectively. However, the hydrated DES samples behave completely differently; the structure at 3–4 Å forms immediately and increases in intensity, while the feature at 1–2 Å diminishes more rapidly. The hydrated DES reactions reached completion more quickly, taking just 6000 s for the 0.20 mol kg−1 sample.
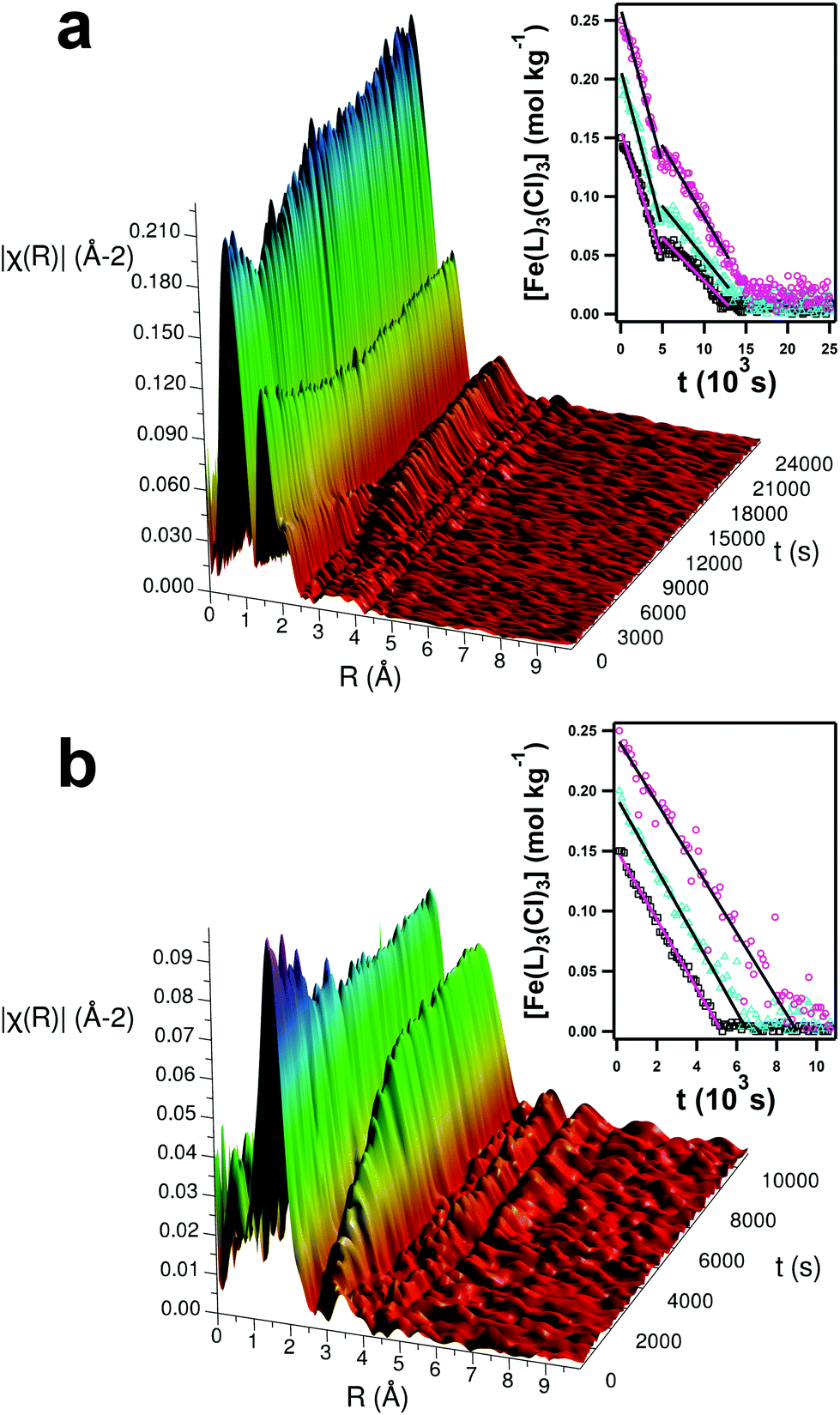 | ||
| Fig. 7 Corrected, time-resolved and Fourier-transformed EXAFS data for 0.15 mol kg−1 of Fe(NO3)3·9H2O in the pure (a) and hydrated (b) DES. (Inset) Analysis of the EXAFS data using CA showing linear regions fitted to the kinetic data for the three starting concentrations of 0.15 (black), 0.20 (teal), and 0.25 (magenta) mol kg−1 of Fe(NO3)3·9H2O, for (a) the pure and (b) the hydrated DES. Kinetic information was linearly fitted in the pre- and post-intermediate region for the pure DES (ca. 4900 s) and across the region from 0–5000 s in the hydrated DES. Fits were performed across the same timescale for each concentration series. For the high-water, high-concentration sample, datapoints were removed which had an unacceptably high level of background, associated with the uncontrollable formation of bubbles during the reaction which was particularly pronounced in this sample. | ||
A two-part component analysis (CA) was performed on the extracted EXAFS signal by analytically fitting the EXAFS signal as a convolution of an ‘initial’ state ([Fe(L)3(Cl)3] complexes) and ‘final’ state (crystalline α-Fe2O3). This analysis provides the population of each state as the reaction progresses and shows the stark systematic differences when water is added. Data and fits for the final nanoparticulate state are shown in the ESI (Fig. S9†), alongside XRD (Fig. S10†) and TEM (Fig. S11 to S14†).56 The CA output was then normalised to the starting material concentrations in order to obtain kinetic reaction data, which is shown in the insets to Fig. 7. It is known that such reactions rely upon thermal hydrolysis of the urea in the DES.39,40 It is noteworthy that EXAFS is not sensitive to this hydrolysis reaction, but only to coordination changes around the Fe centre. Only linear variations in iron complex concentration are observed, other than a discontinuity in the pure DES measurements at 5000 s. The measurements therefore show pseudo-zero-order behavior for iron, suggesting that the iron complex reaction is limited by the rate of urea hydrolysis and diffusion of hydrolysis products; being a component of the solvent, the urea concentration is in a huge excess. Table 4 shows calculated pseudo-zero-order rate constants for Fe. When hydrated, the concentration of iron precursor decreases instantly, quickly and linearly, with minimal concentration effect. This yields rates of 28.6 ± 0.38, 29.6 ± 0.57, and 27.0 ± 1.72 μmol kg−1 s−1 for 0.15, 0.20 and 0.25 mol kg−1 of [Fe(L)3(Cl)3] respectively, meaning that the difference in observed endpoint is almost entirely determined by the quantity of iron which must react, i.e. the reaction is zero-order in iron. The minor differences in rate could be explained by the initial iron concentration also affecting the nitrate and water concentrations, which slightly modify the solution viscosity and ionic strength. As highlighted in the SAS analysis, nanoparticles nucleate and are observable immediately for this system, and then grow rapidly before fusing. The reaction midpoint for each concentration, respectively around 2500 s, 3100 s and 4500 s for 0.15, 0.20 and 0.25 mol kg−1 of [Fe(L)3(Cl)3], aligns with the fusion point where large-scale 1D nanostructures form, as shown in the steep rise in Req in Fig. 5b.
Conversely, the iron in the pure DES system has two linear rate regimes (k1 and k2) across all concentrations, from 0 to 5000 s (faster) and from 5000 to 15000 s (slower). The first region is comparable to the rate seen for the hydrated system, with rates of 22.1 ± 0.45, 27.0 ± 0.76 and 26.7 ± 0.86 μmol kg−1 s−1 for 0.15, 0.20 and 0.25 mol kg−1 of [Fe(L)3(Cl)3] respectively. Slight rate discrepancies between runs are expected due to possible non-uniformity in the cells and heating. There is a clearer concentration effect for the pure DES samples, with the lowest concentration of iron precursor yielding the slowest rate. This result likely reflects the sample viscosity, with the lowest iron concentration giving a water content of 2.3 wt%, as opposed to 3.0 wt% and 3.7 wt% for the higher concentrations. The reactions proceed at this rate until 5000 s, which matches the nucleation point of SAS analysis. Following this event, the reaction slows dramatically and the concentration effect of the [Fe(L)3(Cl)3] becomes stronger; k2 is reduced respectively to 7.3 ± 0.23, 8.8 ± 0.27, and 12.1 ± 0.37 μmol kg−1 s−1 + for 0.15, 0.20 and 0.25 mol kg−1 of [Fe(L)3(Cl)3]. This supports the idea of nucleation at this point, causing a local depletion of iron species following particle formation, slowing the reaction.
3.5. Discussion
The measured initial state of the system has several important implications. Firstly, EPSR models of the data support our interpretation and fitting of the EXAFS data to an octahedral fluxional [Fe(L)3(Cl)3] complex structure, rather than polyanionic species proposed for similar systems which contain only anionic chloride.63 Most of the DES components, especially those containing an oxygen atom, are able to bind iron. As water content is increased, the coordination sphere predictably becomes more water-dominated. It was previously hypothesised that the unusual coordination chemistry of metal ions in DES could lead to potentially useful effects, such as the liquid-phase pre-structuring of reactive compounds, to reduce the activation barrier to reactions.39 This study suggests that this is only true of the pure DES, as even small quantities of water make the complex more like that seen in aqueous solution; the relatively low molar mass of water in the solution means that even small weight-percentages are swamping in molar terms, despite a deceptively small volume or mass fraction. The bulk solution structures were also compared with DES samples without dissolved iron salt. In all cases, the introduction of the iron salt disrupted the DES structure, predominantly because the formed iron complex slightly reduces the bulk Cl− concentration. Further local disruptions in the H-bond network arise due to interactions of the DES with the [Fe(L)3(Cl)3] complex. These two factors cause a series of disruptions in the intermolecular and site–site coordination numbers, which are difficult to predict a priori; the reaction mixtures studied here are already complicated multicomponent mixtures. The negligible Fe–Fe solution interactions prove that the complexes are stable and do not interact prior to the onset of heating. Next we propose a mechanism for the reaction and nanoparticle formation in ChCl:urea DES, from the combined analysis. Initially, a stable [Fe(L)3(Cl)3] complex forms in both solutions, which is fluxional in nature. The ligand L can variably be that of any O-containing donor species in the mixture and is more likely to be H2O in the hydrated mixture. When the mixture is heated above ca. 80 °C, the reaction commences. As has been discussed extensively for such reactions in amide-based DES, above this point the urea in the DES begins to hydrolyse, forming basic products such as ammonia and carbonate ions.11,13,14,39,40 For lanthanides, this caused the formation of insoluble nanometer-scale lanthanide carbonate species.39,83 For iron compounds, this does not occur, and rather it is suggested that the chemistry is fundamentally the same as aqueous iron oxide-forming reactions. Iron salts are hydrolysed under basic conditions to form a gamut of dry and hydrated oxides, hydroxides, and oxyhydroxides, through intermediate [–O–Fe–O–] network structures.82,84 As urea is hydrolysed over time, so too therefore is the initial [Fe(L)3(Cl)3] complex, which may occur as chloride is selectively scavenged by ammonium ions in solution to form solvated NH4Cl, or through reaction of degradation products such as hydroxide with the complex. The limitations of the techniques used do not allow this to be specifically resolved, but suggest pseudo-zero-order kinetics in iron due to the reaction rate being dictated by the hydrolysis of urea, which is in massive excess as an intrinsic component of the solvent. At the constant reaction temperatures studied in the DES, the urea hydrolysis rate is therefore controlled by the water content and viscosity, and thus indirectly by the iron concentration as these are hydrated iron salts. EXAFS suggests that the complex forms Fe(O)x centres, which then form oligomeric [–O–Fe–O–] network structures. Once these exceed the solubility limit, which itself changes over time due to solvent degradation, nanoparticles begin to nucleate and grow. In the hydrated DES this is immediate like in aqueous systems, due to the low solubility of such [–O–Fe–O–] species. However, analysis of EXAFS, SAXS and SANS data all agree that the pure DES system remains homogeneous until 5000 s, at which point nucleation occurs. Therefore, as the reaction progresses, prior to nucleation the oligomeric [–O–Fe–O–] iron complexes that are formed are stabilised and more soluble in the solvation environment of DES. Following this point, the reaction slows markedly in the pure DES. This may be due to several effects, but could simply be the onset of a more strongly diffusion-limited condition as the formation of nanoparticles increases the solution viscosity, and creates a zone depleted in iron species locally where the particles have nucleated. Slow growth of oblate spheroid nanoparticles is then observed in the pure DES, whereas nanoparticles grow rapidly in the hydrated DES into highly elongated 1D structures. For both the pure and hydrated DES, neutron contrast effects are observed which suggest the formation of choline-rich layers near the surface, in agreement with previous experimental observations of extensive structuring at the DES/solid interface.20,23,79,80 This choline-enriched boundary layer may have a nanoparticle surface capping effect, especially in the hydrated system where the growth rate is highest and templated 1D growth of nanoparticles is seen. Cholinium can therefore masquerade as a cationic surfactant due to the structural similarity, inhibiting certain growth axes.78
4. Conclusions
A deep eutectic-solvothermal reaction for the formation of iron oxide nanoparticles has been studied in situ using SANS, SAXS, EXAFS, neutron and X-ray diffraction and atomistic modelling. While this is a complicated multicomponent system with many experimental challenges, the initial and final states of the reaction were determined, and the reaction and nanoparticle formation were followed. Upon dissolution, an initial [Fe(L)3(Cl)3] complex forms, which is remarkably similar in both pure and hydrated DES. This is then broken down over time following thermal hydrolysis of the urea in the DES, precipitating iron oxide nanoparticles which then grow into either small oblate spheroids in the pure DES, or large 1D nanoparticles in the hydrated DES. This process of nanoparticle nucleation and growth begins instantly for the hydrated system, but in the pure DES this is inhibited until a threshold reaction time has elapsed, likely due to the higher solubility of the hydrolysed Fe complex (i.e. [–O–Fe–O–] network structures) in DES. SANS/SAXS analysis provides evidence for a choline-enriched boundary layer at the nanoparticle interface. We have therefore for the first time provided insights across multiple length and time scales into the broad spectrum of processes which occur within DES when used as a medium for nanomaterial synthesis. First, this is a unique study into the inorganic coordination chemistry, speciation and solvation occurring within the DES, in pure and hydrated states. Moreover, this is also the first study of its kind to measure such reactions in DES in situ using small-angle scattering and EXAFS. This helps to enable further understanding of these solvents on a fundamental level, and in the concept of deliberately degrading solvents to drive nanoparticle-forming reactions. Ultimately, this understanding will allow for the rational development and design of DES systems to produce more environmentally sustainable nanomaterials for applications such as energy and pollution remediation, which will hopefully contribute towards ameliorating the cluster of unfolding environmental disasters which threaten humanity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank ANSTO for access to the SANS instrument BILBY under award P6216, STFC ISIS Neutron and Muon source for access to NIMROD under awards RB1620292 and RB1620479 (data can be downloaded from DOI: 10.5286/ISIS.E.RB1620292 and 10.5286/ISIS.E.RB1620479), and Diamond Light Source for access to I20-EDE and I20-Scanning under award SP17574. We thank the STFC ISIS Materials Characterisation Lab for providing access to the Xenocs Nano-inXider SAXS instrument. We thank the UK Science and Technology Facilities Council (STFC) and the UK Engineering and Physical Sciences Research Council (EPSRC) for co-funding a PhD studentship for O. S. H. (EP/L016354/1; STFC Studentship Agreement #3578) and for R. S. A. (EP/L016354/1) in the Centre for Doctoral Training in Sustainable Chemical Technologies at the University of Bath. Data supporting this work is freely accessible in the Bath research data archive system at DOI: 10.15125/BATH-00954. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement No 654000.References
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 13, 6357–6426 CrossRef.
- B. Gurkan, H. Squire and E. Pentzer, J. Phys. Chem. Lett., 2019, 10, 7956–7964 CrossRef CAS.
- R. A. Laudise, Chem. Eng. News, 1987, 65, 30–43 CrossRef CAS.
- W. L. Suchanek and R. E. Riman, Adv. Sci. Technol., 2006, 45, 184–193 CAS.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–701 CrossRef CAS.
- S. Somiya and R. Roy, Bull. Mater. Sci., 2000, 23, 453–460 CrossRef CAS.
- B. Zhang, Y. Xue, A. Jiang, Z. Xue, Z. Li and J. Hao, ACS Appl. Mater. Interfaces, 2017, 9, 7217–7223 CrossRef CAS.
- R. I. Walton, Prog. Cryst. Growth Charact. Mater., 2011, 57, 93–108 CrossRef CAS.
- Y. L. Pang, S. Lim, H. C. Ong and W. T. Chong, Ceram. Int., 2015, 42, 9–34 CrossRef.
- J. Jiang, C. Yan, X. Zhao, H. Luo, Z. Xue and T. Mu, Green Chem., 2017, 19, 3023–3031 RSC.
- E. A. Drylie, D. S. Wragg, E. R. Parnham, P. S. Wheatley, A. M. Z. Slawin, J. E. Warren and R. E. Morris, Angew. Chem.,– Int. Ed., 2007, 46, 7839–7843 CrossRef.
- X. Ge, C. D. Gu, Y. Lu, X. L. Wang and J. P. Tu, J. Mater. Chem. A, 2013, 1, 13454 RSC.
- E. R. Parnham, E. A. Drylie, P. S. Wheatley, A. M. Z. Slawin and R. E. Morris, Angew. Chem.,– Int. Ed., 2006, 45, 4962–4966 CrossRef CAS.
- E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005–1013 CrossRef CAS.
- R. E. Morris, Chem. Commun., 2009, 21, 2990–2998 RSC.
- R. Hayes, S. Imberti, G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 13544–13551 RSC.
- T. Murphy, R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2014, 16, 13182–13190 RSC.
- L. R. Griffin, K. L. Browning, S. M. Clarke, A. M. Smith, S. Perkin, M. W. A. Skoda and S. E. Norman, Phys. Chem. Chem. Phys., 2017, 19, 297–304 RSC.
- J. A. Smith, O. Werzer, G. B. Webber, G. G. Warr and R. Atkin, J. Phys. Chem. Lett., 2010, 1, 64–68 CrossRef CAS.
- J. A. Hammons, F. Zhang and J. Ilavsky, J. Colloid Interface Sci., 2018, 520, 81–90 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- O. S. Hammond, D. T. Bowron, A. J. Jackson, T. Arnold, A. Sanchez-Fernandez, N. Tsapatsaris, V. G. Sakai and K. J. Edler, J. Phys. Chem. B, 2017, 121, 7473–7483 CrossRef CAS.
- O. S. Hammond, H. Li, C. Westermann, F. Endres, A. Y. Al-Murshedi, A. P. Abbott, G. Warr, K. J. Edler and R. Atkin, Nanoscale Horiz., 2019, 4, 158–168 RSC.
- C. R. Ashworth, R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2016, 18, 18145–18160 RSC.
- R. P. Matthews, I. J. Villar-Garcia, C. C. Weber, J. Griffith, F. Cameron, J. P. Hallett, P. A. Hunt and T. Welton, Phys. Chem. Chem. Phys., 2016, 18, 8608–8624 RSC.
- C. F. Araujo, J. A. P. Coutinho, M. M. Nolasco, S. F. Parker, P. J. A. Ribeiro-Claro, S. Rudić, B. I. G. Soares and P. D. Vaz, Phys. Chem. Chem. Phys., 2017, 19, 17998–18009 CAS.
- Z. Tan, Y. Peng, J. Liu, Y. Yang, Z. Zhang, Z. Chen, B. Mao and J. Yan, ChemElectroChem, 2020, 7(22), 4601–4605 CrossRef CAS.
- L. Percevault, A. Jani, T. Sohier, L. Noirez, L. Paquin, F. Gauffre and D. Morineau, J. Phys. Chem. B, 2020, 124, 9126–9135 CrossRef CAS.
- Q. Gao, N. Wu, Y. Qin, A. Laaksonen, Y. Zhu, X. Ji and X. Lu, J. Mol. Liq., 2020, 319, 114298 CrossRef CAS.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS.
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Grätzel, J. Phys. Chem. C, 2009, 113, 772–782 CrossRef CAS.
- J. W. Morgan and E. Anders, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 6973–6977 CrossRef CAS.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS.
- J. Zhang and S. Eslava, Sustainable Energy Fuels, 2019, 3, 1351–1364 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS.
- D. V. Wagle, H. Zhao and G. A. Baker, Acc. Chem. Res., 2014, 47, 2299–2308 CrossRef CAS.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef CAS.
- O. S. Hammond, K. J. Edler, D. T. Bowron and L. Torrente-Murciano, Nat. Commun., 2017, 8, 14150 CrossRef CAS.
- O. S. Hammond, S. Eslava, A. J. Smith, J. Zhang and K. J. Edler, J. Mater. Chem. A, 2017, 5, 16189–16199 RSC.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 9782–9785 CrossRef CAS.
- S. Díaz-Moreno, S. Ramos and D. T. Bowron, J. Phys. Chem. A, 2011, 115, 6575–6581 CrossRef.
- V. Alizadeh, D. Geller, F. Malberg, P. B. Sánchez, A. Padua and B. Kirchner, ChemPhysChem, 2019, 20, 1786–1792 CrossRef CAS.
- A. R. Hillman, R. Barker, R. M. Dalgliesh, V. C. Ferreira, E. J. R. Palin, R. M. Sapstead, E. L. Smith, N.-J. Steinke, K. S. Ryder and A. D. Ballantyne, Faraday Discuss., 2018, 210, 429–449 RSC.
- A. D. Ballantyne, R. Barker, R. M. Dalgliesh, V. C. Ferreira, A. R. Hillman, E. J. Palin, R. Sapstead, E. L. Smith, N. J. Steinke and K. S. Ryder, J. Electroanal. Chem., 2018, 819, 511–523 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- D. T. Bowron, A. K. Soper, K. Jones, S. Ansell, S. Birch, J. Norris, L. Perrott, D. Riedel, N. J. Rhodes, S. R. Wakefield, A. Botti, M.-A. Ricci, F. Grazzi and M. Zoppi, Rev. Sci. Instrum., 2010, 81, 033905 CrossRef CAS.
- A. K. Soper, Mol. Phys., 2001, 99, 1503–1516 CrossRef CAS.
- X. Meng, K. Ballerat-Busserolles, P. Husson and J.-M. Andanson, New J. Chem., 2016, 40, 4492–4499 RSC.
- A. K. Soper , GudrunN and GudrunX: Programs for Correcting Raw Neutron and X-Ray Diffraction Data to Differential Scattering Cross Section, Rutherford Appleton Laboratory Technical Report RAL-TR-2011-013, 2011.
- A. K. Soper, J. Phys.: Condens. Matter, 2007, 19, 335206 CrossRef CAS.
- P. Li, L. F. Song and K. M. Merz, J. Phys. Chem. B, 2015, 119, 883–895 CrossRef CAS.
- A. Sokolova, J. Christoforidis, A. Eltobaji, J. Barnes, F. Darmann, A. E. Whitten and L. de Campo, Neutron News, 2016, 27, 9–13 CrossRef.
- S. Diaz-Moreno, S. Hayama, M. Amboage, A. Freeman, J. Sutter and G. Duller, J. Phys.: Conf. Ser., 2009, 190, 012038 CrossRef.
- S. Hayama, G. Duller, J. P. Sutter, M. Amboage, R. Boada, A. Freeman, L. Keenan, B. Nutter, L. Cahill, P. Leicester, B. Kemp, N. Rubies and S. Diaz-Moreno, J. Synchrotron Radiat., 2018, 25, 1556–1564 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- S. Pascarelli, T. Neisius and S. De Panfilis, J. Synchrotron Radiat., 1999, 6, 1044–1050 CrossRef CAS.
- D. T. Bowron and S. Díaz-Moreno, Anal. Chem., 2005, 77, 6445–6452 CrossRef CAS.
- D. Franke, M. V. Petoukhov, P. V. Konarev, A. Panjkovich, A. Tuukkanen, H. D. Mertens, A. G. Kikhney, N. R. Hajizadeh, J. M. Franklin, C. M. Jeffries and D. I. Svergun, J. Appl. Crystallogr., 2017, 50, 1212–1225 CrossRef CAS.
- M. Apted, G. A. Waychunas and G. Brown, Geochim. Cosmochim. Acta, 1985, 49, 2081–2089 CrossRef CAS.
- A. P. Abbott, S. S. M. Alabdullah, A. Y. M. Al-Murshedi and K. S. Ryder, Faraday Discuss., 2017, 206, 365–377 RSC.
- H. Ohtaki and T. Radnai, Chem. Rev., 1993, 93, 1157–1204 CrossRef CAS.
- J. M. Hartley, C. M. Ip, G. C. H. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, H. L. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 19, 2010–2011 RSC.
- M. E. Di Pietro, O. Hammond, A. van den Bruinhorst, A. Mannu, A. Padua, A. Mele and M. Costa Gomes, Phys. Chem. Chem. Phys., 2021, 23, 107–111 RSC.
- O. S. Hammond and K. J. Edler, Deep Eutectic Solvents: Synthesis, Properties, and Applications, Wiley-VCH, Weinheim, 2019, pp. 25–42 Search PubMed.
- R. Stefanovic, M. Ludwig, G. B. Webber, R. Atkin and A. J. Page, Phys. Chem. Chem. Phys., 2017, 19, 3297–3306 RSC.
- A. K. Soper, Mol. Phys., 2009, 107, 1667–1684 CrossRef CAS.
- M. Gilmore, L. M. Moura, A. H. Turner, M. Swadźba-Kwaśny, S. K. Callear, J. A. McCune, O. A. Scherman and J. D. Holbrey, J. Chem. Phys., 2018, 148, 193823 CrossRef.
- V. Petrouleas, R. M. Lemmon and A. Christensen, J. Chem. Phys., 1978, 68, 2243–2246 CrossRef CAS.
- A. Nath, R. Agarwal and R. M. Lemmon, J. Chem. Phys., 1974, 61, 1542–1547 CrossRef CAS.
- A. K. Soper, E. W. Castner and A. Luzar, Biophys. Chem., 2003, 105, 649–666 CrossRef CAS.
- A. Sanchez-Fernandez, O. S. Hammond, K. J. Edler, T. Arnold, J. Doutch, R. M. Dalgliesh, P. Li, K. Ma and A. J. Jackson, Phys. Chem. Chem. Phys., 2018, 20, 13952–13961 RSC.
- A. Sanchez-Fernandez, O. S. Hammond, A. J. Jackson, T. Arnold, J. Doutch and K. J. Edler, Langmuir, 2017, 33, 14304–14314 CrossRef CAS.
- T. Arnold, A. J. Jackson, A. Sanchez-Fernandez, D. Magnone, A. E. Terry and K. J. Edler, Langmuir, 2015, 31, 12894–12902 CrossRef CAS.
- Z. Chen, B. McLean, M. Ludwig, R. Stefanovic, G. G. Warr, G. B. Webber, A. J. Page and R. Atkin, J. Phys. Chem. C, 2016, 120, 2225–2233 CrossRef CAS.
- S. Kaur, S. Sharma and H. K. Kashyap, J. Chem. Phys., 2017, 147, 194507 CrossRef.
- M. G. Sujana, K. K. Chattopadyay and S. Anand, Appl. Surf. Sci., 2008, 254, 7405–7409 CrossRef CAS.
- J. A. Hammons, T. Muselle, J. Ustarroz, M. Tzedaki, M. Raes, A. Hubin and H. Terryn, J. Phys. Chem. C, 2013, 117, 14381–14389 CrossRef CAS.
- J. A. Hammons, J. Ustarroz, T. Muselle, A. A. J. Torriero, H. Terryn, K. Suthar and J. Ilavsky, J. Phys. Chem. C, 2016, 120, 1534–1545 CrossRef CAS.
- SasView, https://www.sasview.org.
- M. Mohapatra and S. Anand, Int. J. Engg. Sci. Technol., 2010, 2, 127–146 Search PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, ACS Sustainable Chem. Eng., 2019, 7, 4932–4940 CrossRef CAS.
- M. Hanesch, Geophys. J. Int., 2009, 177, 941–948 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM, and small-angle scattering, diffraction, and EXAFS data and analysis. See DOI: 10.1039/D0NR08372K |
| This journal is © The Royal Society of Chemistry 2021 |