
Defect-assisted electronic metal–support interactions: tuning the interplay between Ru nanoparticles and CuO supports for pH-neutral oxygen evolution†
Alexander J.
Porkovich
 *a,
Pawan
Kumar
a,
Zakaria
Ziadi
a,
David C.
Lloyd
a,
Lin
Weng
a,
Nan
Jian
ab,
Toshio
Sasaki
a,
Mukhles
Sowwan
a and
Abheek
Datta
*a
*a,
Pawan
Kumar
a,
Zakaria
Ziadi
a,
David C.
Lloyd
a,
Lin
Weng
a,
Nan
Jian
ab,
Toshio
Sasaki
a,
Mukhles
Sowwan
a and
Abheek
Datta
*a
aOkinawa Institute of Science and Technology (OIST) Graduate University, 1919-1 Tancha, Onna-Son, Okinawa 904-0495, Japan. E-mail: alexander.porkovich@oist.jp; datta.abheek@oist.jp
bInstitute of Nanosurface Science and Engineering, Shenzhen University, Nanhai Ave. 3688, Shenzhen, Guangdong, China
First published on 8th December 2020
Abstract
Electronic metal–support interactions (EMSIs) comprise an area of intense study, the manipulation of which is of paramount importance in the improvement of heterogeneous metal nanoparticle (NP) supported catalysts. EMSI is the transfer of charge from the support to NP, enabling more effective adsorption and interaction of reactants during catalysis. Ru NPs on CuO supports show different levels of EMSI (via charge transfer) depending on their crystal structure, with multiple twinned NPs showing greater potential for EMSI. We use magnetron-assisted gas phase aggregation for the synthesis of batches of Ru NPs with different populations of single crystal and multiple twinned nanoparticles, which were deposited on CuO nanowires (NWs). The surface charging of the Ru-CuO catalysts was investigated by Kelvin probe force microscopy (KPFM) and X-ray photoelectron spectroscopy (XPS). By doubling the population of multiple twinned NPs, the surface potential of the Ru-CuO catalysts increases roughly 4 times, coinciding with a similar increase in the amount of Ru4+. Therefore, tuning the amount of EMSI in a catalyst is possible through changing the population of multiple twinned Ru NPs in the catalyst. Increasing the amount of multiple twin NPs resulted in improved activity in the oxygen evolution reaction (a roughly 2.5 times decrease in the overpotentials when the population of multiple twinned NPs is increased) and better catalyst stability. This improvement is attributed to the fact that the multiple twin NPs maintained a metallic character under oxidation conditions (unlike single crystal NPs) due to the EMSI between the NP and support.
Introduction
Heterogeneous catalysis is a key process necessary to unlock the hydrogen economy. The combinations of noble metal nanoparticles (NPs) with an electron or oxygen rich support has been constantly investigated in the field of electrocatalytic materials research,1–5 with the aim of improving the evolution of hydrogen from water.1,6–11 The research on supported noble metal catalysis has resulted in the discovery of different catalytic mechanisms for different combinations of materials. Early ideas of spill over12 have been influenced by metal support interactions (MSIs),2,4,8–10,13–17 as the understanding of catalytic systems has improved. Strong metal–support interactions (SMSIs)2,10,15 are well studied and have demonstrated evidence of a stoichiometric interaction between the noble metal and support, as a thin layer of the support slowly encapsulates the noble metal during catalysis.15Other systems, however, have been found to show changes to catalysis based on the electronic changes in a system.17 This has led to more focus on electronic metal–support interactions (EMSIs)8,9,13,14,25 which is in principle a charge transfer between noble metal NPs and a reducible oxide support.13
Recent work has attempted to improve catalytic activity via the tuning of MSIs through the support.4,9 Typically, the size and shape of NPs are tuned to improve their catalytic performance,18–22 with multiple twin structures (especially icosahedra) being of interest due to the {111} facets24 and mechanical strain caused by the defective growth of twin boundaries.24,26 However, not much attention has been paid to the effect of NPs on MSI, even though density functional theory (DFT) simulations indicate that the charge transfer between Ru NPs and CuO supports depends on their structure, with the defective twin boundaries of multiple twinned NPs showing higher charge transfer than those shown by single crystal NPs.27 This makes Ru NPs supported on CuO nanowires (NWs) a promising system to demonstrate the tuning of MSIs through the NPs, as CuO is a reducible metal oxide28 and catalytically active in its own right.29
By using magnetron-assisted gas aggregation for tuning the relative populations of single crystal and multiple twinned NPs,30 this study will demonstrate the tuning of the EMSI between Ru NPs and CuO NW supports via the increase of multiple twin populations. PeakForce Kelvin probe force microscopy (formally called PF-KPFM but will be referred to as KPFM) and X-ray photoelectron spectroscopy (XPS) were used to elucidate how the charge transfer between NPs and NWs leads to an improved catalytic performance towards the formation of a more stable catalytic interface.
Experimental section
Deposition and growth of nanoparticles (NPs)
Ru NPs were grown using a dual chamber deposition system (Mantis deposition Ltd) with an aperture separating the water-cooled growth chamber (termed the aggregation zone, kept at a base pressure of ∼10−6 mbar) from a lower pressure chamber containing the substrate (the so-called deposition chamber, with a vacuum of ∼10−8 mbar). The NPs were formed via magnetron assisted gas-aggregation, with sputtered Ru atoms (Target 99.95%, Kurt J. Lesker) being condensed into the NPs due to the higher pressure of unionised Ar and He gas atoms flowing into the aggregation chamber (both gases entered through separate lines). The NPs were eventually forced from the higher-pressure aggregation zone to the lower-pressure deposition chamber, eventually landing on the substrates. To improve the surface uniformity, the substrate was rotated at 2 rpm.For this study, the NPs were formed using different parameters for each sample (see Table 1).
| Sample name | Power applied to target (W) | Pressure of deposition (D) and aggregation (A) zones (mbar) | Ratio of Ar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :He gas flows :He gas flows |
Distance between magnetron and aperture (mm) | Time (min) |
|---|---|---|---|---|---|
| Ru-CuO(A) | 5 | D-7.7 × 10−4 | 16:1 |
125 | 100 |
| A-3.5 × 10−1 | |||||
| Ru-CuO(B) | 7.7 | D-3.5 × 10−4 | 17:1 |
125 | 30 |
| A-1.5 × 10−1 | |||||
| Ru-CuO(C) | 10 | D-1.0 × 10−3 | 21:1 |
125 | 20 |
| A-7.5 × 10−2 |
Growth of CuO nanowires (NWs)
CuO NWs were thermally grown from Cu foils (0.1 mm thick with a purity of 99.9999% from Puratronic) on a hotplate set at 540 °C for 3 hours. The Ru NPs were directly deposited on the oxidised foils. In this case, the Ru decorated CuO foils were directly used for characterisation. In all the other cases, however, Ru decorated CuO NWs were harvested from the foils. These transfers will be highlighted in the following method sections for each characterisation technique.Scanning transmission electron microscopy (STEM) studies
The Ru NPs were deposited directly on carbon film transmission electron microscope (TEM) grids (Cu frame, from Ted-Pella Inc.) for analyzing the coverage, size and crystallinity of differently synthesised samples. As noted in the “Growth of CuO nanowires (NWs)” section, the Ru NPs were directly deposited on the CuO foils when investigating the (as deposited and annealed in dry air) Ru NPs on CuO NWs. The decorated NWs were then transferred mechanically to the TEM grids.For the annealed samples, the Ru decorated CuO foils were placed on a hotplate (controlled by a LakeShore 336 temperature controller) in a cryogenic probe station (ARS). Before annealing, the chamber was purged by vacuuming down to 10−3 hPa (Pfeiffer Vacuum Hi Cube). The chamber was then brought back to atmospheric pressure by flowing 1000 sccm (using mass flow controllers supplied by Bronkhorst) of a mixture of N2 and O2 in a ratio of 80:20 (dry synthetic air). During annealing at 200 °C, the chamber had a constant flow of (1000 sccm) dry synthetic air for 100 min. Then, the annealed Ru decorated CuO NWs were mechanically transferred to a TEM grid as before.
STEM imaging was performed using a JEOL JEM ARM200F microscope (featuring a Cs-probe corrector), with an accelerating voltage of 200 kV.
Kelvin probe force microscopy (PF-KPFM) imaging and sample preparation
Topography and surface potential measurements were performed on a Bruker Multimode 8 scanning probe microscope (Bruker, USA) under ambient atmosphere in the peak force tapping mode. A conducting SCM-PIT-V2 atomic force microscope (AFM) probe (coated with Pt–Ir) from Bruker, USA was used for all PF-KPFM measurements. The SCM-PIT-V2 probe used had a typical resonant frequency of 70 kHz (ω0) with a spring constant of 1.5 N m−1 and tip radius of 25 nm.The CuO NWs and Ru NP decorated CuO NWs were transferred from the CuO foils to an Au film coated glass by direct mechanical transfer. The CuO foils were placed sample side down on the Au coated substrate, and a gentle force was applied to facilitate the transfer of NWs from the foil to substrate. A silver paste was used to make an electrical contact between the Au film and metallic AFM stub. This allowed the application of an AC bias of 5000 mV, at a frequency of 2 kHz (ωm), to the sample.
The topography and surface potential imaging was performed in the PF-KPFM lift mode. When the electric force gradient was modulated, caused by AC bias, the modulation of the resonant frequency gave rise to two pairs of sidebands at ω0 ± ωm and ω0 ± 2ωm. The amplitude of the sideband quantifies the resonant frequency modulation amplitude. Thus, by using the side band amplitude of ω ± ωm for the KPFM feedback, the DC bias (VDC) was adjusted until it disappeared. This led to the point of VDC = VCPD. The minimum optimized lift height of ∼60 nm was used for the PF-KPFM measurements. The topography and surface potential images were captured simultaneously at a scan rate of 0.4 Hz, and the images were further processed and analysed using the Nanoscope Analysis 10 software.
X-ray photo electron spectroscopy (XPS) studies
The XPS measurements of the Ru NP decorated CuO foils were performed directly using an AXIS ultra HSA spectrometer from Kratos Analytical. A monochromated Al Kα (1486.6 eV) source was used under vacuum in the range of 10−9 mbar. A pass energy of 20 eV was used for high-resolution scans. All the data were analysed with the CasaXPS software, using a Shirley background for all peak fitting and the charge reference of C1s adventitious carbon at a binding energy of 284.8 eV.The deconvolution of Ru 3d, C 1s and O 1s peaks was performed using the CasaXPS software. The Ru 3d–C 1s regions were deconvolved according to the literature based on Ru-NP-31 and Ru-RuO2 fitting parameters.32 The positions of the adventitious carbon, carbonic oxides, Ru metal, Ru oxides and Ru satellites and line shapes were also taken from these sources. Oxygen was deconvolved based on the presence of oxides33 and the literature related to the fitting of Cu oxides.34 Annealed samples showed a shift in the position of the adventitious carbon peak but did not show any other notable shifts elsewhere across the XPS spectrum. This was also observed in other literature27,31 based on the annealing of Ru (particularly in the presence of semi-conductors) and was likely due to changes in the work function of the material. As proposed by Greczynski et al.,35 this was due to the position of the adventitious carbon peak being related to the difference between 289.5 eV and the material's work function.
The as-deposited samples of Ru decorated CuO foils were examined by XPS soon after deposition. Annealing of samples was conducted in dry air in a sealed cryongenic probe station directly after deposition. Following this they were investigated using XPS, as mentioned in the STEM section. All the protocols were the same; however, in this case the samples were annealed for 3 h.
Electrochemical measurements
All the electrochemical measurements, except electrochemical impedance spectroscopy (EIS) studies, were performed using a 2-channel CHI 440B electrochemical workstation. Catalyst coated glassy carbon (Ru decorated CuO NWs transferred from the foils of Ru-CuO(A), Ru-CuO(B) and Ru-CuO(C) samples were used as the working electrode; a platinum wire was used as the counter electrode, and an Ag/AgCl (3 M KCl) electrode was used as the reference electrode. All the measurements were performed in a 1 M phosphate-buffered saline (PBS) electrolyte. Linear sweep voltammetric (LSV) measurements were carried out at a scan rate of 5 mV s−1. A Gamry-3000 potentiostat was used for the EIS measurements. The EIS measurements were performed at an applied potential with the scanning frequency values ranging from 30 kHz to 10 MHz at a cell potential of 1.6 V vs. reversible hydrogen electrode (RHE) with a sinusoidal amplitude at 10 mV. Chronoamperometric studies were also carried out at the same potential. All the potentials reported herein were converted against RHE according to the following equation:| ERHE = EAg/AgCl + E°Ag/AgCl + 0.059 pH | (1) |
Results and discussion
Three different samples, labelled Ru-CuO(A), Ru-CuO(B) and Ru-CuO(C), were prepared by varying the deposition parameters of the Ru NPs (see the Experimental section for methodology) used to decorate the CuO NWs (grown thermally as detailed in the Experimental section). Exemplary low-magnification high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images of the Ru NPs used to prepare the Ru-CuO(A), Ru-CuO(B) and Ru-CuO(C) samples are shown in Fig. 1a, b and c, respectively. All the samples showed a mixture of single crystal hcp, multiple twins (particularly fcc icosahedral) and agglomerated Ru NPs.36Fig. 1d shows a typical single crystal hcp Ru NP on CuO NWs, while a similarly supported fcc icosahedral Ru NP is shown in Fig. 1e. In this case, the structure was indexed from the interplanar spacings and the angles between the visible planes. This was determined from the fast-Fourier transforms (FFTs) of the NPs as seen in Fig. S1 (hcp) and S2 (fcc icosahedral).† The hcp nature of the NPs was confirmed by d-spacings of approximately 0.19 nm and 0.12 nm, consistent with the {1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0} and {20} inter planar distances for Ru.37 The fcc icosahedral NP resembled an icosahedron viewed on its 2-fold axis of symmetry. Additionally, the interplanar distances in this case were found to be 0.22 nm and 0.19 nm, matching well with the known {111} and {200} d-spacings of fcc Ru.37
0} and {20} inter planar distances for Ru.37 The fcc icosahedral NP resembled an icosahedron viewed on its 2-fold axis of symmetry. Additionally, the interplanar distances in this case were found to be 0.22 nm and 0.19 nm, matching well with the known {111} and {200} d-spacings of fcc Ru.37
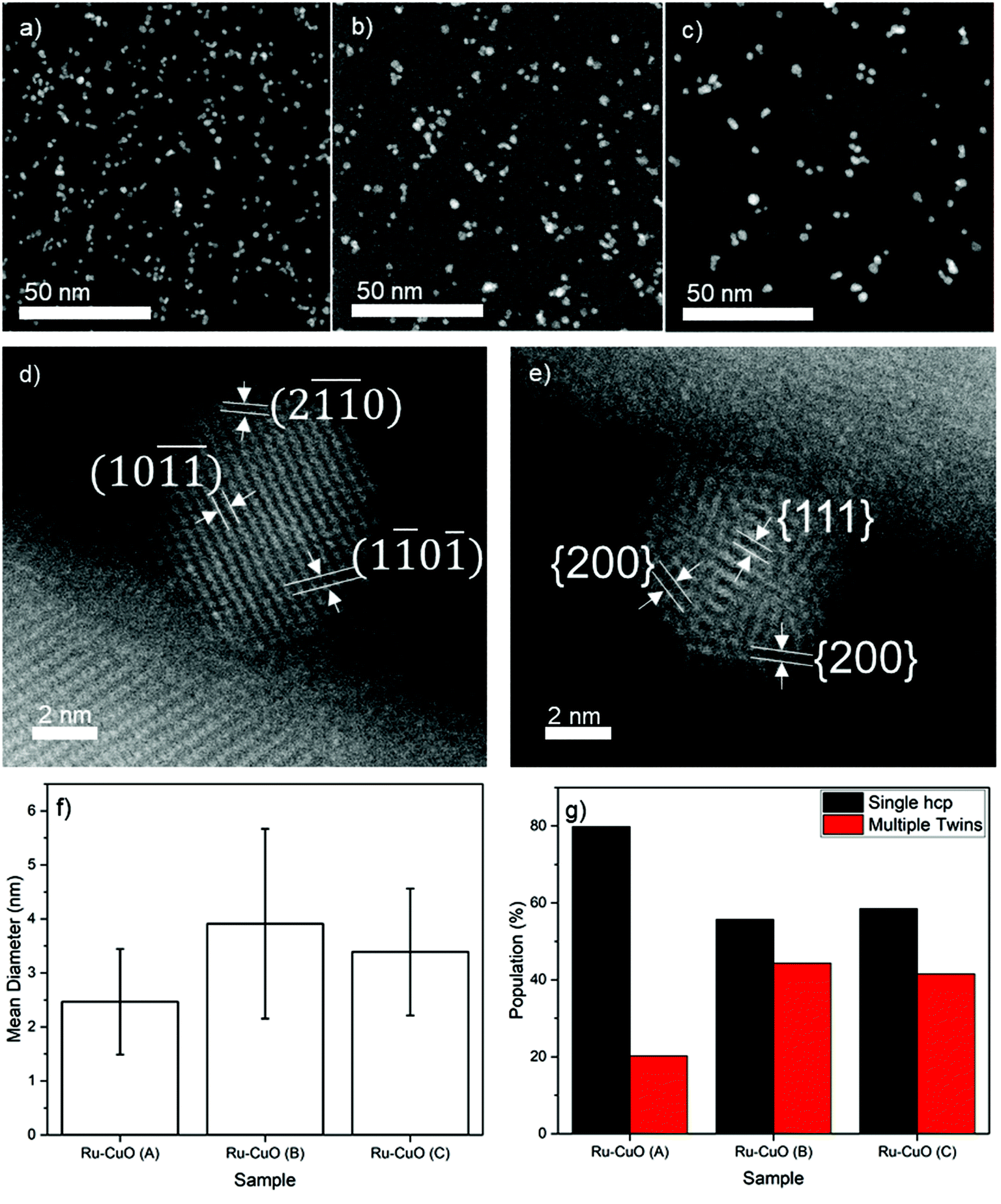 | ||
| Fig. 1 Examples of low magnification images of (a) Ru-CuO(A), (b) Ru-CuO(B) and (c) Ru-CuO(C) samples supported on carbon. High resolution HAADF-STEM images of typical (d) hcp Ru (viewed along the [011] direction) and (e) icosahedral fcc Ru (viewed close to the 2-fold axis of symmetry) NPs supported on CuO NWs. The mean diameter (with error bars showing standard deviation of 1) of each sample can be seen in (f), while (g) shows the relative population of single crystal hcp and multiple twins in each sample. | ||
The size distribution (Fig. S3†) and coverage (Fig. S4†) of these samples showed a degree of variability, as shown in Fig. 1f. Ru-CuO(A) showed the smallest average size of NPs (2.5 nm ± 1.0 nm), while the largest (3.9 nm ± 1.8 nm) average size was found in Ru-CuO(B). Ru-CuO(C) was found to be in the middle (3.4 nm ± 1.2 nm) but did show a developing bi-modality (Fig. S3c†). Most importantly, however, a clear difference was found in the percentage of NPs, which were either single crystal hcp or multiple twins (Fig. 1g). Once again, Ru-CuO(B) was found to have the most multiple twins. Interestingly, while Ru-CuO(C) also showed a high percentage of multiple twins, there was increasing agglomeration of NPs as compared to Ru-CuO(B) (Fig. S5†). There are two generally accepted causes for the aggregation of particles produced by gas-phase synthesis. The first is the coalescence or aggregation on the substrate. This is caused by NPs being deposited on top of each other, or close enough to interact on the surface.38 The other form of aggregation is in the aggregation chamber, where the thermodynamic equilibrium is enough for the formed NPs to interact with each other, but not enough to coalesce into a larger NP, leaving an agglomerate of nanograined materials.36
The subsequent charging behaviour of each of the samples is shown in Fig. 2, with the KPFM images of surface potential and the Ru 3d XPS spectra of Ru-CuO(A) (a and b), Ru-CuO(A) (c and d) and Ru-CuO(C) (e and f). Fig. 2a, c and e show the positive or negative charge, with a significant charge separation, on the NW (topography images of the KPFM signals show the NWs to be intact and uniform as seen in Fig. S6†). All the Ru NP decorated samples resulted in increased surface potential over the pristine CuO NWs (as can be seen in Fig. S7† and by direct comparison with Fig. S8†), as expected, due to the higher work function of CuO (5.3 eV) than that of Ru (4.7 eV).
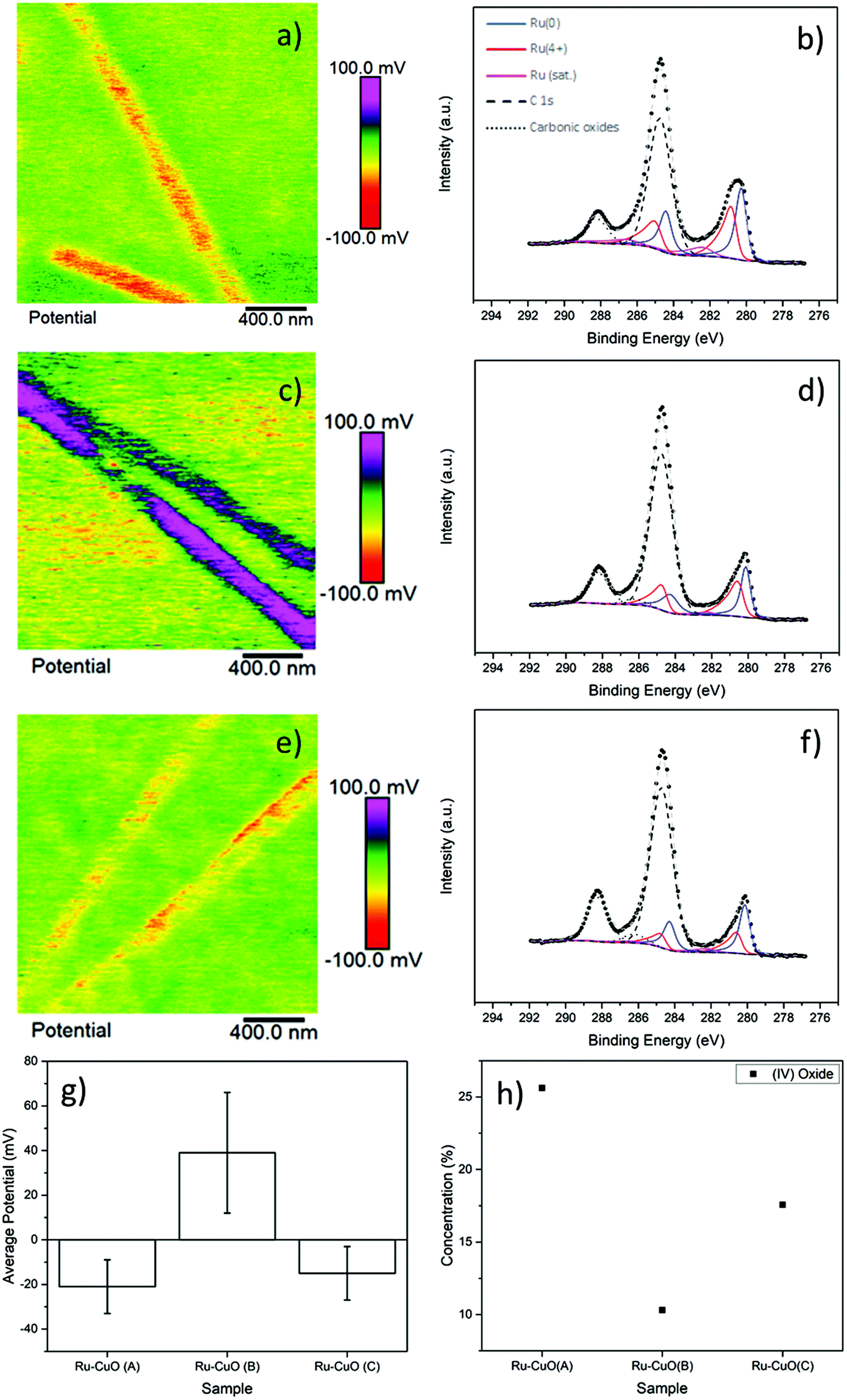 | ||
| Fig. 2 KPFM image and deconvolved high resolution XPS spectra of the Ru 3d and C 1s peaks of the as deposited samples, (a) and (b) Ru-CuO(A), (c) and (d) Ru-CuO(B) and (e) and (f) Ru-CuO(C) samples. The mean surface potential (with error bars showing standard deviation of 1) from each sample can be seen in (g), while (h) shows the concentration of RuO2 for the same samples. | ||
The samples with the most multiple twin NPs (Ru-CuO(B)) showed the greatest change in potential, while the sample with more hcp NPs (Ru-CuO(A)) had the lowest change, with Ru-CuO(C) (more twins than Ru-CuO(A) but less agglomerates than Ru-CuO(B)) falling in the middle, as shown in Fig. 2g (and Fig. S8d†). To normalise for the difference in the coverage fraction between Ru-CuO(A) and Ru-CuO(B) (Fig. S4b†), a sample with similar parameters to Ru-CuO(C) was deposited for 30 min instead of 20 min, showing no increase in potential, (in fact slightly worse, Fig. S9†). Previous DFT studies showed that depositing Ru NPs on CuO surfaces resulted in the transfer of electrons from Ru to CuO, resulting in the polarization of the CuO surface.27 They also showed that multiple twin Ru NPs deposited on the same CuO surface will result in the polarization of the Ru NPs as well, due to the defective twin boundaries. Therefore, in this case it is concluded that this polarization in the multiple twinned NPs (in Ru-CuO(B)) increases the charge transfer between the NP and support. On the surface, the XPS data (Fig. 2b, d and f) appeared contradictory. After deconvoluting the Ru 3d doublet–C 1s XPS region (using fitting parameters and peaks from the literature as described in the Experimental section), it was possible to determine the amount of metallic Ru (Ru0) and charged Ru (Ru4+ and RuO2 satellite) species. A full summary is shown in Fig. S10a and b,† showing that the least amount of Ru0 existed in the Ru-CuO(B) sample (with Ru-CuO(C) > Ru-CuO(A)). This would normally indicate that Ru-CuO(B) was the most oxidized, although in this case there should be a movement of electric charge away from the NP towards the NW. However, this is before the consideration of the RuO2 satellite peak. The reason for the existence of this peak is not well understood in the literature.39–41 What has been shown, is that the peak has an area of ∼50% of Ru4+ in pure RuO232 and is generally undisturbed by other molecules.41 Therefore, this peak is of great importance in determining how much of the charged Ru4+ species is due to oxide formation.
Therefore, concentration of Ru4+ species which are not associated to RuO2 can be determined by the difference in area of the Ru4+ peak, and twice the area of the RuO2 satellite (Fig. S10c†).7 In this case, the Ru4+ in Ru-CuO(B) is then due to the charge transfer phenomena observed previously via DFT.27 The comparison of Fig. 2g and h highlights how the formation of oxide either impedes or indicates the lack of charge transfer, as the pattern of oxide concentration is inversely proportional to the observed charging behaviour (i.e. the trend of oxide concentration follows the order Ru-CuO(B) < Ru-CuO(C) < Ru-CuO(A), while the trend of charge transfer follows the order Ru-CuO(B) > Ru-CuO(C) > Ru-CuO(A)). This is also supported by deconvolution of the O 1s peaks of the samples (Fig. S11a, b and c†). Three main peaks were observed corresponding to the CuO lattice oxide (∼529.6 eV),34 CuO defect oxide and Ru electron screening peak (∼531 eV)32,34 and carbonic oxides (∼533 eV).33 The peak at 531 eV is related to charge transfer in Pt group metals42 and forms the same pattern as that observed in the Ru spectrum (Fig. S11d†).
Metallic Ru NPs are notoriously unstable catalysts, usually requiring oxidation to RuO2 before use in water splitting.4,31 To investigate their stability, we annealed Ru NPs on CuO NW samples as described in the Experimental section and analysed them further by XPS and STEM. The results are shown in Fig. 3. The first thing is that no great change occurred in the Cu 2p region (Fig. S12†) of any of the samples, as expected, since the surface coverage was relatively low. Any changes due to the EMSI reduction of CuO would be extremely localized to the area underneath and very close to the NPs. On the other hand, the Ru 3d region had changed in unique ways for the different samples. There was no Ru0 present in the Ru-CuO(A) sample (Fig. 3a), while a significant reduction of Ru0 also occurred in the Ru-CuO(B) and Ru-CuO(C) samples (Fig. 3b and c, respectively). Moreover, the Ru4+ population in all the cases was now due to the formation of RuO2, given the increase in the RuO2 satellite (for a full summary of species concentration see Fig. S13†). This was further supported by the O 1s XPS region, in which all the CuO defect/Ru screening peaks decreased in value due to the increased oxidation of the particles (Fig. S14†).
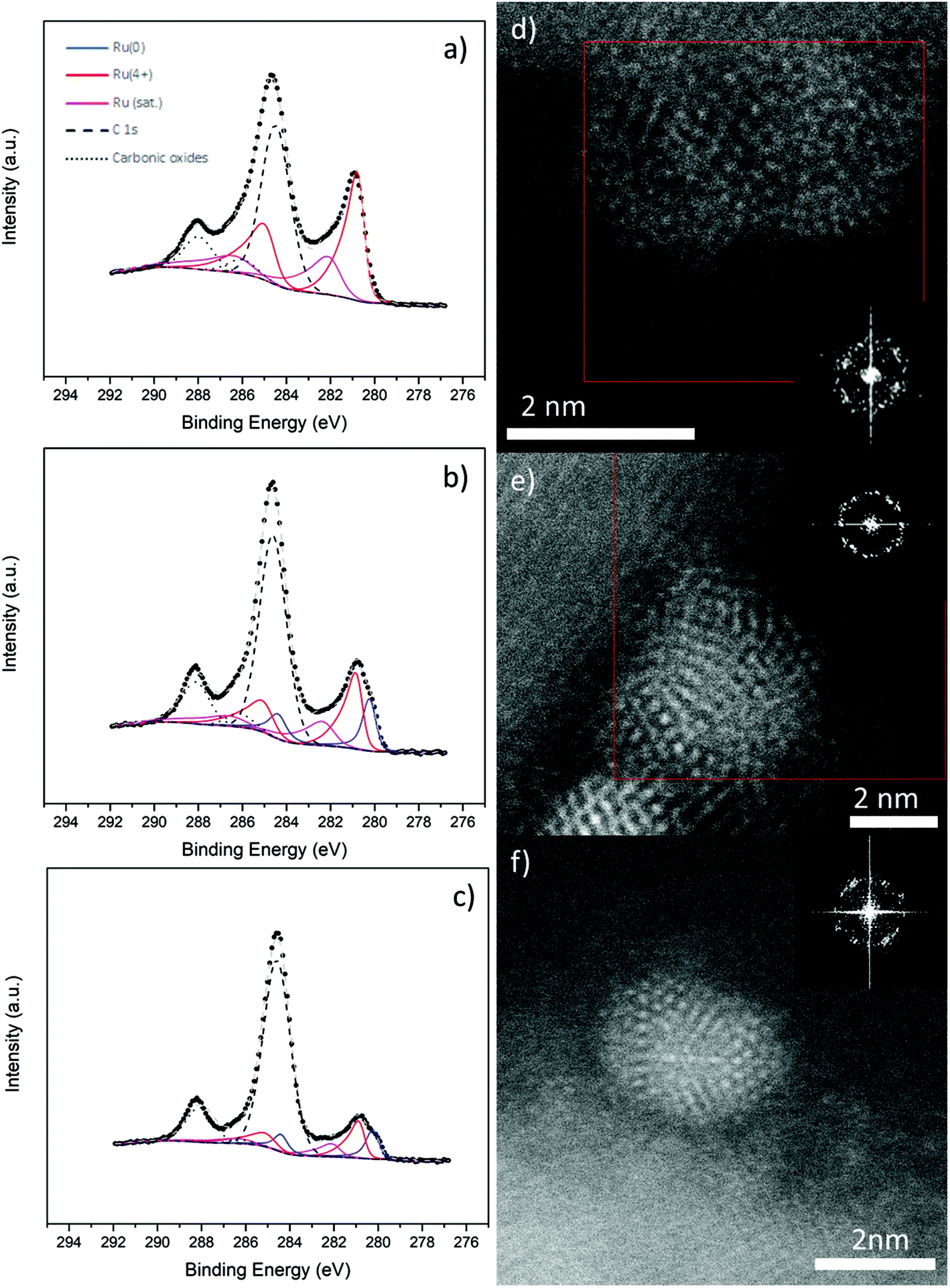 | ||
| Fig. 3 Deconvolved high resolution XPS spectra of the Ru 3d and C 1s peaks of the (a) Ru-CuO(A), (b) Ru-CuO(B) and (c) Ru-CuO(C) samples after annealing at 200 °C in dry air for 3 h. An oxidized Ru hcp NP (with RuO2 tetragonal planes apparent) can be seen in (d), while (e) and (f) show fcc-icosahedral Ru NPs near the 2-fold and 3-fold axes of symmetry, respectively. FFTs of the highlighted red areas (d and e) and full area (f) are the inset in each image. | ||
While XPS provides a global view of the surface, it is important to investigate the samples further by STEM to determine why there is still a considerable metallic character in the Ru-CuO(B) and Ru-CuO(C) samples. Fig. 3d, e and f show typical images of the Ru NPs on CuO NWs found after annealing (Experimental section). Many samples showing RuO2 planes were found, alongside apparent fcc-icosahedral Ru NPs, which still showed a metallic character. Further samples showed evidence of dislocations in metallic hcp planes, which either occurred due to oxidation or allowed oxidation to occur. An example of this is shown in Fig. S15,† where an NP showed significant dislocations across a plane from the hcp {101} family. Simultaneously, a plane with a d-spacing of 0.25 nm (indexed as a {101} plane from the RuO2 tetragonal structure)43 was present within the NP, demonstrating oxide formation.
In Fig. 3d, an image of an NP or a possible agglomerate is shown. In this case, there were two separate sets of RuO2 planes identified (these are highlighted in the enlarged FFT in Fig. S16†). One set was found on the left side of the particle while the other was found on the right side, showing that this was common across most particles. Indeed, this behaviour was observed in the previous studies of Ru NPs on CuO NWs.27 What is interesting, however, is that the NPs of multiple twins (Fig. 3e and f) showed no real signs of oxidation. An fcc-icosahedral Ru NP seen near its 2-fold axis of symmetry is shown in Fig. 3e. The d-spacings determined from the spatial frequencies on the FFT were typically ∼0.22 nm and 0.20 nm, consistent with {111} and {200} fcc-Ru, respectively. These d-spacings were of similar size to that of the as deposited sample seen in Fig. 1. This was far from an isolated phenomenon, as other fcc-icosahedral Ru NPs were found, such as the NP in Fig. 3f. This NP was observed near its 3-fold axis of symmetry and once again showed d-spacings of ∼0.22 nm and 0.2 nm. Clearly then, the higher metallic contents in the XPS measurements of Ru-CuO(B) and Ru-CuO(C) were due to the presence of these multiple twinned NPs, especially those with an fcc-icosahedral structure.
The effect of the tuned EMSI in electrocatalysis was studied by using all the Ru-CuO samples in the oxygen evolution reaction (OER) at neutral pH. The electrocatalytic performances of the materials are presented in Fig. 4. The linear sweep voltammograms (LSVs) in Fig. 4a show all the catalysts to be active towards OER at neutral pH. Ru-CuO(B) proved to be the most efficient of them all with an overpotential of 270 mV @ 10 mA cm−2 (similar to the reported Ru-based catalysts under comparable conditions).11,44 Ru-CuO(C) and Ru-CuO(A) exhibited much higher overpotentials of 410 and 670 mV@10 mA cm−2, respectively. As the overpotential is linked with the energy required to drive the reaction, the significantly lower overpotential of Ru-CuO(B) suggests it functions as a better catalyst (lowering the activation energy of the reaction) than Ru-CuO(A) and Ru-CuO(C). At the same time, Ru-CuO(C) has a lower overpotential than Ru-CuO(A). As the population of multiple twin NPs follows the order Ru-CuO(B) > Ru-CuO(C) > Ru-CuO(A), it follows that the remarkable activity of Ru-CuO(B) can be attributed to the presence of an increased number of multiple twin NPs and to the subsequent significant increase in the EMSI at the nanoparticle–support interface.9,10 Due to the sluggish nature of OER kinetics,45 all the catalysts showed high values of Tafel slopes (Fig. 4b). However, Ru-CuO(B) showed the lowest value of the Tafel slope, i.e. 336 mV dec−1, indicating faster splitting of H2O molecules,10,11 while Ru-CuO(C) and Ru-CuO(A) showed Tafel slopes of 450 and 530 mV dec−1, respectively. Once again, the order (Ru-CuO(B) > Ru-CuO(C) > Ru-CuO(A)) supports the idea that the multiple twinned NPs enhanced the catalytic activity of the system.
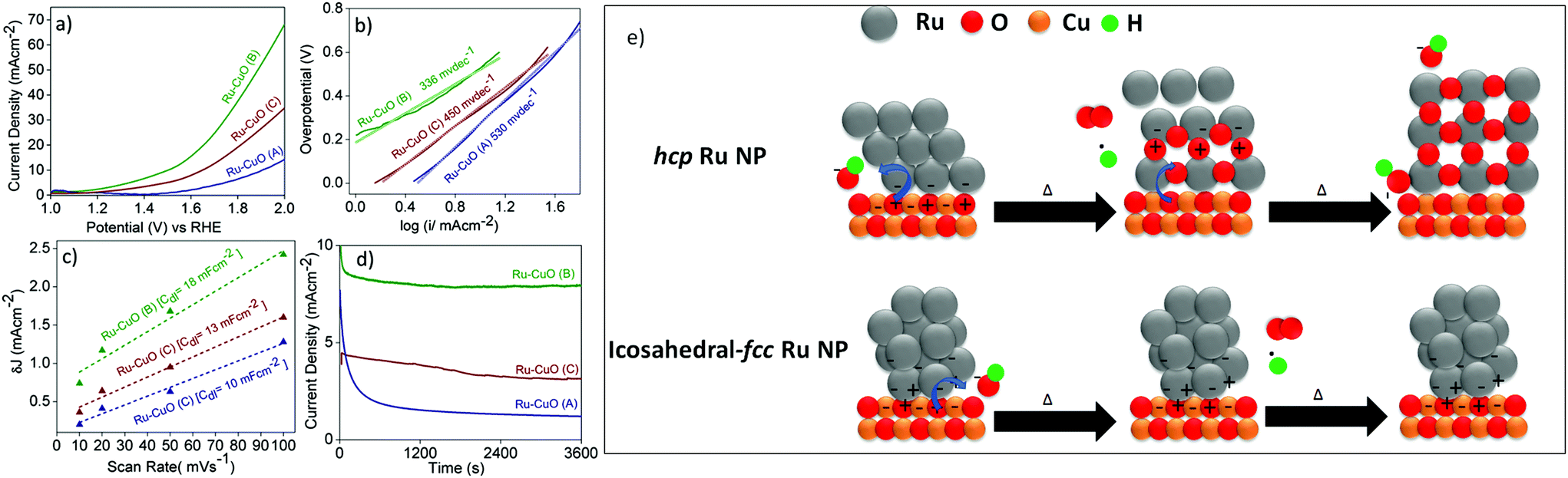 | ||
| Fig. 4 (a) LSV polarization curves, (b) Tafel plots, (c) Cdl measurements and (d) chronoamperometric stability curves for the OER activities of the Ru-CuO catalysts at neutral pH. A schematic representation of the evolution of a hcp (top line) and a fcc-icosahedral (bottom line) Ru NP on CuO NW supports can be seen in (e). Δ represents the energy either through heat or applied voltage bias. Accumulation of an electron is represented by a black dot (●). Sphere colours represent different species: Ru = grey, Cu = copper, O = red and H = green. | ||
The electrochemically active surface area (ECSA), a measure of catalytically actives sites,10 of each catalyst was found by obtaining the double layer capacitance (Cdl) values (Fig. 4c), since Cdl is linearly related to ECSA.46 The Cdl values were calculated from the slopes of straight-line fits of the differences between anodic and cathodic current densities (δJ) versus scan rates from 10 to 100 mV s−1, at a potential of 0.10 V vs. reversible hydrogen electrode (RHE). The δJ values were calculated from CVs recorded in the non-Faradaic region i.e. 0–0.2 V vs. RHE (Fig. S17†). Ru-CuO(B) showed a Cdl of 18 mF cm−2, which is 1.8 and 1.4 times higher than those of Ru-CuO(A) (10 mF cm−2) and Ru-CuO(C) (13 mF cm−2), respectively. The enhanced Cdl value of Ru-CuO(B) indicates the presence of a higher number of catalytically active sites and the result of the electronic interaction between the Ru NPs and CuO NWs. As described earlier, DFT simulations show a greater charge transfer in fcc-icosahedral (multiple twin) NPs, leading to more surface interactions than samples with more single crystal hcp NPs.27 Since Ru-CuO(B) had more multiple twin NPs as discussed previously, it generated a higher number of catalytically active sites through increased charge transfer at the interface.16 This is also in accordance with the KPFM results discussed in the previous section. Ru-CuO(A), having the least number of multiple twin NPs, showed the minimum activity towards OER. The Ru-CuO(C) sample had more multiple twin NPs than Ru-CuO(A) and hence showed better activity. However, the population of Ru NPs in Ru-CuO(C) contained a significant number of agglomerated NPs, and this likely inhibited the charge transfer at the support because it redirected the charge to other NPs within the agglomerate,47 thereby making the catalyst less effective than Ru-CuO(B).
Further support was provided by electrochemical impedance spectroscopy (EIS). The interfacial charge transfer phenomenon in the electrocatalysts was determined through charge transfer resistance (RCT), i.e. the diameter of the semi-circular arc.11,44 The corresponding Nyquist plots are shown in Fig. S18.† The RCT values for Ru-CuO(A), Ru-CuO(B) and Ru-CuO(C) were calculated to be 144, 119 and 361 Ω, respectively, indicating an enhanced interfacial charge transfer kinetics for Ru-CuO(B) at neutral pH, and this again can be related to better EMSI in that sample due to the presence of a higher number of multiple twin NPs. The durability of the catalysts under the reaction conditions was examined using chronoamperometric stability tests, performed at a potential of 1.5 V vs. RHE (Fig. 4d) in all the cases. The same trend, i.e., Ru-CuO(B) > Ru-CuO(C) > Ru-CuO(A), was observed for the activity of the catalysts for a period of 1 h. Thus, it can be concluded that the tuning of EMSI and subsequent electrocatalytic activity is possible via increasing the population of multiple twin Ru NPs in Ru-CuO catalysts. It is also clear that there was some instability over the period. This was likely due to the mixed nature of each sample (i.e. each sample contained single crystals, multiple twins and agglomerated particles). As shown in Fig. 3, single crystal Ru is prone to oxidation, and (while more active in catalysis than RuO2) is known to oxidise during catalysis.23
However, there still remains the question of why multiple twin NPs show no apparent oxidation and better catalytic performance (further evidenced by the post catalytic XPS patterns shown in Fig. S19, 20 and 21,† and by the post catalytic STEM-HAADF image in Fig. S22†). Considering the previous literature on the nature of EMSI13,14,16 and the charge distribution difference between single crystal and multiple twin Ru NPs,27 together with the observations of this study, we propose that the polarization caused by the multiple defective twin boundaries27 allows for the better stabilisation of EMSI by preventing the oxidation of the NPs.
Previous studies highlighted that supported Pt group NPs on CuO supports were generally oxidized from the interface.27,48 The schematic in Fig. 4e is based on the DFT studies of single crystal and icosahedral Ru NPs on a {111} CuO surface.27 It can be seen that the interface of both structures is different with the hcp structure having a large collection of negative charge at the base of the NP (and an overall lack of charge throughout the rest of the NP), while the fcc-icosahedral NP exhibits areas of positive and negative charges at the interface due to the aforementioned polarization (this is also the cause of the increased Ru4+ concentration measured in XPS), with charges extending up into the NP. The interface and the surrounding area are the most active sites due to the accumulation of charge, with this area being where most of the oxygen is evolved.17 Considering the case of the hcp NP, the NP side of the interface is completely negative, while there is polarization on the CuO side. Oxygen radicals would be attracted to the positive areas of the support and then interact with the electrons on the NP. With no polarization within the NP, the oxygen radicals can strip the Ru of electrons, leaving positive holes that other oxygen radicals can react with. This forms a new metal/metal-oxide interface, allowing the process to continue until the NP is RuO2, with the occurrence of catalysis due to the individual activities of RuO2 and CuO. This would explain why single crystal Pt-group NPs have a tendency to oxidise from the interface up.27,48 On the other hand, the fcc-icosahedral NP has a polarized interface, with a balance of positive and negative charges, allowing the NP to continually replenish electrons from the support (effectively using the polarisation of the NP to reverse the flow of negative charge away from the NP) and preventing the binding of Ru and oxygen radicals. This would explain why after oxidation, samples containing multiple twin Ru NPs show a higher concentration of Ru(0) species and a subsequent reduction in the concentration of non-oxide Ru (4+) species.
Conclusions
In conclusion, EMSI tuning in Ru NPs on CuO NW supports was achieved by altering the crystal structure of Ru NPs produced by gas-phase synthesis. This tuning was based on increasing the number of multiple twinned NPs in the catalyst. The KFPM and XPS studies showed a much greater incidence of positive (non-oxide) charge on the Ru NPs. This charge was linked directly to the defective twin boundaries of the multiple twinned NPs and was postulated to prevent the oxidation due to polarizations within the NPs, leading to improved and higher stability catalytic activity for OER at neutral pH.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank the Okinawa Institute of Science and Technology Graduate University for funding and work performed at OIST. The authors thank the OIST Imaging Section for providing access to the JEOL JEM-ARM200F microscope. The authors are grateful for access to the fabrication and characterization facilities run and maintained by the OIST Mech. Eng. and Microfabrication Support Section, Onna, Okinawa, Japan.Notes and references
- Z. Song, T. Cai, J. C. Hanson, J. A. Rodriguez and J. Hrbek, J. Am. Chem. Soc., 2004, 126, 8576–8584 CrossRef CAS.
- X. Liu, M.-H. Liu, Y.-C. Luo, C.-Y. Mou, S. D. Lin, H. Cheng, J.-M. Chen, J.-F. Lee and T.-S. Lin, J. Am. Chem. Soc., 2012, 134, 10251–10258 CrossRef CAS.
- H. Ha, S. Yoon, K. An and H. Y. Kim, ACS Catal., 2018, 8, 11491–11501 CrossRef CAS.
- S. Chen, A. M. Abdel-Mageed, C. Gauckler, S. E. Olesen, I. Chorkendorff and R. J. Behm, J. Catal., 2019, 373, 103–115 CrossRef.
- J. Li, Z. Liu, D. A. Cullen, W. Hu, J. Huang, l. yao, Z. Peng, P. Liao and R. Wang, ACS Catal., 2019, 9, 11088–11103 CrossRef CAS.
- S. D. Tilley, M. Schreier, J. Azevedo, M. Stefik and M. Graetzel, Adv. Funct. Mater., 2014, 24, 303–311 CrossRef CAS.
- V. A. Saveleva, L. Wang, W. Luo, S. Zafeiratos, C. Ulhaq-Bouillet, A. S. Gago, K. A. Friedrich and E. R. Savinova, J. Phys. Chem. Lett., 2016, 7, 3240–3245 CrossRef CAS.
- C. Jackson, G. T. Smith, D. W. Inwood, A. S. Leach, P. S. Whalley, M. Callisti, T. Polcar, A. E. Russell, P. Levecque and D. Kramer, Nat. Commun., 2017, 8, 15802 CrossRef CAS.
- H. He, J. Chen, D. Zhang, F. Li, X. Chen, Y. Chen, L. Bian, Q. Wang, P. Duan, Z. Wen and X. Lv, ACS Catal., 2018, 8, 6617–6626 CrossRef CAS.
- Y. Huang, L. Hu, R. Liu, Y. Hu, T. Xiong, W. Qiu, M.-S. (Jie Tang) Balogun, A. Pan and Y. Tong, Appl. Catal., B, 2019, 251, 181–194 CrossRef CAS.
- Y. Hu, T. Xiong, M.-S. (Jie Tang) Balogun, Y. Huang, D. Adekoya, S. Zhang and Y. Tong, Mater. Today Phys., 2020, 15, 100267 CrossRef.
- W. C. C. Jr. and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef.
- Y. Lykhach, S. M. Kozlov, T. Skála, A. Tovt, V. Stetsovych, N. Tsud, F. Dvorák, V. Johánek, A. Neitzel, J. Myslivecek, S. Fabris, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2015, 15, 284–288 CrossRef.
- T. Binninger, T. J. Schmidt and D. Kramer, Phys. Rev. B, 2017, 96, 165405 CrossRef.
- S. Zhang, P. N. Plessow, J. J. Willis, S. Dai, M. Xu, G. W. Graham, M. Cargnello, F. Abild-Pedersen and X. Pan, Nano Lett., 2016, 16, 4528–4534 CrossRef CAS.
- Y. Peng, B. Lu, N. Wang, L. Li and S. Chen, Phys. Chem. Chem. Phys., 2017, 19, 9336–9348 RSC.
- B. Qiu, C. Wang, N. Zhang, L. Cai, Y. Xiong and Y. Chai, ACS Catal., 2019, 9, 6484–6490 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- M. Andersen, O. Lytken, J. Engbæk, G. Nielsen, N. Schumacher, M. Johansson and I. Chorkendorff, Catal. Today, 2005, 100, 191–197 CrossRef CAS.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS.
- S. H. Joo, J. Y. Park, J. R. Renzas, D. R. Butcher, W. Huang and G. A. Somorjai, Nano Lett., 2010, 10, 2709–2713 CrossRef CAS.
- E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, P. Malacrida, T. W. Hansen, I. Chorkendorff and I. E. L. Stephens, Catal. Today, 2016, 262, 57–64 CrossRef CAS.
- X. Wang, L. Figueroa-Cosme, X. Yang, M. Luo, J. Liu, Z. Xie and Y. Xia, Nano Lett., 2016, 16, 1467–1471 CrossRef CAS.
- P. Zhao, Z. Cao, X. Liu, P. Ren, D.-B. Cao, H. Xiang, H. Jiao, Y. Yang, Y.-W. Li and X.-D. Wen, ACS Catal., 2019, 9, 2768–2776 CrossRef CAS.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef.
- A. Porkovich, Z. Ziadi, P. Kumar, J. Kioseoglou, N. Jian, L. Weng, S. Steinhauer, J. Vernieres, P. Grammatikopoulos and M. Sowwan, ACS Nano, 2019, 13, 12425–12437 CrossRef CAS.
- F. M. Pinto, V. Y. Suzuki, R. C. Silva and F. A. L. Porta, Front. Mater., 2019, 6, 260 CrossRef.
- X. Liu, S. Cui, Z. Sun, Y. Ren, X. Zhang and P. Du, J. Phys. Chem. C, 2015, 120, 831–840 CrossRef.
- S. R. Plant, L. Cao and R. E. Palmer, J. Am. Chem. Soc., 2014, 136, 7559–7562 CrossRef CAS.
- E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, C. Schlaup, M. Malizia, T. W. Hansen, S. Horch, I. E. L. Stephens and I. Chorkendorff, Chem. Sci., 2015, 6, 190–196 RSC.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, ULVAC-PHI, Chigasaki, Japan, 1995 Search PubMed.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- G. Greczynski and L. Hultman, ChemPhysChem, 2017, 18, 1507–1512 CrossRef CAS.
- R. M. Nielsen, S. Murphy, C. Strebel, M. Johansson, I. Chorkendorff and J. H. Nielsen, J. Nanopart. Res., 2010, 12, 1249–1262 CrossRef CAS.
- C. Song, O. Sakata, L. S. R. Kumara, S. Kohara, A. Yang, K. Kusada, H. Kobayashi and H. Kitagawa, Sci. Rep., 2016, 6, 31400 CrossRef CAS.
- M. José-Yacamán, C. Gutierrez-Wing, M. Miki, D.-Q. Yang, K. N. Piyakis and E. Sacher, J. Phys. Chem. B, 2005, 109, 9703–9711 CrossRef.
- K. S. Kim and N. Winograd, J. Catal., 1974, 35, 66–72 CrossRef CAS.
- H. Y. H. Chan, C. G. Takoudis and M. J. Weavery, J. Catal., 1997, 172, 336–345 CrossRef CAS.
- H. Over, A. P. Seitsonen, E. Lundgren, M. Smedh and J. N. Andersen, Surf. Sci., 2002, 504, L196–L200 CrossRef CAS.
- W. D. Ryden, A. W. Lawson and C. C. Sartain, Phys. Rev. B: Solid State, 1970, 1, 1494–1500 CrossRef.
- C.-E. Boman, Acta Chem. Scand., 1970, 24, 116–122 CrossRef CAS.
- H.-S. Park, J. Yang, M. K. Cho, Y. Lee, S. Cho, S.-D. Yim, B.-S. Kim, J. H. Jang and H.-K. Song, Nano Energy, 2019, 55, 49–58 CrossRef CAS.
- A. Datta, A. J. Porkovich, P. Kumar, G. Nikoulis, J. Kioseoglou, T. Sasaki, S. Steinhauer, P. Grammatikopoulos and M. Sowwan, J. Phys. Chem. C, 2019, 123, 26124–26135 CrossRef CAS.
- A. Sahasrabudhe, H. Dixit, R. Majee and S. Bhattacharyya, Nat. Commun., 2018, 9, 2014 CrossRef.
- Y. Park, W. Kim, D. Monllor-Satoca, T. Tachikawa, T. Majima and W. Choi, J. Phys. Chem. Lett., 2013, 4, 189–194 CrossRef CAS.
- S. Steinhauer, J. Zhao, V. Singh, T. Pavloudis, J. Kioseoglou, K. Nordlund, F. Djurabekova, P. Grammatikopoulos and M. Sowwan, Chem. Mater., 2017, 29, 6153–6160 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr06685k |
| This journal is © The Royal Society of Chemistry 2021 |