
Energy band alignment at the heterointerface between a nanostructured TiO2 layer and Au22(SG)18 clusters: relevance to metal-cluster-sensitized solar cells†
Liudmila L.
Larina‡
ab,
Oleksii
Omelianovych‡
a,
Van-Duong
Dao
 a,
Kyunglim
Pyo
c,
Dongil
Lee
c and
Ho-Suk
Choi
*a
a,
Kyunglim
Pyo
c,
Dongil
Lee
c and
Ho-Suk
Choi
*a
aDepartment of Chemical Engineering and Applied Chemistry, Chungnam National University, Daejeon, 34134, Republic of Korea. E-mail: hchoi@cnu.ac.kr
bDepartment of Solar Photovoltaics, Institute of Biochemical Physics, Russian Academy of Sciences, Kosygin St. 4, 119334, Moscow, Russia
cDepartment of Chemistry, Yonsei University, Seoul 03722, Republic of Korea
First published on 27th November 2020
Abstract
This study is the first to quantify energy band alignments at a nanostructured TiO2/Au22(SG)18 cluster interface using X-ray photoelectron spectroscopy. The d-band of Au clusters shows band-like character and occupied states at the Fermi level are not detected. The results provide evidence of the existence of a finite optical energy gap in Au22(SG)18 clusters and the molecular-like nature of these clusters. The pinning position of the Fermi energy level at the interface was determined to be 2.8 and 1.3 eV higher than the top of the TiO2 valence band and the highest occupied molecular orbit level of the Au clusters, respectively. A diffuse reflectance and absorption analysis quantified a 3.2 eV bandgap of the TiO2 layer and a 2.2 eV energy gap between the highest occupied molecular orbit (HOMO) and the lowest unoccupied molecular orbit (LUMO) levels of the Au clusters. Thus, a cliff-like offset of 0.5 eV between the LUMO level and the TiO2 conduction band was determined. The cliff-like offset of 0.5 eV provides room for improving the efficiency of metal-cluster-sensitized solar cells (MCSSC) further by lowering the LUMO level through a change in the cluster size. The offset of 0.5 eV between the HOMO level and the 3I−/I−3 redox level yields a remarkable loss-in-potential, which implies the possibility of increasing the open-circuit voltage further by properly replacing the redox couple in the MCSSCs.
Introduction
To date, the technology for dye-sensitized solar cells (DSCs) is coming into the spotlight as a foundation on which to devise next-generation solar cells. Recently, the use of ligand-stabilized metallic clusters as alternative sensitizers has attracted much attention due to their photoinduced electron-transfer capabilities. Studies in this field, in particular, have focused on the establishment of versatile and reproducible experimental methods to synthesize a series of thiolated Au clusters with well-defined chemical compositions.1 It was proven that sub-nanometer-sized glutathione-protected Au clusters constitute a distinct class of binary system between Au(I)-thiolate complexes and thiolate-protected Au nanocrystals. The decrease in the size of the metal core down to the nanometer scale yields a transition from a metallic to a molecular-like nature with a finite optical energy gap.2–4 Such a change in the electronic structure is related to a quantum size effect.5,6 The existence of the molecule-like electronic states and the correlation between the cluster structure and the corresponding optical absorption spectrum were predicted by time-dependent density functional theory (TDDFT).7,8 This prediction was proven by redox potential and optical absorbance band-edge measurements.6,9,10 Ultraviolet photoelectron spectroscopy (UPS) was applied to probe the size-dependent electronic properties of Cu, Ag, and Au over a wide cluster size distribution.8 The evolution of the d band from the bulk metal to a molecular-like cluster was recorded.11Varnavski et al. reported a systematic investigation of optically excited vibrations in monolayer-protected gold clusters capped with hexane thiolate.9 The appearance of the oscillations from small clusters is ascribed to the emergence of an optical energy gap near the Fermi level. This proves the existence of a finite optical energy gap in the quantum-sized nano-clusters. It is also important to note that the structured optical absorption spectra and related electronic properties are not unique to gold. Dihydrolipoic-acid-stabilized and thiolate-capped silver clusters also exhibit photo-induced electron-transfer properties.10
The application of metal clusters as an alternative sensitizer in DSCs, also known as metal-cluster-sensitized solar cells (MCSSCs), yielded a record conversion efficiency of 3.8% for Au18(SR)14 (SR = glutathione) clusters.11 A pioneering study of the subject was conducted in the Kamat group, resulting in an efficiency of 2.36% for cells which employed Aux–SH metal clusters.12 The recorded open-circuit voltage (Voc) of 832 mV was among the highest values observed for liquid junction solar cells. However, the obtained efficiency is noticeably lower than the efficiency of state-of-the-art DSC. One possible reason for the limited cell efficiency is an unfavorable electronic band structure at the interface between the TiO2 and ligand-stabilized metal clusters. The energy barrier at the interface strongly affects interfacial recombination as well as the charge transport across the junction. Appropriate alignment of the TiO2 conduction band (CB) and the LUMO of the metal clusters at the interface is of primary importance for the achievement of high efficiency. In this regard, ligand-stabilized metal clusters have a significant advantage, i.e., good tunability of the HOMO–LUMO gap along with the alteration of the offset between the TiO2 valence band maximum (VBM) and HOMO level of Au clusters. These parameters of the interface electronic structure can be tuned by proper engineering of the cluster chemical composition. The potential to alter the value of the HOMO–LUMO gap and the interface band alignment with a change of the core structure as well as the ligand configuration13–15 introduces numerous opportunities to design and build a suitable electronic structure of the interface for efficient charge transfer. For this purpose, the substituent effects on the HOMO and LUMO energies as well as doping with metal atoms such as Pd, Ag, and Cd both provide viable options.16 Thus, tailoring of the electronic band structure at the interface between the nanostructured TiO2 layer and ligand-stabilized metal clusters can be exploited to increase the efficiency of MCSSCs.
Despite the abundance of studies that have attempted to gain a fundamental understanding of ligand-stabilized metal clusters and their applications, experimental investigations of electronic structures for solar cell applications are scarce. Several studies have reported the valence band (VB) electronic structure of Au NPs supported on TiO2 using UPS17,18 and hard X-ray photoelectron spectroscopy (XPS).19 However, researchers have addressed the relationship between the electronic structures of ligand-free Au NPs and the quantum size effect on the catalysis process. Taylor et al. examined the evolution of the VB structure of coinage Cu, Ag, and Au clusters in a wide range of sizes using UPS.8 However, their study was devoted to the VB structures of coinage metal clusters and their transition from metallic to molecular-like behavior.
Andersson and co-workers have dedicated numerous efforts on the investigation of the size-related changes in the electronic structure of phenylphosphine (and its derivatives) stabilized Au-clusters as well as their interaction with variations of titania substrates.17,18,20,21 It was suggested that gold-clusters readily agglomerate upon various activation steps, such as washing, calcination, etc.20,21 However, cluster properties vary significantly depending on the protecting ligand,22 pointing out the need for a detailed investigation of each particular cluster depending on the ligand type.
To the best of our knowledge, the electronic structure of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SG clusters have already been briefly studied by XPS.1,23 It is also important to note that the VB was not analyzed despite the fact that doing so is critical for cluster applications in interface-based devices such as MCSSCs. The initial step of the engineering of an interface electronic structure requires precise knowledge of the energy band alignments and corresponding parameters.
:SG clusters have already been briefly studied by XPS.1,23 It is also important to note that the VB was not analyzed despite the fact that doing so is critical for cluster applications in interface-based devices such as MCSSCs. The initial step of the engineering of an interface electronic structure requires precise knowledge of the energy band alignments and corresponding parameters.
In this study, we undertook the quantification of the alignment of HOMO and LUMO states of Au22(SG)18 clusters and the energy levels of TiO2 at the TiO2/Au22(SG)18 cluster interface via an XPS analysis. The obtained results provide evidence of the molecular-like behavior of Au22 clusters protected by glutathione ligands. The scheme of the energy band alignment at the interface of the TiO2/Au22(SG)18 cluster was constructed based on experimental XPS, UV–Vis absorption, and diffuse reflectance data. The scheme provides useful insight for those involved in the construction of efficient MCSSC devices and is essential for further efficiency improvements. It can also be used for the engineering of novel organometallic clusters for MCSSCs.
Experimental section
Materials
Gold(III) chloride trihydrate (HAuCl4·3H2O, reagent grade), reduced L-glutathione (GSH, ≥98%), and sodium borohydride (NaBH4, 99%) were purchased from Sigma-Aldrich. Sodium hydroxide (NaOH, 98%), hydrochloric acid (HCl, 35–37%), and isopropyl alcohol (IPA, 99%) were purchased from Burdick and Jackson. Water was purified using a Millipore Milli-Q system (18.2 MΩ cm). TiO2 paste for the synthesis of mesoporous TiO2 film (Ti-nanoxide, T/SP) was purchased from Solaronix (Switzerland). All chemicals were used as received without further purification.Sample preparation
To fabricate the mesoporous TiO2 layer, TiO2 paste (T/SP, size: 15–20 nm, 100% anatase) was deposited onto the FTO substrate using a screen-printing method (1 cycle). Then the sample was transferred to the furnace for removing organic paste components and forming the nanostructured TiO2 film. The sample was sintered gradually at 325 °C for 5 min, at 375 °C for 5 min, at 450 °C for 15 min, and at 500 °C for 15 min. This is a widely used method for manufacturing mesoporous TiO2 layer for dye-sensitized solar cell.24,25 The heterointerface of the TiO2/Au22(SG)18 clusters was fabricated by drop-casting an 8 μL DI water dispersion of Au22(SG)18 clusters (5 mg mL−1) onto a nanostructured TiO2 layer deposited on an FTO substrate. Finally, the sample was dried in a nitrogen atmosphere.Characterization
The experimental Eg value of the nanostructured TiO2 was determined from the UV–Vis diffuse- reflectance spectrum of the nanostructured TiO2 by applying the Kubelka–Munk method. A UV–Vis spectrophotometer (Varian Cary 300) with an integrating sphere attachment (DRA-CA-30I) for diffuse reflectance measurements was used to establish the optical bandgap. The equipment was calibrated with a Spectralon standard (Labsphere SRS-99-010, 99% reflectance). The diffuse reflectance spectra were measured in the 200–800 nm range.The UV–Vis absorption spectrum of Au22(SG)18 were recorded using a Shimadzu UV–Vis–NIR spectrophotometer (UV-3600). The chemical composition of isolated Au22(SG)18 clusters was identified using electrospray ionization (ESI) mass spectrometry (6230B Accurate Mass TOF LC/MS System, Agilent Technologies) in negative-ion mode (flow rate, 3.0 μL min−1; capillary voltage, 4.0 kV; capillary temperature, 200 °C; m/z range, 1000–20000). The cluster samples were prepared in 0.1 M triethylammonium acetate dissolved in a water and methanol mixture (50:50) at a concentration of 1 mg mL−1 that was directly injected into the mass spectrometer.
The XPS spectra were taken using a Sigma Probe Thermo Fisher VG Scientific spectrometer equipped with a monochromatic Al Kα X-ray source under a base pressure of approximately 10−10 Torr. The samples were held with Cu clips which were grounded onto the sample stage. The binding energy scale was calibrated using the binding energy positions of the Au 4f7/2 core level located at 84.0 eV for a sputtered gold sample stored inside an ultra-high vacuum chamber. This sample is referred to as “Au crystalline film” in this manuscript. The information depth of XPS is less than 5 nm.
The sputter etching was carried out using an EX05 argon ion gun. To prevent the samples from being damaged and to provide gentle etching, the acceleration voltage and ion current were set to 1 kV and 1 μA, respectively.26 The energy of the VBM positions of nanostructured TiO2 and the HOMO peak cutoff position of the ligand-stabilized Au clusters were determined from the emission spectra by linear extrapolation of the leading edge of the valence band emission to the baseline. The core-level binding energy positions were defined as the center of the full width at half maximum (FWHM) of the photoemission peak using the Avantage software package. Detail of the measuring conditions can be found in the literature.27
Fitting deconvolution analysis
Basic constraints for the XPS fitting analysis were applied during the fitting of the raw XPS data. Fitted peaks represent Gaussian–Lorentzian curves, which are commonly used for the fitting of the XPS data. For the fitting analysis, Shirley's background was chosen with the additional constraint that the background should not be of greater intensity than the actual data at any point in the region. The fitting of Au 4f core-level was based on the existence of the surface and bulk states in the gold films,28 nanoparticles29 and, clusters.30–32 FWHM values were kept unchanged ±0.2 eV (related to the accuracy of XPS measurements) for every oxidation state, where applicable. The noticeable broadening of the FWHM could be observed for the gold clusters as compared to the Au-crystallite. XPS line broadening with a decrease of the particle size was widely studied for gold films and nanoparticles and available in the literature.33 For the Ti 2p fitting, the area of the peaks representing Ti oxidation states in Ti 2p3/2 and Ti 2p1/2 were kept close to the ratio of 1:2.
Results and discussion
Synthesis and chemical identification of Au22(SG)18 clusters
The Au22(SG)18 was prepared by previously reported NaBH4 reduction method.34 In brief, an aqueous solution (230 mL) containing 20 mM HAuCl4 (12.5 mL) and 50 mM glutathione (7.5 mL) was prepared and then adjusted to pH 12.0 using 1 mol L−1 NaOH solution; 3.5 mM NaBH4 (0.1 mL) was then added dropwise to this solution for initiating the reaction. The reaction proceeded under vigorous stirring for 30 min and was then quenched by adjusting the pH to 2.5. Then, the solution was stirred under 150 rpm for 6 h. The selective separation of Au22(SG)18 from the initial product was done using a rotary evaporator to remove water and prepare a nearly dry product. The dried product was then dispersed in 10 mL of deionized water, after which 12 mL of isopropyl alcohol was added to induce precipitation. The precipitate was separated by means of centrifugation at 6000 rpm.The synthesized gold clusters were characterized by ESI mass spectrometry. The negative-mode ESI spectra shown in Fig. 1 firmly establish that the synthesized gold clusters are highly monodisperse and that their chemical composition is consistent with Au22(SG)18. The observed peaks in the range of m/z = 2455–2470 Da correspond to Au22(SG)18 ions containing different numbers of H+ and Na+ ions. The experimental isotope pattern for the most intense peak at m/z = 2460–2461 Da matches the simulated peak of [Au22(SG)18-4H]4− well, as shown in the inset of Fig. 1a. The absorption of Au22(SG)18 extended from the ultraviolet to the visible region, up to 580 nm. The absorption shoulder is observed at 500–520 nm. A HOMO–LUMO energy gap of around 2.17 eV was determined from the absorption onset. The obtained value is in reasonably good agreement with the HOMO–LUMO gap of ∼1.89 eV as calculated based on TDDFT.5 Previously Xie's group have reported a study dedicated to the determination of the unambiguous formula of the red-emitting Au22(SG)18.35 In the aforementioned work, the structure of the gold clusters under study theoretically calculated by the means of DFT.
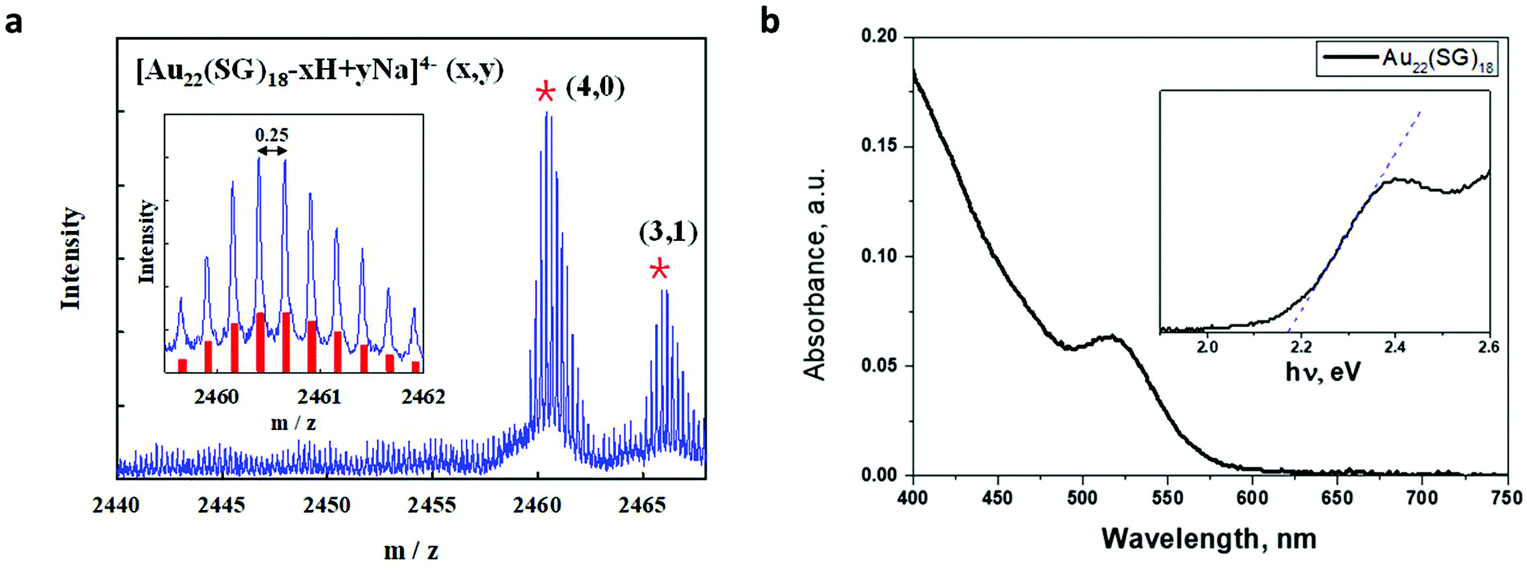 | ||
| Fig. 1 (a) ESI mass spectrum of Au22SG18. The inset shows a comparison between the experimental data (blue line) and the calculated isotope pattern (red bars) of [Au22(SG)18-4H]4−; (b) UV–Vis absorption spectrum of Au22(SG)18 clusters in water. The absorption edge is shown in inset on an expanded scale in the energy range of 2.0–2.6 eV. | ||
Sensibilization of the nanostructured TiO2 and surface morphology
In the current study, we provide an improved TiO2 sensibilization method. In the previous works, the sensibilization is done by dipping the TiO2 electrode into the nanocluster solution. Although it is a proven strategy to produce efficient photoanodes, it involves a tremendous waste of the nanoclusters. The possible alternative way for sensibilization is to do it by the means of drop-casting. As shown in Fig. 2a, by introducing 8 μL we achieved visible changes in the color of the TiO2 layer, after sensibilization it becomes light red. Seeking to probe the surface morphology of the sensitized films, TEM measurements were conducted. High-resolution TEM images are shown in Fig. 2b and c. It is easily seen that gold nanoclusters of sizes around 1 nm are widely distributed around TiO2 nanoparticles of sizes ∼25 nm. Gold nanocluster size is in good agreement with previously reported data for Au22(SG)18.36 Such high density and wide uniform distribution of the Au22(SG)18 indicates the success of the sensibilization method and therefore the establishment of the proper interface between gold nanoclusters and nanostructured TiO2. | ||
| Fig. 2 Sensibilization of nanostructured TiO2. (a) Photographs of the sensibilization process; (b and c) high resolution TEM images of Au22(SG)18 on the nanostructured anatase TiO2. | ||
Electronic structure of Au22(SG)18 clusters
To investigate the electronic structure of the Au22(SG)18 cluster supported on the TiO2 substrate, the core-level emissions of related compounds were monitored. The outcomes of a detailed analysis along with the core-emission spectra of the TiO2 substrate are provided in the ESI (Fig. S1 and Table S1†). Changes in the electronic structure of the Au clusters compared to the sputtered Au films were contrasted with the outcomes of a detailed comparison of the Au 4f spectra.To clarify the modifications of the electronic structure induced by the formation of the Au22(SG)18 cluster, the Au major XPS line, the Au 4f core-level spectrum, from the Au crystallite film was recorded. The Au 4f spectra of Au film and clusters are shown in Fig. 3a. The spectra of both samples have well-separated spin–orbit components. As expected, spin–orbital splitting accounted for 3.7 eV.37 Nevertheless, there is a significant difference between the shapes of the signal peaks. This implies that there is a notable difference in the electronic structure of the Au. As indicated in Fig. 3a, the modifications of the Au electronic structure induced by the formation of the heteronuclear metal–SG ligand bonds affect the Au 4f peak parameters and lead to the broadening of the Au 4f peaks. Next, to identify the difference in the chemical state of the Au in the Au22(SG)18 cluster and the Au crystallite film, peak fitting, and deconvolution of the Au 4f7/2 core-level emissions at spectrum intervals of 82 to 86 eV were conducted for both samples. The fitting results are presented in Fig. 3b and Table 1. The Au 4f7/2 core-level spectrum of the Au crystallite film is decomposed into two components. The peak centered at 84.0 eV originates from the Au atoms in the bulk, and the photoemission from the top surface of the Au atoms is located at 83.7 eV. The narrowing of the valence band of the less coordinated surface atoms resulted in a surface core-level shift of approximately 0.3 eV to lower binding energy. The deconvolution results are in agreement with well-established data for Au crystallite.28
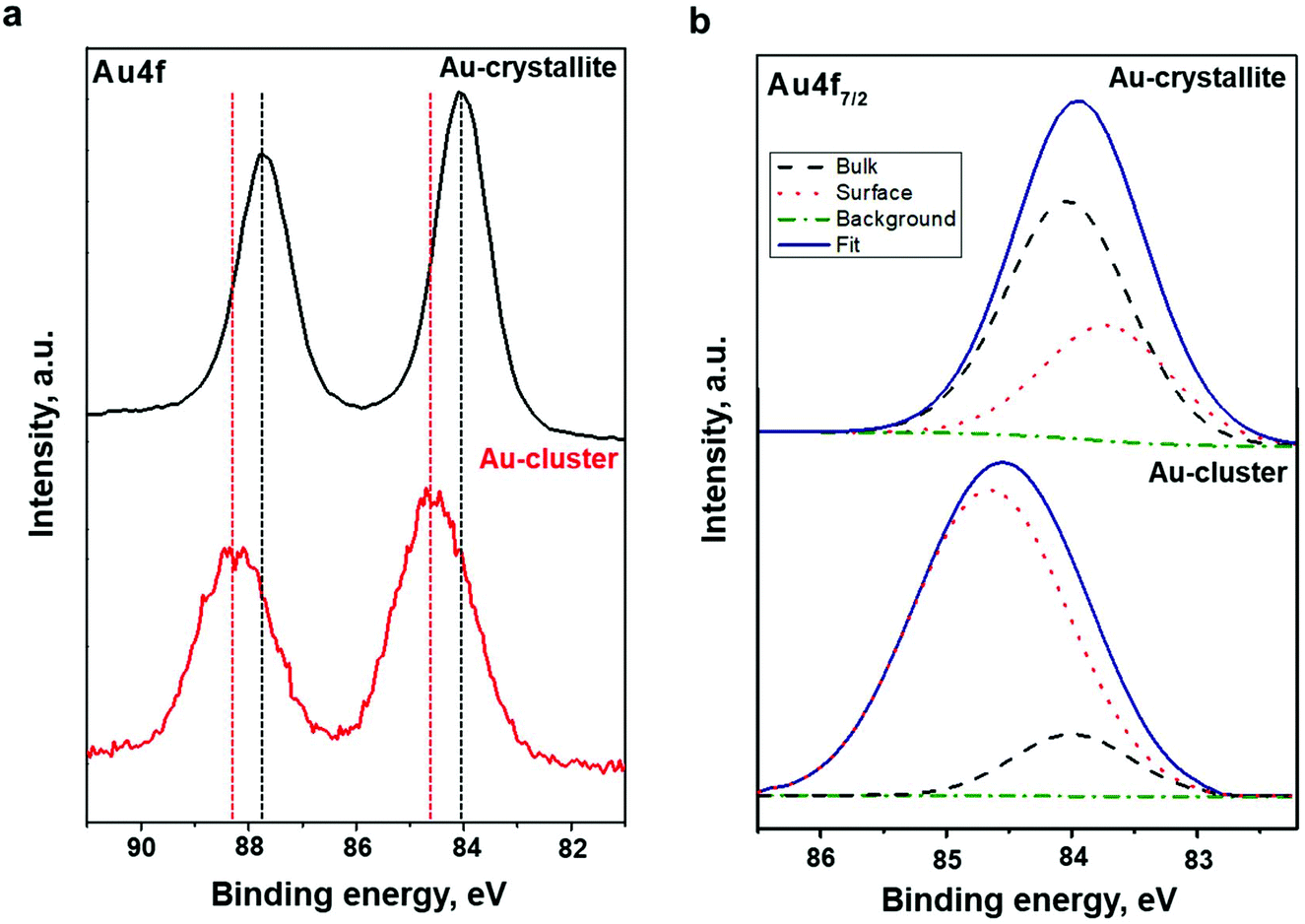 | ||
| Fig. 3 (a) Au 4f7/2 core-level photoemission spectra were taken of Au crystalline film and Au22(SG)18 clusters supported on TiO2. (b) Peak fitting and deconvolution of the Au 4f7/2 core-level emissions for the Au polycrystalline film and Au22(SG)18 clusters supported on TiO2. The binding energy scale is referenced to the Fermi energy level. | ||
| Sample | Compound | Binding energy, eV | At% | FWHM, eV |
|---|---|---|---|---|
| Au-Crystallite | Surface | 83.7 | 33.4 | 0.9 |
| Bulk | 84.0 | 66.6 | 0.9 | |
| Au-Cluster | Surface | 84.6 | 84.3 | 1.7 |
| Bulk | 84.0 | 15.7 | 1.5 | |
As shown in Fig. 3b, the Au 4f7/2 spectrum taken from the Au22(SG)18 clusters were decomposed also into two components, indicating that gold exists in the clusters in the two chemical states. The peak at the binding energy of 84.0 eV can be considered as the standard for the Au atoms in the bulk and can be attributed to the core Au atoms of the Au:SG clusters. The peak at a binding energy of 84.6 eV can be assigned to Au atoms bonded to SG ligands. A similar trend in the Au 4f core-level spectrum was observed for dodecanethiolate-passivated Au nanoparticles supported on highly oriented pyrolytic graphite substrates.29,38 It should be noted that the upward energy shift found by Tanaka et al. for the surface compound was inversely proportional to the size of the Au nanoparticles.29 In our study, the observed shift of the surface compound to higher binding energy relative to the bulk can be explained by the decreased kinetic energy of photo-emitted electrons due to the electron transfer from the surface Au atoms to the SG ligands. The crystal structure of Au22(SG)18 clusters has yet to be revealed. However, TDDFT calculations5 predict a structure of Au22(SG)18 consisting of a bitetrahedron Au7 kernel (core) surrounded by a [Au6(SR)6] ring complex and protected by three [Au3(SR)4] motifs. From the superatom complex model, the Au7 kernel can be considered as Au73+ due to its weak interaction with the [Au6(SR)6] ring.5 Hence, the deconvolution results for the Au 4f7/2 core-level emissions are in line with the theoretically predicted Au22(SG)18 structure. We found that the ratio of the surface compound to the bulk is very high; 84.3% Au exists as a surface compound while only 15.7% is a bulk compound (Table 1).
Aside from core-metallic levels, essential information about the electronic structure of the Au22(SG)18 clusters can be extracted from the valence band spectrum. Fig. 4a shows the valence band spectra from the Au22(SG)18 cluster in the binding energy range of 0–8 eV. As can be seen in this figure, the 5d electrons form a broad band between 2 and 8 eV, and photoemission within the binding energy range of 1–2 eV can be attributed to the 6s electrons. The 6s band is hybridized with the 5d band.39 The d-band drops to zero at around 1.3 eV, below EF, as indicated in Fig. 4a. For comparison, the valence band spectrum from the Au crystalline film is given in Fig. 4b. The spectrum shows the standard gold valence band (d states re-hybridized with s/p states) with occupied states at the Fermi level. The midpoint of the steep slope at the Fermi level is shown on a large scale in the inset of Fig. 4b. A metallic Fermi edge is visible. However, the spectrum recorded for the Au22(SG)18 cluster film (Fig. 4a) did not show the usual metallic Fermi edge observed for Au crystallite. The d-band shows a band-like character and occupied states at the Fermi level are not observed. This type of valence band structure indicates that the Au22(SG)18 clusters are not metallic. The obtained results provide evidence of the existence of a finite optical energy gap in the Au22 cluster protected by glutathione ligands and their molecular-like nature.
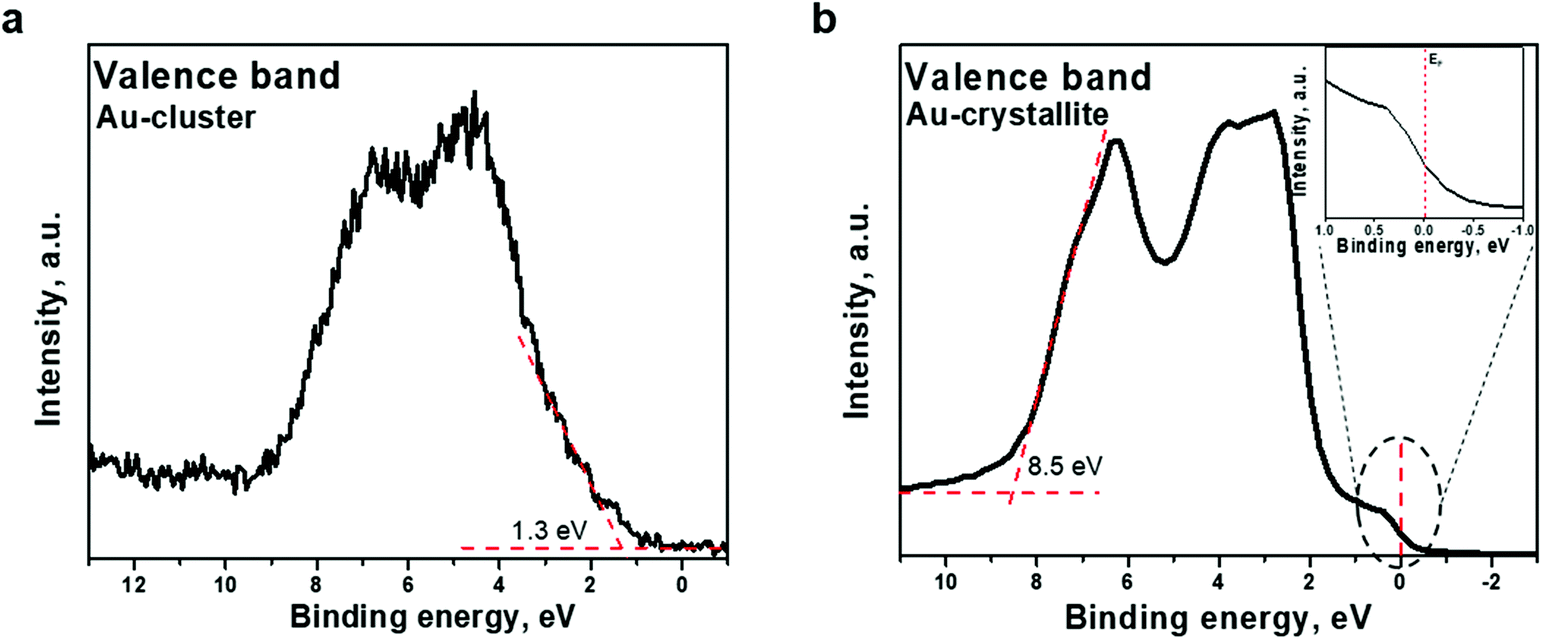 | ||
| Fig. 4 Valence band spectra acquired from (a) Au22(SG)18 clusters supported on TiO2 and (b) Au polycrystalline film. Photoemission from Au crystalline film within the binding energy range of 0–2 eV is shown on an expanded scale. The binding energy scale is referenced to the Fermi energy level. The midpoint of the steep slope at the Fermi level is visible. | ||
Au22(SG)18 clusters: electronic structure in depth
We suggest Ar+ ion etching as an additional tool for the identification of the electronic structure of the clusters. The character of alterations in the Au 4f7/2 core-level emissions as well as in the valence band structure upon Ar+ sputtering can clarify the chemical composition and the electronic structure of the clusters in depth.For this purpose, the Au 4f7/2 core-level spectrum was acquired from the cluster surface before and after mild Ar+ ion sputtering for 30 s. The spectra are shown in Fig. 5. The spectrum acquired from the pristine surface of the Au22(SG)18 cluster (Fig. 5a) is very different from that recorded after short Ar+ ion etching of the surface (Fig. 5b). We found a remarkable change in the spectral weight distribution of the emission intensity in the cluster spectrum. Peak fitting and deconvolution were performed for the Au 4f7/2 core-level emission from both samples. The fitting parameters and atomic percentage of the detected compounds are revealed in Table 2. The relative intensities of the two components were completely changed after Ar ion sputtering (Fig. 5b); the emission intensity at 84.0 eV increased remarkably while the intensity at 84.6 eV decreased. Given that the emission centered at a binding energy of 84.0 eV is ascribed to the core Au atoms while that located at 84.6 eV assigned to the Au atoms bonded to SG ligands, we can conclude that the chemical composition the Au22(SG)18 clusters changed dramatically. The content of the surface compound drops from 84.3 to 30.6%. At the same time, the content of the bulk compounds increases from 15.7 to 69.4% (Table 2).
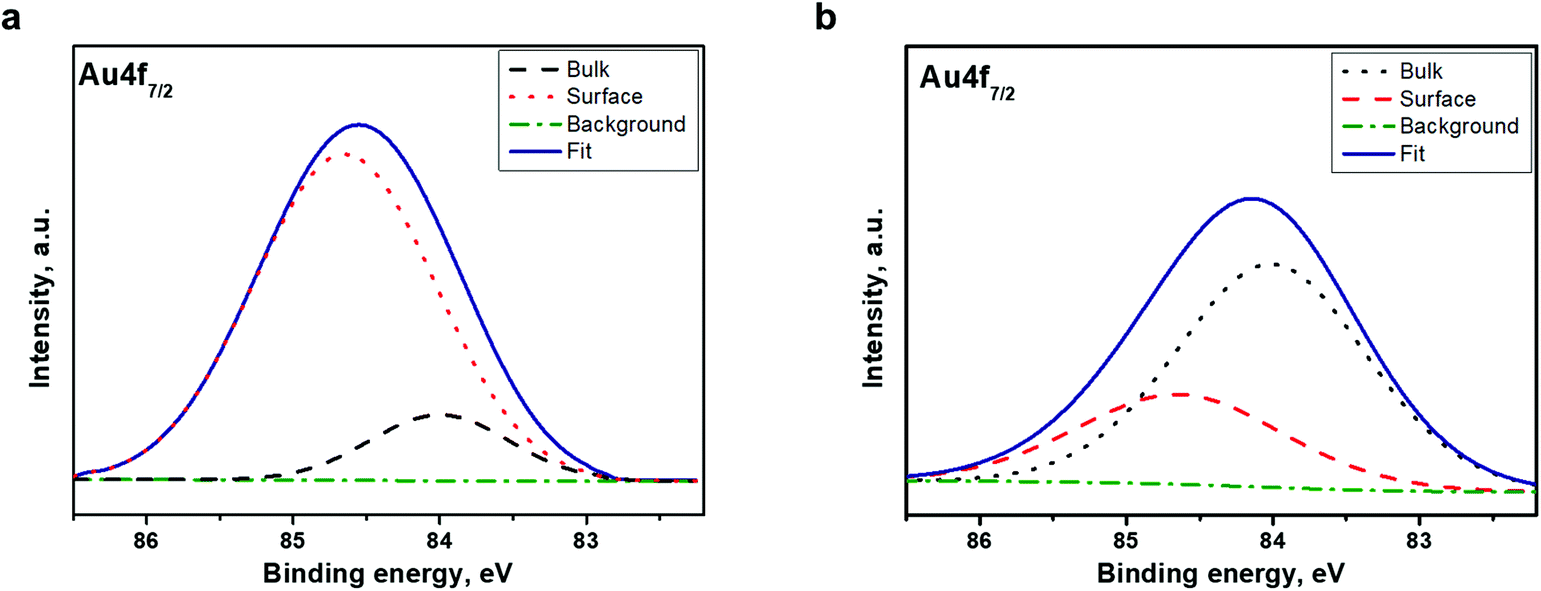 | ||
| Fig. 5 XPS spectra of the Au22(SG)18 clusters in the vicinity of Au 4f7/2 (a) before Ar+ etching and (b) after Ar+ etching. | ||
| Sample | Compound | Binding energy, eV | At% | FWHM, eV |
|---|---|---|---|---|
| Before treatment | Surface | 84.6 | 84.3 | 1.7 |
| Bulk | 84.0 | 15.7 | 1.5 | |
| After treatment | Surface | 84.6 | 30.6 | 1.6 |
| Bulk | 84.0 | 69.4 | 1.7 | |
The effect of Ar+ ion etching on the valence band electronic structure was also observed. As presented in Fig. 6, a shift of the valence band edge of the Au22(SG)18 cluster layer was detected. The onset moves to lower binding energy, from 1.3 to 1.0 eV, upon etching. Although the d-band shows a band-like character, occupied states at the Fermi level are not observed. The shift of the valence band edge toward the Fermi level indicates that clusters trend toward a “more metallic” state. The shift of the valence band edge correlates well with the increase in the percentage of core-Au atoms over shell-Au atoms bonded to SG ligands, as deduced from the Au 4f7/2 core-level deconvolution (Fig. 5b and Table 2). The obtained experimental results prove the existence of two electronic states of Au in the Au22(SG)18 cluster layer, indicating that the clusters exhibit a molecular-like electronic structure.
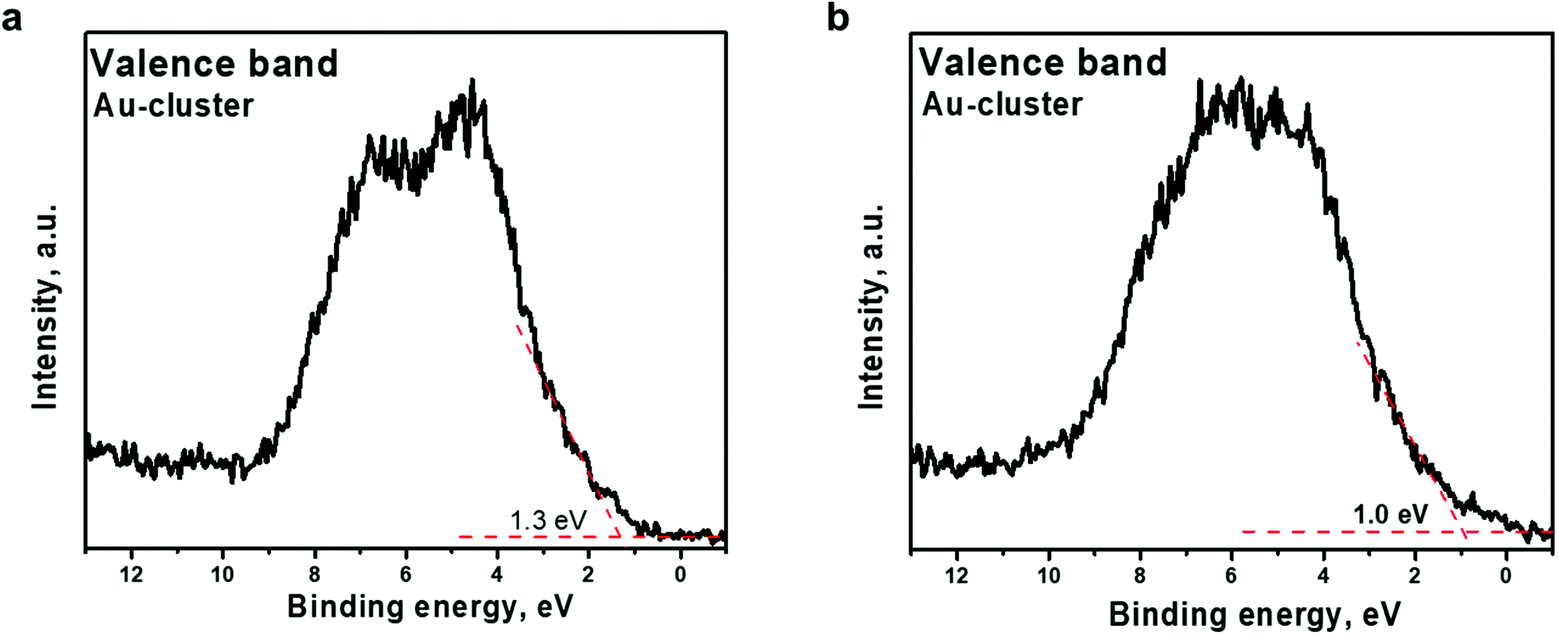 | ||
| Fig. 6 Valence band spectra acquired from Au22(SG)18 clusters supported on TiO2 (a) before Ar+ etching and (b) after Ar+ etching The binding energy scale is referenced to the Fermi energy level. | ||
Electronic band structure at the TiO2/Au22(SG)18 cluster interface
To identify the electronic band structure at the TiO2/Au22(SG)18 cluster interface, the VBM position of the nanostructured TiO2, and the first ionization energy (HOMO) level of the ligand-stabilized Au clusters were determined. For this purpose, the valence band emissions are acquired at the surface of bare nanostructured TiO2 and Au22(SG)18 cluster film coated onto TiO2. Fig. 7 shows the photoemission spectra from the samples in the energy range of −2 to 13 eV. We can clearly distinguish two materials according to the change in the spectral weight distributions of the emission intensities. The TiO2 and Au22(SG)18 cluster spectra show the different valence band electronic structures.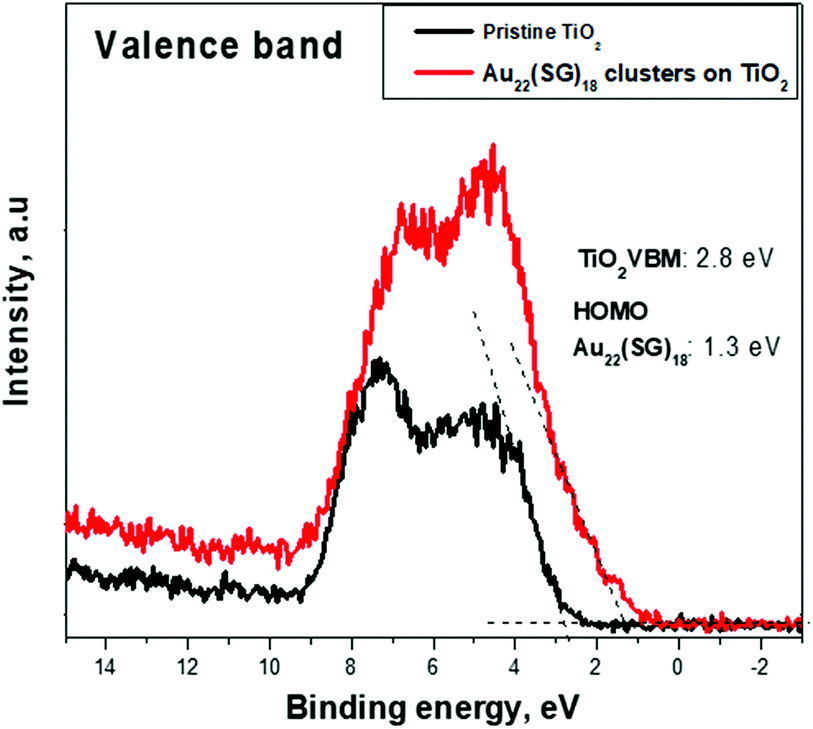 | ||
| Fig. 7 Representative XPS spectra of pristine nanostructured TiO2 and Au22(SG)18 cluster film coated onto TiO2 are shown in the binding energy range of the valence band. A difference in the energy of the valence-band edges is visible for the TiO2 and Au22(SG)18 cluster film coated onto the TiO2. | ||
The broad photoemission at the binding energy of 5 eV observed for TiO2 can be attributed to π (nonbonding) O 2p orbitals, while the peak at 7.5 eV is attributed to σ (bonding) O 2p orbitals.40 A VBM value of 2.8 eV for TiO2 was deduced from the emission spectrum by means of linear extrapolation of the leading edge of the valence band emission to the baseline. The presented analysis of the diffuse reflectance spectrum in terms of indirect optical absorption for nanostructured TiO2 yielded a bandgap value of 3.2 eV (Fig. S2†). Given this result, the estimated VBM of 2.8 eV places EF near the top of the fundamental gap of the TiO2, indicating a high level of n-type doping. Qualitatively different shape of the signal of Au22(SG)18 clusters on TiO2, suggests the significant contribution of Au clusters to the electronic structure in the spectrum range near the valence band. Photoemission forms a broad band between 2 and 8 eV. Photoemission in the binding energy range of 1–2 eV can be attributed to the 6s electrons. The 6s band is strongly hybridized with the 5d band and extends to a higher energy.39 The d band drops to zero at around 1.3 eV, below EF. The HOMO energy level for the Au22(SG)18 clusters were determined from the emission spectrum of the Au22(SG)18 clusters on TiO2 by linear extrapolation of the leading edge of the valence band emission to the baseline. The HOMO positions are determined with respect to EF. For this purpose, we used a sample of the type used in actual devices. Therefore, the energy-level discontinuity of 1.5 eV was deduced between the VBM of TiO2 and the HOMO energy of Au22(SG)18 clusters at the TiO2/cluster interface.
The discontinuity between the conduction band maximum (CBM) of TiO2 and the LUMO level of Au22(SG)18 at the TiO2/cluster interface was calculated from obtained experimental data with the following equation,
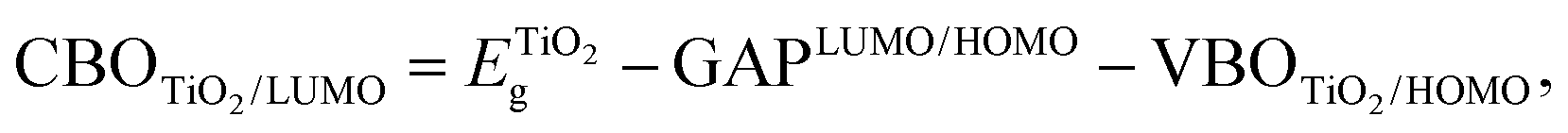 | (1) |
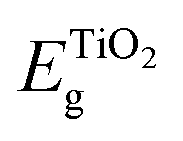 is the bandgap value of TiO2, and GAPLUMO/HOMO is the energy difference between the HOMO level and the LUMO level of the Au22(SG)18 clusters.
is the bandgap value of TiO2, and GAPLUMO/HOMO is the energy difference between the HOMO level and the LUMO level of the Au22(SG)18 clusters.
Fig. 8 displays the scheme of the energy level alignment at the TiO2/cluster interface and the pathways of the electron transfer induced by sub-bandgap photoexcitation of the Au22(SG)18 clusters. The obtained experimental values yield a cliff offset of 0.5 eV between the LUMO level of the Au22(SG)18 and CBM of TiO2 at the TiO2/cluster interface with respect to TiO2. The energy diagram is aligned on the energy scale to the vacuum level. The ionization potential of 7.25 eV for anatase TiO2 was taken from literature.41 A bandgap value of 3.2 eV was obtained from the diffuse reflectance spectrum of the bare TiO2 film shown in Fig. S2.† The energy levels of the standard redox couples used in DSCs as electrolytes also are given in Fig. 8. As indicated, the redox levels of both the Co(II)/Co(III) and 3I−/I−3 couples are suitable to support electron transfer across the interface. Moreover, the relative positions of the HOMO level of the clusters and the Co(II)/Co(III) redox level allow an increase in the open-circuit voltage in MCSSCs. The energy diagram clearly shows the importance of the electronic structure of the configuration of TiO2/LUMO and the HOMO/redox levels for proper solar cell operation. The drawback of the Au22(SG)18 clusters is that the 0.5 eV cliff is not ideal for efficient electron transfers between the LUMO level and the TiO2 CB. Given the similar device configurations, the optimized CBO at the interface is expected to be a small cliff of approximately 0.2 eV,28 and achieving this configuration is crucial for reducing interfacial recombination and increasing the charge injection efficiency. Therefore, the reduction of the interface cliff-like offset will positively be reflected in the photocurrent value. Furthermore, the offset between the HOMO/electrolyte redox level is 0.5 eV (for an iodine-based electrolyte), indicating an opportunity to increase Voc further by lowering the redox electrolyte level through the alteration of the redox couple. The overpotential required for a proper redox reaction in the electrolyte is in the range of 0.2–0.3 eV;42,43 thus, Voc could potentially be improved by ∼0.3 eV. Furthermore, relative band positions of the Au22(SG)18/TiO2 make it possible candidate for photocatalytic water splitting applications. Although, similarly to the MCSSC, the reduction of CBO to −0.2 eV will have beneficial effect on the system performance.
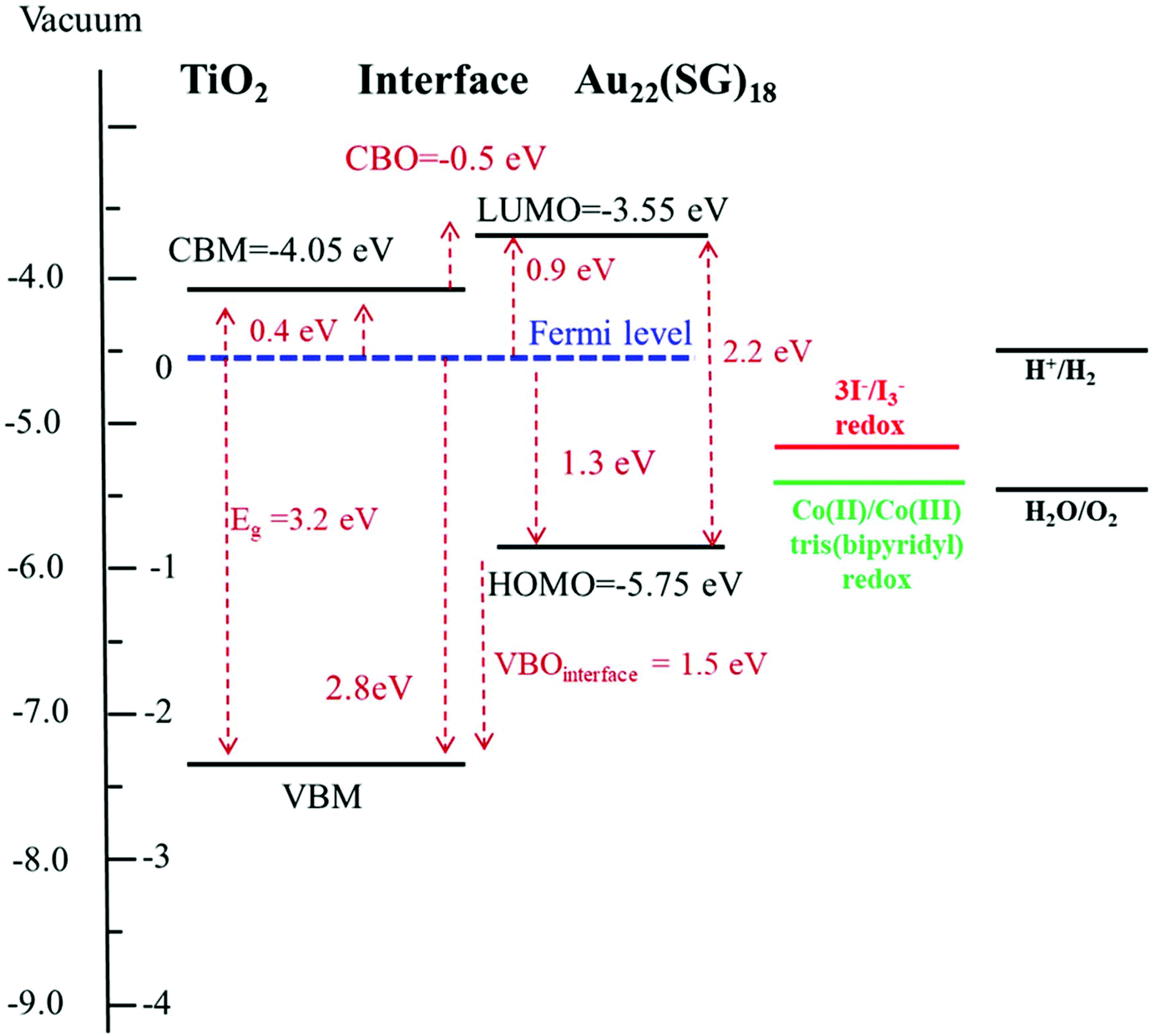 | ||
| Fig. 8 Scheme of the energy band alignment at the TiO2/Au22(SG)18 cluster interface. The energy diagram is aligned on the energy scale to the vacuum level. The diagram is constructed based on the experimental XPS, UV–Vis absorption, and the diffuse reflectance data. | ||
Conclusion
In this work, we investigated the electronic structure of Au22(SG)18 clusters for their application to MCSSCs. The molecular-like structure of the clusters was highlighted through a comparative investigation of the electronic structures of Au22(SG)18 clusters and crystalline Au film using X-ray photoelectron spectroscopy. A valence band edge of 1.3 eV was recorded for the Au22(SG)18 clusters. The lack of occupied states at the Fermi level proved the molecular-like nature of the Au22(SG)18 clusters. We also determined the energy band alignment at the TiO2/Au22(SG)18 cluster interface. The VBM positions of the nanostructured TiO2 and the HOMO level of the ligand-stabilized Au clusters were determined to be 2.8 and 1.3 eV in respect to the Fermi energy level. An energy-level discontinuity of 1.5 eV was deduced between the VBM of the TiO2 and the HOMO energy of the clusters at the TiO2/Au22(SG)18 cluster interface. A cliff-like offset of 0.5 eV between the LUMO level of the clusters and the TiO2 CB was quantified. The determined interface electronic configuration is suitable for electron transfers across the TiO2/Au22(SG)18 interface, but it is not ideal. Considering that an overpotential of the order of 0.2–0.3 eV is required to oxidize the redox electrolyte,42 the determined 0.5 eV offset of the HOMO level with the 3I−/I−3 redox level not only indicates a remarkable loss-in-potential outcome but also opens the possibility of increasing Voc in MCSSCs. Since the very limited experimental information available about the electronic structures of the interfaces of the TiO2/Au22(SG)18 cluster/redox electrolyte, our pioneering results represent an enhancement of understanding of the loss mechanisms in MCSSCs. Because the magnitudes of Voc and photocurrent are strongly affected by the junction properties, an enhancement in the efficiency can be achieved by tuning the energy alignment at the interfaces via cluster size alterations. Furthermore, it is highly suggestible to use Co(II)/Co(III) redox couple with TiO2/Au22(SG)18 interface as it may increase the open-circuit voltage of the operating MCSSC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation (NRF) grants (NRF- 2020H1D3A2A01111061, NRF-2020R1A4A2002590, NRF-2017R1A2B3006651) funded by the Ministry of Science and ICT, Republic of Korea. This work was partially supported by the Russian Science Foundation under Grant No. 20-69-47124.References
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- O. Varnavski, G. Ramakrishna, J. Kim, D. Lee and T. Goodson, J. Am. Chem. Soc., 2010, 132, 16–17 CrossRef CAS PubMed.
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- Y. Pei, J. Tang, X. Tang, Y. Huang and X. C. Zeng, J. Phys. Chem. Lett., 2015, 6, 1390–1395 CrossRef CAS PubMed.
- R. S. Ingram, M. J. Hostetler, R. W. Murray, T. G. Schaaff, J. T. Khoury, R. L. Whetten, T. P. Bigioni, D. K. Guthrie and P. N. First, J. Am. Chem. Soc., 1997, 119, 9279–9280 CrossRef CAS.
- D. Lee, R. L. Donkers, J. M. DeSimone and R. W. Murray, J. Am. Chem. Soc., 2003, 125, 1182–1183 CrossRef CAS PubMed.
- K. J. Taylor, C. L. Pettiette-Hall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 96, 3319–3329 CrossRef CAS.
- O. Varnavski, G. Ramakrishna, J. Kim, D. Lee and T. Goodson, ACS Nano, 2010, 4, 3406–3412 CrossRef CAS PubMed.
- W. T. Chen, Y. J. Hsu and P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 2493–2499 CrossRef CAS PubMed.
- M. A. Abbas, T. Y. Kim, S. U. Lee, Y. S. Kang and J. H. Bang, J. Am. Chem. Soc., 2016, 138, 390–401 CrossRef CAS PubMed.
- Y. S. Chen, H. Choi and P. V. Kamat, J. Am. Chem. Soc., 2013, 135, 8822–8825 CrossRef CAS PubMed.
- J. Akola, K. A. Kacprzak, O. Lopez-Acevedo, M. Walter, H. Grönbeck and H. Häkkinen, J. Phys. Chem. C, 2010, 114, 15986–15994 CrossRef CAS.
- C. M. Aikens, J. Phys. Chem. Lett., 2010, 1, 2594–2599 CrossRef CAS.
- R. Quo and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 12140–12143 CrossRef PubMed.
- C. A. Fields-Zinna, M. C. Crowe, A. Dass, J. E. F. Weaver and R. W. Murray, Langmuir, 2009, 25, 7704–7710 CrossRef CAS PubMed.
- L. Howard-Fabretto and G. G. Andersson, Adv. Mater., 2020, 32, 1–26 CrossRef PubMed.
- G. Krishnan, H. S. Al Qahtani, J. Li, Y. Yin, N. Eom, V. B. Golovko, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2017, 121, 28007–28016 CrossRef CAS.
- B. N. Reinecke, K. P. Kuhl, H. Ogasawara, L. Li, J. Voss, F. Abild-Pedersen, A. Nilsson and T. F. Jaramillo, Surf. Sci., 2016, 650, 24–33 CrossRef CAS.
- D. P. Anderson, J. F. Alvino, A. Gentleman, H. Al Qahtani, L. Thomsen, M. I. J. Polson, G. F. Metha, V. B. Golovko and G. G. Andersson, Phys. Chem. Chem. Phys., 2013, 15, 3917–3929 RSC.
- H. S. Al Qahtani, G. F. Metha, R. B. Walsh, V. B. Golovko, G. G. Andersson and T. Nakayama, J. Phys. Chem. C, 2017, 121, 10781–10789 CrossRef CAS.
- Y. Negishi, W. Kurashige and U. Kamimura, Langmuir, 2011, 27, 12289–12292 CrossRef CAS PubMed.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- O. Omelianovych, V.-D. Dao, L. L. Larina and H.-S. Choi, Electrochim. Acta, 2016, 211, 842–850 CrossRef CAS.
- O. Omelianovych, L. L. Larina, H. J. Oh, E. Park, V. D. Dao and H. S. Choi, Sol. Energy, 2020, 201, 819–826 CrossRef CAS.
- O. Omelianovych, L. L. Larina, V. D. Dao and H. S. Choi, Appl. Surf. Sci., 2018, 457, 381–387 CrossRef CAS.
- L. Larina, D. Shin, J. H. Kim and B. T. Ahn, Energy Environ. Sci., 2011, 4, 3487–3493 RSC.
- P. H. Citrin, G. K. Wertheim and Y. Baer, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 3160–3175 CrossRef CAS.
- A. Tanaka, Y. Takeda, M. Imamura and S. Sato, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 195415 CrossRef.
- M. Quinten, I. Sander, P. Steiner, U. Kreibig, K. Fauth and G. Schmid, Z. Phys. D: At., Mol. Clusters, 1991, 20, 377–379 CrossRef CAS.
- S. Huo, Y. Wu, C. Zhao, F. Yu, J. Fang and Y. Yang, Ind. Eng. Chem. Res., 2020, 59, 14224–14233 CrossRef CAS.
- F. X. Xiao, Z. Zeng and B. Liu, J. Am. Chem. Soc., 2015, 137, 10735–10744 CrossRef CAS PubMed.
- P. Ascarelli, M. Cini, G. Missoni and N. Nisticò, J. de Phys. Colloques, 1977, 38(C2), C2-125–C2-128 Search PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D. E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- C. Zhang, A. Zhang, W. Hou, T. Li, K. Wang, Q. Zhang, J. M. De La Fuente, W. Jin and D. Cui, ACS Nano, 2018, 12, 4408–4418 CrossRef CAS PubMed.
- Leonard J. Brillson, Surfaces and Interfaces of Electronic Materials, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- X. Zheng and L. Zhang, Energy Environ. Sci., 2016, 9, 2511–2532 RSC.
- J. A. Van Bokhoven and J. T. Miller, J. Phys. Chem. C, 2007, 111, 9245–9249 CrossRef CAS.
- R. Sanjinés, H. Tang, H. Berger, F. Gozzo, G. Margaritondo and F. Lévy, J. Appl. Phys., 1994, 75, 2945–2951 CrossRef.
- J. Fujisawa, T. Eda and M. Hanaya, Chem. Phys. Lett., 2017, 685, 23–26 CrossRef CAS.
- L. M. Peter, Phys. Chem. Chem. Phys., 2007, 9, 2630–2642 RSC.
- H. J. Snaith, Adv. Funct. Mater., 2010, 20, 13–19 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr06662a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |