
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChain release mechanisms in polyketide and non-ribosomal peptide biosynthesis
Rory F.
Little
 and
Christian
Hertweck
*
and
Christian
Hertweck
*
Leibniz Institute for Natural Product Research and Infection Biology, HKI, Germany. E-mail: Christian.Hertweck@hki-jena.de
First published on 20th August 2021
Abstract
Review covering up to mid-2021
The structure of polyketide and non-ribosomal peptide natural products is strongly influenced by how they are released from their biosynthetic enzymes. As such, Nature has evolved a diverse range of release mechanisms, leading to the formation of bioactive chemical scaffolds such as lactones, lactams, diketopiperazines, and tetronates. Here, we review the enzymes and mechanisms used for chain release in polyketide and non-ribosomal peptide biosynthesis, how these mechanisms affect natural product structure, and how they could be utilised to introduce structural diversity into the products of engineered biosynthetic pathways.
 Rory F. Little | Rory Little received a Bachelor (Hons) (2013) and Master of Science (2015) degree from Victoria University of Wellington (Te Herenga Waka), New Zealand. He was awarded a Woolf Fisher scholarship to complete a Ph.D. (2019) at the University of Cambridge in the group of Professor Peter Leadlay. He is currently an Alexander von Humboldt postdoctoral fellow in the group of Professor Christian Hertweck at the Leibniz HKI. His research interests are microbial natural product discovery, function, and biosynthesis. |
 Christian Hertweck | Christian Hertweck is head of the Biomolecular Chemistry Department at the Leibniz Institute for Natural Product Research and Infection Biology (HKI) and holds a Chair at the Friedrich Schiller University Jena. After his Ph.D. studies at University of Bonn and the MPI for Chemical Ecology (W. Boland) he was a Feodor Lynen postdoctoral fellow at the University of Washington, Seattle (H. G. Floss and B. S. Moore). His research focuses on microbial natural products, their biosynthesis, and their role in microbial interactions. He is an elected member of the National Academy of Sciences (Leopoldina) and recipient of the Leibniz Award. |
1 Introduction
Polyketides (PKs) and the non-ribosomal peptides (NRPs) are two of the largest natural product families.1,2 Widely produced by bacteria and fungi, members of each family have been turned into important medicines, such as the antibiotics erythromycin (PK) and vancomycin (NRP), the immunosuppressants rapamycin (PK) and cyclosporin (NRP), and the anticancer agents epothilone (PK) and bleomycin (NRP).3–7 One reason for such success is the remarkable diversity of PK and NRP chemical scaffolds.2,8,9 The diversity exhibited by PKs and NRPs is all the more impressive because of the relatively simple chemical units used to construct them. PKs and NRPs are both polymers (often called “chains”) constructed from simple monomers—also referred to as “building blocks” or extension units.1,2 Polyketide synthase (PKS) enzymes condense small carboxylic acids, primarily acetate and propionate, to form PKs, while non-ribosomal peptide synthetases (NRPSs) condense amino acids (and sometimes other organic acids) to form NRPs.1,2 However, these humble beginnings give rise to numerous medicinally important chemical scaffolds including macrolides, polyethers, enediynes, and β-lactams.2,8The mechanisms used for diversifying PK/NRP chains are wide ranging. For example, arsenals of tailoring enzymes can modify the PK/NRP chain after it has been fully processed by a PKS/NRPS enzyme.2,8 However, one of the most important diversification steps often occurs earlier. During its biosynthesis, a PK/NRP chain is covalently tethered to the PKS/NRPS via a 4′-phosphopantetheine (Ppant) group (derived from coenzyme A).10 The free thiol group of the Ppant group forms a thioester bond with the terminal carboxyl group of the growing PK/NRP chain.1 However, this covalent linkage must be broken to release the PK/NRP chain from the PKS/NRPS. A release step is critical both for allowing the product to enter into the cytosol, where it may be modified by additional enzymes and/or exported from the cell, and to enable continuous substrate processing by the PKS/NRPS.1 Nature has not only solved this problem, but also keenly recognised it as an opportunity to profoundly modify the structure of the PK/NRP product.11 For instance, the PK/NRP chain can be released via intra/intermolecular cyclisations, reductions, and fusion to other chemical units, leading to structural diversifications ranging from simple primary alcohols, aldehydes, and carboxylic acids, to more complex tetronates, macrolactones/lactams, and oligomers.11,12 The functionalities created as a result of chain release may themselves undergo subsequent chemical transformations, leading to the creation of complex scaffolds such as spirotetronates and iminopeptides.12,13 In addition, given the impact that the mechanism of chain release can have on PK/NRP structure, an appreciation and understanding of these mechanisms will aid efforts for creating engineered PKS and NRPS enzymes that produce new and diverse products. Here, we review the enzymes and mechanisms used for chain release in PK and NRP biosynthesis pathways, how these mechanisms directly and indirectly affect natural product structure, and their potential to be utilised by synthetic biology to produce structurally diverse products from engineered PKS/NRPS enzymes. It is important to note that, while many of the mechanisms discussed in this review are experimentally well characterised, some lack direct experimental evidence so are only proposed mechanisms. In some cases, obtaining such evidence is stymied by the chain release enzyme acting on a complex, difficult to source, and possibly unstable biosynthetic intermediate. Nevertheless, instances where a given chain release mechanism requires confirming experimental evidence are explicitly stated.
2 PKS and NRPS enzymes
Before discussing chain release mechanisms (sometimes also called “offloading” mechanisms), it is worth briefly covering the biochemistry of PKS and NRPS enzymes. There are multiple classes of both enzymes, differing from one another in characteristics such as whether they are modular, act iteratively, or are composed of multiple standalone proteins.1,2 The most discussed classes in this review are the modular cis-acyltransferase (AT) type I PKSs and Type A NRPSs. In these classes, each protein module is responsible for the incorporation of a single extension unit into the growing PK/NRP chain. Each module of a PKS/NRPS enzyme is itself comprised of discrete catalytic centres called domains, which catalyse the necessary reactions for the chain extension to occur.1,2 In contrast, type II PKSs consist of iteratively acting standalone proteins.14 The minimal type II PKS biosynthesis pathway consists of a standalone ACP and two proteins that resemble KS domains: a KSα and KSβ.14 These two KS proteins are highly similar to one another and together catalyse the necessary chain initiation and extension events required for polyketide biosynthesis.14 Type III PKSs consist of numerous standalone enzymes to catalyse chain elongation without the use a Ppant tether.15 Analogous to PKSs, NRPSs may also act iteratively or be composed of multiple standalone proteins, as will be discussed later.2The number of domains present within a type I PKS or type A NRPS module differs from enzyme to enzyme, except for several essential “core” domains.1,2 To be catalytically active, each module of cis-AT type I PKS must contain an acyltransferase (AT), acyl carrier protein (ACP), and a ketosynthase (KS) domain.1 The ACP of each module serves as an attachment point for a Ppant moiety. Each AT domain selects an extension unit and transfers it to the Ppant group of the adjacent ACP.10 The most common extension units used in PK biosynthesis are acetate (two carbon) and propionate (three carbon) units, typically delivered in their activated forms of malonyl-CoA and (2S)-methylmalonyl-CoA, respectively.1 The KS domain then catalyses C–C bond formation via a decarboxylative Claisen condensation between the nascent PK chain and the extension unit bound to the ACP of the downstream module.10 The PK chain uses the flexible Ppant groups to swing between the different PKS modules of the biosynthetic pathway, each module increasing the chain size by one extension unit.10 The downstream module can be part of the same enzyme as the upstream module, or be part of a separate PKS enzyme altogether.10 Following the Claisen condensation, accessory domains such as ketoreductase (KR), dehydratase (DH), and enoylreductase (ER) domains may reduce the β-keto group.10
Type A NRPS modules contain a different set of domains from type I PKS modules. At a minimum, each module in an NRPS must contain an adenylation (A) domain, a peptidyl carrier protein (PCP, also sometimes called a thiolation (T) domain), and a condensation (C) domain to be functional.2 A domains are responsible for selecting an extension unit, analogous to AT domains in PKS enzymes.2 To achieve this, A domains catalyse an ATP-dependent adenylation reaction of an amino acid, often with a high degree of selectivity.2 The adenylated amino acid is a high energy species with a strong leaving group (AMP), facilitating nucleophilic attack by the thiol of the PCP-linked Ppant group with elimination of AMP.2 The function of C domains is analogous to KS domains, though they catalyse C-N (peptide) bond formation rather than C–C bond formation.2 The PCP-bound NRP chain enters the active site of the C domains, where it is attacked by the α-amino group on the amino acid tethered to the PCP domain of the downstream PCP domain.2
After the final chain extension, the mature chain is left tethered to the ACP/PCP of the terminal module and must be released. Numerous mechanisms exist, utilising both enzymatic domains integrated into the PKS/NRPS or dedicated standalone chain release enzymes. The first class of chain release enzymes to be explored are the α/β hydrolase fold thioesterases.
3 Thioesterase domains (α/β hydrolase fold)
The α/β hydrolase fold thioesterases either catalyse chain release as a discrete domain within a PKS/NRPS (type I thioesterases), or as standalone proteins (type II thioesterases). The use of type I thioesterase (TE) domains to catalyse chain release is common in PK/NRP biosynthesis, to the extent that it is often considered the canonical method.11 TE domains typically catalyse release either by hydrolysis or macrocyclisation, though other mechanisms are also possible, as will be discussed. In the pathways that use them, the TE domain is almost always found on the C-terminus of the final PKS/NRPS module.11 TE domains are between 240–290 amino acid residues in size and possess an α/β hydrolase fold, a conformation consisting of seven to eight parallel β-sheets connected by α-helices (Fig. 1).10 α/β hydrolase folds are commonly found in other enzymes with hydrolytic activity, such as lipases and proteases.16 Between β-sheets six and seven is the “lid” region—a dynamic ca. 40 amino acid element that lines the substrate channel.17 Crystal structures of excised TE domains have revealed that the lid region is either in an apparent “open” state, allowing ready access to the binding pocket, or “closed” state, restricting substrate entry.17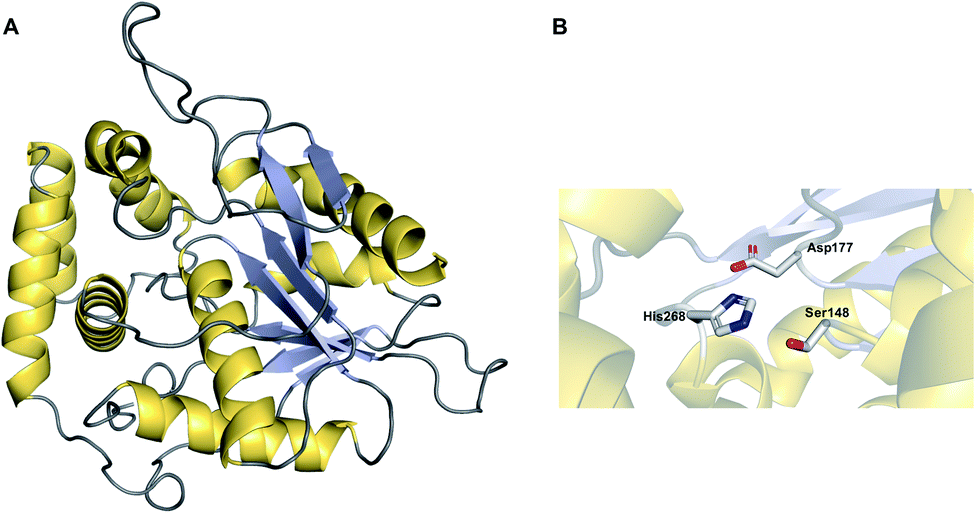 | ||
| Fig. 1 The structure of an α/β hydrolase fold thioesterase domain. (A) The crystal structure of the thioesterase domain from the polyketide pikromycin biosynthesis pathway (PDB: 1MN6). (B) The Ser–Asp–His catalytic triad of the TE domain from pikromycin biosynthesis. | ||
TE domains use a two-step mechanism to catalyse chain release, the first step being a transesterification.17 A the hydroxyl of a catalytic serine residue, typically located at the C-terminus of β-sheet five, attacks the electrophilic carbonyl of the PK/NRP thioester, forming an oxoester.17 The catalytic serine attacks the substrate thioester as it is activated via deprotonation by a conserved histidine (Fig. 2).17 Together, these three residues make up the Ser–Asp–His catalytic triad that is highly conserved in thioesterase domains and other α/β hydrolases.10 The catalytic serine is identifiable by its location in a GxSxG motif (where x is any amino acid).10 In some cases, a cysteine is present instead of a serine residue—in effect using a sulphur nucleophile rather than oxygen.18–20 Why some thioesterase domains select for a catalytic cysteine rather than serine is poorly understood.19 However, the presence of a catalytic cysteine can be an indicator that the TE domain has an unusual activity, as will be discussed in Section 3.2.1.
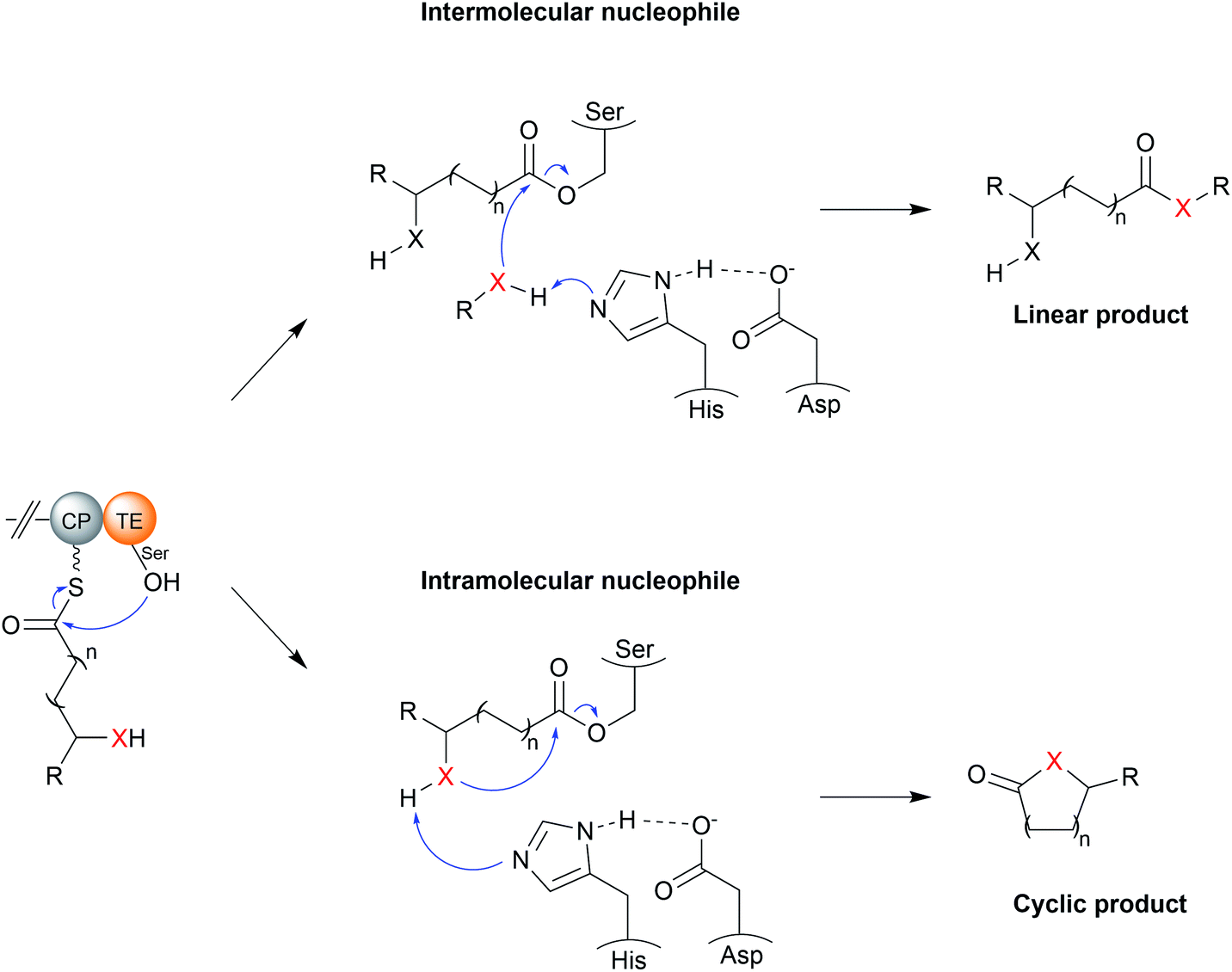 | ||
| Fig. 2 The catalytic mechanism of TE domains. TE domains select either an intermolecular nucleophile (typically water) or an intermolecular nucleophile (resulting in a cyclic product). The X represents a nucleophilic atom (such as an oxygen or nitrogen). The PK/NRP chain is covalently linked to the TE via an oxoester bond to a conserved serine. A conserved His–Asp dyad activates the nucleophile by extracting a proton. The wavey bond depicted from the CP represents a 4′-phosphopantetheine prosthetic group. | ||
The second step of the mechanism is the release of the PK/NRP intermediate from the TE domain itself.17 It is here where the TE domain exerts the greatest influence over the structure of the final product.11,17 During this step, a nucleophile attacks and cleaves the oxoester bond (or thioester bond, in the case of a TE domain with a catalytic cysteine) connecting the PK/NRP chain to the TE domain.17 A tetrahedral oxyanion intermediate forms following nucleophilic attack that is resolved by elimination of the seryl alkoxide/cystyl thiolate, releasing the PK/NRP from the active site.17 Aside from cleavage of oxoester and thioester bonds, there is evidence from synthetic systems that TE domains are also able to cleave the much stronger amide bond, though whether this occurs in nature is unknown.21 Nucleophilic attack may either be intermolecular or intramolecular. A TE domain that selects an intermolecular nucleophile will release a linear product, whereas the selection of an intramolecular nucleophile, i.e., part of the PK/NRP chain itself, will release a cyclic product.11 On top of this, a range of different nucleophilic atoms can be used. While an oxygen nucleophile is the most common, nitrogen, sulphur, and carbanions are also selected by some TE domains.22 Unfortunately, it is not possible to predict what kind of nucleophile a TE domain will select based on protein sequence alone.17,23 Recent evidence suggests that the aforementioned lid region could play a role by altering the conformation of the PK/NRP chain to promote an intramolecular cyclisation rather than attack by an exogenous nucleophile.21 However, in vitro activity assays of TE domains indicate that they are intrinsically capable of catalysing a range of different chain releasing reactions, including hydrolysis, macrocyclisation, dimerisation, and transesterification.23 TE domains may therefore initially serve as a means to increase the diversity of products produced by a biosynthetic pathway, later adapting to favour the production of the product that confers the greatest fitness advantage.23 The following sections discuss the different nucleophiles that TE domains can select in greater detail.
3.1 Intermolecular nucleophiles
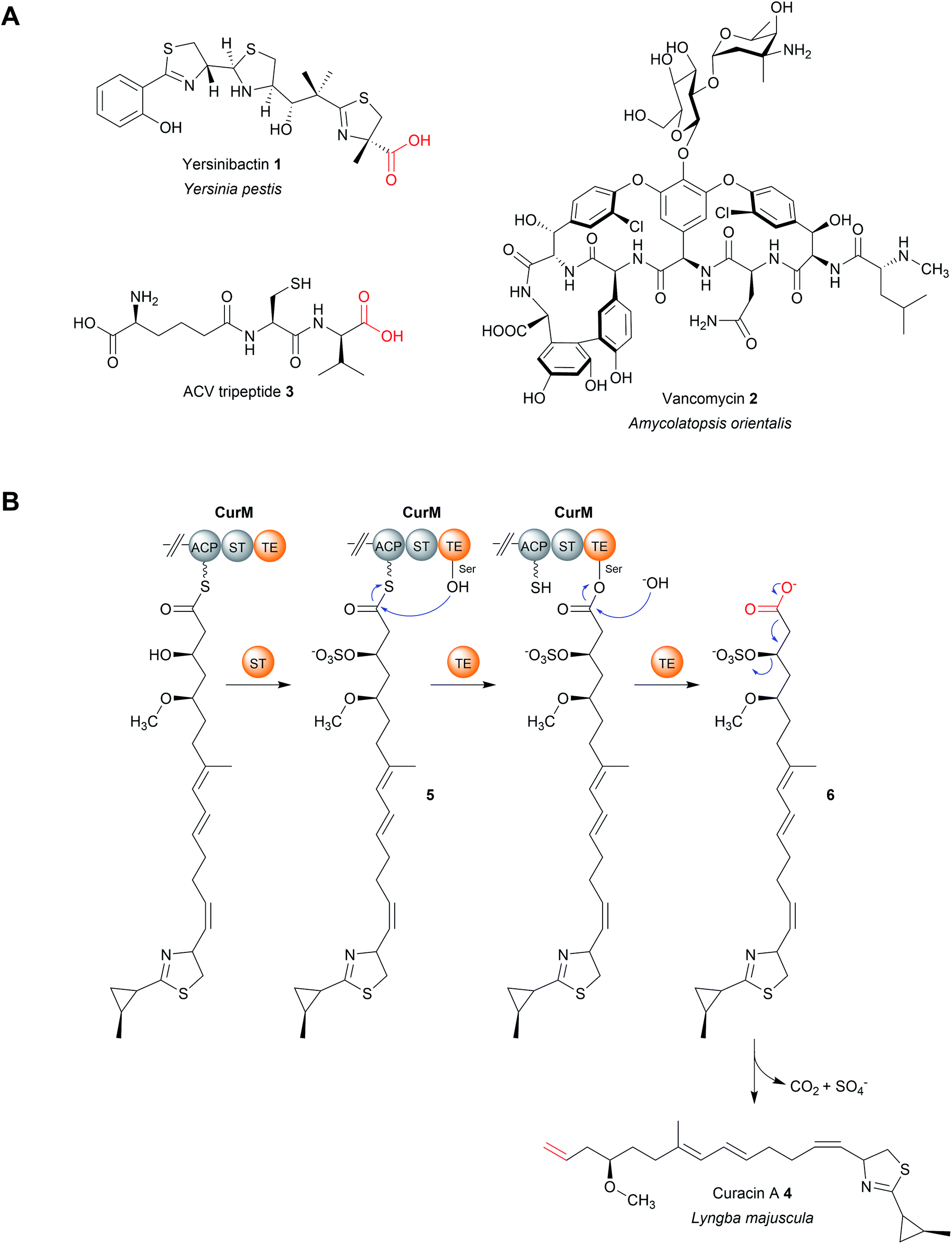 | ||
| Fig. 3 TE-catalysed hydrolytic release. (A) The structures of several natural products released by TE domain-catalysed hydrolysis. (B) Formation mechanism of the terminal double bond in curacin A (4) biosynthesis using an unusual sulphur transferase (ST) domain. | ||
An interesting variation occurs in the biosynthesis of curacin A (4), a PK–NRP hybrid produced by the cyanobacterium Lyngbya majuscule.29 Here, the terminal TE domain in the PKS CurM is preceded by an unusual sulfotransferase (ST) domain that, prior to hydrolytic chain release, sulfonates the β-hydroxyl group of the curacin intermediate (5), creating an excellent β-sulfate leaving group.29 Following TE-catalysed hydrolysis of the PK chain (6), a decarboxylation occurs and the β-sulfate is eliminated, creating a terminal double bond instead of the typical carboxylic acid.29In vitro experiments using a synthetic substrate analogue indicate that, in addition to catalysing hydrolysis, the TE domain also facilitates the decarboxylation reaction (Fig. 3B).29
A TE domain that catalyses an additional reaction besides hydrolysis is also found in the nocardicin A biosynthesis pathway. Nocardicin A is a tripeptide β-lactam antibiotic produced by Nocardia uniformis sp. tsuyamanensis and is comprised of L-para-hydroxyphenylglycine (pHPG), L-serine, and L-arginine.30 In this pathway, the TE domain in the NRPS NocB catalyses the epimerisation of L-para-hydroxyphenylglycine (pHPG) in addition to NRP chain hydrolysis.30 Cocrystallisation of the excised NocB TE domain with a phosphonate substrate mimic indicated that the histidine of the catalytic triad is responsible for extracting the acidic α-proton from pHPG.30 The resultant carbanion could be stabilised by electron delocalisation across the pHPG aromatic ring.30 A proton donor (possibly water) is then proposed to reprotonate the α-carbon of pHPG from the opposite side to complete the epimerisation.31 However, epimerisation and product hydrolysis can only occur after the β-lactam ring has formed,30 again indicating the gatekeeper function TE domains can have (a phenomenon recently reviewed by Horsman et al.17).
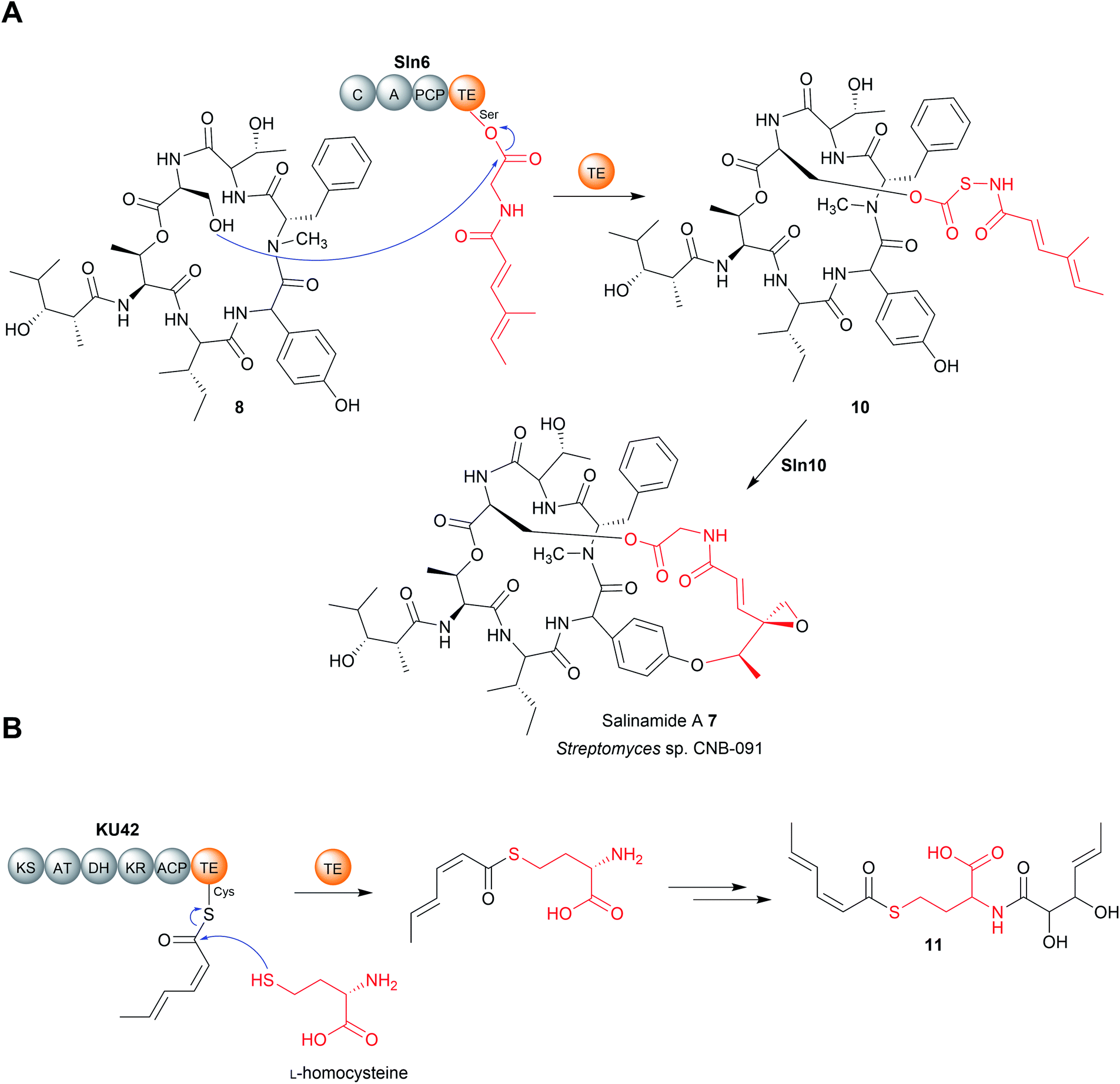 | ||
| Fig. 4 Chain release by TE-catalysed transesterification. (A) TE-catalysed chain release via transesterification in salinamide A (7) biosynthesis. (B) TE-catalysed chain release via transthioesterification by the cryptic fungal PKS KU42. | ||
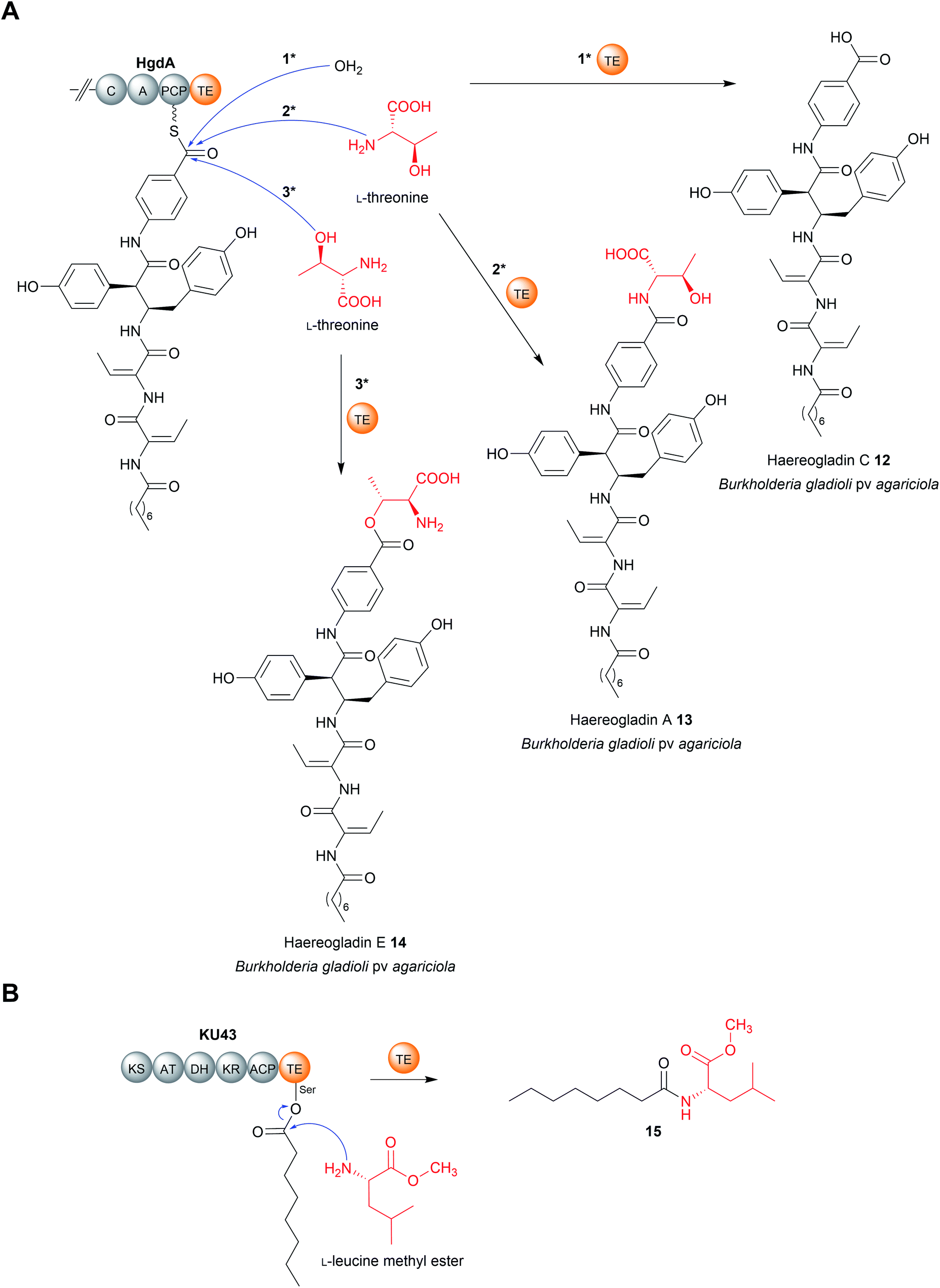 | ||
| Fig. 5 Chain release by TE-catalysed amidation. (A) TE-catalysed chain release in haereogladin biosynthesis. The TE domain of HgdA can select water (i), the amine group of L-threonine (ii), or the hydroxyl group of L-threonine (iii) as the intermolecular nucleophile used for chain release. (B) The KU43-TE domain selects the nitrogen of L-leucine methyl ester to form 15. | ||
Another recent example of a TE domain that catalyses amidation is found in the biosynthesis of a small and, as yet unnamed, polyketide (15) produced by the fungus Hydnomerulius pinastri.33 The PKS responsible, KU43, produces an ACP-bound octanoate unit that is released using the amino group of a L-leucine methyl ester as a nucleophile (Fig. 5B).33 Heterologous expression of the KU43-TE demonstrated it that it selects L-leucine methyl and catalyses chain release via an amination reaction.33
3.2 Intramolecular nucleophiles
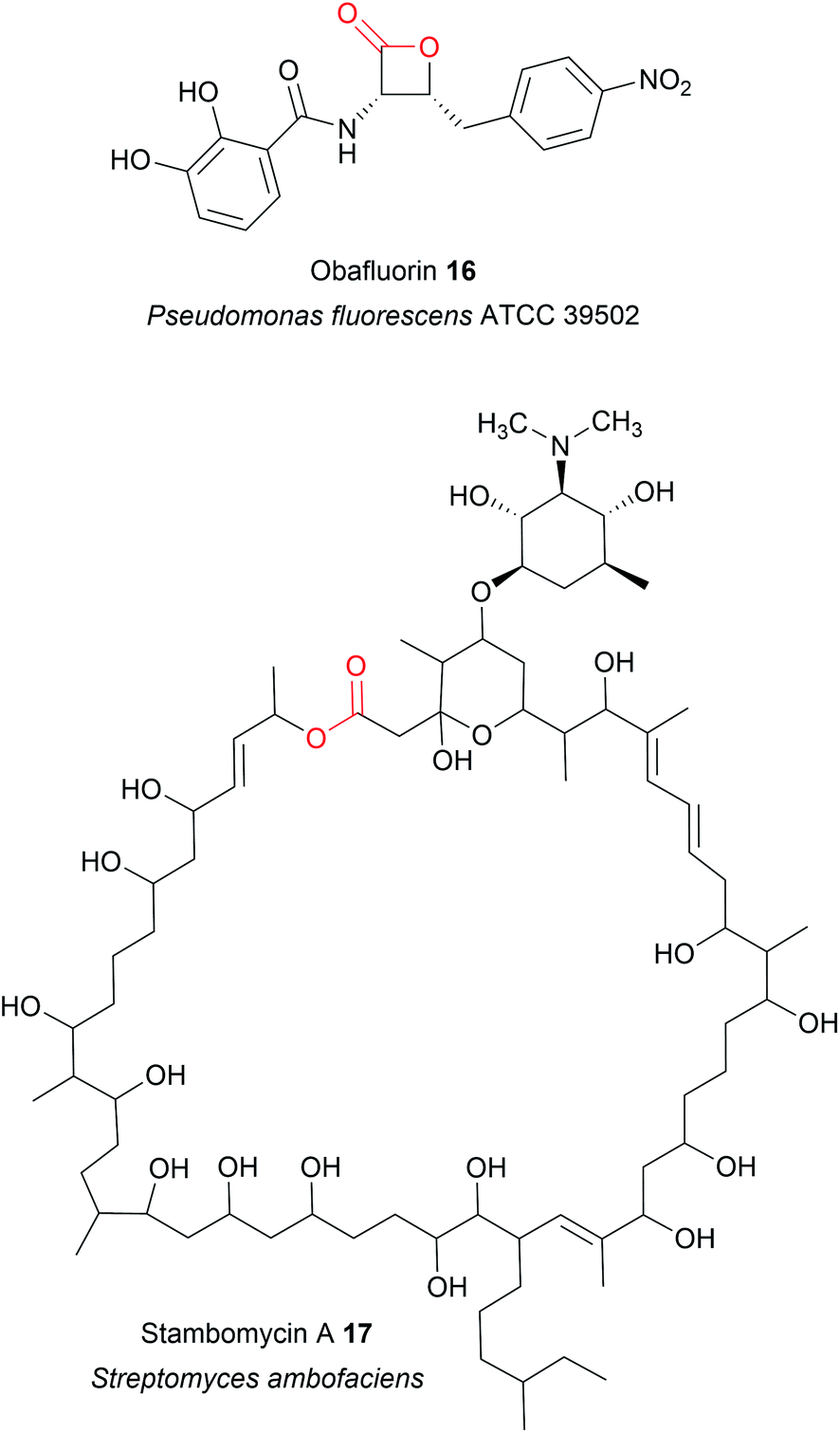 | ||
| Fig. 6 Size range of lactone rings produced by TE domains. The lactone ring of obafluorin (16) is the smallest known to be produced by a TE domain, while the 51-membered stambomycin (17) is the largest. | ||
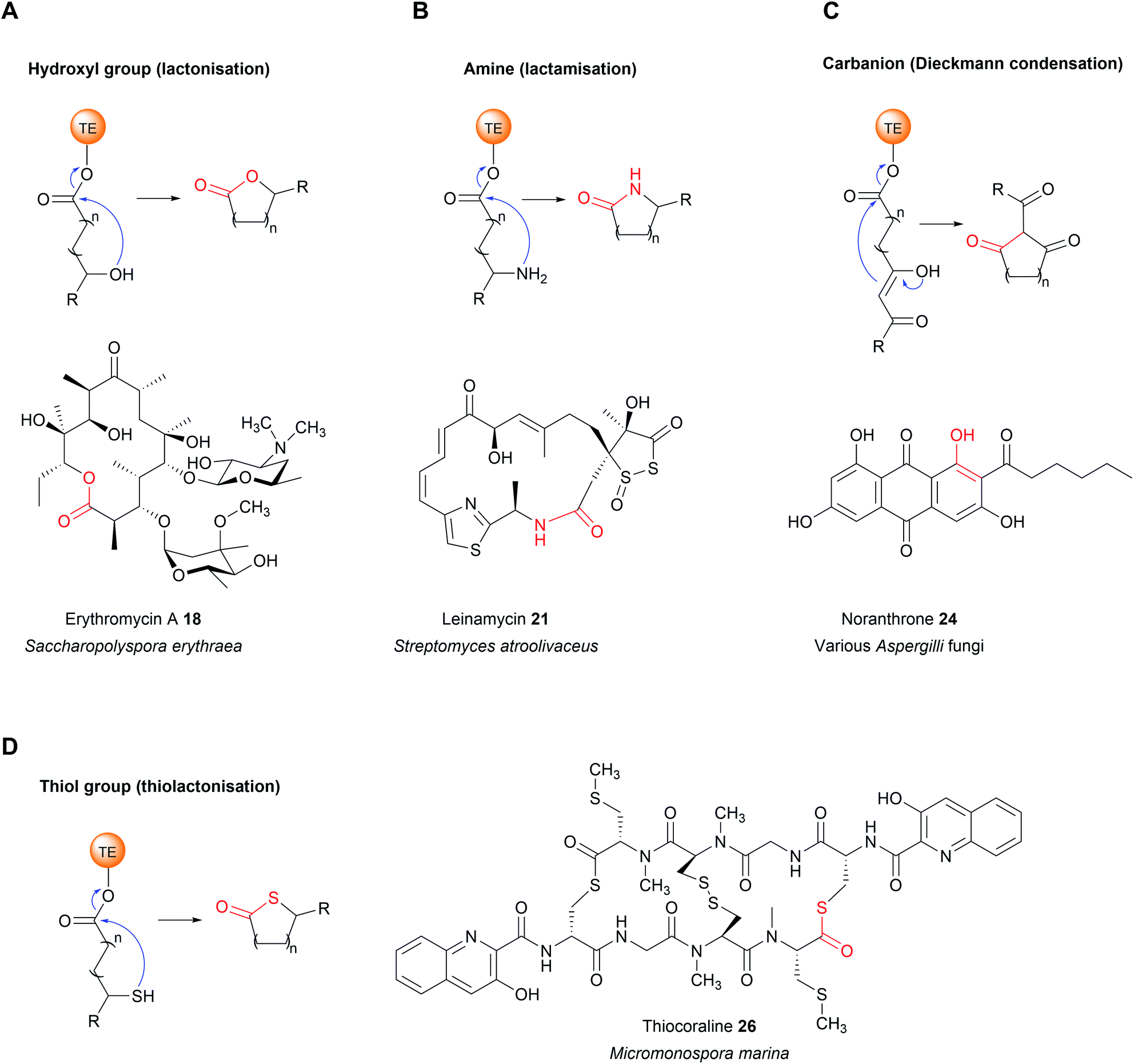 | ||
| Fig. 7 Intermolecular nucleophiles selected by thioesterase domains. (A) A hydroxyl group, as in erythromicin A (18) biosynthesis. (B) An amine, as in leinamycin (21) biosynthesis. (C) A carbanion, as in noranthrone (24) (aflatoxin precursor) biosynthesis. (D) A thiol group, as in thiocoraline (26) biosynthesis. | ||
If the lactone contains 12 or more atoms it is classified as a macrolactone/macrolide.37 The formation of lactones is commonly associated with polyketide biosynthetic pathways, the prototypical example being 6-deoxyerythronolide B (the erythromycin precursor) synthesised by the PKS enzymes DEBS1, DEBS2, and DEBS3.1 Erythromycin A (18), like many macrolides, is an inhibitor of the bacterial ribosome.38 The TE domain of DEBS3 is selective for the C13 hydroxyl over the other hydroxyls in the PK chain, resulting in the exclusive formation of a 14-membered macrolactone.17 TE domains that select hydroxyl/oxygen nucleophiles may also be responsible for the biosynthesis of non-lactone rings, such as the pyrone ring in the cercosporin (PK) biosynthesis or the isochromanone ring in ajudazol (PK) biosynthesis.39,40
TE domains from NRPSs can also catalyse chain release via lactone formation using the hydroxyl group of an amino acid side chain.18,41 One interesting example is the TE domain used in the biosynthesis pathway of obafluorin (16), a tripeptide β-lactone.18 The β-lactone ring of obafluorin (16) is created by an intramolecular cyclisation catalysed by the TE domain of the NRPS ObiF.18 The ObiF-TE domain is unusual for several reasons. For starters, it catalyses formation of a four membered ring, the smallest ring size produced by TE domains.18,22 Also, unlike most TE domains, it is not the most C-terminal domain of ObiF, instead being located between an upstream PCP domain and a downstream A domain.18 Non-terminal TE domains have been identified in only a few other biosynthetic pathways, where their role is unclear or they may catalyse an entirely different reaction (such as cis double bond formation in FR901464 biosynthesis).42–45 Another unusual feature of ObiF-TE is that it has a catalytic cysteine rather than serine.18 Interestingly, using mutagenesis to convert this cysteine to a serine abolished production of obafluorin (16), highlighting the importance of a cysteine thioester linkage to the NRP chain.18 The proposed explanation was that the higher ground state energy of cysteine thioesters compared to oxoesters, coupled with the weaker nature of C–S bonds compared to C–O bonds, make forming a strained β-lactone ring more energetically favourable.18,46 The influence of a catalytic cysteine residue has also been investigated in other TE domains. For instance, replacing the active site serine with a cysteine in the TE domains of the pikromycin (PK) and cilengitide (NRP) biosynthesis pathways converted the domain into catalytically more effective macrolactonisation catalysts, possibly due to the reasons proposed above.19,20 In other cases, however, replacing the serine with cysteine significantly decreased the catalytic activity of the TE domain.47 Why some TE domains select for cysteine while others select for serine is still unclear, but it seems likely there is a fitness trade-off occurring between creating an efficient catalyst for the target ring size and the stability of the TE-bound PK/NRP intermediate.
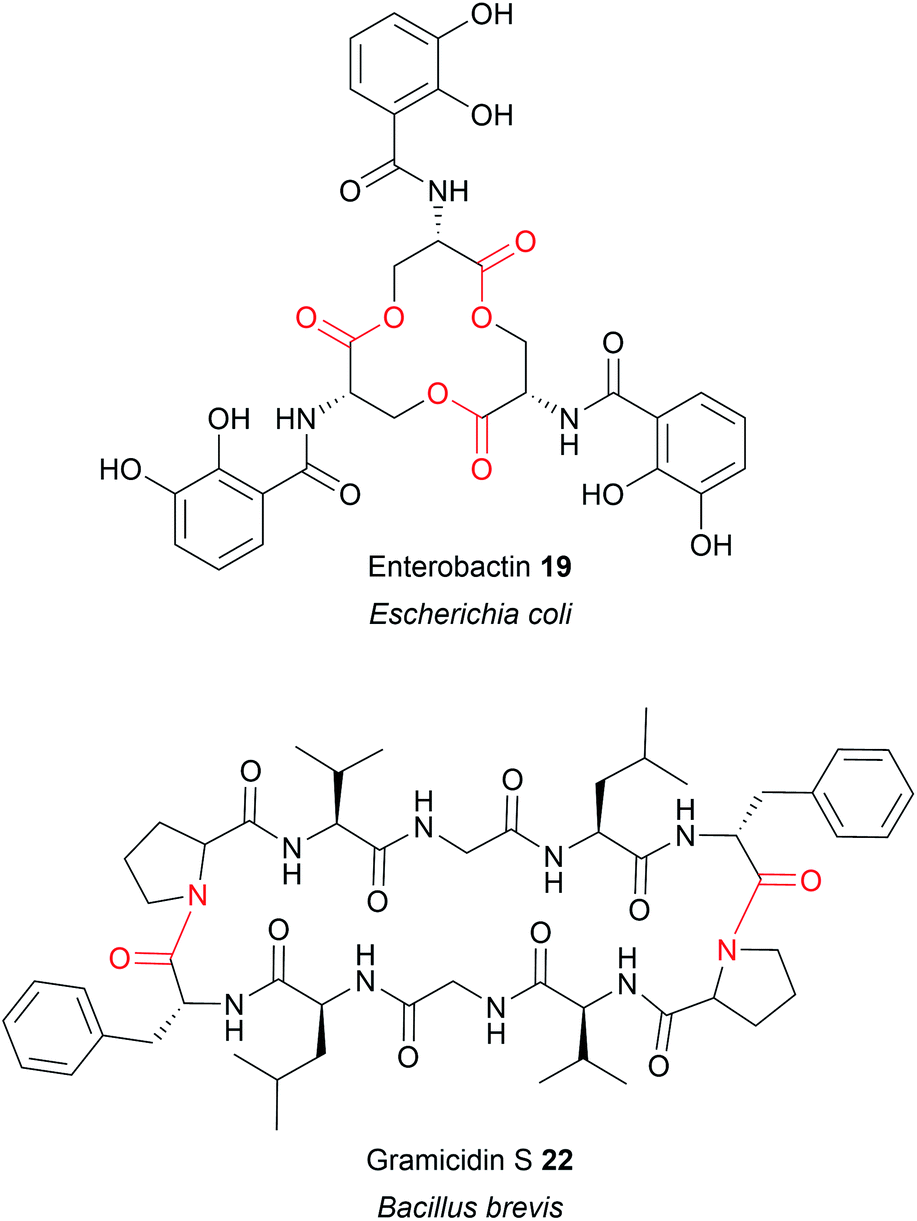
Up to this point, the TE domains discussed all catalyse chain release via a single lactonisation reaction. A variation of this mechanism, exemplified in the biosynthesis of conglobatin (PK),48 enterobactin (NRP) (19),47 elaiophylin (PK) (20),49 cereulide (NRP),50 and valinomycin (NRP)50 is TE-catalysed oligomerisation of two identical PK/NRP chains (Fig. 8). Two different mechanisms were initially proposed for how these oligomerisations occur: a “forward transfer” and a “backwards transfer” mechanism.11 In the forward transfer mechanism, a hydroxyl group of a TE-bound PK/NRP chain is proposed to attacks the thioester of an identical PK/NRP chain tethered to the upstream ACP domain, followed by macrocyclisation/chain release.47 In contrast, in the backwards transfer mechanism an ACP-bound PK/NRP chain is proposed to attack the thioester of an identical PK/NRP chain tethered to the downstream TE domain, sending it “backwards” to the ACP domain.51 The linear dimer is then returned to the active site of the TE domain for macrocyclisation.51 While the forward transfer mechanism was proposed first (to account for the biosynthesis of enterobactin) there is now more evidence that backwards transfer is the true mechanism for all such oligomerisations (Fig. 9).21,47,48,50,52 The ability of TE domains to catalyse both dimerisation and macrolactonisation has been demonstrated in vitro for the C2 symmetrical 16-membered dilactones conglobatin and elaiophylin (20).48,52 Purified TE domains from the conglobatin and elaiophylin (20) biosynthesis pathways were demonstrated to catalyse the dimerisation of two SNAC (N-acetylcysteamine)-substrate analogues,53 forming a linear dimer that is subsequently cyclised to form a macrodilactone.48,52 A similar TE-mediated oligomerisation mechanism is likely occurring in the biosynthesis of the quinoxaline and quinoline chromodepsipeptide natural products echinomycin (NRP) and sandramycin (NRP), respectively, though direct evidence is lacking.54–56
 | ||
| Fig. 8 Examples of natural products produced by TE-catalysed oligomerisation. | ||
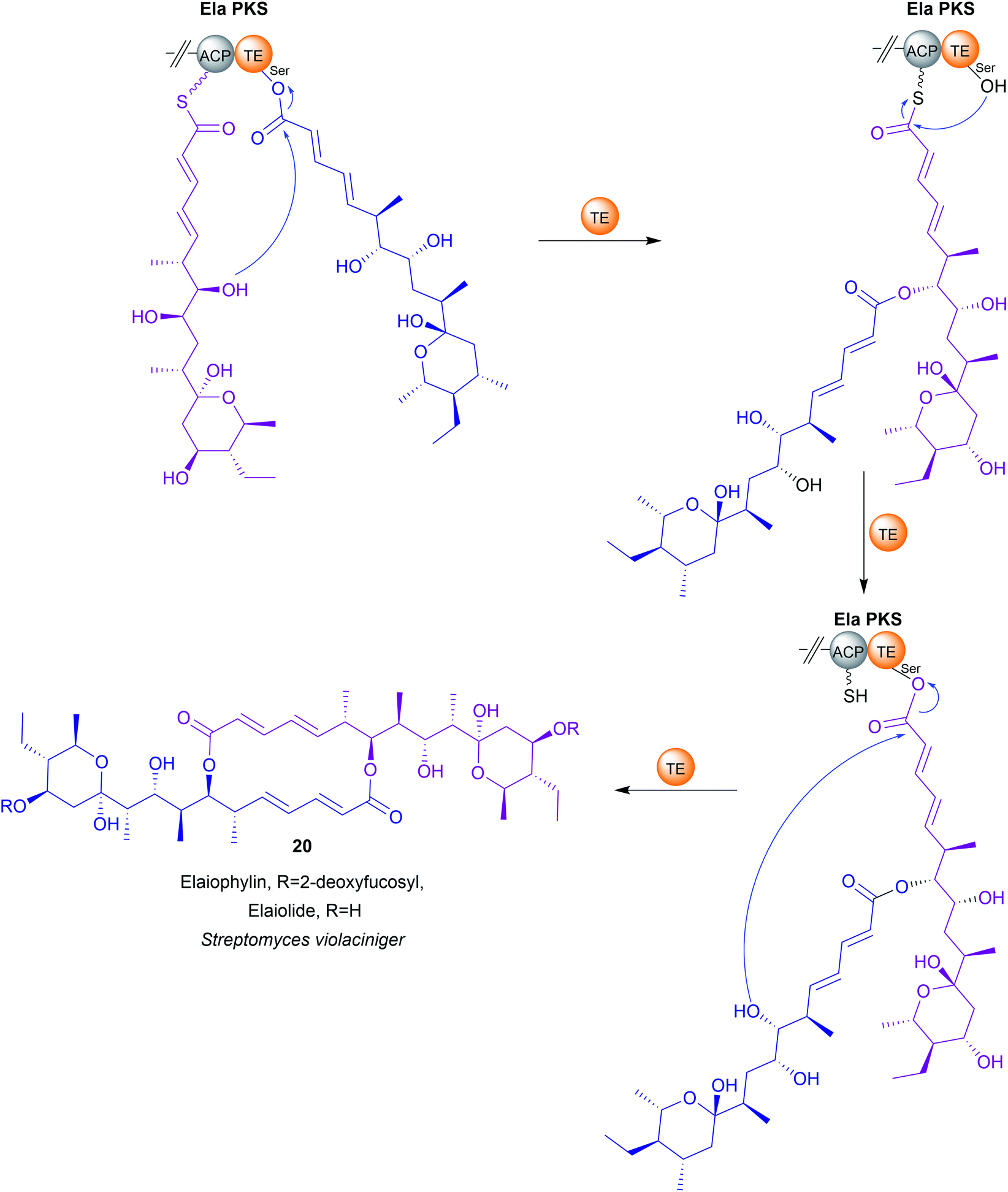 | ||
| Fig. 9 TE-catalysed “backwards transfer” dimerisation mechanism in elaiophylin (20) biosynthesis. | ||
In rare cases, two contiguous TE domains are found on the C-terminus of NPRS proteins. Such “tandem” TE domains are present in the biosynthetic pathways for the cyclic lipopeptides teixobactin,57 arthrofactin,58 malleipeptin,59 syringopeptin,60 massetolide A,61 and the cyclic peptide lysobactin.41 Although not all examples have been biochemically characterised, in general, the first TE domain of the pair is responsible for lactonisation and release. The role of the second TE is less clear and differs from case to case. In the case of teixobactin biosynthesis, only when the active site serine residues of both TE domains were mutated was chain release activity fully abolished, suggesting that the two domain cooperate to release the NRP chain.57 In the case of the arthrofactin biosynthesis pathway, mutating the active site of the second TE decreased arthrofactin production by 95%, indicating that it is important, but not essential, for chain release.58 In contrast, mutating the active site serine in the second TE domain of the lysobactin biosynthesis pathways had no detectable effect on lysobactin production.41 Instead, the second TE domain demonstrated deacetylase activity, making it more akin to the “proofreading” type II TE enzymes discussed later in Section 4. It was even proposed that this TE domain may be post-translationally separated to act as a standalone type II TE, as the two TE domains are readily proteolytically cleaved from one another.58 However, direct evidence for this occurring or being relevant in vivo is lacking.
TE domains that recognise amine groups are also capable of catalysing chain release via oligomerisation reactions. The best characterised example is in the biosynthesis of gramicidin S (NRP), a cyclic dilactam antibiotic produced by Bacillus brevis (Fig. 8).51 The research conducted on gramicidin S (22) biosynthesis provided the first evidence for the “backwards pass” mechanism discussed previously in Section 3.2.1.51
In addition to intramolecular amine groups, the less nucleophilic nitrogen atom of amide groups is also selected by some TE domains. Examples include the biosynthesis of the tetramic acids jamaicamide (PK–NRP hybrid)67 and reutericyclin (PK–NRP hybrid).68 In these cases, the nitrogen of an internal secondary amide attacks the C1 carbon of the thioester, forming a tetramate ring.67,68
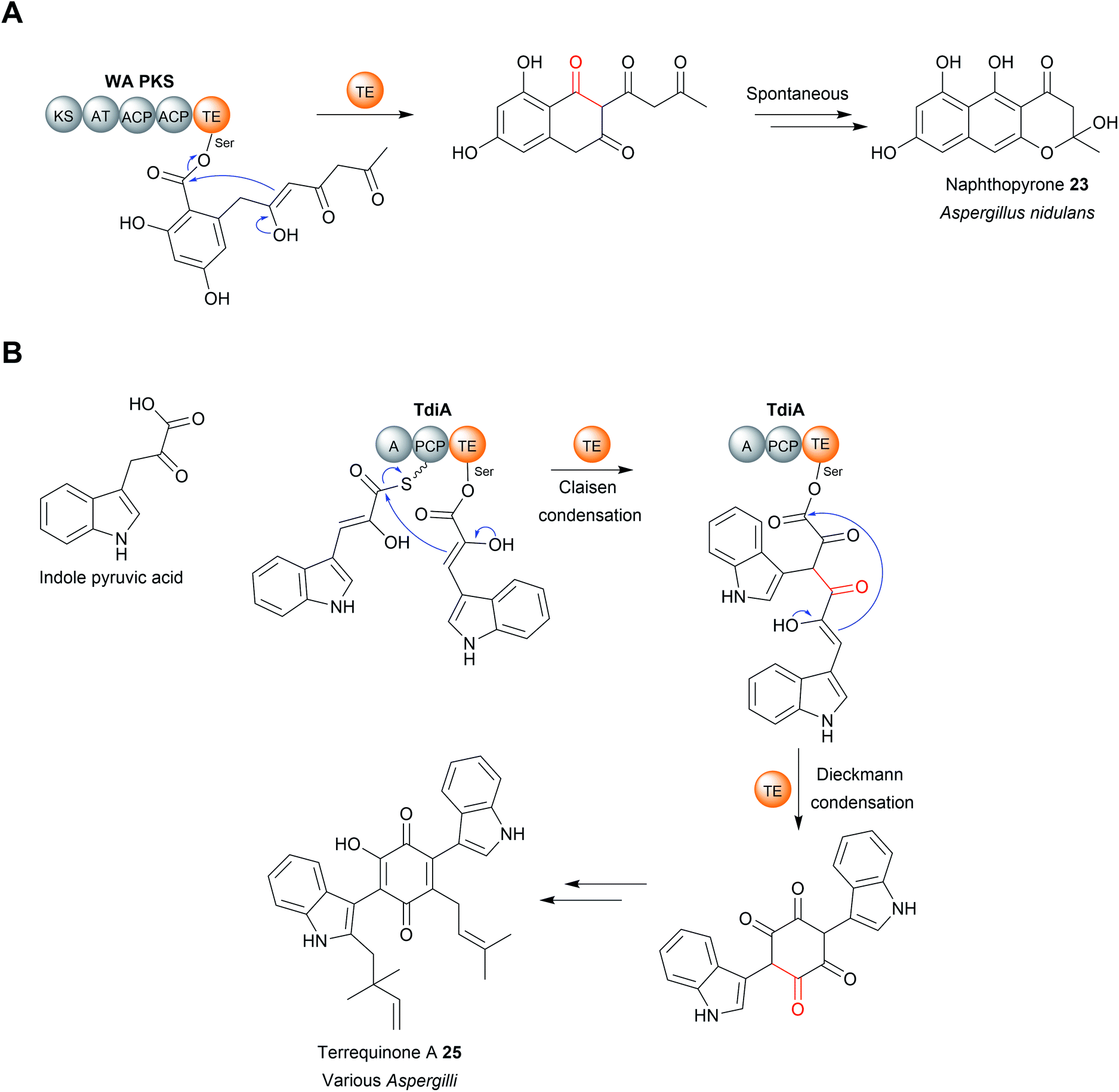 | ||
| Fig. 10 Chain release by a TE-catalysed Dieckmann condensation. (A) TE-catalysed Dieckmann condensation in the biosynthesis of naphthopyrone (23). (B) TE-catalysed Claisen condensation and Dieckmann condensation in terrequinone A (25) biosynthesis. | ||
TE domains are also speculated to catalyse C–C bond formation the cyclisation between C2 and C7 biosynthesis in the polyketides lasalocid, avermectin, and melingmycin, though direct evidence is lacking.22,76–78
The TE domain from the biosynthesis pathway to terrequinone A (25) (Aspergillus) differs in that it catalyses two C–C bond formations: a Claisen condensation and a Dieckmann condensation79 (Fig. 10B). The Claisen condensation first joins two molecules of indole pyruvic acid (derived from L-tryptophan).79 A Dieckmann condensation then occurs to cyclise the indole pyruvic acid dimer, creating the core of terrequinone A (25).79
The dihydromaltophilin (also called HSAF – heat stable antifungal factor) biosynthesis pathway from Lysobacter enzymogenes is one of the few characterised examples of a carbanion selecting TE domain from bacteria (another being in α-lipomycin biosynthesis).80,81 One of the intriguing features of HSAF is that it is comprised of two separate polyketide chains that are linked via an L-ornithine residue. To achieve this linkage, L-ornithine must form an amide bond with each of the polyketide chains.80,82In vitro work with the purified TE from domain indicated that, in addition to catalysing a Dieckmann cyclisation to form the tetramate ring, it also catalyses amide bond formation.80,82
4 Chain release catalysed by type II thioesterases (α/β hydrolase fold)
The TE domains discussed thus far have been discrete domains within a larger PKS or NRPS enzyme. An alternative strategy, however, is to utilise a standalone thioesterase enzyme, called a type II thioesterase (TEII), encoded elsewhere in the biosynthetic gene cluster. Like their domain counterparts, TEIIs also contain an α/β hydrolase fold and a Ser–Asp–His triad.86 TEIIs are commonly encoded in both PK/NRP biosynthetic gene clusters, where they are responsible for hydrolysing small non-reactive thioester intermediates that can stall the PKS/NRPS.86 Examples of such intermediates include ACP/PCP-linked acetyl groups that arise either from the premature decarboxylation of malonyl-CoA or by the loading of acetyl-CoA onto the CP by a PPtase.86–90 TEIIs are therefore often referred to as having an “editing” or “proofreading” role in a biosynthesis pathway.86 As such, mutational inactivation of a gene encoding a TEII often decreases, but does not abolish, natural product production by the biosynthetic gene cluster (BGC).91–93 In several unusual cases, domains that resemble TEIIs are found within a PKS enzyme, as if they were a type I TE domain.39,41,94 TEIIs are phylogenetically distinct from TEIs, and can often be distinguished by containing a conserved methionine adjacent to the catalytic serine (GxSMG).39,94 Like their standalone counterparts, these TEII “domains” appear to have a role in hydrolytic proofreading, helping to maintain the flux of the biosynthetic pathway.39,94While TEIIs are best known for their proofreading function, in some biosynthetic pathways they are responsible for catalysing final product chain release (Fig. 11A). Such examples are found in the biosynthetic pathways of the bacterial polyether ionophores nigericin (27),95 monensin (PK) (28),96 nanchangmycin (PK) (29),97,98 and maduramicin (PK)99 (Fig. 11B). In these cases, after the final extension reaction has occurred the PK chain is transferred to a standalone ACP protein, followed by hydrolytic release catalysed by a TEII enzyme.95,96,98,99
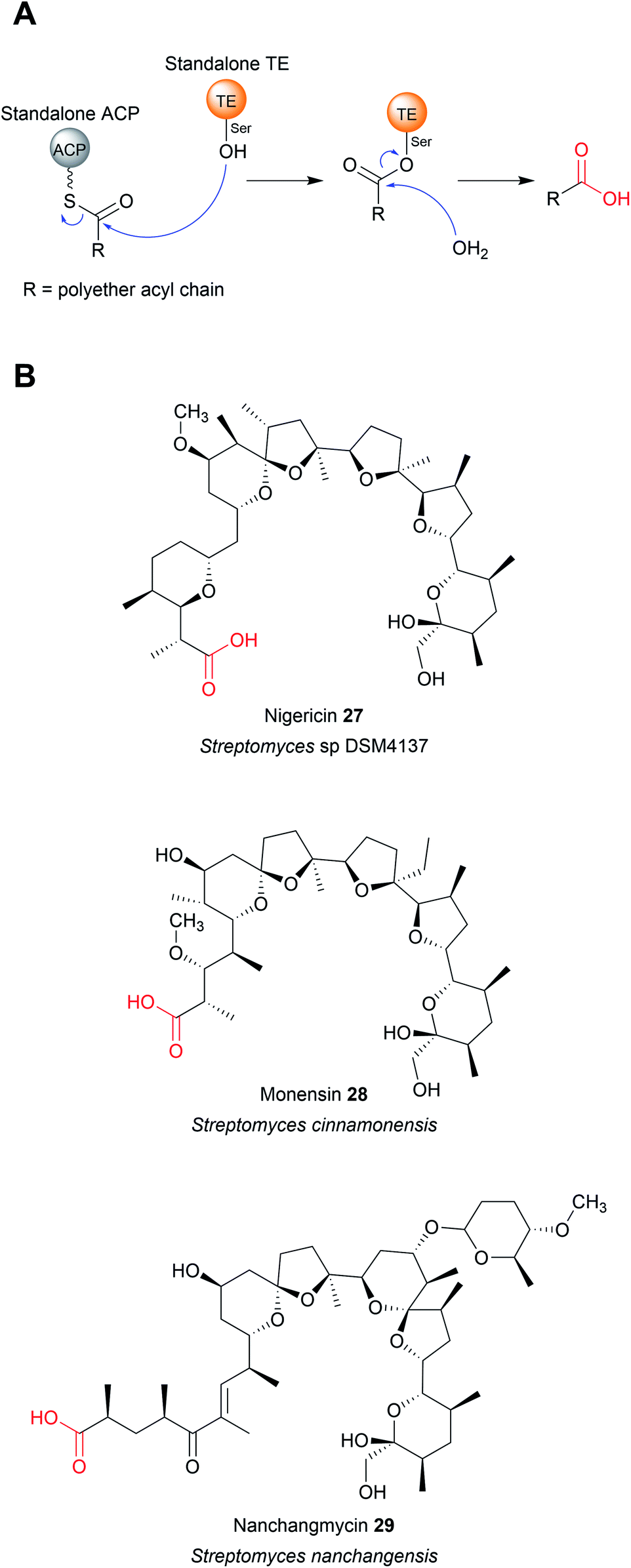 | ||
| Fig. 11 Chain release by TEII enzymes in polyether biosynthesis. (A) Mechanism of polyether chain release from a standalone ACP domain by a TEII enzyme. (B) Examples of polyether polyketides where chain hydrolytic chain release is catalysed by a standalone TEII enzyme. | ||
TEII enzymes also catalyse the hydrolytic chain release of non-polyethers, including zaragozic acid,100 colibactin,101 kinamycin (30),102 and possibly indanomycin.22,103 Kinamycin is noteworthy as it is a type II polyketide. Type II polyketides are aromatic natural products produced by type II PKSs (described in Section 2).14 The TEII enzyme AlpS has been shown to be essential for kinamycin biosynthesis in vivo and its hydrolytic activity with a SNAC-substrate analogue demonstrated in vitro.102 (Fig. 12A). The chain release mechanism in type II polyketide biosynthesis pathways are poorly understood, having even been speculated to occur though spontaneous hydrolysis/aldol reaction.11,14 The discovery that some utilise TEII enzymes is therefore an important discovery for the field.
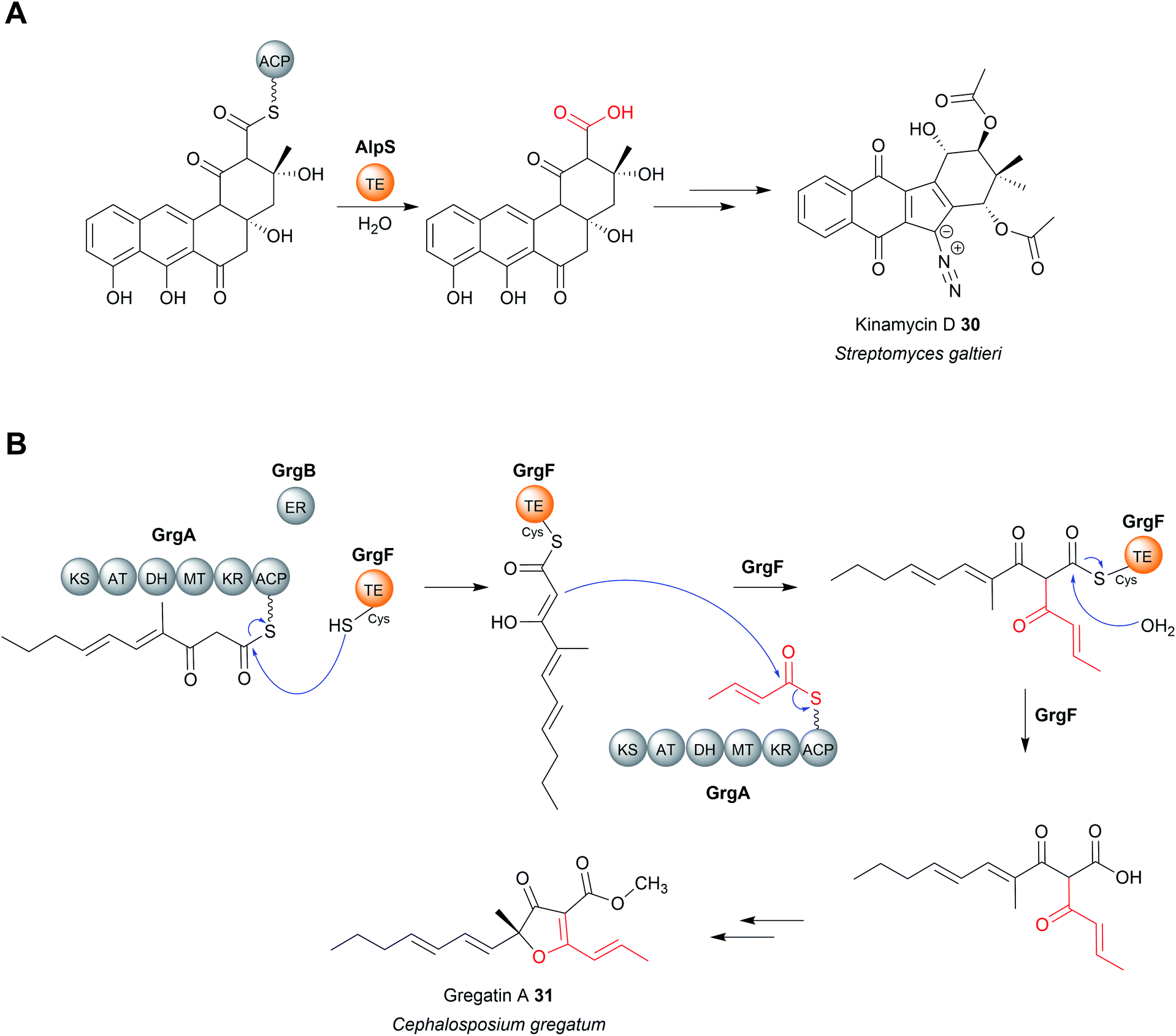 | ||
| Fig. 12 Chain release by TEII enzymes in kinamycin D and gregatin A biosynthesis. (A) The hydrolytic chain release of the type II polyketide kinamycin (30) is catalysed by the standalone TEII AlpS. (B) In gregatin A (31) biosynthesis the TEII GrgF catalyses a Claisen condensation to join two PK chains and hydrolysis of the linear dimer. | ||
Chain release catalysed by TEII enzymes is not always hydrolytic.104 The TEII enzyme in gregatin A (31) biosynthesis catalyses chain release by a Claisen condensation reaction.104 The furanone core of gregatin A is produced by the fusion of two acyl chains. Intriguingly, in vitro reconstitution experiments indicate that the two different chains are both produced by the PKS GrgA.104 The thioesterase GrgF then catalyses fusion of the two PK chains via a Claisen condensation, followed by hydrolytic chain release of the linear dimer (Fig. 12B). The linear dimer is then proposed to undergo a spontaneous cyclisation reaction to produce the furanone-containing gregatin A.104 Aside from gregatin A biosynthesis, a non-hydrolytic TEII enzyme is also encoded in the BGC of pyoluteorin, a PK–NRP hybrid first isolated from P. aeruginosa strains T359 and IFO 3455.105,106 The TEII (PltG) is proposed to catalyse the release of the pyoluteorin intermediate from the PKS PltC the via a Dieckmann condensation, forming a six-membered ring.106,107
5 Chain release catalysed by hot-dog fold thioesterases
Enzymes with thioesterase activity are highly diverse, falling into at least 23 distinct families.108 Furthermore, those that catalyse chain release are not restricted to the α/β hydrolase fold family. One example are the homotetrameric hot-dog fold thioesterases encoded in biosynthetic gene clusters of the enediyne polyketides including calicheamicin (CalE7),109 dynemicin (DynE7),110 and C-1027 (SgcE10) (Fig. 13).111–113 Enediynes are potent DNA-damaging agents synthesised by type I PKSs.114 A hot-dog folds consists of a 5–6 strand curved β-sheet “bun” wrapping around a long central α-helix “hot-dog”.115 Aside from their role in enediyne biosynthesis, hot-dog fold thioesterases are found in both prokaryotes and eukaryotes where they hydrolyse acyl-CoA to release fatty acids and CoA.116 In enediyne biosynthesis, these thioesterases catalyse the hydrolytic release of methylketol hexaene (32) and heptaene (33) (Fig. 14A).117,118 While both products were initially proposed to be enediyne biosynthetic intermediates, they are now believed to be shunt products.117,118 The role of these hot-dog fold thioesterases is therefore akin to the proofreading TEII enzymes discussed in the previous section.11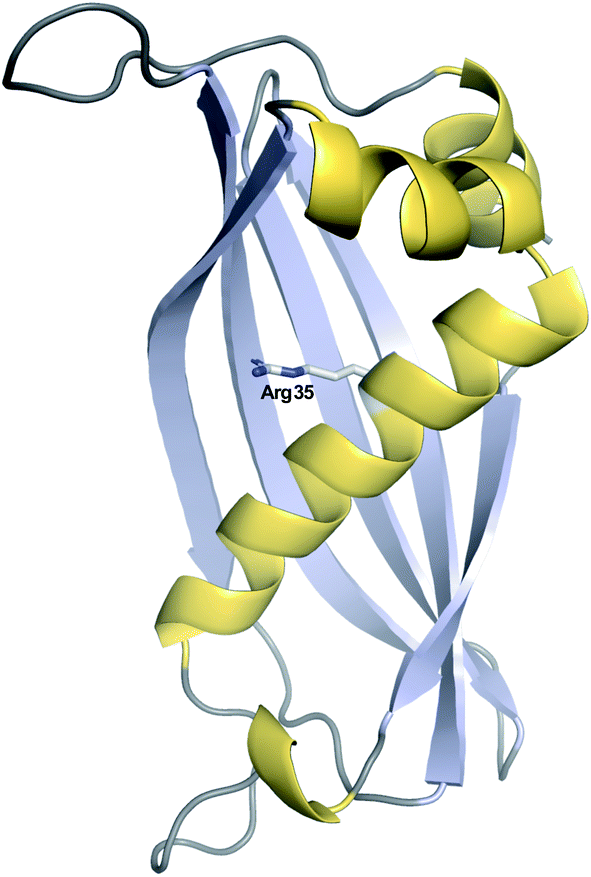 | ||
| Fig. 13 Crystal structure of the hot-dog fold thioesterase DynE3. Displayed is a single monomer of the tetrameric DynE3, a hot-dog fold thioesterase with a proofreading role found in the biosynthesis pathway of the enediyne dynemicin (PDB: 2XEM). The catalytic Arg35 residue, proposed to stabilise the quaternary oxyanion polyketide intermediate, is highlighted. | ||
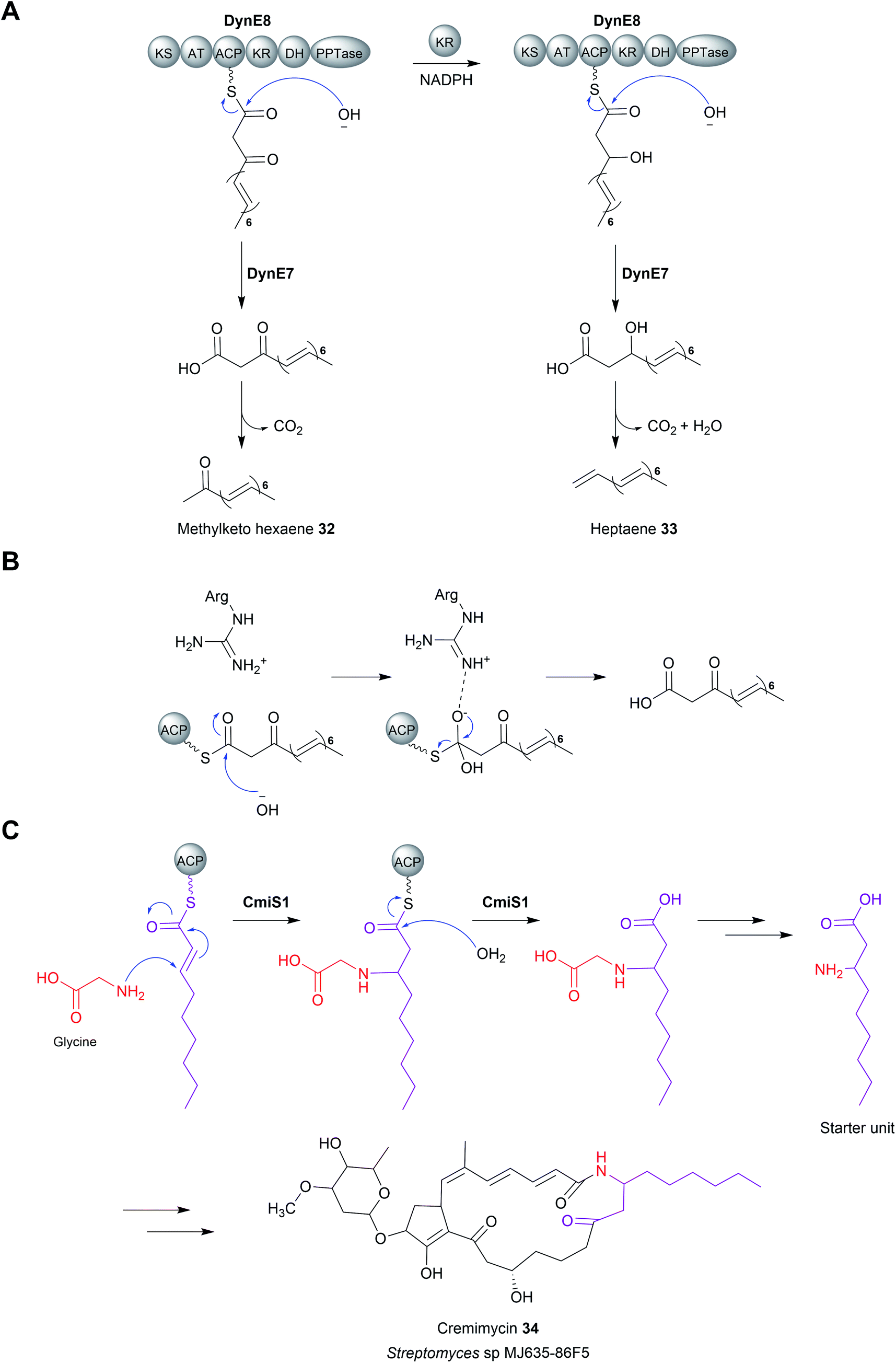 | ||
| Fig. 14 Chain release by hot-dog fold thioesterases (A) the release of methylketo hexanene (32) and heptaene (33) by the hot-dog fold thioesterase DynE8 in dynemicin biosynthesis. These linear products are now believed to be shunt products rather than biosynthetic intermediates. (B) The proposed mechanism of hot-dog fold thioesterases involving an oxyanion-stabilising catalytic arginine. (C) The hot-dog fold thioesterase CmiS1 is responsible for catalysing hydrolytic chain release in cremimycin (34) biosynthesis. CmiS1 also catalyses the addition of glycine to the β-carbon. | ||
Mechanistically, hot-dog fold thioesterases are distinct from α/β-hydrolase fold thioesterases. Unlike the α/β-hydrolase fold thioesterases, the PK chain is never covalently bound to a hot-dog fold thioesterase, instead entering its active site while still tethered to the adjacent ACP domain.109,110 In terms of catalytic residues, a conserved arginine was shown via mutagenesis to be an essential for activity.109,110 The positively charged guanidinium group of the arginine is proposed to stabilise the oxyanion that forms on the C1 carbonyl following attack by a water molecule (Fig. 14B).109,110,119 The tetrahedral intermediate is resolved by loss of the Ppant group and release of the PK chain from the active site. Whether the attacking water molecule is activated via deprotonation (forming an hydroxide ion) is unknown, but could be performed by a conserved glutamic acid or tyrosine residue.110
In contrast to enediyne biosynthesis, in other biosynthetic pathways hot-dog fold thioesterases are responsible for hydrolytic release of the final product itself. A notable example is found in the biosynthesis pathway for the macrolactam cremimycin (34) produced by Streptomyces sp. MJ635-86F5.120 Cremimycin biosynthesis features the incorporation of an unusual extension unit derived from the β-amino acid 3-aminononanoate.120 The biosynthesis of this β-amino fatty acid is performed by the PKS enzymes CmiP4, CmiP3, and CmiP2. The three PKS enzymes produce non-2-enoyl-ACP that is hydrolysed by the standalone hot-dog fold thioesterase CmiS1 (Fig. 14C).120 However, prior to hydrolysis, CmiS1 first catalyses a Michael addition between non-2-enoyl-ACP and glycine, installing what will become the β-amino group.120 The crystal structure of a homologue of CmiS1 from Streptomyces avermitilis MA-4680, SAV606, which catalyses an equivalent reaction has been solved.121 An in vitro activity assay of SAV606 indicated that it also catalyses the Michael addition with glycine in addition to hydrolytic chain release. Analysis of the SAV606 structure led to the proposal that a histidine residue (His59) deprotonates the glycine amine group via a water molecule, thereby promoting its nucleophilic attack on the β-group of the PK chain.121 The same TE-catalysed mechanism for β-amino fatty acid biosynthesis is likely present in the biosynthesis pathways of the macrolactams ML-449 and BE-14106.122,123
6 Chain release catalysed by metallo-β-lactamase (MβL) thioesterases
A third family of standalone thioesterases that catalyse chain release resemble metallo-β-lactamase enzymes (MβL). β-Lactamase enzymes are widespread in bacteria where they have an important role in hydrolysing β-lactam antibiotics.124 MβL enzymes possess an αββα-fold and require a metal cofactor, typically up to two Zn2+ ions, to function.125 Fungal genomes also encode β-lactamases, although their functions are not always clear and can be involved in processes other than xenobiotic degradation.124 In select cases, MβLs are thioesterases (MβL-TEs) that catalyse the hydrolytic chain release of fungal polycyclic polyketides. These polycyclic compounds are synthetised by non-reducing polyketide synthases (nrPKSs). Unlike the modular type I PKSs, nrPKSs act iteratively to synthesise a highly reactive poly-β-keto PK chains that undergo multiple aldol condensations to form aromatic polycyclic compounds.126,127 The regioselectivity of the first aldol condensation is controlled by a specialised product template (PT) domain within the nrPKS, which determines the cyclisation pattern for the compound as a whole.128 In addition, nrPKSs also utilise an N-terminal starter unit acyltransferase (SAT) domain for starter unit selection, which can range from acetyl-CoA to longer fatty acid-derived started units.129,130 While some nrPKSs catalyse hydrolytic chain release using an integrated TE/CLC or reductase domain (discussed later), others lack any apparent chain releasing domain.The nrPKS ACAS (domain architecture SAT–KS–AT–PT–ACP) from Aspergillus terreus is responsible for the biosynthesis of atrochrysone carboxylic acid (35), a precursor to atrochrysone and endocrocin (36) (Fig. 15A).131 While no obvious chain releasing domain is present in ACAS, the BGC also encodes a MβL (ActE) that was demonstrated to catalyse hydrolytic chain release in vitro using a SNAC substrate analogue.131 ActE contains the conserved metal binding site (THxHxDH) characteristic of MβLs likely binding Zn2+.131 Dialysing ActE against a buffer containing the chelating agent EDTA abolished its activity, demonstrating that the metal ions are essential.131 The Zn2+ ion(s) are proposed to stabilise a nucleophilic hydroxide ion and the subsequently formed tetrahedral oxyanion intermediate.125,126 MβL-TEs enzymes are also encoded in the BGCs of the related polyketides, such as asperthecin produced by Aspergillus nidulans, where they likely have the same role in hydrolytic release.132–136
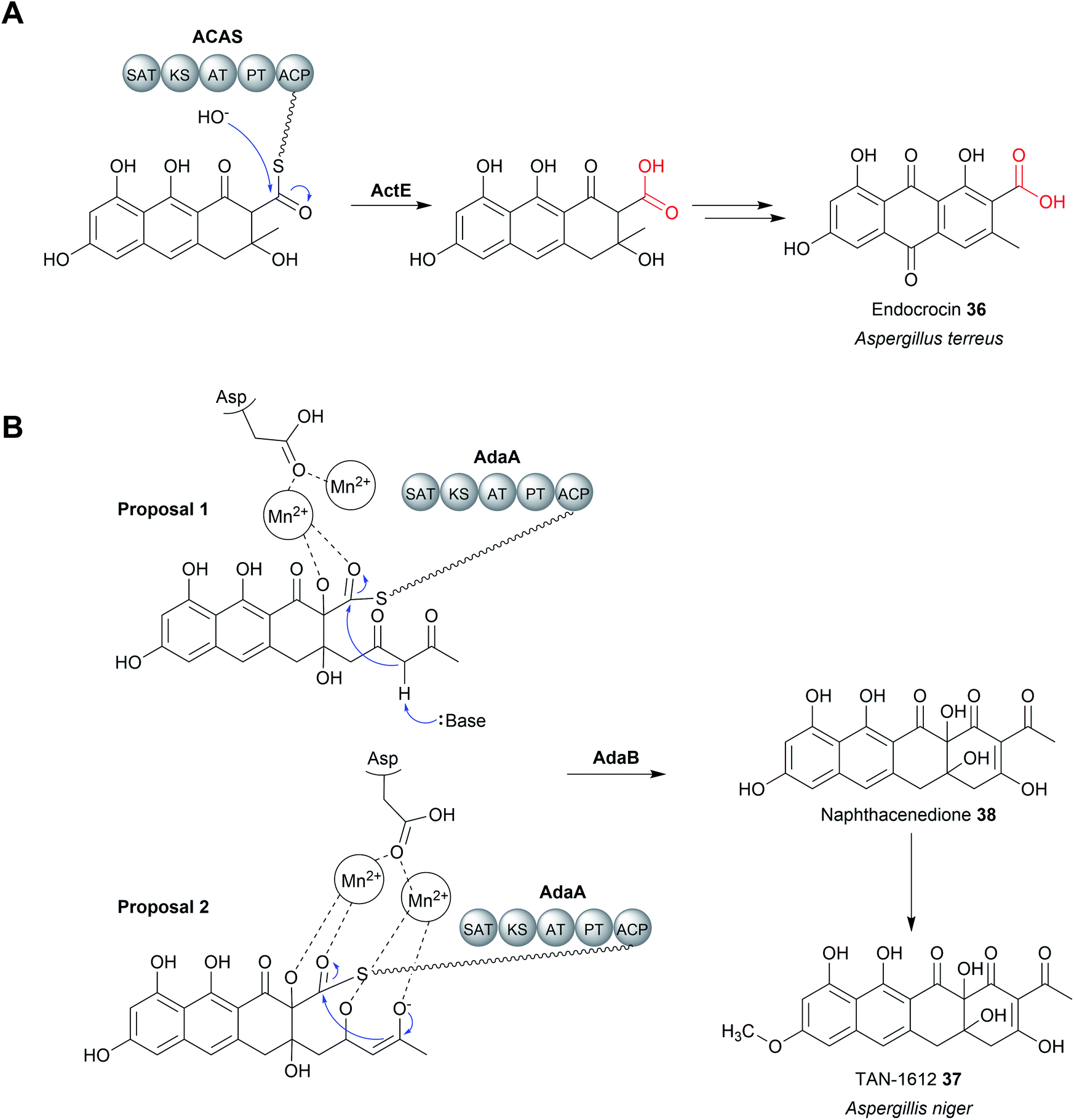 | ||
| Fig. 15 Chain release catalysed by metallo-β-lactamase (MβL) thioesterase. (A) The MβL ActE is responsible for chain release of the endocrocin precursor from the PKS ACAS. (B) The two possible mechanisms have been proposed for the Mn2+ dependent Dieckmann condensation catalysed by AptB/AdaB. | ||
MβL-TEs have also been found that catalyse chain release via a Dieckmann condensation reaction.126 The fungus Aspergillus niger uses a nrPKS to produce the naphthacenedione core of the natural product, TAN-1612 (37).126 The MβL-TE AdaB was demonstrated to catalyse a Dieckmann condensation between C18 (nucleophilic) and C1 (electrophilic) to form the fourth and final ring of the naphthacenedione core (38).126 Interestingly, AdaB was shown to only catalyse the Dieckmann condensation when the tricyclic intermediate is hydroxylated at the C2 position by the monooxygenase AdaC. If no hydroxylation occurred, then AdaB catalyses hydrolytic chain release. A homologue of AdaB from the asperthecin biosynthesis pathway, AptB, could also catalyse the Dieckmann cyclisation to form naphthacenedione.126 Closer biochemical characterisation of AptB revealed it binds two Mn2+ ions, rather than Zn2+. In the proposed mechanism for AdaB/AptB function, the two Mn2+ ions facilitate substrate binding and may assist in the deprotonation of the C18 α-proton, though additional experimental evidence is required (Fig. 15B).126 Given the widespread role of β-lactamases in catalysing hydrolysis reactions, it is likely that the Dieckmann condensation activity is a more recent adaptation.
7 Chain release catalysed by penicillin binding protein (PBP)-like enzymes
A newly discovered class of chain releasing enzyme are homologs of penicillin binding proteins (PBP). The PBPs are peptidases that catalyse the final transpeptidation reaction during bacterial cell wall biosynthesis.137 The ability of PBP-like enzymes to catalyse chain release was first discovered in the biosynthesis pathway of surugamide A–F, a group of linear and cyclic NRPs produced by Streptomyces sp. JAMM992.138 Surugamide A-E are related cyclic NRPs all produced by the NRPS enzymes SurA and SurD.139 In contrast, surugamide F is an unrelated linear NRP carboxylic acid produced by the NRPSs SurB and SurC, the genes for which are encoded adjacent to surA and surD in the same BGC.139 Interestingly, none of the encoded NRPS enzymes contain a thioesterase or another previously characterised chain release domain.138 Just upstream of surA is a small gene encoding a putative 28 kDa penicillin binding protein, SurE.138 The ability of SurE to catalyse a chain releasing lactamisation reaction was demonstrated by incubating an linear SNAC precyclisation precursor of surugamide B with purified SurE, resulting in formation of surugamide B.138 Furthermore, creating an in-frame deletion in surE abolished not the only production of surugamide A–E, but also surugamide F.140–142 SurE is therefore surprisingly responsible for catalysing chain release in both pathways.140–142 An in vitro assay using SNAC-surugamide F revealed that the hydrolysis product is only a minor product, with the major product being a lactam. Surugamide F is therefore proposed to be produced from this lactam by an as yet undiscovered peptidase.142In regards to the enzymatic mechanism of SurE, it contains the conserved Ser–Tyr–His–Lys catalytic tetrad of other PBP peptidases.137,138,140–142 In PBP peptidases the serine acts as a nucleophile while the other catalytic residues are involved in proton transfer/transition state stabilisation.137,141 Mutagenesis of the serine residue in SurE to alanine abolished its activity, consistent with a role in forming an oxoester linkage to the peptide chain (analogous to the catalytic serine of α/β hydrolase thioesterases). The role of the other residues in catalysing lactamisation of the surugamides are still unclear.141 SurE homologues are also encoded in the biosynthetic gene clusters of other NRPs, sometimes even as a dedicated domain within a NRPS enzyme.141 There is therefore still much to be explored in regards to the function and products of PBP-like chain releasing enzymes.
8 Chain release catalysed by reductase (R) domains
8.1 Structure and mechanism of R domains
Aside from TE domains, another chain releasing domain located on the C-termini of some PKS/NRPS enzymes is a reductase (R) domains. R domains catalyse the reductive release of PK/NRP chains as either aldehydes (via a two-electron reduction) or primary alcohols (via a four-electron reduction) (Fig. 16, 17 and 18A).13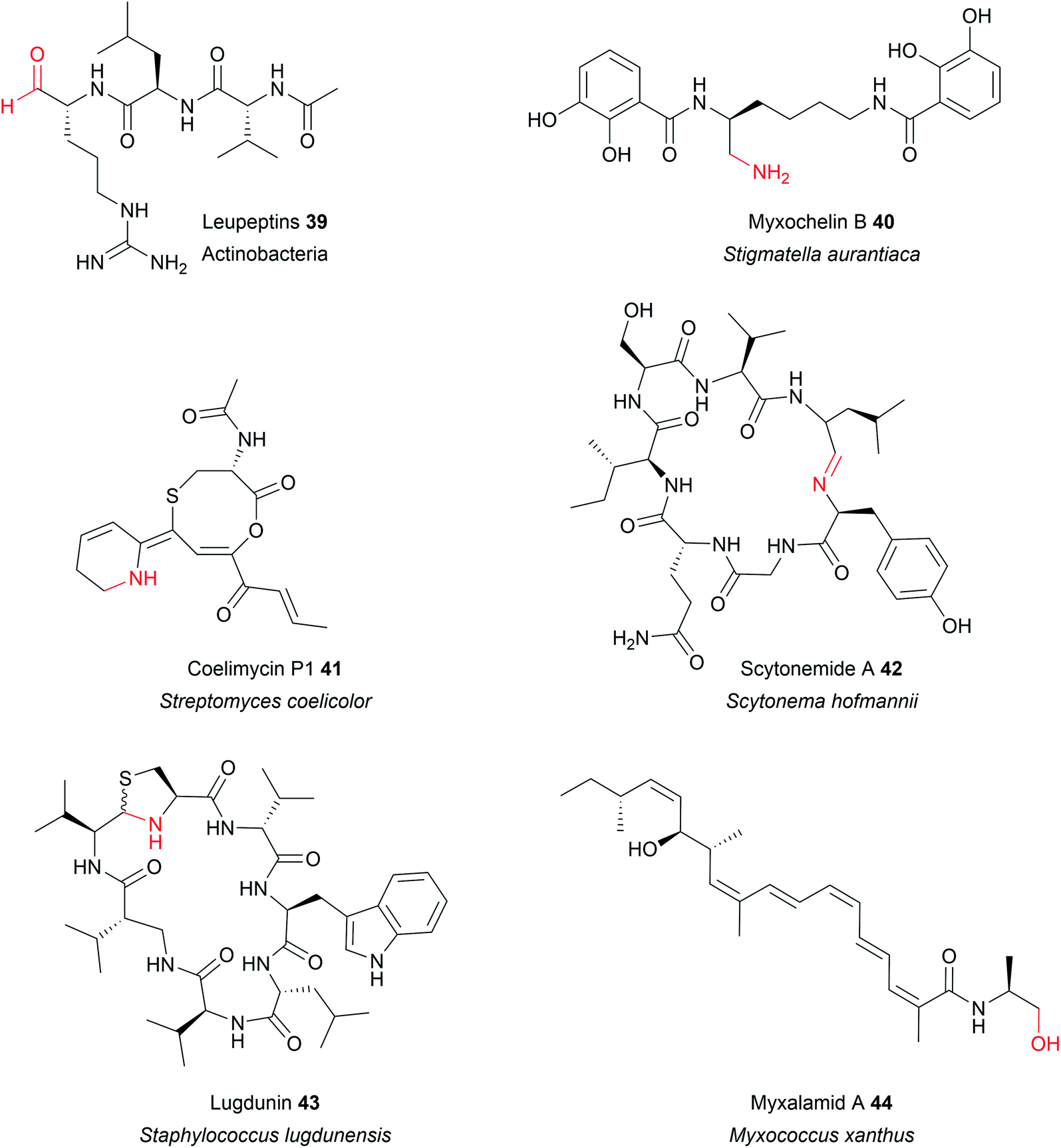 | ||
| Fig. 16 Examples of natural products synthesised by PKS/NRPS and released/modified using R domains. | ||
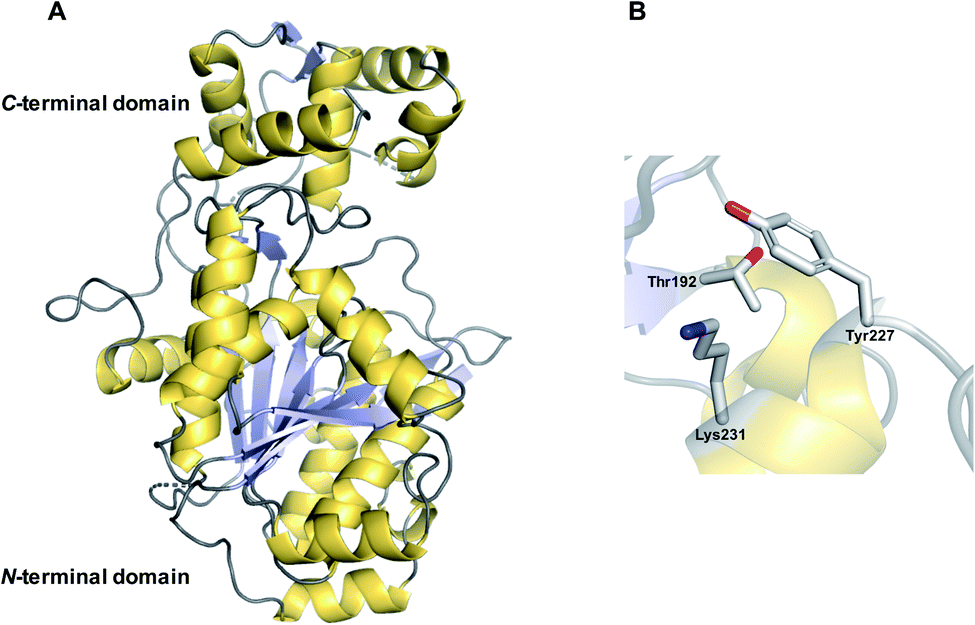 | ||
| Fig. 17 Crystal structure of an R domain. (A) The crystal structure of the R domain from mycobacterial lipopeptide biosynthesis (PDB: 4DQV). The N-terminal domain is responsible for NAD(P)H binding while the C-terminal domain is responsible for substrate binding. (B) The Thr–Lys–Tyr catalytic triad of the R domain located in the N-terminal region. | ||
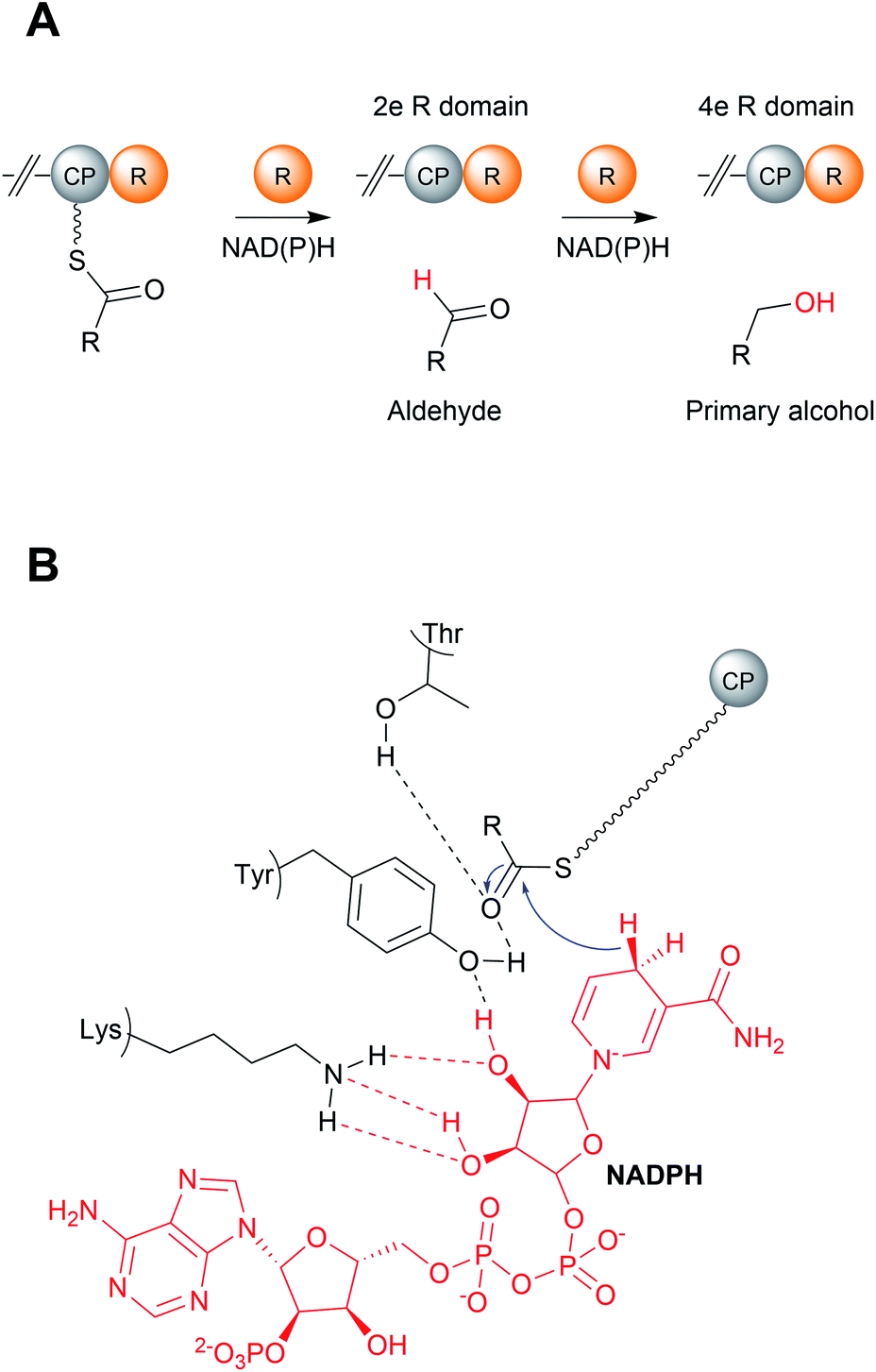 | ||
| Fig. 18 Mechanism of reductase (R) domains. (A) R domains can either catalyse a two-electron reduction to release the PK/NRP chain as an aldehyde or a four-electron reduction to release a primary alcohol. (B) The catalytic mechanism of R domains. The conserved lysine and tyrosine residues bind to NAD(P)H, while the tyrosine binds to the PK/NRP chain. | ||
R domains are mechanistically and structurally distinct from α/β-hydrolase thioesterases, belonging instead to the short-chain dehydrogenases (SDR) family of NAD(P)H dependent oxidoreductases.143 Members of this family all possess an N-terminal Rossmann fold: a sheet of seven parallel β strands flanked by α-helices on either side.144,145 Interestingly, despite their differences, the central β-sheets of TE and R domains have a similar spatial arrangement.144,145
An individual R domain (ca. 400 amino acids in size) can be subdivided into two regions: an N-terminal Rossmann fold region responsible for NAD(P)H binding, and a C-terminal region responsible for substrate binding.13 In contrast to TE domains, where the PK/NRP chain is covalently bonded to the TE domain, R domains act directly on CP-linked PK/NRP chains.13 The diphosphate portion of NAD(P)H interacts with the peptide backbone of a GxxGxxG nucleotide binding motif conserved within the N-terminal region.146 The C-terminal region consists of 4–6 α-helices and 2 β sheets.144–147 The N-terminal region also contains a mobile “gating loop” that interacts with the upstream carrier protein (ACP or PCP) and is proposed to regulate both the binding of NAD(P)H and fix the Ppant arm into a reactive conformation.144,147 Recent work also identified a hydrophobic pocket within the R domain responsible for binding the geminal dimethyl group of the Ppant arm.147 Unlike the highly conserved N-terminus region, the sequence identity of the C-terminal region is highly variable, likely reflecting the diversity of substrates recognised by R domains.144,146,147 Despite this notable sequence diversity, the C-terminal region of R domains are distinguished from other SDR members by containing a short helix-turn-helix motif important for the interface between the R domain and upstream CP.144,146,147
R domains contain a Thr–Tyr–Lys catalytic triad characteristic of SDR family members. The catalytic tyrosine and lysine residues are both critical for binding NAD(P)H, while the Thr stabilises the PK/NRP thioester substrate (Fig. 18B).144,146,148 Hydride transfer by NAD(P)H to the C1 carbon of the PK/NRPS thioester generates a tetrahedral thiohemiacetal intermediate. The intermediate is resolved by loss of the Ppant group, generating a free aldehyde. In the case of four-electron R domains, NAD(P)+ dissociates and is replaced by second molecule of NAD(P)H.13 This second NAD(P)H transfers a hydride to the electrophilic C1 carbon of the aldehyde, resulting in the formation of primary alcohol.13 For several R domains, the reduction to the alcohol occurs faster than the reduction to the aldehyde.144,146 Aldehyde reduction is accompanied with a notable electronic shift, changing an electrophilic aldehyde to a nucleophilic alcohol.13 How some R domains exclusively catalyse a two-electron reduction while others a four-electron reduction is still poorly understood.147,148 A study on NRPS-related carboxylic acid reductases (CARs), indicated that conformational changes in the NAD(P)H binding site control whether this second reduction occurs.145,149 CARs are multidomain enzymes (A–PCP–R structure) that reduce carboxylic acids to their corresponding aldehydes. By comparing the structures of several CAR–R domains, a conformational change in the loop connecting the N and C terminal regions, particularly in an aspartic acid residue, was identified.145,149 One of the loop conformations appeared to facilitate NAD(P)H binding, while the other interfered with it. Based on these observations, it was proposed that the binding of the PK/NRP chain to the R domain promotes NAD(P)H binding, resulting in aldehyde formation.145,149 The aldehyde, however, is unable to maintain the favourable NAD(P)H binding conformation of the R domain, preventing a second molecule of NAD(P)H binding. In support of this theory, mutating the identified Asp residue to glycine enabled the R domain to form the primary alcohol product.149 There are also other factors at play, however, as some R domains contain this Asp residue but still perform four-electron reductions.145,150 Furthermore, biophysical studies have also demonstrated that NAD(P)H binding to the R domain is not dependent on the PK/NRP substrate.148 Further complicating matters are R domains that produce both two-electron and four-electron products, discussed in the following section. The question of how R domains control whether a two-electron or four-electron reduction takes place therefore remains open.147,148
8.2 PK and NRP pathways that use R domains
The natural products produced using an R domain have been extensively covered in an excellent recent review.13 In brief, while the direct products of R domains are either aldehydes or primary alcohols, both functional groups can undergo additional transformations to further diversify the structure of the final product (summarised in Fig. 19).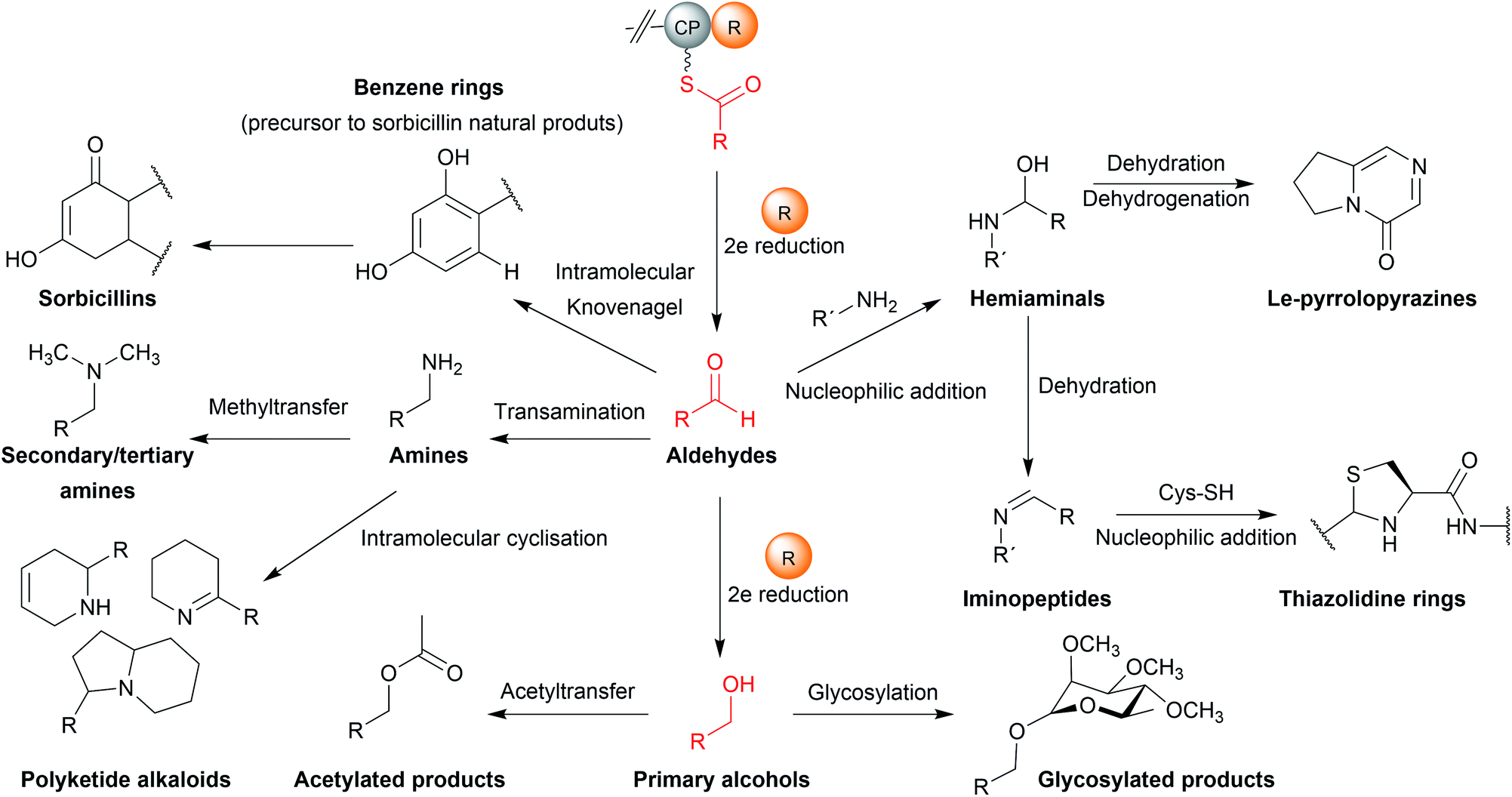 | ||
| Fig. 19 Functional groups and scaffolds derived from R domain products. While aldehydes and primary alcohols are the direct reduction products of R domains, both may be further modified to form other functional groups/moieties. | ||
8.3 Products of two-electron reductase domains
In some cases, the aldehyde generated by a two-electron R domain is retained in the final product. This tends to be uncommon, however, due to the general instability of aldehydes. The electrophilicity of aldehydes often means that the natural products containing them are often potent inhibitors of serine proteases.13 Examples of natural products containing aldehydes produced by two-electron R domains include the NRPs leupeptin (39), linear gramicidin,151 the flavopeptins, and the fellutamides.152–154Alternatively, the aldehyde group can undergo additional transformations to form a range of different functional groups.13 Such transformations can range from a simple transaminations, to more complex reactions that form macrocyclic peptides or heterocycles. If the aldehyde group undergoes a transamination, an amine is formed.13 The first characterised example of this was from the L-lysine biosynthesis pathway in Saccharomyces cerevisiae (also the first example of reductive release by an R domain).155 The tridomain carboxyl acid reductase Lys2 (A–PCP–R) was identified as responsible for lysine biosynthesis, releasing α-aminoadipate that subsequently undergoes a transamination to form L-lysine. Other examples include the siderophores myxochelin B (40),156,157 the zeamine antibiotics,158 and the lipopeptide antibiotic leucinostatin.159 In the case of leucinostatin, the amine of the product is diversified further by methylation.159 The amine can also act as a nucleophile in an intramolecular cyclisation reaction, as occurs in the biosynthesis of the alkaloids cyclizidine160 and coelimycin P1 (41).161
Cyclic iminopeptides, such as the nostocyclopeptides and scytonemide A (42) are formed when the aldehyde generated by a two-electron R domain is spontaneously attacked by an intramolecular amine, followed by a dehydration reaction.162,163 If a dehydration reaction does not occur the final product contains a hemiaminal ring, exemplified by the pyrrolobenzodiazepines anthramycin,164 sibiromycin,165 and tomaymycin.166 In the case of the cyclic peptide lugdunin (43), a peptide antibiotic and immune response modulator produced by the human microbiome, the imide carbon is subsequently attacked by the thiol group of the adjacent cysteine residue, forming a thioazolidine ring.167,168 In the case of the sorbicillin family of natural products, the aldehyde is attacked by a carbanion in a Knoevenagel cyclisation to form a benzene intermediate.169
In the biosynthesis of the fungal indole alkaloid malbrancheamide, a two-electron R domain releases L-Pro–L-Trp dipeptide as an aldehyde.170 The dipeptide aldehyde undergoes spontaneous cyclisation and dehydration to give a dienamine intermediate that is subsequently prenylated and spontaneously oxidised. The prenyl group contains a double bond that acts as a dieneophile in an apparent enzyme catalysed intramolecular [4 + 2] cycloaddition with the proline-derived pyrazinone ring.170 Malbrancheamide biosynthesis highlights an impressive case where an aldehyde generated by an R domain is used to generate a substrate for an intramolecular Diels–Alder reaction.
8.4 Products of four-electron reductase domains
Examples of primary alcohol-containing natural products produced by four-electron R domains include myxochelin A,156,157 myxalamid A (44),146,171 lyngbyatoxin,172 and, recently, a dipeptide produced by the industrially important Clostridium saccharoperbutylacetonicum N1-4.173 Myxochelin A is related to the aldehyde myxochelin B described in the previous section. Myxochelin A is formed if a second reduction reaction occurs before aldehyde transamination can occur.156,157 An R domain that releases both two-electron and four-electron products is also found in the zeamine biosynthesis pathway.158 A four-electron R domain also functions in the biosynthesis of mycobacterial glycolipids, a key component of the mycobacterial cell wall. In this case, the released alcohol is subsequently glycosylated with a molecule of O-methylated rhamnose.174 Other modifications of the primary alcohol are also possible, such as the acetylation that occurs in columbamide A/B biosynthesis.175In addition to the four-electron R domains already described, there are several examples where the reduction of the aldehyde is performed by a separate enzyme. In the case of gramicidin biosynthesis, the R domain releases an aldehyde followed by a second reduction catalysed by the standalone oxidoreductase LgrE, producing a primary alcohol.176 Another example is found in the fungal choline biosynthesis pathway where a CAR-like enzyme contains two contiguous R domains on its C-terminus.177 The A domain of this CAR enzyme adenylates glycine betaine, rather than an amino acid. The tandem R domains then act sequentially, each performing a two-electron reduction, to reduce the glycine betaine thioester to a primary alcohol.177 Whether such tandem R domains are present in PKS/NRPS pathways to generate primary alcohols is unknown.177
8.5 R* domains
A subset of R domains called R* domains catalyse chain release via non-redox Dieckmann condensation (Fig. 20).13,178 While R* domains still contain the Rossmann fold typical of R/SDR family proteins, they do not utilize NAD(P)H and often (but not always) contain a mutation in the Ser/Thr–Tyr-Lys catalytic triad and/or NADPH binding site.13,179,180 Examples of natural products synthesised using R* domains include the PK–NRP hybrid tenellin (45),181 the tetramate equisetin (PK),179 cyclopiazonic acid (PK),180 and the burnettramic acids (PK).182 The NRP quinolactones were also recently shown to use R* domains for chain release, the first example of an R* domain within an exclusively NRPS enzyme (rather than NRPS–PKS hybrid).183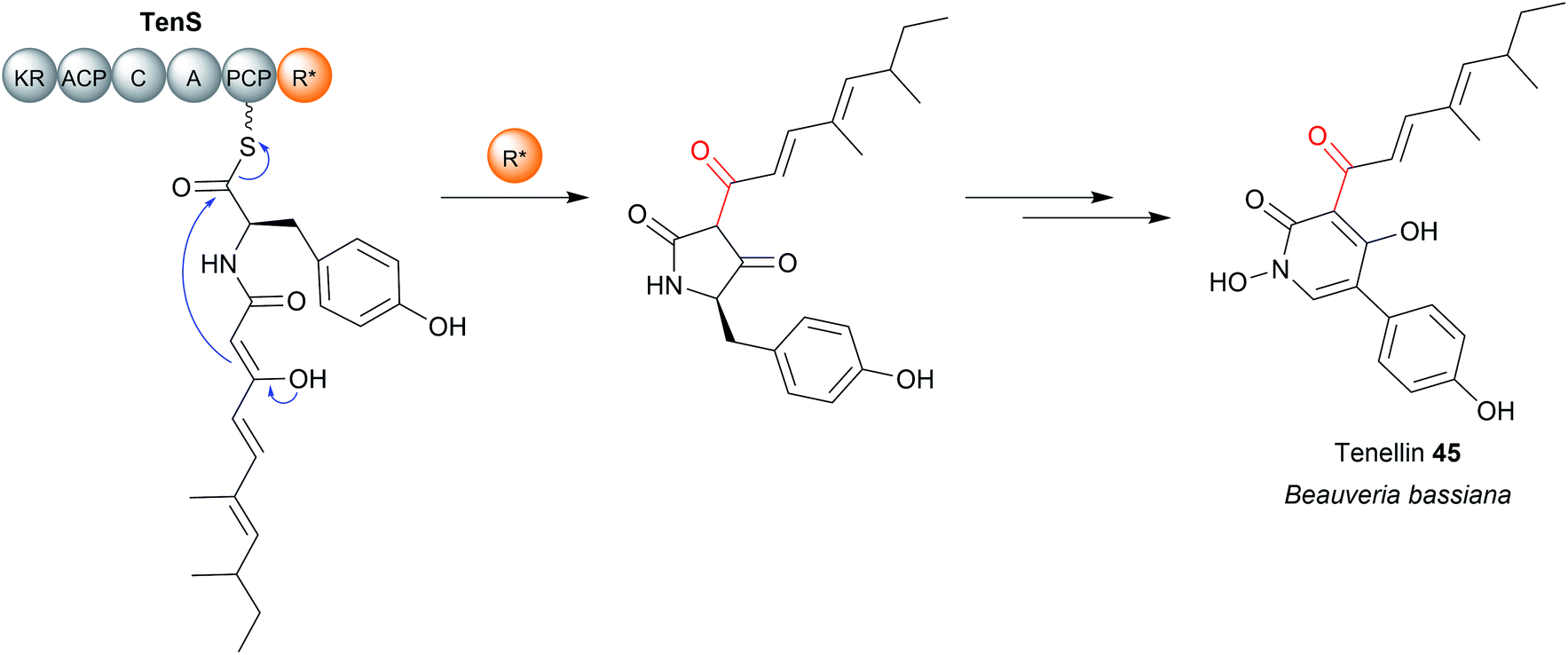 | ||
| Fig. 20 R* domains. R* domains are redox incompetent R domains that catalyse chain release by Dieckmann condensation reactions, such as in tenellin acid (45) biosynthesis. | ||
The enzymatic mechanism of R* domains is unclear. An essential aspartate residue was identified in the R* domain of CpsR, the PKS-NRPS hybrid responsible for cyclopiazonic acid biosynthesis.180 However, an equivalent Asp is also found in redox-competent R domains, where its role has not been determined.180 Whether the Asp plays a specific role in catalysing a Dieckmann condensation, such as extraction of the substrate α-proton to generate a nucleophilic carbanion/enolate, is currently unknown.180
9 Chain release catalysed by aldo-keto reductases
An exception to SDR-family reductive chain release was recently discovered in the closthioamide (46) biosynthesis pathway. Closthioamide is a symmetrical polythioamidated NRP synthesised by the obligate anaerobe Ruminiclostridium cellulolyticum.184 The biosynthesis of closthioamide (46) has been studied extensively, revealing an unusual thiotemplated NRPS-independent pathway.185–188 Closthioamide (46) is assembled from L-aspartate and chorismate, with the growing intermediate tethered to the standalone ACP CtaE (Fig. 21).185,187 Once the intermediate 47 has formed it is taken down two divergent paths, one catalysed by the enzyme CtaJ and the other by CtaK, which ultimately converge to form closthioamide.185 CtaK catalyses a two-electron reduction, releasing intermediate 48 as an aldehyde.185 However, unlike the R domain discussed in the previous section, CtaK belongs to the aldo-keto reductase family rather than the SDR family.185 Unlike SDR R domains, aldo-keto reductases lack a Rossmann fold, instead adopting a (β/α)8 conformation.189 Conserved arginine and lysine residues assist in binding the pyrophosphate backbone of NADH/NADPH, typically favouring the binding of NADPH.189 While aldo-keto reductases are known for catalysing a diverse range of redox reactions in both prokaryotes and eukaryotes, CtaK is the first characterised example of one that catalyses the reductive release of a NRP chain.185,189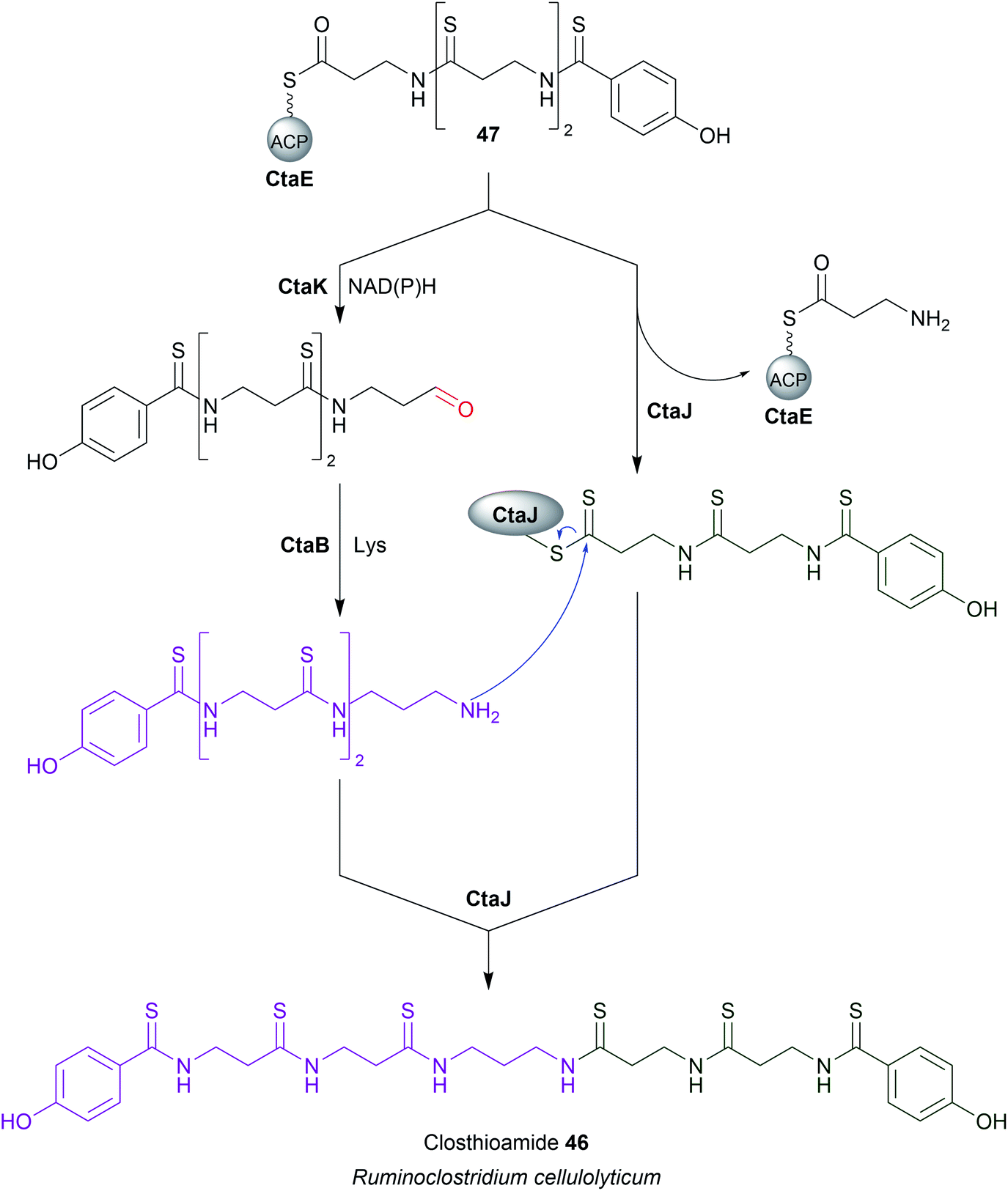 | ||
| Fig. 21 Chain release by an aldo-keto reductase enzyme. The biosynthesis of closthioamide (46) utilises the standalone aldo-keto reductase enzyme CtaK to catalyse chain release. | ||
10 Chain release catalysed by oxygenases
There are several biosynthetic pathways where chain release is proposed to occur through an oxidative mechanism. Examples include the myxothiazole (49),190 melithiazole,190,191 aurafuron,192 and pederin193 biosynthesis pathways. Myxothiazoles are synthesised by a mixture of PKS and PKS-NRPS hybrid enzymes. Myxothiazole A (49) has a terminal amide residue, indicating an unusual chain release mechanism. The final PKS enzyme in the myxothiazole A biosynthesis pathway, MtaG (C–A–MOx–PCP–TE) contains a monooxygenase-like (MOx) domain embedded within its A domain (Fig. 22A).190 MtaG is proposed to catalyse a condensation reaction between L-glycine and the myxothiazole intermediate, followed by hydroxylation of the α-position of L-glycine by the MOx domain.190 Hydroxylation is proposed to lead to spontaneous decomposition of the glycine residue, releasing myxothiazole A (49). The TE domain is then presumed to hydrolyse the residual PCP-bound glyoxylate,190 While plausible, experimental evidence is still required to confirm this mechanism. The related compound melithiazole appears to use the same mechanism of chain release, though the amide is subsequently converted into a methyl ester.190,191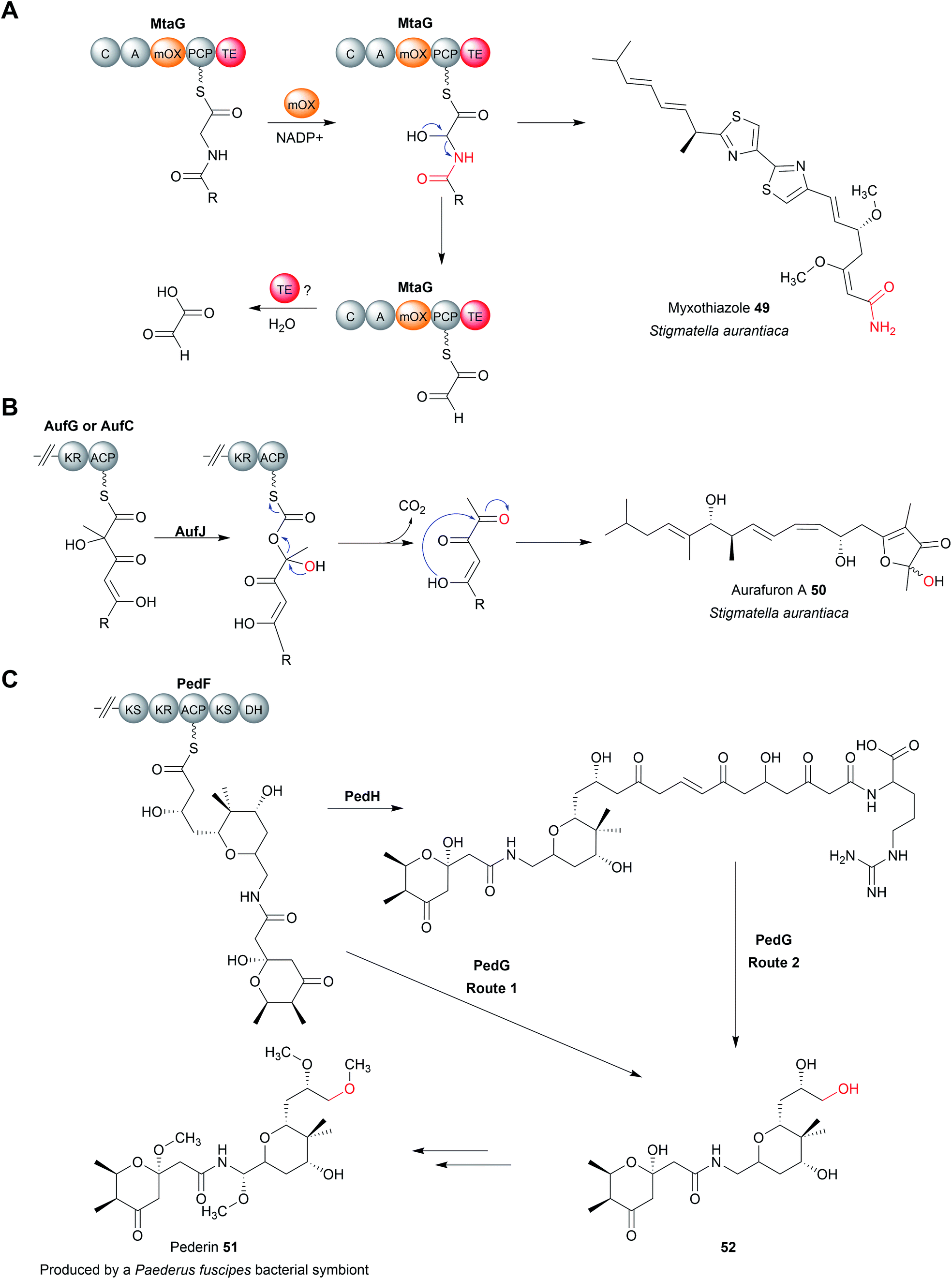 | ||
| Fig. 22 Oxidative chain release. (A) Integrated within the A domain of MtaG is a monooxygenase (MOx) domain proposed to hydroxylate the α-position of the PCP-bound myxothiazole intermediate. Hydroxylation of the α-position is proposed to produce an unstable intermediate that fragments to release myxothiazole (49) with a terminal amide group. The likely role of the TE domain is remove the PCP-bound glyoxylate, though experimental evidence is required. (B) Proposed Baeyer–Villiger monooxygenase (AufJ)-mediated chain release in aurafuron A (50) biosynthesis. Experimental evidence is needed to support this mechanism. (C) Oxidative chain release in proposed pederin (51) biosynthesis. Two different routes have been proposed to release the pederin precursor 52. Experimental evidence is required to determine the true mechanism of chain release. | ||
The aurafurons (50) are synthesised by PKS enzymes, none of which contain a TE domain or any other integrated chain releasing domain.192 The feeding of isotopically labelled precursors demonstrated that the aurafuron backbone is synthesised from three acetate units and four propionate units.192 Interestingly, the C1 carbon derived from the incorporation of the final propionate unit was absent.192 To account for this missing carbon, an oxidative release mechanism was proposed that utilises the putative monooxygenase AufJ encoded in the same BGC (Fig. 22B).192 AufJ bears significant sequence similarity to the Baeyer–Villiger monooxygenase MtmOIV from the mithramycin biosynthesis pathway.192,194 The authors proposed that AufJ catalyses a Baeyer–Villiger reaction (insertion of an oxygen into the α-position) to generate a carbonic acid diester PK intermediate.192 Decarboxylation would then result in chain release with loss of the C1 carbon (consistent with the isotope labelling experiment), though experimental evidence is required to confirm this theory.192 An AufJ homologue is also encoded in the BGCs of the linfuranones, furanones produced by Sphaerimonospora mesophile.192,195 Furanone-containing natural products have also been isolated from molluscs, but whether an AufJ homologue is involved in their biosynthesis is unknown.196
The polyketide pederin (51) is synthesised by trans-AT PKSs by unculturable symbionts within Paederus fuscipes beetles.193 Chain release in pederin biosynthesis is proposed, but not experimentally demonstrated, to occur by oxidative cleavage catalysed by the FAD-dependent monooxygenase PedG, resulting in the formation of a terminal primary alcohol (52) (Fig. 22C).193 Alternatively, the pederin intermediate may be elongated further by the PKS PedH and released via a TE-catalysed intermolecular amidation reaction using L-arginine, releasing a compound resembling onnamide A, a metabolite produced by sponge endosymbionts.197 This extended precursor (53) could also undergo oxidative cleavage by PedG to produce the truncated pederin precursor (52) (Fig. 22C).193 Through the details need to be established, a similar chain release mechanism is likely also occurring in the biosynthesis pathway to the related mycalamide natural products.198
11 Chain release catalysed by FabH-like enzymes
Tetronate natural products are PK or fatty acid chains attached to a tetronic acid (4-hydroxy-[5H]furan-2-one) moiety.12 The tetronates are a diverse natural product family that can exhibit antibiotic, anticancer, antiviral, and antifungal bioactivities (Fig. 23).12 In tetronate biosynthesis pathways, chain release is concomitant with tetronate ring formation itself, catalysed by a single standalone enzyme rather than a catalytic domain within a PKS or NRPS.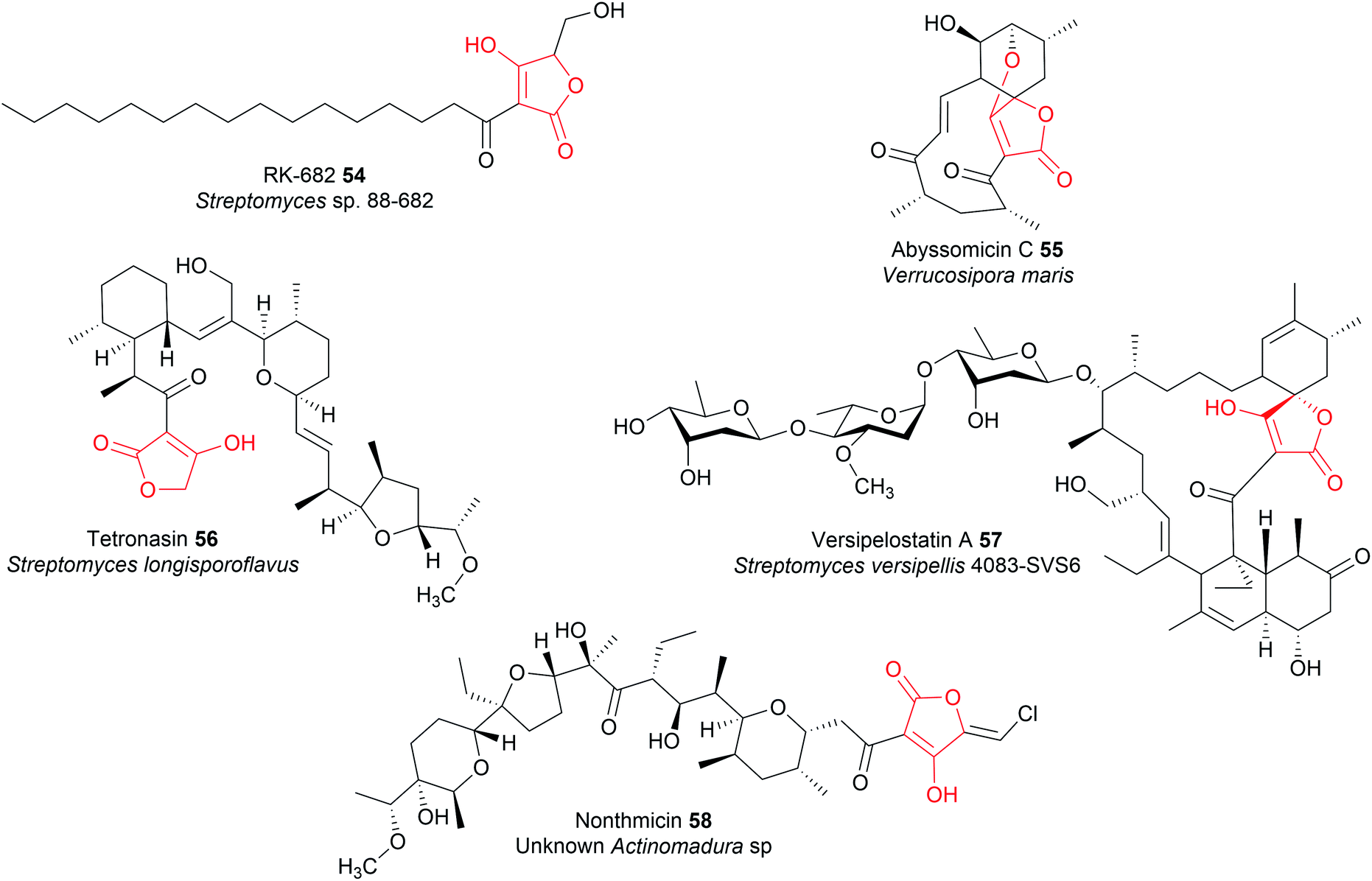 | ||
| Fig. 23 Examples of tetronate natural products. | ||
The mechanism of chain release/tetronate formation was elucidated by reconstituting the biosynthesis of the linear tetronate RK-682 in vitro (54).199 There are three core genes required for tetronate biosynthesis, together called the “glycerate utilisation operon”.12 The three genes encode a FkbH-like enzyme, a FabH like enzyme, and a standalone ACP, respectively. The FkbH-like enzyme is a phosphatase that catalyses the formation of glyceryl-ACP from 1,3-bisphosphoglycerate and the standalone ACP.200 The FabH (β-ketoacyl-ACP synthase III)-like enzyme is sufficient to catalyse both C–O and C–C bond formation between glyceryl-ACP and the β-keto thioester PK/fatty chain, releasing the free tetronate (Fig. 24).12 The order in which these bonds form is unclear, as is whether the FabH enzyme catalyses the formation of both bonds, or whether only one is catalysed and the other forms spontaneously.22 The PK intermediate is likely transferred from the terminal ACP to the conserved cysteine residue of the FabH-like enzyme (part of its Cys–His–His catalytic triad).10,22 The biosynthesis of the polyether tetronate tetronasin appears to be a slight exception to the described process, utilising glycolyl-ACP rather than glyceryl-ACP.201 It is unknown if glycolyl-ACP (or derivatives containing additional functional groups) are tolerated by the FabH enzymes of tetronate pathways that typically accept glyceryl-ACP, if so enabling the creation of novel tetronate compounds.
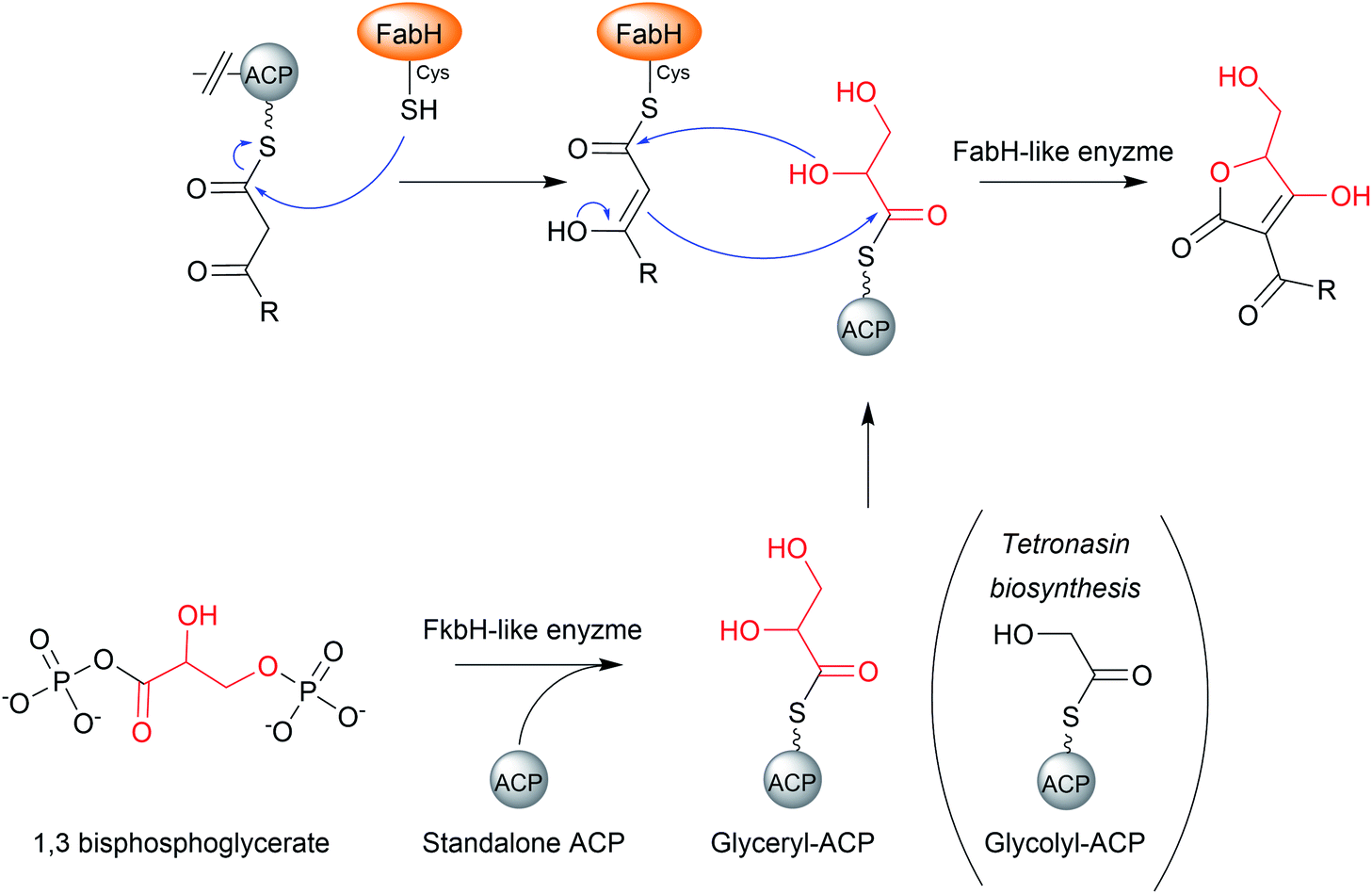 | ||
| Fig. 24 The mechanism of chain release by FabH-like enzymes in the biosynthesis of tetronate natural products. | ||
Polyketide tetronates such as abyssomicin C (55), tetronasin (56), and versipelostatin A (57) are all synthesised by type I PKS enzymes.12 There are several distinguishing characteristics of PK tetronate biosynthetic pathways. To begin with, the terminal PKS module of these pathways contains a C-terminal ACP domain, rather than a C-terminal TE domain or R domain.12 Secondly, in order for chain release to occur, the final ACP-bound PK requires a β-keto group to acidify the α-position for carbanion formation.12 As such, the final PKS module of a polyketide tetronate biosynthesis pathway either lacks a KR domain, or contains one that is catalytically inactive.12,202–205 It would be interesting to test if prematurely forming a β-keto PK intermediate, by mutating the KR of an earlier module, would result in the formation of truncated tetronate products due to premature release by the FabH-like enzyme.
Installing a tetronate ring provides additional opportunities for structural diversification. A common modification is to eliminate the primary alcohol of the tetronate moiety to form an exocyclic double bond.206,207 To achieve this the primary alcohol is first acetylated by an acyltransferase to create a superior leaving group, followed by elimination by a lyase to create the exocyclic double bond itself.206 This exocyclic double bond is an important feature of many tetronates, as it can be used as a dieneophile in both intramolecular or intermolecular [4 + 2] cycloadditions (Diels–Alder reactions) (Fig. 25).208–210 An intramolecular Diels–Alder reaction can produce a spirotetronate moiety, exemplified by compounds such as abyssomicin C (55)210 and versipelostatin C (57).209 Spirotetronates are characterised as two ring structures linked together by a spiroatom (a carbon in the case of the spirotetronates).12 Alternatively, the tetronate may be halogenated via a yet unknown mechanism, as occurs in nonthmicin (58) biosynthesis.211 Exploring the mechanism of this halogenation would be valuable, as the identification of a tetronate halogenase could enable halogens to be enzymatically added to the tetronate groups of other natural products.
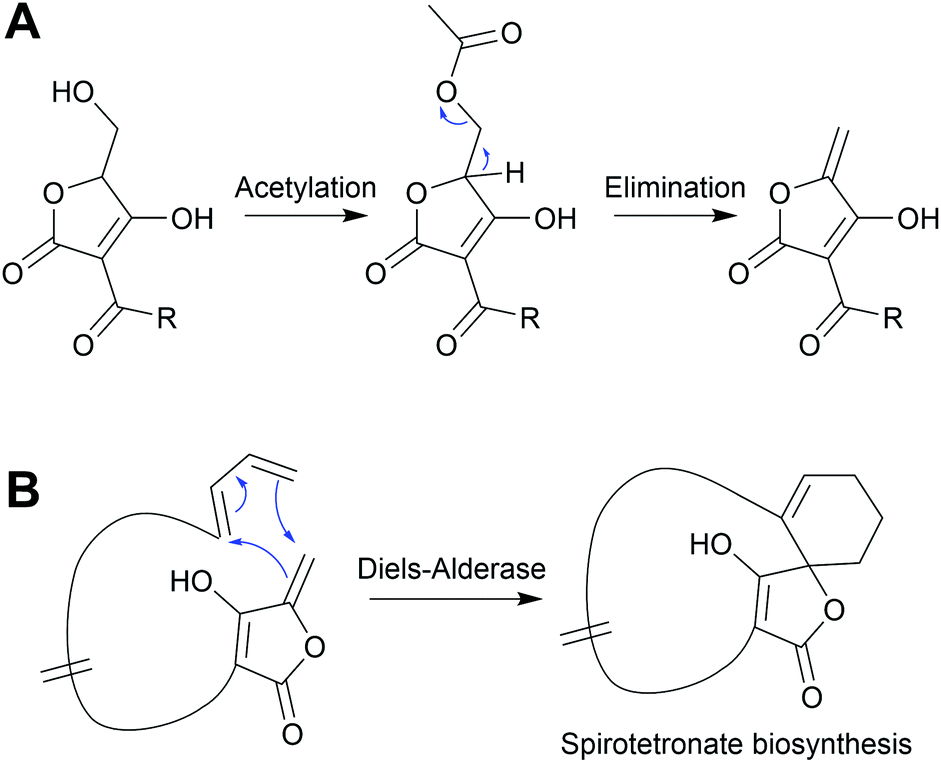 | ||
| Fig. 25 Spirotetronate formation. An exocyclic double bond on the tetronate moiety is formed by an enzyme-catalysed acetylation and elimination reaction. The double bond can then serve as a dieneophile in an intramolecular Diels–Alder reaction ([4 + 2] cycloaddition), forming a spirotetronate. | ||
A FabH-like enzyme (MxnB) is also responsible for chain release in myxopyronin biosynthesis (59).212,213 Analogous to tetronate ring formation, here the central pyrone ring of myxopyronin is formed by the MxnB-catalysed condensation of PK chains tethered to the PKSs MxnJ and MxnK (Fig. 26).212,213 As in tetronate biosynthesis, the order in which the C–C and C–O bonds form is still unclear.206
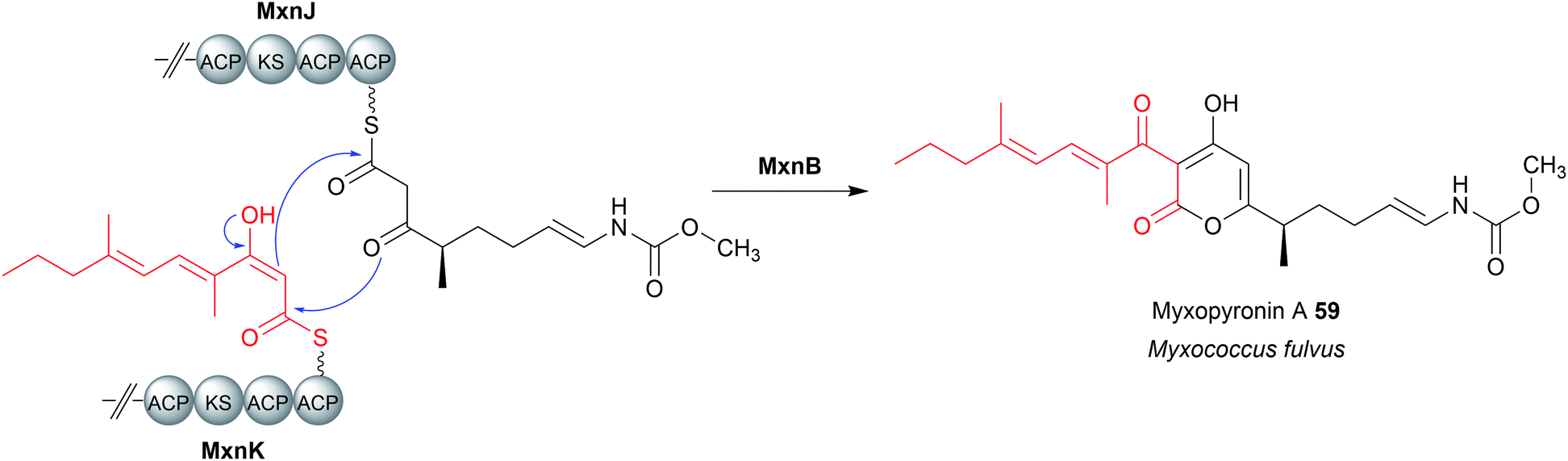 | ||
| Fig. 26 Dimerisation in myxopyronin biosynthesis. Analogous to tetronate biosynthesis, the FabH-like enzyme MxnB is responsible for chain release/ring formation in myxopyronin A (59) biosynthesis. | ||
12 Chain release catalysed by condensation-like (CT) domains
Condensation (C) domains are one of the core domains found in NRPS modules. C domains are responsible for catalysing C–N bond formation between a PCP-bound NRP chain and an amino acid tethered to the PCP domain of the downstream module. C domains are ca. 50 kDa in size and have a V-shaped pseudodimer structure: a single polypeptide forming two subdomains (“monomers”), each of which possesses a chloramphenicol acyltransferase fold.2 At the interface of these two subdomains is the conserved catalytic HxxxDxxS motif (Fig. 27).2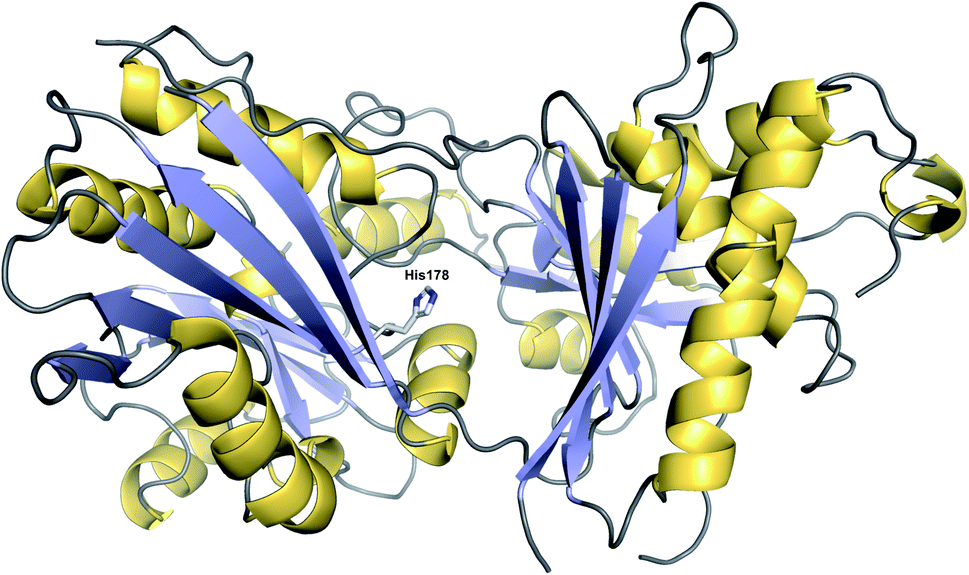 | ||
| Fig. 27 Structure of a CT domain. The crystal structure of the CT domain from fumiquinazoline F biosynthesis (PDB: 5DIJ). The conserved histidine (His178) at the interface between the two pseudomonomers is depicted. | ||
The histidine in this motif was first proposed to act as a general base to deprotonate the α-amine group of the CP-bound amino acid of the downstream module, promoting its nucleophilic attack on the upstream CP-linked thioester (Fig. 28A).214,215 However, deciphering the role of this histidine is not straightforward, as in some C domains it is not essential for activity.216,217 One explanation from recent structural data could be that water can serve as an alternative base in the absence of the histidine.218 Matters complicated further still by calculations indicating that the histidine is protonated (and therefore unable to act as a base) under physiological conditions.219
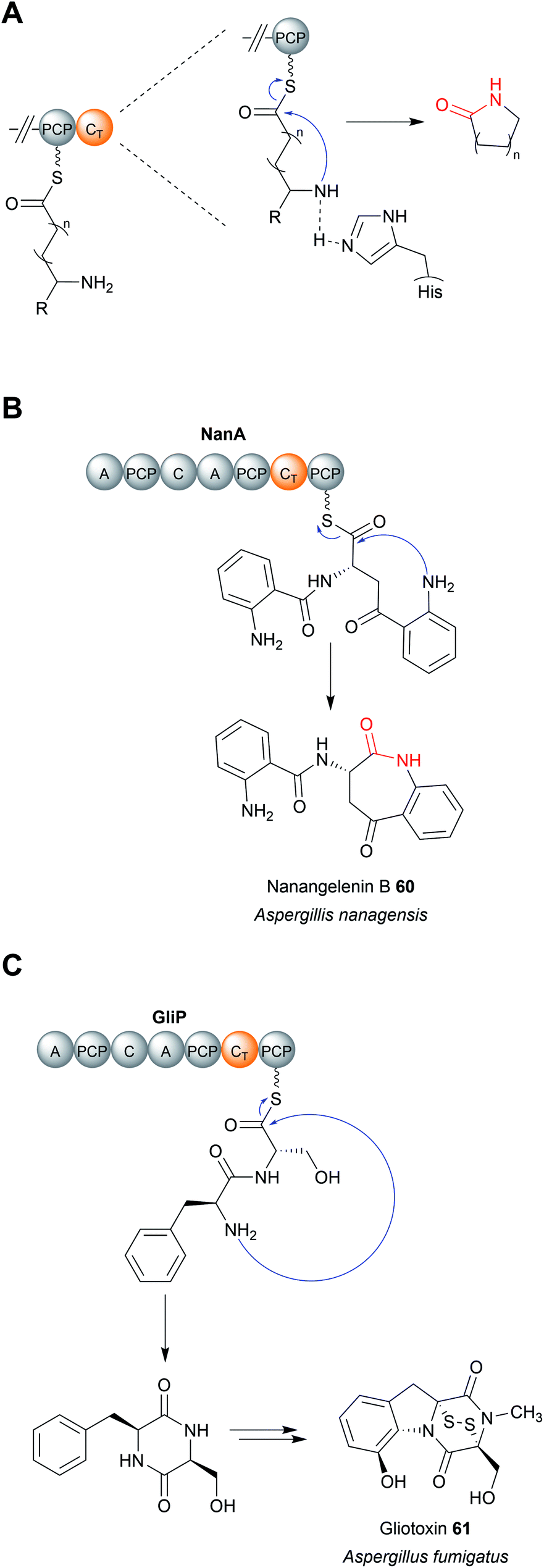 | ||
| Fig. 28 The mechanism of CT domains. (A) The proposed catalytic mechanism of CT domains using the conserved histidine residue as a general base. CT domains typically catalyse chain release via lactam formation. (B) CT-catalysed chain release in nanangelenin B (60) biosynthesis. (C) CT-catalysed chain release in gliotoxin (61) biosynthesis. | ||
Regardless of their enzymatic mechanism, in some biosynthetic pathways use specialised C domains (called CT domains) to catalyse chain release.182,220–223 Like canonical TE domains, CT domains are located on the C-terminus of an NRPS/PKS enzyme. CT domains are especially prevalent in fungi, where 60–90% of all encoded NRPS enzymes contain one on their C-terminus.220 Unlike the fungal TE domains, which typically catalyse hydrolytic chain release, CT domains typically catalyse chain release via macrolactamisation, such as during nanangelenin B (60) biosynthesis (Fig. 28B).182,220,221 However, like TE domains, CT domains can also catalyse chain release by selecting a range of nucleophiles, both intramolecular and intermolecular. For example, CT domains have been found that catalyse chain release via amidation224 (Fig. 29A), Dieckmann condensation (Fig. 29B),225 hydrolysis (Fig. 29C),226 transesterification (Fig. 29D),112,227,228 or lactonisation.221 A notable accomplishment of the research on CT domains is the clarification of chain release in gliotoxin (61) biosynthesis, long proposed to occur spontaneously with diketopiperazine formation.229 However, the second C domain in gliotoxin synthase, GliA (A–PCP–C–A–PCP–CT–PCP), has now been identified as a CT domain, catalysing diketopiperazine formation/chain release (Fig. 28C).230 Interestingly, the PCP downstream of the GliA CT domain is also essential for chain release, indicating that it serves to tether the NRP intermediate specifically for the CT domain.230 The same C-terminal CT–PCP arrangement is found in other NRPSs, including the previously mentioned nanangelenin B synthase NanA, suggesting a conserved function.182,230
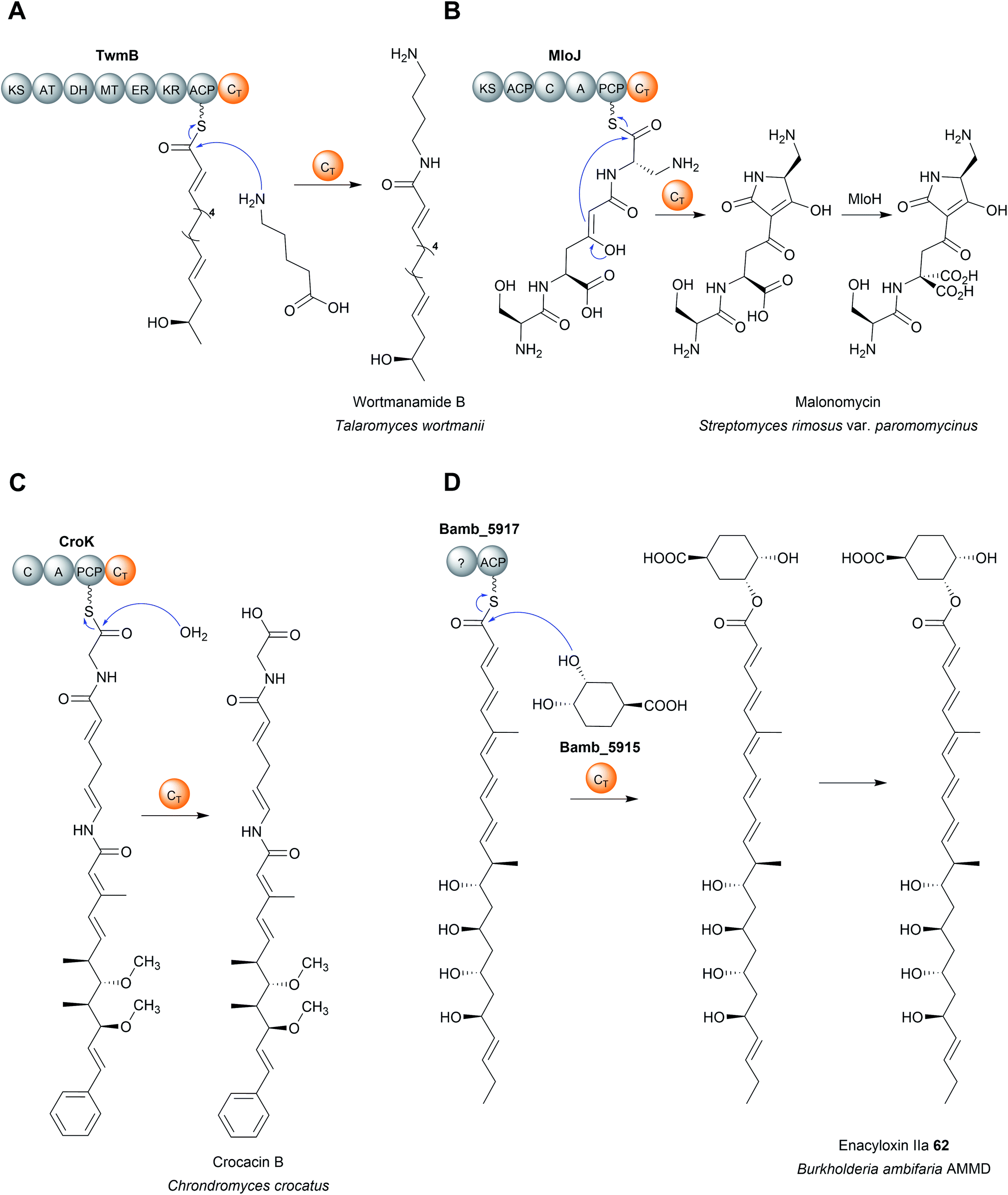 | ||
| Fig. 29 Chain release catalysed by CT domains. (A) CT-domain-catalysed chain release via amidation in wortmanamide B biosynthesis. (B) CT-domain-catalysed chain release via a Dieckmann condensation in malonomycin biosynthesis. (C) CT domain-catalysed chain release via hydrolysis in crocacin B biosynthesis. (D) Chain release via transesterification with (1S,3R,4S)-3,4-dihydroxycyclohexane carboxylic acid (DHCCA) catalysed by the standalone ct enzyme Bamb_5915 in enacyloxin IIa (62) biosynthesis. | ||
Analogous to the TE enzymes discussed in Section 4, CT enzymes are not necessarily confined within larger NRPS/PKS enzymes as domains, but can also catalyse chain release as freestanding enzymes.112,228,231 A recent example of a freestanding CT is the enzyme Bamb_5915 from the enacyloxin (62) biosynthesis pathway in Burkholderia ambifaria AMMD (Fig. 29D).228,231 Bamb_5915 catalyses chain release via a transesterification reaction between the ACP-bound polyketide chain and the 3-position hydroxyl of (1S,3R,4S)-3,4-dihydroxycyclohexane carboxylic acid (DHCCA).216 Chemical analogues of DHCCA were also accepted by Bamb_5915, suggesting the enzyme could be used to create novel enacyloxins.216
In regards to the enzymatic mechanism, like their C domain counterparts, the PK/NRP chain is never covalently bonded to the CT domain (unlike with TE domains).2 Rather, the PK/NRP chain is inserted into the CT active site while remaining tethered to the upstream CP domain (analogous to chain release catalysed by R domains).2 The crystal structure of the CT domain and a T–CT didomain from the Penicillium aethiopicum NRPS TqaA has been solved.232 TqaA is an NRPS responsible for the biosynthesis of the NRP fumiquinazoline F.220 The structure of the C-terminal CT domain revealed a highly similar fold (V-shaped pseudodimer) to canonical C domains.232 The exclusion of other potential nucleophiles, water in particular, was proposed to be accomplished by contacts between the α2 helix with the β11–β13 loop blocking access to the active site, thereby favouring macrocyclisation.232 Mutating the catalytic histidine in the HxxxDxxS motif abolished CT domain activity, proposed to be due to its ability to activate an amine in the peptidyl chain for an intramolecular nucleophilic attack on the C1 thioester.220 However, as discussed in the first paragraph of this section, the role this histidine really plays in C domain catalysis is contentious. Additional experimental evidence is therefore required to elucidate the mechanism of CT domains with confidence.
A key takeaway from activity studies on CT domains is the importance of protein–protein interactions between the CT domain and the upstream PCP domain.220,224 In these studies the isolated CT domains are unable to accept SNAC substrate analogues (which are often used successfully in activity studies on TE domains).53,220,224 CT domain activity could only be detected in vitro when a PCP–CT didomain construct was purified and the substrate loaded onto the PCP domain using a promiscuous PPtase, clearly demonstrating the importance of contacts with the upstream PCP domain.220,224 In the case of the standalone CT domain Bamb_5915, specialised binding domains enable its interaction with the its target ACP protein.231 Such protein–protein interactions will likely be critical to replicate in any engineering efforts where CT domains are excised and transplanted between different biosynthesis pathways.
13 Chain release catalysed by acyltransferase-like enzymes
Acyltransferase (AT) enzymes catalyse the transfer of an acyl group from a donor to an acceptor. A well-known example of acyltransferases in natural product biosynthesis are the AT domains found in type I PKS modules.1 AT domains form a covalent linkage with an acyl group, typically a malonyl or (2S)-methylmalonyl extension unit, via a conserved serine residue (part of the Ser–His catalytic dyad, where the basic His activates the serine nucleophile).10 The AT then catalyses the transfer of the acyl group to the free thiol of the Ppant group attached to the downstream ACP domain.10 AT domains can also be standalone enzymes, as in the case of trans-AT polyketide biosynthesis pathways.233In select cases, enzymes with homology to characterised acyltransferases catalyse chain release in PK biosynthesis pathways.234–238 An early characterised example was the enzyme LovD from the biosynthesis pathway of lovastatin (63), a polyketide produced by Aspergillus terreus.234,235 A key step in the biosynthesis of lovastatin is the attachment of a methylbutyryl diketide sidechain, synthesised by the PKS LovF, to monacolin J acid (64) (Fig. 30A). However, LovF lacks a C-terminal TE, R, or CT domain, making its chain release mechanism initially unclear. The enzyme LovD was later identified as an acyltransferase responsible for releasing the LovF-bound methylbutyryl diketide side chain and transferring it to monacolin J acid.234,235
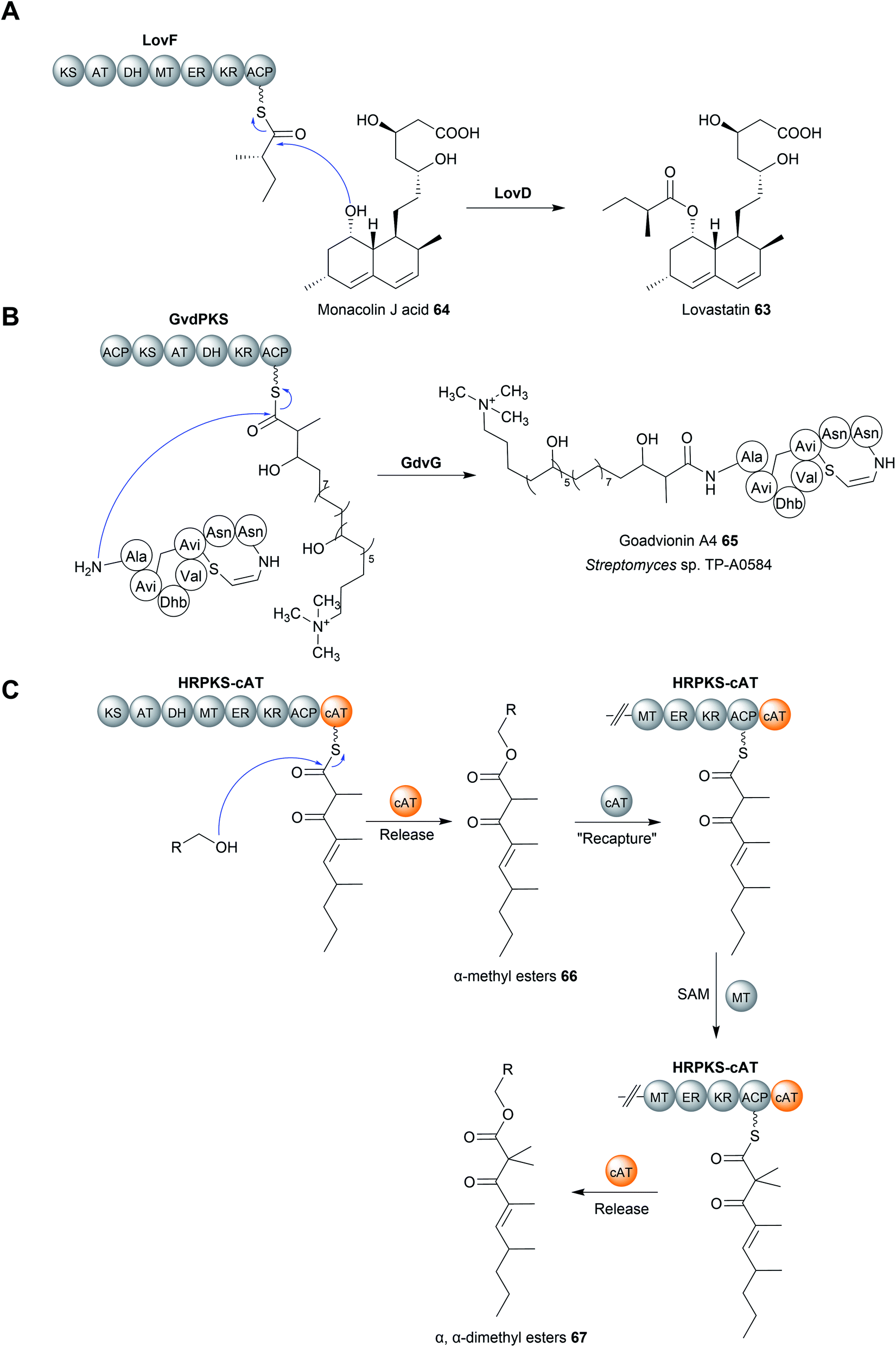 | ||
| Fig. 30 Chain release by acyltransferase enzymes. (A) Transfer of a methylbutyryl diketide unit from the PKS LovF to monacolin J acid (64) catalysed by the acyltransferase LovD. (B) The acyltransferase GdvG is responsible for joining together the PK and RiPP components of goadvionin A4 (65). (C) A carnitine acyltransferase (cAT)-like domain from a fungal HRPKS. A “release and recapture” mechanism was proposed to account for the formation of both α-methyl esters (66) and α,α-dimethyl esters (67). | ||
Another early example was in the biosynthesis pathway of the polyketide rifamycin, where an AT-like amide synthase enzyme (RifF) catalyses chain release via a macroamidation reaction.236 Homologues of RifF are encoded in other BGCs, such as those for geldanamycin and ansamitocin, and catalyse chain release using their conserved Cys–His–Asp catalytic triad (an analogous mechanism to the α/β hydrolase TE domains).22,239–241
A more recent example of an acyltransferase that catalyses chain release is GdvG from the goadvionin (65) biosynthesis pathway. Goadvionin (65) is one of the few known examples of a polyketide–RiPP (ribosomally synthesised and post-translationally modified peptide) hybrid.237 The acyltransferase GdvG, which resembles members of the GNAT acyltransferase family, catalyses the transfer of a PK chain tethered to the polyketide GvdPKS to the N-terminus of the RiPP component (Fig. 30B).237 Homologues of gdvG are present in other polyketide biosynthetic gene clusters, suggesting that this release strategy is used in other biosynthetic pathways to create different PK–RiPP hybrids.237
In addition to catalysing the release of completed PK chains, standalone ATs can also have a proofreading role analogous to the TEII enzymes discussed in Section 3.3.1.242 The acyltransferase enzyme PedC was identified in the biosynthesis pathway for the polyketide pederin, synthesised by trans-AT PKSs (discussed previously in Section 5).242 PedC was demonstrated to catalyse the hydrolytic release of ACP-bound PK chains but not the malonyl-ACP, indicating that it likely plays a proofreading role, removing aberrant or stalled PK chains.242 The release of aberrant chains by such proofreading enzymes in trans-AT PK pathways can be exploited to study biosynthetic intermediates.243,244 Intentionally stalling the biosynthetic pathways of the trans-AT PKs rhizoxin (Mycetohabitans rhizoxinica) and bacillaene (Bacillus amyloliquefaciens) via mutagenesis of their respective TE domains led to the release of stalled pathway intermediates by these proofreading acyltransferases.243,244
Like the thioesterases and CT domains discussed earlier, chain-releasing ATs can either be standalone enzymes, or discrete domains within a module. The only known example of an AT-like domain that catalyses chain release was characterised in the biosynthesis of an as yet-unknown polyketide produced by the fungus Trichoderma virens.245 The genome of this organism encodes a PKS (HRPKS-cAT) that lacks a TE, CT or R domain, instead containing a C-terminal domain resembling a carnitine acyltransferase (cAT).245 When assaying the activity of HRPKS-cAT in vitro, a tetraketide product joined via an ester linkage to Tris or glycerol, both of which were components of the assay buffer, was identified.245 Neither of these esters was produced using a mutated version of HRPKS-cAT where the cAT domain had been deleted, indicating that this domain is responsible for catalysing chain release via a transesterification reaction.245 The nucleophile used in vivo remains unknown.245 The α-position of the tetraketide was either methylated (66) or dimethylated (67) by the methyltransferase (MT) domain of HRPKS-cAT.245 Interestingly, the monomethylated tetraketide ester was converted into the geminal dimethylated form when incubated with HRPKS-cAT and S-adenosyl methionine (SAM).245 However, this additional methylation did not occur if the cAT domain had been deleted.245 From these results, the authors proposed an unprecedented “release and recapture” mechanism, whereby the monomethylated tetraketide ester is transferred back to the terminal ACP of HRPKS-cAT by the cAT domain (Fig. 30C).245 The MT domain then catalyses a second methylation to generate a geminal dimethyl group, followed by chain release by the cAT domain to regenerate the free ester.245
The PK/NRP hybrid paenilamicin appears to utilize an unusual BtrH-like acyltransferase to catalyse chain release.246 The enzyme BtrH was characterised in the butirosin biosynthetic pathway as the acyltransferase responsible for transferring γ-L-Glu-4-amino-2-hydroxybutyrate (AHBA) from a standalone ACP to the aminoglycoside ribostamycin.247 Paenilamicin is produced by Paenibacillus larvae, the pathogen responsible for American Foulbrood in honeybee colonies.248 Interestingly, the C-terminal end of paenilamicin is linked to a molecule of 4,3-spermidine via an amide linkage. The biosynthesis pathway of paenilamicin contains four PKS and seven NRPS modules. The final module in the biosynthesis pathway, PamH NRPS module 7, lacks a thioesterase or another obvious chain release domain.246 However, the biosynthetic gene cluster also encodes a standalone enzyme, PamI, with homology to BrtH.2,246 PamI could therefore catalyse chain release using 4,3-spermidine as an intermolecular nucleophile, resulting in amide formation.2,246 Further biochemical characterisation of BtrH PamI is therefore required to uncover the true mechanism of paenilamicin chain release.2,246
14 Chain release catalysed by AfsA/butenolide synthase (PBS) domains
A recent addition to the stable of domains that catalyse chain release are AfsA-like domains.249,250 The monomeric AfsA protein was first characterised in the biosynthesis pathway for A factor (2-isocapryloyl-3R-hydroxymethyl-γ-butyrolactone) in Streptomyces griseus.251 AfsA catalyses a condensation reaction between dihydroxyacetone phosphate (DHAP) with a β-keto thioester (derived from fatty acid biosynthesis), followed by spontaneous cyclisation to form the γ-butyrolactone ring of A factor (68) (Fig. 31A).251 A factor (68) itself belongs to the γ-butyrolactone family of transcriptional regulators (also referred to as “microbial hormones”) that induce the expression of genes involved in natural product biosynthesis and/or cell differentiation in Gram-positive bacteria.252 In 2020, two groups independently reported the discovery of gladiofungin (69) (syn gladiostatin), an antifungal glutarimide natural product produced by Burkholderia gladioli.249,250 In addition to the glutarimide moiety, gladiofungin (69) also contains a terminal butenolide ring. The biosynthetic gene cluster of gladiofungin (69) revealed that its PK backbone is synthesised by the trans-AT PKS enzymes GlaD and GlaE.249 However, instead of a TE domain, module 10 of GlaE contains the first example of an AfsA-like domain (syn phosphorylated butenolide synthase (PBS) domain) (Fig. 31B).249,250 Deletion of the AfsA domain abolished production of gladiofungin (69), demonstrating it is essential for biosynthesis.249 By purifying an excised ACP–AfsA didomain, the condensation between DHAP and a β-keto SNAC substrate analogue could be reconstituted in vitro, proving the role of the AfsA domain in chain release/butanolide formation.250 Other biosynthetic gene clusters that encode an AfsA domain could be identified by using its amino acid sequence as a bioinformatic handle, indicating that these pathways also produce butanolide/γ-butyrolactone substituted polyketides.249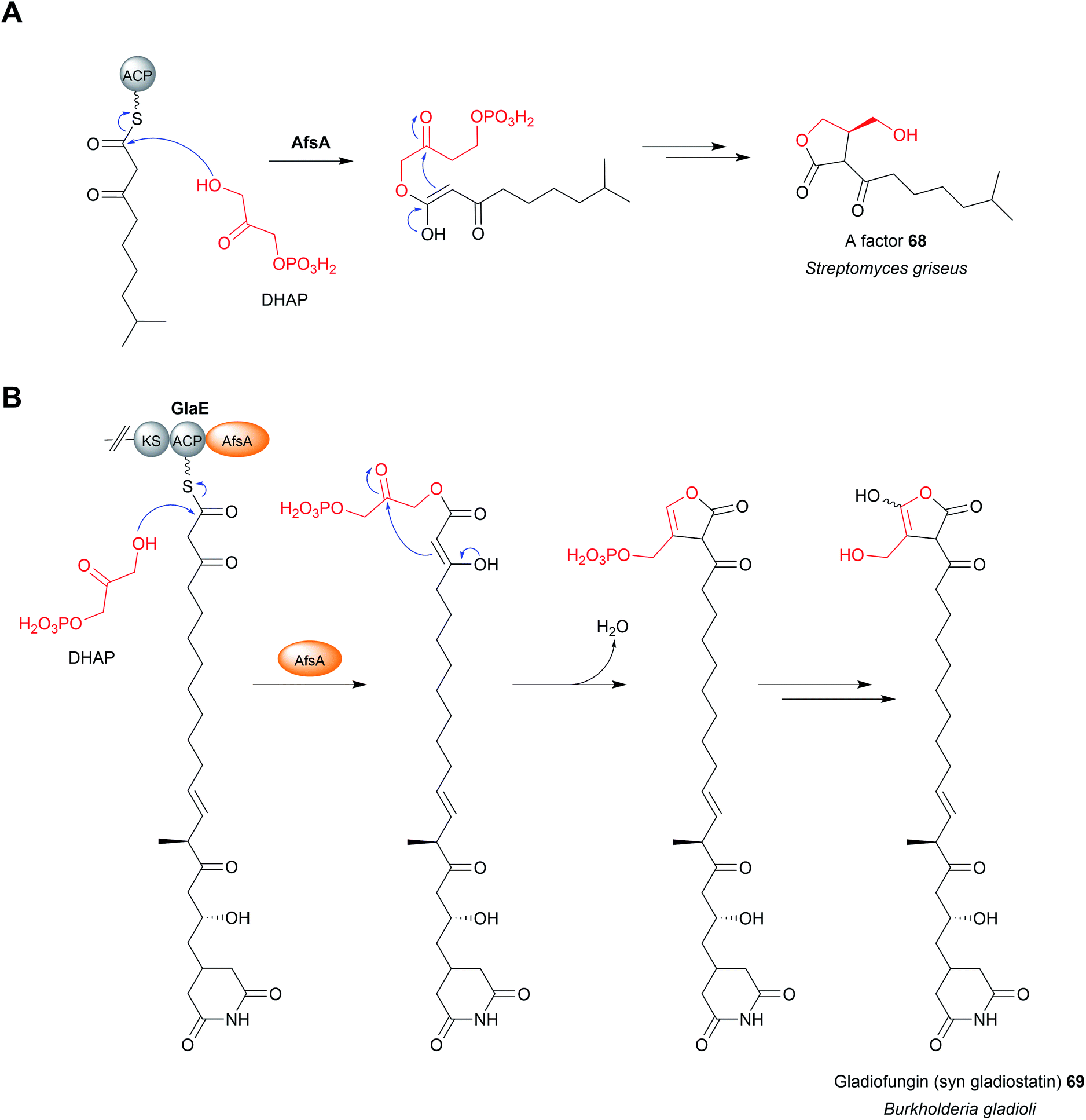 | ||
| Fig. 31 Chain release by AfsA domains. (A) The protein AfsA catalyses the condensation of dihydroxyacetone phosphate (DHAP) and an ACP-bound β-keto fatty acid, leading to a factor (68). (B) An AfsA-like domain is responsible for catalysing chain release using DHAP in gladiofungin (69) biosynthesis. | ||
15 Chain release catalysed by standalone Dieckmann cyclases
Chain release via a Dieckmann condensation has already been discussed in the context of TE, R*, and CT domains. However, other biosynthetic pathways utilize specialised standalone Dieckmann cyclases to catalyse chain release, such as in biosynthesis pathways of the polyketide tetramates pyrroindomycin253 (70) and tirandmycin.254 These examples and more are discussed in detail in a comprehensive recent review.178 To briefly cover the topic here, chain release/tetramate ring formation in pyrroindomycin biosynthesis is catalysed by two enzymes, PyrD3 and PyrD4 (Fig. 32A).253 Surprisingly, both enzymes are independently capable of releasing/cyclising the PCP-bound N-acetoacetyl-L-alanine substrate. Consistent with this result, in vivo deletions of either gene decreased pyrroindomycin production, whereas production was completed abolished only in a ΔpyrD3 ΔpyrD4 double deletion mutant.253 PyrD3 and PyrD4 are unrelated to one another, with PyrD3 resembling a pyruvate dehydrogenase while PyrD4 is predicted to have a α/β hydrolase fold. Whether PyrD3 and PyrD4 act synergistically is unknown, but how two distinct enzyme folds catalyse the same reaction is an intriguing question.253 It would be interesting to know if the BGCs of as yet-undiscovered tetramates related to pyrroindomcyin (70) also encode homologues of both PyrD3 and PyrD4. | ||
| Fig. 32 Chain release catalysed by standalone Dieckmann cyclases. (A) In pyrroindomycin (70) biosynthesis chain release is catalysed via a Dieckmann condensation. Two enzymes, PyrE3 and PyrE4, are both capable of catalysing this reaction. (B) Formation of the tetramate ring in nocamycin I (71) is catalysed by the α/β hydrolase NcmC. | ||
Another family of standalone Dieckmann cyclases are encoded in the gene clusters coding for the biosynthesis of tetramates such as tirandmycin, streptolydigin, and nocamycin (71).254,255 In the case of tirandmycin biosynthesis, a polyketide tetramate produced by Streptomyces sp. 397-9, the enzyme TrdC has been shown to catalyse chain release via a Dieckmann cyclisation.254,256 The crystal structure of the TrdC homologue from the nocamycin (71) biosynthesis pathway, NcmC, has been recently solved (Fig. 32B).255 The structure revealed that NcmC possesses an α/β hydrolase fold like TE domains, but also contains an unusual four-helix bundle inserted between strands β5 and β6.255 A TE domain-like catalytic triad is present in NcmC, except that the catalytic serine is replaced with cysteine (Cys–His–Asp); all three residues were demonstrated to be essential for NcmC activity.255 The essential nature of the catalytic cysteine suggests that the linear nocamycin intermediate is covalently tethered to NcmC via a thioether linkage, though direct evidence is lacking.255
16 Chain release catalysed by ketosynthase domains
Ketosynthase domains are one of the core domains (along with AT domains and ACPs) present in all cis-PKS modules.10 In these systems, KS domains catalyse chain extension via a decarboxylative Claisen condensation reaction between the growing PK chain and an ACP-bound malonyl/(2S)-methylmalonyl unit. KS domains themselves are ca. 430 amino acids in size and possess a α/β/α/β/α thiolase fold (alternating layers of α-helices and β-sheets).10 The active site of KS domains contains a Cys–His–His catalytic triad, with the cysteine acting as a nucleophile to covalently bond the PK chain prior to chain extension. In addition to this well-defined role in chain extension, there is evidence from several pathways that KS domains catalyse chain release. The first (and most convincing) example is from the tenuazonic acid (72) biosynthesis pathway in the fungus Alternaria tenuis.257 The biosynthesis of tenuazonic acid (72) is achieved via the condensation of acetoacetyl-CoA with L-isoleucine followed by a Dieckmann condensation, all of which is catalysed by the PKS-NRPS hybrid Tas1.257 Tas1 is a unimodular PKS-NRPS hybrid with an unusual C–A–PCP–KS domain arrangement. In vitro experimentation demonstrated that the C domain, rather than the terminal KS domain, catalyses the condensation of SNAC-L-isoleucine with acetoacetyl-CoA to form SNAC-N-acetoactyl-L-isoleucine.257 However, incubating SNAC-N-acetoactyl-L-isoleucine with a purified form of the KS domain resulted in tenuazonic acid (72) formation, indicating that the KS domain catalyses chain release via a Dieckmann condensation (Fig. 33).257 | ||
| Fig. 33 Chain release by a ketosynthase domain. The C-terminal KS domain in the NRPs Tas1 catalyses a chain release by a Dieckmann condensation reaction in tenuazonic acid (72) biosynthesis. | ||
The crystal structure of Tas1 KS revealed it was remarkably similar to the KS domains from type I PKS pathways.258 Within its active site was a modified version of the KS Cis–His-His catalytic triad consisting of Cys–His–Asn (Fig. 34).258 Mutagenesis of the catalytic cysteine residue (Cys 179) abolished catalytic activity, consistent with the canonical role of this residue to covalently link the PK chain, while mutagenesis of the catalytic His residue also decreased the relative activity to only 6% of the wild type.258 Molecular docking simulations with N-acetoactyl-L-ile-Cys179 indicated that the likely role of a conserved His is to abstract the α-proton of the β-keto diketide, leading to carbanion attack on the C1 thioester to form the tetramate ring and release the PK chain.258 Given the similarity between the reaction mechanism of ketosynthases and thioesterases (both utilising a serine/cysteine nucleophile and a basic residue), it is unsurprising that KS domains can be repurposed to catalyse chain release. That a Dieckmann condensation is catalysed by Tas1-KS is also fully consistent with the canonical role of KS domains in catalysing C–C bond formation.
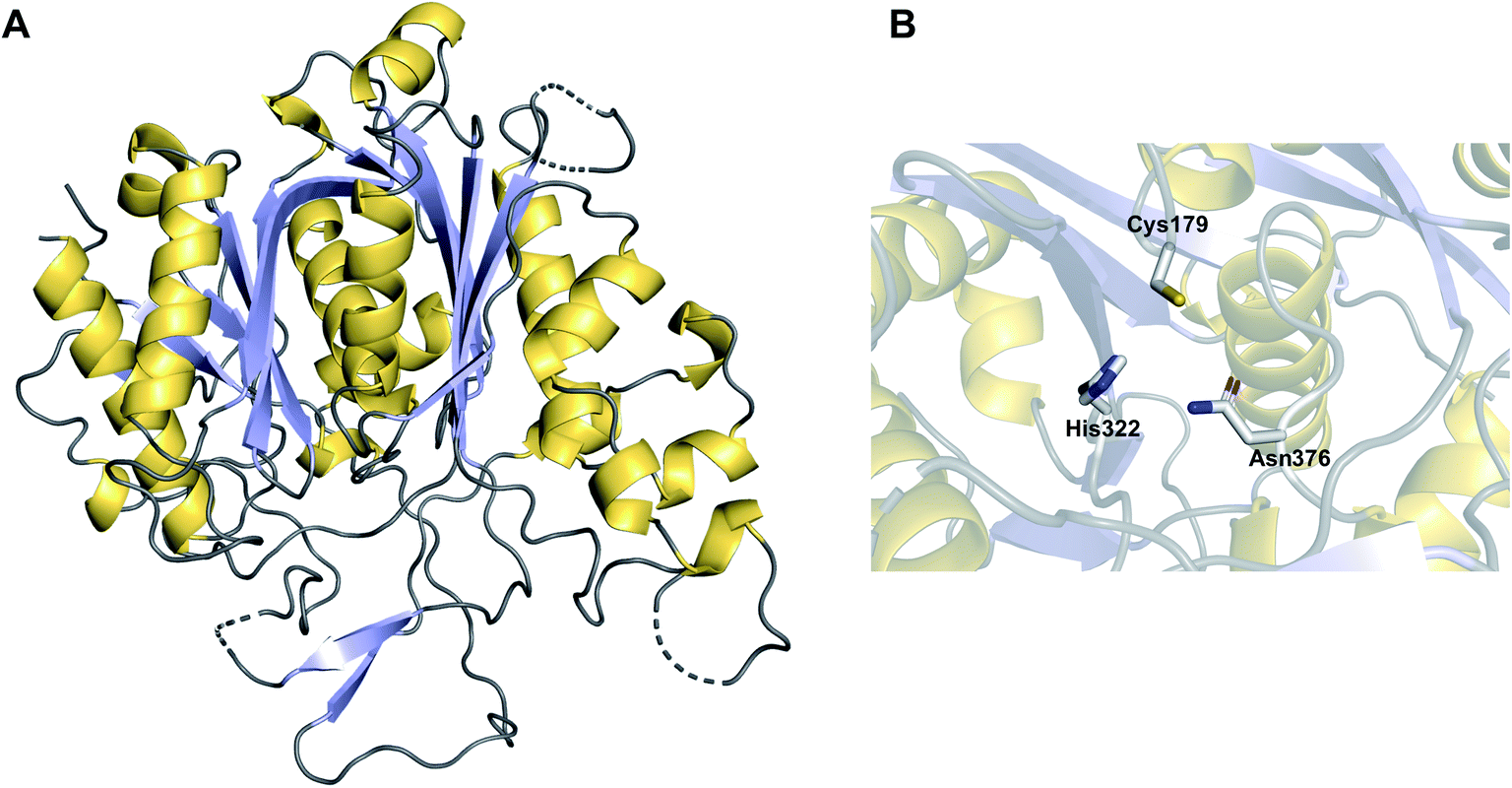 | ||
| Fig. 34 Crystal structure of a ketosynthase domain that catalyses chain release. (A) The crystal structure of C-terminal KS domain from the PKS Tas1, responsible for catalysing chain release in tenuazonic acid (72) biosynthesis (PDB: 6KOG). (B) The catalytic triad of the Tas1 KS domain consists of Cys–His–Asn, a modified version of the typical Cys–His–His catalytic triad present in KS domains. | ||
The β-lactam hexaketide ebelactone is synthesised by seven type I cis-PKS enzymes.259 The final PKS in the pathway, EbeG, contains a C-terminal KS domain, which may be responsible for chain release.259 Chain release is predicted to occur when the β-hydroxyl attacks the C1 thioester carbon, resulting in β-lactone ring formation and concomitant chain release.259 However, the formation of the β-lactone ring was shown to occur spontaneously in aqueous solutions using an SNAC-β-hydroxy-hexaketide substrate analogue.259 Additionally, a homologue to the β-lactone synthase OleC (identified in the biosynthesis pathway of bacterial long-chain olefins) is encoded in the BGC of ebelactone,260 further making the function of the C-terminal KS domain unclear.
17 Chain release catalysed by PLP-dependent enzymes
Several polyketide biosynthesis pathways use pyridoxal 5′-phosphate (PLP)-dependent enzymes to catalyse chain release. PLP is an enzyme cofactor used for many different reactions, notably transaminations.261 The first characterised example of a PLP-dependent chain release mechanism is from the prodiginine biosynthesis pathway in Streptomyces coelicolor.262 The prodiginines are a family of red-coloured tripyrrole antibiotics produced by actinobacteria and other eubacteria.263 The biosynthesis of undecyl prodiginine (73) (a precursor to several of the cyclic prodiginines) is accomplished by the condensation of 4-methoxy-2,2′-bipyrrole-5-carbaldehyde (MBC) (74) with 2-undecylpyrrole (2-UP) (75). 2-UP (75) is synthesised from seven acetate units and one glycine unit.263 Homologues of fatty acid biosynthesis enzymes generate dodecanoic acid that is subsequently transferred to the unusual PKS-NRPS hybrid RedL. RedL has the domain arrangement A–ACP–KS–AT–ACP–OAS, where OAS is an PLP-dependent α-oxoamine synthase domain.263 The ACP-bound dodecanoyl thioester condenses with one molecule of malonyl-ACP to form β-ketomyristoyl-ACP.263 The PLP-dependent OAS domain then catalyses chain release/pyrrole ring formation via a decarboxylative Claisen condensation with L-glycine (Fig. 35A and B).264 In MBC (74) biosynthesis, another OAS domain present in RedN (ACP–ACP–OAS domain arrangement) catalyses a decarboxylative Claisen condensation reaction between ACP-bound β-keto-β-pyrrolyl-propanoyl and L-serine, releasing MBC (74) precursor.265 Homologues of RedN and RedL are also present in the biosynthetic gene cluster of the related marineosin natural products,266 where the OAS domains likely have an identical role.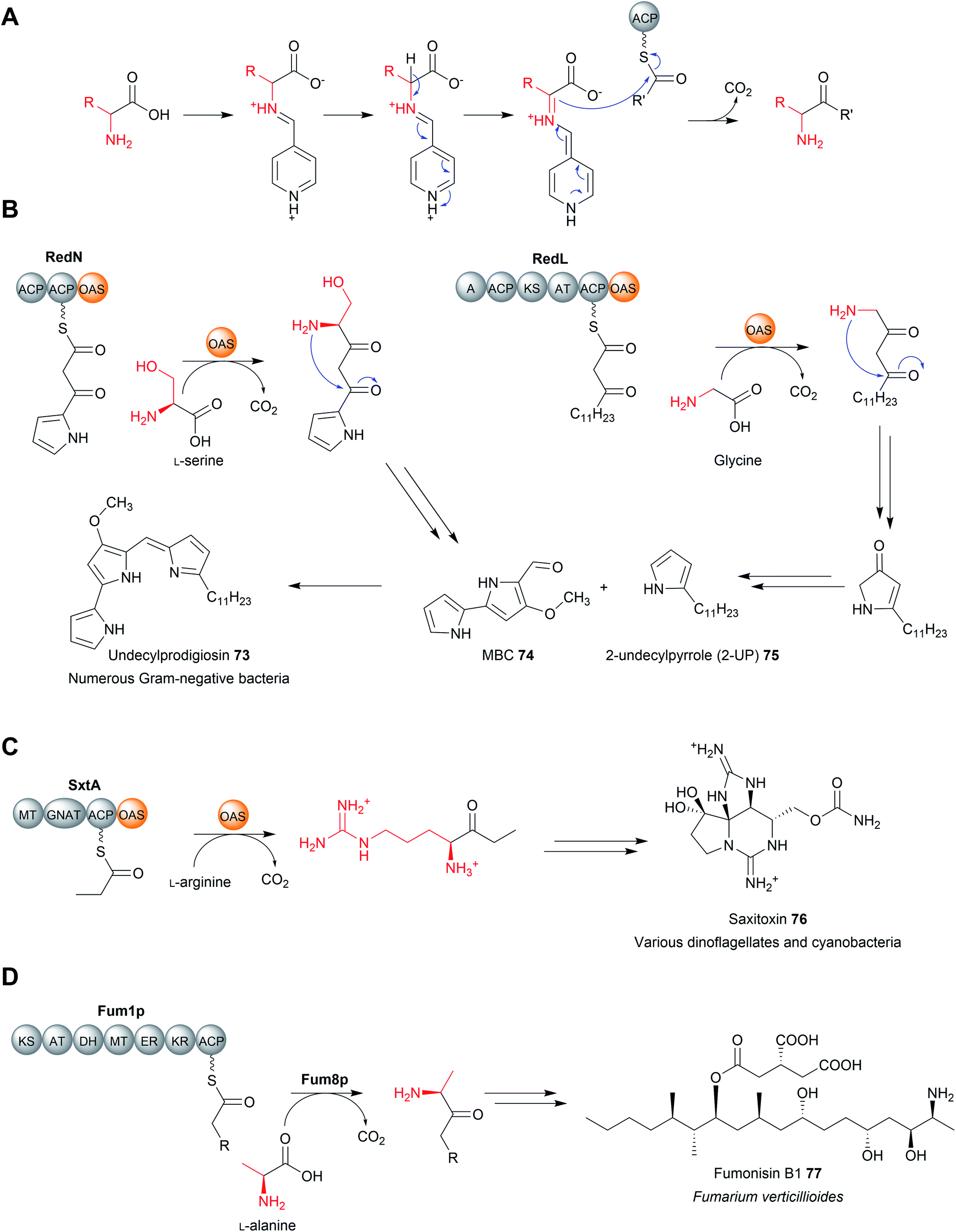 | ||
| Fig. 35 Chain release by PLP-dependent enzymes. (A) The general mechanism of a PLP-dependent Claisen condensation of an amino acid. (B) Chain release catalysed by PLP-dependent OAS domains in the biosynthesis of the prodigiosin precursors 2-UP and MBC (74). (C) In saxitoxin (76) biosynthesis the SxtA OAS domain catalyses chain release by a decarboxylative Claisen condensation reaction with L-arginine. (D) In fumonisin B1 (77) biosynthesis the standalone PLP-dependent enzyme Fum8p catalyses chain release via a decarboxylative Claisen condensation with L-alanine. | ||
The biosynthesis pathway of saxitoxin (76), a tricyclic alkaloid produced by cyanobacteria, also uses a OAS domain to catalyse chain release.267 Saxitoxin (76) is synthesised by the PKS-like enzyme SxtA, which possesses a MT–AT–ACP–OAS domain arrangement.267,268 The AT domain is unusual in that it resembles a GNAT, rather than a canonical PKS AT domain. The AT domain loads a malonyl unit and, together with the methyltransferase (MT) domain, forms propionyl-ACP.268 As in the MBC (74) and 2-UP (75) biosynthesis pathways, the OAS domain then catalyses a decarboxylative Claisen condensation between the acyl-ACP and an amino acid (Fig. 35C). In the case of saxitoxin (76) biosynthesis, L-arginine is selected by the OAS domain to release 4-amino-3-oxo-guandidinoheptane, which undergoes several subsequent oxidative cyclisations to form saxitoxin (76).268
Aside from OAS domains, standalone PLP-dependent enzymes are also capable of catalysing chain release. The only characterised example is Fum8p from the fumonisin B1 (77) biosynthetic pathway in Fusarium verticillioides.269 The iterative PKS Fum1p (KS–AT–DH–MT–ER–KR–ACP) synthesises a 18-carbon PK chain.269 Fum8p, which, like the OAS domains, bears homology to α-oxoamine synthases, then catalyses chain release via a decarboxylative Claisen condensation between L-alanine and the PK chain (Fig. 35D).269
Aside from the MBC (74), 2-UP (75), and saxitoxin (76) biosynthesis pathway, C-terminal PLP-binding domains have also been identified in numerous other PKS and NRPS enzymes, suggesting that more yet-to-be-discovered natural products use a PLP-dependent mechanism of chain release.270 In addition to characterising the unknown products of these pathways, it would be interesting to investigate the substrate tolerance of OAS domains for different amino acids, and whether the OAS domain from one pathway can be transplanted into another to create novel natural product analogues.
18 Final remarks
Our knowledge of the enzymes and mechanisms responsible for PK/NRP chain release has made substantial advances in the past decade. Entirely new enzymatic mechanisms of PK/NRP chain release, including tetronate ring formation, and TE-catalysed transesterification, and AfsA-domain-catalysed butyrolactone formation have now been described. In addition, our understanding of well-known enzymatic domains including TE, R, and CT domains has deepened as additional crystal structures have been obtained. Despite these discoveries, mysteries remain. There are still PK/NRP biosynthesis pathways where the mechanism of chain release is unclear (for instance in squalistatin S1, bongkrekic acid and isoquinoline alkaloid biosynthesis).271–273 In regards to TE domains, the evidence suggests that they are intrinsically able to catalyse a range of different release mechanisms, including hydrolysis, macrocyclization, oligomerisation, and transesterification.23 Given this, how TE domains ultimately specialise to release only a single product is known. The evolutionary pressure(s) that selects for some TE domains to contain a catalytic cysteine rather than serine are also still poorly understood, though this may be influenced by the size of the ring to be formed (for example, smaller, strained rings favouring cysteine over serine).18–20,46,47Regarding R domains, while some progress has been made, why some catalyse two-electron reductions and others catalyse four-electron reductions is still unclear. The answer likely lies in conformation changes that either facilitate or prevent a second molecule of NAD(P)H binding, though experimental evidence is required.13,145 The utility of R domains as synthetic biology tools to release has also not been explored deeply. For instance, can a TE (or CT) domain can be replaced with an R domain to release aldehyde/alcohol products? Given the diversity of functional groups that can arise from aldehydes and primary alcohols (see Fig. 19), such experiments would be of high importance to the field of combinatorial biosynthesis. In a recent step towards this goal, an aldehyde-producing R domain was successfully fused to the C-terminus of a protein construct consisting of the first two modules of the NRPS GxpS, resulting in the release of spontaneously cyclising aldehyde dipeptides.274 However, the general portability and substrate selectivity of R domains in non-native PKS/NRPS enzymes remains to be seen.
Another unsolved mystery is the reason for the largely divergent role of TE and CT domains in fungi, where TEs often catalyse hydrolysis while CT domains catalyse macrocyclisation. Regardless, the fact that distinct protein folds of TE, R*, and CT and KS domains can all catalyse similar/identical chain release reactions is a remarkable example of convergent evolution.
Looking to the future, applying the different chain release mechanisms discussed here to combinatorial biosynthesis systems has the potential to create highly diverse linear and cyclical products. Towards this goal, a recent study tested the effect of replacing the native TE domain from a PKS module with a TE domain from another biosynthesis pathway, followed by assessing its ability to catalyse macrocyclisation.275 The results showed that even when a non-native PKS module was used, if the TE matched the substrate then effective macrocyclisation could still occur, highlighting the importance of the TE domain for the effective processing of non-native substrates by engineered PKSs.275 Ensuring that the mechanism of release is compatible with the product of the engineered PKS/NRPS will be essential for the success of these synthetic assembly lines.
Studying chain release mechanisms has benefits beyond understanding the formation of final product scaffolds. As described in Section 8, disrupting chain release can led to the release of premature PK chains, providing insight into these early and difficult-to-study biosynthetic steps.243,244 We should there look to disrupting chain release mechanisms in the future as a possible means to studying late-stage biosynthetic intermediates. Where such genetic disruption experiments are not possible or unsuccessful, specialised chemical probes have been developed that provide a different means to the same end. These probes, such as methyl 6-decanamido-2-fluoro-3-oxohexanoate, compete with the CP-bound extension unit to accept the polyketide acyl chain during a KS/C domain-catalysed extension reaction.276–280 The result is the premature release of PK/NRP pathway intermediates that can then be analysed by LC-MS. This technique is particularly useful for studying the timing of tailoring reactions, such as cyclisations, occurring when the PK/NRP chain is still bound to the PKS.205,278
The number of characterised chain release mechanisms will likely grow as more genomes are sequenced and new biosynthesis pathways are discovered. Our job will be to characterise these new mechanisms and determine what they can teach us about PK/NRP biosynthesis. The step after that is the exciting prospect of applying these mechanisms in biosynthetic pathways of our own design.
19 Conflicts of interest
There are no conflicts to declare.20 Acknowledgements
The authors would like to acknowledge the financial support of their original work by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 239748522—SFB 1127 ChemBioSys, Cluster of Excellence, Balance of the Microverse, and Leibniz Award. RFL is supported by an Alexander von Humboldt postdoctoral research fellowship.21 References
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef.
- K. H. Altmann, Mini-Rev. Med. Chem., 2003, 3, 149–158 CrossRef CAS.
- U. Galm, M. H. Hager, S. G. Van Lanen, J. Ju, J. S. Thorson and B. Shen, Chem. Rev., 2005, 105, 739–758 CrossRef CAS PubMed.
- D. P. Levine, Clin. Infect. Dis., 2006, 42, S5–S12 CrossRef CAS PubMed.
- J. Li, S. G. Kim and J. Blenis, Cell Metab., 2014, 19, 373–379 CrossRef CAS PubMed.
- J. A. Washington II and W. R. Wilson, Mayo Clin. Proc., 1985, 60, 189–203 CrossRef.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- K. J. Weissman and P. F. Leadlay, Nat. Rev. Microbiol., 2005, 3, 925–936 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2012, 29, 1050–1073 RSC.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- L. Vieweg, S. Reichau, R. Schobert, P. F. Leadlay and R. D. Süssmuth, Nat. Prod. Rep., 2014, 31, 1554–1584 RSC.
- M. W. Mullowney, R. A. McClure, M. T. Robey, N. L. Kelleher and R. J. Thomson, Nat. Prod. Rep., 2018, 35, 847–878 RSC.
- C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat. Prod. Rep., 2007, 24, 162–190 RSC.
- D. Yu, F. Xu, J. Zeng and J. Zhan, IUBMB Life, 2012, 64, 285–295 CrossRef CAS PubMed.
- M. Holmquist, Curr. Protein Pept. Sci., 2000, 1, 209–235 CrossRef CAS.
- M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- J. E. Schaffer, M. R. Reck, N. K. Prasad and T. A. Wencewicz, Nat. Chem. Biol., 2017, 13, 737–744 CrossRef CAS PubMed.
- A. A. Koch, D. A. Hansen, V. V. Shende, L. R. Furan, K. N. Houk, G. Jiménez-Osés and D. H. Sherman, J. Am. Chem. Soc., 2017, 139, 13456–13465 CrossRef CAS.
- L. Qiao, J. Fang, P. Zhu, H. Huang, C. Dang, J. Pang, W. Gao, X. Qiu, L. Huang and Y. Li, Protein J., 2019, 38, 658–666 CrossRef CAS.
- N. Huguenin-Dezot, D. A. Alonzo, G. W. Heberlig, M. Mahesh, D. P. Nguyen, M. H. Dornan, C. N. Boddy, T. M. Schmeing and J. W. Chin, Nature, 2019, 565, 112–117 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Chem. Rev., 2017, 117, 5334–5366 CrossRef CAS PubMed.
- T. P. Hari, P. Labana, M. Boileau and C. N. Boddy, ChemBioChem, 2014, 15, 2656–2661 CrossRef CAS PubMed.
- B. A. Pfeifer, C. C. C. Wang, C. T. Walsh and C. Khosla, Appl. Environ. Microbiol., 2003, 69, 6698–6702 CrossRef CAS PubMed.
- G. Yim, M. N. Thaker, K. Koteva and G. Wright, J. Antibiot., 2014, 67, 31–41 CrossRef CAS.
- E. A. Felnagle, E. E. Jackson, Y. A. Chan, A. M. Podevels, A. D. Berti, M. D. McMahon and M. G. Thomas, Mol. Pharm., 2008, 5, 191–211 CrossRef CAS PubMed.
- V. Rangaswamy, S. Jiralerspong, R. Parry and C. L. Bender, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15469 CrossRef CAS PubMed.
- M. Peschke, C. Brieke, M. Heimes and M. J. Cryle, ACS Chem. Biol., 2018, 13, 110–120 CrossRef CAS PubMed.
- L. Gu, B. Wang, A. Kulkarni, J. J. Gehret, K. R. Lloyd, L. Gerwick, W. H. Gerwick, P. Wipf, K. Håkansson, J. L. Smith and D. H. Sherman, J. Am. Chem. Soc., 2009, 131, 16033–16035 CrossRef CAS.
- N. M. Gaudelli and C. A. Townsend, Nat. Chem. Biol., 2014, 10, 251–258 CrossRef CAS PubMed.
- K. D. Patel, F. B. d'Andrea, N. M. Gaudelli, A. R. Buller, C. A. Townsend and A. M. Gulick, Nat. Commun., 2019, 10, 3868 CrossRef PubMed.
- L. Ray, K. Yamanaka and B. S. Moore, Angew. Chem., Int. Ed., 2016, 55, 364–367 CrossRef CAS.
- M.-C. Tang, C. R. Fischer, J. V. Chari, D. Tan, S. Suresh, A. Chu, M. Miranda, J. Smith, Z. Zhang, N. K. Garg, R. P. S. Onge and Y. Tang, J. Am. Chem. Soc., 2019, 141, 8198–8206 CrossRef CAS.
- T. Thongkongkaew, W. Ding, E. Bratovanov, E. Oueis, M. a. García-Altares, N. Zaburannyi, K. Harmrolfs, Y. Zhang, K. Scherlach, R. Müller and C. Hertweck, ACS Chem. Biol., 2018, 13, 1370–1379 CrossRef CAS.
- Y.-S. Seo, J. Y. Lim, J. Park, S. Kim, H.-H. Lee, H. Cheong, S.-M. Kim, J. S. Moon and I. Hwang, BMC Genomics, 2015, 16, 349 CrossRef.
- L. Laureti, L. Song, S. Huang, C. Corre, P. Leblond, G. L. Challis and B. Aigle, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6258–6263 CrossRef CAS PubMed.
- G. G. Zhanel, M. Dueck, D. J. Hoban, L. M. Vercaigne, J. M. Embil, A. S. Gin and J. A. Karlowsky, Drugs, 2001, 61, 443–498 CrossRef CAS PubMed.
- B. Arsic, J. Barber, A. Čikoš, M. Mladenovic, N. Stankovic and P. Novak, Int. J. Antimicrob. Agents, 2018, 51, 283–298 CrossRef CAS PubMed.
- K. Buntin, K. J. Weissman and R. Müller, ChemBioChem, 2010, 11, 1137–1146 CrossRef CAS.
- A. G. Newman, A. L. Vagstad, K. Belecki, J. R. Scheerer and C. A. Townsend, Chem. Commun., 2012, 48, 11772–11774 RSC.
- J. Hou, L. Robbel and M. A. Marahiel, Chem. Biol., 2011, 18, 655–664 CrossRef CAS PubMed.
- S. Eys, D. Schwartz, W. Wohlleben and E. Schinko, Antimicrob. Agents Chemother., 2008, 52, 1686–1696 CrossRef CAS PubMed.
- D. Schwartz, N. Grammel, E. Heinzelmann, U. Keller and W. Wohlleben, Antimicrob. Agents Chemother., 2005, 49, 4598–4607 CrossRef CAS.
- F. Trottmann, K. Ishida, J. Franke, A. Stanišić, M. Ishida-Ito, H. Kries, G. Pohnert and C. Hertweck, Angew. Chem., Int. Ed., 2020, 59, 13511–13515 CrossRef CAS PubMed.
- H.-Y. He, M.-C. Tang, F. Zhang and G.-L. Tang, J. Am. Chem. Soc., 2014, 136, 4488–4491 CrossRef CAS PubMed.
- D. A. Hansen, A. A. Koch and D. H. Sherman, J. Am. Chem. Soc., 2017, 139, 13450–13455 CrossRef CAS PubMed.
- C. A. Shaw-Reid, N. L. Kelleher, H. C. Losey, A. M. Gehring, C. Berg and C. T. Walsh, Chem. Biol., 1999, 6, 385–400 CrossRef CAS.
- Y. Zhou, P. Prediger, L. C. Dias, A. C. Murphy and P. F. Leadlay, Angew. Chem., Int. Ed., 2015, 54, 5232–5235 CrossRef CAS.
- X. Zhao, Y. Fang, Y. Yang, Y. Qin, P. Wu, T. Wang, H. Lai, L. Meng, D. Wang, Z. Zheng, X. Lu, H. Zhang, Q. Gao, J. Zhou and D. Ma, Autophagy, 2015, 11, 1849–1863 CrossRef CAS.
- G. W. Heberlig and C. N. Boddy, J. Nat. Prod., 2020, 83, 1990–1997 CrossRef CAS.
- K. M. Hoyer, C. Mahlert and M. A. Marahiel, Chem. Biol., 2007, 14, 13–22 CrossRef CAS PubMed.
- Y. Zhou, A. C. Murphy, M. Samborskyy, P. Prediger, L. C. Dias and P. F. Leadlay, Chem. Biol., 2015, 22, 745–754 CrossRef CAS.
- J. Franke and C. Hertweck, Cell Chem. Biol., 2016, 23, 1179–1192 CrossRef CAS.
- C. Zhang, L. Kong, Q. Liu, X. Lei, T. Zhu, J. Yin, B. Lin, Z. Deng and D. You, PLoS One, 2013, 8, e56772 CrossRef CAS.
- A. P. Praseuth, C. C. C. Wang, K. Watanabe, K. Hotta, H. Oguri and H. Oikawa, Biotechnol. Prog., 2008, 24, 1226–1231 CrossRef CAS PubMed.
- J. A. Matson, K. L. Colson, G. N. Belofsky and B. B. Bleiberg, J. Antibiot., 1993, 46, 162–166 CrossRef CAS.
- D. Mandalapu, X. Ji, J. Chen, C. Guo, W.-Q. Liu, W. Ding, J. Zhou and Q. Zhang, J. Org. Chem., 2018, 83, 7271–7275 CrossRef CAS.
- N. Roongsawang, K. Washio and M. Morikawa, ChemBioChem, 2007, 8, 501–512 CrossRef CAS PubMed.
- J. B. Biggins, H.-S. Kang, M. A. Ternei, D. DeShazer and S. F. Brady, J. Am. Chem. Soc., 2014, 136, 9484–9490 CrossRef CAS PubMed.
- B. K. Scholz-Schroeder, J. D. Soule and D. C. Gross, Mol. Plant-Microbe Interact., 2003, 16, 271–280 CrossRef CAS PubMed.
- I. de Bruijn, M. J. D. de Kock, P. de Waard, T. A. van Beek and J. M. Raaijmakers, J. Bacteriol., 2008, 190, 2777 CrossRef CAS PubMed.
- J. W. Trauger, R. M. Kohli, H. D. Mootz, M. A. Marahiel and C. T. Walsh, Nature, 2000, 407, 215–218 CrossRef CAS.
- G.-L. Tang, Y.-Q. Cheng and B. Shen, Chem. Biol., 2004, 11, 33–45 CrossRef CAS.
- R. A. Oliver, R. Li and C. A. Townsend, Nat. Chem. Biol., 2018, 14, 5–7 CrossRef CAS PubMed.
- Y. Ogasawara, K. Katayama, A. Minami, M. Otsuka, T. Eguchi and K. Kakinuma, Chem. Biol., 2004, 11, 79–86 CAS.
- R. Li, R. A. Oliver and C. A. Townsend, Cell Chem. Biol., 2017, 24, 24–34 CrossRef CAS.
- D. J. Edwards, B. L. Marquez, L. M. Nogle, K. McPhail, D. E. Goeger, M. A. Roberts and W. H. Gerwick, Chem. Biol., 2004, 11, 817–833 CrossRef CAS.
- B. Lin Xiaoxi, T. Lohans Christopher, R. Duar, J. Zheng, C. Vederas John, J. Walter, M. Gänzle and M. A. Elliot, Appl. Environ. Microbiol., 2015, 81, 2032–2041 CrossRef CAS.
- H. Zhou, Y. Li and Y. Tang, Nat. Prod. Rep., 2010, 27, 839–868 RSC.
- I. Fujii, A. Watanabe, U. Sankawa and Y. Ebizuka, Chem. Biol., 2001, 8, 189–197 CrossRef CAS PubMed.
- S.-S. Gao, A. Duan, W. Xu, P. Yu, L. Hang, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 4249–4259 CrossRef CAS PubMed.
- J. H. Yu and T. J. Leonard, J. Bacteriol., 1995, 177, 4792–4800 CrossRef CAS PubMed.
- A. L. Vagstad, E. A. Hill, J. W. Labonte and C. A. Townsend, Chem. Biol., 2012, 19, 1525–1534 CrossRef CAS PubMed.
- T. P. Korman, J. M. Crawford, J. W. Labonte, A. G. Newman, J. Wong, C. A. Townsend and S. C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6246–6251 CrossRef CAS PubMed.
- R. E. Minto and C. A. Townsend, Chem. Rev., 1997, 97, 2537–2556 CrossRef CAS PubMed.
- Y. He, Y. Sun, T. Liu, X. Zhou, L. Bai and Z. Deng, Appl. Environ. Microbiol., 2010, 76, 3283–3292 CrossRef CAS PubMed.
- H. Ikeda, T. Nonomiya, M. Usami, T. Ohta and S. Omura, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9509–9514 CrossRef CAS.
- A. Migita, M. Watanabe, Y. Hirose, K. Watanabe, T. Tokiwano, H. Kinashi and H. Oikawa, Biosci., Biotechnol., Biochem., 2009, 73, 169–176 CrossRef CAS PubMed.
- C. J. Balibar, A. R. Howard-Jones and C. T. Walsh, Nat. Chem. Biol., 2007, 3, 584–592 CrossRef CAS PubMed.
- L. Lou, G. Qian, Y. Xie, J. Hang, H. Chen, K. Zaleta-Rivera, Y. Li, Y. Shen, P. H. Dussault, F. Liu and L. Du, J. Am. Chem. Soc., 2011, 133, 643–645 CrossRef CAS PubMed.
- C. Bihlmaier, E. Welle, C. Hofmann, K. Welzel, A. Vente, E. Breitling, M. Müller, S. Glaser and A. Bechthold, Antimicrob. Agents Chemother., 2006, 50, 2113–2121 CrossRef CAS.
- L. Lou, H. Chen, R. L. Cerny, Y. Li, Y. Shen and L. Du, Biochemistry, 2012, 51, 4–6 CrossRef CAS.
- F. Romero, F. Espliego, J. Pérez Baz, T. García de Quesada, D. Grávalos, F. de la Calle and J. L. Fernández-Puentes, J. Antibiot., 1997, 50, 734–737 CrossRef CAS.
- E. Erba, D. Bergamaschi, S. Ronzoni, M. Faretta, S. Taverna, M. Bonfanti, C. V. Catapano, G. Faircloth, J. Jimeno and M. D'Incalci, Br. J. Cancer, 1999, 80, 971–980 CrossRef CAS PubMed.
- H. Okada, H. Suzuki, T. Yoshinari, H. Arakawa, A. Okura, H. Suda, A. Yamada and D. Uemura, J. Antibiot., 1994, 47, 129–135 CrossRef CAS.
- M. Kotowska and K. Pawlik, Appl. Environ. Microbiol., 2014, 98, 7735–7746 CAS.
- E. Yeh, R. M. Kohli, S. D. Bruner and C. T. Walsh, ChemBioChem, 2004, 5, 1290–1293 CrossRef CAS.
- D. Schwarzer, H. D. Mootz, U. Linne and M. A. Marahiel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14083–14088 CrossRef CAS PubMed.
- M. L. Heathcote, J. Staunton and P. F. Leadlay, Chem. Biol., 2001, 8, 207–220 CrossRef CAS.
- A. Koglin, F. Löhr, F. Bernhard, V. V. Rogov, D. P. Frueh, E. R. Strieter, M. R. Mofid, P. Güntert, G. Wagner, C. T. Walsh, M. A. Marahiel and V. Dötsch, Nature, 2008, 454, 907–911 CrossRef CAS.
- A. R. Butler, N. Bate and E. Cundliffe, Chem. Biol., 1999, 6, 287–292 CrossRef CAS.
- Y. Doi-Katayama, Y. J. Yoon, C. Y. Choi, T. W. Yu, H. G. Floss and C. R. Hutchinson, J. Antibiot., 2000, 53, 484–495 CrossRef CAS.
- Y. Xue, L. Zhao, H.-w. Liu and D. H. Sherman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12111 CrossRef CAS.
- H. Nakamura, J. X. Wang and E. P. Balskus, Chem. Sci., 2015, 6, 3816–3822 RSC.
- B. M. Harvey, T. Mironenko, Y. Sun, H. Hong, Z. Deng, P. F. Leadlay, K. J. Weissman and S. F. Haydock, Chem. Biol., 2007, 14, 703–714 CrossRef CAS.
- B. M. Harvey, H. Hong, M. A. Jones, Z. A. Hughes-Thomas, R. M. Goss, M. L. Heathcote, V. M. Bolanos-Garcia, W. Kroutil, J. Staunton, P. F. Leadlay and J. B. Spencer, ChemBioChem, 2006, 7, 1435–1442 CrossRef CAS.
- T. Liu, D. You, C. Valenzano, Y. Sun, J. Li, Q. Yu, X. Zhou, D. E. Cane and Z. Deng, Chem. Biol., 2006, 13, 945–955 CrossRef CAS.
- T. Liu, X. Lin, X. Zhou, Z. Deng and D. E. Cane, Chem. Biol., 2008, 15, 449–458 CrossRef CAS PubMed.
- R. Liu, F. Fang, Z. An, R. Huang, Y. Wang, X. Sun, S. Fu, A. Fu, Z. Deng and T. Liu, J. Ind. Microbiol. Biotechnol., 2020, 47, 275–285 CrossRef CAS PubMed.
- N. Liu, Y.-S. Hung, S.-S. Gao, L. Hang, Y. Zou, Y.-H. Chooi and Y. Tang, Org. Lett., 2017, 19, 3560–3563 CrossRef CAS.
- N. S. Guntaka, A. R. Healy, J. M. Crawford, S. B. Herzon and S. D. Bruner, ACS Chem. Biol., 2017, 12, 2598–2608 CrossRef CAS PubMed.
- K. Hua, X. Liu, Y. Zhao, Y. Gao, L. Pan, H. Zhang, Z. Deng and M. Jiang, mBio, 2020, 11, e01334 CrossRef CAS.
- C. Li, K. E. Roege and W. L. Kelly, ChemBioChem, 2009, 10, 1064–1072 CrossRef CAS PubMed.
- W.-G. Wang, H. Wang, L.-Q. Du, M. Li, L. Chen, J. Yu, G.-G. Cheng, M.-T. Zhan, Q.-F. Hu, L. Zhang, M. Yao and Y. Matsuda, J. Am. Chem. Soc., 2020, 142, 8464–8472 CrossRef CAS.
- R. Takeda, J. Am. Chem. Soc., 1958, 80, 4749–4750 CrossRef CAS.
- P. C. Dorrestein, E. Yeh, S. Garneau-Tsodikova, N. L. Kelleher and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13843 CrossRef CAS.
- H. Gross and J. E. Loper, Nat. Prod. Rep., 2009, 26, 1408–1446 RSC.
- D. C. Cantu, Y. Chen and P. J. Reilly, Protein Sci., 2010, 19, 1281–1295 CrossRef CAS.
- M. Kotaka, R. Kong, I. Qureshi, Q. S. Ho, H. Sun, C. W. Liew, L. P. Goh, P. Cheung, Y. Mu, J. Lescar and Z.-X. Liang, J. Biol. Chem., 2009, 284, 15739–15749 CrossRef CAS PubMed.
- C. W. Liew, A. Sharff, M. Kotaka, R. Kong, H. Sun, I. Qureshi, G. Bricogne, Z.-X. Liang and J. Lescar, J. Mol. Biol., 2010, 404, 291–306 CrossRef CAS PubMed.
- T. Annaval, J. D. Rudolf, C.-Y. Chang, J. R. Lohman, Y. Kim, L. Bigelow, R. Jedrzejczak, G. Babnigg, A. Joachimiak, G. N. Phillips and B. Shen, ACS Omega, 2017, 2, 5159–5169 CrossRef CAS PubMed.
- S. Lin, S. G. Van Lanen and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4183 CrossRef CAS.
- X. Yan, H. Ge, T. Huang, Hindra, D. Yang, Q. Teng, I. Crnovčić, X. Li, J. D. Rudolf, J. R. Lohman, Y. Gansemans, X. Zhu, Y. Huang, L.-X. Zhao, Y. Jiang, F. Van Nieuwerburgh, C. Rader, Y. Duan and B. Shen, mBio, 2016, 7, e02104 CrossRef CAS PubMed.
- B. Shen, W. Liu and K. Nonaka, Curr. Med. Chem., 2003, 10, 2317–2325 CrossRef CAS PubMed.
- J. W. Labonte and C. A. Townsend, Chem. Rev., 2013, 113, 2182–2204 CrossRef CAS PubMed.
- S. C. Dillon and A. Bateman, BMC Bioinf., 2004, 5, 109 CrossRef.
- K. Belecki and C. A. Townsend, J. Am. Chem. Soc., 2013, 135, 14339–14348 CrossRef CAS.
- D. R. Cohen and C. A. Townsend, Nat. Chem., 2018, 10, 231–236 CrossRef CAS PubMed.
- D. C. Cantu, A. Ardèvol, C. Rovira and P. J. Reilly, Chem.–Eur. J., 2014, 20, 9045–9051 CAS.
- K. Amagai, R. Takaku, F. Kudo and T. Eguchi, ChemBioChem, 2013, 14, 1998–2006 CrossRef CAS.
- T. Chisuga, A. Miyanaga, F. Kudo and T. Eguchi, J. Biol. Chem., 2017, 292, 10926–10937 CrossRef CAS.
- H. Jørgensen, K. F. Degnes, A. Dikiy, E. Fjaervik, G. Klinkenberg and S. B. Zotchev, Appl. Environ. Microbiol., 2010, 76, 283–293 CrossRef.
- H. Jørgensen, K. F. Degnes, H. Sletta, E. Fjærvik, A. Dikiy, L. Herfindal, P. Bruheim, G. Klinkenberg, H. Bredholt, G. Nygård, S. O. Døskeland, T. E. Ellingsen and S. B. Zotchev, Chem. Biol., 2009, 16, 1109–1121 CrossRef.
- M. Gao, A. E. Glenn, A. A. Blacutt and S. E. Gold, Front. Microbiol., 2017, 8, 1775 CrossRef.
- C. Bebrone, Biochem. Pharmacol., 2007, 74, 1686–1701 CrossRef CAS PubMed.
- Y. Li, Y.-H. Chooi, Y. Sheng, J. S. Valentine and Y. Tang, J. Am. Chem. Soc., 2011, 133, 15773–15785 CrossRef CAS PubMed.
- Y. Li, W. Xu and Y. Tang, J. Biol. Chem., 2010, 285, 22764–22773 CrossRef CAS PubMed.
- J. M. Crawford, P. M. Thomas, J. R. Scheerer, A. L. Vagstad, N. L. Kelleher and C. A. Townsend, Science, 2008, 320, 243 CrossRef CAS.
- J. M. Crawford, B. C. R. Dancy, E. A. Hill, D. W. Udwary and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16728 CrossRef CAS.
- D. A. Herbst, C. A. Townsend and T. Maier, Nat. Prod. Rep., 2018, 35, 1046–1069 RSC.
- T. Awakawa, K. Yokota, N. Funa, F. Doi, N. Mori, H. Watanabe and S. Horinouchi, Chem. Biol., 2009, 16, 613–623 CrossRef CAS PubMed.
- E. Szewczyk, Y.-M. Chiang, C. E. Oakley, A. D. Davidson, C. C. C. Wang and B. R. Oakley, Appl. Environ. Microbiol., 2008, 74, 7607 CrossRef CAS.
- Y. H. Chooi, R. Cacho and Y. Tang, Chem. Biol., 2010, 17, 483–494 CrossRef CAS.
- L. Liu, Z. Zhang, C.-L. Shao and C.-Y. Wang, Front. Microbiol., 2017, 8, 1685 CrossRef.
- Y.-M. Chiang, E. Szewczyk, A. D. Davidson, R. Entwistle, N. P. Keller, C. C. C. Wang and B. R. Oakley, Appl. Environ. Microbiol., 2010, 76, 2067 CrossRef CAS PubMed.
- T. Taguchi, T. Awakawa, Y. Nishihara, M. Kawamura, Y. Ohnishi and K. Ichinose, ChemBioChem, 2017, 18, 316–323 CrossRef CAS.
- R. F. Pratt, Cell. Mol. Life Sci., 2008, 65, 2138–2155 CrossRef CAS PubMed.
- T. Kuranaga, K. Matsuda, A. Sano, M. Kobayashi, A. Ninomiya, K. Takada, S. Matsunaga and T. Wakimoto, Angew. Chem., Int. Ed., 2018, 57, 9447–9451 CrossRef CAS PubMed.
- A. Ninomiya, Y. Katsuyama, T. Kuranaga, M. Miyazaki, Y. Nogi, S. Okada, T. Wakimoto, Y. Ohnishi, S. Matsunaga and K. Takada, ChemBioChem, 2016, 17, 1709–1712 CrossRef CAS PubMed.
- D. Thankachan, A. Fazal, D. Francis, L. Song, M. E. Webb and R. F. Seipke, ACS Chem. Biol., 2019, 14, 845–849 CrossRef CAS.
- Y. Zhou, X. Lin, C. Xu, Y. Shen, S.-P. Wang, H. Liao, L. Li, H. Deng and H.-W. Lin, Cell Chem. Biol., 2019, 26, 737–744 CrossRef CAS.
- K. Matsuda, M. Kobayashi, T. Kuranaga, K. Takada, H. Ikeda, S. Matsunaga and T. Wakimoto, Org. Biomol. Chem., 2019, 17, 1058–1061 RSC.
- H. Jörnvall, B. Persson, M. Krook, S. Atrian, R. Gonzalez-Duarte, J. Jeffery and D. Ghosh, Biochemistry, 1995, 34, 6003–6013 CrossRef.
- A. Chhabra, A. S. Haque, R. K. Pal, A. Goyal, R. Rai, S. Joshi, S. Panjikar, S. Pasha, R. Sankaranarayanan and R. S. Gokhale, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5681–5686 CrossRef CAS PubMed.
- P. Kinatukara, K. D. Patel, A. S. Haque, R. Singh, R. S. Gokhale and R. Sankaranarayananan, J. Struct. Biol., 2016, 194, 368–374 CrossRef CAS PubMed.
- J. F. Barajas, R. M. Phelan, A. J. Schaub, J. T. Kliewer, P. J. Kelly, D. R. Jackson, R. Luo, J. D. Keasling and S.-C. Tsai, Chem. Biol., 2015, 22, 1018–1029 CrossRef CAS PubMed.
- S. Deshpande, E. Altermann, V. Sarojini, J. S. Lott and T. V. Lee, J. Biol. Chem., 2021, 100432, DOI:10.1016/j.jbc.2021.100432.
- A. S. Haque, K. D. Patel, M. V. Deshmukh, A. Chhabra, R. S. Gokhale and R. Sankaranarayanan, J. Struct. Biol., 2014, 187, 207–214 CrossRef CAS.
- D. Gahloth, M. S. Dunstan, D. Quaglia, E. Klumbys, M. P. Lockhart-Cairns, A. M. Hill, S. R. Derrington, N. S. Scrutton, N. J. Turner and D. Leys, Nat. Chem. Biol., 2017, 13, 975–981 CrossRef CAS PubMed.
- U. R. Awodi, J. L. Ronan, J. Masschelein, E. L. C. de los Santos and G. L. Challis, Chem. Sci., 2017, 8, 411–415 RSC.
- N. Kessler, H. Schuhmann, S. Morneweg, U. Linne and M. A. Marahiel, J. Biol. Chem., 2004, 279, 7413–7419 CrossRef CAS.
- H. Shigemori, S. Wakuri, K. Yazawa, T. Nakamura, T. Sasaki and J. i. Kobayashi, Tetrahedron, 1991, 47, 8529–8534 CrossRef CAS.
- J. C. Albright, A. W. Goering, J. R. Doroghazi, W. W. Metcalf and N. L. Kelleher, J. Ind. Microbiol. Biotechnol., 2014, 41, 451–459 CrossRef CAS.
- T. Aoyagi, T. Takeuchi, A. Matsuzaki, K. Kawamura and S. Kondo, J. Antibiot., 1969, 22, 283–286 CrossRef CAS PubMed.
- D. E. Ehmann, A. M. Gehring and C. T. Walsh, Biochemistry, 1999, 38, 6171–6177 CrossRef CAS PubMed.
- Y. Li, K. J. Weissman and R. Müller, J. Am. Chem. Soc., 2008, 130, 7554–7555 CrossRef CAS.
- N. Gaitatzis, B. Silakowski, B. Kunze, G. Nordsiek, H. Blöcker, G. Höfle and R. Müller, J. Biol. Chem., 2002, 277, 13082–13090 CrossRef CAS.
- J. Masschelein, W. Mattheus, L.-J. Gao, P. Moons, R. Van Houdt, B. Uytterhoeven, C. Lamberigts, E. Lescrinier, J. Rozenski, P. Herdewijn, A. Aertsen, C. Michiels and R. Lavigne, PLoS One, 2013, 8, e54143 CrossRef CAS.
- G. Wang, Z. Liu, R. Lin, E. Li, Z. Mao, J. Ling, Y. Yang, W.-B. Yin and B. Xie, PLoS Pathog., 2016, 12, e1005685 CrossRef PubMed.
- W. Huang, S. J. Kim, J. Liu and W. Zhang, Org. Lett., 2015, 17, 5344–5347 CrossRef CAS.
- J. P. Gomez-Escribano, L. Song, D. J. Fox, V. Yeo, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 2716–2720 RSC.
- T. Golakoti, W. Y. Yoshida, S. Chaganty and R. E. Moore, J. Nat. Prod., 2001, 64, 54–59 CrossRef CAS.
- B. S. Evans, I. Ntai, Y. Chen, S. J. Robinson and N. L. Kelleher, J. Am. Chem. Soc., 2011, 133, 7316–7319 CrossRef CAS PubMed.
- Y. Hu, V. Phelan, I. Ntai, C. M. Farnet, E. Zazopoulos and B. O. Bachmann, Chem. Biol., 2007, 14, 691–701 CrossRef CAS.
- W. Li, A. Khullar, S. Chou, A. Sacramo and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2869 CrossRef CAS.
- W. Li, S. Chou, A. Khullar and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2958 CrossRef CAS PubMed.
- K. Bitschar, B. Sauer, J. Focken, H. Dehmer, S. Moos, M. Konnerth, N. A. Schilling, S. Grond, H. Kalbacher, F. C. Kurschus, F. Götz, B. Krismer, A. Peschel and B. Schittek, Nat. Commun., 2019, 10, 2730 CrossRef.
- A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brötz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511–516 CrossRef CAS.
- A. A. Fahad, A. Abood, K. M. Fisch, A. Osipow, J. Davison, M. Avramović, C. P. Butts, J. Piel, T. J. Simpson and R. J. Cox, Chem. Sci., 2014, 5, 523–527 RSC.
- Q. Dan, S. A. Newmister, K. R. Klas, A. E. Fraley, T. J. McAfoos, A. D. Somoza, J. D. Sunderhaus, Y. Ye, V. V. Shende, F. Yu, J. N. Sanders, W. C. Brown, L. Zhao, R. S. Paton, K. N. Houk, J. L. Smith, D. H. Sherman and R. M. Williams, Nat. Chem., 2019, 11, 972–980 CrossRef CAS PubMed.
- B. Silakowski, G. Nordsiek, B. Kunze, H. Blöcker and R. Müller, Chem. Biol., 2001, 8, 59–69 CrossRef CAS.
- J. A. Read and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 15762–15763 CrossRef CAS.
- J. S. Li, C. C. Barber, N. A. Herman, W. Cai, E. Zafrir, Y. Du, X. Zhu, W. Skyrud and W. Zhang, J. Ind. Microbiol. Biotechnol., 2020, 47, 319–328 CrossRef CAS.
- L. Pang, X. Tian, W. Pan and J. Xie, J. Cell. Biochem., 2013, 114, 1705–1713 CrossRef CAS PubMed.
- N. A. Moss, M. J. Bertin, K. Kleigrewe, T. F. Leão, L. Gerwick and W. H. Gerwick, J. Ind. Microbiol. Biotechnol., 2016, 43, 313–324 CrossRef CAS.
- N. Schracke, U. Linne, C. Mahlert and M. A. Marahiel, Biochemistry, 2005, 44, 8507–8513 CrossRef CAS PubMed.
- Y. Hai, A. M. Huang and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10348 CrossRef CAS.
- X. Mo and T. A. M. Gulder, Nat. Prod. Rep., 2021 10.1039/D0NP00099J.
- J. W. Sims and E. W. Schmidt, J. Am. Chem. Soc., 2008, 130, 11149–11155 CrossRef CAS.
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS PubMed.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS PubMed.
- H. Li, C. L. M. Gilchrist, C.-S. Phan, H. J. Lacey, D. Vuong, S. A. Moggach, E. Lacey, A. M. Piggott and Y.-H. Chooi, J. Am. Chem. Soc., 2020, 142, 7145–7152 CrossRef CAS PubMed.
- F. Zhao, Z. Liu, S. Yang, N. Ding and X. Gao, Angew. Chem., Int. Ed., 2020, 59, 19108–19114 CrossRef CAS.
- T. Lincke, S. Behnken, K. Ishida, M. Roth and C. Hertweck, Angew. Chem., Int. Ed., 2010, 49, 2011–2013 CrossRef CAS.
- K. L. Dunbar, M. Dell, E. M. Molloy, H. Büttner, J. Kumpfmüller and C. Hertweck, Angew. Chem., Int. Ed., 2021, 60, 4104–4109 CrossRef CAS.
- K. L. Dunbar, M. Dell, E. M. Molloy, F. Kloss and C. Hertweck, Angew. Chem., Int. Ed., 2019, 58, 13014–13018 CrossRef CAS.
- K. L. Dunbar, H. Büttner, E. M. Molloy, M. Dell, J. Kumpfmüller and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 14080–14084 CrossRef CAS.
- K. L. Dunbar, M. Dell, F. Gude and C. Hertweck, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8850 CrossRef CAS PubMed.
- O. A. Barski, S. M. Tipparaju and A. Bhatnagar, Drug Metab. Rev., 2008, 40, 553–624 CrossRef CAS.
- S. Weinig, H.-J. Hecht, T. Mahmud and R. Müller, Chem. Biol., 2003, 10, 939–952 CrossRef CAS.
- O. Perlova, J. Fu, S. Kuhlmann, D. Krug, A. F. Stewart, Y. Zhang and R. Müller, Appl. Environ. Microbiol., 2006, 72, 7485 CrossRef CAS.
- B. Frank, S. C. Wenzel, H. B. Bode, M. Scharfe, H. Blöcker and R. Müller, J. Mol. Biol., 2007, 374, 24–38 CrossRef CAS.
- J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14002–14007 CrossRef CAS.
- M. Gibson, M. Nur-e-alam, F. Lipata, M. A. Oliveira and J. Rohr, J. Am. Chem. Soc., 2005, 127, 17594–17595 CrossRef CAS.
- H. Akiyama, C. Indananda, A. Thamchaipenet, A. Motojima, T. Oikawa, H. Komaki, A. Hosoyama, A. Kimura, N. Oku and Y. Igarashi, J. Nat. Prod., 2018, 81, 1561–1569 CrossRef CAS PubMed.
- J. Rovirosa and A. San-Martín, Quim. Nova, 2006, 29, 52–53 CrossRef CAS.
- J. Piel, D. Hui, G. Wen, D. Butzke, M. Platzer, N. Fusetani and S. Matsunaga, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16222 CrossRef CAS PubMed.
- R. A. Mosey and P. E. Floreancig, Nat. Prod. Rep., 2012, 29, 980–995 RSC.
- Y. Sun, F. Hahn, Y. Demydchuk, J. Chettle, M. Tosin, H. Osada and P. F. Leadlay, Nat. Chem. Biol., 2010, 6, 99–101 CrossRef CAS.
- Y. Sun, H. Hong, F. Gillies, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2008, 9, 150–156 CrossRef CAS PubMed.
- D. H. Davies, E. W. Snape, P. J. Suter, T. J. King and C. P. Falshaw, J. Chem. Soc., Chem. Commun., 1981, 1073–1074 RSC.
- R. F. Little, M. Samborskyy and P. F. Leadlay, PLoS One, 2020, 15, e0239054 CrossRef.
- E. M. Gottardi, J. M. Krawczyk, H. von Suchodoletz, S. Schadt, A. Mühlenweg, G. C. Uguru, S. Pelzer, H.-P. Fiedler, M. J. Bibb, J. E. M. Stach and R. D. Süssmuth, ChemBioChem, 2011, 12, 1401–1410 CrossRef CAS.
- Y. Demydchuk, Y. Sun, H. Hong, J. Staunton, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2008, 9, 1136–1145 CrossRef CAS.
- R. Little, F. C. R. Paiva, R. Jenkins, H. Hong, Y. Sun, Y. Demydchuk, M. Samborskyy, M. Tosin, F. J. Leeper, M. V. B. Dias and P. F. Leadlay, Nat. Catal., 2019, 2, 1045–1054 CrossRef CAS.
- C. Kanchanabanca, W. Tao, H. Hong, Y. Liu, F. Hahn, M. Samborskyy, Z. Deng, Y. Sun and P. F. Leadlay, Angew. Chem., Int. Ed., 2013, 52, 5785–5788 CrossRef CAS PubMed.
- N. R. Lees, L.-C. Han, M. J. Byrne, J. A. Davies, A. E. Parnell, P. E. J. Moreland, J. E. M. Stach, M. W. van der Kamp, C. L. Willis and P. R. Race, Angew. Chem., Int. Ed., 2019, 58, 2305–2309 CrossRef CAS.
- H.-Y. He, H.-X. Pan, L.-F. Wu, B.-B. Zhang, H.-B. Chai, W. Liu and G.-L. Tang, Chem. Biol., 2012, 19, 1313–1323 CrossRef CAS.
- T. Hashimoto, J. Hashimoto, K. Teruya, T. Hirano, K. Shin-ya, H. Ikeda, H.-w. Liu, M. Nishiyama and T. Kuzuyama, J. Am. Chem. Soc., 2015, 137, 572–575 CrossRef CAS PubMed.
- M. J. Byrne, N. R. Lees, L.-C. Han, M. W. van der Kamp, A. J. Mulholland, J. E. M. Stach, C. L. Willis and P. R. Race, J. Am. Chem. Soc., 2016, 138, 6095–6098 CrossRef CAS PubMed.
- Y. Igarashi, N. Matsuoka, Y. In, T. Kataura, E. Tashiro, I. Saiki, Y. Sudoh, K. Duangmal and A. Thamchaipenet, Org. Lett., 2017, 19, 1406–1409 CrossRef CAS.
- H. Sucipto, S. C. Wenzel and R. Müller, ChemBioChem, 2013, 14, 1581–1589 CrossRef CAS.
- H. Sucipto, J. H. Sahner, E. Prusov, S. C. Wenzel, R. W. Hartmann, J. Koehnke and R. Müller, Chem. Sci., 2015, 6, 5076–5085 RSC.
- V. Bergendahl, U. Linne and M. A. Marahiel, Eur. J. Biochem., 2002, 269, 620–629 CrossRef CAS.
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef CAS.
- C. G. Marshall, N. J. Hillson and C. T. Walsh, Biochemistry, 2002, 41, 244–250 CrossRef CAS.
- T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Mol. Biol., 2002, 9, 522–526 CAS.
- T. Izoré, Y. T. Candace Ho, J. A. Kaczmarski, A. Gavriilidou, K. H. Chow, D. L. Steer, R. J. A. Goode, R. B. Schittenhelm, J. Tailhades, M. Tosin, G. L. Challis, E. H. Krenske, N. Ziemert, C. J. Jackson and M. J. Cryle, Nat. Commun., 2021, 12, 2511 CrossRef.
- S. A. Samel, G. Schoenafinger, T. A. Knappe, M. A. Marahiel and L.-O. Essen, Structure, 2007, 15, 781–792 CrossRef CAS.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS.
- Y. Hai, M. Jenner and Y. Tang, Chem. Sci., 2020, 11, 11525–11530 RSC.
- D. H. Scharf, T. Heinekamp, N. Remme, P. Hortschansky, A. A. Brakhage and C. Hertweck, Appl. Microbiol. Biotechnol., 2012, 93, 467–472 CrossRef CAS PubMed.
- K. D. Clevenger, J. W. Bok, R. Ye, G. P. Miley, M. H. Verdan, T. Velk, C. Chen, K. Yang, M. T. Robey, P. Gao, M. Lamprecht, P. M. Thomas, M. N. Islam, J. M. Palmer, C. C. Wu, N. P. Keller and N. L. Kelleher, Nat. Chem. Biol., 2017, 13, 895–901 CrossRef CAS.
- Y. Hai and Y. Tang, J. Am. Chem. Soc., 2018, 140, 1271–1274 CrossRef CAS.
- B. J. C. Law, Y. Zhuo, M. Winn, D. Francis, Y. Zhang, M. Samborskyy, A. Murphy, L. Ren, P. F. Leadlay and J. Micklefield, Nat. Catal., 2018, 1, 977–984 CrossRef CAS.
- S. Müller, S. Rachid, T. Hoffmann, F. Surup, C. Volz, N. Zaburannyi and R. Müller, Chem. Biol., 2014, 21, 855–865 CrossRef.
- K. Zaleta-Rivera, C. Xu, F. Yu, R. A. E. Butchko, R. H. Proctor, M. E. Hidalgo-Lara, A. Raza, P. H. Dussault and L. Du, Biochemistry, 2006, 45, 2561–2569 CrossRef CAS.
- J. Masschelein, P. K. Sydor, C. Hobson, R. Howe, C. Jones, D. M. Roberts, Z. Ling Yap, J. Parkhill, E. Mahenthiralingam and G. L. Challis, Nat. Chem., 2019, 11, 906–912 CrossRef CAS.
- C. J. Balibar and C. T. Walsh, Biochemistry, 2006, 45, 15029–15038 CrossRef CAS.
- J. A. Baccile, H. H. Le, B. T. Pfannenstiel, J. W. Bok, C. Gomez, E. Brandenburger, D. Hoffmeister, N. P. Keller and F. C. Schroeder, Angew. Chem., Int. Ed., 2019, 58, 14589–14593 CrossRef CAS.
- S. Kosol, A. Gallo, D. Griffiths, T. R. Valentic, J. Masschelein, M. Jenner, E. L. C. de los Santos, L. Manzi, P. K. Sydor, D. Rea, S. Zhou, V. Fülöp, N. J. Oldham, S.-C. Tsai, G. L. Challis and J. R. Lewandowski, Nat. Chem., 2019, 11, 913–923 CrossRef CAS.
- J. Zhang, N. Liu, R. A. Cacho, Z. Gong, Z. Liu, W. Qin, C. Tang, Y. Tang and J. Zhou, Nat. Chem. Biol., 2016, 12, 1001–1003 CrossRef CAS PubMed.
- E. J. N. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. Richard Hutchinson, Science, 1999, 284, 1368 CrossRef CAS.
- X. Xie, M. J. Meehan, W. Xu, P. C. Dorrestein and Y. Tang, J. Am. Chem. Soc., 2009, 131, 8388–8389 CrossRef CAS PubMed.
- T.-W. Yu, Y. Shen, Y. Doi-Katayama, L. Tang, C. Park, B. S. Moore, C. Richard Hutchinson and H. G. Floss, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9051 CrossRef CAS.
- R. Kozakai, T. Ono, S. Hoshino, H. Takahashi, Y. Katsuyama, Y. Sugai, T. Ozaki, K. Teramoto, K. Teramoto, K. Tanaka, I. Abe, S. Asamizu and H. Onaka, Nat. Chem., 2020, 12, 869–877 CrossRef CAS PubMed.
- R. Fujii, Y. Matsu, A. Minami, S. Nagamine, I. Takeuchi, K. Gomi and H. Oikawa, Org. Lett., 2015, 17, 5658–5661 CrossRef CAS.
- J. F. Castro, V. Razmilic, J. P. Gomez-Escribano, B. Andrews, J. A. Asenjo and M. J. Bibb, Appl. Environ. Microbiol., 2015, 81, 5820–5831 CrossRef CAS.
- A. Rascher, Z. Hu, G. O. Buchanan, R. Reid and C. R. Hutchinson, Appl. Environ. Microbiol., 2005, 71, 4862–4871 CrossRef CAS PubMed.
- A. Stratmann, C. Toupet, W. Schilling, R. Traber, L. Oberer and T. Schupp, Microbiology, 1999, 145(Pt 12), 3365–3375 CrossRef CAS.
- K. Jensen, H. Niederkrüger, K. Zimmermann, A. L. Vagstad, J. Moldenhauer, N. Brendel, S. Frank, P. Pöplau, C. Kohlhaas, C. A. Townsend, M. Oldiges, C. Hertweck and J. Piel, Chem. Biol., 2012, 19, 329–339 CrossRef CAS PubMed.
- B. Kusebauch, B. Busch, K. Scherlach, M. Roth and C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 5001–5004 CrossRef CAS PubMed.
- J. Moldenhauer, X.-H. Chen, R. Borriss and J. Piel, Angew. Chem., Int. Ed., 2007, 46, 8195–8197 CrossRef CAS.
- L. Hang, M.-C. Tang, C. J. B. Harvey, C. G. Page, J. Li, Y.-S. Hung, N. Liu, M. E. Hillenmeyer and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 9556–9560 CrossRef CAS.
- S. Müller, E. Garcia-Gonzalez, A. Mainz, G. Hertlein, N. C. Heid, E. Mösker, H. van den Elst, H. S. Overkleeft, E. Genersch and R. D. Süssmuth, Angew. Chem., Int. Ed., 2014, 53, 10821–10825 CrossRef PubMed.
- N. M. Llewellyn, Y. Li and J. B. Spencer, Chem. Biol., 2007, 14, 379–386 CrossRef CAS.
- E. Genersch, E. Forsgren, J. Pentikäinen, A. Ashiralieva, S. Rauch, J. Kilwinski and I. Fries, Int. J. Syst. Evol. Microbiol., 2006, 56, 501–511 CrossRef CAS PubMed.
- S. P. Niehs, J. Kumpfmüller, B. Dose, R. F. Little, K. Ishida, L. V. Florez, M. Kaltenpoth and C. Hertweck, Angew. Chem., Int. Ed., 2020, 59, 23122 CrossRef CAS PubMed.
- I. T. Nakou, M. Jenner, Y. Dashti, I. Romero-Canelón, J. Masschelein, E. Mahenthiralingam and G. L. Challis, Angew. Chem., Int. Ed., 2020, 59, 23145–23153 CrossRef CAS.
- J.-y. Kato, N. Funa, H. Watanabe, Y. Ohnishi and S. Horinouchi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2378 CrossRef CAS PubMed.
- E. Takano, Curr. Opin. Microbiol., 2006, 9, 287–294 CrossRef CAS PubMed.
- Q. Wu, Z. Wu, X. Qu and W. Liu, J. Am. Chem. Soc., 2012, 134, 17342–17345 CrossRef CAS.
- C. Gui, Q. Li, X. Mo, X. Qin, J. Ma and J. Ju, Org. Lett., 2015, 17, 628–631 CrossRef CAS.
- D. P. Cogan, J. Ly and S. K. Nair, ACS Chem. Biol., 2020, 15, 2783–2791 CrossRef CAS.
- J. C. Carlson, J. L. Fortman, Y. Anzai, S. Li, D. A. Burr and D. H. Sherman, ChemBioChem, 2010, 11, 564–572 CrossRef CAS.
- C.-S. Yun, T. Motoyama and H. Osada, Nat. Commun., 2015, 6, 8758 CrossRef CAS.
- C.-S. Yun, K. Nishimoto, T. Motoyama, T. Shimizu, T. Hino, N. Dohmae, S. Nagano and H. Osada, J. Biol. Chem., 2020, 295, 11602–11612 CrossRef CAS.
- M. A. Wyatt, Y. Ahilan, P. Argyropoulos, C. N. Boddy, N. A. Magarvey and P. H. M. Harrison, J. Antibiot., 2013, 66, 421–430 CrossRef CAS.
- J. K. Christenson, J. E. Richman, M. R. Jensen, J. Y. Neufeld, C. M. Wilmot and L. P. Wackett, Biochemistry, 2017, 56, 348–351 CrossRef CAS PubMed.
- Y.-L. Du and K. S. Ryan, Nat. Prod. Rep., 2019, 36, 430–457 RSC.
- A. M. Cerdeño, M. J. Bibb and G. L. Challis, Chem. Biol., 2001, 8, 817–829 CrossRef.
- N. R. Williamson, P. C. Fineran, F. J. Leeper and G. P. C. Salmond, Nat. Rev. Microbiol., 2006, 4, 887–899 CrossRef CAS.
- S. Mo, P. K. Sydor, C. Corre, M. M. Alhamadsheh, A. E. Stanley, S. W. Haynes, L. Song, K. A. Reynolds and G. L. Challis, Chem. Biol., 2008, 15, 137–148 CrossRef CAS.
- A. E. Stanley, L. J. Walton, M. Kourdi Zerikly, C. Corre and G. L. Challis, Chem. Commun., 2006, 38, 3981–3983 RSC.
- S. M. Salem, P. Kancharla, G. Florova, S. Gupta, W. Lu and K. A. Reynolds, J. Am. Chem. Soc., 2014, 136, 4565–4574 CrossRef CAS.
- R. Kellmann, T. K. Mihali, Y. J. Jeon, R. Pickford, F. Pomati and B. A. Neilan, Appl. Environ. Microbiol., 2008, 74, 4044 CrossRef CAS.
- S. W. Chun, M. E. Hinze, M. A. Skiba and A. R. H. Narayan, J. Am. Chem. Soc., 2018, 140, 2430–2433 CrossRef CAS PubMed.
- R. Gerber, L. Lou and L. Du, J. Am. Chem. Soc., 2009, 131, 3148–3149 CrossRef CAS.
- T. Milano, A. Paiardini, I. Grgurina and S. Pascarella, BMC Struct. Biol., 2013, 13, 26 CrossRef CAS PubMed.
- J. A. Baccile, J. E. Spraker, H. H. Le, E. Brandenburger, C. Gomez, J. W. Bok, J. Macheleidt, A. A. Brakhage, D. Hoffmeister, N. P. Keller and F. C. Schroeder, Nat. Chem. Biol., 2016, 12, 419–424 CrossRef CAS.
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC.
- P. D. Walker, A. N. M. Weir, C. L. Willis and M. P. Crump, Nat. Prod. Rep., 2021, 38, 723–756 RSC.
- A. Tietze, Y.-N. Shi, M. Kronenwerth and H. B. Bode, ChemBioChem, 2020, 21, 2750–2754 CrossRef CAS PubMed.
- A. A. Koch, J. J. Schmidt, A. N. Lowell, D. A. Hansen, K. M. Coburn, J. A. Chemler and D. H. Sherman, Angew. Chem., Int. Ed., 2020, 59, 13575–13580 CrossRef CAS.
- M. Tosin, D. Spiteller and J. B. Spencer, ChemBioChem, 2009, 10, 1714–1723 CrossRef CAS PubMed.
- M. Tosin, L. Betancor, E. Stephens, W. M. Ariel Li, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2010, 11, 539–546 CrossRef CAS.
- M. Tosin, L. Smith and P. F. Leadlay, Angew. Chem., Int. Ed., 2011, 50, 11930–11933 CrossRef CAS PubMed.
- J. S. Parascandolo, J. Havemann, H. K. Potter, F. Huang, E. Riva, J. Connolly, I. Wilkening, L. Song, P. F. Leadlay and M. Tosin, Angew. Chem., Int. Ed., 2016, 55, 3463–3467 CrossRef CAS PubMed.
- Y. T. C. Ho, D. J. Leng, F. Ghiringhelli, I. Wilkening, D. P. Bushell, O. Köstner, E. Riva, J. Havemann, D. Passarella and M. Tosin, Chem. Commun., 2017, 53, 7088–7091 RSC.
| This journal is © The Royal Society of Chemistry 2022 |