
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metabolomics and complementary techniques to investigate the plant phytochemical cosmos†
Hiroshi
Tsugawa
 *abcd,
Amit
Rai
ae,
Kazuki
Saito
ae and
Ryo
Nakabayashi
*a
*abcd,
Amit
Rai
ae,
Kazuki
Saito
ae and
Ryo
Nakabayashi
*a
aRIKEN Center for Sustainable Resource Science, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan. E-mail: htsugawa@go.tuat.ac.jp; roy.nakbayaski@gmail.com
bRIKEN Center for Integrative Medical Sciences, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan
cDepartment of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Nakamachi, Koganei, Tokyo 184-8588, Japan
dGraduate School of Medical Life Science, Yokohama City University, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan
ePlant Molecular Science Center, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8675, Japan
First published on 20th August 2021
Abstract
Covering: up to 2021
Plants and their associated microbial communities are known to produce millions of metabolites, a majority of which are still not characterized and are speculated to possess novel bioactive properties. In addition to their role in plant physiology, these metabolites are also relevant as existing and next-generation medicine candidates. Elucidation of the plant metabolite diversity is thus valuable for the successful exploitation of natural resources for humankind. Herein, we present a comprehensive review on recent metabolomics approaches to illuminate molecular networks in plants, including chemical isolation and enzymatic production as well as the modern metabolomics approaches such as stable isotope labeling, ultrahigh-resolution mass spectrometry, metabolome imaging (spatial metabolomics), single-cell analysis, cheminformatics, and computational mass spectrometry. Mass spectrometry-based strategies to characterize plant metabolomes through metabolite identification and annotation are described in detail. We also highlight the use of phytochemical genomics to mine genes associated with specialized metabolites' biosynthesis. Understanding the metabolic diversity through biotechnological advances is fundamental to elucidate the functions of the plant-derived specialized metabolome.
 Hiroshi Tsugawa | Hiroshi Tsugawa got his PhD from Osaka University in 2012, Japan. His major is systems biology through computational mass spectrometry (CompMS) to deepen the understanding of metabolisms in living organisms. Employing CompMS, he could develop a data processing pipeline for complex MS data and identify unknown metabolites using computational analysis of mass fragmentation. The awards he received are RIKEN BAIHO Award (2020), Top 40 Under 40 of “The Analytical Science” (2018), and RIKEN Incentive Research Award (2016). Since 2021, he is working as an associate professor at the Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology. |
 Amit Rai | Amit Rai got his PhD in 2013 from the National University of Singapore. He is currently working as a Research Scientist at the Metabolomics research group, CSRS, RIKEN, and as a guest lecturer at Chiba University. His research interest includes functional genomics for plant natural products using comparative genomics, metabolomics, integrative multi-omics, and systems biology approach. He has been awarded the Young Scientist award at the National University of Singapore (Nov. 2014), best research poster at “The 4th International Conference on Plant Metabolism”, Dalian, China (2017), and the best original research by The Japanese Society of Pharmacognosy (2018). |
 Kazuki Saito | Kazuki Saito is a Professor Emeritus at Chiba University after retiring as a professor of the Graduate School of Pharmaceutical Sciences. Since April 2020, he has been appointed as the Director of RIKEN Center for Sustainable Resource Science. His research is focused on metabolomics, functional genomics, and biotechnology of plant-specialized metabolism. He is also fond of music (classic and jazz), movies, history, and hiking. |
 Ryo Nakabayashi | Ryo Nakabayashi received his PhD from Chiba University in 2009, Japan. His expertise is natural product chemistry, analytical chemistry, and metabolomics. As a research scientist, he has joined the Metabolomics Research Group (Principal Investigator: Dr Kazuki Saito), RIKEN CSRS, and has developed metabolomics approaches using LC-MS/MS, MALDI-MS, FTICR-MS, and IMS for identifying specialized metabolites and their functions in plants. He has received the RIKEN Incentive Award in 2017 and the JSPCMB Award for Young Scientists in 2020. |
1. Introduction
Plants are predominantly photosynthetic eukaryotes with over 391![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 known species across the planet.1 One of the fascinating characteristics of plants is their unique metabolic system (metabolism), which produces highly complex and bioactive molecules that modulate the cellular activity, microbiome, and phenotype in human health and diseases.2,3 Each plant species produces secondary (specialized) metabolites. According to Afendi et al., the plant metabolite diversity exceeds 1 million, with each plant producing nearly 4.7 structurally unique molecules;4 of these, only ∼300000 structures have been cataloged in the Dictionary of Natural Products (DNP). Moreover, this diversity may be further expanded when human microbial metabolism is considered, where the structure of natural products is modified in the microbiome.5 In fact, the modified molecule often has higher bioactivity than that of the original form. For instance, 4′,7-dihydroxyisoflavan (equol), catalyzed in the microbiome from the plant metabolite 4′,7-dihydroxyisoflavone (daidzein), acts as a superior ligand for estrogen receptors.6
000 known species across the planet.1 One of the fascinating characteristics of plants is their unique metabolic system (metabolism), which produces highly complex and bioactive molecules that modulate the cellular activity, microbiome, and phenotype in human health and diseases.2,3 Each plant species produces secondary (specialized) metabolites. According to Afendi et al., the plant metabolite diversity exceeds 1 million, with each plant producing nearly 4.7 structurally unique molecules;4 of these, only ∼300000 structures have been cataloged in the Dictionary of Natural Products (DNP). Moreover, this diversity may be further expanded when human microbial metabolism is considered, where the structure of natural products is modified in the microbiome.5 In fact, the modified molecule often has higher bioactivity than that of the original form. For instance, 4′,7-dihydroxyisoflavan (equol), catalyzed in the microbiome from the plant metabolite 4′,7-dihydroxyisoflavone (daidzein), acts as a superior ligand for estrogen receptors.6
As many metabolites in plants (phytochemicals) possess biologically active sites for mammalian proteins, nearly half of the commercially available drugs released during 1940–2014 were of plant origin.7 According to the MarketsandMarkets analysis,8 plant extracts as a commercial commodity in industries, including foods, flavors, cosmetic products, and drugs, were estimated to be worth over USD 23.7 billion in 2019 and have been projected to reach USD 59.4 billion by 2025. Plant specialized metabolites play vital roles in disease resistance, interspecific competition, and stress response (e.g., drought, high light, and abnormal metabolism under nutrient deficiency).9 More importantly, these metabolites serve pivotal physiological and biological functions to maintain plant homeostasis, suggesting the widely-held view on the difference between primary and specialized metabolites to be obsolete.10 Therefore, illuminating the structural diversity of plant metabolites is significant for industrial and scientific progress.
Metabolomics, or metabolome analysis, has become popular to explore the biosynthetic pathways of interest and high-throughput screening of natural products.11,12 Moreover, metabolomics is an essential tool to elucidate the synergistic metabolic pathways of plants and their microbiomes, such as the symbiosis between Fabaceae plants and Rhizobium.13 Although next-generation sequencing (NGS) is efficient for obtaining metagenomic information (i.e., the presence of bacterial genes), such static genome information is not always linked to plant phenotypes. In this context, metabolomics provides dynamic information of functional molecules synthesized through the harmonized multi-biomolecule system of metabolites, proteins, RNAs, and epigenomic modifications. Systems biology using multi-omics or transomics data is an active research field to further our understanding of metabolomes.14,15 Therefore, metabolites and their chemical diversity must be comprehensively studied to elucidate the physiological roles of these metabolites and their underlying molecular mechanisms.
Mass spectrometry (MS)-based untargeted metabolomics has the potential to explore the diversity of plant natural products to study plant metabolism and to perform the high-throughput screening of metabolites. Currently, liquid chromatography coupled with high-resolution tandem MS (LC-MS/MS) is a popular technique owing to the (1) scalability of electrospray ionization (ESI), covering a wide array of chemical properties for metabolite ionization;16 (2) high mass accuracy (<100 ppb, 1.79 mDa at m/z 757.52 in 21T FT-ICR) for reliably predicting the molecular formulae of unknown natural products based on the accurate m/z values of the precursors;17 (3) information-rich mass fragmentation (recorded as MS/MS spectrum) that provides information on the substructures of the metabolites;18 and (4) availability of comprehensive metabolite MS/MS spectral libraries (>850000 unique molecular standards).19 Moreover, the methodologies of obtaining the retention index using a series of fatty acid-derived chemicals, as used in gas chromatography (GC)-MS, facilitate systematic annotation in reverse-phase and hydrophilic interaction chromatography.20,21 Furthermore, recent advances in ion mobility (IM) and related informatics tools provide robust and reliable annotation criteria based on the collision cross-section (CCS) values of ∼12 million compounds,22 thus increasing the coverage of metabolic profiling by separating isobars in a drift tube.23,24 However, despite the remarkable advances in MS techniques, databases, and informatics tools, the current metabolomics infrastructure in natural product research is inadequate to unveil the global plant metabolome because of the complexity and diversity of the chemical structures, in addition to the lack of MS/MS spectra for most specialized metabolites, particularly alkaloids.
In this review, we focused on the current strategies to explore highly complex and diverse plant metabolomes. First, we provided an overview of the grand challenges of metabolomics in natural product research. The importance of bottom-up approaches such as isolation and/or enzymatic production of plant metabolites is also discussed to clarify the importance of the MS-based metabolomics approach. Second, we reviewed the current MS-based state-of-the-art top-down approaches. Notably, the computational MS (CompMS) techniques aimed at metabolite annotation from the analysis of raw MS datasets were detailed. Third, functional genomics is considered as a complementary module for metabolome analysis, assisting in the discovery of new metabolites and the elucidation of novel metabolic pathways. Finally, we discussed the prospects of plant metabolomics in the coming decade, emphasizing upon a way forward to meet the limitations in the current approaches. Overall, we highlight the importance of integrating MS informatics (CompMS), bioinformatics, and cheminformatics to accelerate plant natural product research.25
2. Grand challenges in metabolomics: general approaches to natural product chemistry
Many reviews have focused on the challenges in the comprehensive annotation of natural products using advanced analytical and computational techniques (the top-down approach).26–28 Most of these techniques aim to increase the rate of “putatively annotated metabolites”; in other words, they identify metabolites based on indirect evidence but do not validate the results using standard compounds. Meanwhile, identifying novel natural products with explicit validation can unveil biological mechanisms hitherto unknown. This can provide novel opportunities for the contribution of plant natural products biochemistry in the field of human medicine. Moreover, such discoveries have been well supported by general biochemical approaches (bottom-up approach), including the isolation of metabolites and production of target compounds using recombinant enzymes or in vivo gene transfection. Herein, we review both the top-down and the bottom-up approaches in metabolomics and the integration of these approaches.2.1. Annotation in untargeted metabolomics
One of the major challenges in metabolomics is the annotation of diverse molecules.26 Because small molecules (<2000 Da) show a massive physicochemical diversity, metabolites should be identified based on the match of multiple MS properties, including the retention time, CCS, and MS/MS spectra with those of authentic standards.18 As a guideline for annotation, Metabolomics Standards Initiative (MSI) has recommended four confidence levels29 (Fig. 1, Table 1): level 1, identified metabolites using authentic standard compounds; level 2, putatively annotated metabolites using public/commercial spectral libraries; level 3, putatively characterized metabolites based on diagnostic ion and/or partial spectral similarities to known compounds of a chemical class; and level 4, unknown metabolites, although they can still be differentiated and quantified based on the MS profiles. In 2014, Schymanski et al. proposed more reasonable criteria for using high-resolution LC-MS/MS-based metabolomics and exposomics as follows: level 1, same as the MSI level 1 definition; level 2a, putatively annotated metabolites matching literature or library spectra, with unambiguous spectrum structure match; level 2b, putatively annotated metabolites matching diagnostic MS2 fragments and/or ionization behavior, when no other structure fits the experimental information; level 3, tentative candidate metabolites with evidence for possible structures but insufficient information on the exact structure; level 4, metabolites for which an unequivocal molecular formula can be unambiguously assigned using the spectral information; and level 5, metabolites whose exact mass (m/z) can be measured in a biological sample and that are of specific interest for investigation but lack information to assign even a formula.30 Notably, the MSI guidelines are expected to be further revised in the near future to make suitable guidelines in metabolite annotation considered for the latest MS advances including ion mobility.31 While the importance of annotation strategies tackling levels 2, 3, and 4 of Schymanski et al.'s criteria has been reported by several reviews and research articles on metabolomics, level 1 chemical assignment is equally important. Therefore, we firstly summarize the importance of level 1 chemical assignment using a general yet effective approach in natural product chemistry, which facilitates not only the discovery of a novel metabolite structure but also the elucidation of metabolites, which contain the same substructure moieties.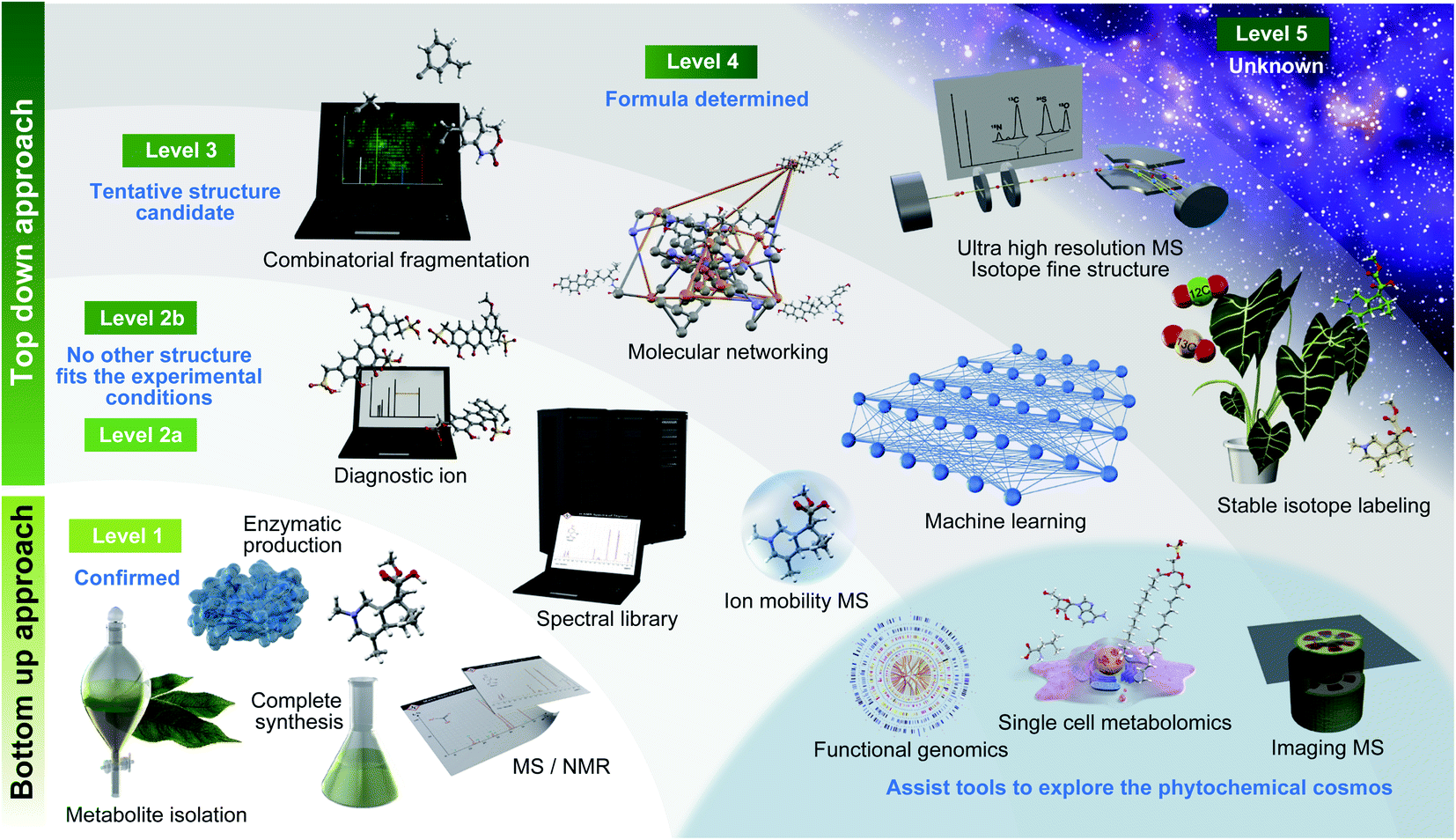 | ||
| Fig. 1 Roadmap to investigate the diversity of phytochemicals. Essential technologies are described along with the definitions of metabolite annotation recommended by Schymanski et al. (2014).30 | ||
| Levels | Metabolomics standards initiative | Levels | Schymanski E. L. et al. (2014) |
|---|---|---|---|
| Level 1 | Confident 2D structure confirmed by the authentic standard confirming the consistency of retention time and MS/MS spectrum | Level 1 | Confirmed structure confirmed via appropriate measurement of a reference standard with MS, MS/MS and retention time matching. If possible, an orthogonal method should also be used |
| Level 2 | Putatively annotated structures matched to literature data, mass spectral databases, or other diagnostic evidences in MS/MS spectrum | Level 2a | Library this involves matching literature or library spectrum data where the spectrum-structure match is unambiguous |
| Level 2b | Diagnostic represents the case where no other structure fits the experimental information. Evidence can include diagnostic MS/MS fragments and/or ionization behaviour, parent compound information and the experimental context | ||
| Level 3 | Putatively characterized compound class (unreached to structure level annotation) requiring at least one piece of metabolite information | Level 3 | Tentative candidate(s) describes a “grey zone”, where evidence exists for possible structure(s), but insufficient information for one exact structure only (e.g., positional isomers) |
| Level 4 | Unequivocal molecular formula is possible when a formula can be unambiguously assigned using the spectral information | ||
| Level 4 | Unknown compounds of interest present in biological samples | Level 5 | Exact mass (m/z) can be measured in a sample and be of specific interest for the investigation. |
2.2. Motivation to analyze authentic standards in untargeted metabolomics
In both definitions, level 1 annotation requires confirmation against authentic standards; however, preparing all chemicals of interest are impractical because of the lack of commercially available compounds, the difficulty of complete organic synthesis, and the high cost of commercial natural products. However, analyzing authentic standards has important implications for the chemical assignment in metabolomics. Once a metabolite is identified, its MS/MS spectrum is deposited in libraries or databases for annotation in public databases, such as MassBank,32 GNPS,33 and MoNA (https://mona.fiehnlab.ucdavis.edu/). Notably, many MS characteristics of metabolites, such as isotopic patterns, m/z values, signal intensities of precursor and product ions, and the fragmentation behavior of the metabolite structure, can be obtained through these resources. Such characteristics are useful to identify the metabolites, even unknown ones, classified in a similar or the same metabolic class based on a unique isotope pattern and/or diagnostic fragment ion matching with a unique substructure. For instance, plants produce multiple isomers of acylsugars, alkaloids, flavonoids (anthocyanins, flavonols, isoflavones, and C-flavones), halogen-containing metabolites, iridoids, lignans, phenolamides, phenylpropanoids, saponins, sulfur-containing metabolites (S-metabolites), and terpenoids, and these can be identified using “feature-centric” approaches. The feature-centric approach employs various systematic protocols and software programs to utilize the MS features, including a unique isotopic pattern, fragment ion, neutral loss, and their combination for metabolite annotation: the example is to trace m/z 285.055 in ESI(+)-MS/MS to grasp metabolites containing kaempferol aglycone.34–36 Feature-centric characterization is also useful for the dereplication of natural products by removing identified, annotated, and characterized metabolites, resulting in the discovery of truly novel metabolite candidates.372.3. Compound isolation from plants to identify novel metabolites
To date, orphan receptor elucidation can accelerate medical research and drug discovery once a ligand is identified.38 Likewise, identifying a novel metabolite structure may lead to the discovery of many metabolites classified in the same metabolite class (or ontology). Therefore, the isolation of truly (dereplicated) unknown compounds from plants of interest is an important step.39–41There are two considerations for unknown metabolite isolation: (1) understanding the physicochemical properties and (2) determining the amount of the starting materials. As described above, MS analysis provides rich information on the physicochemical properties of a metabolite. For instance, when the target ion is highly abundant in either positive or negative ion mode, the chemical property can be considered basic or acidic, respectively. If the ion is equally abundant in both the ion modes, the metabolite is neutral. Notably, at least 100 μg of a compound with high purity must be isolated because structural elucidation often requires nuclear magnetic resonance (NMR) spectroscopy for one- and two-dimensional analyses (up to m/z 1500). If the target metabolite is likely an isomer of an authentic standard compound, it can be quantified using a standard curve, and the amount of the required plant material can be calculated. However, for truly unknown metabolites, a small-scale experiment must be first performed to predict the concentration of metabolites in the plant and then scaled up as needed. Moreover, the discovery of the plant organ that accumulates a high amount of that metabolite is helpful for effectively isolating the molecule of interest.
Recently, combination approaches using both LC-MS/MS and NMR have been developed to maximize the abilities of metabolite detection of LC-MS and structure elucidation of NMR for MSI level 1 identification.25,42–44 The use of LC-MS/MS coupled with solid-phase extraction (SPE)-NMR offers an automated system, starting from sample extraction to high-throughput metabolite annotation. Using this approach, over 100 plant-specific metabolites including previously unknown structures have been characterized from Medicago truncatula.44 Importantly, the required compound amount can be reduced to the order of micrograms for the structure elucidation of metabolite analogues.43 The advantages and limitations of hyphenated MS-NMR systems have been reviewed in detail by several groups.25,42
Subsequently, a fractionation strategy was designed based on the knowledge of predicted physicochemical properties of unknown metabolites. In addition to MS, ultraviolet (UV) detection is the most frequently used modality to confirm the fractionation results. Since many metabolites show polarity that corresponds to the carbon backbone structure and functional moieties, the first step is to use liquid–liquid partition to obtain the fraction containing a high amount of the target metabolite; as such, for a highly polar metabolite, a high-polarity solvent (e.g., n-butanol and water) is recommended. Moreover, in liquid–liquid partition, solubility in organic solvents with a low polarity (e.g., hexane, ethyl acetate, or chloroform) is employed. Next, the combination of cation and anion exchange chromatography is used to separate the metabolites into three groups: acidic, neutral, and basic. Finally, gel filtration chromatography with resins and a single organic solvent or mixed solvents (e.g., methanol and water) are used to separate metabolites according to their molecular weight. In addition, for further purification, size-based fractions of the metabolites can be separated by reverse-phase chromatography. An efficient way to isolate the target metabolites is to use preparative LC-MS, which can trace the metabolites and isolate it simultaneously by separating a large number of samples injected into the system. Thus, based on the physicochemical characteristics of the target metabolite, a higher rate of isolation can be achieved.
2.4. Enzymatic approaches to obtain natural compounds of interest
The high sensitivity of the MS approach also provides information on the metabolite at trace concentrations in plants. Flavonol glycosides specific to flowers were detected by flavonol profiling using LC-MS but not validated against authentic standards.45 Flavonols were enzymatically synthesized using UGT78D3 identified by reverse genetics and transcriptomic analysis. A T-DNA insertion mutant for three possible flavonol glycosides was screened, and two of these, namely, kaempferol and quercetin 3-O-arabinoside-7-O-rhamnoside, were isolated by chromatography using in vitro reaction products with the glycosyltransferase UGT78D3. Genome sequencing provides precise information on gene functions but this method is feasible when precursors are commercially available for enzymatic reactions.2.5. Purposes of metabolite annotation in MSI level 2, 3, and 4 confidences
We summarized the advantages of level 1 identification by a general yet critical plant biochemical approaches in the previous sections. Improving the annotation rates of MSI levels 2 and 3 are an emerging need in metabolomics for the (1) dereplication of natural products, (2) discovery of novel metabolite structures, and (3) elucidation of metabolism in living organisms. The dereplication process can be accelerated by advanced analytical and CompMS46 techniques that efficiently classify MS ion features to known and expectable (known–unknown) metabolites of MSI levels 1, 2, and 3 and unknown metabolites of MSI level 4. The discovery of new metabolites with the information of a known molecular backbone (aglycone) is also facilitated by untangling the mass spectra, which contain the information of the substructures. In addition, the use of metabolic profiles of level 1 confidence alone is inadequate to understand plant metabolism because <10% of the MS raw data can be annotated by authentic standard-centric annotation.47,48 Although the isotope- and fragment feature-centric metabolite annotation, which is a knowledge propagation technique,49 is possible for level 2b and 3 of Schymanski et al.'s criteria, such metabolite abbreviated information [e.g., flavonol (C15H10O6) O-hex] cannot be utilized directly in either metabolic pathway analysis or integrated genomic, transcriptomic, and proteomic analysis. Therefore, developing bioinformatic tools that can use such structure descriptions, such as plant metabolite-set enrichment analysis,50 is also an emerging need in plant biology. Such methods have recently been proposed in lipidomics (e.g., lipid ontology enrichment analysis).51 Recently, a related methodology, where the metabolites are categorized by metabolic pathways and shared tandem MS patterns, has been developed for the interpretation of plant metabolomics data.52 We believe that the current cheminformatics and CompMS platforms do not meet the requirements of bioinformatics researchers in plant biology compared with the available lipidomics, proteomics, and transcriptomics platforms. Cutting-edge technologies for unknown metabolite annotations have been developed thus far, and the significance of such top-down approaches can be maximized by harmonized integration with general bottom-up approaches.3. Cutting-edge technologies to identify unknown metabolites
This section focuses on the annotation in metabolomics for natural product chemistry. Here, we describe the current methodologies to enable the annotation of levels 2 and 3 of the MSI guidelines and levels 2a, 2b, 3, and 4 of Schymanski et al.'s criteria.3.1. Technological advances in biology, chemistry, and instrumentation
In this section, technological advances, including stable isotope labeling (SIL), ultrahigh-resolution mass spectrometry (UHRMS), imaging mass spectrometry (IMS), and single-cell metabolomics, are highlighted to show how technology can be used to elucidate unknown metabolites in plants.Nakabayashi et al. showed the successful integration of the modern top-down metabolomics approach and the general bottom-up biochemical approach.64 Asparagus, one of the staple vegetables, is a perennial plant, and it biosynthesizes S-containing metabolites, including asparagusic acid in addition to many unknowns. Sulfur has the stable isotope 34S, which is abundant in nature (4.29%), suggesting that exploration using the differences in the m/z values between 32S and 34S (1.9958 Da) can easily reveal unknown S-metabolites. In this study, the metabolome data using LC-FTICR-MS were acquired, and 34S-specific isotope features were obtained for all the detected ions. The ion at m/z 307.08931 ([M + H]+, calculated for C10H19N4O3S2, 307.08930) was detected in these data. The search in the databases returned no results for C10H18N4O3S2, suggesting that the ion is a new metabolite. Because the metabolite was not detected by UV chromatography, no chromophore in its structure was expected. Moreover, the metabolite was well detected in both positive and negative ionization modes with a high signal intensity, leading to the hypothesis that the metabolite forms a zwitterion. Raw asparagus tissues (970.7 g) were prepared for the isolation of this metabolite as a small-scale experiment. Liquid–liquid partition, excluding low-polarity metabolites and reverse-phase chromatography, successfully obtained a rich fraction including the targeted metabolite. After purifying 61 mg of the compound by preparative LC-MS, a new asparagus metabolite, named asparaptine, was identified by NMR spectroscopy and acid hydrolysis.
Recent IMS analyses have revealed the tissue specificity of plant metabolites.66 Segmentation analysis,67 an unsupervised spatial pattern analysis of the detected metabolites, provides a unique localization pattern for a group of metabolites, which are highly accumulated in certain tissues or organs of plants. This indicates that the tissue accumulating the target metabolite of interest can mainly be used for isolation. In fact, the spatial multi-omics approach integrating spatial transcriptomics,68–70 proteomics,71 and metabolomics accelerates the understanding of tissue-specific molecular mechanisms, metabolic pathways across tissues, and physiological roles in a plant phenotype.
SIL-assisted spatial metabolomics also facilitates the elucidation of unknown metabolites. Theoretically, both labeled and non-labeled metabolites are localized in the same tissue or organ of the labeled and non-labeled plants, respectively. The m/z values of the non-labeled metabolites should not be detected in the labeled plant and vice versa. The accumulation pattern of the labeled and non-labeled metabolites can be utilized to decrease the false positive annotations and facilitate molecular formula predictions by identifying the number of elements in the IMS data.
3.2. Cheminformatics and CompMS
Computational science is essential to accelerate research in biology. The term “bioinformatics” is popular and widely used when researchers (particularly biologists) state the importance of informatics in biology. However, metabolomics is an interdisciplinary area integrating biology, physics (MS), and chemistry. Therefore, the use of bioinformatics alone is not sufficient to properly represent each computational science in metabolomics. In this context, three terminologies, namely, MS informatics (CompMS), cheminformatics (computational chemistry), and bioinformatics (computational biology) should be used when considering the informatics of metabolomics (Fig. 2). In this section, we focus on the advances in CompMS and cheminformatics.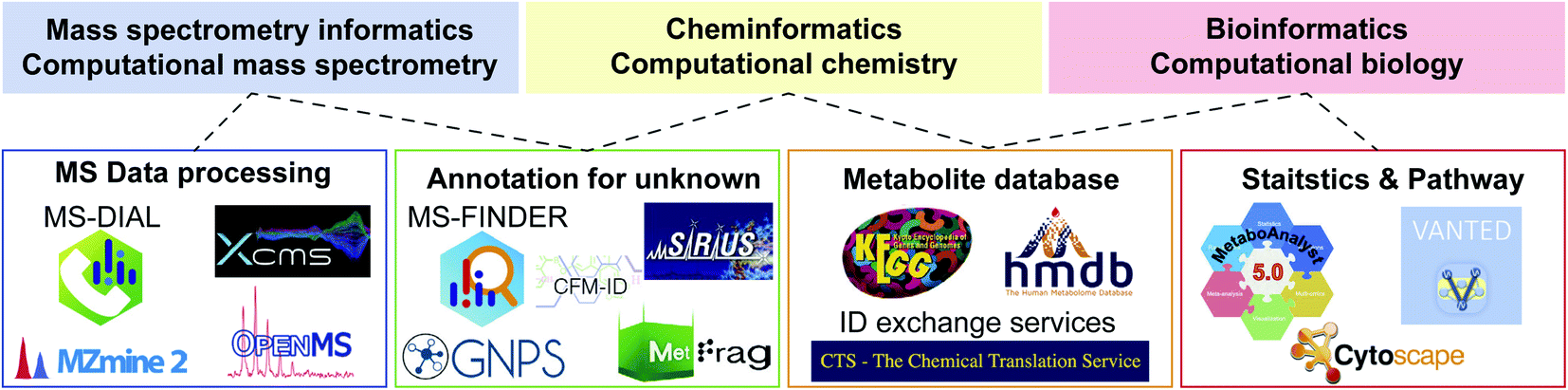 | ||
| Fig. 2 Links among various informatics areas and the related computational programs in metabolomics. Not all tools are described, and advances are warranted. | ||
| Software | Platform | Description as of 2020 |
|---|---|---|
| GNPS | Web | Supporting annotation of unknown EI-MS and MS/MS spectra by many molecular networking techniques. Sharing natural product mass spectra is accelerated. The result of data processing programs from e.g. MS-DIAL and MZmine 2 can directly be analyzed in GNPS environment |
| MetaboAnalyst | Web/local server | Supporting statistics, visualization, and multi-omics analyses for metabolomics data. The metabolome data table for MetaboAnalyst is often prepared by other data processing software tools while it can also be performed in the web application |
| MS-DIAL | OS free based on C#.net standard environment | The most recently developed of the programs presented. Supporting many data processing pipelines for GC-MS, LC-MS, ion mobility, and data independent acquisition in addition to the intuitive GUI environment and statistics analyses. Functions for curating the results of annotation and alignment are substantial, and many third-party programs including GNPS and MetFamily support the MS-DIAL output |
| MZmine 2 | OS free based on Java Environment | Supporting many data processing functions for GC-MS and LC-MS data in addition to data visualization and basic statistics analyses. The parameter optimization can easily be performed in each of processing module, and the direct links to other software programs like GNPS and Sirius have also been supported by many developers |
| OpenMS | OS free based on C++ and python environment | Providing an infrastructure of metabolomics and proteomics data analysis workflow by a wide range of customizable tools and functions. With KNIME and Galaxy environments, flexible and scalable workflow can be built. In addition, many data visualization and statistics approaches are supported |
| XCMS | OS free based on R environment | The first platform of metabolomics data processing. Many informatics researchers contribute to the function developments. Because it is an R package program, the biggest advantage of XCMS is that the result can easily be incorporated to well-maintained bioinformatics tools in the R environment |
| XCMS-online | Web | Providing an easy-to-use environment for metabolomics data processing. Many default parameter settings are available as a starter for each vendor’s machine data. With Metlin, statistics, and pathway platforms maintained in Scripps, it provides the state-of-the-art systems biology platform using metabolomics data |
304633 ESI(+)-MS/MS and 367612 ESI(−)-MS/MS spectra of 36876 unique structures by the first layer of InChIKey.100 In the metabolite structure databases, we used information in MS-FINDER,85 which includes 354438 unique structures, also curated by the first layer of InChIKey, from HMDB,101 SMPDB,102 LipidMAPS,103 YMDB,104 ECMDB,105 BMDB,106 DrugBank,107 FooDB (https://foodb.ca/), PlantCyc,108 ChEBI,109 T3DB,110 STOFF-IDENT (https://www.lfu.bayern.de/stoffident/#!home), Blood Exposome DB,111 Natural Products Atlas,112 KNApSAcK,4 NANPDB,113 UNPD,114 and biomolecule subspace of PubChem115 (of note, at the time of writing this review, structures of the COCONUT116 database were imported to MS-FINDER, and the total number of structures increased to 555975 in the latest version). The chemical ontology of these structures was defined by the superclass and direct parent terms of ClassyFire.117 The statistics are shown in Fig. 3a. Importantly, the curated databases for metabolomics do not cover all of the metabolites reported in the literature. Moreover, the metabolite database of MS-FINDER does not cover all the metabolites that have been reported in scientific papers. To perfectly perform statistics of database coverages in the future, the community-based approach,112 in addition to the database collection approach,118 are needed, while the important statement of this review is evidenced by the statistics of Fig. 3a.
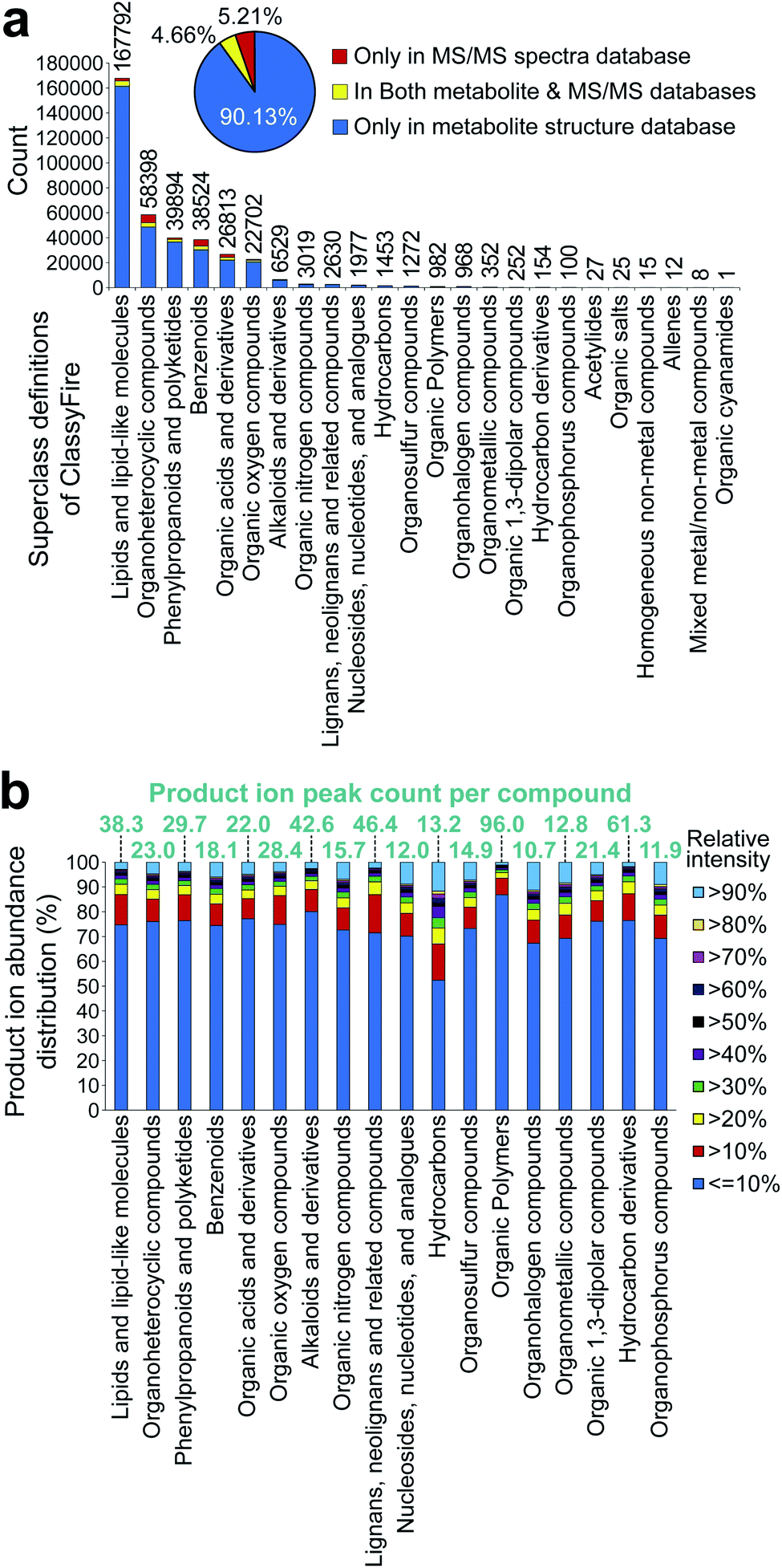 | ||
| Fig. 3 Statistics of metabolite structure and tandem mass spectral database. (a) The record statistics of the metabolite structure and mass spectra were examined. The metabolite structures implemented in MS-FINDER were used for the statistics, and the detail of spectral records was described in the main text. The chemical ontology was defined by the superclass term of ClassyFire. The number above each bar chart shows the count of unique structures defined by the first layer of InChIkey. The red, yellow, and blue colors indicate structures only contained in the MS/MS databases, structures contained in both spectral and metabolite databases, and structures only contained in the metabolite structure databases, respectively. (b) Statistics of the MS/MS peak count and the relative abundance per compound were examined. Statistics was performed using all the spectral records described in (a). The chemical ontology was defined by the superclass term of ClassyFire. | ||
The results showed the heterogeneity of the chemical cosmos, with only six chemical classes covering 95% of the known metabolites. This may be because (1) the top six groups are the major components of animal and plant cells, (2) the minor chemical classes (<5% of the entire chemical space) are chemically complex and difficult to elucidate, and (3) the definition of chemical classes used is the chemical ontology superclass, which is often broadly defined. For (2), the statistics of the product ion peak abundances were examined (Fig. 3b). Although the inconsistency of the spectral record numbers should be considered, the average product ion peak count per compound was mostly equal among the chemical classes. The only exception is alkaloids (42.6 peaks per compound), lignans (46.4 peaks per compound), organic polymers (96.0 peaks per compound), and hydrocarbon derivatives (61.3 peaks per compound). Our investigation showed that in these four chemical classes, the characteristic ion features to define the aglycone and/or unique substructure moiety were not observed, and the mass fragmentation patterns were difficult to interpret, resulting in the difficulty of comprehensive structure elucidation. Moreover, the statistics of relative intensities, assuming that a high intensity can help us efficiently elucidate and curate structures, also showed no major difference among the chemical classes. This result would indicate that the number of spectral records for minor chemical classes is not sufficient to elucidate the entire chemical space of each chemical class by (1) mass spectrometry specialist-centric manual assignment; (2) characteristic ion-centric knowledge propagation technique, as used in GNPS33 and MS-DIAL55 environments; (3) machine learning techniques, as used in SIRIUS,119 CANOPUS,120 and CFM-ID;121,122 and (4) other combinatorial techniques, such as MS-FINDER85 and MetFrag.123,124 Importantly, even though authentic standards for a certain chemical class are not available, the spectra would become more informative if the records of available standards are acquired under several conditions, resulting in the better performance of existing tools even for minor chemical classes.
868 compounds, benzenylium (m/z 77.0386; C6H5+) ions were observed in 5648 compounds. Because there are 13587 compounds containing the benzene moiety in the spectral database, this result indicates that 41.6% benzene-containing molecules generate benzenylium ions in the positive ion mode. As a nitrogen-containing substructure, anilinium (m/z 93.0573; C6H7N+) ion was observed in 1052 compounds and the frequency was calculated to be 20.2% (1052 in 5197 compounds). The most frequently observed neutral loss in the positive ion mode was water loss (18.01 Da; H2O), indicating the neutral loss of water in 54.2% molecules (7496 in 13830) containing hydroxyl moiety denoted as “–[OH]” in the simplified molecular-input line-entry system (SMILES) arbitrary target specification (SMART) format. Interestingly, neutral loss of a lysine moiety (146.1055 Da; C6H14N4O2) was observed in 83% compounds with a lysine moiety. Moreover, phenolate (m/z 93.034; C6H5O−) and phosphate (m/z 78.959; PO3−) ions were observed in the substructures of 74.4% and 47.4% molecules, respectively.
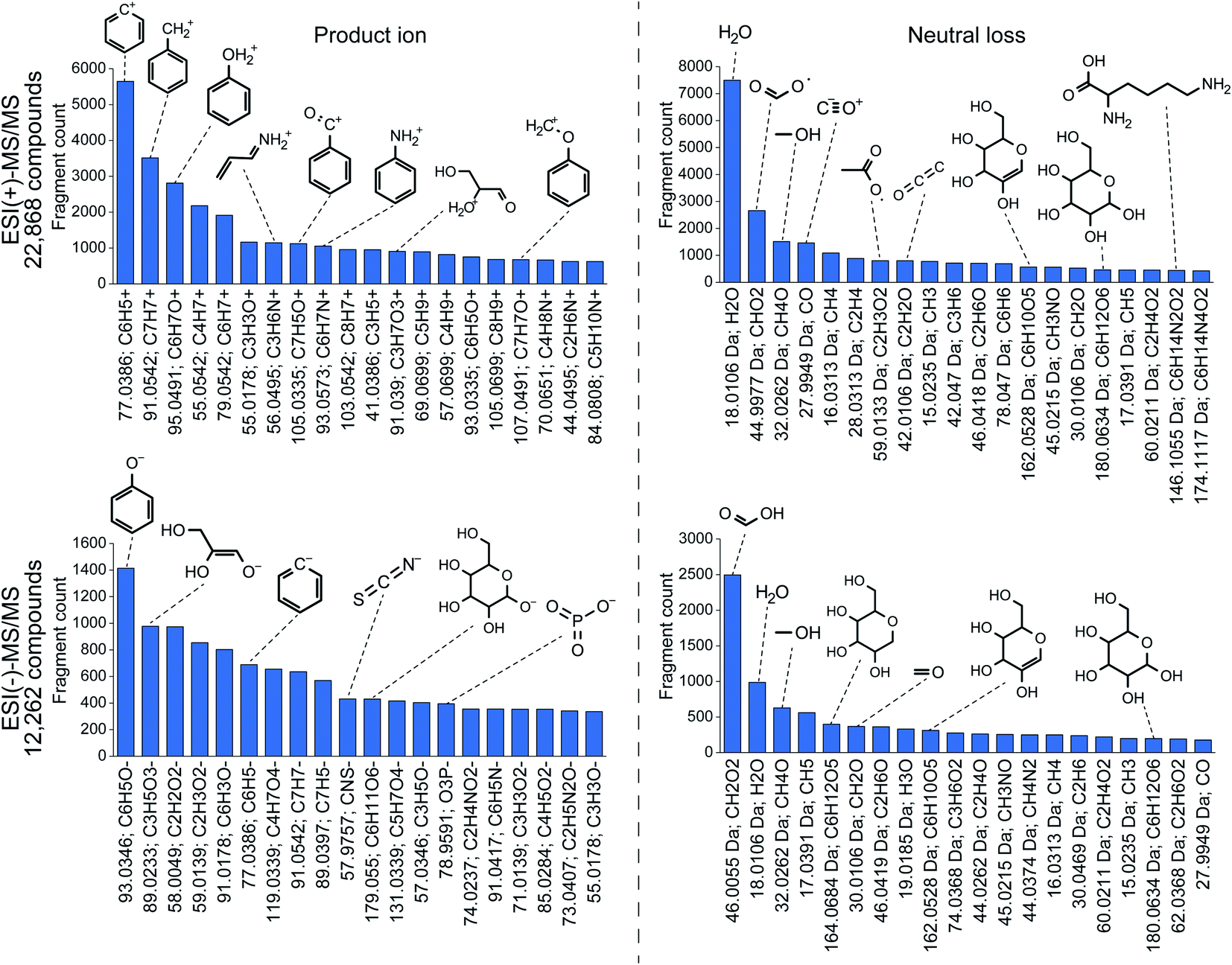 | ||
| Fig. 4 Statistics of fragment ions and neutral losses. The molecular formula and substructure were assigned using MS-FINDER. Before assignment, multiple records for the same metabolite were merged as a single query. | ||
These statistics are particularly useful to elucidate unknown mass spectraI addition, the specificity of the fragment ion-based substructure elucidation can be increased by the co-existence of related fragment ions, suggesting the existence of a targeted substructure; for instance, the observation of both m/z 96.969 (HPO4−) and m/z 78.959 (PO3−) strongly suggests the existence of a phosphate substructure, although the product ion of m/z 96.96 is also detected in compounds containing a sulfate moiety (m/z 96.960; HSO4−). The importance of the co-existence of fragment ions during metabolite annotation has also been demonstrated and discussed previously by the implementation of topic modelling for metabolomics data with MS2LDA.36 Furthermore, the relevance of the substructure (even the entire structure) and fragment ions (both product ion and neutral loss) can be investigated by machine learning techniques, such as deep learning and support vector machine (SVM), to increase the specificity, as used in SIRIUS 4119 and CFM-ID.121,122 Machine learning to elucidate unknown spectra is currently an active research field as the training set of the substructure-fragment pairs can be easily obtained using combinatorial tools such as MetFrag,123,124 MAGMa,125 and MS-FINDER.85 Meanwhile, the accuracy of structure elucidation tools may be saturated in the near future unless the spectral databases are expanded. Moreover, we emphasize the importance of improving the accuracy of fragment annotation tools rather than structure elucidation tools to increase the quality of the training dataset; in other words, the reliability of the substructure-fragment ion pairs should be improved to increase the accuracy of structure elucidation tools.
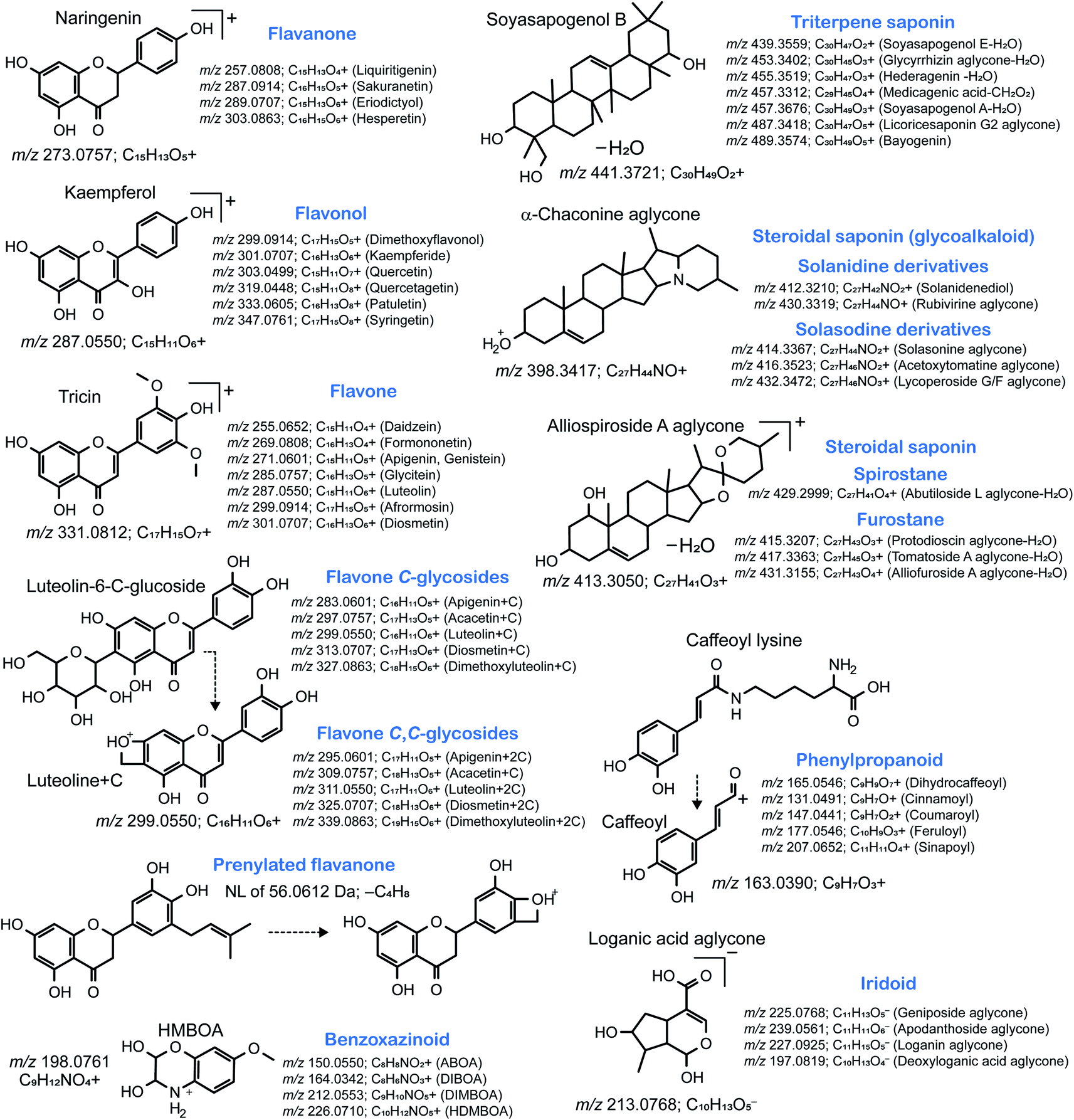
In this review, we performed molecular networking for alkaloids, a chemical class that is difficult to elucidate because of the highly complex mass fragmentation behaviors (Fig. 6): the source data is available as ESI Data.† We used the tandem mass spectra of the PlaSMA database to derive the molecular networks, the methodology of which together with the database details have been described in the previous report.56 A total of 135 alkaloids were mapped in a network, where metabolites were linked by the similarity of structures (red color edge) and the MS/MS spectra (blue color edge). All the MS/MS spectra analyzed for Fig. 6 were acquired under the same analytical condition. The MS/MS similarity was calculated in the MS-DIAL program using the modified Bonanza score.127 The source code and the MS/MS database are freely available at the RIKEN PRIMe website (http://prime.psc.riken.jp/). The structure similarity (blue edge) was calculated by the Tanimoto (Jaccard) index to the fingerprints of MACCS,128 PubChem (https://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt), CDK,129 and Klekota-Roth.130 Interestingly, many unique MS/MS patterns were identified in each alkaloid subclass. For instance, tropane alkaloids, ornithine metabolites biosynthesized in Solanaceae and Erythroxylaceae, generate a product ion at m/z 124.1121 (C8H14N+), which matches the tropane backbone. Indolizidine alkaloids generated the product ion at m/z 160.0757 (C10H10NO+), and yohimbine and corynanthean alkaloids generated product ions at m/z 174.0913 (C11H12NO+). Moreover, many monoterpene indole alkaloids (MIAs) generated product ions at m/z 144.0808 (C10H10N+) matched to the indole moiety, and the fragment ion was detected in the MS/MS spectra of >50% of MIAs, denoted by circles in Fig. 6. Furthermore, the molecular network suggested that the ion mobility-centric diagnostic criteria described by the CCS value of the structure would increase the confidence of metabolite annotations. In Fig. 6, the CCS value is reflected by the size of the node. Many metabolites with similar MS/MS spectra have distinct CCS values. The CCS values were obtained from the AllCCS22 and PNNL CCS databases.131 For structures lacking CCS information, the value was predicted using AllCCS (http://allccs.zhulab.cn/). However, this network also revealed that many alkaloids do not show similar MS/MS spectra and distinct CCS values, even though they are part of the same biosynthetic pathway (e.g., strictosidine, strictosamide, and camptothecin). Further integrative approaches and database accumulation are warranted to elucidate the yet unknown total alkaloid chemical cosmos.
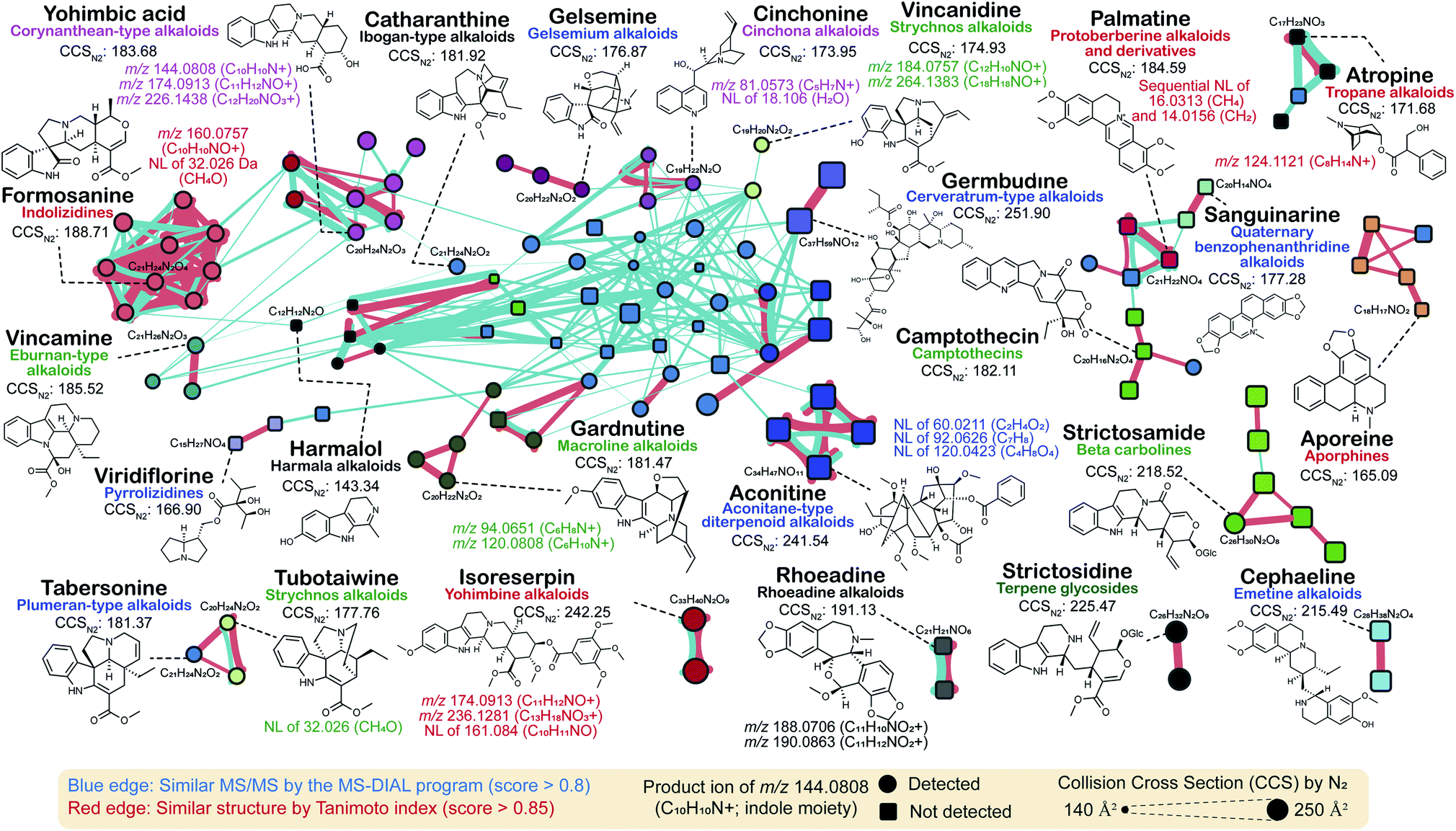 | ||
| Fig. 6 Molecular networking of 135 alkaloids in the PlaSMA database. The MS/MS spectra were obtained under the same analytical condition. The MS/MS similarity (red edge) was calculated in the MS-DIAL program. The structure similarity (blue edge) was calculated by the Tanimoto index based on the structure fingerprints. The metabolite classification follows the definitions of the direct parent terms in ClassyFire. The same node color means the same direct parent term of ClassyFire. | ||
4. Phytochemical genomics in a supporting role to explore the metabolite cosmos
The advances in the form of next-generation sequencing technologies, emerging approaches for assembly scaffolding, and innovative computational tools to achieve chromosome-scale genome assemblies for even a highly repetitive genome content have begun the genomic era for non-model plant species rich in metabolite diversity. Genomics-metabolomics together complement each other to prioritize genes and metabolites for functional and chemical annotations, respectively. In this section, we briefly describe the impact and potential of genomics to complement the advances in new metabolite discovery.4.1. Importance of genome mining for plant natural product discovery
The emergence and expansion of plant functional genomics are often attributed to multi-omics analyses, which have enabled the prioritization and functional characterization of thousands of genes to date.132–134 Genes associated with the biosynthetic pathways of specialized metabolites are often coexpressed and strongly correlated with metabolite accumulation, forming a basis for this analytical approach.135 Nevertheless, the number of genes identified through this strategy is in hundreds, if not in thousands. Therefore, additional criteria to narrow down the candidate genes for functional characterization are essential to predict possible intermediates of specialized metabolite biosynthetic pathways. Genome mining is vital for expanding the microbial natural product discoveries.28 In addition to the added advantage of a smaller genome size with hundreds of thousands of published microbial genomes as a resource for comparative genome analyses, gene clusters have been established as the key feature of microbial natural product biosynthesis. Thus, an identified gene cluster not only associates genes with biosynthesis but also provides clues into metabolite intermediates based on the enzymatic properties of the genes in that cluster. However, compared with the microbial systems, plant genome-based natural product discoveries remain limited because of the near negligible genomic resources available owing to the sheer vastness and diversity of the plant kingdom, their enormous genome sizes, prevalence of repeat-rich genomes, and polyploidy.132,134In recent years, tremendous progress has been achieved in the handling some of the challenges mentioned above owing to a significant reduction in the per-base sequencing cost, advances in long-read sequencing technologies, increased sequencing throughput, and a rapidly expanding toolbox. RNA-Seq-based de novo transcriptome assembly has allowed for the generation of genome resources for thousands of plant species in the last few years.133,134 Consortium-based efforts, such as the 1KP Project, have reported genome resources for 1124 plant species, covering several diverse plant species from distant lineages.136 The established high-quality genome assemblies further complements these efforts.135 In addition to individual genome projects targeting specific plants of interest, consortium efforts, such as Earth Biogenome Project137 and Darwin Tree of Life project (https://www.darwintreeoflife.org/), are aimed at establishing whole-genome assemblies for thousands of diverse plant species in the next decade. These high-quality genome assemblies are particularly valuable for understanding the roles of gene clusters and structural variants, which would be beneficial for modern functional genomics and deep learning tools to predict the functions of unknown components. Here, we briefly elaborate on the current genome-mining approaches and integrative omics approaches to explore natural product biosynthesis (Fig. 7).
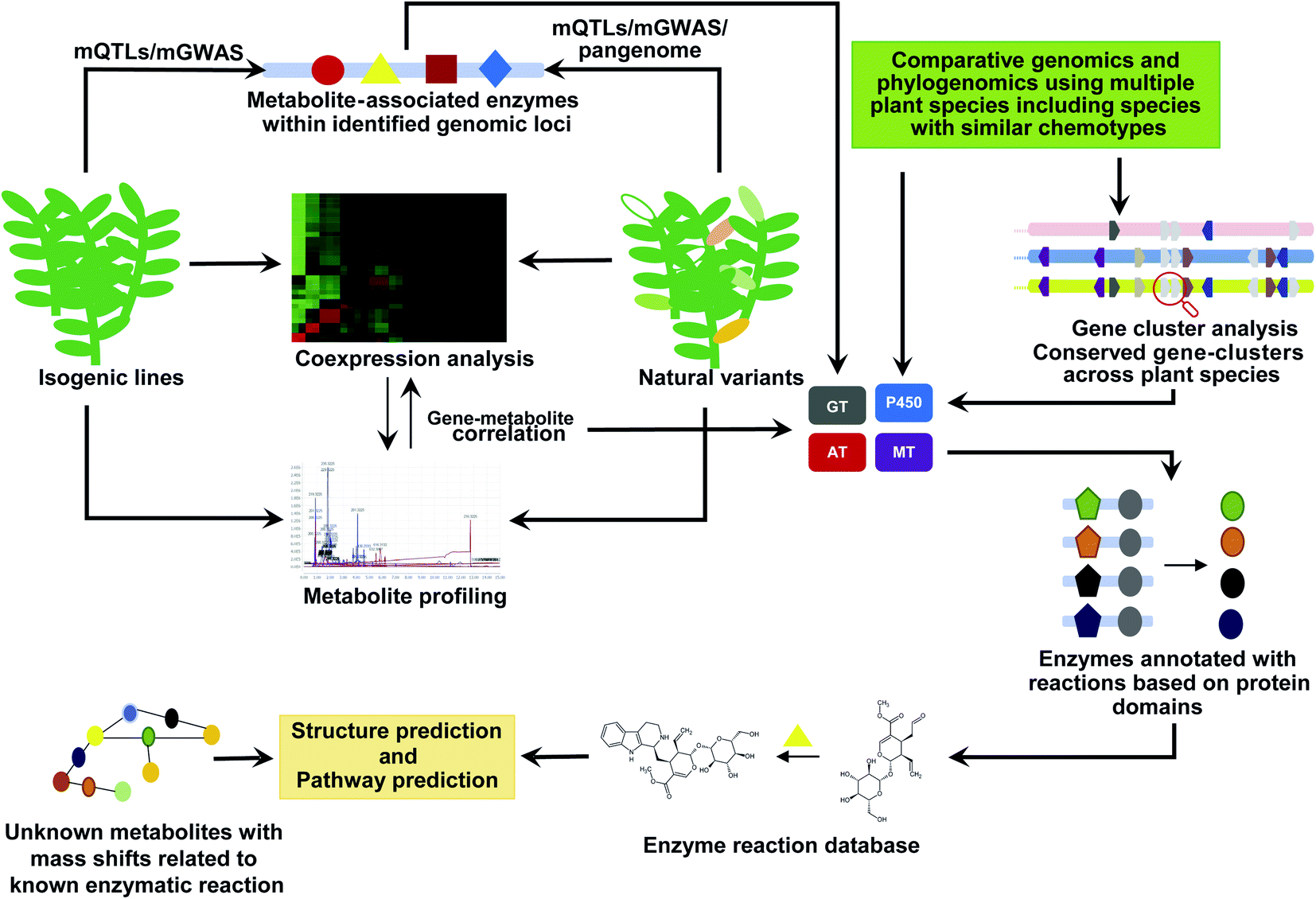 | ||
| Fig. 7 Genome mining to discover and annotate new metabolites. Multi-omics analysis using isogeneic lines and natural variants of a given plant species could be used to identify new metabolites and putatively associated genes involved in its biosynthesis. Using comparative genomics, phytochemical genomics, gene cluster analysis, and using the prior known biochemical reactions will allow to improve the annotation of new metabolites taking into consideration the observed mass-shift and the identified enzyme families across species with similar chemotypes. GT: glycosyltransferases; AT: acyltransferase; MT: methyltransferases; P450: cytochrome P450. | ||
4.2. Near-isogenic lines (NILs), recombinant inbred lines (RILs), and chromosome segment substitution lines (CSSLs) for metabolome-assisted functional genomics
NILs, RILs, and CSSLs are valuable genetic resources for identifying genes associated with a given trait. Typically, these lines serve as powerful tools for genetic analysis and characterization of donor varieties or quantitative traits of the species against the genetic background of a recurrent parent138–146 as well as for the identification of minor-effect quantitative trait loci (QTLs), resulting in the acquisition of novel properties/traits of the donor genotype and identifying the genomic segments and potential genes responsible for a specific trait.140,141,143,147–149 Moreover, NILs, RILs, and CSSLs are vital genetic resources for identifying the genes associated with novel agronomical properties and specialized metabolism.144–146,149,150 QTLs associated with metabolites have been identified to detect polygenic regions and genes associated with biosynthetic pathways.148,150–155 Using 210 RILs, Kang et al. identified 4681 putative metabolites associated with QTLs and used in silico analysis to characterize 35 candidate genes associated with the biosynthesis of 30 structurally identified metabolites, including genes responsible for the variation in the feruloyl serotonin and L-asparagine content across populations.156 Using a large cross-population of maize and its wild ancestor, teosinte, Xu et al. identified genetic factors controlling the metabolic divergence responsible for maize domestication.157 The authors used integrative omics approaches to identify the candidate genes contributing to metabolite divergence and verified the roles of flavanone 3-hydroxylase1, purple aleurone1, and maize terpene synthase1 in the divergence of their related biosynthetic pathways. Using RILs created by crossing Arabidopsis Col-0 and C24, Knoch et al. identified 786 metabolic QTLs on the short arm of chromosome 4 responsible for a major proportion of metabolic variation, including potential genes involved in the biosynthetic pathways.158Inbred-line genomics has been particularly successful in the discovery of new metabolites and associated biosynthetic genes in tomatoes. Metabolic QTL analysis across 76 introgression lines of tomato identified 679 genomic regions associated with the specialized metabolism in the fruit pericarp;150 multi-omics analysis identified the candidate genes associated with the key QTLs. Subsequently, Solyc06g062290 and Solyc10g085230, which are involved in glycoalkaloid biosynthesis, were functionally characterized. Schilmiller et al. used CSSLs and a forward genetics approach to identify the diversity of mono- and sesquiterpene biosynthesis and associated QTLs in the secreting glandular trichomes of tomato.159 The authors identified genomic regions including potential candidate acyltransferases, which regulate the accumulation of total trichome terpenes or acyl sugars, alteration of sesquiterpenes with intact monoterpene moieties, accumulation of the monoterpene α-thujene, and acylsucrose lacking an acetyl group, and shifts in the length of the acyl chains in acyl sucrose. Furthermore, Schilmiller et al. functionally characterized the BAHD family of acyltransferases (Solyc01g105580 or SlAT2), encoding an acetyl-CoA-dependent acyltransferase, and found the addition of acetyl groups to the major detectable tetra-acylsucrose.160 Moreover, Alseekh et al. identified 338 putative metabolite QTLs associated with flavonoids, steroidal glycoalkaloids, and other specialized metabolites using the seeds of Solanum pennellii introgression lines. Authors experimentally validated flavonoid-associated QTLs, including Solyc12g098600 and Solyc12g096870, which encode seed-specific uridine 5-diphosphate-glycosyltransferases.161 In a comprehensive multi-omics analysis using a population of hundreds of diverse tomato accessions, Zhu et al. identified thousands of genetic regions associated with metabolism.162 They showed that the alleles of the genes associated with large fruit were linked to metabolism and identified five major loci that reduced the accumulation of anti-nutritional steroidal glycoalkaloids in ripe fruit. Using an introgression population developed from the wild Peruvian accession of Solanum pennellii (LA0716 or PI246502) and the Solanum lycopersicum cultivar M82, Szymanski et al. established the genetic basis of chemical variations accompanying the transfer of wild-type fruit traits.163 In this study, integrated genome-transcript-metabolite-phenotype QTL analysis was used to elucidate the biosynthesis of esculeosides and lycoperosides from α-tomatine during fruit development and ripening.
These and several other exceptional studies on Arabidopsis, tomato, rice, wheat, soybean, pepper, maize, and potato, among other plant species, have shown a strong association of specialized metabolites with QTLs.157,158,164–166 These lines also offer a potential resource for identifying the intermediates of a given metabolic pathway. However, the generation of such lines is challenging and requires time and resources. Nevertheless, the advantages of discovering new properties and establishing new metabolites through such lines are promising. Artificially generated genetic variability within a given species through fast-neutron or gamma-ray bombardments and ethyl methanesulfonate (EMS) mutagenesis is an alternative to screen and select lines based on the desired phenotype for further characterization.167
4.3. Natural variants for metabolome-assisted functional genomics
Within a given plant species, ecosystem changes result in spontaneous mutations driven by evolutionary processes, such as natural or artificial selection (i.e., domestication), thus deriving natural intraspecific variation (hereafter, natural variation).133,168 These natural variants are also the main toolsets for plant breeders to establish inbred lines with desired agronomical traits.169 Natural variants include single-gene (monogenic) allelic variants and, in many cases, even massive changes through transposon-based genome expansions, deletion/expansion of enzymes, and altered regulation of enzymes involved in biosynthetic pathways. The natural variants of Arabidopsis have served to identify over 100 genes associated with the adaptation of plants to different natural environments, including transcription factors, hormones, and primary and biosynthetic enzymes.132,170 In natural variants of Arabidopsis, untargeted metabolite profiling identified 18 unknown mass features, including novel flavonol derivative saiginol A, which shows enhanced UV-B absorbent properties compared with other phenylpropanoids.41 With over 400000 rice germplasm accessions stored in gene banks worldwide, the metabolite profiling of rice natural variants identified differential levels of aromatic and bioactive metabolites across accessions, contributing to typical characteristics and phenotypes.152,171,172 Natural variants of crops such as tomato, soybean, maize, potato, peanut, strawberry, and cucumber are being collected and maintained worldwide, offering an excellent resource for identifying new metabolites and their association with the phenotype.162,169,173,174 The availability of high-quality genome assemblies allows researchers to use natural variants for accumulating molecular evidence, resulting in colossal chemodiversity.
Genome-wide association studies (GWASs) have gained prominence in achieving a high resolution (to the single nucleotide level) and dissect the genetic architecture with the associated traits.132,174 The advantages of GWAS coupled with metabolomics for large-scale interactive gene-metabolite annotation and identification and metabolic pathway elucidation are well-known.175,176 The combination of GWAS with untargeted metabolomics of 440 Arabidopsis natural variants identified the novel specialized metabolite N-malonyl-D-alloisoleucine.177 GWAS, combined with metabolomics of 529 rice accessions, enabled the identification and functional characterization of 36 candidate genes associated with the specialized metabolism of physiological and nutritional importance.175 Furthermore, unknown metabolites, including sakuranetin, pyridoxine O-glycoside, and phytocassane D, and 166 other metabolites were identified using associations with functionally related genes with this approach. GWAS combined with QTL analysis identified genes involved in specialized metabolism.178,179 GWAS is increasingly being applied in the combination of QTLs to identify and validate potential single-nucleotide polymorphisms (SNPs) associated with a given trait180,181 as it allows the precise identification of the genes and genomic regions associated with the trait of interest. Relatively fewer loci often control the metabolic traits with significant effects, making the combination of GWAS with QTL and coexpression analysis a powerful functional genomics approach. Metabolome-associated GWAS analysis can assign unknown metabolites to a specific genomic region, which can be further used in prioritization for further structural validation.182
One of the limitations of GWAS, which mainly uses short-read sequencing for mapping to a single reference genome, is the loss of genetic information from highly polymorphic regions due to its dependence on the sequence similarity. A single reference genome for a plant species, given the huge natural variants, also means that it may not include some of the vital genomic segments or genes responsible for specific features.183,184 Therefore, pangenomes are essential to understand the extent of genomic variation and overall molecular features that characterize a species. A pangenome for a given species mainly includes the core genome, shared across natural variants, and the dispensable genome, genetic part that varies across the participating accessions, including its chemodiversity.183 The comparison of genomes between the inbred grapevine variety Pinot Noir (PN40024; sequenced in 2007; reference genome) and the grapevine variant Uruguayan Tannat clone (UY11) containing high polyphenol levels in the berry skin and seed showed that 1873 genes were absent in the reference genome.185 UY11 expressed 141 novel unique genes encoding 19 different enzymes associated with polyphenol biosynthesis, including cultivar-specific genes regulating polyphenol accumulation. The pangenome for tomato, constructed using 725 phylogenetically and geographically distinct accessions, identified 4873 genes absent from the reference genome.186 Moreover, TomLoxC (Solyc01g006540) was revealed to be involved in the production of apocarotenoid, which contributes to the desirable tomato flavor. Pangenome for Brachypodium dystachyon showed that the core genome was rich in genes associated with essential processes such as primary metabolite synthesis, while the dispensable genome was rich in genes associated with disease resistance and abiotic stress response.187 Most importantly, the dispensable genome showed higher synonym substitution compared to the core genome, suggesting ongoing active evolution within these natural variants through these gene sets, including genes associated with metabolism. Pangenomes for Arabidopsis and some of the key crops, including maize,188 soybean,189 rice,190,191 medicago,192 tomato,186,193Brassica napus,194 sunflower,195 wheat,196 and Brassica oleracea,197 have been constructed in the past few years and are a valuable resource for identifying various genes and their functions, which confer a characteristic phenotype to a given cultivar. The analysis of natural variants can help understand the role of genetic diversity to derive the evolution of specialized metabolites' biosynthesis through means such as SNPs, small insertions/deletions, structure, gene presence/absence, gene copy number, and other miscellaneous genomic features. Pangenome, GWAS, and QTL analysis combined with metabolomics in natural variants provide a means to link unknown metabolites to genes of known function, which can be used to predict and annotate additional metabolites and genes.
4.4. Comparative genomics and phylogenetic approaches to elucidate the metabolites' diversity and role in speciation
Comparative genomics and phylogenomics approaches allow for tracing back and speculate regarding events that drove the evolution of specialized metabolites, including the identification of key genes and metabolites.179 Evolutionary forces, including (1) localized gene duplication, sub-genome duplication, or whole-genome duplication, followed by sub- or neo-functionalization of specific enzymes; (2) allelic variation; (3) gene loss; and (4) catalytic promiscuity, work cohesively under the influence of positive natural selection to bring large structural diversity in specialized metabolism.168,198 A generalized scenario for the evolution of specialized metabolites involves the emergence of new enzymes through local, sub-genome, or whole-genome duplications, thus providing gene pools to evolve new functions responsible for chemodiversity.199,200 In the dynamic evolutionary process, enzyme catalytic promiscuity allows the divergence of the metabolic stream toward enhanced chemodiversity. Gene duplication with enzyme promiscuity, followed by changes in the substrate specificity, has been identified as the mechanism underlying the evolution of the glucosinolate biosynthetic pathway.201 The catalytic promiscuity of the enzymes, such as acyltransferases, a diverse enzyme family catalyzing O-acylation and N-acylation of structurally diverse acceptor substrates including alkaloids, phenylpropanoids, terpenoids, and acylsugars, is one of the key driving forces of metabolite diversity.202 By expressing a bifunctional lysine/ornithine decarboxylase enzyme, L/ODC, in Arabidopsis, Shimizu et al. showed the emergence of non-native specialized metabolites, including alkaloid-like metabolites.203 The authors used the core chemical structure of cadaverine to identify the metabolic intermediates and enzymes involved in artificially established chemodiversity in Arabidopsis; they demonstrated the role of promiscuous enzymes in deriving the metabolite diversity and described the emergence of metabolite scaffolds as the key event.Analyzing the genomes of multiple plant species has provided evidence of a rather surprising prevalent convergent evolution for different metabolite classes across the plant kingdom.204 Comparative genomics using genomes of Nicotiana attenuata and Nicotiana obtusifolia showed the association of genome evolution with the establishment of nicotine biosynthetic pathways.205 Caffeine and other purine alkaloid biosynthetic pathways in plants have evolved from several unrelated gene families.206 The biosynthesis of MIAs, one of the most diverse and economically valuable metabolite classes, has evolved convergently.207 Using comparative genomics and phylogenomics, Rai et al. showed the importance of strictosidine biogenesis in the evolution of camptothecin and other MIA biosynthetic pathways.207 They compared the genomes of MIA-producing plants and showed that enzymes associated with MIA biosynthesis emerged after the evolution of functional strictosidine synthase (STR). STR loss was associated with the loss of the ability to evolve the cellular components essential for MIA biosynthesis. They also identified the parallel evolution of CPT biosynthesis in distant plant species. Convergent evolution through substrate promiscuity has also been reported for Lys-derived alkaloids, sesterterpenoids, glucosinolates, benzylisoquinoline alkaloids, and tropane alkaloids.204
A general analytical pipeline to discover candidate genes involved in specialized metabolism begins with synteny analysis (intra- and inter-species) to identify gene sets that are conserved across the plant species, produce similar classes of metabolites, and have undergone duplication. Such gene sets can be further analyzed for gene family classification, followed by synonymous substitution analysis to identify genes with recent modifications or specialization. Phylogenetic analysis and hypothesis testing using various (HyPhy) tools208 with prior knowledge of the type of specialized metabolite produced in the target plant species allow the identification of positively selected candidate genes. Comparative genomics and phylogenomics with homology-based annotation predict the potential enzymatic activity and spatial expression patterns in tissues accumulating specialized metabolites as the criteria can further assist in narrowing down the candidate genes for functional characterization.207 Phylogeny-based enzyme classification allows predicting potential functions of candidate genes, and in extension, metabolite intermediates of the associated enzymatic reactions. Phylogenomics-based plant metabolite structure prediction is not new but the analytical scale is limited to a few candidate structures and requires time and resources. In a recent study, Defossez et al. constructed a framework to predict the landscape-scale phytochemical diversity of known and unclassified molecules using an untargeted metabolomics approach on 416 grassland vascular plant species with phylogenetic information, species distribution modeling, and ensemble machine learning.209 The authors showed that the functional phytochemical diversity and identity could be predicted from phylogenetic branching and ecological characteristics, offering an approach to discover bioactive molecules outside the well-established biodiversity hotspot. The association of phylogeny with phytochemical diversity suggests the advantage of combining genomics with metabolomics to identify the genes and unknown metabolite intermediates associated with specialized metabolism.
4.5. Gene cluster analysis to link the genome architect with the metabolome
Until recently, plant metabolic gene clusters were regarded as unreal, given the complexity of the genome structure and the compartmentalized biosynthesis of specialized metabolites. However, recent studies have identified the physical proximity of genes associated with specialized metabolism, highlighting the possibility of loose or partial gene clusters in plant genomes.210 For well-characterized biosynthetic pathways of specialized metabolites, such as anthocyanins, carotenoids, and glucosinolates, genes are not clustered and are rather scattered throughout the genome. Nevertheless, with the increased number of available high-quality genomes, genome mining has shown clear evidence of clustered genes associated with different specialized metabolic pathways. The significant proximity of genes associated with specialized metabolism on the chromosomes of Arabidopsis has been identified.211 Since the first report on metabolic gene clusters associated with benzoxazinoid biosynthesis in maize, over 20 clusters associated with the biosynthesis of diverse classes of specialized metabolites, including diterpenes, triterpenes, polyketides, steroidal alkaloids, monoterpene indole alkaloids, benzylisoquinoline alkaloids, and cyanogenic glycosides, have been identified and validated across different plant species.132,133 The unexpected phenomenon of prevalent specialized metabolite gene clusters across different plant species offers a unique opportunity to discover and characterize genes and metabolites associated with specialized metabolism. One of the most remarkable gene clusters identified was in the Opium (poppy) genome; the noscapine gene cluster included the (S)- to (R)-reticuline (STORR) gene fusion and four genes associated with morphine alkaloid biosynthesis, representing a total of 28 genes localized in the 584 Kb region of chromosome 11.212 The further analysis of this genome revealed that all functionally characterized BIA biosynthetic genes are part of the gene clusters, including several potential functional genes such as PS1126530.1 (cytochrome P450) and PS1126590.1 (methyltransferase) co-expressed with 15 other genes from the BIA biosynthetic pathway.Recently developed toolsets such as PlantClusterFinder,213 PhytoClust,214 and plantiSMASH215 allow users to select the gene segment length, co-expression pattern, similarity with previously identified plant metabolic gene clusters, number of tandem repeats, and type of member enzymes as screening criteria to predict the plant gene clusters. PhytoClust and plantiSMASH offer gene co-expression as one of the criteria to identify the gene clusters. PlantClusterFinder relies on the assigned genes to a given pathway and uses the knowledge of previously identified gene clusters to identify new gene clusters. With relatively fewer functional gene clusters being identified, whether the origin of gene clusters in plants is to provide the advantage of co-expression by shared promoter elements and the local chromatin environment, as in the case of microorganisms, remains debatable. Based on comparative genomics for known gene clusters, coinheritance has been proposed as the central driver of cluster formation.207 Comparisons of the thalianol gene cluster at the species level revealed differences in cluster organization and auxiliary gene involvement, with the interplay between core and unlinked auxiliary genes elucidating a mechanism underlying diversification across plant species.216 The analysis of gene clusters in the O. pumila genome identified 357 potential gene clusters, including 30 gene clusters associated with MIA biosynthesis,207 conserved across plant species. Remarkably, while most gene clusters were conserved and collinear between the O. pumila and coffee genomes, a single gene encoding STR was lost from the functional gene cluster C1541 of the coffee genome. Further comparative genomics and phylogenetic analysis showed that retaining STR was vital for the evolution of MIA biosynthesis—an opportunity that was lost for coffee, resulting in a completely different chemotype of this species. Similarly, gene clusters associated with various other metabolic pathways are also heterogeneous, with genes in the morphine and SG pathways being scattered and genes in thebaine and noscapine pathways being closely clustered.217 The conserved nature of the gene clusters reported thus far in the plant species producing similar specialized metabolites and the associated dynamics within the genomic region suggests gene clusters as metabolic modules for the evolution and maintenance of chemodiversity. The reduced rate of recombination between the genes at proximity explains the reason for conserved gene clusters across species.210 Simultaneously, this could serve as a positive selection force for genes related to local adaptation. During evolution, the tandem duplication of genes within a gene cluster and sub-/neo-functionalization could offer a means to expand the metabodiversity, thus offering a site for the active evolution and expansion of chemodiversity. Conversely, the loss of critical genes or the entire gene cluster would result in a loss of the ability to retain or evolve an entire family of specialized metabolites and, ultimately, the dominance of other metabolite families to expand within the plant species.207
The physical proximity of gene clusters conserved across plant families does offer a case to expect the production of similar metabolite classes, as observed in the case of widespread localization of C-terminal trans-prenyltransferase and N-terminal terpene synthase involved in the biosynthesis of a large sesterterpene repertoire in Brassicaceae.218 While the number of identified gene clusters across plant genomes is growing, there is no clear approach to rationalize, that is, to select genes for functional characterization, restricting one's ability to take full advantage of these discoveries. A gene cluster in which members are strongly coexpressed can be prioritized for functional characterization, and encoded enzymes can be tailored to provide clues into the prediction of new metabolites. In cases of conserved gene clusters across plant species that produce similar classes of specialized metabolites, gene information can be tailored to predict metabolite intermediates, which can be further validated using NMR or MS/MS-based approaches. Combining coexpression analysis, integrative omics, comparative genomics, and phylogenomics for gene sets assigned to a given gene cluster offers an exciting avenue for resolving the structures of some unknown mass features identified through metabolomics. This would provide a means to prioritize the mass features for further characterization and validation.
4.6. Machine learning and genomics to explore cellular components involved in specialized metabolism
The discovery of new metabolites, including their structural features, has always been a challenge. Exploiting the advantages of the increasing number of high-quality genomes and approaches such as machine learning and deep learning has the potential to drive genome-assisted natural product discovery in plants, and future efforts in this direction are warranted. Machine learning approaches rely on large datasets to avoid data overfitting. Machine learning in genomics is not new and has been at the core of gene prediction, protein domain prediction based on sequence information, and promoter motif prediction.132,219,220 Since recent genomics analyses have generated massive data, the application of deep learning-based neural network approaches in biology has offered opportunities of using genomics to predict molecular signatures, including transcription factors, epigenetic markers, chromatin state, histone binding state, and gene expression.219 Almost all these tools have been developed, tested, and applied to human or animal genomics, and the direct transfer for plants is possible. However, specific properties that characterize the plant genome219 must be considered. For instance, modeling the gene expression levels for maize must consider the tetraploid nature of its genome, which would lead to the biased quantification of the gene expression, resulting in the poor quality of the test and training datasets.221 Advances in deep learning have been successfully exploited to develop applications and tools for plant identification, species distribution modeling, weed detection, plant disease, and pest forecasts, and crop yield predication based on images, thus playing significant roles in advancing the field of plant phenology and functional trait biology.222,223 The application of deep and machine learning for predicting miRNAs and their targets (miTAR,224 DeepMirTar,225 miRAW226), tRNA annotation (tRNA-DL227), polyadenylation sites (DeepPASTA228), transcription factor binding sites (DeFine,229 DeepBind,230 DeepSEA231), long non-coding RNAs (IncRNA-LSTM232), genomic methylation cites (DeepCpG233), RNA-binding protein binding sites (pysster234), protein–protein interactions (DPPI,235 AutoCorrelation,236 and SigProd237), essential genes (DeepHE238), and phenotype based on genotype (DeepGS239) underscores the potential future roles of these approaches to extract meaningful knowledge from genomic data.One of the exciting applications of deep learning for image processing is the development of Google DeepVariant.240 DeepVariant views mapped sequenced data as an image and treated variants as image classification to identify structural variants, including SNPs, short-indels, and inversions, and outperformed conventional mapping-based approaches in terms of accurate calling.241 Several tools based on the DeepVariant framework have been developed, including DeepSV,242 for the accurate identification of genomic deletions, and DeepTrio,243 for predicting the parental structural variants, thus allowing diploid genome phasing. DeepVariant combined with other tools, particularly highly accurate long-read sequencing platforms such as PacBio, allows for the further investigation of highly repetitive regions to identify variants with roles in the evolution of desired traits and natural product biosynthetic pathways.244 The machine- or deep learning application for predicting protein functions and tertiary structures has achieved remarkable success. For instance, the deep learning tool AlphaFold allows for the accurate prediction of protein tertiary structure based on an input gene sequence.245,246 In this study, authors established computational method to predict protein structures de novo with atomic accuracy. In another related study, a three-track neural network-based integrated approach was used to transform information at the 1D sequence level, the 2D distance map level, and the 3D coordinate level to achieve relative accuracy of the predicted protein structure as achieved by AlphaFold approach.247 Using this approach, the authors managed to generate accurate protein–protein complex models from sequence information alone, which offers key insights into the protein function of unknown structures. Compared with years of hard work put into accurately predicting protein structures using a combination of X-ray crystallography and cryo-imaging, the computational prediction of the protein structure is much faster with comparable accuracy, highlighting the possibility of predicting the functions of unknown enzymes in the future. High-quality and high-throughput prediction of enzyme commission (EC) numbers using DeepEC248 and DEEPre249 is another important approach in genomics-based metabolomics. Accurate EC number prediction allows to identify enzyme catalytic functions and establish gene–protein interaction relationships. Therefore, these tools are the key for building genome-scale metabolic networks and designing novel metabolic pathways. Recently, Moore et al. described a machine learning approach to evaluate the features associated with genes involved in specialized metabolism.250 Although established and tested for Arabidopsis, the model can be transferred to other plant species for predicting genes involved in natural product biosynthesis.
The extraction of accurate genomic features is valuable for building a genome-scale metabolic model, representing a template for describing the overall ongoing metabolic processes. Modeling approaches, such as metabolism with gene expression (i.e., ME model), biochemical systems approach, and kinetic modeling, are widely used, together with careful manual curation to reconstruct a metabolic model.251,252 Genome-scale metabolic models rely on accurate gene prediction and functional annotation; therefore, advances in accurate gene prediction and annotation are valuable for expanding the scope of metabolic networks in plant natural product discovery. Genome-scale metabolic models have been successfully created for several plant species, including Arabidopsis, maize, oilseed rape, rice, soybean, and crassulacean acid metabolism (CAM) plants, as well as for Chlamydomonas.132,251,253–255 Annotation and manual curation of metabolic network resources such as plant metabolic network (PMN),213,256 which includes the reference database PlantCyc (https://plantcyc.org/) and 126 species/taxon-specific databases, are valuable for providing an overview of possible metabolic processes across different plant species based on a standard metabolic framework adopted from MetaCyc. For instance, PlantCyc provides access to manually curated and/or computationally predicted information on enzymes, biochemical reactions, and processes shared among or unique to over 500 plant species. Seaver et al. established an algorithm to streamline automated plant genome annotation and used a curated template of metabolite compartmentalization for over 100 metabolic subsystems to reconstruct metabolic models for 39 plant species.255,257 Using this PlantSEED network, authors reconstructed plant primary metabolic model with improved compartmentalization and comparative consistency. Although the presence of a gene is not sufficient to predict whether it is functional, the annotation-based genome-scale model offers a framework to further refine metabolic models using mass balance as constraints for variables such as conditional or time-based gene expression and metabolic flux analysis, among others, resulting in reconstructed metabolic models for the accurate prediction of the metabolic state under a given condition.251,258 Advances in predicting gene expression using gene sequences, phylogenic considerations, and chromatin state using machine learning approaches could be used to generate datasets that may serve as constraints to further refine the metabolic models. The scope of machine learning for gene feature annotation and multi-omics analysis, together with constraint-based metabolic modeling, is vast and could drive future discoveries of new metabolites.132,258,259
The annotation and prediction of gene function and its associated features are imperative, and much effort has been put into this area. Adopting profile-hidden Markov models (pHMMs) with homology-based gene annotation enables reliable representation based on conserved functional subunits of proteins and accurate gene functional annotation.219 The second and third critical assessment of functional annotation (CAFA), a timed challenge to assess the computational methods for automatically assigning protein functions, has shown progress in accurately predicting molecular functionals and biological annotations, which is encouraging for future applications of genomics to detect new metabolites.260 As the number of sequenced plant genomes is increasing and the sequencing of complex genomes is becoming more accessible, comparative genomics and phylogenetics-based identification of phytochemodiversity hotspots is feasible.209 Integrative omics has been the core of plant functional genomics efforts to date, and by incorporating comparative genomics, a multicriteria-based approach can be used to prioritize genes and unknown metabolites for in-depth characterization.261,262 Applying deep learning-based models to predict system responses using multi-omics datasets has seen tremendous progress; nevertheless, it is difficult to interpret these models. More interpretable models can be built using alternates such as SHAP263 and DeepLIFT,264 which assign importance or contribution values to the final model outcome. Future advances in accurate constraint-based genome-scale metabolic models for specialized metabolism and their integration with deep learning models would offer more avenues to discover and predict unknown metabolites.
5. Prospects of metabolomics
So far, we have discussed in detail the widely used LC-MS/MS and genomics approach based on cutting-edge techniques in metabolomics. In the following sections, several advanced and emerging technologies for metabolite annotation using other fragmentation techniques, ion mobility, and mass spectrometry imaging are highlighted. Moreover, we discuss the perspective of metabolomics and elaborate on how this area can be expanded with advances such as “virtual metabolomics” and how the AI research meets with mass spectrometry data in metabolomics to accelerate the discovery of unknown metabolites and the increase in annotation confidence.5.1. Structure elucidation using other mass fragmentation technologies
Low-energy CID (<100 eV)-based mass fragmentation is the gold standard of LC-MS/MS-based metabolomics to elucidate the structure of metabolites. However, the MS/MS spectra do not contain all the structural information since some of the weakest chemical bonds in a molecular ion are preferentially cleaved by the accumulation of the collision energy (i.e., excitation of molecular vibrations). Therefore, alternative approaches are required to complement CID-centric MS/MS spectral information and further elucidate unknown metabolite structures. Recently, several conventional and practical methodologies to determine double bond positions, cis (Z)/trans (E) isomers, and acyl chain positional isomers have become popular in lipidomics because of their biological relevance. In addition to classic ozone-induced dissociation (OzID),265 which requires prolonged reaction time for fragmentation, the Paterno–Buchi (PB) reaction is used to determine the position of the double bonds in free fatty acids and complex lipids, such as glycero(phospho)lipids.266,267 Following the lipid double bond reaction with acetone or 2-acetylpyridine under UV irradiation, the produced oxetane (four-membered ring ether) moiety is preferentially fragmented by low-energy CID. Combined with 2-acetylpyridine and MSn analysis, the PB reaction can be used to determine the double bond positions and sn-positional isomers.268 mCPBA epoxidation for lipid double-bond identification (MELDI)269 can be used together with LC-MS/MS and DESI-MSI. In addition, radical-induced dissociation techniques, such as oxygen attachment dissociation (OAD),270 electron impact excitation of ions from organics (EIEIO),271 and ultraviolet photodissociation (UVPD),272 are used for the same purposes but require no derivatization/reaction. In OAD, gas-phase hydroxyl radicals are introduced into the collision cell, and this radical binds to the double bond moiety, leading to odd electron-centric double bond-specific cleavage as charge remote fragmentation. EIEIO combined with EID using electron energy of 0–20 eV has been proposed, which can generate sequential fragment ions, as acquired in EI-MS. UVPD is used together with MSn fragmentation, in which one of the fragment ions from low-energy CID is isolated and irradiated. All the above techniques can be used in combination with ion mobility separation and IMS, although the required computational support is not as adequate at the moment. Although these approaches are currently applied only in lipidomics and proteomics, they may be useful in natural product chemistry to elucidate complex molecular structures and accelerate the discovery of new molecules.5.2. Spatial metabolomics with ion mobility (IM) spectrometry and toward spatial multi-omics
IM spectrometry has become a popular technique in metabolomics and lipidomics to increase (1) the peak capacity in MS data, (2) the purity of the MS/MS spectra, and (3) the confidence in metabolite annotation. IM spectrometry coupled with tandem MS, such as parallel accumulation-serial fragmentation (PASEF),273 is also an attractive approach to increase the reliability of metabolite annotations. Microbial lipidome has been illustrated using LC-IM-MS/MS.57 IM can be maximized with mass spectrometry imaging (MSI) since IM complements MSI in terms of isobaric separation and annotation confidence. Therefore, spatial metabolomics with MALDI-IM-MS274 is expected to become popular in the coming future. Although the accuracy criteria for metabolite annotation in MSI have been reported without CCS information,275 false discovery rate (FDR)-controlled spatial metabolomics analysis can be performed in combination with comprehensive CCS databases. In the future, data-independent MS/MS acquisition techniques may be coupled to MALDI-IM-MS to further accelerate spatial omics; therefore, CompMS tools such as METASPACE (https://metaspace2020.eu/) supporting a common data format (imzML)276 should be developed to accelerate spatial metabolomics research.In the coming decade, spatial multi-omics is expected to become a challenging research area of molecular biology. Various biotechniques for spatial transcriptomics have been actively developed. For instance, using Visium,277,278 Slide-seqV2,279 or photo-isolation chemistry (PIC),280 RNA expression can be determined at a spatial resolution of 10–200 μm. In addition, spatial proteomics is executable using mass cytometry281 and DNA-tagged antibody sequencing,282 among other techniques.283 In addition, even in spatial metabolomics, the existence of “purinosome metabolon”, which is a molecule reactor machine consisting of multiple enzymes for purine biosynthesis in a single cell, has been discovered at a spatial resolution of <1 μm using gas cluster ion beam secondary ion MS (GCIB-SIMS).284 Advances in the relevant informatics field can further support these biotechniques. Importantly, the information on metabolite, protein, and RNA localization facilitates the study of molecular mechanisms and elucidation of plant metabolic pathways.
5.3. Plant kingdom-wide annotation
The development of MS technologies has enabled us to obtain large-scale metabolomics data with extraordinary sensitivity and accuracy at high resolution and in great detail. A certain number of metabolites in Arabidopsis thaliana are biosynthesized via roughly 27416 genes (https://www.arabidopsis.org/index.jsp) in its genome. These metabolites show organ, tissue, and cell specificity. This spatial metabolite diversity suggests the diverse roles of metabolites in different tissues. Artificial intelligence-based approaches are expected to allow rapid and accurate chemical categorization of unknown metabolites. Biosynthetic genes of the assigned metabolites can be narrowed down by a combination of other omics approaches with metabolomics, and their functions can be revealed using genome-editing approaches, such as CRISPR-Cas9.285 The comparative analyses of these transgenic plants lacking metabolites, as a result of genome editing, can reveal the physiological roles of these metabolites.
The next step is to expand these analyses beyond a single species. Other omics studies have shown the direction, in which metabolomics should head, i.e., plant kingdom-wide annotation. There are approximately 391000 vascular plants on the Earth,1 and they likely produce as many metabolites as there are stars in our galaxy, earning the moniker “metabolite cosmos”. To accomplish this plant kingdom-wide annotation, the performance of metabolomics must be improved in terms of chemical annotation. A key to this improvement is to add additional dimensions to the MS/MS data, such as the data acquired using SIL, isotopic fine structure analysis, and IM spectrometry. Another key is to focus on highly segmented structures (e.g., functional moieties or partial structures). The strategies for N- and S-metabolites indicate that such approaches of profiling do not rely on structures but simply on the elements. Further segmentation can promote the identification of unknown metabolites with double or triple bonds; ketones; hydroxyl, amino, and carboxylic moieties; and halogens. Focusing on segmented moieties can also promote the elucidation of the complete structure based on the MS/MS spectra. However, approaches to segment the reconstruction are inadequate. Using accumulated MS/MS spectra, fragmentation patterns can be observed in certain structures. The construction of a partial structure, which can be searched in databases, will prove to be a breakthrough in finding novel metabolites.286,287
5.4. Virtual platform-based annotation
Two foremost problems in metabolomics are (1) the difficulty of chemical annotation and (2) the cost of instruments and software. While the former problem has been gradually solved, as described above, the latter remains unsolved in academia. To solve these problems, database or platform-based projects are ongoing. These efforts have achieved outstanding results, advancing the field of metabolomics. However, researchers still must perform chemical annotation manually. A solution is to virtually share the whole metabolome data with complete chemical annotation. Although there is a risk to the providers, it is worth actively conducting metabolomics. The providers must stipulate evidence and reference to detect incorrect annotations. Once the data are obtained from a database, researchers can perform “virtual metabolomics” using free academic software programs, such as MS-DIAL, MS-FINDER, GNPS, or currently available analytical tools. All data, free software/programs, and annotation results render access to metabolomics easier when new metabolites are explored. To reduce data, most data can be centered at particular organizations that have the required infrastructure or can reliably perform chemical annotation based on data procured from companies. An NCBI-like resource for metabolome is highly desired to promote new discoveries. Nonetheless, owing to its sheer vastness, we cannot expect to deal with phytochemodiversity even with sophisticated infrastructures, technologies, and approaches.5.5. Artificial intelligence for mass spectrometry data (AIMS) research
Currently, 1.5 million spectral records of over 40000 metabolites are available as the CID-MS/MS training dataset; the statistics are based on the databases explained in the section “3.2.2. Mass spectral databases for annotation”. Moreover, 0.7 million records of over 570000 compounds are available as the EI-MS training dataset; the statistics are based on the spectral records of MassBank,32 MoNA, NIST20, and Wiley10. Given that the machine learning of face recognition has been carried out by 0.1 M–300 M face images of 1 K to 4 M identities,288 where K and M denote kilo and million, respectively, we believe that the size of the mass spectral records is not small for machine learning research. For example, a simple question in AIMS research is to ask whether the highly accurate prediction for the existence of hydroxy moiety (–OH) is possible or not by the MS properties. Because water loss is observed in >50% molecules containing the hydroxyl moiety (see Section 3.2.3.), the accuracy would be increased when multiple properties are considered. Likewise, the prediction model for each of the 881 PubChem fingerprints and others can be constructed. Moreover, the predicted fingerprints can be used to search the structures for unknown spectra, and the idea is firstly implemented in CSI:FingerID.289 Although the fragmentation tree (FT)290 and kernel support vector machine (KSVM) have been used as the model parameters and machine learner, respectively, in CSI:FingerID, further AIMS research would be needed to facilitate annotation in metabolomics.
In lipidomics, the in silico tandem mass spectral libraries or rule-based annotations are used for the annotation pipeline,57 in which the information of the product ion abundances are not considered, although the ion abundances can be used to predict the sn1/sn2 positional isomers.291 Because the m/z values (qualitative information) can be theoretically generated in silico, the next step in lipidomics annotation is to consider and predict the intensities: deep learning studies have been performed in shotgun proteomics.292 In this light, the AIMS research for natural products is not enough yet because even the m/z values are unpredictable although the issue has been tackled by the developers of CFM-ID.121 The important note in this area is that the number of informatics researchers in metabolomics is very small compared to that in the genomics research field. The data for AIMS researches are available, as mentioned above. Moreover, over 50 TB of raw MS data are available at Metabolomics Workbench293 and MetaboLights294 as the test dataset. Further stimulation of this AIMS research field will undoubtedly contribute to an increase in the accuracy and precision for predicting the molecular formula, metabolite class, substructure, molecular backbone, and even the stereochemistry of structures.
6. Conclusion
Over two decades have passed since Oliver et al. first used the term “metabolome” in their article.295 Advances in metabolomics have changed plant biochemical approaches from empirical (bottom-up) to computational (top-down) methods. At present, various protocols for metabolome analysis have been proposed based on empirical experiences, and reliable computational approaches for sample preparation and MS analysis have been well established. In omics research (metabolomics, lipidomics, glycomics, proteomics, transcriptomics, and genomics), where massive data are generated every day, reproducibility, reusability, and transparency have become fundamental. Recently, many journals have encouraged to submit raw MS data (primary data) in respective repositories293,294 and metabolite profile data (secondary data) generated from these raw data, which is a welcome step to accelerate open science.We believe that in the coming era of computational (metabolomics) science, it will be pivotal to extract metabolites and their biological information from large-scale data with high accuracy to facilitate the reuse of metabolomics datasets and efficiently elucidate the molecular mechanisms driving the observed phenotypes. Metabolomics and the complementary techniques described in Fig. 1 require computational sciences and/or novel approaches to improve the annotation rate and to deepen the understanding of metabolisms. In addition to the role of storage of mass spectral records, the supports of fragment ion curation, spectra search engine, and knowledge conversion from chemical to biology (metabolite) will maximize the value of level 1 identification of metabolites. Moreover, processing tools, databases, and repositories for mass spectrometry imaging (MSI) should be further developed since the MSI spatial data of newly identified metabolites offer information on metabolite localization, thereby enhancing the discovery of the associated genes and proteins that are active in the same environment. Moreover, functional genomics, single cell metabolomics and transcriptomics, and spatial multi-omics techniques such as MALDI/DESI-MS for spatial metabolomics, and Visium,277 Slide-seqV2,279 or PIC280 for spatial transcriptomics should be integrated efficiently with the hypothesis generated by metabolomics to mine the candidate genes that regulate novel metabolites and/or elucidate the association of the metabolites with the plant phenotype.
Moreover, increasing the annotation rate by computational sciences in combination with ion mobility, spectral library, stable isotope labeling, and ultra-high-resolution MS will enhance the discovery in metabolite-based genome-wide association study (mGWAS)296,297 and illuminate the diversity of metabolites in the plant kingdom. Although the advances in genomics enable one to discover the gene clusters and to decode the plant revolution from the viewpoint of metabolisms, the information of metabolomics is essential not only to validate the gene functions but also to offer new opportunities for genome mining that meets with novel metabolites. In this context, computer-assisted smart metabolite annotation of data is warranted to advance the research in natural product chemistry, elucidate the true diversity of plant specialized metabolites, and identify novel drug targets. Though we have introduced a number of available spectral records and MS raw data, these numbers are still growing. Importantly, computational technologies are advancing at an astounding pace. Given the indispensability of metabolomics in biology, we hope that the present review will be helpful not only to the researchers active in this field but also to students and/or beginners undertaking metabolomics research.
7. Author contributions
K. S. initiated the project, and H. T. and R. N. coordinated the writing of this review. H. T., A. R., and R. N. wrote the parts of computational mass spectrometry, functional genomics, and cutting-edge metabolomics technique, respectively. All authors thoroughly discussed this project and helped to improve the manuscript.8. Conflicts of interest
There are no conflicts to declare.9. Acknowledgements
We would like to thank Editage (https://www.editage.com) and Dr Megha Rai (Chiba University, Japan) for English language editing.10. Notes and references
- M. Vellend, L. Baeten, A. Becker-Scarpitta, V. Boucher-Lalonde, J. L. McCune, J. Messier, I. H. Myers-Smith and D. F. Sax, Annu. Rev. Plant Biol., 2017, 68, 563–586 CrossRef CAS PubMed.
- S. Bernardini, A. Tiezzi, V. Laghezza Masci and E. Ovidi, Nat. Prod. Res., 2018, 32, 1926–1950 CrossRef CAS PubMed.
- X. M. Wu and R. X. Tan, Nat. Prod. Rep., 2019, 36, 788–809 RSC.
- F. M. Afendi, T. Okada, M. Yamazaki, A. Hirai-Morita, Y. Nakamura, K. Nakamura, S. Ikeda, H. Takahashi, M. Altaf-Ul-Amin, L. K. Darusman, K. Saito and S. Kanaya, Plant Cell Physiol., 2012, 53, e1 CrossRef CAS PubMed.
- M. R. Wilson, L. Zha and E. P. Balskus, J. Biol. Chem., 2017, 292, 8546–8552 CrossRef CAS PubMed.
- B. Mayo, L. Vazquez and A. B. Florez, Nutrients, 2019, 11, 2231 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- Market reports by MarketsandMarkets, Plant Extracts Market – Forecast to 2026, https://www.marketsandmarkets.com/Market-Reports/plant-extracts-market-942.html Search PubMed.
- T. Isah, Biol. Res., 2019, 52, 39 CrossRef PubMed.
- M. Erb and D. J. Kliebenstein, Plant Physiol., 2020, 184, 39–52 CrossRef CAS PubMed.
- M. M. Rinschen, J. Ivanisevic, M. Giera and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2019, 20, 353–367 CrossRef CAS PubMed.
- C. H. Johnson, J. Ivanisevic and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2016, 17, 451–459 CrossRef CAS PubMed.
- P. Trivedi, J. E. Leach, S. G. Tringe, T. Sa and B. K. Singh, Nat. Rev. Microbiol., 2020, 18, 607–621 CrossRef CAS PubMed.
- I. N. Jamil, J. Remali, K. A. Azizan, N. A. Nor Muhammad, M. Arita, H. H. Goh and W. M. Aizat, Front. Plant Sci., 2020, 11, 944 CrossRef PubMed.
- K. Yugi, H. Kubota, A. Hatano and S. Kuroda, Trends Biotechnol., 2016, 34, 276–290 CrossRef CAS PubMed.
- Y. Wang, S. Y. Liu, Y. J. Hu, P. Li and J. B. Wan, RSC Adv., 2015, 5, 78728–78737 RSC.
- A. P. Bowman, G. T. Blakney, C. L. Hendrickson, S. R. Ellis, R. M. A. Heeren and D. F. Smith, Anal. Chem., 2020, 92, 3133–3142 CrossRef CAS PubMed.
- T. Kind, H. Tsugawa, T. Cajka, Y. Ma, Z. J. Lai, S. S. Mehta, G. Wohlgemuth, D. K. Barupal, M. R. Showalter, M. Arita and O. Fiehn, Mass Spectrom. Rev., 2018, 37, 513–532 CrossRef CAS PubMed.
- J. C. Xue, C. Guijas, H. P. Benton, B. Warth and G. Siuzdak, Nat. Methods, 2020, 17, 953–954 CrossRef CAS PubMed.
- Q. F. Zhu, T. Y. Zhang, L. L. Qin, X. M. Li, S. J. Zheng and Y. Q. Feng, Anal. Chem., 2019, 91, 6057–6063 CrossRef CAS PubMed.
- S. J. Zheng, S. J. Liu, Q. F. Zhu, N. Guo, Y. L. Wang, B. F. Yuan and Y. Q. Feng, Anal. Chem., 2018, 90, 8412–8420 CrossRef CAS PubMed.
- Z. Zhou, M. Luo, X. Chen, Y. Yin, X. Xiong, R. Wang and Z. J. Zhu, Nat. Commun., 2020, 11, 4334 CrossRef PubMed.
- K. Giles, J. Ujma, J. Wildgoose, S. Pringle, K. Richardson, D. Langridge and M. Green, Anal. Chem., 2019, 91, 8564–8573 CrossRef CAS PubMed.
- A. C. Schrimpe-Rutledge, S. D. Sherrod and J. A. McLean, Curr. Opin. Chem. Biol., 2018, 42, 160–166 CrossRef CAS PubMed.
- J. L. Wolfender, J. M. Nuzillard, J. J. J. van der Hooft, J. H. Renault and S. Bertrand, Anal. Chem., 2019, 91, 704–742 CrossRef CAS PubMed.
- H. Tsugawa, Curr. Opin. Biotechnol., 2018, 54, 10–17 CrossRef CAS PubMed.
- K. Uppal, D. I. Walker, K. Liu, S. Z. Li, Y. M. Go and D. P. Jones, Chem. Res. Toxicol., 2016, 29, 1956–1975 Search PubMed.
- J. J. J. van Der Hooft, H. Mohimani, A. Bauermeister, P. C. Dorrestein, K. R. Duncan and M. H. Medema, Chem. Soc. Rev., 2020, 49, 3297–3314 RSC.
- L. W. Sumner, A. Amberg, D. Barrett, M. H. Beale, R. Beger, C. A. Daykin, T. W. Fan, O. Fiehn, R. Goodacre, J. L. Griffin, T. Hankemeier, N. Hardy, J. Harnly, R. Higashi, J. Kopka, A. N. Lane, J. C. Lindon, P. Marriott, A. W. Nicholls, M. D. Reily, J. J. Thaden and M. R. Viant, Metabolomics, 2007, 3, 211–221 CrossRef CAS PubMed.
- E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff, H. P. Singer and J. Hollender, Environ. Sci. Technol., 2014, 48, 2097–2098 CrossRef CAS PubMed.
- R. A. Spicer, R. Salek and C. Steinbeck, Sci. Data, 2017, 4, 170138 CrossRef PubMed.
- H. Horai, M. Arita, S. Kanaya, Y. Nihei, T. Ikeda, K. Suwa, Y. Ojima, K. Tanaka, S. Tanaka, K. Aoshima, Y. Oda, Y. Kakazu, M. Kusano, T. Tohge, F. Matsuda, Y. Sawada, M. Y. Hirai, H. Nakanishi, K. Ikeda, N. Akimoto, T. Maoka, H. Takahashi, T. Ara, N. Sakurai, H. Suzuki, D. Shibata, S. Neumann, T. Iida, K. Tanaka, K. Funatsu, F. Matsuura, T. Soga, R. Taguchi, K. Saito and T. Nishioka, J. Mass Spectrom., 2010, 45, 703–714 CrossRef CAS PubMed.
- M. X. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W. T. Liu, M. Criisemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderon, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C. C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C. C. Liaw, Y. L. Yang, H. U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Q. Dai, R. Neupane, J. Gurr, A. M. C. Rodriguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P. M. Allard, P. Phapale, L. F. Nothias, T. Alexandrovr, M. Litaudon, J. L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D. T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Muller, K. M. Waters, W. Y. Shi, X. T. Liu, L. X. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutierrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- F. Qiu, D. D. Fine, D. J. Wherritt, Z. Lei and L. W. Sumner, Anal. Chem., 2016, 88, 11373–11383 CrossRef CAS PubMed.
- K. Morreel, Y. Saeys, O. Dima, F. Lu, Y. Van de Peer, R. Vanholme, J. Ralph, B. Vanholme and W. Boerjan, Plant Cell, 2014, 26, 929–945 CrossRef CAS PubMed.
- J. J. van der Hooft, J. Wandy, M. P. Barrett, K. E. Burgess and S. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13738–13743 CrossRef PubMed.
- H. Mohimani, A. Gurevich, A. Shlemov, A. Mikheenko, A. Korobeynikov, L. Cao, E. Shcherbin, L. F. Nothias, P. C. Dorrestein and P. A. Pevzner, Nat. Commun., 2018, 9, 4035 CrossRef PubMed.
- T. Ngo, A. V. Ilatovskiy, A. G. Stewart, J. L. J. Coleman, F. M. McRobb, R. P. Riek, R. M. Graham, R. Abagyan, I. Kufareva and N. J. Smith, Nat. Chem. Biol., 2021, 17, 501 CrossRef CAS PubMed.
- R. Nakabayashi, M. Kusano, M. Kobayashi, T. Tohge, K. Yonekura-Sakakibara, N. Kogure, M. Yamazaki, M. Kitajima, K. Saito and H. Takayama, Phytochemistry, 2009, 70, 1017–1029 CrossRef CAS PubMed.
- Z. G. Yang, R. Nakabayashi, Y. Okazaki, T. Mori, S. Takamatsu, S. Kitanaka, J. Kikuchi and K. Saito, Metabolomics, 2014, 10, 543–555 CrossRef CAS PubMed.
- T. Tohge, R. Wendenburg, H. Ishihara, R. Nakabayashi, M. Watanabe, R. Sulpice, R. Hoefgen, H. Takayama, K. Saito, M. Stitt and A. R. Fernie, Nat. Commun., 2016, 7, 12399 CrossRef CAS PubMed.
- R. M. Boiteau, D. W. Hoyt, C. D. Nicora, H. A. Kinmonth-Schultz, J. K. Ward and K. Bingol, Metabolites, 2018, 8, 8 CrossRef PubMed.
- J. J. van der Hooft, M. Akermi, F. Y. Unlu, V. Mihaleva, V. G. Roldan, R. J. Bino, R. C. de Vos and J. Vervoort, J. Agric. Food Chem., 2012, 60, 8841–8850 CrossRef CAS PubMed.
- A. Bhatia, S. J. Sarma, Z. Lei and L. W. Sumner, Methods Mol. Biol., 2019, 2037, 113–133 CrossRef CAS PubMed.
- K. Yonekura-Sakakibara, T. Tohge, F. Matsuda, R. Nakabayashi, H. Takayama, R. Niida, A. Watanabe-Takahashi, E. Inoue and K. Saito, Plant Cell, 2008, 20, 2160–2176 CrossRef CAS PubMed.
- K. Scheubert, F. Hufsky and S. Bocker, J. Cheminf., 2013, 5, 12 CAS.
- R. R. da Silva, P. C. Dorrestein and R. A. Quinn, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12549–12550 CrossRef CAS PubMed.
- P. M. Seitzer and B. C. Searle, J. Proteome Res., 2019, 18, 791–796 CrossRef CAS PubMed.
- A. T. Aron, E. C. Gentry, K. L. McPhail, L. F. Nothias, M. Nothias-Esposito, A. Bouslimani, D. Petras, J. M. Gauglitz, N. Sikora, F. Vargas, J. J. J. van Der Hooft, M. Ernst, K. Bin Kang, C. M. Aceves, A. M. Caraballo-Rodriguez, I. Koester, K. C. Weldon, S. Bertrand, C. Roullier, K. Y. Sun, R. M. Tehan, C. A. Boya, M. H. Christian, M. Gutierrez, A. M. Ulloa, J. A. T. Mora, R. Mojica-Flores, J. Lakey-Beitia, V. Vasquez-Chaves, Y. Zhang, A. I. Calderon, N. Tayler, R. A. Keyzers, F. Tugizimana, N. Ndlovu, A. A. Aksenov, A. K. Jarmusch, R. Schmid, A. W. Truman, N. Bandeira, M. X. Wang and P. C. Dorrestein, Nat. Protoc., 2020, 15, 1954–1991 CrossRef CAS PubMed.
- J. Xia and D. S. Wishart, Nucleic Acids Res., 2010, 38, W71–W77 CrossRef CAS PubMed.
- M. R. Molenaar, A. Jeucken, T. A. Wassenaar, C. H. A. van de Lest, J. F. Brouwers and J. B. Helms, Gigascience, 2019, 8, giz061 CrossRef PubMed.
- K. McLuskey, J. Wandy, I. Vincent, J. J. J. van der Hooft, S. Rogers, K. Burgess and R. Daly, Metabolites, 2021, 11, 103 CrossRef CAS PubMed.
- R. Nakabayashi and K. Saito, Curr. Opin. Plant Biol., 2020, 55, 84–92 CrossRef CAS PubMed.
- R. Nakabayashi, T. Mori, N. Takeda, K. Toyooka, H. Sudo, H. Tsugawa and K. Saito, Anal. Chem., 2020, 92, 5670–5675 CrossRef CAS PubMed.
- H. Tsugawa, T. Cajka, T. Kind, Y. Ma, B. Higgins, K. Ikeda, M. Kanazawa, J. VanderGheynst, O. Fiehn and M. Arita, Nat. Methods, 2015, 12, 523–526 CrossRef CAS PubMed.
- H. Tsugawa, R. Nakabayashi, T. Mori, Y. Yamada, M. Takahashi, A. Rai, R. Sugiyama, H. Yamamoto, T. Nakaya, M. Yamazaki, R. Kooke, J. A. Bac-Molenaar, N. Oztolan-Erol, J. J. B. Keurentjes, M. Arita and K. Saito, Nat. Methods, 2019, 16, 295–298 CrossRef CAS PubMed.
- H. Tsugawa, K. Ikeda, M. Takahashi, A. Satoh, Y. Mori, H. Uchino, N. Okahashi, Y. Yamada, I. Tada, P. Bonini, Y. Higashi, Y. Okazaki, Z. W. Zhou, Z. J. Zhu, J. Koelmel, T. Cajka, O. Fiehn, K. Saito, M. Arita and M. Arita, Nat. Biotechnol., 2020, 38, 1159–1163 CrossRef CAS PubMed.
- X. Huang, Y. J. Chen, K. Cho, I. Nikolskiy, P. A. Crawford and G. J. Patti, Anal. Chem., 2014, 86, 1632–1639 CrossRef CAS PubMed.
- C. Bueschl, B. Kluger, N. K. N. Neumann, M. Doppler, V. Maschietto, G. G. Thallinger, J. Meng-Reiterer, R. Krska and R. Schuhmacher, Anal. Chem., 2017, 89, 9518–9526 CrossRef CAS PubMed.
- R. Nakabayashi, Y. Sawada, Y. Yamada, M. Suzuki, M. Y. Hirai, T. Sakurai and K. Saito, Anal. Chem., 2013, 85, 1310–1315 CrossRef CAS PubMed.
- C. J. Thompson, M. Witt, S. Forcisi, F. Moritz, N. Kessler, F. H. Laukien and P. Schmitt-Kopplin, J. Am. Soc. Mass Spectrom., 2020, 31, 2025–2034 CrossRef CAS PubMed.
- R. Nakabayashi, K. Hashimoto, K. Toyooka and K. Saito, Anal. Chem., 2017, 89, 2698–2703 CrossRef CAS PubMed.
- R. Nakabayashi and K. Saito, Curr. Opin. Biotechnol., 2017, 43, 8–16 CrossRef CAS PubMed.
- R. Nakabayashi, Z. G. Yang, T. Nishizawa, T. Mori and K. Saito, J. Nat. Prod., 2015, 78, 1179–1183 CrossRef CAS PubMed.
- P. M. Kumara, R. U. Shaanker and T. Pradeep, Phytochemistry, 2019, 159, 20–29 CrossRef PubMed.
- Y. H. Dong, P. Sonawane, H. Cohen, G. Polturak, L. Feldberg, S. H. Avivi, I. Rogachev and A. Aharoni, New Phytol., 2020, 228, 1986–2002 CrossRef CAS PubMed.
- R. Nakabayashi, K. Hashimoto, K. Toyooka and K. Saito, Metabolomics, 2019, 15, 24 CrossRef PubMed.
- S. Giacomello, F. Salmen, B. K. Terebieniec, S. Vickovic, J. F. Navarro, A. Alexeyenko, J. Reimegard, L. S. McKee, C. Mannapperuma, V. Bulone, P. L. Stahl, J. F. Sundstrom, N. R. Street and J. Lundeberg, Nat. Plants, 2017, 3, 17061 CrossRef CAS PubMed.
- Y. Shinozaki, P. Nicolas, N. Fernandez-Pozo, Q. Y. Ma, D. J. Evanich, Y. N. Shi, Y. M. Xu, Y. Zheng, S. I. Snyder, L. B. B. Martin, E. Ruiz-May, T. W. Thannhauser, K. S. Chen, D. S. Domozych, C. Catala, Z. J. Fei, L. A. Mueller, J. J. Giovannoni and J. K. C. Rose, Nat. Commun., 2018, 9, 364 CrossRef PubMed.
- S. Giacomello and J. Lundeberg, Nat. Protoc., 2018, 13, 2425–2446 CrossRef CAS PubMed.
- Y. H. Liu, S. Lu, K. F. Liu, S. Wang, L. Q. Huang and L. P. Guo, Plant Methods, 2019, 15, 135 CrossRef PubMed.
- T. Fujii, S. Matsuda, M. L. Tejedor, T. Esaki, I. Sakane, H. Mizuno, N. Tsuyama and T. Masujima, Nat. Protoc., 2015, 10, 1445–1456 CrossRef CAS PubMed.
- T. Nakashima, H. Wada, S. Morita, R. Erra-Balsells, K. Hiraoka and H. Nonami, Anal. Chem., 2016, 88, 3049–3057 CrossRef CAS PubMed.
- K. Yamamoto, K. Takahashi, H. Mizuno, A. Anegawa, K. Ishizaki, H. Fukaki, M. Ohnishi, M. Yamazaki, T. Masujima and T. Mimura, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3891–3896 CrossRef CAS PubMed.
- K. Yamamoto, K. Takahashi, L. Caputi, H. Mizuno, C. E. Rodriguez-Lopez, T. Iwasaki, K. Ishizaki, H. Fukaki, M. Ohnishi, M. Yamazaki, T. Masujima, S. E. O'Connor and T. Mimura, New Phytol., 2019, 224, 848–859 CrossRef CAS PubMed.
- C. Rich-Griffin, A. Stechemesser, J. Finch, E. Lucas, S. Ott and P. Schafer, Trends Plant Sci., 2020, 25, 186–197 CrossRef CAS PubMed.
- R. Spicer, R. M. Salek, P. Moreno, D. Canueto and C. Steinbeck, Metabolomics, 2017, 13, 106 CrossRef PubMed.
- H. Y. Chang, S. M. Colby, X. Du, J. D. Gomez, M. J. Helf, K. Kechris, C. R. Kirkpatrick, S. Li, G. J. Patti, R. S. Renslow, S. Subramaniam, M. Verma, J. Xia and J. D. Young, Anal. Chem., 2021, 93, 1912–1923 CrossRef CAS PubMed.
- Z. J. Lai, H. Tsugawa, G. Wohlgemuth, S. Mehta, M. Mueller, Y. X. Zheng, A. Ogiwara, J. Meissen, M. Showalter, K. Takeuchi, T. Kind, P. Beal, M. Arita and O. Fiehn, Nat. Methods, 2018, 15, 53–56 CrossRef CAS PubMed.
- C. A. Smith, E. J. Want, G. O'Maille, R. Abagyan and G. Siuzdak, Anal. Chem., 2006, 78, 779–787 CrossRef CAS PubMed.
- R. Tautenhahn, G. J. Patti, D. Rinehart and G. Siuzdak, Anal. Chem., 2012, 84, 5035–5039 CrossRef CAS PubMed.
- T. Pluskal, S. Castillo, A. Villar-Briones and M. Oresic, BMC Bioinf., 2010, 11, 395 CrossRef PubMed.
- H. L. Rost, T. Sachsenberg, S. Aiche, C. Bielow, H. Weisser, F. Aicheler, S. Andreotti, H. C. Ehrlich, P. Gutenbrunner, E. Kenar, X. Liang, S. Nahnsen, L. Nilse, J. Pfeuffer, G. Rosenberger, M. Rurik, U. Schmitt, J. Veit, M. Walzer, D. Wojnar, W. E. Wolski, O. Schilling, J. S. Choudhary, L. Malmstrom, R. Aebersold, K. Reinert and O. Kohlbacher, Nat. Methods, 2016, 13, 741–748 CrossRef CAS PubMed.
- J. Chong, O. Soufan, C. Li, I. Caraus, S. Z. Li, G. Bourque, D. S. Wishart and J. G. Xia, Nucleic Acids Res., 2018, 46, W486–W494 CrossRef CAS PubMed.
- H. Tsugawa, T. Kind, R. Nakabayashi, D. Yukihira, W. Tanaka, T. Cajka, K. Saito, O. Fiehn and M. Arita, Anal. Chem., 2016, 88, 7946–7958 CrossRef CAS PubMed.
- O. Fraisier-Vannier, J. Chervin, G. Cabanac, V. Puech, S. Fournier, V. Durand, A. Amiel, O. Andre, O. A. Benamar, B. Dumas, H. Tsugawa and G. Marti, Anal. Chem., 2020, 92, 9971–9981 CrossRef CAS PubMed.
- B. C. DeFelice, S. S. Mehta, S. Samra, T. Cajka, B. Wancewicz, J. F. Fahrmann and O. Fiehn, Anal. Chem., 2017, 89, 3250–3255 CrossRef CAS PubMed.
- X. Shen, R. Wang, X. Xiong, Y. Yin, Y. Cai, Z. Ma, N. Liu and Z. J. Zhu, Nat. Commun., 2019, 10, 1516 CrossRef PubMed.
- H. Treutler, H. Tsugawa, A. Porzel, K. Gorzolka, A. Tissier, S. Neumann and G. U. Balcke, Anal. Chem., 2016, 88, 8082–8090 CrossRef CAS PubMed.
- A. Smirnov, Y. Qiu, W. Jia, D. I. Walker, D. P. Jones and X. Du, Anal. Chem., 2019, 91, 9069–9077 CrossRef CAS PubMed.
- M. Wang, A. K. Jarmusch, F. Vargas, A. A. Aksenov, J. M. Gauglitz, K. Weldon, D. Petras, R. da Silva, R. Quinn, A. V. Melnik, J. J. J. van der Hooft, A. M. Caraballo-Rodriguez, L. F. Nothias, C. M. Aceves, M. Panitchpakdi, E. Brown, F. Di Ottavio, N. Sikora, E. O. Elijah, L. Labarta-Bajo, E. C. Gentry, S. Shalapour, K. E. Kyle, S. P. Puckett, J. D. Watrous, C. S. Carpenter, A. Bouslimani, M. Ernst, A. D. Swafford, E. I. Zuniga, M. J. Balunas, J. L. Klassen, R. Loomba, R. Knight, N. Bandeira and P. C. Dorrestein, Nat. Biotechnol., 2020, 38, 23–26 CrossRef CAS PubMed.
- A. K. Jarmusch, M. Wang, C. M. Aceves, R. S. Advani, S. Aguirre, A. A. Aksenov, G. Aleti, A. T. Aron, A. Bauermeister, S. Bolleddu, A. Bouslimani, A. M. Caraballo Rodriguez, R. Chaar, R. Coras, E. O. Elijah, M. Ernst, J. M. Gauglitz, E. C. Gentry, M. Husband, S. A. Jarmusch, K. L. Jones 2nd, Z. Kamenik, A. Le Gouellec, A. Lu, L. I. McCall, K. L. McPhail, M. J. Meehan, A. V. Melnik, R. C. Menezes, Y. A. Montoya Giraldo, N. H. Nguyen, L. F. Nothias, M. Nothias-Esposito, M. Panitchpakdi, D. Petras, R. A. Quinn, N. Sikora, J. J. J. van der Hooft, F. Vargas, A. Vrbanac, K. C. Weldon, R. Knight, N. Bandeira and P. C. Dorrestein, Nat. Methods, 2020, 17, 901–904 CrossRef CAS PubMed.
- J. J. J. van der Hooft, J. Wandy, M. P. Barrett, K. E. V. Burgess and S. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13738–13743 CrossRef PubMed.
- Y. Sawada, R. Nakabayashi, Y. Yamada, M. Suzuki, M. Sato, A. Sakata, K. Akiyama, T. Sakurai, F. Matsuda, T. Aoki, M. Y. Hirai and K. Saito, Phytochemistry, 2012, 82, 38–45 CrossRef CAS PubMed.
- I. Tada, H. Tsugawa, I. Meister, P. Zhang, R. Shu, R. Katsumi, C. E. Wheelock, M. Arita and R. Chaleckis, Metabolites, 2019, 9, 251 CrossRef CAS PubMed.
- E. L. Schymanski, C. Ruttkies, M. Krauss, C. Brouard, T. Kind, K. Duhrkop, F. Allen, A. Vaniya, D. Verdegem, S. Bocker, J. Rousu, H. Shen, H. Tsugawa, T. Sajed, O. Fiehn, B. Ghesquiere and S. Neumann, J. Cheminf., 2017, 9, 22 Search PubMed.
- Z. T. Lei, L. Jing, F. Qiu, H. Zhang, D. Huhman, Z. Q. Zhou and L. W. Sumner, Anal. Chem., 2015, 87, 7373–7381 CrossRef CAS PubMed.
- S. Lee, S. Hwang, M. Seo, K. B. Shin, K. H. Kim, G. W. Park, J. Y. Kim, J. S. Yoo and K. T. No, Phytochemistry, 2020, 177, 112427 CrossRef CAS PubMed.
- R. Aoyagi, K. Ikeda, Y. Isobe and M. Arita, J. Lipid Res., 2017, 58, 2229–2237 CrossRef CAS PubMed.
- S. R. Heller, A. McNaught, I. Pletnev, S. Stein and D. Tchekhovskoi, J. Cheminf., 2015, 7, 23 Search PubMed.
- D. S. Wishart, Y. D. Feunang, A. Marcu, A. C. Guo, K. Liang, R. Vazquez-Fresno, T. Sajed, D. Johnson, C. Li, N. Karu, Z. Sayeeda, E. Lo, N. Assempour, M. Berjanskii, S. Singhal, D. Arndt, Y. Liang, H. Badran, J. Grant, A. Serra-Cayuela, Y. Liu, R. Mandal, V. Neveu, A. Pon, C. Knox, M. Wilson, C. Manach and A. Scalbert, Nucleic Acids Res., 2018, 46, D608–D617 CrossRef CAS PubMed.
- A. Frolkis, C. Knox, E. Lim, T. Jewison, V. Law, D. D. Hau, P. Liu, B. Gautam, S. Ly, A. C. Guo, J. Xia, Y. Liang, S. Shrivastava and D. S. Wishart, Nucleic Acids Res., 2010, 38, D480–D487 CrossRef CAS PubMed.
- E. Fahy, S. Subramaniam, R. C. Murphy, M. Nishijima, C. R. Raetz, T. Shimizu, F. Spener, G. van Meer, M. J. Wakelam and E. A. Dennis, J. Lipid Res., 2009, 50(suppl), S9–S14 CrossRef PubMed.
- T. Jewison, C. Knox, V. Neveu, Y. Djoumbou, A. C. Guo, J. Lee, P. Liu, R. Mandal, R. Krishnamurthy, I. Sinelnikov, M. Wilson and D. S. Wishart, Nucleic Acids Res., 2012, 40, D815–D820 CrossRef CAS PubMed.
- A. C. Guo, T. Jewison, M. Wilson, Y. Liu, C. Knox, Y. Djoumbou, P. Lo, R. Mandal, R. Krishnamurthy and D. S. Wishart, Nucleic Acids Res., 2013, 41, D625–D630 CrossRef CAS PubMed.
- A. Foroutan, C. Fitzsimmons, R. Mandal, H. Piri-Moghadam, J. Zheng, A. Guo, C. Li, L. L. Guan and D. S. Wishart, Metabolites, 2020, 10, 233 CrossRef CAS PubMed.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. F. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon, C. Knox and M. Wilson, Nucleic Acids Res., 2018, 46, D1074–D1082 CrossRef CAS PubMed.
- P. Zhang, K. Dreher, A. Karthikeyan, A. Chi, A. Pujar, R. Caspi, P. Karp, V. Kirkup, M. Latendresse, C. Lee, L. A. Mueller, R. Muller and S. Y. Rhee, Plant Physiol., 2010, 153, 1479–1491 CrossRef CAS PubMed.
- J. Hastings, G. Owen, A. Dekker, M. Ennis, N. Kale, V. Muthukrishnan, S. Turner, N. Swainston, P. Mendes and C. Steinbeck, Nucleic Acids Res., 2016, 44, D1214–D1219 CrossRef CAS PubMed.
- D. Wishart, D. Arndt, A. Pon, T. Sajed, A. C. Guo, Y. Djoumbou, C. Knox, M. Wilson, Y. J. Liang, J. Grant, Y. F. Liu, S. A. Goldansaz and S. M. Rappaport, Nucleic Acids Res., 2015, 43, D928–D934 CrossRef CAS PubMed.
- D. K. Barupal and O. Fiehn, Environ. Health Perspect., 2019, 127, 97008 CrossRef PubMed.
- J. A. van Santen, G. Jacob, A. L. Singh, V. Aniebok, M. J. Balunas, D. Bunsko, F. C. Neto, L. Castano-Espriu, C. Chang, T. N. Clark, J. L. C. Little, D. A. Delgadillo, P. C. Dorrestein, K. R. Duncan, J. M. Egan, M. M. Galey, F. P. J. Haeckl, A. Hua, A. H. Hughes, D. Iskakova, A. Khadilkar, J. H. Lee, S. Lee, N. LeGrow, D. Y. Liu, J. M. Macho, C. S. McCaughey, M. H. Medema, R. P. Neupane, T. J. O'Donnell, J. S. Paula, L. M. Sanchez, A. F. Shaikh, S. Soldatou, B. R. Terlouw, T. A. Tran, M. Valentine, J. J. J. van der Hooft, D. A. Vo, M. X. Wang, D. Wilson, K. E. Zink and R. G. Linington, ACS Cent. Sci., 2019, 5, 1824–1833 CrossRef CAS PubMed.
- F. Ntie-Kang, K. K. Telukunta, K. Doring, C. V. Simoben, A. M. AF, Y. I. Malange, L. E. Njume, J. N. Yong, W. Sippl and S. Gunther, J. Nat. Prod., 2017, 80, 2067–2076 CrossRef CAS PubMed.
- J. Gu, Y. Gui, L. Chen, G. Yuan, H. Z. Lu and X. Xu, PLoS One, 2013, 8, e62839 CrossRef CAS PubMed.
- S. Kim, J. Chen, T. J. Cheng, A. Gindulyte, J. He, S. Q. He, Q. L. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2019, 47, D1102–D1109 CrossRef PubMed.
- M. Sorokina and C. Steinbeck, J. Cheminf., 2020, 12 Search PubMed.
- Y. Djoumbou Feunang, R. Eisner, C. Knox, L. Chepelev, J. Hastings, G. Owen, E. Fahy, C. Steinbeck, S. Subramanian, E. Bolton, R. Greiner and D. S. Wishart, J. Cheminf., 2016, 8, 61 Search PubMed.
- M. Sorokina, P. Merseburger, K. Rajan, M. A. Yirik and C. Steinbeck, J. Cheminf., 2021, 13, 2 Search PubMed.
- K. Duhrkop, M. Fleischauer, M. Ludwig, A. A. Aksenov, A. V. Melnik, M. Meusel, P. C. Dorrestein, J. Rousu and S. Bocker, Nat. Methods, 2019, 16, 299–302 CrossRef PubMed.
- K. Duhrkop, L. F. Nothias, M. Fleischauer, R. Reher, M. Ludwig, M. A. Hoffmann, D. Petras, W. H. Gerwick, J. Rousu, P. C. Dorrestein and S. Bocker, Nat. Biotechnol., 2020, 39, 462–471 CrossRef PubMed.
- Y. Djoumbou-Feunang, A. Pon, N. Karu, J. Zheng, C. Li, D. Arndt, M. Gautam, F. Allen and D. S. Wishart, Metabolites, 2019, 9 CrossRef CAS PubMed.
- F. Allen, A. Pon, M. Wilson, R. Greiner and D. Wishart, Nucleic Acids Res., 2014, 42, W94–W99 CrossRef CAS PubMed.
- S. Wolf, S. Schmidt, M. Muller-Hannemann and S. Neumann, BMC Bioinf., 2010, 11, 148 CrossRef PubMed.
- C. Ruttkies, E. L. Schymanski, S. Wolf, J. Hollender and S. Neumann, J. Cheminf., 2016, 8, 3 Search PubMed.
- L. Ridder, J. J. van der Hooft and S. Verhoeven, Mass Spectrometry, 2014, 3, S0033 CrossRef PubMed.
- J. J. J. van der Hooft, J. Vervoort, R. J. Bino, J. Beekwilder and R. C. H. de Vos, Anal. Chem., 2011, 83, 409–416 CrossRef CAS PubMed.
- J. A. Falkner, J. W. Falkner, A. K. Yocum and P. C. Andrews, J. Proteome Res., 2008, 7, 4614–4622 CrossRef CAS PubMed.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed.
- E. L. Willighagen, J. W. Mayfield, J. Alvarsson, A. Berg, L. Carlsson, N. Jeliazkova, S. Kuhn, T. Pluskal, M. Rojas-Cherto, O. Spjuth, G. Torrance, C. T. Evelo, R. Guha and C. Steinbeck, J. Cheminf., 2017, 9, 33 Search PubMed.
- J. Klekota and F. P. Roth, Bioinformatics, 2008, 24, 2518–2525 CrossRef CAS PubMed.
- S. M. Colby, D. G. Thomas, J. R. Nunez, D. J. Baxter, K. R. Glaesemann, J. M. Brown, M. A. Pirrung, N. Govind, J. G. Teeguarden, T. O. Metz and R. S. Renslow, Anal. Chem., 2019, 91, 4346–4356 CrossRef CAS PubMed.
- A. Rai, M. Yamazaki and K. Saito, Current Opinion in Systems Biology, 2019, 15, 58–67 CrossRef.
- A. Rai, K. Saito and M. Yamazaki, Plant J., 2017, 90, 764–787 CrossRef CAS PubMed.
- M. Yamazaki, A. Rai, N. Yoshimoto and K. Saito, Plant Biotechnology Reports, 2018, 12, 69–75 CrossRef.
- M. Mutwil, Curr. Opin. Plant Biol., 2020, 55, 38–46 CrossRef CAS PubMed.
- J. H. Leebens-Mack, M. S. Barker, E. J. Carpenter, M. K. Deyholos, M. A. Gitzendanner, S. W. Graham, I. Grosse, Z. Li, M. Melkonian, S. Mirarab, M. Porsch, M. Quint, S. A. Rensing, D. E. Soltis, P. S. Soltis, D. W. Stevenson, K. K. Ullrich, N. J. Wickett, L. DeGironimo, P. P. Edger, I. E. Jordon-Thaden, S. Joya, T. Liu, B. Melkonian, N. W. Miles, L. Pokorny, C. Quigley, P. Thomas, J. C. Villarreal, M. M. Augustin, M. D. Barrett, R. S. Baucom, D. J. Beerling, R. M. Benstein, E. Biffin, S. F. Brockington, D. O. Burge, J. N. Burris, K. P. Burris, V. Burtet-Sarramegna, A. L. Caicedo, S. B. Cannon, Z. Çebi, Y. Chang, C. Chater, J. M. Cheeseman, T. Chen, N. D. Clarke, H. Clayton, S. Covshoff, B. J. Crandall-Stotler, H. Cross, C. W. dePamphilis, J. P. Der, R. Determann, R. C. Dickson, V. S. Di Stilio, S. Ellis, E. Fast, N. Feja, K. J. Field, D. A. Filatov, P. M. Finnegan, S. K. Floyd, B. Fogliani, N. García, G. Gâteblé, G. T. Godden, F. Goh, S. Greiner, A. Harkess, J. M. Heaney, K. E. Helliwell, K. Heyduk, J. M. Hibberd, R. G. J. Hodel, P. M. Hollingsworth, M. T. J. Johnson, R. Jost, B. Joyce, M. V. Kapralov, E. Kazamia, E. A. Kellogg, M. A. Koch, M. Von Konrat, K. Könyves, T. M. Kutchan, V. Lam, A. Larsson, A. R. Leitch, R. Lentz, F.-W. Li, A. J. Lowe, M. Ludwig, P. S. Manos, E. Mavrodiev, M. K. McCormick, M. McKain, T. McLellan, J. R. McNeal, R. E. Miller, M. N. Nelson, Y. Peng, P. Ralph, D. Real, C. W. Riggins, M. Ruhsam, R. F. Sage, A. K. Sakai, M. Scascitella, E. E. Schilling, E.-M. Schlösser, H. Sederoff, S. Servick, E. B. Sessa, A. J. Shaw, S. W. Shaw, E. M. Sigel, C. Skema, A. G. Smith, A. Smithson, C. N. Stewart, J. R. Stinchcombe, P. Szövényi, J. A. Tate, H. Tiebel, D. Trapnell, M. Villegente, C.-N. Wang, S. G. Weller, M. Wenzel, S. Weststrand, J. H. Westwood, D. F. Whigham, S. Wu, A. S. Wulff, Y. Yang, D. Zhu, C. Zhuang, J. Zuidof, M. W. Chase, J. C. Pires, C. J. Rothfels, J. Yu, C. Chen, L. Chen, S. Cheng, J. Li, R. Li, X. Li, H. Lu, Y. Ou, X. Sun, X. Tan, J. Tang, Z. Tian, F. Wang, J. Wang, X. Wei, X. Xu, Z. Yan, F. Yang, X. Zhong, F. Zhou, Y. Zhu, Y. Zhang, S. Ayyampalayam, T. J. Barkman, N.-p. Nguyen, N. Matasci, D. R. Nelson, E. Sayyari, E. K. Wafula, R. L. Walls, T. Warnow, H. An, N. Arrigo, A. E. Baniaga, S. Galuska, S. A. Jorgensen, T. I. Kidder, H. Kong, P. Lu-Irving, H. E. Marx, X. Qi, C. R. Reardon, B. L. Sutherland, G. P. Tiley, S. R. Welles, R. Yu, S. Zhan, L. Gramzow, G. Theißen, G. K.-S. Wong and I. One, Thousand Plant Transcriptomes, Nature, 2019, 574, 679–685 CrossRef PubMed.
- H. A. Lewin, G. E. Robinson, W. J. Kress, W. J. Baker, J. Coddington, K. A. Crandall, R. Durbin, S. V. Edwards, F. Forest, M. T. P. Gilbert, M. M. Goldstein, I. V. Grigoriev, K. J. Hackett, D. Haussler, E. D. Jarvis, W. E. Johnson, A. Patrinos, S. Richards, J. C. Castilla-Rubio, M. A. van Sluys, P. S. Soltis, X. Xu, H. Yang and G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4325–4333 CrossRef CAS PubMed.
- G. Yan, H. Liu, H. Wang, Z. Lu, Y. Wang, D. Mullan, J. Hamblin and C. Liu, Front. Plant Sci., 2017, 8, 1786 CrossRef PubMed.
- A. Habib, J. J. Powell, J. Stiller, M. Liu, S. Shabala, M. Zhou, D. M. Gardiner and C. Liu, Theor. Appl. Genet., 2018, 131, 613–624 CrossRef CAS PubMed.
- A. J. Monforte and S. D. Tanksley, Genome, 2000, 43, 803–813 CrossRef CAS PubMed.
- K. W. Broman, Genetics, 2005, 169, 1133–1146 CrossRef CAS PubMed.
- D. Balakrishnan, M. Surapaneni, V. R. Yadavalli, K. R. Addanki, S. Mesapogu, K. Beerelli and S. Neelamraju, Sci. Rep., 2020, 10, 7766 CrossRef CAS PubMed.
- Z. H. Sun, J. Du, X. Y. Pu, M. K. Ali, X. M. Yang, C. L. Duan, M. R. Ren, X. Li and Y. W. Zeng, Agronomy, 2019, 9, 40 CrossRef.
- M. L. Ali, P. L. Sanchez, S. B. Yu, M. Lorieux and G. C. Eizenga, Rice, 2010, 3, 218–234 CrossRef.
- D. Balakrishnan, M. Surapaneni, S. Mesapogu and S. Neelamraju, Theor. Appl. Genet., 2019, 132, 1–25 CrossRef CAS PubMed.
- X. Li, W. Wang, Z. Wang, K. Li, Y. P. Lim and Z. Piao, Front. Plant Sci., 2015, 6, 432 Search PubMed.
- H. J. Klee and D. M. Tieman, Nat. Rev. Genet., 2018, 19, 347–356 CrossRef CAS PubMed.
- M. S. Mia, H. Liu, X. Wang and G. Yan, Front. Plant Sci., 2019, 10, 271 CrossRef PubMed.
- R. Z. Yuan, N. Zhao, B. Usman, L. Luo, S. Y. Liao, Y. F. Qin, G. Nawaz and R. B. Li, Genes, 2020, 11, 980 CrossRef CAS PubMed.
- S. Alseekh, T. Tohge, R. Wendenberg, F. Scossa, N. Omranian, J. Li, S. Kleessen, P. Giavalisco, T. Pleban, B. Mueller-Roeber, D. Zamir, Z. Nikoloski and A. R. Fernie, Plant Cell, 2015, 27, 485–512 CrossRef CAS PubMed.
- D. Jaganathan, A. Bohra, M. Thudi and R. K. Varshney, Theor. Appl. Genet., 2020, 133, 1791–1810 CrossRef CAS PubMed.
- M. Kusano, Z. G. Yang, Y. Okazaki, R. Nakabayashi, A. Fukushima and K. Saito, Mol. Plant, 2015, 8, 58–67 CrossRef CAS PubMed.
- D. Wang, W. Sun, Z. Yuan, Q. Sun, K. Fan, C. Zhang and S. Yu, Sci. Rep., 2021, 11, 189 CrossRef CAS PubMed.
- F. Matsuda, Y. Okazaki, A. Oikawa, M. Kusano, R. Nakabayashi, J. Kikuchi, J. I. Yonemaru, K. Ebana, M. Yano and K. Saito, Plant J., 2012, 70, 624–636 CrossRef CAS PubMed.
- L. Gong, W. Chen, Y. Q. Gao, X. Q. Liu, H. Y. Zhang, C. G. Xu, S. B. Yu, Q. F. Zhang and J. Luo, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20320–20325 CrossRef CAS PubMed.
- K. Li, D. H. Wang, L. Gong, Y. Y. Lyu, H. Guo, W. Chen, C. Jin, X. Q. Liu, C. Y. Fang and J. Luo, Plant J., 2019, 100, 908–922 CrossRef CAS PubMed.
- G. H. Xu, J. J. Cao, X. F. Wang, Q. Y. Chen, W. W. Jin, Z. Li and F. Tian, Plant Cell, 2019, 31, 1990–2009 CrossRef CAS PubMed.
- D. Knoch, D. Riewe, R. C. Meyer, A. Boudichevskaia, R. Schmidt and T. Altmann, J. Exp. Bot., 2017, 68, 1655–1667 CrossRef CAS PubMed.
- A. Schilmiller, F. Shi, J. Kim, A. L. Charbonneau, D. Holmes, A. Daniel Jones and R. L. Last, Plant J., 2010, 62, 391–403 CrossRef CAS PubMed.
- A. L. Schilmiller, A. L. Charbonneau and R. L. Last, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16377–16382 CrossRef CAS PubMed.
- S. Alseekh, I. Ofner, Z. Liu, S. Osorio, J. Vallarino, R. L. Last, D. Zamir, T. Tohge and A. R. Fernie, Plant J., 2020, 103, 2007–2024 CrossRef CAS PubMed.
- G. Zhu, S. Wang, Z. Huang, S. Zhang, Q. Liao, C. Zhang, T. Lin, M. Qin, M. Peng, C. Yang, X. Cao, X. Han, X. Wang, E. van der Knaap, Z. Zhang, X. Cui, H. Klee, A. R. Fernie, J. Luo and S. Huang, Cell, 2018, 172, 249–261 e212 CrossRef CAS PubMed.
- J. Szymanski, S. Bocobza, S. Panda, P. Sonawane, P. D. Cardenas, J. Lashbrooke, A. Kamble, N. Shahaf, S. Meir, A. Bovy, J. Beekwilder, Y. Tikunov, I. Romero de la Fuente, D. Zamir, I. Rogachev and A. Aharoni, Nat. Genet., 2020, 52, 1111–1121 CrossRef CAS PubMed.
- T. T. Shi, A. N. Zhu, J. Q. Jia, X. Hu, J. Chen, W. Liu, X. F. Ren, D. F. Sun, A. R. Fernie, F. Cui and W. Chen, Plant J., 2020, 103, 279–292 CrossRef CAS PubMed.
- N. Carreno-Quintero, A. Acharjee, C. Maliepaard, C. W. B. Bachem, R. Mumm, H. Bouwmeester, R. G. F. Visser and J. J. B. Keurentjes, Plant Physiol., 2012, 158, 1306–1318 CrossRef CAS PubMed.
- A. Maharijaya, B. Vosman, K. Pelgrom, Y. Wahyuni, R. C. H. de Vos and R. E. Voorrips, Arthropod-Plant Interactions, 2019, 13, 1–9 CrossRef.
- C. Addo-Quaye, E. Buescher, N. Best, V. Chaikam, I. Baxter and B. P. Dilkes, G3: Genes, Genomes, Genet., 2017, 7, 413–425 CrossRef CAS PubMed.
- C. Fang, A. R. Fernie and J. Luo, Trends Plant Sci., 2019, 24, 83–98 CrossRef CAS PubMed.
- S. D. Turner-Hissong, M. E. Mabry, T. M. Beissinger, J. Ross-Ibarra and J. C. Pires, Curr. Opin. Plant Biol., 2020, 54, 93–100 CrossRef CAS PubMed.
- D. Weigel, Plant Physiol., 2012, 158, 2–22 CrossRef CAS PubMed.
- V. D. Daygon, M. Calingacion, L. C. Forster, J. J. Voss, B. D. Schwartz, B. Ovenden, D. E. Alonso, S. R. McCouch, M. J. Garson and M. A. Fitzgerald, Sci. Rep., 2017, 7, 8767 CrossRef PubMed.
- F. Matsuda, R. Nakabayashi, Z. Yang, Y. Okazaki, J. Yonemaru, K. Ebana, M. Yano and K. Saito, Plant J., 2015, 81, 13–23 CrossRef CAS PubMed.
- C. Fang and J. Luo, Plant J., 2019, 97, 91–100 CrossRef CAS PubMed.
- X. Huang and B. Han, Annu. Rev. Plant Biol., 2014, 65, 531–551 CrossRef CAS PubMed.
- W. Chen, Y. Gao, W. Xie, L. Gong, K. Lu, W. Wang, Y. Li, X. Liu, H. Zhang, H. Dong, W. Zhang, L. Zhang, S. Yu, G. Wang, X. Lian and J. Luo, Nat. Genet., 2014, 46, 714–721 CrossRef CAS PubMed.
- R. R. Fuentes, D. Chebotarov, J. Duitama, S. Smith, J. F. De la Hoz, M. Mohiyuddin, R. A. Wing, K. L. McNally, T. Tatarinova, A. Grigoriev, R. Mauleon and N. Alexandrov, Genome Res., 2019, 29, 870–880 CrossRef CAS PubMed.
- R. C. Strauch, E. Svedin, B. Dilkes, C. Chapple and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11726–11731 CrossRef CAS PubMed.
- C. Sauvage, V. Segura, G. Bauchet, R. Stevens, P. T. Do, Z. Nikoloski, A. R. Fernie and M. Causse, Plant Physiol., 2014, 165, 1120–1132 CrossRef CAS PubMed.
- P. D. Sonawane, A. Jozwiak, S. Panda and A. Aharoni, Curr. Opin. Plant Biol., 2020, 55, 118–128 CrossRef CAS PubMed.
- H. Sonah, L. O'Donoughue, E. Cober, I. Rajcan and F. Belzile, Plant Biotechnol. J., 2015, 13, 211–221 CrossRef CAS PubMed.
- B. Brachi, N. Faure, M. Horton, E. Flahauw, A. Vazquez, M. Nordborg, J. Bergelson, J. Cuguen and F. Roux, PLoS Genet., 2010, 6, e1000940 CrossRef PubMed.
- S. Zhou, K. A. Kremling, N. Bandillo, A. Richter, Y. K. Zhang, K. R. Ahern, A. B. Artyukhin, J. X. Hui, G. C. Younkin, F. C. Schroeder, E. S. Buckler and G. Jander, Plant Cell, 2019, 31, 937–955 CrossRef CAS PubMed.
- P. E. Bayer, A. A. Golicz, A. Scheben, J. Batley and D. Edwards, Nat. Plants, 2020, 6, 914–920 CrossRef PubMed.
- M. F. Danilevicz, C. G. Tay Fernandez, J. I. Marsh, P. E. Bayer and D. Edwards, Curr. Opin. Plant Biol., 2020, 54, 18–25 CrossRef CAS PubMed.
- C. Da Silva, G. Zamperin, A. Ferrarini, A. Minio, A. Dal Molin, L. Venturini, G. Buson, P. Tononi, C. Avanzato, E. Zago, E. Boido, E. Dellacassa, C. Gaggero, M. Pezzotti, F. Carrau and M. Delledonne, Plant Cell, 2013, 25, 4777–4788 CrossRef CAS PubMed.
- L. Gao, I. Gonda, H. Sun, Q. Ma, K. Bao, D. M. Tieman, E. A. Burzynski-Chang, T. L. Fish, K. A. Stromberg, G. L. Sacks, T. W. Thannhauser, M. R. Foolad, M. J. Diez, J. Blanca, J. Canizares, Y. Xu, E. van der Knaap, S. Huang, H. J. Klee, J. J. Giovannoni and Z. Fei, Nat. Genet., 2019, 51, 1044–1051 CrossRef CAS PubMed.
- S. P. Gordon, B. Contreras-Moreira, D. P. Woods, D. L. Des Marais, D. Burgess, S. Shu, C. Stritt, A. C. Roulin, W. Schackwitz, L. Tyler, J. Martin, A. Lipzen, N. Dochy, J. Phillips, K. Barry, K. Geuten, H. Budak, T. E. Juenger, R. Amasino, A. L. Caicedo, D. Goodstein, P. Davidson, L. A. J. Mur, M. Figueroa, M. Freeling, P. Catalan and J. P. Vogel, Nat. Commun., 2017, 8, 2184 CrossRef PubMed.
- G. Haberer, N. Kamal, E. Bauer, H. Gundlach, I. Fischer, M. A. Seidel, M. Spannagl, C. Marcon, A. Ruban, C. Urbany, A. Nemri, F. Hochholdinger, M. Ouzunova, A. Houben, C. C. Schon and K. F. X. Mayer, Nat. Genet., 2020, 52, 950–957 CrossRef CAS PubMed.
- Y. Liu, H. Du, P. Li, Y. Shen, H. Peng, S. Liu, G. A. Zhou, H. Zhang, Z. Liu, M. Shi, X. Huang, Y. Li, M. Zhang, Z. Wang, B. Zhu, B. Han, C. Liang and Z. Tian, Cell, 2020, 182, 162–176 e113 CrossRef CAS PubMed.
- Q. Zhao, Q. Feng, H. Lu, Y. Li, A. Wang, Q. Tian, Q. Zhan, Y. Lu, L. Zhang, T. Huang, Y. Wang, D. Fan, Y. Zhao, Z. Wang, C. Zhou, J. Chen, C. Zhu, W. Li, Q. Weng, Q. Xu, Z. X. Wang, X. Wei, B. Han and X. Huang, Nat. Genet., 2018, 50, 278–284 CrossRef CAS PubMed.
- W. Wang, R. Mauleon, Z. Hu, D. Chebotarov, S. Tai, Z. Wu, M. Li, T. Zheng, R. R. Fuentes, F. Zhang, L. Mansueto, D. Copetti, M. Sanciangco, K. C. Palis, J. Xu, C. Sun, B. Fu, H. Zhang, Y. Gao, X. Zhao, F. Shen, X. Cui, H. Yu, Z. Li, M. Chen, J. Detras, Y. Zhou, X. Zhang, Y. Zhao, D. Kudrna, C. Wang, R. Li, B. Jia, J. Lu, X. He, Z. Dong, J. Xu, Y. Li, M. Wang, J. Shi, J. Li, D. Zhang, S. Lee, W. Hu, A. Poliakov, I. Dubchak, V. J. Ulat, F. N. Borja, J. R. Mendoza, J. Ali, J. Li, Q. Gao, Y. Niu, Z. Yue, M. E. B. Naredo, J. Talag, X. Wang, J. Li, X. Fang, Y. Yin, J. C. Glaszmann, J. Zhang, J. Li, R. S. Hamilton, R. A. Wing, J. Ruan, G. Zhang, C. Wei, N. Alexandrov, K. L. McNally, Z. Li and H. Leung, Nature, 2018, 557, 43–49 CrossRef CAS PubMed.
- P. Zhou, K. A. Silverstein, T. Ramaraj, J. Guhlin, R. Denny, J. Liu, A. D. Farmer, K. P. Steele, R. M. Stupar, J. R. Miller, P. Tiffin, J. Mudge and N. D. Young, BMC Genomics, 2017, 18, 261 CrossRef PubMed.
- M. Alonge, X. Wang, M. Benoit, S. Soyk, L. Pereira, L. Zhang, H. Suresh, S. Ramakrishnan, F. Maumus, D. Ciren, Y. Levy, T. H. Harel, G. Shalev-Schlosser, Z. Amsellem, H. Razifard, A. L. Caicedo, D. M. Tieman, H. Klee, M. Kirsche, S. Aganezov, T. R. Ranallo-Benavidez, Z. H. Lemmon, J. Kim, G. Robitaille, M. Kramer, S. Goodwin, W. R. McCombie, S. Hutton, J. Van Eck, J. Gillis, Y. Eshed, F. J. Sedlazeck, E. van der Knaap, M. C. Schatz and Z. B. Lippman, Cell, 2020, 182, 145–161 e123 CrossRef CAS PubMed.
- J. M. Song, Z. Guan, J. Hu, C. Guo, Z. Yang, S. Wang, D. Liu, B. Wang, S. Lu, R. Zhou, W. Z. Xie, Y. Cheng, Y. Zhang, K. Liu, Q. Y. Yang, L. L. Chen and L. Guo, Nat. Plants, 2020, 6, 34–45 CrossRef CAS PubMed.
- S. Hubner, N. Bercovich, M. Todesco, J. R. Mandel, J. Odenheimer, E. Ziegler, J. S. Lee, G. J. Baute, G. L. Owens, C. J. Grassa, D. P. Ebert, K. L. Ostevik, B. T. Moyers, S. Yakimowski, R. R. Masalia, L. Gao, I. Calic, J. E. Bowers, N. C. Kane, D. Z. H. Swanevelder, T. Kubach, S. Munos, N. B. Langlade, J. M. Burke and L. H. Rieseberg, Nat. Plants, 2019, 5, 54–62 CrossRef CAS PubMed.
- J. D. Montenegro, A. A. Golicz, P. E. Bayer, B. Hurgobin, H. Lee, C. K. Chan, P. Visendi, K. Lai, J. Dolezel, J. Batley and D. Edwards, Plant J., 2017, 90, 1007–1013 CrossRef CAS PubMed.
- A. A. Golicz, P. E. Bayer, G. C. Barker, P. P. Edger, H. Kim, P. A. Martinez, C. K. Chan, A. Severn-Ellis, W. R. McCombie, I. A. Parkin, A. H. Paterson, J. C. Pires, A. G. Sharpe, H. Tang, G. R. Teakle, C. D. Town, J. Batley and D. Edwards, Nat. Commun., 2016, 7, 13390 CrossRef CAS PubMed.
- G. D. Moghe and R. L. Last, Plant Physiol., 2015, 169, 1512–1523 CAS.
- R. C. Moore and M. D. Purugganan, Curr. Opin. Plant Biol., 2005, 8, 122–128 CrossRef CAS PubMed.
- B. R. Lichman, G. T. Godden and C. R. Buell, Curr. Opin. Plant Biol., 2020, 55, 74–83 CrossRef CAS PubMed.
- Y. He, A. Galant, Q. Y. Pang, J. M. Strul, S. F. Balogun, J. M. Jez and S. X. Chen, J. Biol. Chem., 2011, 286, 28794–28801 CrossRef CAS PubMed.
- B. J. Leong and R. L. Last, Curr. Opin. Struct. Biol., 2017, 47, 105–112 CrossRef CAS PubMed.
- Y. Shimizu, A. Rai, Y. Okawa, H. Tomatsu, M. Sato, K. Kera, H. Suzuki, K. Saito and M. Yamazaki, Plant J., 2019, 100, 505–521 CrossRef CAS PubMed.
- E. Pichersky and E. Lewinsohn, Annu. Rev. Plant Biol., 2011, 62, 549–566 CrossRef CAS PubMed.
- S. Xu, T. Brockmoller, A. Navarro-Quezada, H. Kuhl, K. Gase, Z. Ling, W. Zhou, C. Kreitzer, M. Stanke, H. Tang, E. Lyons, P. Pandey, S. P. Pandey, B. Timmermann, E. Gaquerel and I. T. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6133–6138 CrossRef CAS PubMed.
- R. Huang, A. J. O'Donnell, J. J. Barboline and T. J. Barkman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10613–10618 CrossRef CAS PubMed.
- A. Rai, H. Hirakawa, R. Nakabayashi, S. Kikuchi, K. Hayashi, M. Rai, H. Tsugawa, T. Nakaya, T. Mori, H. Nagasaki, R. Fukushi, Y. Kusuya, H. Takahashi, H. Uchiyama, A. Toyoda, S. Hikosaka, E. Goto, K. Saito and M. Yamazaki, Nat. Commun., 2021, 12, 405 CrossRef CAS PubMed.
- S. Weaver, S. D. Shank, S. J. Spielman, M. Li, S. V. Muse and S. L. Kosakovsky Pond, Mol. Biol. Evol., 2018, 35, 773–777 CrossRef CAS PubMed.
- E. Defossez, C. Pitteloud, P. Descombes, G. Glauser, P. M. Allard, T. W. N. Walker, P. Fernandez-Conradi, J. L. Wolfender, L. Pellissier and S. Rasmann, Proc. Natl. Acad. Sci. U. S. A., 2021, 118 Search PubMed.
- H. W. Nutzmann, C. Scazzocchio and A. Osbourn, Annu. Rev. Genet., 2018, 52, 159–183 CrossRef CAS PubMed.
- J. H. Wisecaver, A. T. Borowsky, V. Tzin, G. Jander, D. J. Kliebenstein and A. Rokas, Plant Cell, 2017, 29, 944–959 CrossRef CAS PubMed.
- L. Guo, T. Winzer, X. Yang, Y. Li, Z. Ning, Z. He, R. Teodor, Y. Lu, T. A. Bowser, I. A. Graham and K. Ye, Science, 2018, 362, 343–347 CrossRef CAS PubMed.
- P. Schlapfer, P. Zhang, C. Wang, T. Kim, M. Banf, L. Chae, K. Dreher, A. K. Chavali, R. Nilo-Poyanco, T. Bernard, D. Kahn and S. Y. Rhee, Plant Physiol., 2017, 173, 2041–2059 CrossRef CAS PubMed.
- N. Topfer, L. M. Fuchs and A. Aharoni, Nucleic Acids Res., 2017, 45, 7049–7063 CrossRef CAS PubMed.
- S. A. Kautsar, H. G. Suarez Duran, K. Blin, A. Osbourn and M. H. Medema, Nucleic Acids Res., 2017, 45, W55–W63 CrossRef CAS PubMed.
- Z. Liu, J. Cheema, M. Vigouroux, L. Hill, J. Reed, P. Paajanen, L. Yant and A. Osbourn, Nat. Commun., 2020, 11, 5354 CrossRef CAS PubMed.
- Q. Li, S. Ramasamy, P. Singh, J. M. Hagel, S. M. Dunemann, X. Chen, R. Chen, L. Yu, J. E. Tucker, P. J. Facchini and S. Yeaman, Nat. Commun., 2020, 11, 1190 CrossRef CAS PubMed.
- A. C. Huang, S. A. Kautsar, Y. J. Hong, M. H. Medema, A. D. Bond, D. J. Tantillo and A. Osbourn, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6005–E6014 CrossRef CAS PubMed.
- H. Wang, E. Cimen, N. Singh and E. Buckler, Curr. Opin. Plant Biol., 2020, 54, 34–41 CrossRef CAS PubMed.
- J. Zou, M. Huss, A. Abid, P. Mohammadi, A. Torkamani and A. Telenti, Nat. Genet., 2019, 51, 12–18 CrossRef CAS PubMed.
- J. D. Washburn, M. K. Mejia-Guerra, G. Ramstein, K. A. Kremling, R. Valluru, E. S. Buckler and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5542–5549 CrossRef CAS PubMed.
- S. Esposito, D. Carputo, T. Cardi and P. Tripodi, Plants, 2019, 9, 34 CrossRef PubMed.
- Y. Jiang and C. Li, Plant Phenomics, 2020, 2020, 4152816 CrossRef PubMed.
- T. Gu, X. Zhao, W. B. Barbazuk and J.-H. Lee, BMC Bioinf., 2021, 22, 96 CrossRef CAS PubMed.
- M. Wen, P. Cong, Z. Zhang, H. Lu and T. Li, Bioinformatics, 2018, 34, 3781–3787 CrossRef CAS PubMed.
- A. Pla, X. Zhong and S. Rayner, PLoS Comput. Biol., 2018, 14, e1006185 CrossRef PubMed.
- X. Gao, Z. Wei and H. Hakonarson, Hum. Hered., 2018, 83, 163–172 CrossRef CAS PubMed.
- A. Arefeen, X. Xiao and T. Jiang, Bioinformatics, 2019, 35, 4577–4585 CrossRef CAS PubMed.
- M. Wang, C. Tai, W. E and L. Wei, Nucleic Acids Res., 2018, 46, e69 CrossRef PubMed.
- B. Alipanahi, A. Delong, M. T. Weirauch and B. J. Frey, Nat. Biotechnol., 2015, 33, 831–838 CrossRef CAS PubMed.
- J. Zhou and O. G. Troyanskaya, Nat. Methods, 2015, 12, 931–934 CrossRef CAS PubMed.
- J. Meng, Z. Chang, P. Zhang, W. Shi and Y. Luan, lncRNA-LSTM: Prediction of Plant Long Non-coding RNAs Using Long Short-Term Memory Based on p-nts Encoding, in Intelligent Computing Methodologies, ICIC 2019, Lecture Notes in Computer Science, ed. D. S. Huang, Z. K. Huang and A. Hussain, Springer, Cham, 2019, vol. 11645, DOI:10.1007/978-3-030-26766-7_32.
- C. Angermueller, H. J. Lee, W. Reik and O. Stegle, Genome Biol., 2017, 18, 67 CrossRef PubMed.
- S. Budach and A. Marsico, Bioinformatics, 2018, 34, 3035–3037 CrossRef CAS PubMed.
- S. Hashemifar, B. Neyshabur, A. A. Khan and J. Xu, Bioinformatics, 2018, 34, i802–i810 CrossRef CAS PubMed.
- Y. Guo, L. Yu, Z. Wen and M. Li, Nucleic Acids Res., 2008, 36, 3025–3030 CrossRef CAS PubMed.
- S. Martin, D. Roe and J. L. Faulon, Bioinformatics, 2005, 21, 218–226 CrossRef CAS PubMed.
- X. Zhang, W. Xiao and W. Xiao, PLoS Comput. Biol., 2020, 16, e1008229 CrossRef CAS PubMed.
- W. Ma, Z. Qiu, J. Song, J. Li, Q. Cheng, J. Zhai and C. Ma, Planta, 2018, 248, 1307–1318 CrossRef CAS PubMed.
- R. Poplin, P. C. Chang, D. Alexander, S. Schwartz, T. Colthurst, A. Ku, D. Newburger, J. Dijamco, N. Nguyen, P. T. Afshar, S. S. Gross, L. Dorfman, C. Y. McLean and M. A. DePristo, Nat. Biotechnol., 2018, 36, 983–987 CrossRef CAS PubMed.
- T. Yun, H. Li, P. C. Chang, M. F. Lin, A. Carroll and C. Y. McLean, Bioinformatics, 2020, 36, 5582–5589 CrossRef CAS PubMed.
- L. Cai, Y. Wu and J. Gao, BMC Bioinf., 2019, 20, 665 CrossRef PubMed.
- E. K. K. Ip, C. Hadinata, J. W. K. Ho and E. Giannoulatou, Bioinformatics, 2020, 36, 3549–3551 CrossRef CAS PubMed.
- R. Luo, F. J. Sedlazeck, T. W. Lam and M. C. Schatz, Nat. Commun., 2019, 10, 998 CrossRef PubMed.
- M. AlQuraishi, Bioinformatics, 2019, 35, 4862–4865 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021 DOI:10.1038/s41586-021-03819-2.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021 DOI:10.1126/science.abj8754.
- J. Y. Ryu, H. U. Kim and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13996–14001 CrossRef CAS PubMed.
- Y. Li, S. Wang, R. Umarov, B. Xie, M. Fan, L. Li and X. Gao, Bioinformatics, 2018, 34, 760–769 CrossRef CAS PubMed.
- B. M. Moore, P. Wang, P. Fan, B. Leong, C. A. Schenck, J. P. Lloyd, M. D. Lehti-Shiu, R. L. Last, E. Pichersky and S. H. Shiu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2344–2353 CrossRef CAS PubMed.
- A. Rai and K. Saito, Curr. Opin. Biotechnol., 2016, 37, 127–134 CrossRef CAS PubMed.
- L. M. Dersch, V. Beckers and C. Wittmann, Metab. Eng., 2016, 34, 1–24 CrossRef CAS PubMed.
- T. J. Clark, L. Guo, J. Morgan and J. Schwender, Annu. Rev. Plant Biol., 2020, 71, 303–326 CrossRef CAS PubMed.
- C. G. Dal'Molin, L. E. Quek, R. W. Palfreyman, S. M. Brumbley and L. K. Nielsen, Plant Physiol., 2010, 154, 1871–1885 CrossRef PubMed.
- S. M. D. Seaver, C. Lerma-Ortiz, N. Conrad, A. Mikaili, A. Sreedasyam, A. D. Hanson and C. S. Henry, Plant J., 2018, 95, 1102–1113 CrossRef CAS PubMed.
- K. Dreher, Methods Mol. Biol., 2014, 1083, 151–171 CrossRef CAS PubMed.
- S. M. Seaver, S. Gerdes, O. Frelin, C. Lerma-Ortiz, L. M. Bradbury, R. Zallot, G. Hasnain, T. D. Niehaus, B. El Yacoubi, S. Pasternak, R. Olson, G. Pusch, R. Overbeek, R. Stevens, V. de Crecy-Lagard, D. Ware, A. D. Hanson and C. S. Henry, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9645–9650 CrossRef CAS PubMed.
- G. Zampieri, S. Vijayakumar, E. Yaneske and C. Angione, PLoS Comput. Biol., 2019, 15, e1007084 CrossRef PubMed.
- D. M. Camacho, K. M. Collins, R. K. Powers, J. C. Costello and J. J. Collins, Cell, 2018, 173, 1581–1592 CrossRef CAS PubMed.
- N. Zhou, Y. Jiang, T. R. Bergquist, A. J. Lee, B. Z. Kacsoh, A. W. Crocker, K. A. Lewis, G. Georghiou, H. N. Nguyen, M. N. Hamid, L. Davis, T. Dogan, V. Atalay, A. S. Rifaioglu, A. Dalkiran, R. Cetin Atalay, C. Zhang, R. L. Hurto, P. L. Freddolino, Y. Zhang, P. Bhat, F. Supek, J. M. Fernandez, B. Gemovic, V. R. Perovic, R. S. Davidovic, N. Sumonja, N. Veljkovic, E. Asgari, M. R. K. Mofrad, G. Profiti, C. Savojardo, P. L. Martelli, R. Casadio, F. Boecker, H. Schoof, I. Kahanda, N. Thurlby, A. C. McHardy, A. Renaux, R. Saidi, J. Gough, A. A. Freitas, M. Antczak, F. Fabris, M. N. Wass, J. Hou, J. Cheng, Z. Wang, A. E. Romero, A. Paccanaro, H. Yang, T. Goldberg, C. Zhao, L. Holm, P. Toronen, A. J. Medlar, E. Zosa, I. Borukhov, I. Novikov, A. Wilkins, O. Lichtarge, P. H. Chi, W. C. Tseng, M. Linial, P. W. Rose, C. Dessimoz, V. Vidulin, S. Dzeroski, I. Sillitoe, S. Das, J. G. Lees, D. T. Jones, C. Wan, D. Cozzetto, R. Fa, M. Torres, A. Warwick Vesztrocy, J. M. Rodriguez, M. L. Tress, M. Frasca, M. Notaro, G. Grossi, A. Petrini, M. Re, G. Valentini, M. Mesiti, D. B. Roche, J. Reeb, D. W. Ritchie, S. Aridhi, S. Z. Alborzi, M. D. Devignes, D. C. E. Koo, R. Bonneau, V. Gligorijevic, M. Barot, H. Fang, S. Toppo, E. Lavezzo, M. Falda, M. Berselli, S. C. E. Tosatto, M. Carraro, D. Piovesan, H. Ur Rehman, Q. Mao, S. Zhang, S. Vucetic, G. S. Black, D. Jo, E. Suh, J. B. Dayton, D. J. Larsen, A. R. Omdahl, L. J. McGuffin, D. A. Brackenridge, P. C. Babbitt, J. M. Yunes, P. Fontana, F. Zhang, S. Zhu, R. You, Z. Zhang, S. Dai, S. Yao, W. Tian, R. Cao, C. Chandler, M. Amezola, D. Johnson, J. M. Chang, W. H. Liao, Y. W. Liu, S. Pascarelli, Y. Frank, R. Hoehndorf, M. Kulmanov, I. Boudellioua, G. Politano, S. Di Carlo, A. Benso, K. Hakala, F. Ginter, F. Mehryary, S. Kaewphan, J. Bjorne, H. Moen, M. E. E. Tolvanen, T. Salakoski, D. Kihara, A. Jain, T. Smuc, A. Altenhoff, A. Ben-Hur, B. Rost, S. E. Brenner, C. A. Orengo, C. J. Jeffery, G. Bosco, D. A. Hogan, M. J. Martin, C. O'Donovan, S. D. Mooney, C. S. Greene, P. Radivojac and I. Friedberg, Genome Biol., 2019, 20, 244 CrossRef CAS PubMed.
- A. Rai, M. Rai, H. Kamochi, T. Mori, R. Nakabayashi, M. Nakamura, H. Suzuki, K. Saito and M. Yamazaki, DNA Res., 2020, 27 Search PubMed.
- L. Sun, A. Rai, M. Rai, M. Nakamura, N. Kawano, K. Yoshimatsu, H. Suzuki, N. Kawahara, K. Saito and M. Yamazaki, J. Nat. Med., 2018, 72, 867–881 CrossRef CAS PubMed.
- S. Lundberg and S. I. Lee, Advances in Neural Information Processing Systems, 2017, abs/1705.07874 Search PubMed.
- A. Shrikumar, P. Greenside and A. Kundaje, Presented in part at the Proceedings of the 34th International Conference on Machine Learning, Proceedings of Machine Learning Research, 2017 Search PubMed.
- M. C. Thomas, T. W. Mitchell, D. G. Harman, J. M. Deeley, J. R. Nealon and S. J. Blanksby, Anal. Chem., 2008, 80, 303–311 CrossRef CAS PubMed.
- J. Zhao, X. B. Xie, Q. H. Lin, X. X. Ma, P. Su and Y. Xia, Anal. Chem., 2020, 92, 13470–13477 CrossRef CAS PubMed.
- L. Chong, R. Tian, R. Shi, Z. Ouyang and Y. Xia, Front. Chem., 2019, 7, 807 CrossRef CAS PubMed.
- W. Cao, S. Cheng, J. Yang, J. Feng, W. Zhang, Z. Li, Q. Chen, Y. Xia, Z. Ouyang and X. Ma, Nat. Commun., 2020, 11, 375 CrossRef CAS PubMed.
- T. H. Kuo, H. H. Chung, H. Y. Chang, C. W. Lin, M. Y. Wang, T. L. Shen and C. C. Hsu, Anal. Chem., 2019, 91, 11905–11915 CrossRef CAS PubMed.
- H. Takahashi, Y. Shimabukuro, D. Asakawa, S. Yamauchi, S. Sekiya, S. Iwamoto, M. Wada and K. Tanaka, Anal. Chem., 2018, 90, 7230–7238 CrossRef CAS PubMed.
- T. Baba, J. L. Campbell, J. C. Y. Le Blanc, P. R. S. Baker and K. Ikeda, J. Lipid Res., 2018, 59, 910–919 CrossRef CAS PubMed.
- J. S. Brodbelt, L. J. Morrison and I. Santos, Chem. Rev., 2020, 120, 3328–3380 CrossRef CAS PubMed.
- F. Meier, A. D. Brunner, S. Koch, H. Koch, M. Lubeck, M. Krause, N. Goedecke, J. Decker, T. Kosinski, M. A. Park, N. Bache, O. Hoerning, J. Cox, O. Rather and M. Mann, Mol. Cell. Proteomics, 2018, 17, 2534–2545 CrossRef CAS PubMed.
- J. Soltwisch, B. Heijs, A. Koch, S. Vens-Cappell, J. Hohndorf and K. Dreisewerd, Anal. Chem., 2020, 92, 8697–8703 CrossRef CAS PubMed.
- A. Palmer, P. Phapale, I. Chernyavsky, R. Lavigne, D. Fay, A. Tarasov, V. Kovalev, J. Fuchser, S. Nikolenko, C. Pineau, M. Becker and T. Alexandrov, Nat. Methods, 2017, 14, 57–60 CrossRef CAS PubMed.
- T. Schramm, A. Hester, I. Klinkert, J. P. Both, R. M. A. Heeren, A. Brunelle, O. Laprevote, N. Desbenoit, M. F. Robbe, M. Stoeckli, B. Spengler and A. Rompp, J. Proteomics, 2012, 75, 5106–5110 CrossRef CAS PubMed.
- S. Maniatis, T. Aijo, S. Vickovic, C. Braine, K. Kang, A. Mollbrink, D. Fagegaltier, Z. Andrusivova, S. Saarenpaa, G. Saiz-Castro, M. Cuevas, A. Watters, J. Lundeberg, R. Bonneau and H. Phatnani, Science, 2019, 364, 89–93 CrossRef CAS PubMed.
- P. L. Stahl, F. Salmen, S. Vickovic, A. Lundmark, J. F. Navarro, J. Magnusson, S. Giacomello, M. Asp, J. O. Westholm, M. Huss, A. Mollbrink, S. Linnarsson, S. Codeluppi, A. Borg, F. Ponten, P. I. Costea, P. Sahlen, J. Mulder, O. Bergmann, J. Lundeberg and J. Frisen, Science, 2016, 353, 78–82 CrossRef CAS PubMed.
- R. R. Stickels, E. Murray, P. Kumar, J. L. Li, J. L. Marshall, D. J. Di Bella, P. Arlotta, E. Z. Macosko and F. Chen, Nat. Biotechnol., 2021, 39, 313–319 CrossRef CAS PubMed.
- M. Honda, S. Oki, R. Kimura, A. Harada, K. Maehara, K. Tanaka, C. Meno and Y. Ohkawa, Nat. Commun., 2021, 12, 4416 CrossRef CAS PubMed.
- I. Paul, C. White, I. Turcinovic and A. Emili, FEBS J., 2021 DOI:10.1111/febs.15685.
- W. Timp and G. Timp, Sci. Adv., 2020, 6, eaax8978 CrossRef CAS PubMed.
- E. Lundberg and G. H. H. Borner, Nat. Rev. Mol. Cell Biol., 2019, 20, 285–302 CrossRef CAS PubMed.
- V. Pareek, H. Tian, N. Winograd and S. J. Benkovic, Science, 2020, 368, 283–290 CrossRef CAS PubMed.
- X. Liu, S. R. Wu, J. Xu, C. Sui and J. H. Wei, Acta Pharm. Sin. B, 2017, 7, 292–302 CrossRef PubMed.
- K. B. Kang, M. Ernst, J. J. J. van der Hooft, R. R. da Silva, J. Park, M. H. Medema, S. H. Sung and P. C. Dorrestein, Plant J, 2019, 98, 1134–1144 CAS.
- S. Rogers, C. W. Ong, J. Wandy, M. Ernst, L. Ridder and J. J. J. van der Hooft, Faraday Discuss, 2019, 218, 284–302 RSC.
- Z. Zhu, G. Huang, J. Deng, Y. Ye, J. Huang, X. Chen, J. Zhu, T. Yang, J. Lu, D. Du and J. Zhou, arXiv, 2103, 04098, 2021.
- K. Duhrkop, H. Shen, M. Meusel, J. Rousu and S. Bocker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12580–12585 CrossRef PubMed.
- S. Bocker and K. Duhrkop, J. Cheminf., 2016, 8, 5 Search PubMed.
- J. Hartler, A. Triebl, A. Ziegl, M. Trotzmuller, G. N. Rechberger, O. A. Zeleznik, K. A. Zierler, F. Torta, A. Cazenave-Gassiot, M. R. Wenk, A. Fauland, C. E. Wheelock, A. M. Armando, O. Quehenberger, Q. Zhang, M. J. O. Wakelam, G. Haemmerle, F. Spener, H. C. Kofeler and G. G. Thallinger, Nat. Methods, 2017, 14, 1171–1174 CrossRef CAS PubMed.
- S. Gessulat, T. Schmidt, D. P. Zolg, P. Samaras, K. Schnatbaum, J. Zerweck, T. Knaute, J. Rechenberger, B. Delanghe, A. Huhmer, U. Reimer, H. C. Ehrlich, S. Aiche, B. Kuster and M. Wilhelm, Nat. Methods, 2019, 16, 509–518 CrossRef CAS PubMed.
- M. Sud, E. Fahy, D. Cotter, K. Azam, I. Vadivelu, C. Burant, A. Edison, O. Fiehn, R. Higashi, K. S. Nair, S. Sumner and S. Subramaniam, Nucleic Acids Res., 2016, 44, D463–D470 CrossRef CAS PubMed.
- N. S. Kale, K. Haug, P. Conesa, K. Jayseelan, P. Moreno, P. Rocca-Serra, V. C. Nainala, R. A. Spicer, M. Williams, X. Li, R. M. Salek, J. L. Griffin and C. Steinbeck, Curr. Protoc. Bioinf., 2016, 53, 14 13 11–14 13 18 Search PubMed.
- S. G. Oliver, M. K. Winson, D. B. Kell and F. Baganz, Trends Biotechnol., 1998, 16, 373–378 CrossRef CAS PubMed.
- J. Luo, Curr. Opin. Plant Biol., 2015, 24, 31–38 CrossRef CAS PubMed.
- M. L. Slaten, A. Yobi, C. Bagaza, Y. O. Chan, V. Shrestha, S. Holden, E. Katz, C. Kanstrup, A. E. Lipka, D. J. Kliebenstein, H. H. Nour-Eldin and R. Angelovici, Plant Physiol., 2020, 183, 483–500 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1np00014d |
| This journal is © The Royal Society of Chemistry 2021 |