
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe application potential of machine learning and genomics for understanding natural product diversity, chemistry, and therapeutic translatability†
David
Prihoda‡
 ab,
Julia M.
Maritz‡
c,
Ondrej
Klempir
a,
David
Dzamba
a,
Christopher H.
Woelk
c,
Daria J.
Hazuda
c,
Danny A.
Bitton
a and
Geoffrey D.
Hannigan
*c
ab,
Julia M.
Maritz‡
c,
Ondrej
Klempir
a,
David
Dzamba
a,
Christopher H.
Woelk
c,
Daria J.
Hazuda
c,
Danny A.
Bitton
a and
Geoffrey D.
Hannigan
*c
aR&D Informatics Solutions, MSD Czech Republic s.r.o., Prague, Czech Republic
bDepartment of Informatics and Chemistry, Faculty of Chemical Technology, University of Chemistry and Technology, Prague, Czech Republic
cExploratory Science Center, Merck & Co., Inc., Cambridge, MA, USA. E-mail: geoffrey.hannigan@merck.com
First published on 27th November 2020
Abstract
Covering: up to the end of 2020.
The machine learning field can be defined as the study and application of algorithms that perform classification and prediction tasks through pattern recognition instead of explicitly defined rules. Among other areas, machine learning has excelled in natural language processing. As such methods have excelled at understanding written languages (e.g. English), they are also being applied to biological problems to better understand the “genomic language”. In this review we focus on recent advances in applying machine learning to natural products and genomics, and how those advances are improving our understanding of natural product biology, chemistry, and drug discovery. We discuss machine learning applications in genome mining (identifying biosynthetic signatures in genomic data), predictions of what structures will be created from those genomic signatures, and the types of activity we might expect from those molecules. We further explore the application of these approaches to data derived from complex microbiomes, with a focus on the human microbiome. We also review challenges in leveraging machine learning approaches in the field, and how the availability of other “omics” data layers provides value. Finally, we provide insights into the challenges associated with interpreting machine learning models and the underlying biology and promises of applying machine learning to natural product drug discovery. We believe that the application of machine learning methods to natural product research is poised to accelerate the identification of new molecular entities that may be used to treat a variety of disease indications.
1. Introduction
Natural products have long been a rich source of bioactive entities that enabled drug discovery. Natural products can be broadly defined as chemical entities that are produced by a living organism, and these often take the form of primary metabolites (those required for life, or unique to a subset of life/conditions) and secondary metabolites (those that are advantageous but not required; these are the focus of this review). Natural products represent a source of therapeutic opportunity and microbial mechanistic insights. In recent years, the natural product field has benefitted from contributions from the machine learning (ML) field, both chemically and biologically. In this review we will discuss how ML advances are being applied to natural products and natural product drug discovery. We will focus on how ML is being used to understand biological and chemical “languages” (i.e. genomes and molecular structures) and how this uniquely informs insights into natural product chemistry and diversity. We will further review how genome mining and related techniques are being used to understand links between microbiome natural products and human health. We will discuss how the identification of disease-associated novel metabolites and/or targets from the microbiome show promise in empowering pharmaceutical drug discovery.2. Relevant advances in machine learning
2.1 Recent advances in machine learning
ML is a long-established field enjoying a resurgence of interest and activity in recent years. The ML field can be defined as the study and application of algorithms that perform prediction or exploration tasks through pattern recognition instead of explicitly defined rules.1 Such algorithms may be further classified into groups such as supervised and unsupervised learning, with the former referring to algorithms that use and map data to known groups for classification, and the latter referring to algorithms that do not use pre-defined or known groups (e.g. “clustering” samples by similarity). Algorithms can, for example, be further classified into parametric algorithms (those that make assumptions about the data distribution) and non-parametric algorithms (those that do not make distribution assumptions). Overall ML is a rich field, and for a more complete review of ML background and concepts we recommend Tarca et al.2ML's current surge in activity is largely the result of increasingly powerful computational resources and the availability of large datasets. One primary motivation for using ML techniques is the performance scalability offered by the ability of the algorithms to enhance themselves as they adjust and “learn” from datasets. Such approaches also allow their users to extract useful information from large, complex datasets at scale.
The ML field has continued to grow in its breadth of applications and approaches. Some impactful ML applications have included speech and image recognition/classification,3–5 media recommendation engines,6,7 and geographical mapping and navigation.8 In these applications, ML is leveraged as a way to gain valuable information from very complex data types and very large datasets. These applications, in which ML is used to gain signal from complex data, have also extended into the field of natural product biology and chemistry. In biology and chemistry, ML can leverage its propensity for complex pattern recognition to gain novel insights into genomic signatures, chemical activities, compound diversity, and therapeutic associations.
2.2 Advances in deep learning
Deep Learning (DL) is a sub-discipline of ML that is increasingly relevant to natural product biology and chemistry. DL refers to the area of ML that utilizes deeply-layered neural networks that conceptually behave like the networks of neurons found within the human brain. DL approaches have been gaining popularity, powering many methods relevant to computational biology including sequence alignment,9 protein structure prediction,10,11 and decoding of splicing signals.12,13 DL has also been applied in chemistry, such as informing high throughput screening (HTS), Quantitative Structure Activity Relationship (QSAR) analyses, and others.14–16One important application of biological and chemical DL is in understanding natural product diversity and chemistry. This is especially evident in the applications of DL natural language processing (NLP) methods. To date, the majority of NLP applications have been centered around understanding spoken and written languages, although scientists are increasingly re-purposing these techniques to provide an understanding of genomic and molecular “languages”. Much like NLP approaches can be used to represent and define words by their sentence context, scientists are using NLP to represent and define genomic elements, such as genes, by their genomic context.10,17,18 Likewise NLP approaches are being applied to represent and define molecules and fragments as new mathematical structures; an approach that provides new insights and analytical opportunities for understanding chemical relationships and activities.19,20 When applied to natural product chemistry and genome mining, these and other DL and NLP approaches are providing new insights into natural product diversity, chemical properties, and therapeutic potential. Below we pursue a more detailed discussion of these natural product insights, as they are empowered by DL, NLP, and other ML approaches.
3. Understanding natural product chemistry through ML & genome mining
A key approach to understanding natural product chemistry and biology is through understanding the genomes in which their synthesis pathways are encoded. Natural products are largely the products of microbial biosynthesis, whose processes are encoded by biosynthetic gene clusters (BGCs) within their genomes. A BGC is a group of genes in close genomic proximity, which together orchestrate the synthesis of secondary metabolites via a complex pathway of enzymatic reactions and regulatory switches. In addition to advances in traditional molecular biology methods (e.g. heterologous expression and knock-out studies), the advent of next generation sequencing technologies and the increasing abundance of complete microbial genomes, ML genome mining approaches have provided a profound opportunity to understand natural product chemistry and diversity through their BGC “genomic language”. The process of identifying BGCs with ML techniques can be broken down into a series of biological and chemical steps, which we define as annotation, feature representation, BGC identification, structural prediction, and activity profiling (Fig. 1).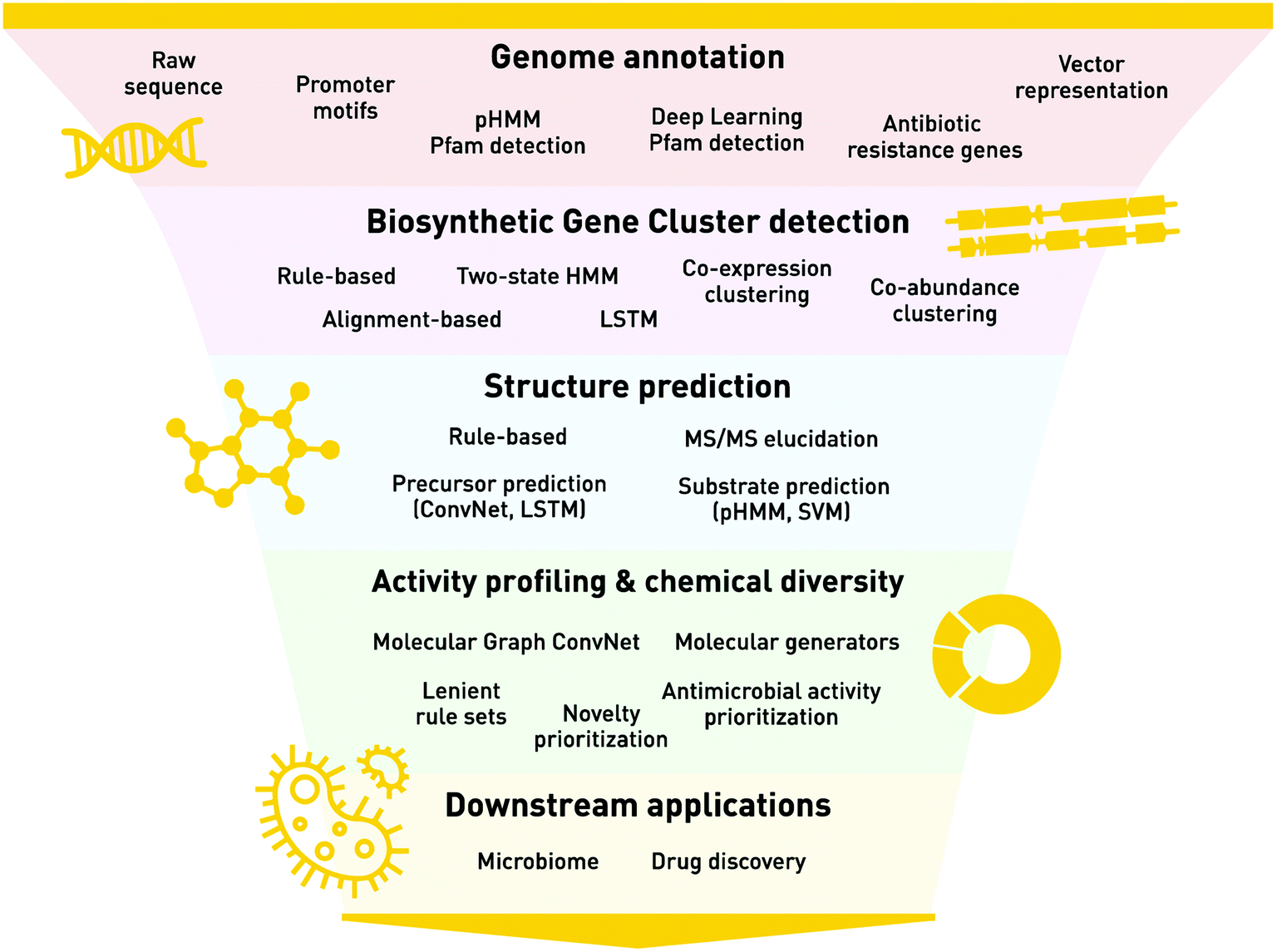 | ||
| Fig. 1 The biological and chemical steps for identifying BGCs with ML techniques. This flow chart highlights the steps of genome annotation (e.g. how we represent genomes, using values such as DNA sequences), biosynthetic gene cluster detection from genomes, structure prediction of the natural product from a given biosynthetic gene cluster, activity profiling and chemical diversity of the natural products, and the downstream applications including microbiome studies and drug discovery. Each section lists the important high-level methods used, including the machine learning applications. Abbreviations are as follows: pHMM = profile Hidden Markov Model, HMM = Hidden Markov Model, Pfam = Protein Family, LSTM = Long Short-Term Memory Network, MS/MS = Tandem Mass Spectrometry, SVM = Support Vector Machine. A detailed reference of biosynthetic gene clusters & natural product prediction and analysis methods is provided in ESI Table 1.† | ||
3.1 Genome annotation
The first step of the natural product genome mining process, genome annotation, can include representing the genome as a data string as simple as the raw DNA or amino acid sequence of the genome, or can include more sophisticated information such as genes. An example of a major leap beyond these genomic annotations, such as raw DNA sequences, was the adoption of profile Hidden Markov Models21,22 (pHMMs) which provide a high-level feature representation by annotating conserved functional subunits of proteins. Sets of pHMMs curated by the first BGC detection approaches were greatly supplemented by databases such as Pfam23 or CATH.24 While future approaches will likely benefit from improved speed and accuracy of protein domain detection using DL methods [UniLanguage, Google ProtCNN], pHMMs remain a foundation of feature annotation for many BGC detection methods.After genomes are annotated, they can further be represented as naïve or pre-trained numeric vectors using DL and NLP approaches. Vector representation approaches can provide unique functional insights into the input feature space as well as improved downstream performance. The utility of this approach was recently highlighted by the tools Pfam2Vec, Mod2Vec, and others,25–27 where protein family domains (Pfams) and functional modules were converted to numeric vectors based on their genomic context. Many other vectorization methods are directly influenced by (and in some cases are derivatives of) the revolutionary Word2Vec algorithm, although many have yet to be incorporated into the BGC space.28 Biological interpretations of Word2Vec include DNA2Vec (DNA vectorization),29 Gene2Vec (gene vectorization),18 and ProtVec (protein and protein family vectorization).30 While these latter vector-based representation approaches are not currently being leveraged for natural products, we expect them to be used more in coming years.
3.2 Biosynthetic gene cluster detection
Once a genome has been annotated and potentially converted to numeric vectors, BGCs are identified. Early approaches employed relatively simple intergenic distance thresholds on top of BLAST or profile Hidden Markov Model (pHMM) hits to detect biosynthetic areas of interest (NP.searcher,21 SMURF22). This approach was extended by two commonly used BGC detection methods PRISM31 and antiSMASH,32 which devised an extendable framework of rules for the detection of numerous biosynthetic product classes based on presence and absence of specific protein domains. While the predefined rules in PRISM and antiSMASH improved performance, they are by definition not ML approaches because many aspects are hard coded.As a supplement to rule-based methods such as PRISM and antiSMASH, ML approaches have been introduced with the goal of detecting novel BGCs independently of known biosynthesis mechanisms. ClusterFinder33 uses a second two-state HMM on top of a chain of pHMM-detected Pfam tokens, where each Pfam token contributes a specific positive or negative weight to BGC or non-BGC likeness. This approach was further supplemented by DeepBGC,26 which used a bi-directional Long Short-Term Memory (LSTM) network fed with a sequence of Pfam tokens represented as meaningful vectors (Pfam2Vec) generated by pre-training on a corpus of bacterial genomes. While these approaches have offered some analytical advantages (i.e. potential to identify completely novel BGCs and thus NPs), it is important to note that they do have higher false positive rates than many rule-based approaches. Other notable methods use promoter motif hits or expression microarrays in fungi and RNA sequencing in plants, including CASSIS34 and FunGeneClusterS35 for detection and PlantClusterFinder36 for validation. Building upon this idea, MetaBGC employed co-abundance clustering to group biosynthetic short reads into putative BGCs.37
3.3 Chemical structure prediction
After BGCs have been identified by these genome mining strategies, BGC information can be used to better understand natural product chemistry. Predicting natural product structure from a genome sequence is a daunting challenge that still requires development, however some approaches are being utilized to tackle this. AntiSMASH and PRISM are tools that use a variety of curated rules to predict structural scaffolds of natural products from BGCs.31 Other tools focus on single BGC classes as ways to predict natural product structures from precursors, such as DeepRiPP (which relies on rule-based RiPP-Prism instead of ML),38 BAGEL (which relies on database matches to inform potential structures),39 RODEO40 and RiPPMiner (which performs ML predictions but is based on a relatively small dataset that may be prone to overfitting)41 for ribosomally synthesized, post-translationally modified peptide (RiPP) precursor peptides. SANDPUMA42 is a tool for non-ribosomal peptide-synthetase (NRPS) substrate specificity, although its ML approach can have some shortcomings in that it’s NRPS domain substrate training set can yield erroneous predictions. Another important tool, NRPSpredictor,43 largely pioneered the use of support vector machines to predict NRPS substrate specificity. Because structural prediction from BGCs is complex, BGC product structure is often elucidated using experimental techniques such as heterologous expression, purification, and accompanied MS/MS.44,45 Metabolomics approaches, in which targeted and untargeted MS/MS profiles are taken, is also an increasingly useful approach used for structural elucidation.3.4 Activity profiling & chemical diversity
In studies where BGC-derived natural product structures are elucidated, many ML techniques are providing further information around activity, mechanistic targets, toxicity, and other features. Graph neural networks46 are promising tools for working with chemical structures, and can be applied to antibiotic discovery16 or predicting drug–target interactions.47 Deep neural networks can also be used for generation of drugs, natural products and metabolites, for example with the tool MOSES.48 Together these advances are allowing us to better understand natural product biology and chemistry.There are many ML opportunities for analyzing genomic and chemical diversity of predicted BGCs and their natural products. At the genomic level, BGCs can be clustered and compared to their putative orthologs, for example using the BiG-SCAPE/CORASON toolkit.49 On the chemical level, putative RiPP precursor structures can be aligned to a database of known chemical structures in order to assign a novelty index and dereplicate known products, as implemented in BARLEY.38 Finally, natural products can be prioritized using their predicted activity. Leveraging the fact that antibiotic BGCs often contain a second resistant copy of their antibiotic target gene, ARTS50 performs resistant target gene genome mining for antibiotics with novel targets.
While some ML methods provide scaffold predictions and other “clues” to natural product structures, the promise of discovering accurate yet novel chemistry remains elusive. Rule-based approaches still remain the backbone of BGC detection and natural product chemistry research, as they create a community-driven feed-forward discovery loop by continuous extension of their rule sets based on novel insights gained from applying them to unexplored genomes and communities. Since version 5.1.12, antiSMASH has also allowed for the detection of putative BGCs using more lenient rule sets. However, the prospect of detecting a whole novel subclass of natural products requires a more data-driven approach. In our view, the ultimate limitation of current approaches is the fact that they use the same underlying protein functional annotation methods such as pHMMs (SANDPUMA, RRE-Finder51), they do not pre-process the gene sequences features (NeuRiPP, NRPSPredictor43) or take an intermediate approach (DeepBGC). Contrastingly, machine translation, speech recognition, and image processing applications have shown that the true potential of DL does not lie in a single-layer architecture of aggregated hand-crafted features, but rather in a comprehensive multi-layered architecture. Such multi-layered architectures utilize low-levels to ingest a raw genomic sequence through layers pre-trained on available data, as well as final layers that predict the desired target. Finally, irrespective of detection methods, validation with unseen datasets is needed in the field, instead of validation using random splits which fail to account for the highly conserved nature of BGCs. For example, this was illustrated by NeuRiPP's and DeepBGC's ability to re-discover biosynthetic classes hidden during training.26,52
3.5 Data availability for BGC and NP ML models
A critical aspect of all ML methods discussed above is data, as the choice of ML algorithm is associated with data quality and quantity and different kinds of data are suitable for different applications. For example, if one wanted to use a supervised learning method to predict BGCs or the bioactivity of NPs one would need a high-quality validated database such as MIBiG or one of the ones summarized in the recent review.53 Unfortunately, the lack of large quantities of high-quality data and standardized databased for ML models are a great challenge for the field.4. Exploring the natural product landscape of the human microbiome
4.1 Natural products of the environmental microbiome
The natural environment has long been a source of natural products and anti-microbial compounds. In particular, soil and marine microorganisms have been the source of numerous important natural products.54–56 Traditionally, the discovery process has required the collection of soil and marine microorganisms, followed by culturing, extracting, and then screening compounds from these cultures. Although the majority of organisms in these environments are not culturable, several antibiotics have been discovered without any knowledge of the enzymes involved in the biosynthesis or the corresponding genes (for review, see ref. 57). These traditional, top-down approaches have been complemented by more recent bottom-up approaches, which leverage advances in genomics and bioinformatics, such as high throughput sequencing, genome mining, and ML to identify and activate gene clusters in the host organism.584.2 Understanding natural products of the human microbiome
The scalability of microbial mining advances described above have begun being used in the complex human microbiome genomics systems. The ClusterFinder algorithm has been used to identify >14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 putative small-molecule encoding BGCs in human-associated bacterial genomes, most of which were from the oligosaccharide and the RiPP natural product classes.45 One sub-class of RiPPs, thiopeptides, were found to be widely distributed in the genomes and metagenomes of human gut, oral and vaginal microbiota.45 Experimental interrogation of this group of BGCs revealed a novel antibiotic with activity against several Gram-positive vaginal pathogens.45 Another tool, MetaBGC, has been applied to metagenomic data to identify type II polyketide synthases from gut, skin and oral samples.37 Although some of these gene clusters are associated with particular small molecules of known/predicted activity, the vast majority of these gene clusters remain uncharacterized.59 Despite the recent advances in the search for natural products in the human microbiome, the field is currently facing several challenges, such as how to prioritize the numerous identified and predicted BGCs for further discovery, or how to establish whether these microbes actually produce particular secondary metabolites in humans.60
000 putative small-molecule encoding BGCs in human-associated bacterial genomes, most of which were from the oligosaccharide and the RiPP natural product classes.45 One sub-class of RiPPs, thiopeptides, were found to be widely distributed in the genomes and metagenomes of human gut, oral and vaginal microbiota.45 Experimental interrogation of this group of BGCs revealed a novel antibiotic with activity against several Gram-positive vaginal pathogens.45 Another tool, MetaBGC, has been applied to metagenomic data to identify type II polyketide synthases from gut, skin and oral samples.37 Although some of these gene clusters are associated with particular small molecules of known/predicted activity, the vast majority of these gene clusters remain uncharacterized.59 Despite the recent advances in the search for natural products in the human microbiome, the field is currently facing several challenges, such as how to prioritize the numerous identified and predicted BGCs for further discovery, or how to establish whether these microbes actually produce particular secondary metabolites in humans.60
Although the majority of BGCs and natural products in the human microbiome remain uncharacterized, studies are now beginning to associate BGCs with disease states, potentially leading to discoveries of novel natural products, novel druggable targets, novel lead compounds, and other important drug discovery advancements. By mapping metagenomic data to databases of known BGCs (such as MIBiG61 or IMG-ABC) or using tools (such as antiSMASH, DeepBGC or MetaBGC) to predict novel BGCs from metagenomic data we can begin to describe the BGC landscape of a given population. Differential abundance and/or ML algorithms can then be applied to link BGCs to disease phenotypes. For example, a recent paper used a combination of differential abundance testing and random forest classifiers to identify 43 BGCs found in the human gut that could discriminate patients with Parkinson's disease from healthy controls.62 Other recent work has used similar methods to identify over a thousand oral BGCs that were differentially represented between healthy subjects, dental caries and periodontitis.63
4.3 Methodological challenges & opportunities
The detection of BGCs and their products in human and environmental data have been pursued by diverse bioinformatic strategies. While these novel bioinformatic methods are becoming more popular and offer promising steps forward, they suffer from several challenges including a lack of large publicly available datasets, lack of dataset-associated metadata, shallow sequencing depth, small sample sizes, and an widespread inability to validate findings across multiple studies (largely due to the aforementioned limitations). Another limitation is the availability of sequenced bacterial genomes from diverse samples as many tools rely on these to predict the initial sets of BGCs. However as larger, deeper, and more diverse metagenomic datasets become available, and other tools like metagenome assemblers become more precise, our ability to leverage ML approaches in this field will improve.Finally, even though these methods allow the identification of BGC signatures that are associated with a particular disease, determining if the BGC of interest is expressed and what metabolite/small molecule it produces remain difficult, as discussed above. The incorporation of other ‘omics’ technologies can help resolve these issues. Comparative transcriptomics of cultured bacterial strains from the human microbiome can be used to determine which BGCs are transcriptionally active.44 However, as most bacteria in the human microbiome are unculturable, other methods, such as metatranscriptomics, will be required to determine BGCs that are active and differentially expressed between healthy and diseased microbiomes. Other technologies such as metabolomics63 or synthetic biology64 can be combined with these approaches to help associate BGCs of interest to the small molecules they encode. Lastly, the application of long-read sequencing not only to bacterial genomes from the human microbiome but also to the microbiome samples themselves offers the ability to increase the sensitivity of BGC prediction and annotation using ML tools.65
5. Translating machine learning to natural product drug discovery
An exciting aspect of applying ML to genomics and natural products is the promise of empowering new avenues of drug discovery. With the FDA approval of nine natural product-based drugs in 2019 alone, natural products continue to be a fruitful source for small molecule drug discovery and target identification.66 Despite this, classic screening approaches can suffer from inefficiencies, such as redundant discoveries, which can yield low returns on investment. In the context of the microbiome, BGCs and their natural products provide unique insights into potential underlying mechanisms of microbe–host and microbe–microbe interactions, and indeed many natural products have been associated with therapeutically relevant activities including antibiotic activity66 and immunomodulatory activity.67,68 By elucidating mechanisms and the underlying chemistry by which microbiome metabolic products interact with human cells in disease states, researchers are better able to design small molecule screens and identify putative targets and drug candidates, which could lead to better therapies. These approaches are enabling our ability to more completely understand the biosynthetic potential of organisms through their genomic languages, allowing us to more accurately predict structures and activity features of natural products such as toxicity, and equipping us to develop improved therapeutics by associating these natural products with disease phenotypes.5.1 Functional BGC screening for drug discovery
ML models and BGCs are becoming increasingly valuable in drug discovery, especially for target identification. At a high level, traditional small molecule drug discovery is a process in which therapeutic targets (e.g. receptors in a pathway) are identified and “targeted” with a molecule (e.g. a receptor agonist) to modify a biological process and confer a therapeutic benefit. This process involves extensive experimentation and refinement before the final molecule is an acceptable drug candidate for clinical evaluation. In the context of BGCs and the microbiome, BGCs and their resulting natural products can be considered a starting point in which therapeutic targets (e.g. a receptor associated with a natural product) can be identified as promising, due to its association with disease states through data analysis (i.e. reverse translation) (Fig. 2). The analytically identified natural products can inform target identification and follow up experimentation can validate such in silico findings. Together this set of prediction and validation steps could be considered a “functional BGC” screen for targets that warrant further development.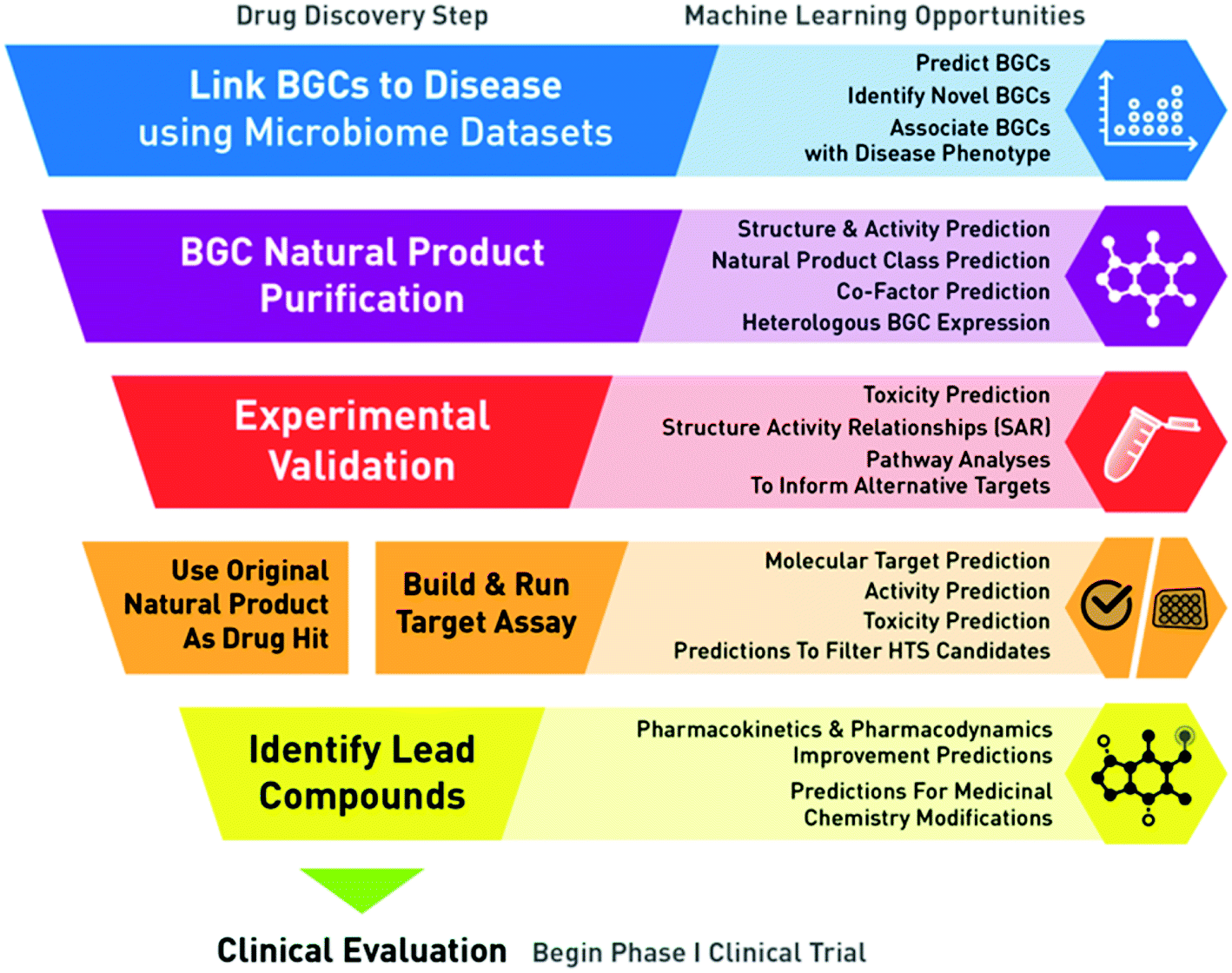 | ||
| Fig. 2 Machine learning applications within a “functional biosynthetic gene cluster screen” to facilitate drug discovery. This workflow outlines the process of taking a biosynthetic gene cluster (BGC) and resulting natural product to clinical evaluation as a lead compound. The process begins by identifying BGCs associated with disease states in microbiome datasets (blue). The BGC-derived natural product(s) are purified (purple) for use in experimental validation of whether the natural product is associated with the predicted disease phenotype (red). The validated natural product molecule can be carried forward as a drug hit, or if the natural product is not a reasonable drug hit, it can serve as a starting point for building and running target assays to identify appropriate molecule drug hits (orange). Finally the molecular hits are developed into lead compounds (yellow) to be tested in a clinical setting. This diagram highlights the high-level drug discovery steps of the process (left), as well as the areas of opportunity for machine learning to be applied and improved (right). | ||
Once a promising BGC-associated drug target has been identified (and further validated in vitro and in vivo), a high throughput screen (HTS) can be run to test millions of molecules against an assay designed to identify which molecules confer the desired activity associated with that target. The molecules that confer the desired therapeutic activity are considered “hits” whose activity and feasibility are then further optimized through medical chemistry. The final optimized molecule from this process is considered a lead, which is the best candidate for progressing into early-stage clinical trials for safety, and then efficacy.
As discussed above, one way in which ML can impact natural product drug discovery pipelines is by aiding in activity prediction, such as toxicity or potential for clinical advancement.16,26 The high throughput screening approach is very time consuming and expensive, and ML algorithms are beginning to enable more refined hit and lead identification by flagging chemicals that are potentially toxic, unstable, or likely to be ineffective in the clinic, which are based on their training data.16
5.2 Existing challenges & future opportunities for machine learning
One of the challenges with leveraging ML methods in natural product identification is the lack of interpretability. The ability of ML algorithms, especially DL algorithms, to measure signal from complex and high dimensional data makes them incredibly useful for finding new signals in data, but this comes at a cost. The complexity and dimensionality make it difficult to interpret what key features are predictive. In the context of BGC identification, this lack of interpretability can make it difficult or impossible to identify a single mechanism or target of interest. Said another way, there is often a tradeoff between model predictability and interpretability, where one comes at the cost of the other. When leveraging these techniques, it is becoming increasingly critical for scientists to balance the predictability and interpretability of their models, so as to achieve an outcome that meets their needs of either high predictive performance or interpretability of key predictive features. An important area of future development will be continued improvements in interpretable models (including components such as feature importance calculations), as well as continued effective integration of ML models with rule-based approaches, so as to improve scientific interpretability.Another challenge for leveraging ML with natural product identification is establishing the appropriate validation of findings. While ML techniques can guide our scientific exploration and development, experiments are still required to protect against false positive discoveries. Such validation will include in vitro activity assays and in vivo phenotypic studies. Just as it is important for the field to continue developing new and improved models, it is also important for us to continue working toward more effective and fit-for-purpose validation approaches (e.g. cell-based in vitro activity assays for validation screening).
Yet another challenge remains the lack of training data, especially in the natural product space which involved complex genomic and chemical information. This lack of data in turn limits researchers' abilities to build strong ML models and tools. An area of future development for the natural product field will likely include publications and resources of structured data such as BGCs, their natural products (including chemical structures), their RNA and protein expression conditions, any associated disease states, and their activity classes. As this information grows, we likewise expect to see improvements in ML space.
5.3 Future outlook
Overall, we expect ML and microbial genomics to continue playing important roles in drug discovery and development. By leveraging tools from other fields such as NLP, we are able to view our biological and chemical systems through a new lens, and glean new insights into the underlying systems. By applying ML techniques to biological experiments, we can predict which molecules might have therapeutic benefits. By using ML for tasks such as activity and toxicity prediction, we can make our drug discovery and development processes less resource intensive and more accurate. With this being said, we also clarify that ML and DL alone are not the only route forward for the natural product field, and diverse approaches (including rule-based methods and others) will continue to be critical for incorporating human expertise and addressing ML shortcomings such as high false positive rates due to limited training data and other issues. Together ML, genomics, and natural products have great potential for improving drug discovery and impacting human health.6. Conflicts of interest
The authors are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.7. Acknowledgements
We thank our colleagues Todd Black and Lee Roberts for discussions and feedback while preparing this work.8. References
- M. W. Libbrecht and W. S. Noble, Nat. Rev. Genet., 2015, 16, 321–332 CrossRef CAS.
- A. L. Tarca, V. J. Carey, X. W. Chen, R. Romero and S. Draghici, PLoS Comput. Biol., 2007, 3, e116 CrossRef.
- A. Graves, A.-r. Mohamed and G. E. Hinton, 2013 IEEE International Conference on Acoustics, Speech and Signal Processing, 2013, pp. 6645–6649 Search PubMed.
- A. Krizhevsky, I. Sutskever and G. E. Hinton, in Advances in Neural Information Processing Systems 25, ed. F. Pereira, C. J. C. Burges, L. Bottou and K. Q. Weinberger, Curran Associates, Inc., 2012, pp. 1097–1105 Search PubMed.
- M. Raghu and E. W. Schmidt, 2020, arXiv: abs/2003.11755.
- M. P. O'Mahony, P. Cunningham and B. Smyth, An Assessment of Machine Learning Techniques for Review Recommendation, Berlin, Heidelberg, 2010 Search PubMed.
- I. Portugal, P. Alencar and D. Cowan, Expert Syst. Appl., 2018, 97, 205–227 CrossRef.
- P. Mirowski, M. K. Grimes, M. Malinowski, K. M. Hermann, K. Anderson, D. Teplyashin, K. Simonyan, K. Kavukcuoglu, A. Zisserman and R. Hadsell, 2018, CoRR: abs/1804.00168.
- R. Jafari, M. M. Javidi and M. Kuchaki Rafsanjani, SN Appl. Sci., 2019, 1, 592 CrossRef.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Zidek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Nature, 2020, 577, 706–710 CrossRef CAS.
- M. Torrisi, G. Pollastri and Q. Le, Comput. Struct. Biotechnol. J., 2020, 18, 1301–1310 CrossRef CAS.
- K. Jaganathan, S. Kyriazopoulou Panagiotopoulou, J. F. McRae, S. F. Darbandi, D. Knowles, Y. I. Li, J. A. Kosmicki, J. Arbelaez, W. Cui, G. B. Schwartz, E. D. Chow, E. Kanterakis, H. Gao, A. Kia, S. Batzoglou, S. J. Sanders and K. K. Farh, Cell, 2019, 176, 535–548 CrossRef CAS.
- Y. Xu, Y. Wang, J. Luo, W. Zhao and X. Zhou, Nucleic Acids Res., 2017, 45, 12100–12112 CrossRef CAS.
- G. Gini, F. Zanoli, A. Gamba, G. Raitano and E. Benfenati, SAR QSAR Environ. Res., 2019, 30, 617–642 CrossRef CAS.
- S. Hu, P. Chen, P. Gu and B. Wang, IEEE J. Biomed. Health Inform., 2020, 10, 3020–3028 Search PubMed.
- J. M. Stokes, K. Yang, K. Swanson, W. Jin, A. Cubillos-Ruiz, N. M. Donghia, C. R. MacNair, S. French, L. A. Carfrae, Z. Bloom-Ackermann, V. M. Tran, A. Chiappino-Pepe, A. H. Badran, I. W. Andrews, E. J. Chory, G. M. Church, E. D. Brown, T. S. Jaakkola, R. Barzilay and J. J. Collins, Cell, 2020, 180, 688–702 CrossRef CAS.
- M. L. Bileschi, D. Belanger, D. Bryant, T. Sanderson, B. Carter, D. Sculley, M. A. DePristo and L. J. Colwell, 2019, bioRxiv: 626507, DOI:10.1101/626507.
- J. Du, P. Jia, Y. Dai, C. Tao, Z. Zhao and D. Zhi, BMC Genomics, 2019, 20, 82 CrossRef CAS.
- S. K. Chakravarti, ACS Omega, 2018, 3, 2825–2836 CrossRef CAS.
- H. Öztürk, A. Özgür, P. Schwaller, T. Laino and E. Ozkirimli, Drug Discovery Today, 2020, 25, 689–705 CrossRef.
- M. H. Li, P. M. Ung, J. Zajkowski, S. Garneau-Tsodikova and D. H. Sherman, BMC Bioinf., 2009, 10, 185 CrossRef.
- N. Khaldi, F. T. Seifuddin, G. Turner, D. Haft, W. C. Nierman, K. H. Wolfe and N. D. Fedorova, Fungal Genet. Biol., 2010, 47, 736–741 CrossRef CAS.
- S. El-Gebali, J. Mistry, A. Bateman, S. R. Eddy, A. Luciani, S. C. Potter, M. Qureshi, L. J. Richardson, G. A. Salazar, A. Smart, E. L. L. Sonnhammer, L. Hirsh, L. Paladin, D. Piovesan, S. C. E. Tosatto and R. D. Finn, Nucleic Acids Res., 2019, 47, D427–D432 CrossRef CAS.
- I. Sillitoe, N. Dawson, T. E. Lewis, S. Das, J. G. Lees, P. Ashford, A. Tolulope, H. M. Scholes, I. Senatorov, A. Bujan, F. Ceballos Rodriguez-Conde, B. Dowling, J. Thornton and C. A. Orengo, Nucleic Acids Res., 2018, 47, D280–D284 CrossRef.
- D. S. Chiriac, Genomic and Metabolic Guided Discovery of Bacterial Natural Products, MS thesis, Queen's University, 2019.
- G. D. Hannigan, D. Prihoda, A. Palicka, J. Soukup, O. Klempir, L. Rampula, J. Durcak, M. Wurst, J. Kotowski, D. Chang, R. Wang, G. Piizzi, G. Temesi, D. J. Hazuda, C. H. Woelk and D. A. Bitton, Nucleic Acids Res., 2019, 47, e110 CrossRef CAS.
- A. Viehweger, S. Krautwurst, D. H. Parks, B. König and M. Marz, 2019, bioRxiv: 524280, DOI:10.1101/524280.
- T. Mikolov, I. Sutskever, K. Chen, G. Corrado and J. Dean, 2013, CoRR: abs/1310.4546.
- P. Ng, 2017, arXiv: abs/1701.06279.
- E. Asgari and M. R. K. Mofrad, 2015, arXiv: abs/1503.05140.
- M. A. Skinnider, N. J. Merwin, C. W. Johnston and N. A. Magarvey, Nucleic Acids Res., 2017, 45, W49–W54 CrossRef CAS.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS.
- T. Wolf, V. Shelest, N. Nath and E. Shelest, Bioinformatics, 2016, 32, 1138–1143 CrossRef CAS.
- T. C. Vesth, J. Brandl and M. R. Andersen, Synth. Syst. Biotechnol., 2016, 1, 122–129 CrossRef.
- P. Schlapfer, P. Zhang, C. Wang, T. Kim, M. Banf, L. Chae, K. Dreher, A. K. Chavali, R. Nilo-Poyanco, T. Bernard, D. Kahn and S. Y. Rhee, Plant Physiol., 2017, 173, 2041–2059 CrossRef CAS.
- Y. Sugimoto, F. R. Camacho, S. Wang, P. Chankhamjon, A. Odabas, A. Biswas, P. D. Jeffrey and M. S. Donia, Science, 2019, 366, 1–11 CrossRef.
- N. J. Merwin, W. K. Mousa, C. A. Dejong, M. A. Skinnider, M. J. Cannon, H. Li, K. Dial, M. Gunabalasingam, C. Johnston and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 371–380 CrossRef CAS.
- A. J. van Heel, A. de Jong, C. Song, J. H. Viel, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2018, 46, W278–W281 CrossRef CAS.
- J. I. Tietz, C. J. Schwalen, P. S. Patel, T. Maxson, P. M. Blair, H. C. Tai, U. I. Zakai and D. A. Mitchell, Nat. Chem. Biol., 2017, 13, 470–478 CrossRef CAS.
- P. Agrawal, S. Khater, M. Gupta, N. Sain and D. Mohanty, Nucleic Acids Res., 2017, 45, W80–W88 CrossRef CAS.
- M. G. Chevrette, F. Aicheler, O. Kohlbacher, C. R. Currie and M. H. Medema, Bioinformatics, 2017, 33, 3202–3210 CrossRef CAS.
- M. Rottig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef.
- G. C. A. Amos, T. Awakawa, R. N. Tuttle, A. C. Letzel, M. C. Kim, Y. Kudo, W. Fenical, B. S. Moore and P. R. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E11121–E11130 CrossRef CAS.
- M. S. Donia, P. Cimermancic, C. J. Schulze, L. C. Wieland Brown, J. Martin, M. Mitreva, J. Clardy, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 1402–1414 CrossRef CAS.
- X. Wang, Z. Li, M. Jiang, S. Wang, S. Zhang and Z. Wei, J. Chem. Inf. Model., 2019, 59, 3817–3828 CrossRef CAS.
- W. Torng and R. B. Altman, J. Chem. Inf. Model., 2019, 59, 4131–4149 CrossRef CAS.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. I. Nikolenko, A. n. Aspuru-Guzik and A. Zhavoronkov, 2018, arXiv: abs/1811.12823.
- J. C. Navarro-Muñoz, N. Selem-Mojica, M. W. Mullowney, S. A. Kautsar, J. H. Tryon, E. I. Parkinson, E. L. C. De Los Santos, M. Yeong, P. Cruz-Morales, S. Abubucker, A. Roeters, W. Lokhorst, A. Fernandez-Guerra, L. T. D. Cappelini, A. W. Goering, R. J. Thomson, W. W. Metcalf, N. L. Kelleher, F. Barona-Gomez and M. H. Medema, Nat. Chem. Biol., 2020, 16, 60–68 CrossRef.
- M. D. Mungan, M. Alanjary, K. Blin, T. Weber, M. H. Medema and N. Ziemert, Nucleic Acids Res., 2020, 48, W546–W552 CrossRef CAS.
- A. M. Kloosterman, K. E. Shelton, G. P. van Wezel, M. H. Medema and D. A. Mitchell, 2020, bioRxiv: 2020.2003.2014.992123, DOI:10.1101/2020.03.14.992123.
- E. L. C. de Los Santos, Sci. Rep., 2019, 9, 13406 CrossRef.
- R. Zhang, X. Li, X. Zhang, H. Qin and W. Xiao, Nat. Prod. Rep., 2020 10.1039/d0np00043d.
- L. Laureti, L. Song, S. Huang, C. Corre, P. Leblond, G. L. Challis and B. Aigle, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6258–6263 CrossRef.
- F. Pereira and J. Aires-de-Sousa, Mar. Drugs, 2018, 16, 236 CrossRef.
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2019, 36, 122–173 RSC.
- L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 CrossRef CAS.
- Y. Luo, R. E. Cobb and H. Zhao, Curr. Opin. Biotechnol., 2014, 30, 230–237 CrossRef CAS.
- M. S. Donia and M. A. Fischbach, Science, 2015, 349, 1254766 CrossRef.
- M. R. Wilson, L. Zha and E. P. Balskus, J. Biol. Chem., 2017, 292, 8546–8552 CrossRef CAS.
- S. A. Kautsar, K. Blin, S. Shaw, J. C. Navarro-Munoz, B. R. Terlouw, J. J. J. van der Hooft, J. A. van Santen, V. Tracanna, H. G. Suarez Duran, V. Pascal Andreu, N. Selem-Mojica, M. Alanjary, S. L. Robinson, G. Lund, S. C. Epstein, A. C. Sisto, L. K. Charkoudian, J. Collemare, R. G. Linington, T. Weber and M. H. Medema, Nucleic Acids Res., 2020, 48, D454–D458 Search PubMed.
- S. Wang, N. Li, H. Zou and M. Wu, Neurosci. Lett., 2019, 696, 93–98 CrossRef CAS.
- G. Aleti, J. L. Baker, X. Tang, R. Alvarez, M. Dinis, N. C. Tran, A. V. Melnik, C. Zhong, M. Ernst, P. C. Dorrestein and A. Edlund, mBio, 2019, 10, e00321-19 CrossRef.
- K. Yamanaka, K. A. Reynolds, R. D. Kersten, K. S. Ryan, D. J. Gonzalez, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1957–1962 CrossRef CAS.
- S. Goldstein, L. Beka, J. Graf and J. L. Klassen, BMC Genomics, 2019, 20, 23 CrossRef.
- B. G. de la Torre and F. Albericio, Molecules, 2020, 25, 745 CrossRef CAS.
- F. Mahmoudi, B. Baradaran, A. Dehnad, D. Shanehbandi, L. Mohamed Khosroshahi and M. Aghapour, Br. J. Biomed. Sci., 2016, 73, 97–103 CrossRef CAS.
- J. Hrdy, L. Sukenikova, P. Petraskova, O. Novotna, D. Kahoun, M. Petricek, A. Chronakova and K. Petrickova, Microorganisms, 2020, 8, 621 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0np00055h |
| ‡ Equal first author contribution. |
| This journal is © The Royal Society of Chemistry 2021 |