
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
When crown ethers finally click: novel, click-assembled, fluorescent enantiopure pyridino-crown ether-based chemosensors – and an N-2-aryl-1,2,3-triazole containing one†
Balázs
Szemenyei
 a,
Mira
Malmosi
a,
Dávid
Pál
a,
Péter
Baranyai
b,
László
Drahos
c,
Ildikó
Móczár
*a and
Péter
Huszthy
*a
a,
Mira
Malmosi
a,
Dávid
Pál
a,
Péter
Baranyai
b,
László
Drahos
c,
Ildikó
Móczár
*a and
Péter
Huszthy
*a
aDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, PO Box 91, H-1521 Budapest, Hungary. E-mail: moczar.ildiko@vbk.bme.hu; huszthy.peter@vbk.bme.hu
bWigner Research Centre for Physics, PO Box 49, H-1525 Budapest, Hungary
cInstitute of Organic Chemistry, Research Centre for Natural Sciences, PO Box 286, H-1519 Budapest, Hungary
First published on 3rd November 2021
Abstract
Three novel, click-assembled fluorescent pyridino-18-crown-6 ethers have been synthesized for enantiomeric sensing. We also prepared new azide- and ethynyl-substituted pyridino-18-crown-6 ethers as their precursors, which open the way for further interesting applications using click chemistry. An optically active pyridino-18-crown-6 ether containing an N-2-aryl-1,2,3-triazole type fluorophore unit was also synthesized via post-triazole arylation. These four fluorescent sensor molecules were studied in terms of their optical properties as well as their enantiomeric recognition abilities toward the hydrogen perchlorate salts of 1-phenylethylamine, 1-(1-naphthyl)ethylamine, phenylglycine methyl ester and phenylalanine methyl ester in acetonitrile.
Introduction
Enantiomeric differentiation of chiral primary ammonium salts is of great importance,1,2 because organic molecules with similar structures are often drug molecules. Many times only one enantiomer of these molecules gives the biologically desired effect, and the other one might be responsible for dangerous side effects.3 As purity requirements in the pharmaceutical and agrochemical industries are becoming stricter,4 sensor molecules which enable selective and sensitive recognition of enantiomers have gained much interest.5 Fluorescence spectroscopy is a highly useful tool for sensing enantiomers, because of its selectivity and high sensitivity;6 therefore, fluorophore units are widely used in chiral chemosensors,5,7–12 including fluorescent optically active crown ethers.13–17 Chiral crown ethers containing a pyridine moiety were also prepared by Izatt and Bradshaw et al., and these macrocycles showed excellent selectivity toward the enantiomers of protonated primary amines, amino acid esters and amino alcohols.18–20 Therefore, they are widely used as sensor molecules21 as well as selector molecules in chiral chromatography.22,23We set the goal to synthesize fluorescent enantiopure pyridino-crown ether-based sensor molecules. We have already prepared and examined optically active pyridino-crown ethers having anthracene,24 and more recently, benzothiazole14 fluorophore units connected directly or through a linker, respectively, to position 4 of the pyridine ring. Herein, we report the introduction of azide and ethynyl groups to position 4 of the pyridine ring, as a preparative novelty. These results opened the way for click chemistry, which is not only useful for incorporating new kinds of fluorophore moieties, but also enables other interesting applications in the future.
There is a wide range of reported fluorophores which can be assembled by a click reaction.25–30 Fluorescence can be induced via a click reaction from two non-fluorescent precursors.27 Attaching an electron-donating group to the electron-deficient pyridine unit through a triazole moiety creates push–pull fluorophores,30 which can, in some cases, even be utilized as ratiometric sensors.25 Furthermore, these fluorophores are more easily synthesized than 5-aryl-2-(4-pyridyl)oxazoles (e.g., PYMPO), which bear similar photophysical properties.25 In this work, we synthesized pyridino-crown ethers (S,S)-1 and (S,S)-2 (Fig. 1) having an N-1-aryl-1,2,3-triazole unit, where fluorescence was installed by click chemistry itself. Also, there have been reported examples, where azides or alkynes bearing already fluorescent moieties were connected to macrocycles by a click reaction to obtain chemosensors having various fluorophores,29e.g., 1,8-naphthalimide among others.31,32 Our interest turned to 1,8-naphthalimides for their advantageous properties for sensing, easy synthesis and derivatization,33 thus we prepared pyridino-crown ether (S,S)-3 (Fig. 1) into which a naphthalimide unit was introduced via a click reaction. It is known, however, that N-2-aryl-1,2,3-triazoles have much stronger fluorescence than N-1-aryl-1,2,3-triazoles,34–38 making the incorporation of the former type fluorophores into chemosensors advantageous.36 Motivated by this, we also synthesized fluorescent sensor molecule (S,S)-4 (Fig. 1), which contains an N-2-aryl-1,2,3-triazole type fluorophore.
 | ||
| Fig. 1 Schematics of novel fluorescent pyridino-crown ether type chemosensors containing a triazole unit. | ||
We studied the optical properties, especially fluorescence changes, of these four ligands [(S,S)-1–(S,S)-4] upon complexation with the enantiomers of protonated chiral primary amines and amino acid esters, focusing on their ability to discriminate between the enantiomers of these ammonium guests.
Results and discussion
Synthesis
The synthesis of pyridino-crown ether type sensor molecules (S,S)-1–(S,S)-4 containing a triazole unit is outlined in Schemes 1–3.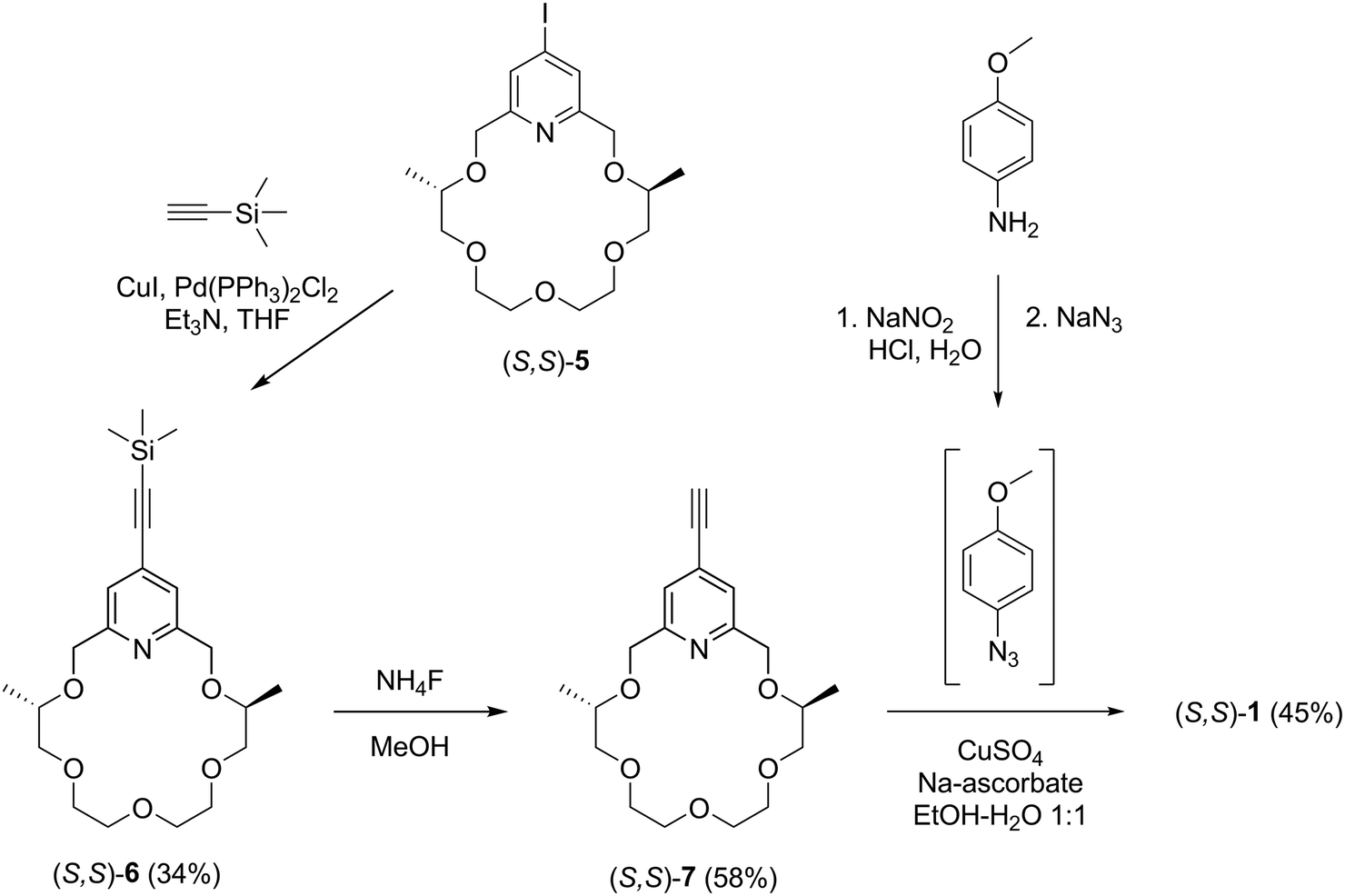 | ||
| Scheme 1 Synthesis of sensor molecule (S,S)-1. | ||
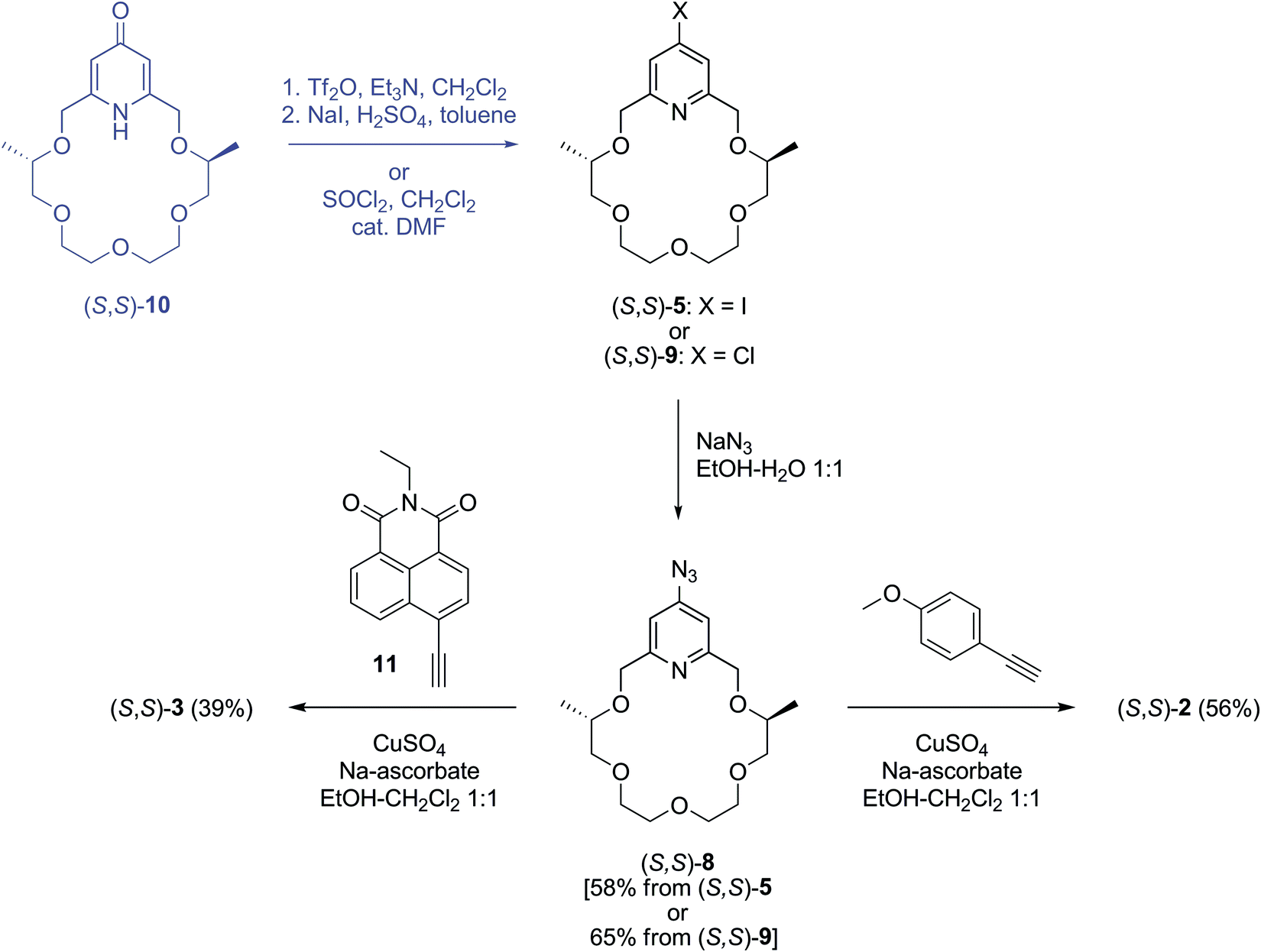 | ||
| Scheme 2 Synthesis of sensor molecules (S,S)-2 and (S,S)-3. | ||
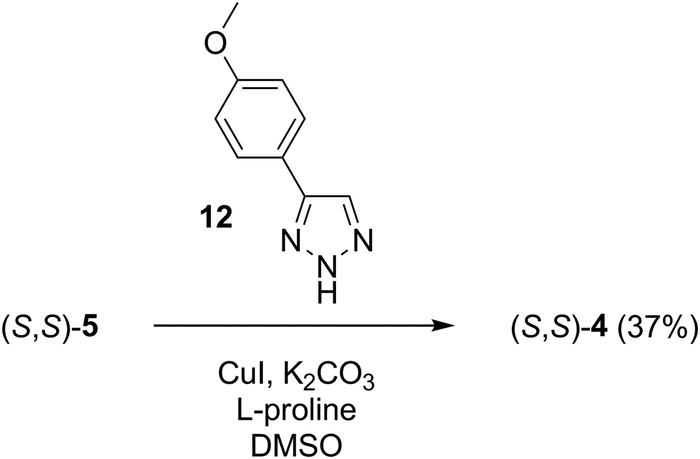 | ||
| Scheme 3 Synthesis of sensor molecule (S,S)-4. | ||
Iodopyridino-crown ether (S,S)-522 was reacted with ethynyltrimethylsilane by Sonogashira coupling according to the reported methods31,39 to give crown ether derivative (S,S)-6 (Scheme 1). The trimethylsilyl protecting group was removed by ammonium fluoride in methanol to yield ethynyl-substituted pyridino-crown ether (S,S)-7. Chemosensor (S,S)-1 was synthesized from ethynyl derivative (S,S)-7 by a click reaction in a mixture of ethanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)40 using a copper(II) sulfate–sodium ascorbate system as a catalyst and 4-methoxyphenyl azide as a reagent, which was prepared41in situ from 4-methoxyaniline by diazotization.
:1)40 using a copper(II) sulfate–sodium ascorbate system as a catalyst and 4-methoxyphenyl azide as a reagent, which was prepared41in situ from 4-methoxyaniline by diazotization.
Azidopyridino-crown ether (S,S)-8 was prepared from iodopyridino-crown ether (S,S)-522 or, alternatively, from chloropyridino-crown ether (S,S)-922 with sodium azide in a mixture of ethanol–water (1:1) (Scheme 2) according to the method reported40 for the synthesis of 4-azidopyridine. We obtained azide (S,S)-8 with a slightly better yield from chloro compound (S,S)-9 than from iodo compound (S,S)-5. This is in accordance with the well-known fact that chlorine is displaced easier by an aromatic nucleophilic substitution than iodine. Moreover, if we take into account that chloro compound (S,S)-9 can be prepared from pyridono-crown ether (S,S)-10 with a better yield (74%) than using two steps to get iodo compound (S,S)-5 with 56% yield,22 the overall yield of azide (S,S)-8 from macrocycle (S,S)-10 is much better for the “chloro compound” route (48% in two steps) than for the “iodo compound” one (32% in three steps).
Chemosensors (S,S)-2 and (S,S)-3 were synthesized in click reactions by reacting azide derivative (S,S)-8 with 4-ethynylanisole and naphthalimide derivative 11, respectively, in a mixture of ethanol–dichloromethane (1:1)28 using a copper(II) sulfate–sodium ascorbate system as a catalyst (Scheme 2). Naphthalimide derivative 11 was obtained as described in the literature starting from 4-bromo-1,8-naphthalic anhydride.31
The synthesis of triazole 12 was performed according to the reported procedures starting from 4-hydroxybenzaldehyde.42–44 The direct triazole arylation reaction35 of the former compound by iodopyridino-crown ether (S,S)-522 in DMSO in the presence of potassium carbonate as a base afforded sensor molecule (S,S)-4 (Scheme 3). The formation of some chemosensor (S,S)-2 as a byproduct was also observed in this reaction, of which separation by chromatography was possible.
Structure elucidation
Sensor molecules (S,S)-1–(S,S)-4 were characterized by 1H NMR, 13C NMR, IR and HRMS methods (see the Experimental section and ESI†). The connectivity of the triazole nitrogen atom for sensor molecule (S,S)-4 was proved by a 1H–1H ROESY experiment. In this case, one of the three possible isomers is ligand (S,S)-2, which formed as a byproduct, and its physical and spectroscopic properties are different from those of ligand (S,S)-4. The absence of a ROE cross-peak between the pyridine proton (7.99 ppm) and phenyl proton (7.84 ppm) excludes the third isomer (Fig. 2). It is well-known that Cu(I)-catalysed azide–alkyne cycloaddition reactions yield 1,4-disubstituted 1,2,3-triazoles with exquisite selectivity.45–47 The 2D NMR spectra taken for click-derived ligands (S,S)-1–(S,S)-3 gave the expected results (see the ESI†).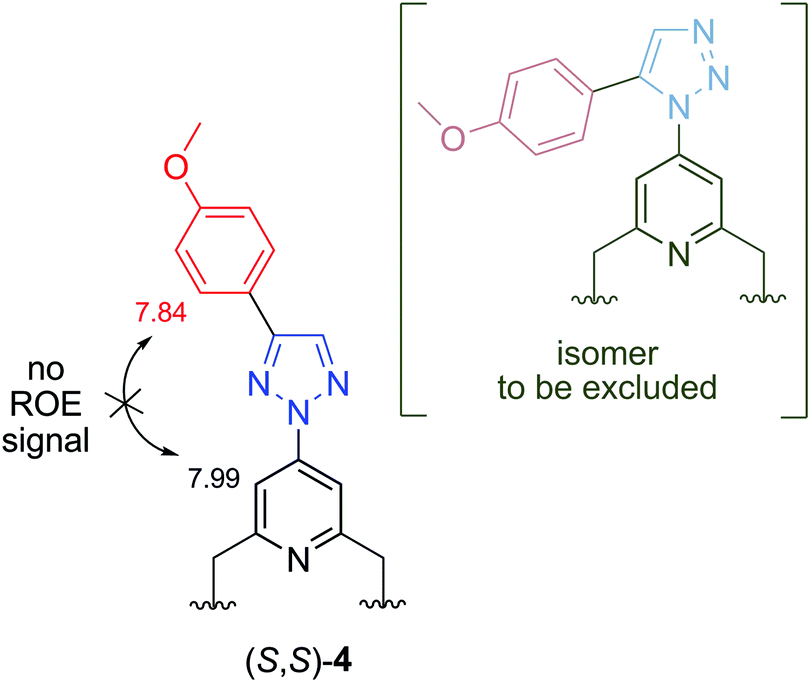 | ||
| Fig. 2 Part of the structure of sensor molecule (S,S)-4 with proton signal values (ppm) and an alternative structure excluded by a 2D ROESY experiment. | ||
Optical properties
First, we studied the optical (absorption and fluorescence) properties of the sensor molecules, which are summarized in Table 1.| (S,S)-1 | (S,S)-2 | (S,S)-3 | (S,S)-4 | |
|---|---|---|---|---|
| λ abs,max (nm) | 271 | 257 | 240, 355 | 289, 312 |
| ε max (× 104 M−1 cm−1) | 2.7 | 2.4 | 4.4, 1.8 | 2.0, 2.5 |
| λ em,max (nm) | 384 | 405 | 428 | 378 |
| Φ f | 0.018 | 0.022 | 0.81 | 0.93 |
N-1-Aryl triazoles (S,S)-1 and (S,S)-2 have absorption maxima at shorter wavelengths than N-2-aryl triazole (S,S)-4. Shi et al. found that while 1,4-disubstituted N-1-aryl-1,2,3-triazoles prefer twisted conformations around the C–N bond, 2,4-disubstituted N-2-aryl-1,2,3-triazoles favor a co-planar conformation with the possibility for a higher degree of conjugation,34,36 which can be an explanation for the latter statement. The most red-shifted absorption band was recorded in the case of ligand (S,S)-3 having a naphthalimide moiety with a more extended π system. Regarding the position of the absorption bands, reported fluorescent pyridino-crown ethers having a benzothiazole unit (λabs,max = 300–320 nm)14 are similar to ligand (S,S)-4, and those bearing an anthracene moiety (λabs,max around 365 nm)24 resemble sensor molecule (S,S)-3. Among 1,2,3-triazole-, benzothiazole-14 and anthracene-containing24 pyridino-crown ethers, the latter have by far the smallest molar absorption coefficients at the lowest energy absorption maxima.
It can be observed that while the fluorescence quantum yields of N-1-aryl triazoles (S,S)-1 and (S,S)-2 are quite low, that of N-2-aryl triazole (S,S)-4 is significantly higher, in accordance with a series of reported examples.26,34–37 It can also be stated that the difference in the connection of aryl groups to N-1 and C-4 atoms in triazoles (S,S)-1 and (S,S)-2 hardly influenced the fluorescence quantum yield. For comparison, benzothiazole-containing pyridino-crown ethers have fluorescence quantum yields of 0.007–0.135 (in acetonitrile),14 and those with an anthracene unit showed 0.58 and 0.60 (in acetonitrile).24 Therefore, the strongest fluorescence (and brightness) was achieved with ligands (S,S)-4 and (S,S)-3 among the new and reported14,24 fluorescent pyridino-crown ether-based enantioselective sensors.
Enantiomeric recognition studies
The chiral recognition abilities of pyridino-crown ethers (S,S)-1–(S,S)-4 toward the enantiomers of 1-phenylethylamine hydrogen perchlorate (PEA), 1-(1-naphthyl)ethylamine hydrogen perchlorate (NEA), phenylglycine methyl ester hydrogen perchlorate (PGME) and phenylalanine methyl ester hydrogen perchlorate (PAME) (Fig. 3) were studied in acetonitrile by UV–vis and fluorescence spectroscopies. | ||
| Fig. 3 Chiral primary ammonium salts used in the enantiomeric recognition studies. | ||
The absorption spectra showed no [in the case of (S,S)-3] or little to moderate [in the cases of (S,S)-1, (S,S)-2 and (S,S)-4] changes upon the addition of the chiral ammonium guests (Fig. 4), apart from the partial to total overlap of the longest wavelength bands of macrocycles (S,S)-1, (S,S)-2 and (S,S)-4 with that of NEA. Based on the absorption changes, crown ethers (S,S)-1 and (S,S)-4 also revealed slight protonation (besides the complexation) with PGME and PAME, which could be neglected during the evaluation of fluorescence titration data (see below).
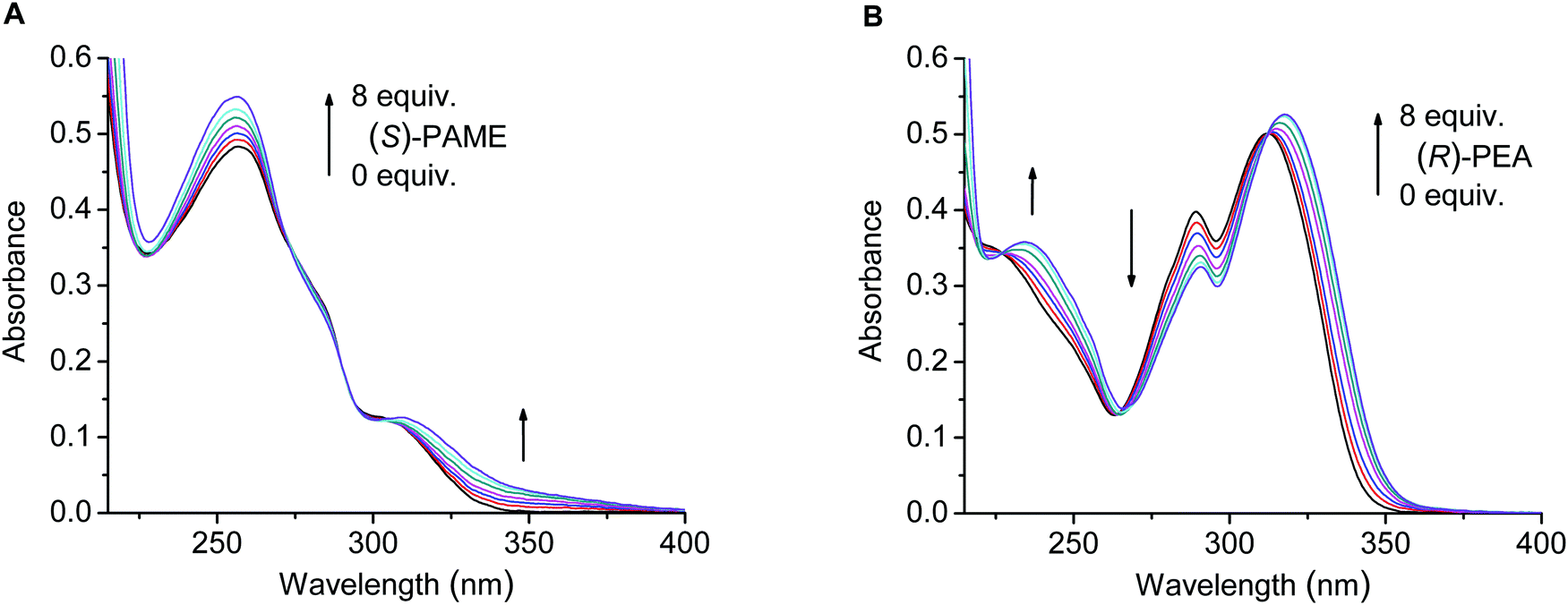 | ||
| Fig. 4 Series of absorption spectra upon the titration of (S,S)-2 (20 μM) with (S)-PAME (0, 0.2, 0.5, 1, 2, 4, 8 equiv.) (A) and (S,S)-4 (20 μM) with (R)-PEA (0, 0.2, 0.5, 1, 2, 4, 8 equiv.) (B) in MeCN. | ||
Compared to the above spectral changes, the fluorescence ones were much more pronounced in most cases, which were used to determine the stability constants of the complexes by global nonlinear regression analysis. The titration series of emission spectra could be fitted satisfactorily using a 1:1 complex formation model.
In the case of sensor molecule (S,S)-2, significant fluorescence quenching was observed upon the addition of the optically active salts (Fig. 5A), rendering appreciable enantioselectivity toward NEA and PEA, but only slight recognition to the enantiomers of PGME and practically no for PAME (Table 2).
 | ||
| Fig. 5 Series of fluorescence emission spectra upon the titration of (S,S)-2 (5 μM) with (S)-PEA (0, 0.4, 0.8, 1.6, 2.4, 3.2, 4.4, 6.4, 9.6, 16, 24, 48, 80, 144 equiv.), λex = 300 nm (A), (S,S)-3 (2 μM) with (R)-NEA (0, 1.5, 4, 6, 10, 20, 60 equiv.), λex = 355 nm (B), (S,S)-4 (5 μM) with (S)-PAME (0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 20, 36, 76, 156 equiv.), λex = 300 nm (C), and (S,S)-1 (2 μM) with (S)-PEA (0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 17, 27, 47, 87 equiv.), λex = 265 nm (D) in MeCN. | ||
| (S,S)-1 | (S,S)-2 | (S,S)-3 | (S,S)-4 | |||||
|---|---|---|---|---|---|---|---|---|
| logK |
ΔlogK |
logK |
ΔlogK |
logK |
ΔlogK |
logK |
Δ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) logK logK |
|
| a Complex stability constants (K) are given in M−1. b Complex stability constants (K) could not be determined because of the total spectral overlap of the host and the guest. c No spectral changes occurred. | ||||||||
| (R)-PEA | 5.48 ± 0.03 | 0.28 | 4.83 ± 0.02 | 0.28 | 5.25 ± 0.02 | 0.28 | ||
| (S)-PEA | 5.20 ± 0.02 | 4.55 ± 0.03 | 4.97 ± 0.02 | |||||
| (R)-NEA | 5.01 ± 0.03 | 0.37 | 5.13 ± 0.03 | 0.45 | 5.37 ± 0.02 | 0.44 | ||
| (S)-NEA | 4.64 ± 0.03 | 4.68 ± 0.04 | 4.93 ± 0.02 | |||||
| (R)-PGME | 5.67 ± 0.04 | −0.13 | 4.74 ± 0.03 | −0.07 | 5.30 ± 0.03 | −0.11 | ||
| (S)-PGME | 5.80 ± 0.04 | 4.81 ± 0.03 | 5.41 ± 0.04 | |||||
| (R)-PAME | 5.48 ± 0.03 | −0.03 | 4.23 ± 0.05 | −0.05 | 4.90 ± 0.02 | 0.01 | ||
| (S)-PAME | 5.51 ± 0.03 | 4.28 ± 0.06 | 4.89 ± 0.03 | |||||
Fluorescence spectral changes did not occur when ligand (S,S)-3 was titrated with PEA, PGME and PAME, but interestingly about 20% quenching could be detected upon the addition of the enantiomers of NEA (Fig. 5B), which enabled the determination of the complex stability constants (Table 2). Probably, the electronic signal transmission between the electron-deficient pyridine and similarly electron-deficient naphthalimide units is hindered, thereby resulting in no fluorescence changes by the addition of PEA, PGME and PAME (presumably complexation with these salts also took place similarly to other pyridino-crown ethers14,24). However, the extended aromatic moiety of NEA may show a little overlap with the triazole ring, so a limited emission change can be caused by a direct π–π interaction between the fluorophore unit and the guest molecule. Appreciable selectivity between the enantiomers was also experienced in this case (Table 2).
Titrations of pyridino-crown ether (S,S)-4 with all the four chiral salts induced significant fluorescence quenching (Fig. 5C) similarly to crown ether (S,S)-2. The extent of enantiomeric differentiation was appreciable for NEA and PEA, moderate toward PGME, and practically zero for PAME (Table 2).
Free sensor molecule (S,S)-1 showed an emission peak at 384 nm, which decreased to a great extent when the ammonium salts were added; however, simultaneously, a new, much more intense band emerged with a maximum of around 580 nm (Fig. 5D). The latter spectral shape corresponds to the emission spectrum of protonated ligand (S,S)-1 (Fig. 6A). Since these guests caused no (PEA and NEA) or negligible (PGME and PAME) protonation in the ground state (Fig. 6B), the protonation of ligand (S,S)-1 took place only in the excited state resulting in emission from the protonated species (Fig. 6A). Presumably, the protonation process occurred from the hydrogen-bonded complexes, because the series of spectra for fluorescence titration could be fitted satisfactorily using a 1:1 complexation model (Table 2). It should be noted here that this phenomenon (i.e., proton transfer in the complex upon excitation) has also been observed in the cases of pyridino-crown ethers having a benzothiazole unit.14 Additionally, it is also worth mentioning that host (S,S)-1 has a similar behavior to its reported25 analogous pyridine derivative having no crown ether macroring, regarding the appearance of a new, red-shifted, high-intensity emission band upon protonation.
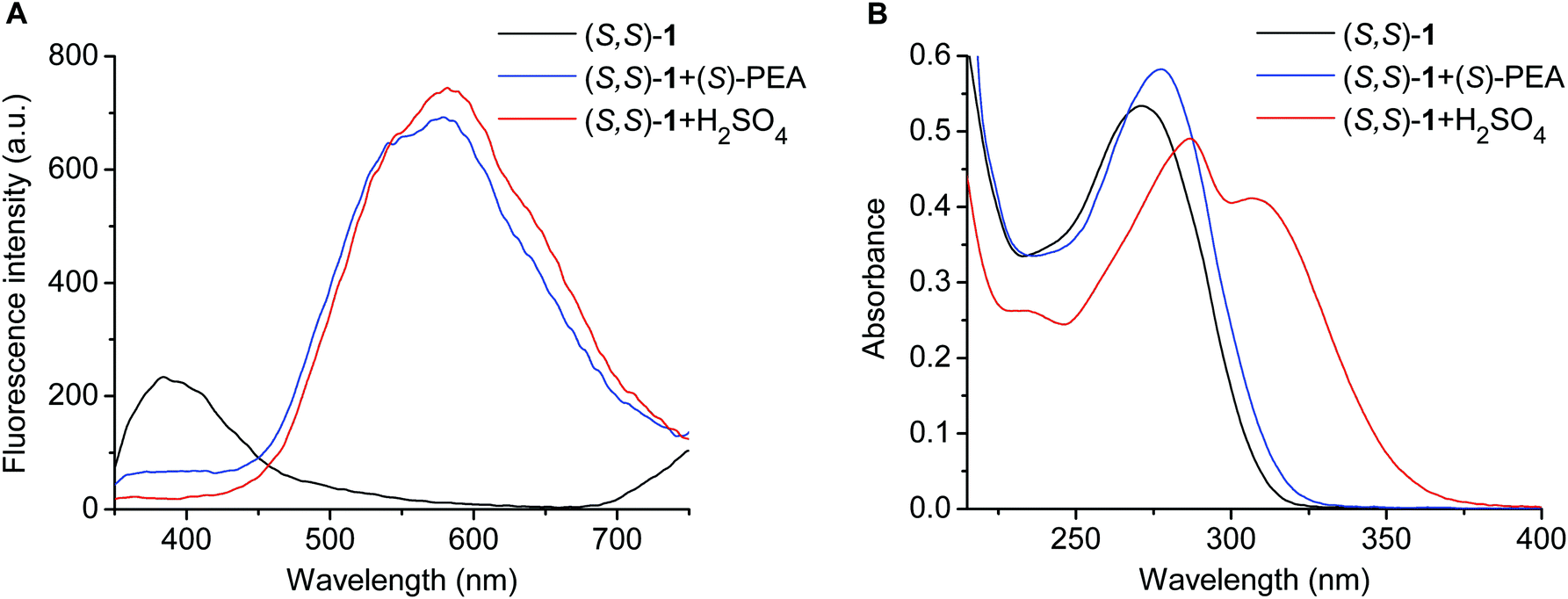 | ||
| Fig. 6 Fluorescence emission spectra of (S,S)-1 alone (5 μM) and the end-points of its titrations with (S)-PEA and H2SO4 in MeCN, λex = 265 nm (A). Absorption spectra of (S,S)-1 alone (20 μM) and the end-points of its titrations with (S)-PEA and H2SO4 in MeCN (B). | ||
A problem which was encountered with chemosensor (S,S)-1 is that it does not absorb light above 325 nm, which means that it could not be selectively excited when titrated with NEA, which absorbs all the way up to 325 nm. As a consequence of this, distorted titration curves were obtained, which did not allow the determination of stability constants. Macrocycle (S,S)-1 showed moderate and no enantioselectivity toward PGME and PAME, respectively, but it exhibited considerable recognition toward the enantiomers of PEA (Table 2).
Considering the results for chemosensors (S,S)-1–(S,S)-4 in general, the highest enantioselectivities were found toward NEA (Table 2), which can be attributed to the extended π system of the guest conferring stronger π–π interactions.21 In the cases of crown ethers (S,S)-3 and (S,S)-4, these ΔlogK values (0.45 and 0.44, Table 2) are slightly or significantly higher than those for similar dimethyl-substituted pyridino-crown ethers having an anthracene (0.38)24 or a benzothiazole unit (0.30, 0.25).14
Regarding analogous ligands (S,S)-1, (S,S)-2 and (S,S)-4, it can be seen that the connectivity of the triazole moiety to the pyridine ring had an influence on the stability of complexes (Table 2). Namely, the equilibrium constants for each optically active guest increase in the order of macrocycles (S,S)-2, (S,S)-4 and (S,S)-1. This phenomenon may be explained by different conformational and electronic effects in the aromatic parts of the chemosensors, e.g., different dihedral angles between the pyridine and triazole rings34,36 or different electron densities in the pyridine rings. However, the enantioselectivities were quite similar for a given chiral salt [where it could be determined and including (S,S)-3 also], and only ligand (S,S)-2 showed slightly smaller recognition for the enantiomers of NEA and PGME (Table 2).
A pair of titration curves of sensor molecule (S,S)-4 with the enantiomers of NEA is shown in Fig. 7A. Larger fluorescence quenching when adding (R)-NEA is clearly visible, indicating a heterochiral preference. Similarly, the complex formation of host (S,S)-1 with (R)-PEA caused more significant fluorescence enhancement (on the basis of the excited state proton transfer in the complex) than with the (S)-enantiomer (Fig. 7B). In the latter case, the quenching processes at 384 nm were similar to those in Fig. 7A, but exhibited a smaller difference.
 | ||
| Fig. 7 Titration curves of (S,S)-4 (5 μM) with the enantiomers of NEA (0, 0.5, 1, 2, 4, 5, 10, 20 equiv.) at 378 nm, λex = 330 nm (A) and (S,S)-1 (2 μM) with the enantiomers of PEA (0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 17, 27 equiv.) at 580 nm, λex = 265 nm (B) in MeCN. | ||
Toward the enantiomers of PGME, apparently inverse discrimination was observed than for NEA or PEA. However, this is because the absolute configuration of PGME is reversed according to the CIP rules. Thus, (S)-PGME has the same spatial arrangement of substituents (aromatic, amino and the third groups) around the chiral center as (R)-NEA or (R)-PEA.14,24 The enantiomers of PAME were not differentiated by the triazolo macrocycles, similar to the earlier reported pyridino-crown ethers.14,24 It may be explained by the presence of a methylene spacer between the aromatic group and the stereogenic center.14,24
Conclusions
The synthesis of crown ether-based fluorescent sensors (S,S)-1–(S,S)-4 containing a triazole moiety has been achieved. Macrocycles (S,S)-1–(S,S)-3 were obtained by a click reaction, enabled by the synthesis of new pyridino-crown ether derivatives having an ethynyl or an azido group [(S,S)-7 and (S,S)-8]. Pyridino-crown ether (S,S)-4 was prepared by direct N-arylation.Enantiomeric recognition of crown ethers (S,S)-1–(S,S)-4 toward the enantiomers of chiral protonated primary aralkylamines and amino acid esters was studied in acetonitrile by fluorescence spectroscopy. Sensor molecules (S,S)-2–(S,S)-4 exhibited fluorescence quenching upon complexation, but in the case of ligand (S,S)-3, just a moderate decrease in emission was experienced and only with NEA. Titrations of crown ether (S,S)-1 were accompanied by the appearance of a second emission band, due to the excited state proton transfer in the complexes. Appreciable enantioselectivities were observed for NEA and PEA; however, PGME was differentiated only moderately.
Experimental
General
The starting materials were purchased from Sigma-Aldrich, Merck and Alfa Aesar. Aluminum oxide 60 F254 neutral type E plates (Merck) were used for thin-layer chromatography (TLC). Aluminum oxide (neutral, activated, Brockmann I, 40–160 μm) was used for column chromatography. The ratios of solvents for the eluents are given in volumes (mL/mL). Solvents were dried and purified according to well-established methods.48 Evaporations were carried out under reduced pressure.Melting points were taken on a Boetius micro-melting point apparatus and are uncorrected. Optical rotations were taken on a PerkinElmer 241 polarimeter that was calibrated by measuring the optical rotations of both enantiomers of menthol. IR spectra were recorded using a Bruker Alpha-T Fourier transform infrared (FT-IR) spectrometer. 1H (500 MHz) and 13C (125 MHz) NMR spectra were obtained using a Bruker DRX-500 Avance spectrometer. 1H (300 MHz) and 13C (75.5 MHz) NMR spectra were recorded using a Bruker 300 Avance spectrometer. 2D NMR spectra were obtained using a Bruker DRX-500 Avance spectrometer. HRMS analyses were performed using a Waters Q-TOF Premier or a Thermo Velos Pro Orbitrap Elite system mass spectrometer in positive ESI mode.
UV–vis spectra were recorded using a Unicam UV4-100 spectrophotometer. Quartz cuvettes with a path length of 1 cm were used. Fluorescence emission spectra were recorded using a PerkinElmer LS 50B luminescent spectrometer and were corrected by the spectrometer software. Quartz cuvettes with a path length of 1 cm were used. Fluorescence quantum yields were determined relative to quinine sulfate (Φf = 0.53 in 0.1 M H2SO4).6 Enantiomers of PEA, NEA, PGME and PAME were prepared as reported49 in our laboratory. For fluorescence titrations, the concentrations were 2 μM for (S,S)-1 and (S,S)-3, and 5 μM for (S,S)-2 and (S,S)-4. In order to determine the stability constants of complexes by global nonlinear regression analysis, SPECFIT/32™ software was used.
Syntheses and characterizations of new compounds
To the resulting crude 4-methoxyphenyl azide, ethynylpyridino-crown ether (S,S)-7 (156 mg, 0.447 mmol) was added, followed by CuSO4·5H2O (1.0 mg, 0.0040 mmol), sodium ascorbate (5.3 mg, 0.027 mmol), ethanol (1.25 mL) and water (1.25 mL), and the mixture was stirred at rt for 20 h. The solvent was removed at rt, and the residue was dissolved in a mixture of ethyl acetate (50 mL) and water (50 mL). The phases were shaken well and separated. The aqueous phase was further extracted with ethyl acetate (2 × 50 mL). The combined organic phase was dried over anhydrous MgSO4, filtered, and evaporated. The crude product was purified by column chromatography on alumina using ethanol–toluene 1:80 mixture as an eluent, followed by recrystallization from toluene–hexane to give (S,S)-1 (76 mg, 34%) as an off-white powder.
R
f: 0.30 (alumina TLC, EtOH–toluene 1:40); m.p.: 101–104 °C; [α]25D = +24.2 (c = 1.15, CHCl3); IR (KBr) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max (cm−1) 3137, 3089, 2967, 2894, 2862, 1618, 1595, 1567, 1521, 1471, 1446, 1392, 1369, 1353, 1331, 1306, 1259, 1195, 1165, 1115, 1098, 1068, 1042, 1029, 1007, 922, 882, 865, 836, 817, 798, 697, 624, 541, 524, 496, 465, 407; 1H NMR (500 MHz, acetone-d6) δ (ppm) 1.16 (d, J = 6 Hz, 6H), 3.39–3.58 (m, 12H), 3.79–3.87 (m, 2H), 3.89 (s, 3H), the diastereotopic benzylic type –CH2– protons give an AB quartet: δA 4.79 and δB 4.82 (JAB = 13 Hz, 4H), 7.16 (d, J = 9 Hz, 2H), 7.87 (s, 2H), 7.88 (d, J = 9 Hz, 2H), 9.09 (s, 1H); 13C NMR (75.5 MHz, acetone-d6) δ (ppm) 17.75, 56.06, 71.17, 71.31, 72.54, 74.43, 76.44, 115.69, 117.13, 121.41, 122.80, 131.34, 139.87, 146.63, 160.50, 160.93; HRMS m/z (M + H+) found: 499.2541, C26H35N4O6+ requires 499.2551.
max (cm−1) 3137, 3089, 2967, 2894, 2862, 1618, 1595, 1567, 1521, 1471, 1446, 1392, 1369, 1353, 1331, 1306, 1259, 1195, 1165, 1115, 1098, 1068, 1042, 1029, 1007, 922, 882, 865, 836, 817, 798, 697, 624, 541, 524, 496, 465, 407; 1H NMR (500 MHz, acetone-d6) δ (ppm) 1.16 (d, J = 6 Hz, 6H), 3.39–3.58 (m, 12H), 3.79–3.87 (m, 2H), 3.89 (s, 3H), the diastereotopic benzylic type –CH2– protons give an AB quartet: δA 4.79 and δB 4.82 (JAB = 13 Hz, 4H), 7.16 (d, J = 9 Hz, 2H), 7.87 (s, 2H), 7.88 (d, J = 9 Hz, 2H), 9.09 (s, 1H); 13C NMR (75.5 MHz, acetone-d6) δ (ppm) 17.75, 56.06, 71.17, 71.31, 72.54, 74.43, 76.44, 115.69, 117.13, 121.41, 122.80, 131.34, 139.87, 146.63, 160.50, 160.93; HRMS m/z (M + H+) found: 499.2541, C26H35N4O6+ requires 499.2551.
:80 mixture as an eluent, followed by recrystallization from toluene–hexane to give (S,S)-2 (64 mg, 56%) as a white powder.
R
f: 0.34 (alumina TLC, EtOH–toluene 1:40); m.p.: 99–103 °C; [α]25D = +19.0 (c = 1.00, CHCl3); IR (KBr) max (cm−1) 3143, 3124, 3083, 3044, 2965, 2907, 2868, 1619, 1600, 1567, 1505, 1476, 1448, 1416, 1368, 1353, 1330, 1309, 1287, 1260, 1100, 1068, 1055, 1020, 944, 922, 870, 838, 818, 796, 736, 696, 636, 617, 553, 541, 528, 495, 404; 1H NMR (500 MHz, CDCl3) δ (ppm) 1.19 (d, J = 6 Hz, 6H), 3.43–3.65 (m, 12H), 3.82–3.90 (m, 2H), 3.83 (s, 3H), the diastereotopic benzylic type –CH2– protons give an AB quartet: δA 4.89 and δB 4.93 (JAB = 14 Hz, 4H), 6.97 (d, J = 8 Hz, 2H), 7.71 (s, 2H), 7.82 (d, J = 8 Hz, 2H), 8.25 (s, 1H); 13C NMR (125 MHz, CDCl3) δ (ppm) 17.16, 55.48, 70.79, 71.02, 71.54, 74.31, 76.33, 110.10, 114.52, 116.28, 122.50, 127.44, 144.18, 148.85, 160.17, 161.50; HRMS m/z (M + H)+ found: 499.2572, C26H35N4O6+ requires: 499.2551.
:80 mixture as an eluent, followed by recrystallization from toluene–hexane to give (S,S)-3 (60 mg, 39%) as yellowish crystals.
R
f: 0.34 (alumina TLC, EtOH–toluene 1:40); m.p.: 158–159 °C; [α]25D = +15.0 (c = 0.91, CHCl3); IR (KBr) max (cm−1) 3344, 3308, 3170, 3088, 2973, 2926, 2874, 1700, 1665, 1588, 1543, 1510, 1475, 1462, 1445, 1383, 1371, 1342, 1244, 1222, 1110, 1065, 1048, 1025, 893, 867, 819, 785, 693, 659, 584, 490, 446, 415; 1H NMR (500 MHz, CD3CN) δ (ppm) 1.18 (d, J = 6 Hz, 6H), 1.25 (t, J = 7 Hz, 3H), 3.39–3.52 (m, 12H), 3.76–3.86 (m, 2H), 4.08 (q, J = 7 Hz, 2H), 4.73–4.82 (m, 4H), 7.70 (t, J = 8 Hz, 1H), 7.77 (s, 2H), 7.93 (d, J = 7 Hz, 1H), 8.38 (d, J = 7 Hz, 1H), 8.41 (d, J = 7 Hz, 1H), 8.82 (s, 1H), 8.88 (d, J = 8 Hz, 1H); 13C NMR (125 MHz, CD3CN) δ (ppm) 13.52, 17.14, 36.11, 71.09, 71.29, 71.80, 74.91, 76.29, 111.64, 123.66, 123.88, 128.35, 128.41, 129.36, 129.61, 130.99, 131.69, 133.11, 134.04, 134.21, 145.01, 147.15, 161.92, 164.19, 164.50; HRMS m/z (M + H)+ found: 616.2752, C33H38N5O7+ requires 616.2766.
:6 and then dimetoxyethane–toluene 1:4 mixtures as eluents to give (S,S)-4 (84 mg, 37%) as a pale brown oil.
R
f: 0.38 (alumina TLC, EtOH–toluene 1:40); [α]25D = +6.4 (c = 1.00, CHCl3); IR (neat) max (cm−1) 3115, 2967, 2865, 1613, 1595, 1547, 1490, 1460, 1436, 1349, 1292, 1250, 1176, 1107, 1084, 1030, 991, 976, 967, 915, 878, 832, 797, 729, 653, 616, 529, 490; 1H NMR (500 MHz, CDCl3) δ (ppm) 1.22 (d, J = 6 Hz, 6H), 3.45–3.66 (m, 12H), 3.85–3.92 (m, 2H), 3.86 (s, 3H), 4.92 (s, 4H), 7.00 (d, J = 9 Hz, 2H), 7.84 (d, J = 9 Hz, 2H), 7.99 (s, 2H), 8.05 (s, 1H); 13C NMR (125 MHz, CDCl3) δ (ppm) 17.23, 55.52, 70.81, 71.06, 71.77, 74.26, 76.30, 109.33, 114.59, 122.09, 127.86, 133.68, 146.83, 149.96, 160.67 (two 13C signals together); HRMS m/z (M + H)+ found: 499.2542, C26H35N4O6+ requires 499.2551.
:7 and then ethanol–toluene 1:140 mixtures as eluents to give (S,S)-6 (90 mg, 34%) as a brown oil.
R
f: 0.39 (alumina TLC, EtOH–toluene 1:40); [α]25D = +12.3 (c = 1.00, CHCl3); IR (neat) max (cm−1) 2966, 2931, 2866, 1597, 1551, 1452, 1411, 1372, 1350, 1339, 1250, 1110, 954, 923, 843, 759, 699, 663, 588; 1H NMR (300 MHz, CDCl3) δ (ppm) 0.24 (s, 9H), 1.16 (d, J = 6 Hz, 6H), 3.36–3.67 (m, 12H), 3.71–3.86 (m, 2H), 4.77 (s, 4H), 7.26 (s, 2H and solvent signal together); 13C NMR (75.5 MHz, CDCl3) δ (ppm) −0.23, 17.05, 70.68, 70.89, 71.54, 73.89, 76.10, 99.18, 102.70, 122.40, 131.87, 158.67; HRMS m/z (M + H)+ found: 422.2347, C22H36NO5Si+ requires 422.2357.
:120 mixture as an eluent to give (S,S)-7 (38 mg, 58%) as an off-white solid.
R
f: 0.39 (alumina TLC, EtOH–toluene 1:40); m.p.: 74–77 °C; [α]25D = +27.7 (c = 1.00, CHCl3); IR (KBr) max (cm−1) 3418, 3243, 2971, 2901, 2871, 2782, 2108, 1759, 1598, 1551, 1490, 1460, 1411, 1376, 1352, 1337, 1305, 1283, 1251, 1214, 1167, 1110, 1042, 1004, 976, 958, 932, 915, 898, 856, 806, 758, 683, 637, 585, 579, 545, 532, 509, 463; 1H NMR (500 MHz, CDCl3) δ (ppm) 1.18 (d, J = 6 Hz, 6H), 3.25 (s, 1H), 3.41–3.68 (m, 12H), 3.74–3.89 (m, 2H), 4.81 (s, 4H), 7.32 (s, 2H); 13C NMR (75.5 MHz, CDCl3) δ (ppm) 17.08, 70.70, 70.92, 71.60, 73.97, 76.18, 81.06, 81.65, 122.53, 130.82, 158.97; HRMS m/z (M + H)+ found: 350.1956, C19H28NO5+ requires 350.1962.
Starting from chloropyridino-crown ether (S,S)-9. Chloro compound (S,S)-922 (97.5 mg, 0.271 mmol), NaN3 (1.0 g, 15 mmol), ethanol (1 mL) and water (1 mL) were mixed in a round-bottom flask, and stirred at 80 °C for a week. The reaction mixture was washed into a separatory funnel with ethyl acetate (50 mL) and water (50 mL). The phases were shaken well and separated. The aqueous phase was extracted with ethyl acetate (2 × 50 mL). The combined organic phase was dried over anhydrous MgSO4, filtered, and the solvent was removed. The crude product was purified by column chromatography on alumina using first ethanol–toluene 1
:60 and then dioxane–hexane 1:6 mixtures as eluents to give (S,S)-8 (64 mg, 65%) as a yellow oil.
R
f: 0.38 (alumina TLC, EtOH–toluene 1:40); [α]25D = +30.7 (c = 1.00, CHCl3); IR (neat) max (cm−1) 2968, 2930, 2864, 2107, 1592, 1572, 1448, 1371, 1339, 1319, 1225, 1104, 921, 854, 763, 719, 670, 572, 538; 1H NMR (300 MHz, CDCl3) δ (ppm) 1.16 (d, J = 6 Hz, 6H), 3.38–3.68 (m, 12H), 3.72–3.89 (m, 2H), 4.79 (s, 4H), 6.90 (s, 2H); 13C NMR (75.5 MHz, CDCl3) δ (ppm) 17.24, 70.84, 71.02, 71.68, 74.13, 76.29, 110.58, 149.71, 160.82; HRMS m/z (M + H)+ found: 367.1980, C17H27N4O5+ requires: 367.1976.
Starting from iodopyridino-crown ether (S,S)-5. Iodo compound (S,S)-522 (69 mg, 0.15 mmol) and NaN3 (1.0 g, 15 mmol) were reacted in a mixture of ethanol (0.5 mL) and water (0.5 mL), in the same way as described above for chloro compound (S,S)-9, to give azide (S,S)-8 (32 mg, 58%).
All the properties of (S,S)-8 prepared this way were identical to those of the one prepared from (S,S)-9.
Author contributions
Conceptualization: B. S., I. M. and P. H.; methodology: B. S. and M. M.; formal analysis: P. B.; investigation: B. S., M. M., D. P. and L. D.; writing – original draft: B. S.; writing – review and editing: P. B., I. M. and P. H.; visualization: B. S. and I. M.; supervision: I. M. and P. H.; project administration: I. M. and P. H.; funding acquisition: I. M. and P. H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Research, Development and Innovation Office (NKFIH No. K128473, K112289 and PD104618) is gratefully acknowledged. B. Szemenyei is grateful for the fellowship provided by the Gedeon Richter's Talentum Foundation (1103 Budapest, Gyömrői út 19–21). We acknowledge the Governmental Agency for IT Development (KIFÜ) for awarding us access to resource based in Hungary. The authors thank Dr. György Tibor Balogh for recording some of the high-resolution mass spectra and Áron Szigetvári for elucidation of the 2D NMR spectra.References
- X. X. Zhang, J. S. Bradshaw and R. M. Izatt, Chem. Rev., 1997, 97, 3313–3361 CrossRef CAS PubMed.
- V. N. Khose, M. E. John, A. D. Pandey, V. Borovkov and A. V. Karnik, Symmetry, 2018, 10, 34 CrossRef.
- L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2006, 2, 85–100 CAS.
- M. C. Núñez, M. E. García-Rubiño, A. Conejo-García, O. Cruz-López, M. Kimatrai, M. A. Gallo, A. Espinosa and J. M. Campos, Curr. Med. Chem., 2009, 16, 2064–2074 CrossRef PubMed.
- L. Pu, Acc. Chem. Res., 2017, 50, 1032–1040 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science + Business Media, New York, NY, 3rd edn, 2006 Search PubMed.
- L. Pu, Chem. Rev., 2004, 104, 1687–1716 CrossRef CAS PubMed.
- A. Accetta, R. Corradini and R. Marchelli, Top. Curr. Chem., 2011, 300, 175–216 CrossRef CAS PubMed.
- L. Pu, Acc. Chem. Res., 2012, 45, 150–163 CrossRef CAS PubMed.
- X. Zhang, J. Yin and J. Yoon, Chem. Rev., 2014, 114, 4918–4959 CrossRef CAS PubMed.
- S. Yu and L. Pu, Tetrahedron, 2015, 71, 745–772 CrossRef CAS.
- O. K. Grigorova, D. I. Gusev, A. D. Averin, O. A. Maloshitskaya and I. P. Beletskaya, Russ. Chem. Bull. Int. Ed., 2019, 68, 848–854 CrossRef CAS.
- I. Móczár and P. Huszthy, Chirality, 2019, 31, 97–109 CrossRef PubMed.
- D. Pál, I. Móczár, B. Szemenyei, D. Marczona, I. Kocsis, G. Prikler, P. Vezse, P. Baranyai and P. Huszthy, Tetrahedron, 2019, 75, 2900–2909 CrossRef.
- D. Pál, M. Gede, I. Móczár, P. Baranyai, P. Bagi and P. Huszthy, Period. Polytech., Chem. Eng., 2020, 64, 20–36 CrossRef.
- Á. Golcs, B. Á. Ádám, V. Horváth, T. Tóth and P. Huszthy, Molecules, 2020, 25, 2571 CrossRef PubMed.
- H. Szabó-Szentjóbi, A. Márton, D. Pál, G. Dargó, Á. Szigetvári, C. Szántay, G. T. Balogh, T. Tóth and P. Huszthy, Period. Polytech., Chem. Eng., 2020, 64, 37–45 CrossRef.
- B. A. Jones, J. S. Bradshaw and R. M. Izatt, J. Heterocycl. Chem., 1982, 19, 551–556 CrossRef CAS.
- R. B. Davidson, J. S. Bradshaw, B. A. Jones, N. K. Dalley, J. J. Christensen, R. M. Izatt, F. G. Morin and D. M. Grant, J. Org. Chem., 1984, 49, 353–357 CrossRef CAS.
- J. S. Bradshaw, P. K. Thompson, R. M. Izatt, F. G. Morin and D. M. Grant, J. Heterocycl. Chem., 1984, 21, 897–901 CrossRef CAS.
- R. M. Izatt, T. Wang, J. K. Hathaway, X. X. Zhang, J. C. Curtis, J. S. Bradshaw, C. Y. Zhu and P. Huszthy, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 17, 157–175 CrossRef CAS.
- J. Kupai, S. Lévai, K. Antal, G. T. Balogh, T. Tóth and P. Huszthy, Tetrahedron: Asymmetry, 2012, 23, 415–427 CrossRef CAS.
- T. Németh, S. Lévai, A. Kormos, J. Kupai, T. Tóth, G. T. Balogh and P. Huszthy, Chirality, 2014, 26, 651–654 CrossRef PubMed.
- B. Szemenyei, I. Móczár, D. Pál, I. Kocsis, P. Baranyai and P. Huszthy, Chirality, 2016, 28, 562–568 CrossRef CAS PubMed.
- J. Shi, L. Liu, J. He, X. Meng and Q. Guo, Chem. Lett., 2007, 36, 1142–1143 CrossRef CAS.
- D. Schweinfurth, K. I. Hardcastle and U. H. F. Bunz, Chem. Commun., 2008, 2203–2205 RSC.
- C. Le Droumaguet, C. Wang and Q. Wang, Chem. Soc. Rev., 2010, 39, 1233–1239 RSC.
- S. S. Bag and R. Kundu, J. Org. Chem., 2011, 76, 3348–3356 CrossRef CAS PubMed.
- Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC.
- P. Kautny, D. Bader, B. Stöger, G. A. Reider, J. Frölich and D. Lumpi, Chem. – Eur. J., 2016, 22, 18887–18898 CrossRef CAS PubMed.
- S. Ast, P. J. Rutledge and M. H. Todd, Eur. J. Inorg. Chem., 2012, 5611–5615 CrossRef CAS.
- J. K.-H. Wong, M. H. Todd and P. J. Rutledge, Molecules, 2017, 22, 200 CrossRef PubMed.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- W. Yan, Q. Wang, Q. Lin, M. Li, J. L. Petersen and X. Shi, Chem. – Eur. J., 2011, 17, 5011–5018 CrossRef CAS PubMed.
- Y. Liu, W. Yan, Y. Chen, J. L. Petersen and X. Shi, Org. Lett., 2008, 10, 5389–5392 CrossRef CAS PubMed.
- Q. Lai, Q. Liu, Y. He, K. Zhao, C. Wei, L. Wojtas, X. Shi and Z. Song, Org. Biomol. Chem., 2018, 16, 7801–7805 RSC.
- Q. Lai, Q. Liu, K. Zhao, C. Shan, L. Wojtas, Q. Zheng, X. Shi and Z. Song, Chem. Commun., 2019, 55, 4603–4606 RSC.
- Y. Zhang, X. Ye, J. L. Petersen, M. Li and X. Shi, J. Org. Chem., 2015, 80, 3664–3669 CrossRef CAS PubMed.
- A. Petrosyan, P. Ehlers, A.-E. Surkus, T. V. Ghochikyan, A. S. Saghyan, S. Lochbrunner and P. Langer, Org. Biomol. Chem., 2016, 14, 1442–1449 RSC.
- Z. Jia and Q. Zhu, Bioorg. Med. Chem. Lett., 2010, 20, 6222–6225 CrossRef CAS PubMed.
- O. Berger, A. Kaniti, C. T. van Ba, H. Vial, S. A. Ward, G. A. Biagini, P. G. Bray and P. M. O’Neill, ChemMedChem, 2011, 6, 2094–2108 CrossRef CAS PubMed.
- S. P. Chavan, S. Garai and K. P. Pawar, Tetrahedron Lett., 2013, 54, 2137–2139 CrossRef CAS.
- X.-J. Quan, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2014, 16, 5728–5731 CrossRef CAS PubMed.
- H. Wang, P. Mi, W. Zao, R. Kumar and X. Bi, Org. Lett., 2017, 19, 5613–5616 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, J. Org. Chem., 2006, 71, 7546–7557 CrossRef CAS PubMed.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed.
- J. A. Riddick, W. B. Bunger and T. K. Sakano, in Techniques of Chemistry, ed. A. Weissberger, Wiley-Interscience, New York, NY, 4th edn, 1986, vol. 2 Search PubMed.
- Z. Köntös, P. Huszthy, J. S. Bradshaw and R. M. Izatt, Tetrahedron: Asymmetry, 1999, 10, 2087–2099 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj04173h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |