
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled reduction of aromaticity of alkylated polyaromatic compounds by selective oxidation using H2WO4, H3PO4 and H2O2: a route for upgrading heavy oil fractions†
Ewa
Nowicka
 *a,
Meenakshisundaram
Sankar
a,
Robert L.
Jenkins
a,
David W.
Knight
a,
David J.
Willock
a,
Graham J.
Hutchings
a,
Manuel
Francisco‡
b and
Stuart H.
Taylor
*a
*a,
Meenakshisundaram
Sankar
a,
Robert L.
Jenkins
a,
David W.
Knight
a,
David J.
Willock
a,
Graham J.
Hutchings
a,
Manuel
Francisco‡
b and
Stuart H.
Taylor
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: nowickae@cardiff.ac.uk; taylorsh@cardiff.ac.uk
bExxonMobil Research & Engineering Company, 1545 Route 22 East, Annandale, New Jersey 08801, USA
First published on 13th July 2021
Abstract
Heavy crude oil fractions which form the residues from fractional distillation are a significant proportion of current hydrocarbon reserves. However, processing residues for use as chemicals or fuels is hampered by the high polyaromatic content of this material. Selective reduction of aromaticity by targeted ring opening and the preservation of alkylated chain side groups are key requirements for the upgrading of alkylated polynuclear aromatic hydrocarbons (PAHs) to more easily processed and higher value molecules. In this study, a H2WO4 catalyst combined with H3PO4 and H2O2 oxidant is applied to the selective oxidation of PAHs containing different lengths of substituent alkylated chain, and different numbers of rings in the fused aromatic core. For a model substrate of 2-ethylnaphthalene, using traditional organic solvents, aliphatic carbon was oxidized more readily compared to aromatic carbon. However, it was found that the oxidation to desired products can be specifically controlled as the selectivity is directed by the choice of solvent, with reactions carried out in acetonitrile giving oxidation only in the aromatic region of the molecule. With larger polyaromatic molecules, a biphasic solvent system is used with Aliquat 336 as a phase transfer agent. Even for this more complex reaction system high conversion to the corresponding alkylated ring opened compounds was obtained.
1. Introduction
Heavy crude oil fractions, residues and bitumens represent between 4 wt% and 16 wt% of current processed crude oil across global production sites.1 As such, this represents a significant and largely under-used feedstock in the petrochemicals industry. Currently, these fractions can only be used in low value applications such as binders for road surfaces and as a waterproofing adhesive in construction. The use of this complex mix of high molecular weight compounds through further upgrading to higher value products would help maximise productivity and enable us to meet an increasing demand for fuels without developing a large number of new reserves.2 The petroleum industry uses three major methods for upgrading: deep oil fluid catalytic cracking, thermal cracking, including delayed coking and hydrocracking with desulfurization. Fluid catalytic cracking converts a high boiling feedstock to more useful petroleum compounds, with the use of high temperature, moderate pressure, and most importantly a fluidized powdered catalyst. Thermal cracking and delayed coking uses no catalyst or additional chemicals to achieve cracking at very high temperatures (450–470 °C).3,4 Hydrocracking on the other hand requires high pressure hydrogen, which can result in over-cracking and formation of low value gas.5 A very low selectivity and high energy demand are the major drawbacks for all these methods.Oxidation offers a more facile route for petroleum residue transformation, and potentially could become a core technology. These heavy fractions consist mostly of alkylated polynuclear aromatic hydrocarbons, (PAHs), so the challenge is to control the oxidation of aromatic carbon whilst preserving alkyl chain side groups.6–9 The fact that PAH molecules are difficult to oxidise is apparent when it is realised that they are also a major component of anthropogenic atmospheric pollution from combustion and industrial activity.10 Their chemical removal in this context is also an area of intense research but with the goal of complete reaction to CO2 and water. We have recently reported that Ruthenium Ion Catalyzed Oxidation (RICO) of alkylated PAH molecules can have the required selectivity for residue processing.11 RICO chemistry finds a wide range of applications in C–H bond activation and oxygen transfer reactions.12 By judicious choice of experimental conditions and solvent system we were able to demonstrate oxidation of aromatic regions of model alkylated PAH compounds, whilst aliphatic carbons were unreacted.13 In RICO chemistry RuCl3 is transformed into a RuO4 oxidising species using NaIO4 as oxidant. The high oxidation state RuO4 moiety shows a strong affinity for double bonds in the aromatic system of PAH molecules that are chemically isolated, i.e. regioselectivity favours the formation of a [3+2] intermediate in positions that lead to minimal loss of aromaticity.14 This provides a route to selectively oxidise aromatic carbon producing predominantly aldehyde and carboxylic acid functionality. However, this ruthenium-based catalytic system relies on an expensive metal and requires a strong oxidant which produces a stoichiometric equivalent of salt waste, therefore a cheaper catalyst and more environmentally benign oxidant would be preferred.
As early as 1959, Payne and Williams demonstrated that a combination of a tungsten based catalyst with H3PO4 and H2O2 as oxidant can give a highly selective oxidative catalytic system for organic synthesis.15 This system later became known as the Ishii-Venturello oxidation system.16,17 This combination of relatively cheap catalyst and high efficiency oxidant has been reported to be active for the oxidation of alcohols,18 aldehydes,19 olefins,16 as well as aromatic hydrocarbons.20,21 A number of studies have been published demonstrating the use of W containing compounds as oxidation catalysts, for example as Keggin type polyoxometylates.22,23 However, tungsten in the form of H2WO4 is a more potent oxidation catalyst, mediating the conversion of PAH molecules such as phenanthrene and pyrene to the corresponding diacids when combined with H3PO4 and using H2O2 as oxidant.20,24 The use of hydrogen peroxide as the primary oxidant is also attractive as it removes the production of salt waste that would occur with the RICO approach. These observations suggest that aqueous H2WO4/H2O2 could be suitable for selective oxidation of alkylated-PAHs to reduce aromaticity. However, oxidation of alkylated PAH molecules is more challenging than the parent PAH compounds exemplified by phenanthrene and pyrene. It is known that the α-carbon, the first aliphatic carbon bound to the aromatic system, is potentially more susceptible to oxidation than the aromatic core itself due to the relatively low C–H bond energy for carbon atoms in the benzyl position. To date there are no studies to establish the influence of alkyl substituents in the oxidation of alkylated-PAH substrates with this catalyst system.
The high molecular weight alkylated PAH molecules found in residues are very hydrophobic requiring a non-polar solvent. In contrast, a polar solvent is needed for the H2O2 oxidant and H2WO4 catalyst so that reactions will usually take place in a biphasic system. While this has advantages in terms of product separation25 it can also result in poor mixing between the substrate and oxidising species. Reaction efficiency can be improved using a phase transfer catalyst and the addition of an acid. Barrio et al.26 have combined experimental findings and theoretical calculations to propose that oxidation in this case begins with the reaction of H2WO4 with H2O2 to form an anionic bisperoxotungstate complex in the aqueous phase. This complex reacts further with phosphonic acid, to give a form that can be transported via the phase transfer catalyst to the organic phase, where oxidation of the substrate takes place. Following substrate oxidation, the monoperoxo tungstate ion moves back to the aqueous layer where it can again react with H2O2 to reform the anion and the cycle repeats itself.
Herein, we investigate the use of the H2WO4/H2O2 catalytic system for the oxidation of a range of alkylated polynuclear aromatic hydrocarbons, as model compounds for upgrading of crude oil residues. For larger molecules we employ a biphasic solvent system with phase transfer agent to improve catalytic efficiency. Our fundamental focus is to examine oxidation catalysed by H2WO4 as an alternative to the previously discussed RICO Chemistry.
2. Experimental
2.1. Oxidation reactions
It is believed that after 6 hours reaction, no oxidant remained in the reaction mixture as it was either consumed in the oxidation reaction or alternatively decomposed.
Comparison of the integrated areas for protons A and D between the 0 and X h samples gives a measure of the percentage of products in which the aliphatic chain remains intact. The detailed methodology of analysing 1H NMR peaks using this technique has been described in a separate publication.11 An example of a 1H NMR spectrum used for the calculation of percentage of preserved alkyl chain is shown in Fig. S1 (ESI†).
In the oxidation of 9-octadecylphenanthrene, the following conditions were used. First the substrate (0.043 g, 0.1 mmol) was dissolved in C6H5Cl (3 ml), H2WO4 (0.038 g, 0.154 mmol), H3PO4 (10% 0.02 ml) and Aliquat 336 (0.0024 g in toluene) were added and stirred for a few minutes. Subsequently H2O2 (50%, 4 ml, 65 mmol) was added dropwise. The reaction was performed at 110 °C. Post reaction analysis steps were the same as above.
Conversion in oxidation reactions of 2-nonylphenanthrene, 9-octadecylphenanthrene and 2-octadecylpyrene was determined using GC, according to the methodology described above for 2-ethylnaphthalene.
2.2. Product analysis
1H NMR, 13C NMR and 2D NMR spectra were recorded on a Bruker DPX 500 MHz instrument. For 13C NMR and 2D NMR analysis, firstly the solvent used in the reaction was evaporated, the sample was then placed under vacuum until it was completely dry and a volume of approx. 1 ml of deuterated solvent added to the dry residue to dissolve it. The solution (0.5–0.7 ml) was subsequently transferred to the NMR tube for analysis.Gas Chromatography–Mass Spectrometry (GC-MS) analysis was performed using a Waters GCT premier instrument fitted with an Agilent HP-5MS column. Unless otherwise stated in the earlier part of the experimental section, samples were analyzed as received from the reaction mixture. A sample of 1 ml of reaction mixture (from both or single solvent layer) was transferred to a GC-MS vial and analyzed without further treatment.
Mass Spectrometry analysis (MS) was performed using a Waters LCT Premier XE (ES), while EI data was generated using a Waters GCT Premier EI (EI) instrument. Similarly to GC-MS analysis, samples were analyzed after the reaction was completed. A volume of approximately 1 ml was transferred to a GCMS vial and analyzed without further treatment.
Infrared (IR) analysis were performed using a Jasco FT/IR-660 spectrometer. Compounds dissolved in dichloromethane solution were deposited on the surface of a NaCl plate. The film formed on the plate was analysed directly.
3. Results and discussion
3.1. Oxidation of 2-ethylnaphthalene
Since our aim was to investigate the selectivity trends for alkylated aromatics, this study focused primarily on 2-ethylnaphthalene, (1), as it is a simple alkylated polynuclear aromatic compound with the advantage that monophasic and biphasic solvent environments can be compared. In previous reports we have shown that the oxidation of 2-ethylnaphthalene using RICO chemistry leads to oxidation of the aromatic carbon region of the molecule, whilst there was little or no oxidation of the alkyl side chain.11 This observation was independent of the solvent system used, as in both monophasic and biphasic solvent systems aromatic carbons were predominantly oxidized. In this work, a range of solvents, which differ in their polarity index,27 were evaluated for oxidation of 2-ethylnaphtalene using H2WO4/H2O2 and the main products observed are presented in Table 1.| Entry | Organic solvent | Polarity indexa (εr) | React. Tb/°C | Con.c /% | Major products |
|---|---|---|---|---|---|
| Note:a εr = dielectric constant.b Reaction temperature.c Conversion.Reaction conditions: 2-ethylnaphthalene 18.8 mmol, H2WO4 0.153 g (0.614 mmol), H2O2 35% 12 ml (137 mmol), Aliquat 336 0.24 ml, H3PO4 10% 0.14 ml, Solvent 8 ml, t = 6 h. | |||||
| 1 | Toluene C6H5CH3 | 2.4 (2.38) | 80 | 30 |

|
| 2 | Chlorobenzene C6H5Cl | 2.7 (5.62) | 80 | 52 |
|
| 3 | Propionitrile C2H5CN | 3.9 (27.7) | 75 | 16 |

|
| 4 | Acetonitrile CH3CN | 5.8 (37.5) | 65 | 12 |

|
Oxidation of 2-ethylnaphthalene using a biphasic system with toluene as the organic solvent (the lowest polarity index) resulted in formation of acetonaphthone, (2), with high selectivity (91%, Table 1, entry 1, Fig. S2, ESI†), alongside other minor products. This shows that in a biphasic system with very different solvent polarities oxidation of the α-carbon of the ethyl side chain of the molecule is the preferred reaction pathway.
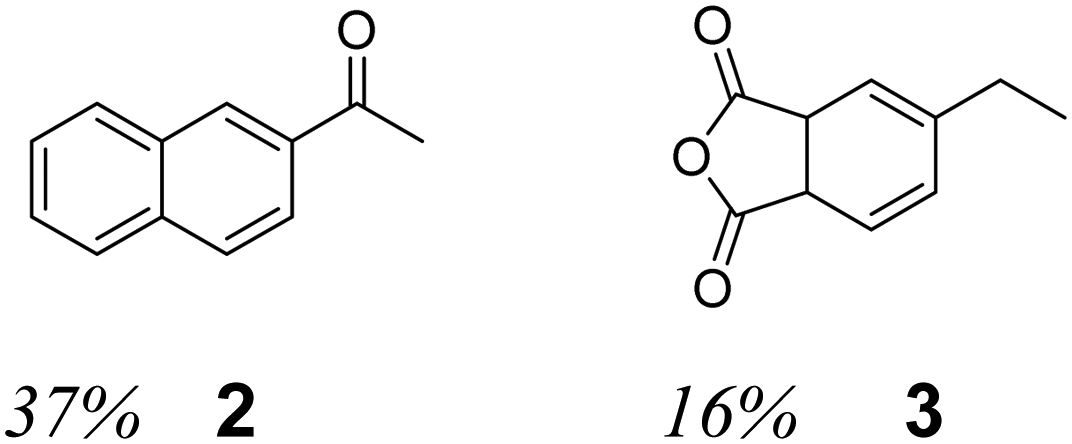
When chlorobenzene is used as the organic phase solvent, GC-MS analysis (Fig. S3, ESI†), shows the formation of two major products (Table 1, entry 2); acetonaphthone, (2), 37% and 5-ethyl phthalic acid anhydride, (3), 16%, suggesting that both aromatic and aliphatic carbon can be oxidized.
The oxidation of 2-ethylnaphthalene with proponitrile as the organic solvent of the biphasic reaction mixture was carried out at a lower temperature than those used for the less polar solvents (75 °C cf. 80 °C), due to its lower boiling point. A notably lower conversion is seen compared to the toluene and chlorobenzene systems and the major product, naphthalene-2-yl-ethanol (4), is only oxidized at the α-carbon of the alkyl chain. No acetonapthone was detected (Table 1, entry 3 and Fig. S4, ESI†), as would be expected from the further oxidation of (4).
The highest polarity solvent considered for the organic phase was acetonitrile (Table 1, entry 4 and Fig. S5, ESI†). Acetonitrile has been previously reported to have a positive influence on the selectivity in RICO chemistry,28 and so it was investigated in more detail for this new work with H2WO4/H2O2. The high polarity index of acetonitrile meant that attempts to use it as the organic solvent led to a monophasic reaction mixture and its low boiling point (82 °C) required the reaction temperature to be further reduced to 65 °C. A range of products were formed from 2-ethylnaphthalene oxidation using H2WO4/H2O2 in acetonitrile. The major products were: 5-ethyl phthalic acid anhydride (3), 6-ethylnaphthalene-1,4-dione (5) and 6-ethyl-2-hydroxynaphthalene-1,4-dione (6), all of these preserved the ethyl chain, with only one aromatic ring oxidized as confirmed by 13C NMR analysis of the product mixture. Indeed, a comparison of 13C NMR spectra for the product mixture following 20 h of reaction with that of a pure standard of 2 showed no peak corresponding to the ketone group of acetonaphthone (2). This is the expected alkyl oxidation product that had been observed for reaction in toluene and chlorobenzene following oxidation at the α-carbon of the alkyl side chain (Fig. S6, ESI†). This observation suggests that acetonitrile can provide a solvent environment able to direct oxidation to the aromatic region. These results differ from previously reported oxidation of 2-ethylnaphthalene in acetonitrile, as products in which unreacted and oxidized ethyl chain products were observed. However, that earlier study used tungsten polyoxometalates rather than tungstic acid and also employed a different ratio of substrate, catalyst and oxidant.22
The selectivity for the reaction catalysed with H2WO4/H2O2 also differed significantly from the oxidation of 2-ethylnaphtalene using RICO chemistry. RICO gave a range of products for which the unreacted ethyl chain was observed (e.g. phthalaldehyde, isobenzofuran-1,3-dione, 5-ethylisobenzofuran-1,3-dione).11 In our RICO work, selectivity was not affected by the choice of solvent for 2-ethylnaphtahlene oxidation (CH3CN, H2O or CH3CN, DCM, H2O).11,14 However, it was shown that, using RICO, the solvent affects the selectivity for oxidation of molecules with three or more aromatic rings, giving a lower degree of aliphatic carbon oxidation in the presence of acetonitrile.13
A possible explanation for the change of selectivity with solvent for 2-ethylnaphtalene using the H2WO4/H2O2 system could be the distribution of the tungsten complex in the reaction medium. Introduction of acetonitrile allows the reaction to occur in a single homogeneous phase, presumably improving mixing of the catalyst, substrate and oxidant. The bisperoxotungstate complex, that forms initially from reaction of H2WO4 with H2O2, can more easily move through the reaction medium without the need for a phase transfer agent. A similar observation was reported by Noyori et al.29 Tungsten is reduced as the substrate is oxidised, and so, once it has oxidized the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or C–H bond in a biphasic system the tungsten complex has to transfer back to the aqueous phase. In the monophasic system provided by acetonitrile the additional phase transfer steps are eliminated.30 Even so, the use of acetonitrile as a solvent raises a problem of substrate solubility for higher molecular weight alkylated PAH molecules and limits the reaction temperature in an open reaction vessel because of the relatively low solvent boiling point. Therefore, for the more general problem of residue upgrading, the use of non-polar solvents is difficult to avoid. To address this point, further experiments with alkylated aromatic hydrocarbons containing more fused aromatic rings were performed in chlorobenzene.
C or C–H bond in a biphasic system the tungsten complex has to transfer back to the aqueous phase. In the monophasic system provided by acetonitrile the additional phase transfer steps are eliminated.30 Even so, the use of acetonitrile as a solvent raises a problem of substrate solubility for higher molecular weight alkylated PAH molecules and limits the reaction temperature in an open reaction vessel because of the relatively low solvent boiling point. Therefore, for the more general problem of residue upgrading, the use of non-polar solvents is difficult to avoid. To address this point, further experiments with alkylated aromatic hydrocarbons containing more fused aromatic rings were performed in chlorobenzene.
3.2. Oxidation of polyaromatics with larger aromatic systems and longer alkyl chains
Various factors, such as the size of the aromatic ring system and substitution pattern of alkyl substituted PAHs can influence the regioselectivity in oxidation reactions between the aromatic and aliphatic regions of the molecule.31 In our studies we focus not only on the effect of aromatic system size, but also on understanding how different substituent chain lengths and substitution positions influence the selectivity of the reaction.Three different compounds were chosen for this study, the substituted three ring PAH molecules; 2-nonylphenanthrene (7) and 9-octadecylphenanthrene (8), and the substituted four ring system 2-octadecylpyrene (9). To determine the selectivity of the oxidation reaction between aromatic and aliphatic regions, the 1H NMR quantification methodology developed previously and summarised in the Experimental section, was applied (Fig. S1, ESI†).11
Table 2 summarises the conversion and proportion of preserved aliphatic hydrogen for each of the substrates. It is clear that conversion is dependent upon the number of fused aromatic rings present in the substrate, with higher conversion for the larger ring system of 2-octadecylpyrene (9). The alkyl chain length has relatively little influence on the conversion as 7 and 8 show very similar values. However, comparison of the measured percentage preserved aliphatic hydrogen for substrates 7 and 8 does suggest that the location of the alkyl chain plays a crucial role in reaction selectivity. Aliphatic carbon at position 9 of the aromatic phenanthrene system in 8 is more susceptible to oxidation than carbon at position 2 of the same aromatic system (standard atom numberings are shown in Fig. S7, ESI†). It has been found from DFT calculations for the RICO oxidation system, that positions 9 and 10 in the phenanthrene ring system are the most energetically favourable positions for oxidation as the removal of these carbons from the aromatic system has only a limited effect on the resonance structures available.14 The α-carbon of an alkyl chain substituted at these positions is likely to be placed close to the oxidant making it more susceptible to oxidation. The equivalent positions in pyrene are 4, 5, 9 and 10 so that the high level of preserved aliphatic H for 9 can be understood in a similar way.
| Substrate | Conversion/% | Preserved aliphatic H/% |
|---|---|---|
| Reaction conditions: substrate 0.1 mmol, H2WO4 0.019 g (0.077 mmol), H2O2 35% 2 ml(22.8 mmol), Aliquat 336 0.002 g (in 1 ml C6H5CH3 solution), H3PO4 10% 0.01 ml, C6H5Cl 3 ml, T = 80 °C, t = 16 h. | ||
| 2-Nonylphenanthrene, (7) | 48 | 78 |
| 9-Octadecylphenanthrene, (8) | 46 | 40 |
| 2-Octadecylpyrene, (9) | 95 | 93 |
Quantification of the extent of preserved alkyl chain provides a key indicator of catalyst efficacy, but it is still important to positively identify the reaction products. We have used a combination of analytical techniques to suggest the most likely dominant product species in these oxidation reactions. Fig. S8 (ESI†) gives the IR spectrum for the product mixture from the organic layer following 2-octadecylpyrene (9) oxidation. This shows broad bands at 1698.5 cm−1 and 1735.6 cm−1, characteristic of the CO stretching modes associated with saturated non-aromatic aldehydes and aromatic carboxylic acids, respectively. Additionally, the broad peak observed in the –OH region suggests the presence of alcohol or carboxylic acid groups. MS analysis identified a product with m/z = 581.31, with a suggested formula of C34H45O8, (Fig. S9, ESI†). In pyrene we have noted that the positions 4, 5, 9 and 10 are the most easily oxidised CC bonds, so that a likely candidate for this oxidation product is 4-octadecylbiphenyl-2,2′,6,6′-tetracarboxylic acid (10). Fig. S1 (ESI†) shows the 1H NMR spectra of 9 and that of the oxidation product mixture. Regarding protons in position α to the aromatic ring; oxidation of the α-C–H bond to give CO is a known reaction and would reduce the intensity of the 3.20 ppm resonance (marked A in Fig. S1, ESI†). A new resonance would also be expected between 2.90–3.00 ppm, for the C–H protons in the aliphatic chain that become α to the ketone group and β to the aromatic ring. Some new features are seen in the 1H NMR spectrum of reaction products but these could also be caused by changes in the chemical shifts of protons α to the aromatic ring system due to oxidation of aromatic carbon. To clarify the situation, Heteronuclear Multiple Bond Correlation (HMBC) 2D NMR spectroscopy was used, which allows the correlation of chemical shifts of two types of nuclei separated from each other by two or more chemical bonds. Using this technique it was possible to observe aromatic protons which were correlated to C from the CO region, even when those carbons did not give visible signals during standard 13C NMR analysis. Fig. 1 shows the HMBC 2D spectra with features correlating the CO region, of the 13C NMR spectrum with the aromatic protons (C–H) marked confirming that oxidation takes place in the aromatic region of the substrate away from the alkyl substituent, as would be expected for the proposed main product, 10.
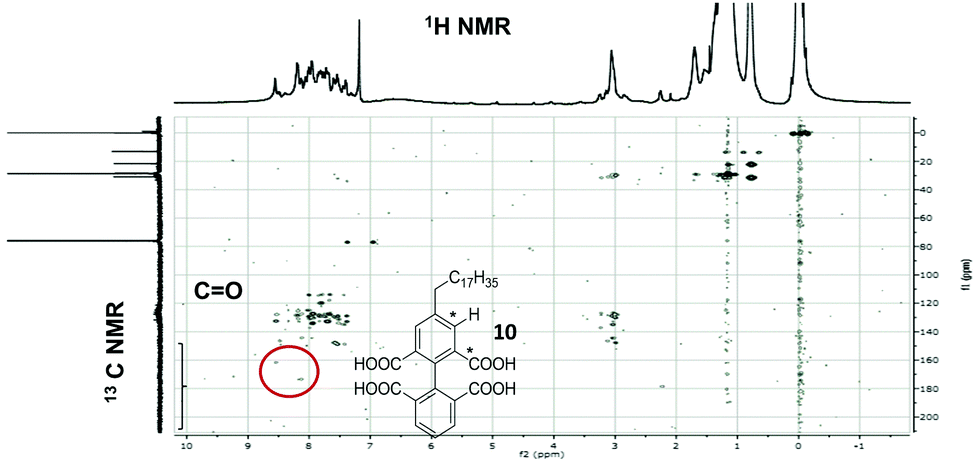 | ||
| Fig. 1 HMBC (500 MHz, CDCl3) spectrum of 2-octadecylpyrene oxidation products (organic layer). The structure of the main product-octadecylbiphenyl-2,2′,6,6′-tetracarboxylic acid, and the correlating elements circled in the spectra, are marked by an * in the chemical structure. | ||
The combined evidence from IR, MS, 1H NMR and HMBC shows that the major product of 2-octadecylpyrene oxidation has a preserved aliphatic chain. The product contains four carboxylic acid groups and this, coupled with the knowledge of the regioselectivity preference for oxidation of pyrene, strongly suggests that 4-octadecylbiphenyl-2,2′,6,6′-tetracarboxylic acid (10) is the major product (Scheme 1). Even without specific product assignment, these observations confirm that the H2WO4/H2O2 system delivers the desired reaction selectivity, of ring opening and reducing aromaticity while preserving aliphatic carbon side chains.
 | ||
| Scheme 1 2-Octadecylpyrene and its major oxidation product. | ||
For phenanthrene positions 9 and 10 (Fig. S7, ESI†) are the most susceptible to oxidation.11 In the case of 2-octadecylpyrene, (7) the corresponding positions (4, 5, 9 and 10) were not hindered by the substituent, and the aromatic region of the molecule was readily oxidized with a high conversion of the substrate (95%, Table 1) with 93% of the products retaining the aliphatic substituent intact. When the alkyl substituent is at a position that favours aromatic oxidation, such as the 9 position of 9-octadecylphenanthrene, (8), 1H NMR quantification analysis suggests that only 40% of molecules in the post reaction mixture had a preserved alkyl chain. However, only 46% of 8 was converted, so the amount of preserved alkyl chain in the oxidised products will be masked by unreacted substrate. Fig. S10 (ESI†) shows the MS spectrum for the reaction mixture after oxidation of 8. This confirmed a signal at m/z = 430.32 corresponding to unreacted substrate, 8, and two strong peaks at higher m/z values consistent with the addition of one oxygen atom (m/z = 446.36) and two oxygen atoms (m/z = 462.34). The second peak corresponds to 2-nonadecanoylbiphenyl-2-carbaldehyde (11) (Scheme 2), as an oxidation product. As further support for this assignment 1H NMR spectroscopy confirmed the presence of the aldehyde group (9.75 ppm, Fig. S11, ESI†).
 | ||
| Scheme 2 9-Octadecylphenanthrene and a possible product of oxidation consistent with MS analysis. | ||
The likely presence of (11) suggests that the aromatic CC bond was cleaved by oxidation at the C9–C10 bond position with high selectivity, so that the aromatic–aliphatic C–C bond was preserved with a ketone formed at the new α-carbon position. These data provide new insight into the selectivity achievable using the H2WO4/H2O2 oxidation system and demonstrates that the chemistry is potentially useful for upgrading of polyaromatic residuals with minimal loss of carbon.
3.3. Kinetic studies
The oxidation reactions were also monitored as a function of time to compare the oxidation rates of molecules possessing different numbers of aromatic rings, and varying lengths of alkyl chain. Previously, using RICO chemistry, we found that the number of aromatic rings in a substituted PAH determines the reaction rate, with larger aromatic systems reacting faster than smaller ones.13 In the case of the tungsten/peroxide catalyst system, when naphthalene, (12), phenanthrene, (13), and pyrene, (14), were used as substrates, it was also found that the reaction rate increased with the increasing number of fused aromatic rings (Fig. 2). This observation means that aromaticity in higher molecular weight polyaromatics can be reduced preferentially in the presence of lower molecular weight ones. | ||
| Fig. 2 Comparison of amount of substrate remaining as a function of time for polyaromatic hydrocarbons during competitive reaction between naphthalene (■), phenanthrene (●) and pyrene (▲). | ||
Experiments to determine the competitive rates of oxidation of alkylated aromatics, using 1-decylnaphthalene (15), 2-nonylphenanthrene (7) and 2-octadecylpyrene (9) were also performed (Fig. 3). The difference in reaction rate between 1-decylnaphthalene and 2-nonylphenanthrene was small but it is clear that the phenanthrene derivative reacts faster than the substituted naphthalene example. The difference in alkyl chain length is only one methylene group, so the alkyl chain length would not be expected to be a controlling factor. Moreover, 2-octadecylpyrene was oxidized at a higher rate than other substrates, confirming that the number of fused rings is the key factor determining the rate of oxidation. The same trend was observed when RICO chemistry was used in oxidation of alkylated PAHs.13 This shows that the H2WO4/H2O2 catalytic system, similarly to RICO, possesses the ability to selectively reduce the aromaticity, it is also a more attractive approach due to the lower cost of both catalyst and oxidant.
 | ||
| Fig. 3 Comparison of oxidation rates for the competitive reaction between 1-decylnaphthalene (■), 2-nonylphenanthrene (●), and 2-octadecylpyrene (▲). | ||
4. Conclusions
The results presented clearly indicate that the H2WO4/H2O2 catalyst system is promising for the selective oxidation of polynuclear aromatic hydrocarbons to reduce aromaticity. It has been shown that the selectivity of oxidation can be tuned by changing the solvent employed in the reaction. Acetonitrile was the best solvent tested in terms of the selectivity for oxidation of the aromatic over the aliphatic region of 2-ethylnaphthalene. This monophasic reaction resulted in the preservation of the alkyl substituent, while reducing the aromatic content. In the homogeneous solvent phase, aromatic oxidation appears more favoured and ring opening to acids can be achieved. For PAHs containing more fused rings the use of acetonitrile is not practical, and a biphasic system, with a less polar organic solvent is required to solubilise the PAH compounds. In these cases, it is still possible to oxidise aromatic regions of the molecules without affecting the alkyl side chains. The aliphatic side chain does not significantly affect the rate of the reaction, but the position of substitution on the aromatic core has an affect on both conversion and degree of oxidation at the α-carbon position of the alkyl side groups. The relative reaction rates for molecules with different numbers of fused rings was also established, with larger ring systems oxidised at a higher rate than molecules with smaller ones. In general, the H2WO4/H2O2 oxidation system can be considered to have demonstrated promising chemistry with respect to valorisation of crude oil residues. Judicious choice of solvent gives an oxidation system capable of achieving the required activity and selectivity to produce higher value products with only minor loss of carbon.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge research funding from ExxonMobil.References
- M. S. Rana, V. Sámano, J. Ancheyta and J. A. I. Diaz, Fuel, 2007, 86, 1216–1231 CrossRef CAS.
- J. G. Speight, The Chemistry and Technology of Petroleum, Taylor and Francis Group, 4th edn, 2006 Search PubMed.
- S. A. Treese, P. R. Pujado and D. S. J. Jones, Handbook of Petroleum Processing, Springer International Publishing, 2nd edn, 2015 Search PubMed.
- A. N. Sawarkar, A. B. Pandit, S. D. Samant and J. B. Joshi, Can. J. Chem. Eng., 2007, 85, 1–24 CrossRef CAS.
- C. Leyva, M. S. Rana, F. Trejo and J. Ancheyta, Ind. Eng. Chem. Res., 2007, 46, 7448–7466 CrossRef CAS.
- C. Djerassi and R. R. Engle, J. Am. Chem. Soc., 1953, 75, 3838–3840 CrossRef CAS.
- F. Shi, M. K. Tse and M. Beller, Adv. Synth. Catal., 2007, 349, 303–308 CrossRef CAS.
- J. Hu, D. Zhang and F. W. Harris, J. Org. Chem., 2005, 70, 707–708 CrossRef CAS PubMed.
- J. W. Cook and R. Schoental, J. Chem. Soc., 1948, 170–173 RSC.
- A. Mehra, Y. Wang, J. E. Krechmer, A. Lambe, F. Majluf, M. A. Morris, M. Priestley, T. J. Bannan, D. J. Bryant, K. L. Pereira, J. F. Hamilton, A. R. Rickard, M. J. Newland, H. Stark, P. Croteau, J. T. Jayne, D. R. Worsnop, M. R. Canagaratna, L. Wang and H. Coe, Atmos. Chem. Phys., 2020, 20, 9783–9803 CrossRef CAS.
- E. Nowicka, M. Sankar, R. L. Jenkins, D. W. Knight, D. J. Willock, G. J. Hutchings, M. Francisco and S. H. Taylor, Chem. – Eur. J., 2015, 21, 4285–4293 CrossRef CAS PubMed.
- B. Plietker, Synthesis, 2005, 2453–2472 CrossRef CAS.
- E. Nowicka, T. J. Clarke, M. Sankar, R. L. Jenkins, D. W. Knight, S. Golunski, G. J. Hutchings, D. J. Willock, M. Francisco and S. H. Taylor, Chem. – Eur. J., 2018, 24, 655–662 CrossRef CAS PubMed.
- E. Nowicka, N. W. Hickey, M. Sankar, R. L. Jenkins, D. W. Knight, D. J. Willock, G. J. Hutchings, M. Francisco and S. H. Taylor, Chem. – Eur. J., 2018, 24, 12359–12369 CrossRef CAS PubMed.
- G. B. Payne and P. H. Williams, J. Org. Chem., 1959, 24, 54–55 CrossRef CAS.
- C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831–3833 CrossRef CAS.
- Y. Ishii and Y. Sakata, Dioxygen Activation and Homogeneous Catalytic Oxidation, Elsevier Science Publishers B. V., 1991, pp. 411–416 Search PubMed.
- S. E. Jacobson, D. A. Muccigrosso and F. Mares, J. Org. Chem., 1979, 44, 921–924 CrossRef CAS.
- K. Sato, M. Hyodo, J. Takagi, M. Aoki and R. Noyori, Tetrahedron Lett., 2000, 41, 1439–1442 CrossRef CAS.
- Y. Saito, S. Araki, Y. Sugita and N. Kurata, US Pat., 4831189, 1989 Search PubMed.
- D. C. Duncan, R. C. Chambers, E. Hecht and C. L. Hill, J. Am. Chem. Soc., 1995, 117, 681–691 CrossRef CAS.
- A. C. Estrada, M. M. Q. Simðes, I. C. M. S. Santos, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, ChemCatChem, 2011, 3, 771–779 CrossRef CAS.
- R. Tayebee, J. Korean Chem. Soc., 2008, 52, 23–29 CrossRef CAS.
- E. R. R. Young and R. L. Funk, J. Org. Chem., 1998, 63, 9995–9996 CrossRef CAS.
- G. B. Payne and C. W. Smith, J. Org. Chem., 1957, 22, 1682–1685 CrossRef CAS.
- L. Barrio, M. Campos-Martın and L. G. Fierro, J. Phys. Chem. A, 2007, 111, 2166–2171 CrossRef CAS PubMed.
- I. M. Smallwood, Handbook of Organic Solvent Properties, Butterworth-Heinmann, Oxford, 1996 Search PubMed.
- P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936–3938 CrossRef CAS.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310–8311 CrossRef CAS PubMed.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- Z. Jing, S. Rodrigues, E. Strounina, M. Li, B. Wood, J. R. Underschultz, J. S. Esterle and K. M. Steel, Int. J. Coal Geol., 2019, 201, 1–13 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj01986d |
| ‡ Deceased March 9, 2016. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |