
Chalcogen bonding interactions in chelating, chiral bis(selenocyanates)†
Huu-Tri
Huynh
a,
Olivier
Jeannin
a,
Emmanuel
Aubert
b,
Enrique
Espinosa
b and
Marc
Fourmigué
 *a
*a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: marc.fourmigue@univ-rennes1.fr
bUniversité de Lorraine, CNRS, CRM2, F-54000 Nancy, France
First published on 24th November 2020
Abstract
Introduction of methyl substituents on the achiral 1,2-bis(selenocyanatomethyl)benzene leads to a novel chelating ChB donor, namely 1,2-bis(1-selenocyanatoethyl)benzene (1), as a mixture of three diastereomers, the two anti enantiomers and the syn (meso) form. Structure determinations show the recurrent formation of short Se⋯N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C ChB interactions in both the anti (racemic mixture) and syn isomers. Co-crystallization of anti-1 with 4,4′-bipyridine affords a 2
C ChB interactions in both the anti (racemic mixture) and syn isomers. Co-crystallization of anti-1 with 4,4′-bipyridine affords a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct, (anti-1)2(bipy), with one very short Se⋯NPy ChB (RR = 0.87). Co-crystallization of anti-1 with tetraphenylphosphonium halides (Cl−, Br−, I−) provides 1:1 adducts while a 2:1 adduct is isolated between syn-1 and Et4NCl, formulated as Et4N+[(syn-1)2Cl−]. Comparison of chloride chelation with anti-1 and syn-1 shows much shorter (NC)Se⋯Cl− ChB interactions with the syn isomer, tentatively rationalized on the basis of theoretical calculations of (i) the electrostatic surface potential of neutral ChB donors and (ii) the chloride BSSE complexation energy.
:1 adduct, (anti-1)2(bipy), with one very short Se⋯NPy ChB (RR = 0.87). Co-crystallization of anti-1 with tetraphenylphosphonium halides (Cl−, Br−, I−) provides 1:1 adducts while a 2:1 adduct is isolated between syn-1 and Et4NCl, formulated as Et4N+[(syn-1)2Cl−]. Comparison of chloride chelation with anti-1 and syn-1 shows much shorter (NC)Se⋯Cl− ChB interactions with the syn isomer, tentatively rationalized on the basis of theoretical calculations of (i) the electrostatic surface potential of neutral ChB donors and (ii) the chloride BSSE complexation energy.
Introduction
Non-covalent interactions1 are currently developing as a cross-field area encompassing many different topics found in crystal engineering, anion recognition and transport, catalysis, biochemistry and material science. Following the huge development of halogen bonding (XB) in the last 25 years,2 it became clear that elements of groups 14, 15 and 16 of the periodic table were also prone to exhibit electrophilic sites able to interact with a charge-concentrated area, in a way very similar to that shown by halogens where the development of an electron-deficient region (also called σ-hole)3 in the prolongation of the covalent bond to the halogen allows for strong and directional interactions with Lewis bases. These newcomers to the fields, described as tetrel (TrB),4 pnictogen (PnB)5 and chalcogen bonds (ChB),6 albeit much less developed than XB, present some specificities and offer new possibilities particularly on the field of supramolecular self-assembly and crystal engineering,6a,7 catalysis,8 anion recognition9 and transport,10 and biochemistry.11 If we focus on ChB, one main difference with XB is the presence, on the chalcogen atom when properly activated, of not one but two electron-depleted regions, located approximately in the extension of the two covalent bonds to the chalcogen.3,12,13 This added element of complexity has several important structural implications such as: (i) possible deviations between the Ci–Ch and Ch⋯X (X = Lewis base) axes, and (ii) possibility for dissymmetry of chalcogen substitution implying a dissymmetry of the two σ-hole area. As a consequence, the very strong predictability of XB interactions finds in the analogous ChB systems some limitations, which have hampered its extensive use in crystal engineering strategies. Several prominent examples have however successfully overcome these problems, such as the self-assembly of 1,2-tellurazole 2-oxides into a variety of supramolecular aggregates,14 the use of bis-(selenophene/tellurophene) derivatives as chelating systems toward anions (Scheme 1a),10,15,16 or double chalcogen bonding interactions exhibited by benzo-1,3-chalcogenazoles17 or chalcogenadiazoles.18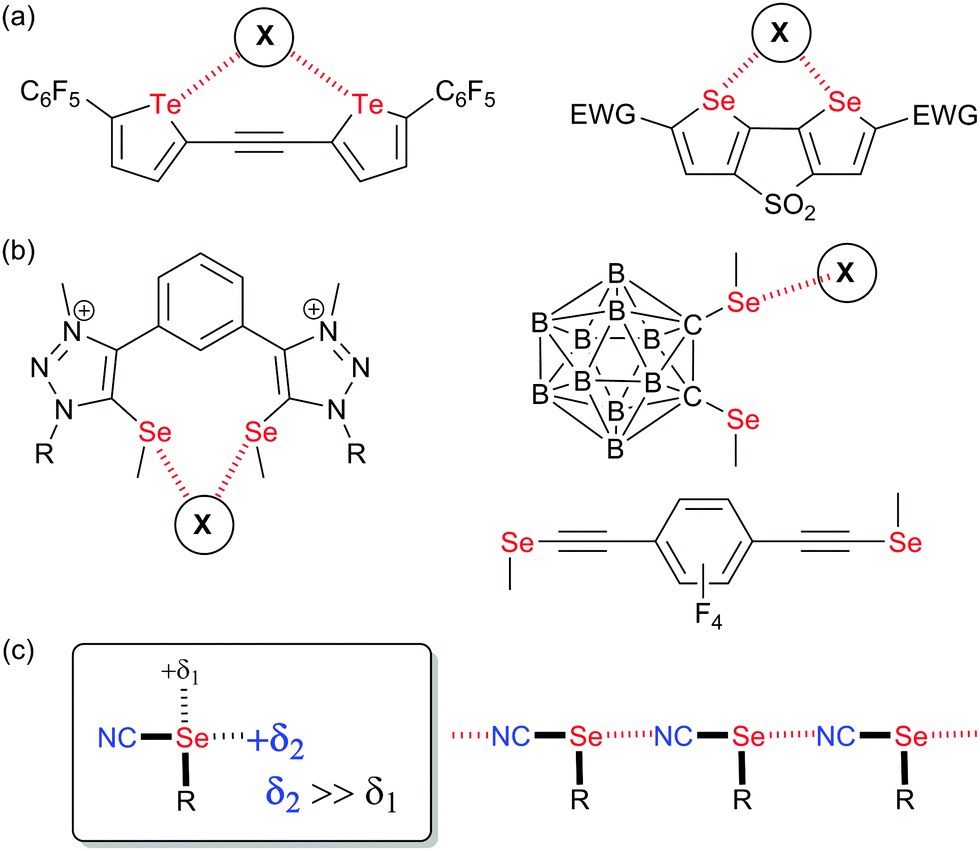 | ||
| Scheme 1 (a) Structures of reported ChB donors chelating halides. (b) Systems with strongly asymmetric selenium activation (c) Organic selenocyanates: Sigma-holes and solid-state association. | ||
Along these lines, one attempt to restore in ChB systems the strong directionality offered by halogens consists in functionalizing the chalcogen atom with only one strong electron-withdrawing substituent, as for example in rotaxanes incorporating selenomethyltriazolium moieties,19 in selenomethyl- or telluromethyl-acetylenes,20 in icosahedral ortho-carboranes substituted with two methylseleno or methyltelluro groups (Scheme 1b),21 or in selenocyanate derivatives R–SeCN (Scheme 1c).22,23 In such compounds, the electron-withdrawing character of the nitrile substituents strongly activates one of the two σ-holes, in the prolongation of the NC–Se bond, allowing to recover the predictability of interaction with Lewis bases. Indeed, selenocyanates themselves most often crystalize into chains ⋯NC–(R)Se⋯NC–(R)Se⋯ where the lone pair of the nitrogen atom of the nitrile interacts through ChB with the selenium atom showing Se⋯N distances around 3.0 Å, that is a reduction ratio RR (defined as the actual interatomic distance over the sum of the van der Waals radii) around 0.86. Recently, we reported several bis- or tetrakis-substituted selenocyanate derivatives such as A–D in Scheme 2a,23 easily prepared from the corresponding benzylic bromides and KSeCN, which were shown to organize in the solid state with these recurrent chain-like motifs. These ChB interactions are notably enhanced when the selenocyanate is faced with stronger Lewis bases such as pyridines24 or halide anions.25 A remarkable feature of the ortho derivative A (or the 1,2,4,5-tetrakis derivative D) is its ability to chelate one single atom through the two selenium atoms, giving rise to seven-membered rings with either neutral (DMF) or anionic (halides) Lewis bases (Scheme 2b). These interactions were also confirmed in solution by 13C and 77Se NMR.26 With the ChB donors A and D, two specific conformations of the ortho-selenocyanatomethyl arms were observed on the solid state upon halide chelation, namely syn or anti, depending on the halide anion, its coordination number and the associated counter ion, without possibility to evaluate the relative stability of both conformations. In order to clarify this point and possibly favor one over the other conformation in these halide recognition processes, we designed an ortho bis(selenocyanate) derivative 1 analogous to A, but bearing an extra methyl group on each benzylic bridge (Scheme 2c). Compound 1 does thus exist as three diastereomers, the two anti enantiomers and the syn (meso) form. We describe here its synthesis, separation of the anti and syn forms and their association with neutral (4,4′-bipyridine) and charged (halide anion) Lewis bases. This novel ChB donor provides also a rare example of introduction of chirality in chalcogen-bonded systems.27 Indeed, only a few examples of selenylation reagents were reported where an intramolecular Se⋯N or Se⋯O interaction rigidifies the chiral reagent and thus allows for stereochemical control,6c,28 while recently a planar chiral ferrocenyl plateform was functionalized with iodomethyl- and selenomethyl-ethynyl moieties29 for evaluation in the Ritter reaction.30
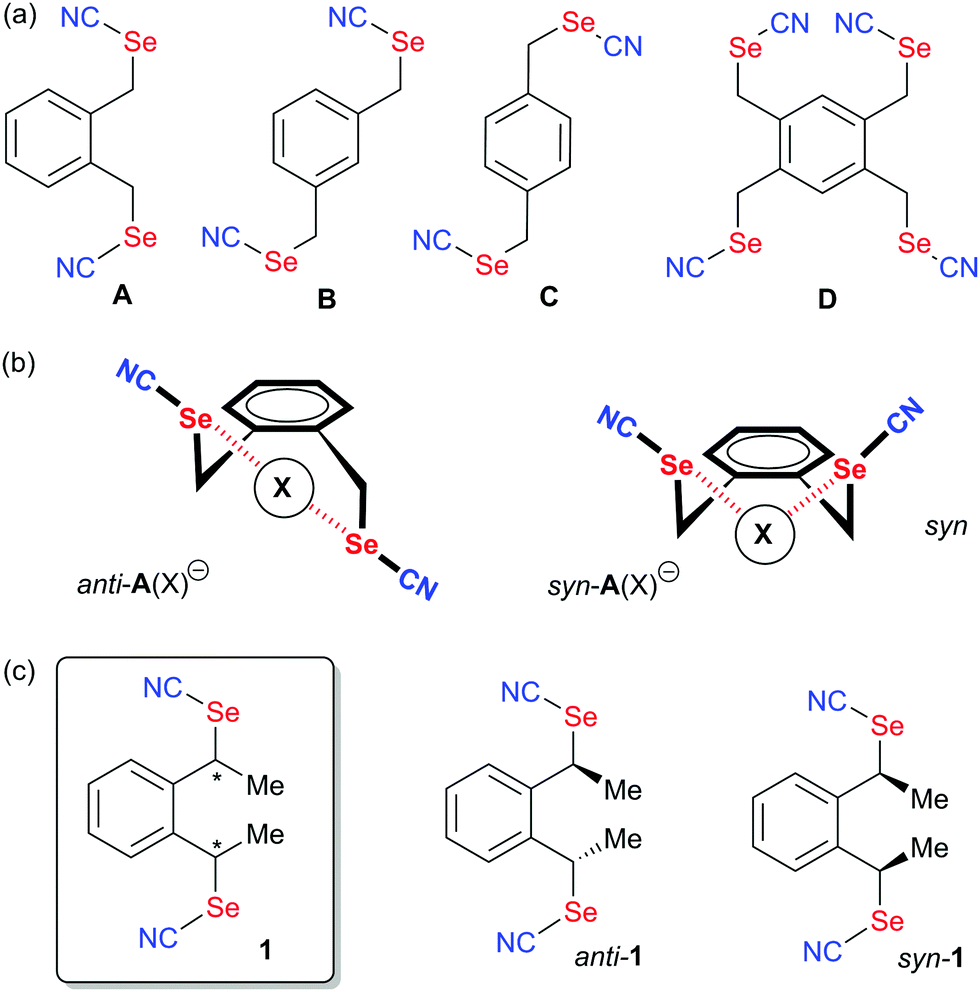 | ||
| Scheme 2 Benzylic selenocyanates and their halide chelates. | ||
Results and discussion
Syntheses
The preparation of 1 is based on the nucleophilic substitution of 1,2-bis(1-bromoethyl)benzene 2 with KSeCN (Scheme 3). The preparation of 2 from reaction of 1,2-diethylbenzene with NBS has been reported to afford a diastereoisomeric mixture in a 3:1 ratio.31 Recrystallization was reported to yield a crystalline material composed essentially of the majority compound, whose stereochemistry was not assigned at that time. Based on the product obtained from the majority dibromo compound when reacted with glycine,32 the main diastereoisomer was later shown to be the anti one.
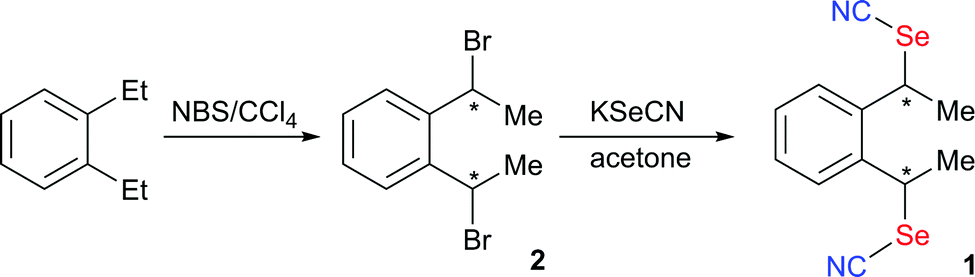 | ||
| Scheme 3 Synthetic path to 1. | ||
We have performed the bromination reaction of 1,2-diethylbenzene in the same conditions and isolated indeed a 70:30 anti-syn mixture. Recrystallization from hexane afforded the pure anti isomer whose stereochemistry was confirmed here by single crystal X-ray diffraction. Concentration of the mother liquors gives a 23:77 anti-syn mixture based on NMR. The dibromo derivative anti-2 crystallizes in the monoclinic system, S. G. P21/n, with one molecule in general position (Fig. 1a). No short intermolecular Br⋯Br interactions are identified. Nucleophilic substitution with KSeCN was not stereochemically conservative and afforded a mixture of anti- and syn-1, in 3:1 ratio when performed from pure anti-2. Recrystallization from acetone allowed to isolate anti-1 in a pure form. It crystallizes in the monoclinic system, S. G. P21/a, with one molecule in general position (Fig. 1b) showing one of the SeCN moieties disordered over two positions with 60:40 refined occupancy. From the concentrated mother liquors, some crystals of syn-1 were isolated with difficulties after hexane diffusion. The syn isomer was found to crystallize in the monoclinic system, S. G. P21/c, with one molecule in general position (Fig. 1c).
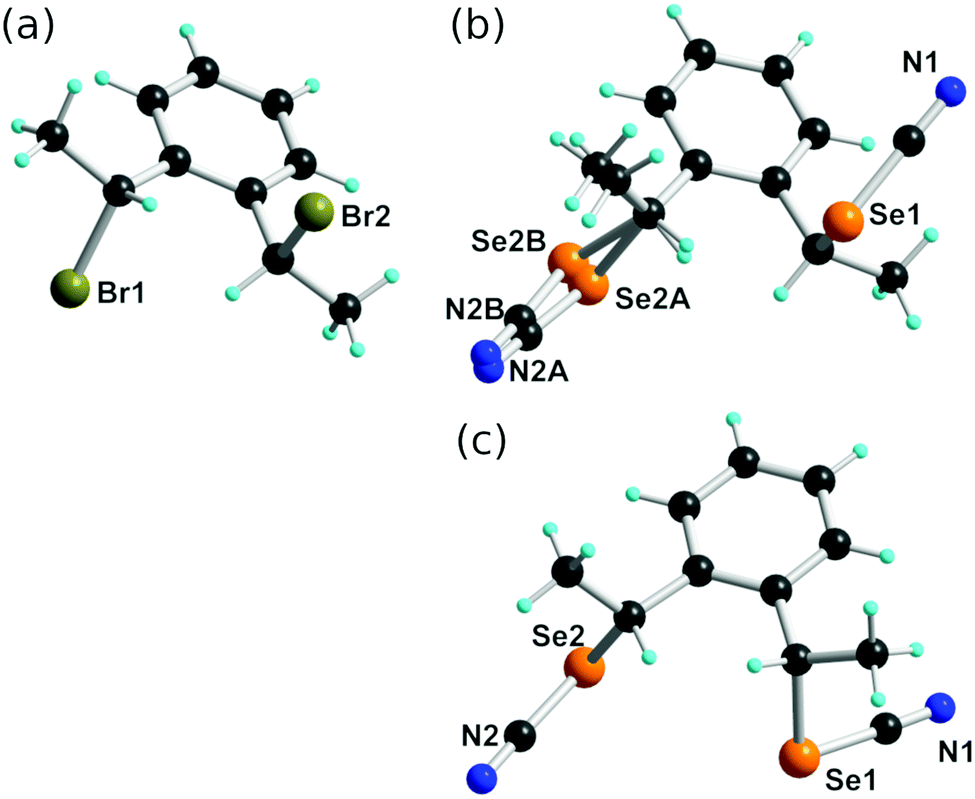 | ||
| Fig. 1 Detail of the molecular structures of (a) anti-2, (b) anti-1, (c) syn-1. | ||
The solid-state organization of anti-1 exhibits characteristic ChB interactions found in organic selenocyanates. As shown in Fig. 2, selenium atom Se1 acts as a twofold ChB donor, through its two σ-holes, with one very short interaction (RR = 0.86) in the prolongation of the NC–Se(1) bond, toward N2A(−x, −y, −z), and one longer contact (RR = 0.93) in the prolongation of the CH2–Se(1) bond toward N1(−0.5 + x, 0.5 − y, z). On the other hand, selenium atom Se2 in not engaged at all in such a Se⋯N ChB but makes an inversion-centered Se(2A)⋯Se(2A) motif (3.391(6) Å, RR = 0.89) analogous to the so-called type I halogen-bonded motifs found in halogenated molecules. This complex behavior contrasts with that found for the less-substituted achiral derivatives such as the bis(selenocyanato) o-, m-, p-xylylenes where ⋯NC–Se(R)⋯⋯NC–Se(R)⋯ chains are systematically observed. It probably comes as a consequence of the steric constraint brought by the two extra methyl substituents in anti-1. The situation is different in syn-1. As shown in Fig. 3, each of the two selenium atoms acts here as ChB donor toward the nitrogen atom of the selenocyanate of neighboring molecules, giving the recurrent chains mentioned above, with however larger intermolecular distances (RR = 0.89, 0.95) and poorer linearity (the NC–Se⋯N angles here are only 162 and 127°).
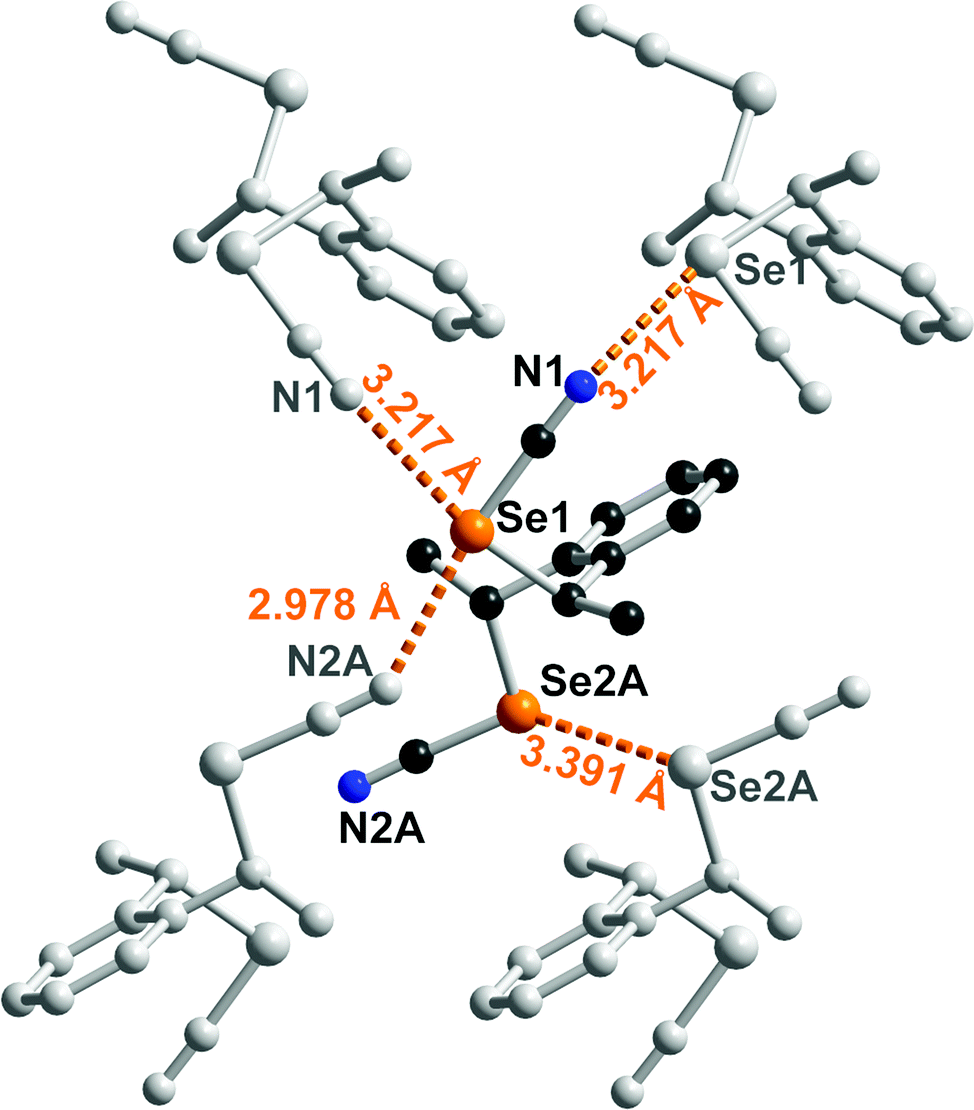 | ||
| Fig. 2 Details of the ChB interactions in anti-1. Only the major component of the disordered SeCN group is shown. Relevant bond distances and angles: Se(1)⋯N(2A)i: 2.978(25) Å, C(9)–Se(1)⋯N(2)i: 172.7(5)°; Se(1)–N(1)ii: 3.217(3) Å, C(7)–Se(1)⋯N(1)ii: 167.9(8)° (i: −x, −y, −z; ii: −0.5 + x, 0.5−y, z). | ||
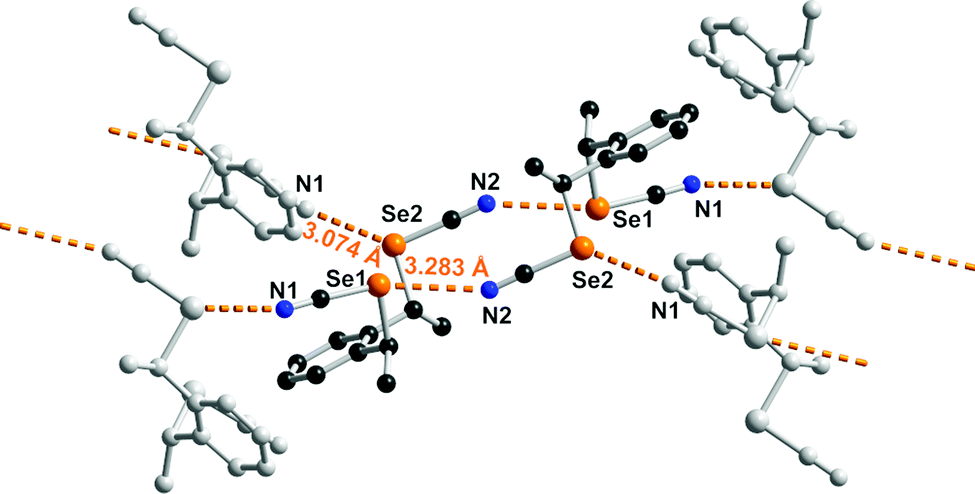 | ||
| Fig. 3 Details of the ChB interactions in syn-1. Relevant bond distances and angles: Se(1)⋯N(2)i: 3.283(7) Å, C(9)–Se(1)⋯N(2)i: 162.6(2)°; Se(2)⋯N(1)ii: 3.074(7) Å, C(12)–Se(2)–N(1): 127.05(25)° (i: 1−x, −1−y, 1−z; ii: 1−x, −0.5+y, 0.5−z). | ||
Co-crystal formation with anti-1 was investigated with both neutral Lewis bases such as 4,4′-bipyridine and anionic halide salts such as Ph4PX (X = Cl−, Br−, I−). With 4,4′-bipyridine, co-crystallization afforded a chalcogen-bonded structure of 2:1 stoichiometry formulated as (anti-1)2(bipy). It crystallizes in the monoclinic system, S. G. P21/a, with the 4,4′-bipyridine lying on an inversion center while linking the inversion-related enantiomers of anti-1 (Fig. 4). Both selenium atoms act here as ChB donors toward the pyridinic nitrogen atom, with one very short ChB, for Se(1)⋯N(3) (RR = 0.87) whereas the Se(2)⋯N(3) interaction is essentially a van der Waals contact distance, with a marked directionality for both interactions as the NC–Se⋯N(3) angles amount to 173.62(13) and 168.66(11)° respectively. Besides, the Se2 selenium atom acts as a ChB donor in a Se(2)⋯N(2) contact through its second weaker σ-hole located in the prolongation of the CH2–Se bond, while Se(1) forms an inversion-centered Type I Se⋯Se motif. Note that the co-crystals formed from 4,4’-bipyridine and the unsubstituted meta- or para- bis(selenocyanato)xylylenes are characterized with notably stronger Se⋯N ChB interactions, with RR values in the range 0.82–0.84.24a
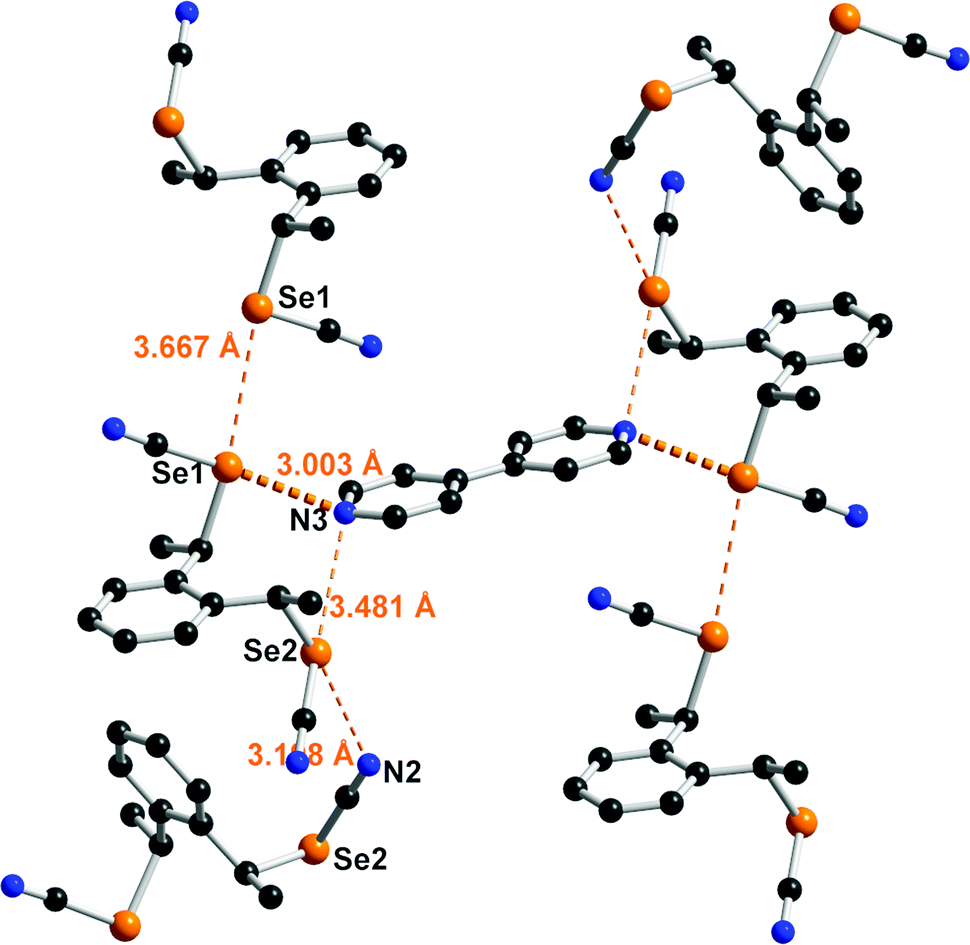 | ||
| Fig. 4 Detail of the trimolecular adduct between anti-1 and 4,4′-bipyridine. The strongest interaction is highlighted with a thicker dotted line. Relevant bond distances and angles: Se(1)⋯N(3): 3.003(4) Å, C(9)–Se(1)⋯N(3): 173.62(13)°; Se(2)⋯N(3): 3.481(3) Å, C(12)–Se(2)⋯N(3): 168.66(11)°; Se(2)⋯N(2)i: 3.198(3) Å, C(11)⋯Se(2)⋯N(2)i: 141.06(8)°; Se(1)⋯Se(1)ii: 3.667(1) (i: 0.5+x, 0.5−y, z; ii: −x, −y, 2−z). | ||
Co-crystals of anti-1 with the three tetraphenyl phosphonium halides, i.e. Ph4PCl, Ph4PBr and Ph4PI, all crystallize in a 1:1 stoichiometry with the halide in a μ2 environment, chelated by the ditopic ChB donor. The chloride and bromide salts are isostructural, and crystallize with Et2O solvate in the triclinic system, space group P1, with the anionic complex in general position. As shown in Fig. 5a and b, the environment of the Cl− and Br− anions is asymmetric with one (NC)–Se⋯X− ChB slightly shorter than the other. In both cases, the overall Se⋯X− distance corresponds to a reduction ratio (considering the ionic radius of the halide rather than the van der Waals radius of the neutral atom, i.e. Cl−: 1.81 Å, Br−: 1.96 Å, I−: 2.20 Å) in the range 0.88–0.90. These values are notably larger than those reported earlier in the unsubstituted achiral analogs chelating Cl− or Br− anions, which exhibit RR values as small as 0.84.25
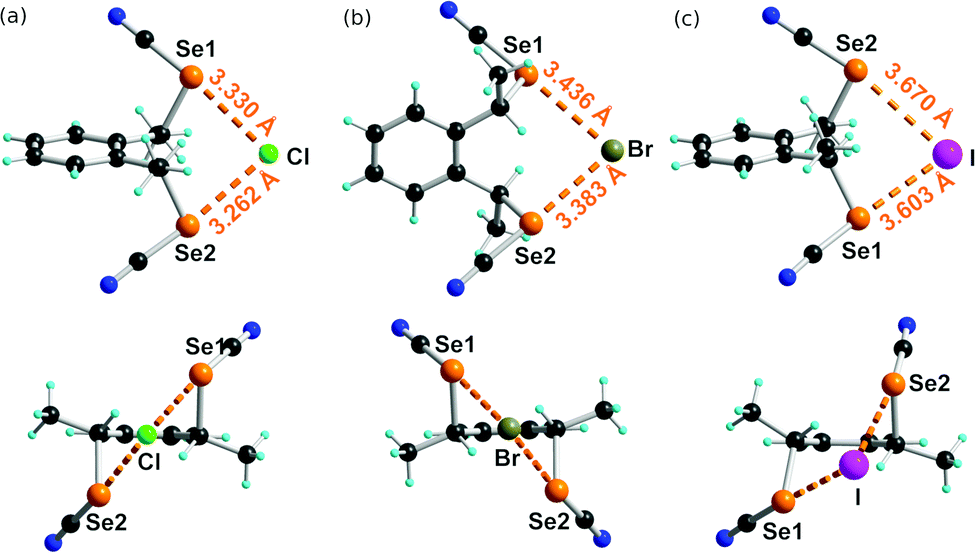 | ||
| Fig. 5 Detail of the three halide adducts with structural characteristics in: (a) Ph4P+[(anti-1)Cl−]·(Et2O)0.5, (b) Ph4P+[(anti-1)Br−]·(Et2O)0.5,and (c) Ph4P+[(anti-1)I−]. Relevant bond distances and angles: Se(1)⋯Cl: 3.330(7) Å, (N)C–Se(1)⋯Cl: 173.75(13)°, Se(2)⋯Cl: 3.262(6) Å, (N)C–Se2⋯Cl: 173.67(13)°; Se(1)⋯Br: 3.436(4) Å, (N)C–Se(1)⋯Br: 175.12(13)°, Se(2)⋯Br: 3.383(3) Å, (N)C–Se2⋯Br: 174.48(13)°; Se(1)⋯I: 3.603(1) Å, (N)C–Se(1)⋯I: 177.6(2)°, Se(2)⋯I: 3.670(1) Å, (N)C–Se(2)⋯I: 167.8(2)°. | ||
The situation is more complex in the iodide adduct (Fig. 5c). It crystallizes without solvent in the orthorhombic system, space group P212121, with the anionic complex in general position. The Se⋯I− distances are here associated with RR values of 0.88–0.89 and one (N)C–Se⋯I− angle deviates notably from 180° (167°).
These three structures and the relatively long Se⋯X− distances seem to indicate that the introduction of the methyl substituents on the benzylic positions induces, in the anti-isomer at least, an unfavorable effect on the ChB donor ability of this chelating system. At first sight, this effect might have two origins, (i) the electron-donating effect of the methyl groups might decrease the overall ChB donor ability of the selenium atoms, and/or (ii) the steric constraints brought by the methyl groups do not favor the optimal “coordination” of the halide anion, at least in the anti conformation of the chelate.
One first element of answer can be already found in the crystal structure obtained from the association of Et4NCl and the syn isomer, using a syn-enriched sample. A 2:1 association formulated as (Et4N+)[(syn-1)2Cl−] is indeed isolated, which crystallizes in the monoclinic system, space group P21/a, with the Cl− anion lying on an inversion center in a μ4 square-planar environment (Fig. 6). The Se⋯Cl− distances are very short (3.11–3.16 Å, RR = 0.84-0.85), even shorter than those reported in the chloride adduct of the achiral chelating ortho-bis(selenocyanate)xylylene A (3.17–3.20 Å, RR = 0.85-0.86) or the 1,2,4,5-tetrakis(selenocyanoatomethyl)benzene E (3.16–3.22 Å, RR = 0.85-0.87).25
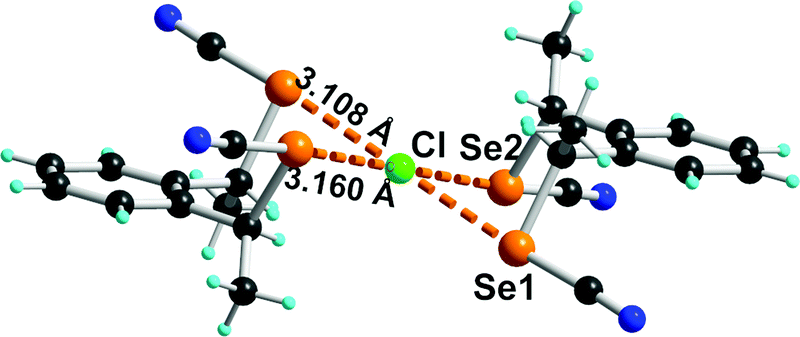 | ||
| Fig. 6 Detail of the anionic moiety [(syn-1)2Cl−] in its Et4N+ salt. Relevant bond distances and angles: Se(1)⋯Cl: 3.108(3) Å, C–Se(1)⋯Cl: 176.2(2)°; Se(2)⋯Cl: 3.160(1) Å, C–Se(2)⋯Cl: 176.0(3)°. | ||
In order to rationalize these differences, theoretical calculations of the relative energies of the molecules (anti vs. syn) and their Cl− adducts were performed. The total energy was calculated for the syn and anti forms of 1 [B3LYP 6-311++G(d,p)] after geometry optimization, keeping the C2 geometry for the anti form, and the Cs geometry for the syn form (Fig. 7). Under these conditions, the anti form is more stable by 2.31 kcal mol−1 (9.7 kJ mol−1) than the syn form. Without symmetry constraints, the syn form is able to find a more stable conformation through an intramolecular Se⋯Se ChB interaction (3.958 Å), reducing the difference to 0.68 kcal mol−1 (2.8 kJ mol−1).
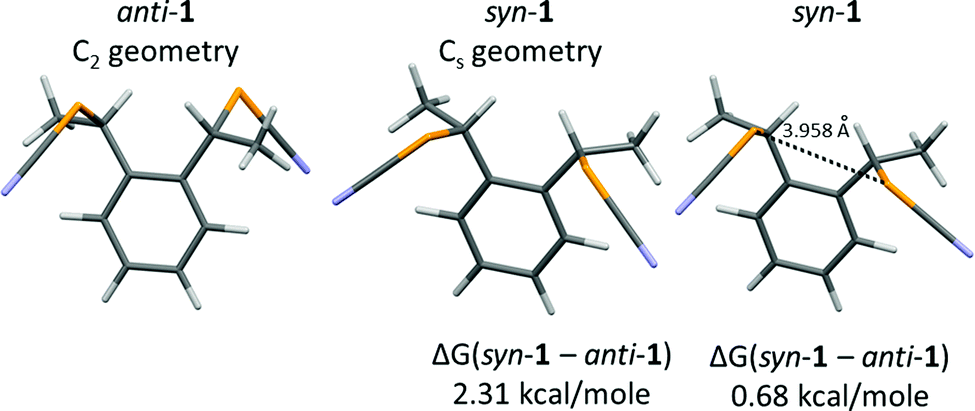 | ||
| Fig. 7 Optimized geometries of anti-1 and syn-1 with and without symmetry constraints. | ||
The evolution of the σ-hole amplitude has been determined for three molecules, bearing either a methyl group on each benzylic carbon (that is in anti-1 and syn-1) or a hydrogen atom (that is the unsubstituted molecule A in Scheme 1) or, for comparison purposes, a fluorine atom (instead of a methyl group) in the fluorine-substituted derivatives of A (that is anti-3 and syn-3). As shown in Fig. 8a, we note a very small difference between anti-1 and syn-1, with Vs,max values of 46.26 and 47.05 kcal mol−1 respectively, i.e. a slightly larger Vs,max value for syn-1. Such a small difference cannot explain the much shorter ChB interaction experienced with the Cl− anion with syn-1 (see above). The evolution in the series of substituents Me2/H2/F2 follows the expected trend, with a strengthening of the Vs,max in the order F2 > H2 > Me2 associated with the electron withdrawing effect of the fluorine atoms. Furthermore, an even stronger electropositive area (54.3 kcal mol−1) is identified in the fluorine substituted syn-3 in-between the two activated benzylic hydrogen atoms located in α position with respect to the fluorine atoms. These calculations demonstrate that, whatever the substituents, the syn form exhibits systematically a slightly stronger σ-hole than the anti form. However, they are unable to explain the much shorter ChB interaction with Cl−.
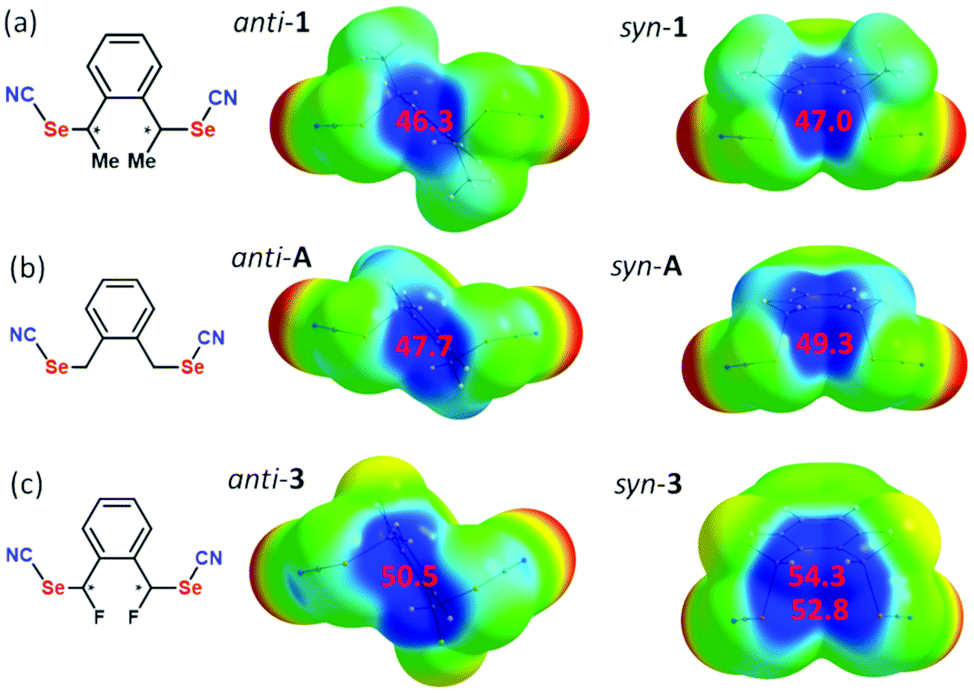 | ||
| Fig. 8 Details of the ESP maps of (a) methyl-substituted anti-1 and syn-1, (b) unsubstituted A and (c) fluorine-substituted anti-3 and syn-3, at optimized geometries, plotted on the 0.001 a.u. isosurface of the electronic density. The extrema values Vs,max (kcal mol−1) of the electropositive (blue) area are indicated in red numbers. Potential scale ranges from −25.1 kcal mol−1 (red) to +37.7 kcal mol−1. | ||
Calculations performed on 1:1 adducts with the syn and anti forms of 1 and a chloride anion give geometries very close to those observed in the crystal structures of Ph4P+[(anti-1)Cl−]·(Et2O)0.5 and Et4N+[(syn-1)2Cl−] (Fig. 9). The notably shorter Se⋯Cl− distances with syn-1 (3.11-3.16 Å, vs. 3.26-3.33 Å with anti-1) is very well reproduced by the calculations, which give a Se⋯Cl− distance of 3.139 Å in the syn-1·Cl− adduct (Cs geometry) that is notably smaller than the 3.210 Å distance calculated in anti-1·Cl− adduct (C2 geometry). The overall energy is very similar in both adducts ΔG(syn-1·Cl− − anti-1·Cl−) = 1.29 kcal mol−1, being slightly more stable with anti- adducr 1. On the other hand, the BSSE complexation energy (−35.36 kcal mol−1 for anti-1·Cl−, −35.55 kcal mol−1 for syn-1·Cl−) gives a small advantage to the syn adduct. It appears therefore that the shorter Se⋯Cl− ChB interaction experienced with the syn-1 ChB donor is not associated with a sizeable stabilization of this chloride adduct, when compared with its anti analog. As shown in Fig. 8, the electropositive area (in blue) interacting with Cl− exceeds that delineated with the selenium atoms only and include also the two benzylic hydrogen atoms. The overall interactions in both adducts thus implies also CBz–H⋯Cl− contacts, whose geometrical features (for both experimental and calculated structures) are collected in Table 1. These contacts are quite short (2.79-2.86 Å vs. a van der Waals contact distance of 3.01 Å) even if they deviate notably from linearity (C–H⋯Cl− angles in the range 123–128°). Their contribution to the overall stabilization of the adducts can probably not be omitted.
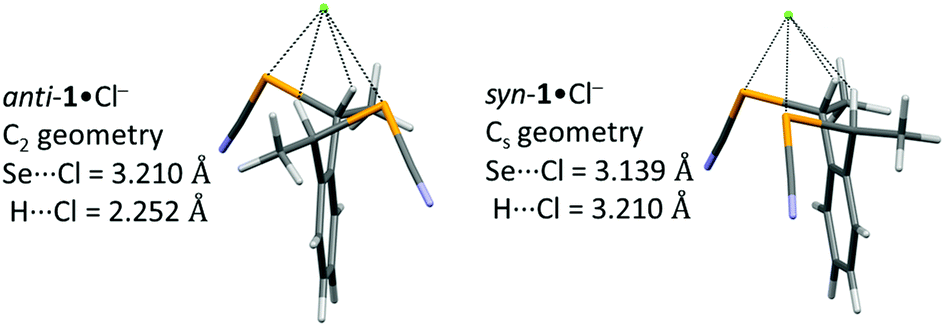 | ||
| Fig. 9 Optimized geometries of the anti-1·Cl− and syn-1·Cl− adducts with indicated symmetry constraints. | ||
:1 adducts (in italics)
| H⋯Cl− dist (Å) | C–H⋯Cl– ang. (°) | ||
|---|---|---|---|
| (anti-1)Cl− | X-ray | 2.863(6) | 128.1(2) |
| 2.800(4) | 127.0(2) | ||
| Calcd. | 2.522 | 138.1 | |
| (syn-1)Cl− | X-ray | 2.794(2) | 125.2(3) |
| 2.811(2) | 123.0(4) | ||
| Calcd. | 2.573 | 134.9 |
Conclusions
Introduction of methyl substituents on the achiral 1,2-bis(selenocyanatomethyl)benzene (A) leads to diastereoisomers of a novel chelating, chiral ChB donor, namely 1,2-bis(1-selenocyanatoethyl)benzene (1), as a mixture of the two anti enantiomers and the syn (meso) form. The anti (racemic) mixture was isolated in a pure form by recrystallization. We were able to isolate crystals of both anti-1 and syn-1. Structure determinations show the recurrent formation of short Se⋯N≡C ChB interactions (RR = 0.88-0.95). Co-crystallization of anti-1 with 4,4’-bipyridine affords a 2:1 adduct, i.e. (anti-1)2(bipy), with one very short Se⋯NPy ChB at 3.003 Å (RR = 0.87). Co-crystallization of anti-1 with tetraphenylphosphonium halides (Cl−, Br−, I−) provides the 1:1 adducts, while a 2:1 adduct is isolated between syn-1 and Et4NCl, formulated as Et4N+[(syn-1)2Cl−]. Comparison of chloride chelation with anti-1 and syn-1 shows much shorter (NC)Se⋯Cl− ChB interactions with the syn isomer. Calculations of (i) the electrostatic surface potential of neutral ChB donors for σ-hole amplitude determination and (ii) the Cl− BSSE complexation energy, cannot explain these differences even if the geometry optimizations well reproduce them. Besides, the concomitant contribution of C–H⋯Cl− hydrogen bonds involving benzylic hydrogen atoms α to the SeCN moieties is highlighted. Altogether, the notably shorter Se⋯Cl− distances found with the syn isomer appear to be a mere consequence of the overall relative orientation of both selenocyanate moieties together with contribution of CBz–H⋯Cl− hydrogen bonds, as illustrated by the broader spatial expansion of the electropositive area observed in the syn isomer. This modulation of the ChB donor ability, combined with the introduction of chirality, provides novel chelating ChB donors which will be of interest in crystal engineering, anion recognition processes and catalysis. We are pursuing our works along these lines.
Experimental section
General remarks
NMR spectra were recorded at room temperature using CDCl3 unless otherwise noted. Chemical shifts are reported in ppm and 1H NMR spectra were referenced to residual CHCl3 (7.26 ppm) and 13C NMR spectra were referenced to CHCl3 (77.2 ppm). All reagents are commercially available and were used without further purification. Melting points were measured on a Kofler hot-stage apparatus and are uncorrected. Elemental analysis were performed at BioCIS laboratory, UMR-8076-CNRS-University Paris-Saclay. All the reactions were performed under an argon atmosphere. Methanol, acetonitrile and dichloromethane were dried using inert pure solvent column device.Syntheses
:30 (based on 1H NMR). Pure anti-2 is obtained by recrystallization in hexane, while the mother liquors are enriched in syn isomer in a ratio anti/syn of 23:77.
anti-2 m.p. 89 °C. 1 H NMR (300 MHz, CDCl3) δ 2.15 (6H, d, J3 = 6.84 Hz); 5.71 (2H, q, J3 = 6.84); 7.34–7.40 (2H, m); 7.58–7.63 (2H, m)
syn-21 H NMR (300 MHz, CDCl3) δ 2.10 (6H, d, J3 =6.90 Hz); 5.61 (2H, q, J3 = 6.90); 7.32-7.38 (2H, m); 7.58-7.63 (2H, m).
:24 (based on NMR). Pure sample of anti-1 is obtained from recrystallization in acetone. The same reaction performed with 2 in a anti/syn ratio of 23:77 afforded 1 in a anti/syn ratio of 66:34.
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H–CH3); 101.86; 127.24; 129.65, 137.06. Elem. Anal calcd for C12H12N2Se2: C, 42.12; H, 3.54; N, 8.19; found: C, 41.92; H, 3.57; N, 8.20. Mother liquors are enriched in syn compound with a syn:anti distribution of 67:33. Their dissolution in acetone and diffusion of hexane afforded a few crystals of syn-1 suitable for X-ray diffraction. syn-1: 1H NMR (300 MHz, CDCl3) δ 2.15 (6H, d, J3 = 6.90 Hz); 5.17 (2H, q, J3 = 6.90); 7.40–7.45 (2H, m); 7.54–7.59 (2H, m). 13C NMR: 24.03 (CH3); 40.11 (H-CH3); 101.97; 127.58; 129.69, 136.84.
H–CH3); 101.86; 127.24; 129.65, 137.06. Elem. Anal calcd for C12H12N2Se2: C, 42.12; H, 3.54; N, 8.19; found: C, 41.92; H, 3.57; N, 8.20. Mother liquors are enriched in syn compound with a syn:anti distribution of 67:33. Their dissolution in acetone and diffusion of hexane afforded a few crystals of syn-1 suitable for X-ray diffraction. syn-1: 1H NMR (300 MHz, CDCl3) δ 2.15 (6H, d, J3 = 6.90 Hz); 5.17 (2H, q, J3 = 6.90); 7.40–7.45 (2H, m); 7.54–7.59 (2H, m). 13C NMR: 24.03 (CH3); 40.11 (H-CH3); 101.97; 127.58; 129.69, 136.84.
| Compound | anti-1 | syn-1 | anti-2 | (anti-1)2·bipy |
|---|---|---|---|---|
| Formula | C12H12N2Se2 | C12H12N2Se2 | C10H12Br2 | C17H16N3Se2 |
| FW (g mol−1) | 342.16 | 342.16 | 292.02 | 420.25 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/a | P21/n | P21/n | P21/a |
| a (Å) | 7.5686(4) | 8.5502(6) | 8.2203(8) | 7.1221(4) |
| b (Å) | 21.7486(11) | 7.0090(4) | 14.6643(16) | 28.6165(13) |
| c (Å) | 8.2075(4) | 22.6261(15) | 9.4380(9) | 8.6600(5) |
| α (°) | 90.00 | 90.00 | 90.00 | 90.00 |
| β (°) | 105.783(2) | 90.121(3) | 110.312(3) | 105.763(2) |
| γ (°) | 90.00 | 90.00 | 90.00 | 90.00 |
| V (Å3) | 1300.07(11) | 1355.94(15) | 1066.96(19) | 1698.62(16) |
| T (K) | 150(2) | 296(2) | 150(2) | 296(2) |
| Cryst. dim. (mm3) | 0.16 × 0.12 × 0.05 | 0.23 × 0.17 × 0.02 | 0.12 × 0.09 × 0.02 | 0.26 × 0.21 × 0.18 |
| Z | 4 | 4 | 4 | 4 |
| D calc (g cm−3) | 1.748 | 1.676 | 1.818 | 1.643 |
| μ (mm−1) | 5.663 | 5.429 | 7.544 | 4.353 |
| Abs. corr. | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| T min, Tmax | 0.447, 0.753 | 0.344, 0.897 | 0.447, 0.860 | 0.347, 0.457 |
| Total refls. | 20013 | 22207 | 19035 | 12513 |
| Uniq. refls. (Rint) | 2993 (0.0572) | 3062 (0.0753) | 2439 (0.0571) | 3912 (0.0291) |
| Unique refls. | 2631 | 1862 | 1943 | 3041 |
| (I > 2s(I))R1 | 0.027 | 0.0495 | 0.0220 | 0.0367 |
| wR2 (all data) | 0.066 | 0.1128 | 0.0513 | 0.0717 |
| GoF | 1.067 | 1.076 | 1.052 | 1.085 |
| Res. dens (e Å−3) | 0.50, −0.65 | 0.41, −0.52 | 0.43, −0.78 | 0.39, −0.64 |
| Compound | Ph4PCl(anti-1) ·0.5 Et2O | Ph4PBr(anti-1) ·0.5 Et2O | Ph4PI(anti-1) | Et4NCl(syn-1)2 |
|---|---|---|---|---|
| CCDC | ||||
| Formula | C38H37ClN2O0.50PSe2 | C38H37BrN2O0.50PSe2 | C36H32IN2PSe2 | C16H22Cl0.50N2.50Se2 |
| FW (g mol−1) | 754.03 | 798.49 | 808.42 | 425 |
| Crystal system | Triclinic | Triclinic | Orthorhombic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P212121 | P21/c |
| a (Å) | 9.2775(11) | 9.3752(5) | 9.2747(2) | 9.0534(5) |
| b (Å) | 13.5469(16) | 13.5967(6) | 10.6749(2) | 13.1176(6) |
| c (Å) | 14.3091(15) | 14.3500(7) | 34.3002(6) | 16.4261(7) |
| α (°) | 82.498(4) | 82.051(2) | 90.00 | 90.00 |
| β (°) | 88.218(4) | 87.549(2) | 90.00 | 112.672(2) |
| γ (°) | 83.497(4) | 83.173(2) | 90.00 | 90.00 |
| V (Å3) | 1771.3(4) | 1798.15(15) | 3395.94(11) | 1800.00(15) |
| T (K) | 296(2) | 296(2) | 296(2) | 296(2) |
| Cryst. dim. (mm3) | 0.23 × 0.11 × 0.10 | 0.28 × 0.16 × 0.13 | 0.22 × 0.10 × 0.08 | 0.25 × 0.21 × 0.01 |
| Z | 2 | 2 | 4 | 4 |
| D calc (g·cm−3) | 1.414 | 1.475 | 1.581 | 1.568 |
| μ (mm−1) | 2.238 | 3.242 | 3.161 | 4.179 |
| Abs. corr. | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| T min, Tmax | 0.744, 0.799 | 0.542, 0.656 | 0.691, 0.777 | 0.366, 0.959 |
| Total refls. | 43521 | 29306 | 24731 | 4122 |
| Uniq. refls. (Rint) | 8141 (0.0375) | 8249 (0.0298) | 7781 (0.0247) | 4121 |
| Unique refls. (I > 2s(I)) | 5829 | 5578 | 6512 | 2069 |
| R 1 | 0.0434 | 0.0371 | 0.0327 | 0.0624 |
| wR 2 (all data) | 0.1222 | 0.091 | 0.070 | 0.1591 |
| GoF | 1.142 | 1.059 | 1.02 | 1.027 |
| Res. dens (e Å−3) | 0.652, −0.951 | 0.898, −0.596 | 0.557, −0.391 | 0.568, −0.561 |
| Flack param. | — | — | 0.64(1) | — |
Theoretical calculations
Theoretical calculations were performed using the Gaussian09 software36 at the DFT level employing the B3LYP functional and the 6-311 + +G(d,p) basis set. Molecular structures of anti-1, syn-1, anti-A, syn-A, fluorine-substituted anti-3 and syn-3 were optimized and frequency calculations were performed in order to check that true energy minima were obtained. Electrostatic potential (ESP) mapped on the ρ = 0.001 a.u. isodensity surface were then computed with the AIMAll software package;37 the maximum of ESP Vs,max in the region of the (NC)Se σ-holes associated with Se atoms were located with MultiWFN software.38Whereas anti-1 optimized under C2 symmetry toward a true energy minimum, syn-1 optimized toward a Cs molecular structure associated with a small imaginary frequency (−20 cm−1). Further optimization of that structure without symmetry constrain led to a true energy minimum characterized by an intramolecular Se⋯Se interaction [d(Se⋯Se) = 3.958 Å, RR = 1.04; α(C–Se⋯Se) = 154.6°)], with the (NC)Se σ-hole of one of the two selenium atoms pointing toward one lone pair of the second chalcogen atom. The Cs constrained syn-1 molecular structure is then reported for comparison purposes, since in that structure the two chalcogen (NC)Se σ-holes are oriented as in the halide adducts. The same situation occurs for syn-A which was calculated under constrained Cs symmetry (imaginary frequency = −12 cm−1; the two σ-holes pointing outward the molecule) and also under C1 symmetry [intramolecular Se⋯Se interaction d(Se⋯Se) = 3.692 Å, RR = 0.97; α(C–Se⋯Se) = 169.4°]. For fluorine-substituted syn-3 molecule, the Cs symmetry constrained structure corresponds also to a saddle point (imaginary frequency = −9 cm−1; the two σ-holes pointing outward the molecule) but the unconstrained C1 symmetry structure does not present the previously observed intramolecular Se⋯Se interaction due to an unfavorable relative NCSe orientations [d(Se⋯Se) = 4.044 Å, RR = 1.06; α(C–Se⋯Se) = 128.4°].
Molecular structures of the (anti-1)Cl− and (syn-1)Cl− were also optimized using the same calculation conditions and true energy minima were obtained. Basis Set Superposition Error corrected structures and complexation energies were obtained through the counterpoise method of Boys & Bernard.39 More details are given in the Supporting Information file (Cartesian coordinates of optimized molecular structures).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Agency for Research (ANR 17-CE07-0025-01 and ANR 17-CE07-0025-02. The EXPLOR mesocentre is thanked for computing facilities (Project 2019CPMXX0984).References
- (a) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (b) Y. Zhao, Y. Cotelle, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 4270–4277 CrossRef CAS.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS.
- (a) A. Bauza, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef CAS; (b) A. Bauza, S. K. Seth and A. Frontera, Coord. Chem. Rev., 2019, 384, 107–125 CrossRef CAS.
- (a) L. M. Lee, M. Tsemperoli, A. I. Poblador-Bahamonte, S. Benz, N. Sakai, K. Sugihara and S. Matile, J. Am. Chem. Soc., 2019, 141, 810–814 CrossRef CAS; (b) P. Scilabra, G. Terraneo and G. Resnati, J. Fluorine Chem., 2017, 203, 62–74 CrossRef CAS.
- (a) P. Scilabra, G. Terraneo and G. Resnati, Acc. Chem. Res., 2019, 52, 1313–1324 CAS; (b) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC; (c) L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS; (d) N. Biot and D. Bonifazi, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS; (e) C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CAS.
- N. Biot and D. Bonifazi, Chem. – Eur. J., 2020, 26, 2904–2913 CrossRef CAS.
- (a) P. Wonner, L. Vogel, M. Düser, L. Gomes, F. Kniep, B. Mallick, D. B. Werz and S. M. Huber, Angew. Chem., Int. Ed., 2017, 56, 12009–12012 CrossRef CAS; (b) P. Wonner, L. Vogel, F. Kniep and S. M. Huber, Chem. – Eur. J., 2017, 23, 16975 CrossRef; (c) S. Benz, J. Mareda, C. Besnard, N. Sakai and S. Matile, Chem. Sci., 2017, 8, 8164–8169 RSC; (d) S. E. Reisman, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 7198–7199 CrossRef CAS; (e) W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141, 9175–9179 CrossRef CAS.
- (a) M. S. Taylor, Coord. Chem. Rev., 2020, 213, 213270 CrossRef; (b) E. Navarro-Garcia, B. Galmés, M. D. Velasco, A. Frontera and A. Caballero, Chem. – Eur. J., 2020, 26, 4706–4713 CrossRef CAS; (c) J. Y. C. Lim, J. Y. Liew and P. D. Beer, Chem. – Eur. J., 2018, 24, 14560–14566 CrossRef CAS.
- S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef CAS.
- M. Macchione, A. Goujon, K. Strakova, H. V. Humeniuk, G. Licari, E. Tajkhorshid, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2019, 58, 15752–15756 CrossRef CAS.
- (a) M. E. Brezgunova, J. Lieffrig, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, J. G. Angyan, M. Fourmigué and E. Espinosa, Cryst. Growth Des., 2013, 13, 3283–3296 CrossRef CAS; (b) H. Wang, J. Liu and W. Z. Wang, Phys. Chem. Chem. Phys., 2018, 20, 5227–5234 RSC.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kübel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299 CrossRef CAS.
- G. E. Garrett, E. I. Carrera, D. S. Seferos and M. S. Taylor, Chem. Commun., 2016, 52, 9881–9884 RSC.
- M. Macchione, M. Tsemperouli, A. Goujon, A. R. Mallia, N. Sakai, K. Sugihara and S. Matile, Helv. Chim. Acta, 2018, 101, e1800014 CrossRef.
- N. Biot and D. Bonifazi, Chem. – Eur. J., 2018, 24, 5439–5443 CrossRef CAS.
- (a) A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmoudkhani, J. Am. Chem. Soc., 2005, 127, 3184–3190 CrossRef CAS; (b) V. Kumar, Y. Xu and D. L. Bryce, Chem. Eur. J., 2020, 26, 3275–3286 CrossRef CAS; (c) G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef CAS.
- J. Y. C. Lim, I. Marques, A. L. Thompson, K. E. Christensen, V. Felix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 3122–3133 CrossRef CAS.
- A. Dhaka, O. Jeannin, I.-R. Jeon, E. Aubert, E. Espinosa and M. Fourmigué, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202011981.
- M. Beau, S. Lee, S. Kim, W.-S. Han, O. Jeannin, M. Fourmigué, E. Aubert, E. Espinosa and I.-R. Jeon, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202010462.
- J. George, V. L. Deringer and R. Dronskowski, J. Phys. Chem. A, 2014, 118(17), 3193–3200 CrossRef CAS.
- O. Jeannin, H.-T. Huynh, A. M. S. Riel and M. Fourmigué, New J. Chem., 2018, 42, 10502–10509 RSC.
- (a) H.-T. Huynh, O. Jeannin and M. Fourmigué, Chem. Commun., 2017, 53, 8467–8469 RSC; (b) A. M. S. Riel, O. Jeannin, O. B. Berryman and M. Fourmigué, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 34–38 CrossRef CAS.
- A. M. S. Riel, H.-T. Huynh, O. Jeannin, O. Berryman and M. Fourmigué, Cryst. Growth Des., 2019, 19, 1418–1425 CrossRef CAS.
- V. Kumar, C. Leroy and D. L. Bryce, CrystEngComm, 2018, 20, 6406–6411 RSC.
- R. Weiss, E. Aubert, P. Peluso, S. Cossu, P. Pale and V. Mamane, Molecules, 2019, 24, 4484 CrossRef CAS.
- (a) K. Fujita, M. Iwaoka and S. Tomoda, Chem. Lett., 1994, 923–926 CrossRef CAS; (b) T. Wirth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1726–1728 CrossRef CAS.
- V. Mamane, P. Peluso, E. Aubert, R. Weiss, E. Wenger, S. Cossu and P. Pale, Organometallics, 2020, 39, 3936–3950 CrossRef CAS.
- S. M. Walter, F. Kniep, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2011, 50, 7187–7191 CrossRef CAS.
- E. Eru, G. E. Hawkes, J. H. P. Utley and P. B. Wyatt, Tetrahedron, 1995, 51, 3033–3044 CrossRef CAS.
- D.-R. Hou, M.-S. Wang, M.-W. Chung, Y.-D. Hsieh and H.-H. G. Tsai, J. Org. Chem., 2007, 72, 9231–9239 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, A71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, C71, 3–8 CrossRef.
- J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- AIMAll (Version 19.10.12), T. A. Keith, TK Gristmill Software, Overland Park KS, USA, 2019 (ahttp://im.tkgristmill.com).
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS; (b) T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details. CCDC 2040289–2040296. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0nj05293k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |