
π-Conjugation effect on the aggregation-induced emission of extended viologens†
Murat
Tonga‡

Department of Chemistry, University of Massachusetts, Amherst, MA 01003, USA. E-mail: murattonga@gmail.com
First published on 10th December 2020
Abstract
This work focuses on the AIE properties of extended viologens (EVs), specifically the effect of π-conjugation by employing several aryl groups including phenyl, naphthyl, and anthryl. The DFT calculations show that these EVs with larger aryl groups adopt a twisted geometry due to steric hindrance between the aryl and vinylene–pyridinium units, while the EV with a phenyl ring adopted a planar one.
Recent decades have witnessed the evolution of luminescent materials which have been widely investigated in numerous areas such as electronics, optics, and biotechnology.1 Most of the applications typically require strong emission in the solid-state. However, many traditional luminescent materials easily undergo an undesired aggregation-caused quenching (ACQ) due to the formation of unfavorable species such as excimers as a result of strong π–π interactions in the solid-state.2 This causes huge limitations in practical applications. With the discovery of an opposite effect –aggregation-induced emission (AIE) – which is an enhancement of emission in the aggregate/solid-state, a new route has emerged for the development of advanced functional materials.3 The AIE luminescent materials show a quenched or weak emission in the solution state but emit strongly in the aggregate/solid-state. In general, the AIE molecules attain a twisted conformation which prevents the molecule from having a strong π–π stacking. Although the cause is still unknown, the mainly accepted mechanism is the restriction of intramolecular motion (RIM) which stems from structural planarization, J-aggregates, excited-state intramolecular proton transfer (ESIPT), twisted intramolecular charge transfer (TICT), and E/Z isomerization.4 In the aggregate state, the RIM process blocks the excimer decay through the non-radiative pathways, which causes the radiative channel to become the main deactivation pathway, thus leading to an intense emission.
Extended viologens, 1,1′-disubstituted-4,4′-bipyridinium organic salts, are a class of organic compounds made by the insertion of π-conjugated segments into the bipyridinium backbone. Their distinctive features including reversible redox, electrochromic and electron-withdrawing properties have found versatility in various applications such as electrochromic devices,5 molecular machines,6 memory devices,7 gas storage,8 herbicides,9 solar cells,10etc. Despite the remarkable accomplishments observed with extended viologens, the investigation of their AIE properties is still rare.11 In an earlier article, we reported the synthesis and photophysical properties of some A–π–D–π–A type extended viologens.12 In the present study, the AIE behaviors of the extended viologens shown in Scheme 1 were studied, particularly the effect of π-conjugation (i.e. phenyl, naphthyl, and anthryl). The AIE effects could benefit from the restriction of rotational and vibrational motions and E/Z isomerization of the vinylene–pyridinium units in the solid-state. Moreover, the vinylene units can cause a twisted geometry with the larger aryl backbones, which is beneficial for achieving an AIE property. Besides, the electron-accepting property of the pyridinium units in the acceptor–donor–acceptor type design allowed accomplishing desirable AIE characteristics, especially the emission color (blue, green, and red emission) in a binary solvent system of acetonitrile and toluene.
 | ||
| Scheme 1 Structures of the EV luminogens. | ||
The synthesis and characterization of the EV luminogens, P-EV (phenylene-EV), NAP-EV (naphthalene-EV), and ANT-EV (anthracene-EV) were reported in our previous study.12 Hexyl groups on the pyridinium termini were sufficient to dissolve these ionic luminogens in polar solvents such as methanol, acetonitrile (MeCN), and DMF. But, they are insoluble in nonpolar solvents like benzene, toluene, and diethyl ether.
The UV-vis absorption and photoluminescence (PL) spectra of the EV luminogens in MeCN were demonstrated in Fig. 1 and Table 1. The ICT process generally observed in the donor–acceptor systems are well-studied with solution absorption spectroscopy to examine the excited state characteristics of these compounds, particularly π-conjugation extension. The similarities in their absorption bands imply that they are structural analogues (Fig. 1a): the absorptions at ∼240–340 nm (π–π* transition of the π-conjugated backbone) and ∼340–560 nm (an ICT transition from donor to acceptor). The ICT peak maxima of the EV luminogens followed the increasing trend of π-conjugation: P-EV < NAP-EV < ANT-EV. Also, different ICT strength was noticed. For instance, NAP-EV has the highest ICT strength while ANT-EV has the lowest one. The weak ICT character of ANT-EV suggested its poor electronic communications due to the large distortion with the central anthryl core, which could be confirmed by computation modeling discussed below.13 The maximum absorption peak of NAP-EV located at 411 nm was red-shifted by 18 nm relative to P-EV. The luminogen ANT-EV showed 69 nm redshift, due to the larger π-extension of anthryl compared to naphthyl. Fig. 1b shows the normalized PL spectra of the EV luminogens. The PL spectra exhibited a similar trend as was observed for their absorbance spectra. Red-shifted PL spectra were observed with an increase in the π-conjugation: P-EV (79 nm), NAP-EV (128 nm), and ANT-EV (163 nm). The large Stokes shift implies a large change in the dipole moment at the excited state.14
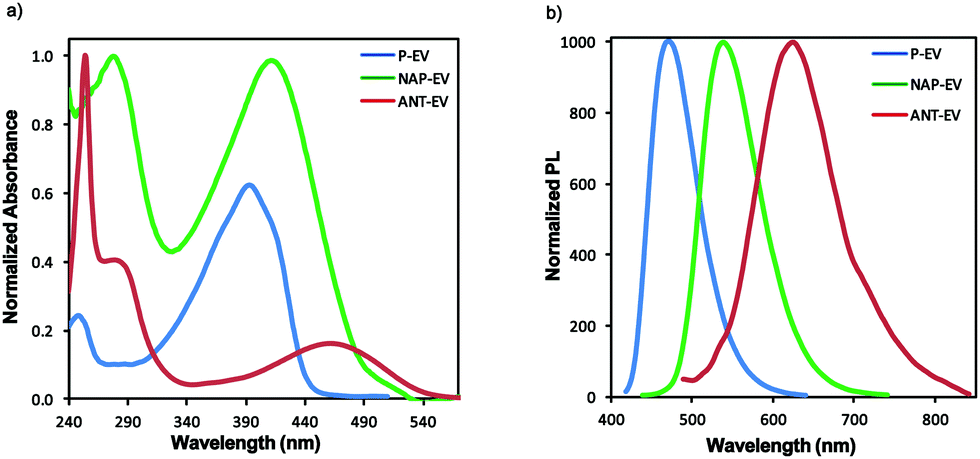 | ||
| Fig. 1 Normalized absorption (a) and photoluminescence spectra (b) of the EV luminogens in MeCN. | ||
| Absmax (π–π*) (nm) | Absmax (ICT) (nm) | PLmax (nm) | Absonset (nm) | Stokes shift (nm) | |
|---|---|---|---|---|---|
| P-EV | 248 | 393 | 472 | 450 | 79 |
| NAP-EV | 278 | 411 | 539 | 537 | 128 |
| ANT-EV | 254 | 462 | 625 | 624 | 163 |
The AIE features of the EV luminogens were studied by precipitating the MeCN (5 μM) solution with the addition of different toluene fractions (ftoluene = 10–90%). P-EV showed a moderate emission in the pure MeCN (Fig. 2a). The PL intensity initially increased drastically at ftoluene = 10% due to the change in solvent polarity. It is possible that P-EV molecules can form randomly packed amorphous aggregates which usually have weak π–π interactions, thereby boosting up the PL intensity.15 However, the PL intensity starts to diminish with the added ftoluene = 30–50% and it almost gave the intensity of the pure MeCN solution at ftoluene = 50%. Further increasing ftoluene to 70–90% nearly caused a complete quenching in the emission, which could be ascribed to the formation of crystalline aggregates. At higher ftoluene, the crystalline aggregates of P-EV built in a well-ordered fashion can have strong π–π stacking which can cause the formation of detrimental species, thus decreasing the emission intensity. The overall PL intensity is determined by the collective effects of the two kinds of aggregates. However, it is difficult to control the formation of aggregates in high toluene content since toluene is not a suitable solvent for these organic salt molecules. Thus, the PL intensity might demonstrate irregularities in high toluene content. Mie or the light scattering effect obstructs the accurate determination of quantum yields.16 Therefore, the change in the PL peak intensity (I/I0 ratio, where I is the peak intensity in the MeCN–toluene mixture and I0 is in the pure MeCN) with added ftoluene was calculated to evaluate the AIE effect. At ftoluene = 10%, the emission intensity was enhanced more than 3 times compared to the pure MeCN solution and about 20 times compared to the aggregate state formed at ftoluene = 90%, which is the most quenched state. Then, it demonstrated a downward trend by acquiring a 2 times enhancement in the intensity until ftoluene reaches 50%, which nearly showed the same intensity as the pure MeCN solution. It was also noticed that the PL peak maxima of P-EV aggregates remained almost steady when ftoluene = 10–50% and then they showed 22–32 nm redshifts beyond ftoluene > 50% (Fig. 2b). Under illumination with a 365 nm UV lamp, a blue emission located at 475 nm was observed for the aggregates formed at ftoluene = 10–50% (Fig. 2b-inset). Hence, it can be inferred that P-EV is AIE-active.
 | ||
| Fig. 2 AIE results of P-EV in MeCN–toluene solvent systems. | ||
Similarly, the AIE properties of the EV luminogen NAP-EV were also investigated in the MeCN–toluene solvent system (Fig. 3a). The AIE effect arises from the RIM of the naphthyl backbone and the vinylene–pyridinium units. The PL intensity decreased gradually with the added ftoluene = 30%, which could be explained by the formation of crystalline aggregates. Then, it intensifies noticeably when the ftoluene reaches 50%, presumably by forming amorphous aggregates. Further increasing the ftoluene completely quenched the emission at ftoluene = 90%. Many AIE designs also had a similar issue, but the reason is not clear yet. It is speculated that these aggregates could be produced by the reorganization of the aggregates into well-organized structures at higher fractions of a precipitating solvent.17 At ftoluene = 50%, the PL intensity increased by ∼1.2 times compared to the pure MeCN solution and ∼45 times compared to the aggregate state formed with added ftoluene = 90%, which is the most quenched phase (Fig. 3b). A steady trend in the position of the emission maxima was noted until ftoluene reaches 70% and then it was red-shifted by 93 nm at ftoluene = 90%. Thus, AIE-active NAP-EV showed a green emission (λPL = 543 nm) at ftoluene = 50%.
 | ||
| Fig. 3 AIE results of NAP-EV in MeCN–toluene solvent systems. | ||
The EV luminogen ANT-EV in the pure MeCN solution had a weak emission compared to P-EV and NAP-EV. Fig. 4a demonstrates that the PL intensity remained relatively constant when the ftoluene = 10–70%. Then, it increased dramatically at ftoluene = 90%. At higher toluene fractions, the twisted nature of the anthryl core and the vinylene groups restricts π–π interactions, which leads to an intense emission.18 The PL peak intensity (I/I0 ratio) at ftoluene = 90% was determined as ∼4.5 times higher than that of pure MeCN solution (Fig. 4b). The improvement in the emission intensity was also accompanied by some shifts in the PL peak as ftoluene varies. Initially, it was quite stable when ftoluene ≤ 50%, then it red-shifted by 8 nm for ftoluene = 70%, and finally it blue-shifted by ∼20 nm at ftoluene = 90%. It was noteworthy that accomplishing an AIE effect with a blue shift in the emission is an atypical event and it is called aggregation-induced blue-shifted emission (AIBSE). This has been observed in some 9,10-substituted anthracene molecules.19 The cause is still unknown; however, it might be the change in the conformation during the aggregation process suppressing the formation of charge transfer state.20 The aggregates of ANT-EV generated with added ftoluene = 90% exhibited a red emission located at 606 nm as shown under illumination with a 365 nm UV lamp (Fig. 4b-inset).
 | ||
| Fig. 4 AIE results of ANT-EV in MeCN–toluene solvent systems. | ||
Color-tunable luminescent materials are advantageous for the advancement of optoelectronic devices, particularly for the generation of white light. These EV luminogens accomplished tunable emissions of blue (P-EV), green (NAP-EV), and red color (ANT-EV) at the aggregate states by simply altering the amount of nonpolar solvent toluene in the mixed solvent system. The CIE 1931 coordinates of the EV luminogens are shown in the chromaticity diagram in Fig. S1 (ESI†). The white light emission is typically produced by mixing two or more complementary colors (blue and green or red, green, and blue) that span the full visible region (400–700 nm).21 Interestingly, the resulting emission maxima of the aggregate states of the EV luminogens cover the required RGB spectrum (Fig. S1, ESI†). Therefore, these EV luminogens could be good candidates to produce white light. Additional experiments would be required to investigate this further.
The optimized geometries could be useful to gain insight into the relationship between the structures and AIE features of the EV luminogens in the absence of the crystal structures. To investigate this, density functional theory (DFT) was studied using the Spartan 04 program at the B3LYP/6-311G* level. Table S1 (ESI†) presents their dihedral angles and the frontier orbital energy levels. The dihedral angles at specific sites can help to understand the conformation of the EV luminogens. Therefore, the dihedral angles α, β, and γ were thoroughly investigated; where α is described as the angle between the central aromatic groups (phenyl, naphthyl, and anthryl) and the vinylene unit, β is the angle between the pyridinium unit and the vinylene unit, and γ is the angle between the pyridinium unit and the central aromatic groups.
As shown in Fig. 5, the luminogens NAP-EV and ANT-EV adopted a twisted geometry where the vinylene and pyridinium units are forced out of the central aryl core due to steric hindrance. However, this steric hindrance is almost canceled out in the luminogen P-EV by adopting a highly planar geometry with the dihedral angles of α = 1.3°, β = 3.4°, and γ = 4.7°. Although P-EV has less distortion (or a quite planar) according to computation, it showed a stronger PL intensity than NAP-EV having a more twisted conformation. This may stem from the isomerization of the double bonds through E/Z conversion upon excitation, which could consume the excited state energy via non-radiative decay. However, this conversion process is hindered in the aggregate state, which causes an AIE effect.22 The photoisomerization is most likely difficult for the luminogens NAP-EV and ANT-EV since they have large backbones. The dihedral angles were much higher for NAP-EV and ANT-EV which stems from a large distortion between the central aryl core and the vinylene–pyridinium segments. The RIM in the solution phase gives rise to non-radiative pathways which quench the PL. However, in the aggregate state, the restriction of such torsional angles blocks the non-radiative channels, which yields an AIE characteristic with a strong emission. Moreover, the anion–π+ interactions in which the π-conjugated backbone intrinsically contains positive charges and counter anions might also cause the AIE behavior. Due to the position of the anions relative to the π systems, the anion–π+ interactions could hinder π–π stacking by acting as a separator between the molecules.23 In particular, the distances between bromide anions and the pyridinium cations presented in Table S1 (ESI†) show that this distance was larger for the π-extended luminogens NAP-EV and ANT-EV (in the range of 3.3–3.9 Å) compared to P-EV (3.3 Å). It is known that an emission quenching occurs when the π–π stacking distance is smaller than 3.5 Å.24 Thus, the anion–π+ interactions might also allow yielding the AIE effect. Further experiments are required to prove this.
 | ||
| Fig. 5 The optimized geometries of the EV luminogens. The small red dots represent the bromide ions. | ||
Conclusions
All in all, this contribution presents the AIE properties of the extended viologens by investigating the impact of π-conjugation. The AIE aspects resulted from not only the restraint of the twisted conformation formed due to the free rotations of the double bond but also E/Z isomerization of the double bonds in the solid-state. As a result of the electron-withdrawing aspect of the pyridinium units (with a typical ICT transition) and variable π-extension in the acceptor–donor–acceptor design, the diverse PL colors (RGB colors) were readily obtained in the MeCN–toluene solvent systems. Therefore, this work could lead to developing new designs to attain efficient RGB and white color systems in optoelectronic applications.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Guo, Z. Zhao and B. Z. Tang, Adv. Opt. Mater., 2018, 6, 1800264 CrossRef; (b) X. Y. Zhang, K. Wang, M. Y. Liu, X. Q. Zhang, L. Tao, Y. W. Chen and Y. Wei, Nanoscale, 2015, 7, 11486 RSC; (c) Y. Chen, Y. Bai, Z. Han, W. He and Z. Guo, Chem. Soc. Rev., 2015, 44, 4517 RSC; (d) C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174 RSC.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS.
- (a) A. Beneduci, S. Cospito, M. L. Deda and G. Chidichimo, Adv. Funct. Mater., 2015, 25, 1240 CrossRef CAS; (b) L. Veltri, V. Maltese, F. Auriemma, C. Santillo, S. Cospito, M. L. Deda, G. Chidichimo, B. Gabriele, C. D. Rosa and A. Beneduci, Cryst. Growth Des., 2016, 16, 5646 CrossRef CAS; (c) A. N. Woodward, J. M. Kolesar, S. R. Hall, N.-A. Saleh, D. S. Jones and M. G. Walter, J. Am. Chem. Soc., 2017, 139, 8467 CrossRef CAS; (d) I. Pibiri, A. Beneduci, M. Carraro, V. Causin, G. Casella, G. A. Corrente, G. Chidichimo, A. Pace, A. Riccobono and G. Saielli, J. Mater. Chem. C, 2019, 7, 7974 RSC; (e) W. W. Porter, T. P. Vaid and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 16559 CrossRef CAS.
- S. Cantekin, A. J. Markvoort, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2015, 137, 3915 CrossRef CAS.
- (a) C. Zhang, Y. T. Tai, J. Shang, G. Liu, K. L. Wang, C. Hsu, X. Yi, X. Yang, W. Xue, H. Tan, S. Guo, L. Pan and R. W. Li, J. Mater. Chem. C, 2016, 4, 3217 RSC; (b) G. Liu, C. Wang, W. Zhang, L. Pan, C. Zhang, X. Yang, F. Fan, Y. Chen and R.-W. Li, Adv. Electron. Mater., 2016, 2, 1500298 CrossRef.
- O. Buyukcakir, S.-H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934 RSC.
- (a) S. J. Yuan, F. J. Xu, S. O. Pehkonen, Y. P. Ting, K. G. Neoh and E. T. Kang, Biotechnol. Bioeng., 2009, 103, 268 CrossRef CAS; (b) Z. Shi, K. G. Neoh and E. T. Kang, Langmuir, 2004, 20, 6847 CrossRef CAS.
- S. W. Boettcher, J. M. Spurgeon, M. C. Putnam, E. L. Warren, D. B. Turner-Evans, M. D. Kelzenberg, J. R. Maiolo, H. A. Atwater and N. S. Lewis, Science, 2010, 327, 185 CrossRef CAS.
- (a) S. K. Samantaa and S. Bhattacharya, J. Mater. Chem., 2012, 22, 25277 RSC; (b) J. Sun, Y. Lu, L. Wang, D. Cheng, Y. Sun and X. Zeng, Polym. Chem., 2013, 4, 4045 RSC; (c) M. Olesińska, G. Wu, S. Gómez-Coca, D. Antón-García, I. Szabó, E. Rosta and O. A. Scherman, Chem. Sci., 2019, 10, 8806 RSC; (d) F. Qiao, Z. Yuan, Z. Lian, C.-Y. Yan, S. Zhuo, Z.-Y. Zhou and L.-B. Xing, Dyes Pigm., 2017, 146, 392–397 CrossRef CAS.
- M. Tonga and P. M. Lahti, Synth. Met., 2019, 254, 75 CrossRef CAS.
- J. Zhang, J. Chen, B. Xu, L. Wang, S. Ma, Y. Dong, B. Li, L. Ye and W. Tian, Chem. Commun., 2013, 49, 3878 RSC.
- S. S. M. Fernandes, M. C. R. Castro, A. I. Pereira, A. Mendes, C. Serpa, J. Pina, L. L. G. Justino, H. D. Burrows and M. M. M. Raposo, ACS Omega, 2017, 7, 9268 CrossRef.
- (a) X. Q. Zhang, Z. G. Chi, B. J. Xu, C. J. Chen, X. Zhou, Y. Zhang, S. W. Liu and J. R. Xu, J. Mater. Chem., 2012, 22, 18505 RSC; (b) W. Huang, H. Zhou, B. Lib and J. Su, RSC Adv., 2013, 3, 3038 RSC.
- Y. Liu, X. T. Tao, F. Wang, J. Shi, J. Sun, W. Yu, Y. Ren, D. Zou and M. H. Jiang, J. Phys. Chem. C, 2007, 111, 6544 CrossRef CAS.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- B. Xu, J. T. He, Y. J. Dong, F. P. Chen, W. L. Yu and W. J. Tian, Chem. Commun., 2011, 47, 6602 RSC.
- (a) J. Huang, N. Sun, P. Chen, R. Tang, Q. Li, D. Ma and Z. Li, Chem. Commun., 2014, 50, 2136 RSC; (b) L. Bu, M. Sun, D. Zhang, W. Liu, Y. Wang, M. Zheng, S. Xue and W. Yang, J. Mater. Chem. C, 2013, 1, 2028 RSC.
- S. Banerjee, A. K. Both and M. Sarkar, ACS Omega, 2018, 3(11), 15709 CrossRef CAS.
- (a) M. Chen, Y. Zhao, L. Yan, S. Yang, Y. Zhu, I. Murtaza, G. He, H. Meng and W. Huang, Angew. Chem., Int. Ed., 2016, 56, 722 CrossRef; (b) A. Krishna, E. Varathan, P. Sreedevi, V. Subramanian, V. Karunakaran and R. L. Varma, Dyes Pigm., 2018, 159, 77 CrossRef CAS.
- J.-S. Ni, H. Liu, J. Liu, M. Jiang, Z. Zhao, Y. Chen, R. T. K. Kwok, J. W. Y. Lam, Q. Peng and B. Z. Tang, Mater. Chem. Front., 2018, 2, 1498 RSC.
- (a) J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974 CrossRef CAS; (b) K. Leduskrasts and E. Suna, RSC Adv., 2019, 9, 460 RSC.
- E. R. Jimenez and H. Rodríguez, J. Mater. Sci., 2020, 55, 1366 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj05075j |
| ‡ Present address: Polnox Corporation, 225 Stedman St # 23, Lowell, MA 01851, USA. E-mail: mtonga@Polnox.com |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |