
Facile synthesis of pyronin-9-thione via a trisulfur radical anion mechanism†
Yu-Zhuo
Mai
a,
Yu-Zhong
Xie
*ab,
Ming-Hua
Zheng
ab,
Xin
Zhou
ac and
Jing-Yi
Jin
 *a
*a
aResearch Centre of Chemical Biology, Yanbian University, Yanji 133002, China. E-mail: whyjs@ybu.edu.cn; jyjin-chem@ybu.edu.cn
bDepartment of Chemistry, Yanbian University, Yanji 133002, China
cDepartment of Chemistry, Qingdao University, Qingdao 266071, China
First published on 27th November 2020
Abstract
Pyronin-9-thione (PY-S) is a key synthetic intermediate to develop various fluorescent probes for biological labeling and imaging. We developed a facile synthesis of PY-S from pyronin with a high yield up to 90%. The investigation of the reaction mechanism disclosed that the trisulfur radical anion (S3˙−) played a key role in the formation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond. The new established methodology was proved to have a short reaction time, operation easiness, and low toxicity and is suitable for the gram-scale synthesis of PY-S.
S bond. The new established methodology was proved to have a short reaction time, operation easiness, and low toxicity and is suitable for the gram-scale synthesis of PY-S.
Introduction
Pyronin (PY) is a common dye widely used in biological labeling and imaging.1–3 Due to its excellent photophysical properties, such as high fluorescence quantum yield and good photostability, a great deal of PY-based fluorescent probes have been thus developed by the introduction of various recognition groups at the meso-position of PY.4–6 Pyronin-9-one (PY-O) and pyronin-9-thione (PY-S) are generally used as the key synthetic intermediates to obtain PY-based probes (Fig. 1a). In comparison to PY-O, PY-S has been proved to be an ideal intermediate because of its higher reactivity. PY-S shows a high yield up to 75% in the following nucleophilic substitution reaction.5PY-S can be synthesized directly under the ButOK-S8 conditions but with an undesired yield of 55%.5 On the other hand, PY-O can be synthesized with a high yield of 97% by a base-catalyzed direct oxidation,7 which can be further converted to PY-S with Lawesson's reagent (Fig. 1b).8 Nevertheless, this two-step combination strategy is too tedious and toxic, making it unacceptable for large-scale synthesis. Therefore, the development of an effective and green strategy for the one-step synthesis of PY-S remains highly desirable.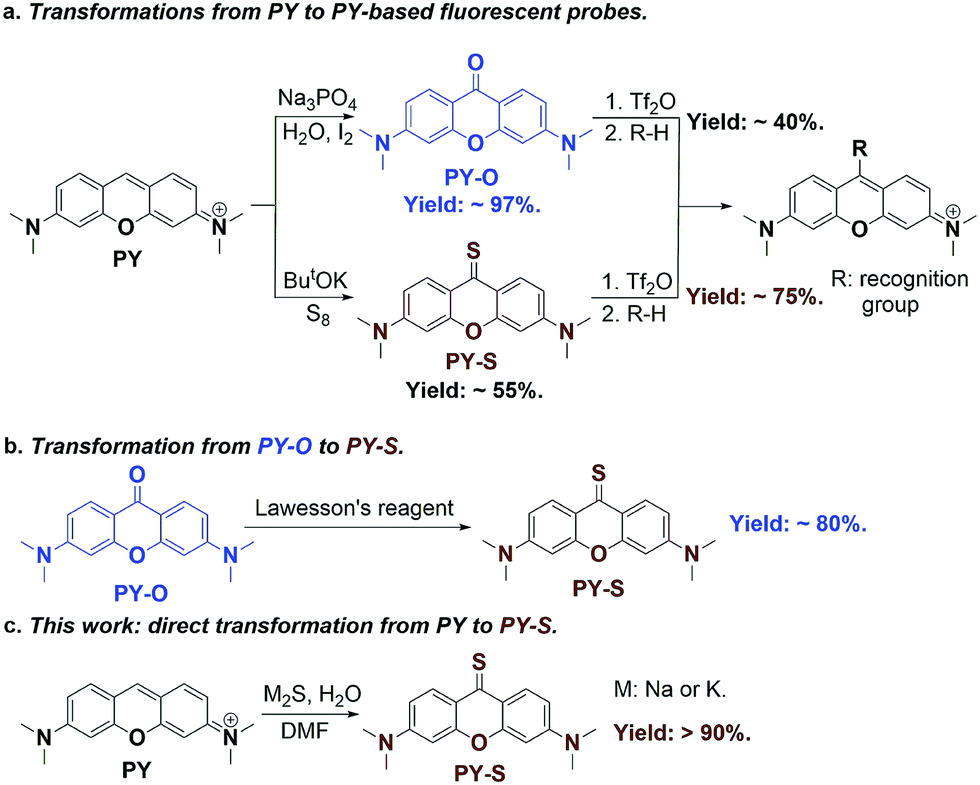 | ||
| Fig. 1 (a) Preparation of PY-based fluorescent probes through PY-O or PY-S as synthetic intermediates. (b) Transformation from PY-O to PY-S using Lawesson's reagent as a sulfur donor. (c) One-step transformation from PY to PY-S using M2S as a sulfur donor. | ||
To this end, we set to report here a new one-step strategy for the synthesis of PY-S from PY with a high yield up to 90% (Fig. 1c). Metal sulfide (Na2S or K2S) is used as a cheap sulfur donor. Both experimental and theoretical calculation results support that the trisulfur radical anion (S3˙−) plays a key role in the formation of the CS bond. The new established methodology was proved to be facile and suitable for the gram-scale synthesis of PY-S.
Results and discussion
We first explored the possibility of the preparation of PY-S using sodium sulfide hydrate as a sulfur donor. PY was added to an aqueous solution of Na2S and heated to 80 °C for 5 hours until PY completely disappeared upon TLC detection (Table 1, entry 1). Both PY-S and PY-O were obtained with the isolated yields as 36% and 22%, respectively. We ascribed the production of PY-O to the presence of a large amount of OH− (H2S: pK1 = 6.97; pK2 = 12.90) in the aqueous system. We then examined the reaction in various polar organic solvents, including ethanol (EtOH), 1,4-dioxane (dioxane), dimethoxyethane (DME), dimethyl sulfoxide (DMSO), N-methyl pyrrolidone (NMP), and N,N-dimethylformamide (DMF). The results are collected in Table 1 (entry 2–7). The production of PY-O was significantly depressed as found in polar organic solvents, while the best yield of PY-S was obtained in DMF as the solvent (yield: 80%). The results also implied that the polarity of the solvents was not the key factor for the reaction. Thus, the promoted dissolution of sodium sulphide using dipolar solvents should not play the key roles in the reaction system. Effects of the reaction temperature on the transformation in DMF were then investigated, and it was found that the PY-S yield could reach up to 90% at 140 °C in 1 hour (Table 1, entries 7–13). Further studies disclosed that the amounts of Na2S·9H2O also impacted the yields of PY-S. When the amounts of Na2S·9H2O were less than 3 equiv., the yields were lower than 50% (Table 2, entries 1 and 2). Obviously enhanced yields were obtained when the amounts of the hydrate were 3 or 4 equiv. (Table 2, entries 3 and 4). More addition of Na2S·9H2O slightly improved the yields of PY-S (Table 2, entries 5–8). Considering that the sulfide hydrate contained 9 equiv. H2O, it was thus necessary to evaluate the effects of water quantities on the conversion. We then used the anhydrous sulfides (M2S, M = Na or K) as the sulfur donors. It was surprising to observe that the PY-S yields were significantly decreased in the absence of H2O (Table 3, entries 1 and 2). A little amount of water was essential for the facile synthesis of PY-S (Table 3, entries 3–8).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Time (h) | Yieldb (%) | |
| PY-S | PY-O | ||||
| a General reaction procedure: Na2S·9H2O (288 mg, 1.2 mmol) and 8 mL of solvent were first heated at the set temperature for 5 minutes and then 2 mL of DMF solution containing PY (50 mg, 0.12 mmol) was added maintaining the temperature. The subsequent work-up could be found in the ESI. b Yields were calculated after purification by flash column chromatography. | |||||
| 1 | H2O | 80 | 5 | 36 | 22 |
| 2 | EtOH | 80 | 5 | 28 | Trace |
| 3 | Dioxane | 80 | 5 | 21 | Trace |
| 4 | DME | 80 | 5 | 25 | Trace |
| 5 | DMSO | 80 | 5 | 20 | Trace |
| 6 | NMP | 80 | 4 | 53 | Trace |
| 7 | DMF | 80 | 4 | 80 | Trace |
| 8 | DMF | 90 | 1.5 | 86 | Trace |
| 9 | DMF | 100 | 1.5 | 89 | Trace |
| 10 | DMF | 110 | 1 | 89 | Trace |
| 11 | DMF | 120 | 1 | 88 | Trace |
| 12 | DMF | 130 | 1 | 88 | Trace |
| 13 | DMF | 140 | 1 | 90 | Trace |

|
|
||
|---|---|---|
| Entry | Na2S·9H2O (equiv.) | Yield (%) |
| 1 | 1 | 34 |
| 2 | 2 | 47 |
| 3 | 3 | 67 |
| 4 | 4 | 84 |
| 5 | 5 | 85 |
| 6 | 6 | 86 |
| 7 | 8 | 89 |
| 8 | 10 | 90 |

|
|
|||
|---|---|---|---|
| Entry | M2S | H2O (equiv.) | Yield (%) |
| 1 | Na2S | 0 | 43 |
| 2 | K2S | 0 | 39 |
| 3 | Na2S | 1 | 91 |
| 5 | K2S | 1 | 92 |
| 5 | Na2S | 5 | 91 |
| 6 | K2S | 5 | 92 |
| 7 | Na2S | 10 | 91 |
| 8 | K2S | 10 | 92 |

As observed in our experiments, the DMF suspension of Na2S was blue at room temperature. In a continuous heating process, however, it was found that the blue color of the mixture faded slowly with the temperature being increased gradually, and completely disappeared at around 50 °C. The color was recovered beginning at 70 °C and turned deep-blue after 100 °C (Fig. S3, ESI†). The UV-Vis spectra of the mixtures clearly disclosed that the amounts of one species with absorbance at around 616 nm were responsible for the color change with the temperature (Fig. S4, ESI†). The same species could be observed in the K2S–H2O–DMF system (Fig. S5, ESI†), and the yields were proportional to the concentrations of the species (Fig. 2). The blue species should be ascribed to the trisulfur radical anion S3˙− as described by T. Chivers in 1974,9 which has been recently utilized in the formation of various C–S bonds.10–14 We assumed that S3˙− was the key initiator of the reaction. To examine our conjunction, TEMPO was introduced as the radical scavenger. With increasing amounts of TEMPO (1.0–4.0 equiv.), the yields of PY-S decreased (Table S1, ESI†). Addition of 4.0 equiv. TEMPO to the reaction system resulted in an instant fading of the blue color to colorless (Fig. S6, ESI†), and the PY-S yield significantly lowered to 8.4%. Liquid chromatography-mass spectroscopy (LC-MS) was also utilized to analyze the mixture of the K2S–H2O–DMF system and TEMPO (Fig. S7, ESI†). Two molecular ion peaks of 112.89 and 236.15 were observed, which should be ascribed to HOS3− (112.92) and the adduct of TEMPO and S3˙− (236.06).
 | ||
| Fig. 2 Normalized UV-Vis spectra of K2S–H2O in DMF at different temperatures. In preparation, K2S (1.2 mmol, 10.0 equiv.) and H2O (2.2 μL, 1.0 equiv.) were added to DMF (8 mL) followed by heating at different temperatures for 5 min. The obtained mixture was then cooled and diluted 4 folds with DMF for the subsequent measurement of the absorption spectra. | ||
To further check the oxidative role of DMF in the formation of S3˙− from M2S,10 we designed the other reaction conditions, in which DME was used as solvents and benzoyl peroxide (BPO) was used as an oxidant (Table 4). We found that the addition of 1.0 equiv. BPO could result in a similar yield compared with the case of DMF as oxidants.
|
|
||
|---|---|---|
| Entry | BPO (equiv.) | Yield (%) |
| a Reaction conditions: K2S (132 mg, 1.2 mmol), H2O (2.2 μL, 1.0 equiv.) and BPO in 8 mL of DME were first heated at 80 °C for 5 minutes and then 2 mL of DMSO solution containing PY (50 mg, 0.12 mmol) was added in refluxing. | ||
| 1 | 0.1 | 48 |
| 2 | 0.2 | 68 |
| 3 | 0.3 | 79 |
| 4 | 0.4 | 84 |
| 5 | 0.5 | 87 |
| 6 | 1.0 | 90 |
It should be noted that our finding was the first case of the S3˙− radical anion involved in the construction of the CS bond. We then carried out theoretical calculations to construct the basic pathway of the reaction using the Gaussian 16 program.15 The density functional theory UB3LYP method was first used to optimize the molecular geometries of the reactants (PY and S3˙− radical anion), intermediates, transition states and products at the level of 6-31G(d), and vibration frequency analysis was performed on them at the same level.16 Intrinsic reaction coordinate (IRC) analysis was carried out to confirm that all stationary points are smoothly connected to each other (Fig. S8, ESI†).17 After confirming the involved intermediate and the transition state, the zero point energies were obtained for each stationary point. On the basis of the predicted stable point configurations, single point energy calculations were then performed at the UM06-2X/6-311G(d,p) level.18 The results suggested that the addition of the S3˙− radical anion with PY should occur spontaneously and result in a radical intermediate (PY-S-I) because of the decreased Gibbs free energy. It was found that the decreased value of free energy was small, which implied that the high concentrations of S3˙− were vital to the reactions. The transition state with high energy was predicted as a five-member cyclic structure (PY-S-T). A concerted formation of the CS bond was expected with the simultaneous elimination of the HSS˙ radical (Fig. 3). We thus suggested the reaction mechanism as shown in Fig. 4. It should be noted that the presence of 1.0 equiv. H2O was essential to the reaction as observed in the experiments. As mentioned above, the pKa,2 of H2S was 12.90, which implied that little water could exist in the reaction mixture but both SH− and OH− should be anticipated. Here, we speculated that SH− was vital to the production of the S3˙− radical anion on the basis of the experimental observations. Further, OH− was also involved in the recycling of the HSS˙ radical to the reaction pathway and the generated S2˙− would be further converted to S3˙− taking part in the reaction (Fig. 4).11
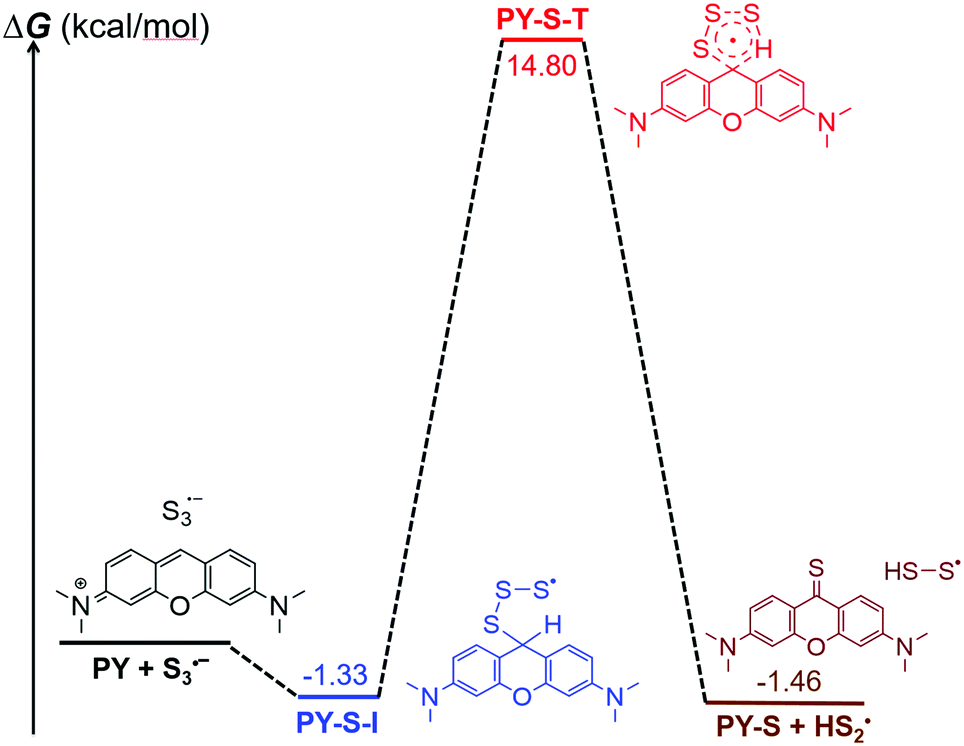 | ||
| Fig. 3 Theoretically predicted reaction pathway. | ||
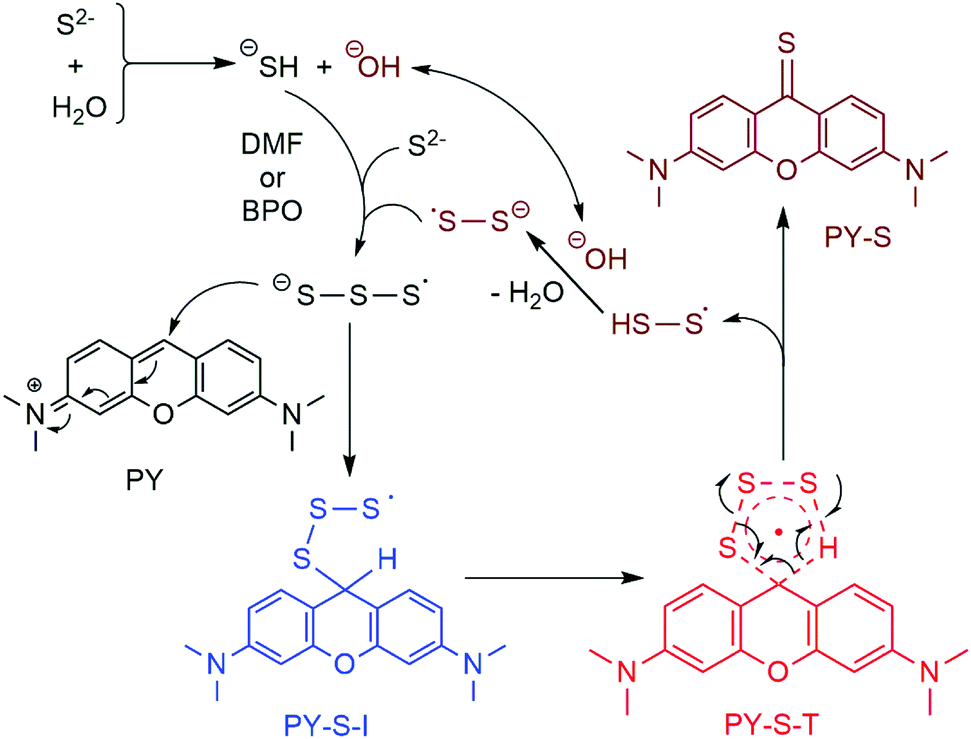 | ||
| Fig. 4 Proposed reaction mechanism using H2O as additives. | ||
To further examine the suggested mechanism, we used HCOOH as additives instead of water in the K2S–DMF system to carry out the reaction. The corresponding mechanism was assumed as follows. The acidic proton of HCOOH could react with the sulfide anion to produce SH− and formate HCOO−. Decarboxylation of the formate could provide hydride to interact with the HSS• radical and afford S2˙−. HCOOH was thus expected as a perfect replacement of H2O as additives in the transformation. In fact, we did find that the introduction of 1.0 equiv. HCOOH instead of water as additives to the K2S–DMF system could also afford PY-S in 91% yield. Finally, we conducted a gram-scale preparation of PY-S under the optimized conditions, i.e., potassium sulfide (2.674 g, 242 mmol) and H2O (44 μL, 24.2 mmol) were added to 8 mL of DMF and then heated at 140 °C for 5 minutes. And then a DMF solution (4 mL) containing pyronin (1.0 g, 24.2 mmol) was added. The reaction was completed in 2 hours and the yield reached up to 88%.
Conclusions
In conclusion, we developed a facile synthetic method of PY-S from PY using cheap and low toxic sulfide (Na2S or K2S) as sulfur donors. The S3− radical anion was the key initiator of the reaction. The mechanism was proposed according to the experimental observations and theoretical calculations. The gram-scale synthesis of PY-S was readily fulfilled by the approach.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21768004, 21662037, and 21762045).Notes and references
- K. Lang and J. W. Chin, Nat. Chem., 2013, 5, 81–82 CrossRef CAS.
- A. Morozumi, M. Kamiya, S. Uno, K. Umezawa, R. Kojima, T. Yoshihara, S. Tobita and Y. Urano, J. Am. Chem. Soc., 2020, 142, 9625–9633 CAS.
- R. R. Sauers, S. N. Husain, A. P. Piechowski and G. R. Bird, Dyes Pigm., 1987, 8, 35–53 CrossRef CAS.
- Y.-Q. Sun, J. Liu, H. Zhang, Y. Huo, X. Lv, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 12520–12523 CrossRef CAS.
- X. Zhou, Y. Zeng, C. Liyan, X. Wu and J. Yoon, Angew. Chem., Int. Ed., 2016, 55, 4729–4733 CrossRef CAS.
- J. Tang, Z. Guo, Y. Zhang, B. Bai and W.-H. Zhu, Chem. Commun., 2017, 53, 10520–10523 RSC.
- J. L. Bachman, P. R. Escamilla, A. J. Boley, C. I. Pavlich and E. V. Anslyn, Org. Lett., 2019, 21, 206–209 CrossRef CAS.
- J. Tang, M. A. Robichaus, K. L. Wu, J. Pei, N. T. Nguyen, Y. Zhou, T. G. Wensel and H. Xiao, J. Am. Chem. Soc., 2019, 141, 14699–14704 CrossRef CAS.
- T. Chivers, Nature, 1974, 5478, 32–33 CrossRef.
- Z.-Y. Gu, J.-J. Cao, S.-Y. Wang and S.-J. Ji, Chem. Sci., 2016, 7, 4067–4072 RSC.
- B.-B. Liu, H.-W. Bai, H. Liu and S.-J. Ji, J. Org. Chem., 2018, 83, 10281–10288 CrossRef CAS.
- J.-H. Li, Q. Huang, S.-Y. Wang and S.-J. Ji, Org. Lett., 2018, 20, 4704–4708 CrossRef CAS.
- J.-H. Li, Q. Huang, W. Rao, S.-Y. Wang and S.-J. Ji, Chem. Commun., 2019, 55, 7808–7811 RSC.
- F. Su, S. Chen, X. Mo, K. Wu, J. Wu, W. D. Lin, Z. Lin, J. Lin, H.-J. Zhang and T.-B. Wen, Chem. Sci., 2020, 11, 1503–1509 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- H. B. Schlegel, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS.
- B.-W. Li, M.-Y. Wang and J.-Y. Liu, Mol. Catal., 2020, 492, 11028 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and data available. See DOI: 10.1039/d0nj04808a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |