
Spatial confinement of chemically engineered cancer cells using large graphene oxide sheets: a new mode of cancer therapy†
Tianshu
Chen
ab,
Qianqian
Zhang
c,
Yuchen
Song
c,
Albertina N
Isak
c,
Xiaochen
Tang
ab,
Hao
Wang
d,
Zhongliang
Ma
c,
Fenyong
Sun
d,
Qiuhui
Pan
*ab and
Xiaoli
Zhu
 *d
*d
aDepartment of Clinical Laboratory Medicine, Shanghai Children's Medical Center, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, P. R. China. E-mail: panqiuhui@scmc.com.cn
bShanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics, Shanghai 200127, P. R. China
cSchool of Life Sciences, Shanghai University, Shanghai 200444, P. R. China
dDepartment of Clinical Laboratory Medicine, Shanghai Tenth People's Hospital of Tongji University, Shanghai 200072, P. R. China. E-mail: xiaolizhu@shu.edu.cn
First published on 11th September 2021
Abstract
Treating cancer with high efficacy while eliminating side effects has been the holy grail of cancer research. The challenge, however, arises from the similarity in molecular traits of cancer cells and normal cells because truly specific cancer biomarkers are extremely scarce if not entirely unavailable. Often, biomarkers serving as the therapeutic targets are present on both healthy cells and cancers, but at different levels, causing not only off-target side effects but also on-target side effects. This work has reported a new concept of cancer treatment, spatial confinement of cells to inhibit cell migration and invasion, which directly addresses the defining trait of cancer on the cellular level, unchecked division. Using large sized graphene oxide (LS-GO), cell surfaces can be patched. Unlike conventional chemotherapy, this spatial confinement does not affect the viability of non-dividing cells but significantly inhibits tumor cell migration and invasion in vitro and in vivo. This new concept has the potential to become a general therapeutic for many cancer types with reduced side effects.
New conceptsThe size of graphene sheets ranges from 10 nm to 1000 μm, covering the size of general cells (usually tens of μm). Small-sized graphene whose size is much smaller than cells has been confirmed to be able to enter cells through endocytosis and has been widely explored as a drug carrier; while the large-sized graphene whose size is equal to or larger than cells cannot enter and has been ignored in biomedicine. Here, we realized the interaction between large-sized graphene and cells for the first time through a DNA engineering strategy, and on this basis, realized the unique biomedical application of large-sized graphene. By interacting with DNA-engineered cell membrane, large-sized graphene as a “cage” was imposed on cancer cells and was interestingly observed to limit spatial behavior of cancer cells, including cell migration and invasion. This new concept of cancer treatment was also put into practice by implanting the large-sized graphene in the region of solid tumors and developing a corresponding strategy for tumor treatment, which showed ideal therapeutic effects in animal models. |
Introduction
As early as the 1950s, cancer was recognized as the abnormal proliferation, migration, and invasion of mutant cells, namely cancer cells.1 Today, although considerable progress has been made in the understanding of the initiation and progression of cancer,2 this cognition is still the widely accepted way to define cancer cells.3 But unfortunately, this cognition at the cellular level has not brought major changes to the treatment of cancer. Only after the understanding of cancer reached the molecular level, a variety of drugs targeting specific molecules have been developed and become the most important approach for cancer treatment.4 In this context, optimists believe that a complete cure for cancer is not far away. However, recent in-depth research on the mechanism of cancer seems to have poured cold water on this beautiful vision.5,6 Specifically, the molecular phenotypes of cancer cells are observed to vary greatly.7 To this day, it is still impossible to define cancer cells by specific molecular populations, and may never be achieved. This leads to a frustrating fact that drug combos targeting single or multiple molecules cannot cover all cancers, or even different subtypes of a certain cancer. Furthermore, during the treatment of cancer cells, acquired resistant mutations similar to the drug resistance of bacteria may appear with administration,8 which makes it almost impossible to treat cancer with specific drugs once and for all.On the basis of deeper understanding of cancer at the molecular level, the essential characteristics of cancer cells at the cellular level can be re-examined from a new perspective with a question being posed again: “Is there a method to directly inhibit the spatial behavior of cancer cells?”. One possible strategy emerges with the progress of material science: to impose a “cage” on cancer cells to limit their spatial migration and invasion behavior. After considering the characteristics of various materials, we realized that large-sized graphene oxide (LS-GO) is a competitive alternative to work as the “cage”. GO is a two-dimensional nanomaterial composed of carbon, oxygen and hydrogen,9 the three most important elements of living beings. Currently, GO, usually referring to small-sized graphene oxide (SS-GO), has been widely reported in the construction of nano-drug delivery systems (nano-DDS) for tumor therapy,10,11 whereas LS-GO whose size exceeds the capacity of cells to swallow is ignored. But the structural characteristics of LS-GO seem to be very suitable as a “cage” for cancer cells. Specifically, the monolayer thickness of LS-GO allows it to be easily inserted between cells, while the micrometer size in the other two dimensions allows LS-GO to cover rather than enter cancer cells. The dense and hexagonal honeycomb lattice structure of LS-GO is also expected to block the connection between cells. Furthermore, the excellent performance in terms of biocompatibility and mechanical strength of LS-GO are also favorable.12
To achieve the above concept, LS-GO with an average area of 18 μm2 is adopted in this work. Chemically engineered cancer cells, whose membrane proteins are conjugated with oligonucleotides are adopted to stabilize the spatial confinement of LS-GO via π–π stacking interaction between the oligonucleotides and the LS-GO. In this way, it is exciting to observe that the LS-GO does not affect the basic physiological activity of cells, but can significantly inhibit the migration and invasion of cancer cells. On the basis of the comprehensive study on the effect of LS-GO on cancer cells, we implant LS-GO in the region of solid tumors and develop a corresponding strategy for tumor treatment, which shows ideal therapeutic effects in animal models. The concept and successful applications of LS-GO working as a “cage” to confine cancer cells provide broad prospects for the treatment of cancer at the cellular level.
Results and discussion
Chemically engineered cancer cells are obtained through a two-step strategy shown in Fig. 1a. Briefly, after being treated with tetra-acylated N-azidoacetylmannosamine (Ac4ManNAz), cancer cells can express azido-labeled membrane proteins through glycometabolism pathways.13,14 By using a click chemical reaction between the azido group and a dibenzocyclooctyne (DBCO)-labeled oligonucleotide, the membrane proteins will be further conjugated with the oligonucleotide. Here, to ensure that the oligonucleotide is not degraded by the exonuclease or endonuclease that may exist outside the cells, the L-configuration (LDNA) was adopted.15 Western blotting and fluorescence imaging were performed separately to check whether click chemistry occurred and whether the LDNA was successfully conjugated to the cells. For the formation, DBCO-biotin instead of the DBCO-LDNA was used so as to generate chemiluminescent signals through binding with and subsequent catalysis of streptavidin-horseradish peroxidase (HRP). The western blotting result shows that the proteins extracted from Ac4ManNAz-treated A549 cells, a commonly used human non-small cell lung cancer (NSCLC) cell line, present clear bands in the presence of DBCO-biotin and streptavidin-HRP (Fig. 1b). Without any Ac4ManNAz or DBCO-biotin, no band can be obtained, suggesting the occurrence of the click chemistry. For the fluorescence imaging analysis, the other terminal of the DBCO-LDNA was labeled with a fluorophore Cy5 so as to make it visible under a confocal laser scanning microscope. Fig. 1c shows that the membrane of A549 cells exhibits the expected red fluorescence, which cannot be observed in the control groups lacking either Ac4ManNAz or DBCO-LDNA-Cy5. In the third row of Fig. 1c, when the A549 cells have not been treated with Ac4ManNAz, the cell membrane protein will not be labeled with azido, resulting in the failure of DBCO-LDNA-Cy5 to bind to the cell membrane or enter the cell. The DBCO-LDNA-Cy5 that is not bound to the cells will be washed away by DPBS. Therefore, the red fluorescence will not be detected on the A549 cells in this case. Zeta potential analysis also shows that attributed to the negatively charged LDNA, the negative charges of the cells increase significantly after the cells are treated with Ac4ManNAz and DBCO-LDNA successively (Fig. 1d). The above results together indicate that chemically engineered cancer cells conjugated to LDNA on the surface have been obtained.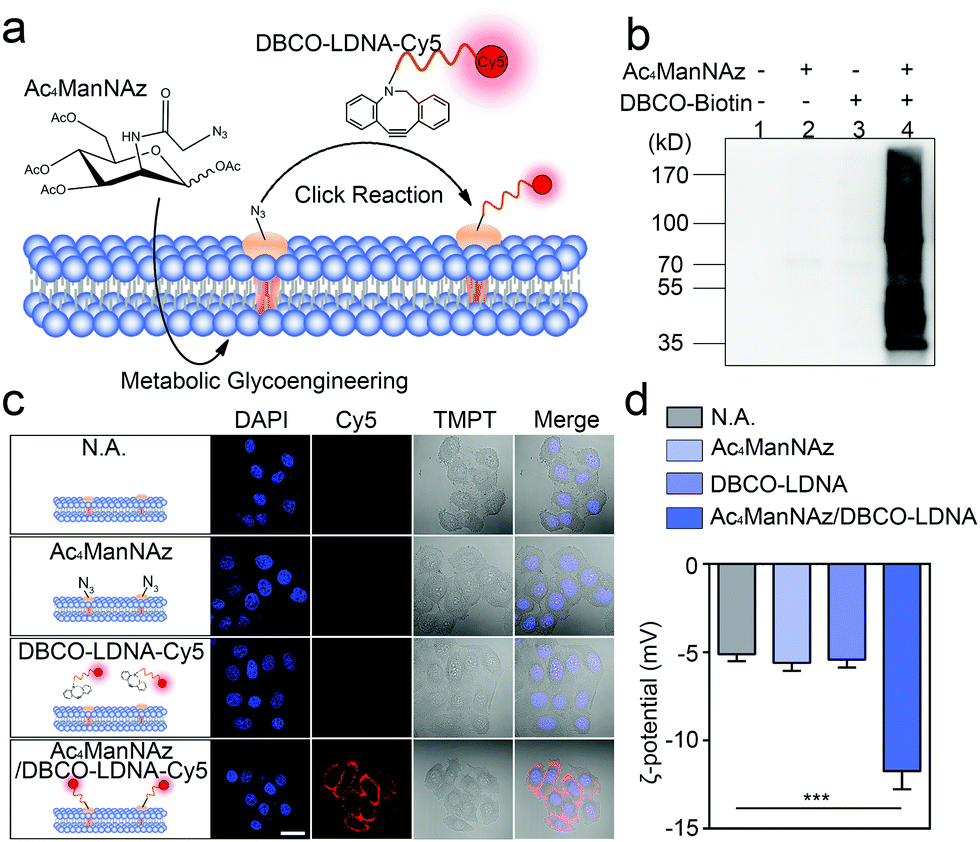 | ||
| Fig. 1 (a) Schematic illustration of the preparation of chemically engineered cancer cells. (b) Western blotting results of the proteins extracted from Ac4ManNAz-treated A549 cells. The proteins were labeled with DBCO-biotin and streptavidin-HRP successively before chemiluminescence imaging. (c) Fluorescence images of A549 cells treated with/without Ac4ManNAz and DBCO-LDNA-Cy5. The nucleus was stained with DAPI. All images were obtained using a confocal laser scanning microscope. Scale bar = 20 μm. (d) Zeta potential of A549 cells treated with/without Ac4ManNAz and DBCO-LDNA. n = 3. p values were calculated by the Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
LS-GO was allowed to interact with the chemically engineered cancer cells, while SS-GO and unengineered cancer cells were adopted as controls. The diameter of LS-GO and SS-GO was in the range of 1 to 5 μm and 0.2 to 0.5 μm, respectively (Fig. 2a and Fig. S1, ESI†); and the average area of LS-GO and SS-GO was calculated to be 18 μm2 and 0.033 μm2, respectively (Fig. 2b). To study the performance of GO presented to the cancer cells, green fluorescent protein (GFP) was adopted to label and visualize GO (Fig. 2c). Dil, a widely used fluorescent membrane dye, was used to label the cell membrane. Fig. 2d shows that when LS-GO is presented to LDNA-conjugated A549 cells, a clear green fluorescence of LS-GO appears outside the cell membrane. The three-dimensional image of the A549 cells after chemical engineering treatment and exposure to LS-GO also showed the coverage of LS-GO on the cell membrane (Fig. S2, ESI†). In the control groups using A549 cells without the treatment of N-azidoacetylmannosamine (Ac4ManNAz), or dibenzocyclooctyne (DBCO)-LDNA, LS-GO cannot be observed on cells. Moreover, if the LDNA conjugated on the surface of A549 cells was hybridized with a complementary oligonucleotide to form a double-stranded LDNA (dsLDNA) before exposure to the LS-GO, the presence of LS-GO was also not observed. The above results suggest that LS-GO covers the surface of chemically engineered cancer cells via the interaction with the single-stranded LDNA. Then the coverage rate of LS-GO on the chemically engineered cells was explored. Fluorescence images and statistical results show that the amount of LS-GO on the surface of the cells increases as the concentration of DBCO-LDNA increases (Fig. S3, ESI†). When SS-GO instead of LS-GO is adopted, it is interesting that SS-GO tends to be endocytosed by the cells rather than covering the cell membrane. This result indicates that the size of GO plays a crucial role in its interaction with cells, and the size of LS-GO has exceeded the limit of endocytosis by cells.
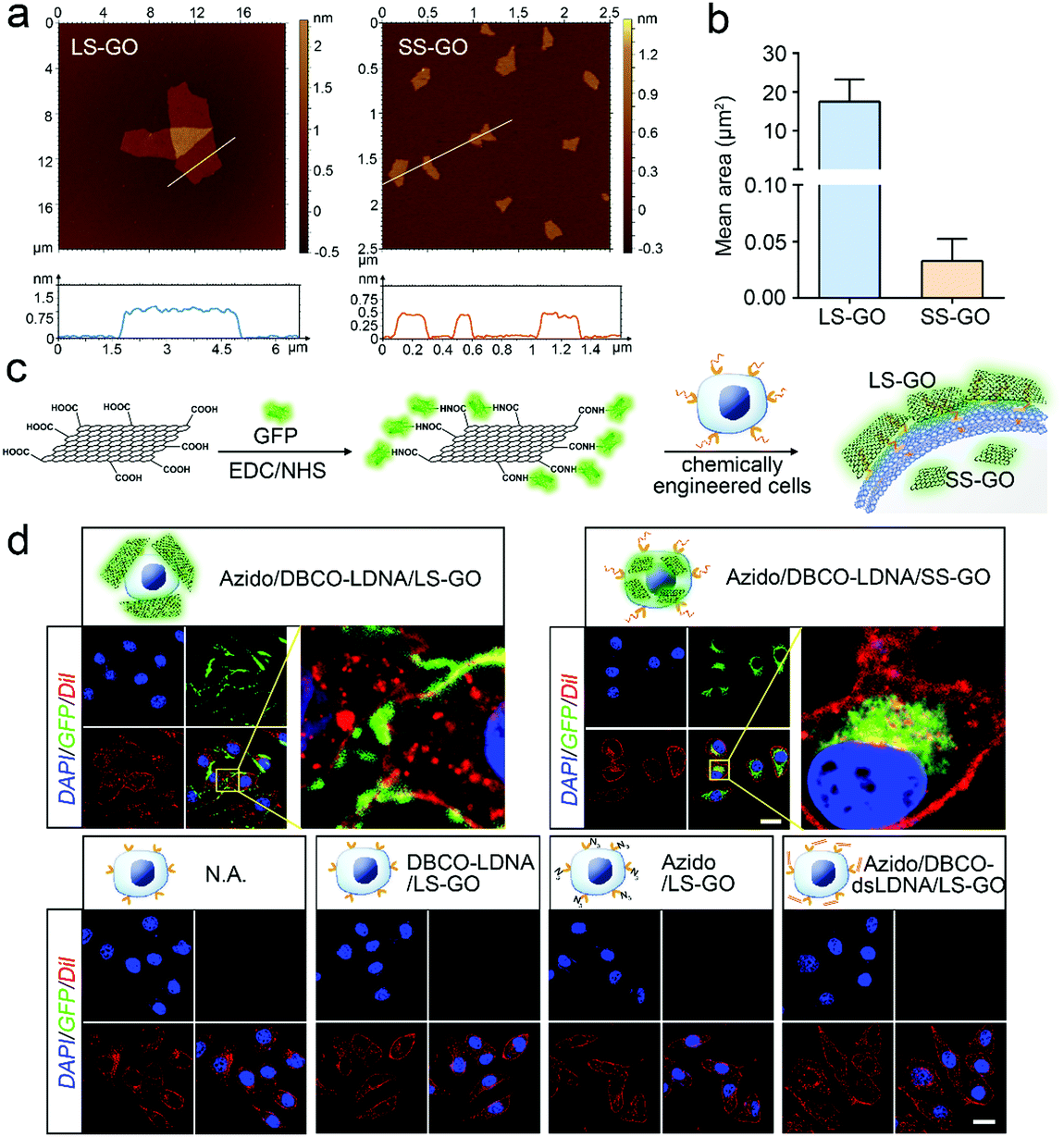 | ||
| Fig. 2 Interaction of graphene oxide with chemically engineered cells. (a) Atomic force microscopic (AFM) images of LS-GO and SS-GO. (b) Area of LS-GO and SS-GO obtained from Image J analysis of the AFM images shown in (a) and Fig. S1 (ESI†). For LS-GO, n = 20; for SS-GO, n = 163. (c) Schematic illustration of the preparation of GFP-labeled GO. (d) Fluorescence images of chemically engineered A549 cells exposed to GFP-labeled GO. The nucleus was stained with DAPI, and the membrane was stained with Dil. All images were obtained using a confocal laser scanning microscope. Scale bar = 20 μm. | ||
Next, the effect of LS-GO on the chemically engineered cells was studied. The results show that the cell viability, apoptosis and proliferation of A549 cells are not affected after chemical engineering treatment and exposure to LS-GO (Fig. S4–S6, ESI†). In these respects, SS-GO also has no significant effect on unengineered or engineered cells. For some other cancer cell lines (MDA-MB-231 cells: human breast cancer cells; and HeLa cells: human cervical cancer cells), the results show that LS-GO also has no significant effect on the above aspects (Fig. S7–S9, ESI†). These results suggest that GO has good biocompatibility, which is also consistent with some reported literature reports.16
But it is interesting that LS-GO has a significant effect on chemically engineered cells in terms of migration and invasion. For migration, as shown in Fig. 3a and b, the results of the wound healing assay show that exposure of the chemically engineered cells to LS-GO greatly reduces the wound healing rate (43.6% reduction compared to untreated cells, p ≤ 0.001), which is barely affected in the control groups using SS-GO or unengineered cells. These results indicate that LS-GO can inhibit cell migration through the interaction with the LDNA conjugated on the cell membrane. This phenomenon is further confirmed by the results of the transwell migration assay (Fig. 3c–e), which shows a similar reduction in the migration rate (59.0% reduction compared to untreated cells, p ≤ 0.001). By staining the cytoskeletal protein F-actin with phalloidin-iFluor 488, it is observed from the cell fluorescence images that the LS-GO covered LDNA-conjugated cells have fewer pseudopods than the control groups (Fig. 3f), which should be the direct cause of the decreased cell migration ability.17 Cell migration is controlled by polymerization of F-actin and formation of pseudopods.18 With the coverage of LS-GO on the chemically engineered cells, some signaling pathways that regulate the formation of pseudopods were probably affected, which will be explored in the last part of this work. Here, these signaling pathways may be the reason that cause F-actin distributed in the cytoplasm to be different from other control groups. The cell invasion ability was then studied by using a transwell matrigel invasion assay. As shown in Fig. 3g–i, exposure of the LDNA-conjugated cells to LS-GO also greatly reduced the invasion rate (53.6% reduction compared to untreated cells, p ≤ 0.001). Similar effects of LS-GO on the migration and invasion of chemically engineered MDA-MB-231 cells and HeLa cells were also observed (Fig. S10–S12, ESI†). Therefore, in summary, here it can be concluded from the above results that LS-GO inhibits the migration and invasion of chemically engineered cells via the interaction with the LDNA on the cell surface.
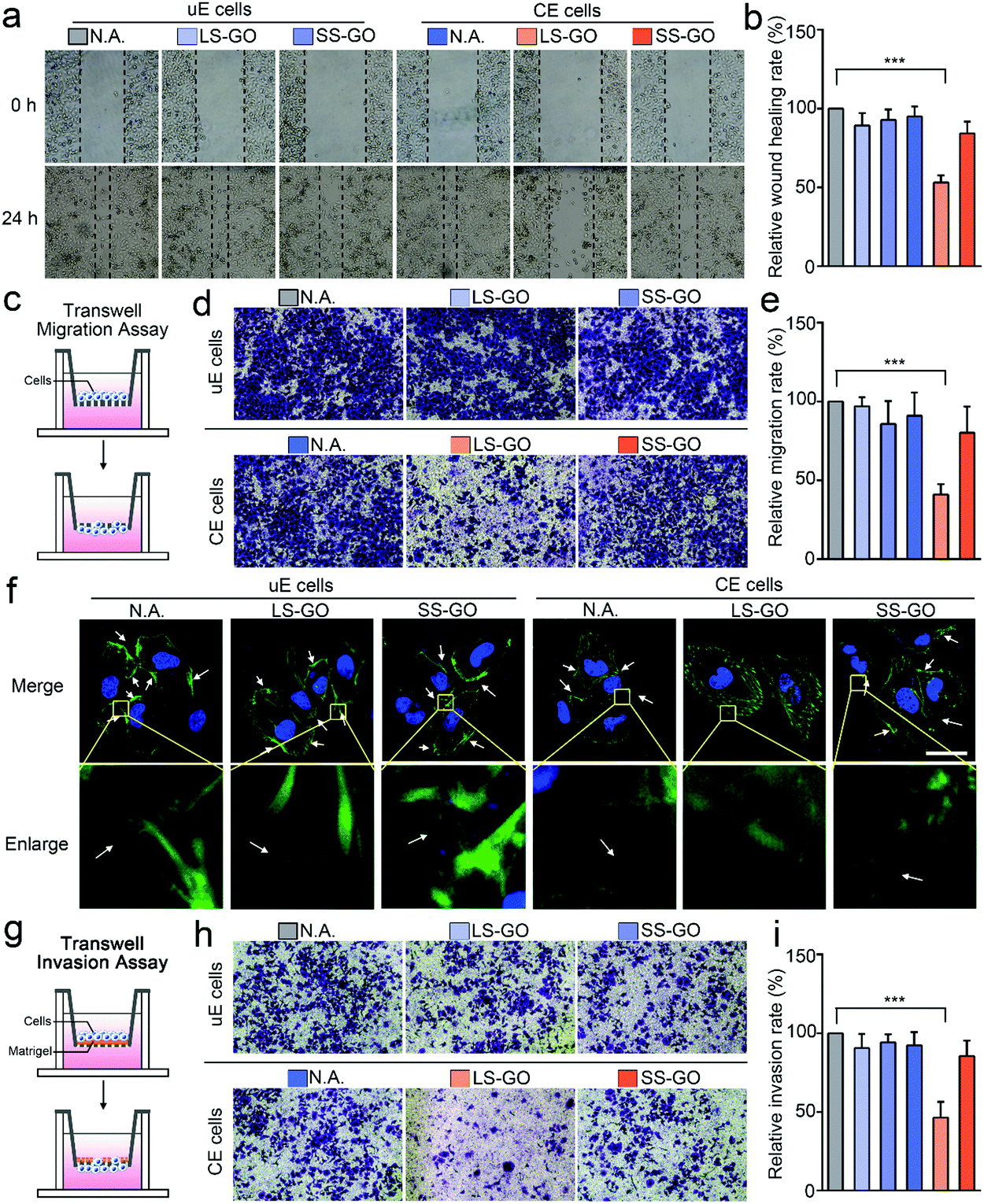 | ||
| Fig. 3 Effect of LS-GO on the migration and invasion of chemically engineered cells. (a) Wound healing assay of the effect of GO on the migration of A549 cells. (b) Statistical results of the wound healing rate obtained from (a). n = 3. (c) Schematic of transwell migration assay. (d) Transwell migration assay of the effect of GO on the migration of A549 cells. (e) Statistical results of the migration rate obtained from (d). n = 3. (f) Fluorescence images of A549 cells exposed to GO. The nucleus was stained with DAPI, and cytoskeletal protein F-actin was stained with phalloidin-iFluor 488. All images were obtained using a confocal laser scanning microscope. The white arrows point to the pseudopods. Scale bar = 20 μm. (g) Schematic of the transwell matrigel invasion assay. (h) Transwell matrigel invasion assay of the effect of GO on the invasion of A549 cells. (i) Statistical results of the invasion rate obtained from (h). n = 3. uE cells: unengineered cells. CE cells: chemically engineered cells. p values were calculated by the Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Considering that tumor metastasis is the clinical manifestation of the migration and invasion of cancer cells and is the critical factor in the death of cancer patients,19 a tumor metastasis model was further constructed to explore if the LS-GO can inhibit the metastasis of chemically engineered tumors in vivo. The model was constructed by planting A549 cells subcutaneously in BALB/c nude mice, followed by studying the lung metastases of the subcutaneous tumors. As shown in Fig. 4a, the subcutaneous tumor bearing mice were treated with Ac4ManNAz and DBCO-LDNA successively to produce a chemically engineered tumor, after which LS-GO was implanted in the region of the subcutaneous tumor. In the course of treatment, there was no significant change in the weight of mice, demonstrating the biosecurity of all the materials (Fig. S13, ESI†). Then, the tumor metastasis was monitored using an in vivo fluorescence imaging system. Fig. 4b shows that no obvious lung metastases were observed in the presence of LS-GO, whereas both subcutaneous tumors and lung metastases could be observed in the control groups without LS-GO or with SS-GO instead. The major parenchyma organs and tumor tissues of the mice were further taken for imaging ex vivo, and the results were consistent with that of integral imaging (Fig. 4c and d). It is noted that in all cases, fluorescence can be detected in the kidneys, which is due to the metabolic excretion of the fluorescence tracer DBCO-Cy5 through the kidneys.20,21 Next, the lung tissues were collected for histopathological observation (H&E staining) (Fig. 4e). The images show that in the presence of LS-GO, there was little high-density variation in the lungs, that is, lung metastasis is significantly inhibited; while in the control groups, there were partial high-density variations, suggesting that the tumor has metastasized to the lungs. The statistics of the number of lung nodules in mice show that there was an average of 7.8 lung metastatic nodules in the presence of LS-GO, while the control groups had an average of 26.8 lung metastatic nodules. Thus, it can be calculated that the inhibition rate of LS-GO on tumor lung metastasis is about 71%. H&E staining results of other parenchyma organs show that there were no obvious side effects in all the groups (Fig. S14, ESI†). As for the subcutaneous tumors, it is also observed that the tumors of the LS-GO group were slightly smaller than those of other control groups (Fig. S15, ESI†), which indicates that due to the inhibition of invasion of cancer cells by LS-GO, tumor growth has also been inhibited to some extent. In summary, the above in vivo results suggest that the interaction between LS-GO and the chemically engineered cancer cells still works in vivo and inhibits tumor metastasis at the animal level. Also considering the good biocompatibility of the materials used and the possible degradability of GO,22 this strategy by implanting LS-GO in chemically engineered tumors provides a promising approach for the treatment of tumors.
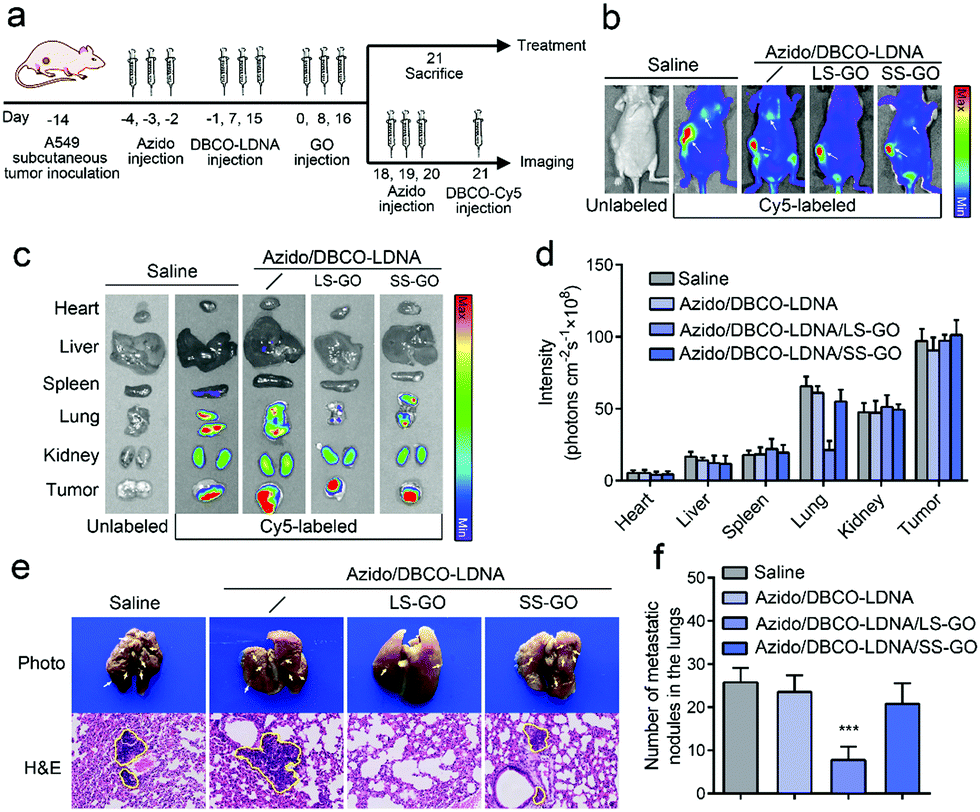 | ||
| Fig. 4 Inhibition of tumor metastasis by implanting LS-GO in vivo. (a) Treatment and imaging schedule in the tumor metastasis model. (b) Integral imaging of tumor bearing mice treated with saline, or Ac4ManNAz/LDNA, or Ac4ManNAz/LDNA/LS-GO, or Ac4ManNAz/LDNA/SS-GO. After the above different treatments, except for the leftmost group, Ac4ManNAz and DBCO-Cy5 were adopted as the fluorescence tracer to image the tumors. n = 4. (c) Ex vivo imaging of major parenchyma organs and subcutaneous tumors after different treatments. n = 4. (d) The bar chart of the fluorescence intensities calculated from (c). (e) Photos of lung tissues (upper panel) and H&E staining images of lung tissues (lower panel) collected from the mice after different treatments. The white arrows and yellow circles point to the lung metastatic nodules. Original magnification: 15×. (f) The number of lung metastatic nodules in mice after different treatments. n = 4. p values were calculated by the Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Finally, the mechanism of the inhibition on the migration and invasion of chemically engineered cells by the LS-GO was studied. As shown in Fig. 5a, the volcano plots of transcriptome analysis show that compared with the control group, there are 318 genes differentially expressed in the LS-GO-treated chemically engineered cells, in which the expression of 143 genes increased and the expression of 175 genes decreased. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was further performed (Fig. 5b). The results show that compared with the control group, LS-GO induced significant changes in the MAPK signaling pathway, which is known to be closely related to migration and invasion by regulating the rolling cycle of the microfilament skeleton and the growth of pseudopods during cell migration.23 Tracing back to the results of transcriptome analysis, several migration-related genes in the MAPK signaling pathway were observed to be up-regulated (including DUSP6) or down-regulated (including EGR1, FOS and JUN) to varying degrees (Fig. 5c), which were further confirmed by quantitative real-time PCR measurement of the mRNAs (Fig. 5d) and western blotting analysis of their corresponding expressed proteins (Fig. 5e and f). The above results collectively indicate that the MAPK signaling pathway is inhibited, which precisely explains the results shown in Fig. 3f that the growth of the pseudopods is inhibited in the presence of LS-GO.
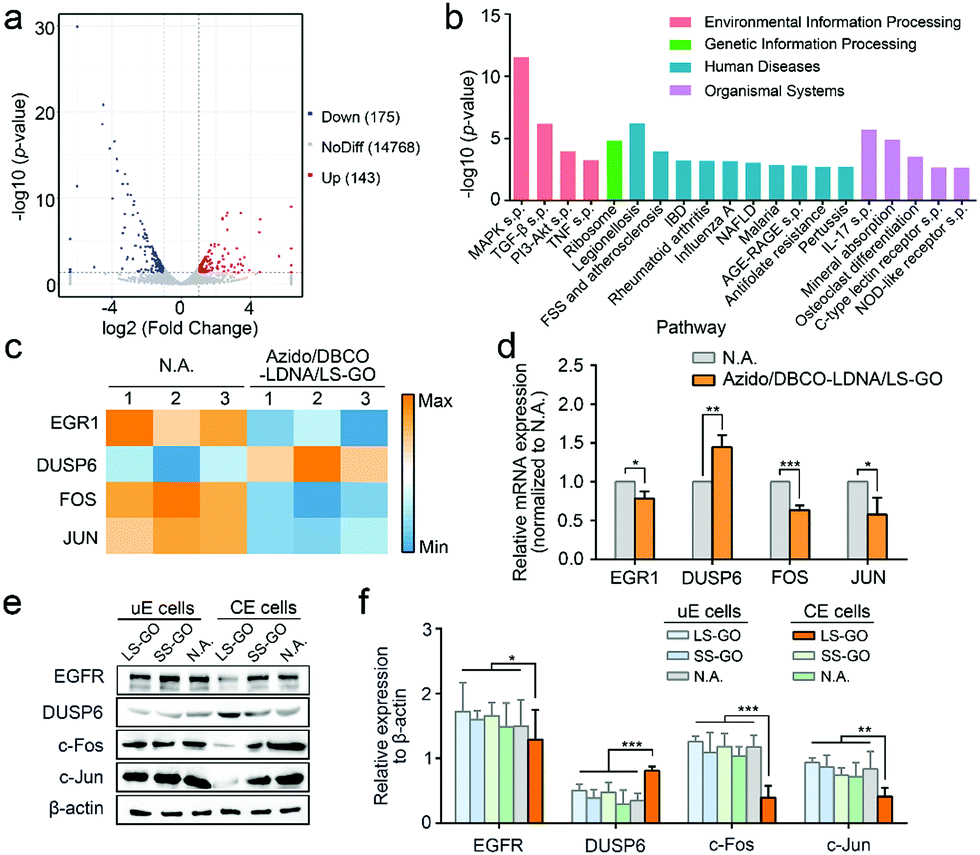 | ||
| Fig. 5 Transcriptome analysis of LS-GO-treated chemically engineered cells. (a) Volcano plot displaying gene expression between unengineered A549 cells and LS-GO-treated chemically engineered A549 cells. The red and blue dots indicate up-regulated and down-regulated genes, respectively. (b) The significantly enriched KEGG pathway (Top 20). The horizontal axis displays the different signal pathway. s.p.: signaling pathway; FSS: Fluid shear stress; IBD: Inflammatory bowel disease; NAFLD: Non-alcoholic fatty liver disease. (c) Heatmap of EGR1, DUSP6, FOS and JUN in the MAPK signaling pathway between unengineered A549 cells and LS-GO-treated chemically engineered A549 cells. (d) Relative quantification of EGR1, DUSP6, FOS and JUN in the MAPK signaling pathway between unengineered A549 cells and LS-GO-treated chemically engineered A549 cells by qRT-PCR. n = 3. (e) Western blotting results of EGFR, DUSP6, c-Fos and c-Jun expression in A549 cells. (f) Statistical results of expression obtained from (e). n = 3. uE cells: unengineered cells. CE cells: chemically engineered cells. p values were calculated by the Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
The proteins (DUSP6, EGFR, c-Fos and c-Jun) expressed by the four genes mentioned above (DUSP6, EGR1, FOS and JUN) are known to be included in the MAPK/ERK signaling cascade pathway,24–26 which is the most widely studied branch pathway of the MAPK signaling pathway. Therefore, to specify if this branch pathway is exactly the target of LS-GO, more signal proteins in this branch pathway were studied, including EGFR, Ras, bRaf, cRaf, mitogen-activated protein kinase kinase (MEK), extracellular signal-regulated kinase (ERK) and matrix metalloproteinase-9 (MMP-9). Western blotting results of the expression of these proteins are observed to support the speculation that the branch pathway is inhibited (Fig. 6a and b). In detail, the MAPK/ERK signaling cascade pathway involves the phosphorylation of multiple signal proteins (including EGFR, bRaf, cRaf, MEK and ERK). The results show that the expression of these proteins barely changes, but their phosphorylation levels are greatly inhibited, which is a typical manifestation of the inhibition of this pathway.27 In addition, it is noted that the expression of matrix metalloproteinase-9 (MMP-9) was down-regulated with the down-regulation of its transcription factors c-Fos and c-Jun. MMP-9 is an enzyme that can degrade various proteins in the extracellular matrix, destroy the histological barrier of tumors, and thus promote tumor metastasis.28,29 Therefore, down-regulation of MMP-9 takes direct responsibility at the molecular level for the inhibition of the migration and invasion of chemically engineered cells. The above results confirm that LS-GO inhibits cell migration and invasion through the inhibition on the branch MAPK/ERK signaling cascade pathway.
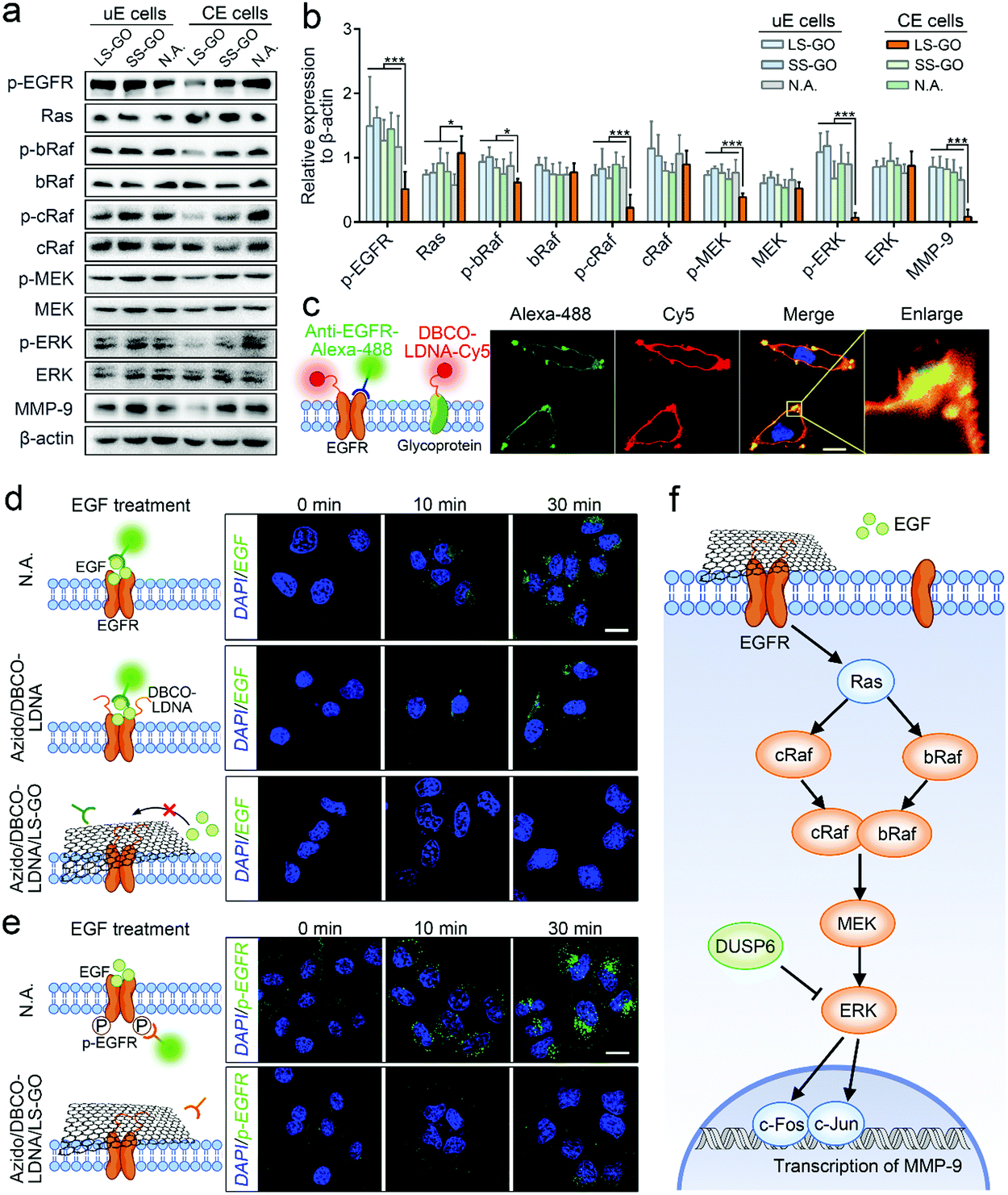 | ||
| Fig. 6 Inhibition of the branch MAPK/ERK signaling cascade pathway. (a) Western blotting results of proteins and phosphorylated proteins in the MAPK/ERK signaling pathway in A549 cells. (b) Statistical results of expression obtained from (a). n = 3. (c) Fluorescence images of EGFR (green) and LDNA (red) on chemically engineered A549 cells. The nucleus was stained with DAPI. All images were obtained using a confocal laser scanning microscope. Scale bar = 5 μm. (d) Fluorescence images of EGF on unengineered, chemically engineered and LS-GO-treated chemically engineered A549 cells after EGF treatment for 0, 10, and 30 min. The nucleus was stained with DAPI. All images were obtained using a confocal laser scanning microscope. Scale bar = 20 μm. (e) Fluorescence images of p-EGFR on unengineered and LS-GO-treated chemically engineered A549 cells after EGF treatment for 0, 10, and 30 min. Scale bar = 20 μm. (f) Proposed mechanism of the effects of LS-GO on the MAPK/ERK signaling cascade pathway. p values were calculated by the Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Because EGFR is the only membrane protein in the MAPK/ERK signaling cascade pathway studied above, it is speculated that LS-GO inhibits this pathway by directly acting on EGFR. To verify this speculation, we first confirmed that EGFR can be conjugated with LDNA in the processing of chemically engineered cells. The fluorescence images in Fig. 6c show that the red fluorescence of protein-conjugated LDNA covers most of the green fluorescence of EGFR with the overlapping part presented in yellow, suggesting that most of the EGFR has been conjugated with LDNA. This result is reasonable and expected, because EGFR is a glycoprotein with sialic acid residues,30 which can theoretically be labeled with azido and DBCO-LDNA through glycometabolism pathways and click chemistry during chemical engineering. In parallel, other glycoproteins on the cell membrane can also be conjugated with LDNA, showing red fluorescence other than the EGFR sites. We next studied the effect of LS-GO on the interaction between EGFR and its ligand EGF, a polypeptide that activates the MAPK/ERK signaling cascade pathway through binding with EGFR.31 By incubating cells with EGF followed by washing the cells, it can be observed that EGF exists on the surface of unengineered cells and engineered cells (Fig. 6d). But in the presence of LS-GO, the fluorescence of EGF in engineered cells almost disappears, suggesting that LS-GO has blocked the binding of EGF with EGFR. Further immunofluorescence results show that the phosphorylation of EGFR caused by the binding of EGF was inhibited (Fig. 6e), which is consistent with the western blotting results shown in Fig. 6a.
Taking all of the above results shown in Fig. 5 and 6, a clear mechanism can be obtained (Fig. 6f): The surface of chemically engineered cells carries a large amount of single-stranded LDNA, which can interact with LS-GO through π–π stacking and thus facilitate the covering of LS-GO on the cell surface. As a glycoprotein on the cell membrane, EGFR is one of the sites carrying LDNA in the chemical engineering processing, and therefore also participates in the interaction with LS-GO, which leads to the blocking of the binding of EGFR with its ligand EGF. The downstream MAPK/ERK signaling cascade pathway is therefore inhibited, which ultimately leads to the inhibition of cell migration and invasion. It should be noted that EGFR is only one of the sites that interacts with LS-GO. The molecular-level impact of LS-GO on the spatial confinement of cancer cells may be broad-spectrum, and needs to be further explored.
Conclusions
In conclusion, LS-GO was adopted to cover and confine chemically engineered cancer cells spatially. As a result, it is found that the migration and invasion of cancer cells was greatly inhibited, which is partly ascribed to the blocking of the interaction between membrane proteins and external ligands and the consequent inhibition of downstream pathways. This effect has been validated at the cell level and in animal models. By implanting LS-GO into subcutaneous tumors, it was observed that the tumors shrunk to a certain extent and the lung metastases were significantly inhibited. In this way, a novel treatment strategy for tumor metastasis was developed, which targets the essential behavioral characteristics of cancer cells directly and thus may have universality for tumors with different molecular phenotypes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22074090, 81871727), the Natural Science Foundation of Shanghai (Grant 19ZR1474200), Three-year action plan of Shanghai Shenkang Hospital Development Center for promoting clinical skills and clinical innovation ability of municipal hospitals (Grant SHDC2020CR2061B), Program of Shanghai Academic Research Leader (Grant 18XD1402600), Pudong New Area Municipal Health Commission of Shanghai (Grant PW2019D-10) and Shanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics (Grant 20dz2260900).Notes and references
- J. Cairns, Nature, 1975, 255, 197–200 CrossRef CAS PubMed.
- M. Gerstung, C. Jolly, I. Leshchiner, S. C. Dentro and S. Gonzalez, et al. , Nature, 2020, 578, 122–128 CrossRef CAS PubMed.
- C. Tomasetti, L. Li and B. Vogelstein, Science, 2017, 355, 1330–1334 CrossRef CAS.
- P. L. Bedard, D. M. Hyman, M. S. Davids and L. L. Siu, Lancet, 2020, 395, 1078–1088 CrossRef CAS.
- M. Loda, Cancer Res., 2016, 76, 4924–4930 CrossRef CAS PubMed.
- A. Brock and S. Huang, Cancer Res., 2017, 77, 6473–6479 CrossRef CAS PubMed.
- I. Dagogo-Jack and A. T. Shaw, Nat. Rev. Clin. Oncol., 2018, 15, 81–94 CrossRef CAS.
- M. Russo, G. Crisafulli, A. Sogari, N. M. Reilly and S. Arena, et al. , Science, 2019, 366, 1473–1480 CrossRef CAS PubMed.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS.
- Y. W. Chen, Y. L. Su, S. H. Hu and S. Y. Chen, Adv. Drug Delivery Rev., 2016, 105, 190–204 CrossRef CAS PubMed.
- Z. J. Gu, S. Zhu, L. Yan, F. Zhao and Y. L. Zhao, Adv. Mater., 2019, 31, 1800662 CrossRef PubMed.
- L. Dong, J. Yang, M. Chhowalla and K. P. Loh, Chem. Soc. Rev., 2017, 46, 7306–7316 RSC.
- H. Wang, R. B. Wang, K. M. Cai, H. He and Y. Liu, et al. , Nat. Chem. Biol., 2017, 13, 415–424 CrossRef CAS PubMed.
- J. Y. Li, S. Y. Liu, L. Q. Sun, W. Li, S. Y. Zhang, S. Yang, J. Li and H. H. Yang, J. Am. Chem. Soc., 2018, 140, 16589–16595 CrossRef CAS PubMed.
- M. Schubert, R. Bergmann, C. Förster, W. Sihver and S. Vonhoff, et al. , Bioconjugate Chem., 2017, 28, 1176–1188 CrossRef CAS PubMed.
- K. Yang, L. Z. Feng, X. Z. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- A. R. Houk, A. Jilkine, C. O. Mejean, R. Boltyanskiy, E. R. Dufresne, S. B. Angenent, S. J. Altschuler, L. F. Wu and O. D. Weiner, Cell, 2012, 148, 175–188 CrossRef CAS PubMed.
- A. Bisaria, A. Hayer, D. Garbett, D. Cohen and T. Meyer, Science, 2020, 368, 1205–1210 CrossRef CAS PubMed.
- H. Hamidi and J. Ivaska, Nat. Rev. Cancer, 2018, 18, 532–547 CrossRef PubMed.
- A. Wurzer, J. Pollmann, A. Schmidt, D. Reich and H. J. Wester, Mol. Pharming, 2018, 15, 4296–4302 CrossRef CAS PubMed.
- F. Hu, D. Mao, Kenry, X. L. Cai, W. B. Wu, D. L. Kong and B. Liu, Angew. Chem., Int. Ed., 2018, 57, 10182–10186 CrossRef CAS PubMed.
- K. Lu, S. Dong, T. Xia and L. Mao, ACS Nano, 2021, 15, 396–409 CrossRef CAS PubMed.
- L. Ge, Y. Ly, M. Hollenberg and K. DeFea, J. Biol. Chem., 2003, 278, 34418–34426 CrossRef CAS PubMed.
- T. Harada, T. Morooka, S. Ogawa and E. Nishida, Nat. Cell Biol., 2001, 3, 453–459 CrossRef CAS PubMed.
- Z. F. Zhang, S. Kobayashi, A. C. Borczuk, R. S. Leidner, T. LaFramboise, A. D. Levine and B. Halmos, Carcinogenesis, 2010, 31, 577–586 CrossRef CAS PubMed.
- Y. Zhang, X. H. Feng and R. Derynck, Nature, 1998, 394, 909–913 CrossRef CAS PubMed.
- W. Kolch, Nat. Rev. Mol. Cell Biol., 2005, 6, 827–837 CrossRef CAS PubMed.
- M. Mishra, J. Flaga and R. A. Kowluru, J. Cell. Physiol., 2016, 231, 1709–1718 CrossRef CAS PubMed.
- S. Hiratsuka, K. Nakamura, S. Iwai, M. Murakami, T. Itoh, H. Kijima, J. M. Shipley, R. M. Senior and M. Shibuya, Cancer Cell, 2002, 2, 289–300 CrossRef CAS PubMed.
- Y. C. Liu, H. Y. Yen, C. Y. Chen, C. H. Chen and P. F. Cheng, et al. , Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11332–11337 CrossRef CAS PubMed.
- C. Huang, K. Jacobson and M. D. Schaller, J. Cell Sci., 2004, 117, 4619–4628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00350j |
| This journal is © The Royal Society of Chemistry 2021 |