
Synthesis of monodisperse high entropy alloy nanocatalysts from core@shell nanoparticles†
Yifan
Chen
 ab,
Xun
Zhan
c,
Sandra L. A.
Bueno
a,
Ibrahim H.
Shafei
a,
Hannah M.
Ashberry
a,
Kaustav
Chatterjee
a,
Lin
Xu
b,
Yawen
Tang
*b and
Sara E.
Skrabalak
*a
ab,
Xun
Zhan
c,
Sandra L. A.
Bueno
a,
Ibrahim H.
Shafei
a,
Hannah M.
Ashberry
a,
Kaustav
Chatterjee
a,
Lin
Xu
b,
Yawen
Tang
*b and
Sara E.
Skrabalak
*a
aDepartment of Chemistry, Indiana University, 800 E. Kirkwood Ave., Bloomington, IN 47405, USA. E-mail: sskrabal@indiana.edu
bJiangsu Key Laboratory of New Power Batteries, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China. E-mail: tangyawen@njnu.edu.cn
cElectron Microscopy Center, Indiana University, 800 E. Kirkwood Ave, Bloomington, IN 47405, USA
First published on 22nd January 2021
Abstract
High-entropy alloy (HEA) nanoparticles (NPs) hold great promise in electrocatalysis because of their nearly unlimited compositions, tailorable active sites, and high durability. However, the synthesis of these compositionally complex structures as monodisperse NPs remains a challenge by colloidal routes because the different rates of metal precursor reduction lead to phase separation. Here, we report the conversion of core@shell NPs into HEA NPs through annealing, with conservation of sample monodispersity. This potentially general route for high-quality HEA NPs was demonstrated by preparing PdCu@PtNiCo NPs via seed-mediated co-reduction, wherein Pt, Ni, and Co were co-deposited on PdCu seeds in solution. These multimetallic NPs were then converted to single-crystalline and single-phase PdCuPtNiCo NPs through annealing. On account of their small particle size, highly dispersed Pt/Pd content, and low elemental diffusivity, these HEA NPs were found to be a highly efficient and durable catalyst for the oxygen reduction reaction. They were also highly selective for the four-electron transfer pathway. We expect that this new synthetic strategy will facilitate the synthesis of new HEA NPs for catalysis and other applications.
New conceptsWe present a new synthetic strategy for monodisperse high-entropy alloy (HEA) nanoparticles (NPs). First, core@shell NPs, in this example consisting of 5 metals, are prepared by seed-mediated co-reduction. Then, these “precursor” NPs are annealed to facilitate interdiffusion and HEA formation, with the monodispersity of the original precursor NPs being transferred to the final sample. Only recently have HEAs been prepared in NP form and they are typically achieved by rapid heating and quenching of metal precursors that facilitate the atomic level mixing that is a necessity for HEA formation. We break from these conventions with the demonstration of this NP conversion strategy, where we hypothesize that the atomic level mixing is promoted by the high interfacial strain in the core@shell precursor NPs. Given the variety of core@shell NPs that can be prepared by colloidal methods, we anticipated this NP conversion strategy to be general to many compositions and will open up avenues for shape and architectural control of HEA NPs. |
Recently, high-entropy alloys (HEAs) have been recognized as a new class of promising electrocatalysts. These materials are single-phase solid solutions consisting of 5 or more elements in nearly equal proportions,1 which brings together many metal combinations that are traditionally inaccessible as binary and ternary alloys in accordance with the Hume-Rothery Rules.2,3 The multi-element nature of these materials leads to high mixing entropy, which facilitates formation of solid solutions in simple crystal structures.4 These features mean that a near-continuum of surface sites may be provided by their compositional complexity.5 Moreover, the low atomic diffusivity that originates from the high entropy mixing of HEAs may also provide high durability in corrosive catalytic environments.6 These qualities provide many opportunities for catalyst design with HEAs. In fact, there are recent studies showing that HEA nanoparticles (NPs) with low Pt content can exhibit mass activities much greater than Pt/C for the oxygen reduction reaction (ORR), along with high durability.7,8 However, synthetic routes for HEAs as nanostructured materials with high surface areas are required.
HEAs were initially fabricated by thermal methods in which metal ingots were liquefied and rapidly cooled to produce bulk materials.9 More recently, Jin and co-workers fabricated the HEA AlNiCoIrMo in nanoporous form by first melting the precursors together, followed by chemical dealloying in alkaline solution.10 Also, a carbothermal shock method was used to synthesize HEAs as size-controlled NPs, where flash heating and cooling of metal precursors in the presence of a NP support was central to their formation.11 Downsizing HEAs to the nanoscale has been synthetically challenging as most colloidal routes to metal NPs rely on reduction of metal salts, which will occur at different rates in the case of HEAs prepared from 5 or more metal precursors.12 Bondesgaard and co-workers reported a colloidal route for HEA nanocatalysts by exploiting novel solvothermal conditions, where metal acetylacetonate precursors were dissolved in an acetone–ethanol mixture to control the reduction kinetics.13 Still, a general colloidal route for HEA NPs is lacking but could provide new opportunities in catalysis and beyond.
Here, we demonstrate a controllable strategy for the synthesis of HEA NPs based on the conversion of colloidally prepared core@shell NPs into single-phase HEA NPs. Most commonly, core@shell metal NPs have a binary composition, wherein the core will be of one metal and the shell will be of a different metal. Depending on the miscibility of the two metals, such core@shell NPs can be converted to single-phase alloy NPs. For example, Xia and co-workers studied the thermal stability of shape-controlled core@shell Pd@Pt nanocrystals by in situ transmission electron microscopy (TEM) and found that alloying between the core and the shell metals occurs at high temperatures, with the specific temperature depending on nanocrystal shape.14 Likewise, Yang and co-workers found that core@shell Ni@Co NPs can be transformed into alloy NiCo NPs at 600 °C, which occurred through a procedure removing surface oxide and metal segregation.15 Here, the synthesis of two-phase core@shell NPs consisting of 5 metals is reported and then converted to single-phase HEA NPs. Specifically, intermetallic PdCu NPs served as seeds upon which Pt, Ni, and Co were co-deposited by seed-mediated co-reduction (SMCR), producing core@shell PdCu B2@PtNiCo NPs that could be converted to HEA PdCuPtNiCo NPs. The deposition of Pt, Ni, and Co was selected because prior work with SMCR indicated that precursors of these metals could be co-reduced under similar synthetic conditions,16 while Pt and Pd are known ORR electrocatalysts to which alloying them with Ni, Cu, and Co has proven to enhance activity in bimetallic systems. The intermetallic PdCu NPs were selected as seeds to provide a difference in crystal structure between the core (ordered, B2, Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), shell (random alloy, face-centered cubic (FCC), Fmm), and annealed sample (random alloy, FCC, Fmm), making characterization by powder X-ray diffraction (XRD) and electron microscopy more straightforward.
m), shell (random alloy, face-centered cubic (FCC), Fmm), and annealed sample (random alloy, FCC, Fmm), making characterization by powder X-ray diffraction (XRD) and electron microscopy more straightforward.
The initial preparation of core@shell NPs is central to the formation of monodisperse HEA NPs as a one-step co-reduction method in which typical metal precursors – palladium(II) bromide (PdBr2), copper(II) acetate (Cu(ac)2), platinum bromide (PtBr2), nickel acetylacetonate (Ni(acac)2), and cobalt(II) acetate (Co(ac)2) – are reduced in heated 1-octadecene (ODE) and oleylamine (OLA) produces multiphase NPs (Fig. S1, see ESI,† for full experimental details). A representative TEM image (Fig. S1a, ESI†) shows a diversity of NP shapes and their sizes are not uniform. The XRD pattern shows the formation of three phases at least: Pt, intermetallic PdCu (B2), and an alloy (Fig. S1b, ESI†). High-resolution TEM (HRTEM) indicates some NPs are core@shell (Fig. S1c, ESI†), which is further confirmed by the corresponding scanning TEM-energy dispersive X-ray spectroscopy (STEM-EDS) elemental mapping images (Fig. S1d, ESI†) and line scan profile (Fig. S1e, ESI†). Pt, Ni, and Co are located as a shell, where Pd and Cu are located at the core of the NP. The different reduction potentials of each precursor and the tendency to form intermetallic PdCu preferentially under these reaction conditions account for the formation of core@shell NPs.17,18 Unfortunately, the Pt impurities as well as NPs of different shapes are difficult to separate from the core@shell NPs by centrifugation or modification of the reaction conditions. That is, one-step colloidal pathways in which so many precursors are required are likely to yield complex mixtures rather than a single NP type such as HEA NPs.
Instead, core@shell PdCu B2@PtNiCo NPs were intentionally prepared using SMCR to improve the monodispersity of the precursor NPs. This method has been used in our group to prepare core@shell NPs with bimetallic surfaces, wherein two metal precursors are co-reduced in the presence of seeds to deposit an alloy onto the seeds.19 Recently, we showed that random alloy PtM (M = Ni, Co, Cu, or Fe) shells could be deposited onto intermetallic PdCu seeds via SMCR, producing NPs composed of 4 metals.16 Using this strategy, we hypothesized that an additional metal could be added to create core@shell NPs composed of 5 metals that could then be annealed at high temperature to promote diffusion, mixing, and the formation of HEA NPs. The overall strategy is summarized in Fig. 1. Specifically, intermetallic PdCu B2 seeds (Fig. S2 (ESI†); 7.5 ± 0.7 nm in size) were prepared and PtBr2, Ni(acac)2, and Co(ac)2 were co-reduced in the presence of the seeds in a heated solution of ODE and OLA. The obtained PdCu B2@PtNiCo NPs (Fig. 2a and b) had an average size of (10.4 ± 0.4) nm. The bulk composition of the PdCu B2@PtNiCo NPs evaluated by scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDS) (i.e., measurement obtained over an area containing a large amount of NPs) indicates the coexistence of Pd (20 at%), Cu (17 at%), Pt (22 at%), Ni (21 at%), and Co (20 at%) in the NPs, nearly equimolar amounts of each metal (Table S1, ESI†). The XRD pattern for the PdCu B2@PtNiCo NPs displays two sets of diffraction patterns, which can be indexed to the intermetallic PdCu B2 phase (the shoulders at lower 2θ) and a random alloy FCC structure (Fig. 2c). The STEM-EDS elemental mapping images (Fig. 2d) and line scan profile (Fig. 2e) shows that Pd and Cu are located inside the NPs, whereas the Pt, Ni, and Co are distributed on the surface, which is consistent with the SMCR approach.
 | ||
| Fig. 1 Scheme of the synthetic process of PdCuPtNiCo HEAs, where intermetallic PdCu B2 seeds are prepared first. Then Pt, Ni, and Co are deposited on the intermetallic PdCu B2 seeds using SMCR, resulting in core@shell PdCu B2@PtNiCo NPs. These core@shell NPs are then annealed to convert them to single-phase HEA PdCuPtNiCo NPs. | ||
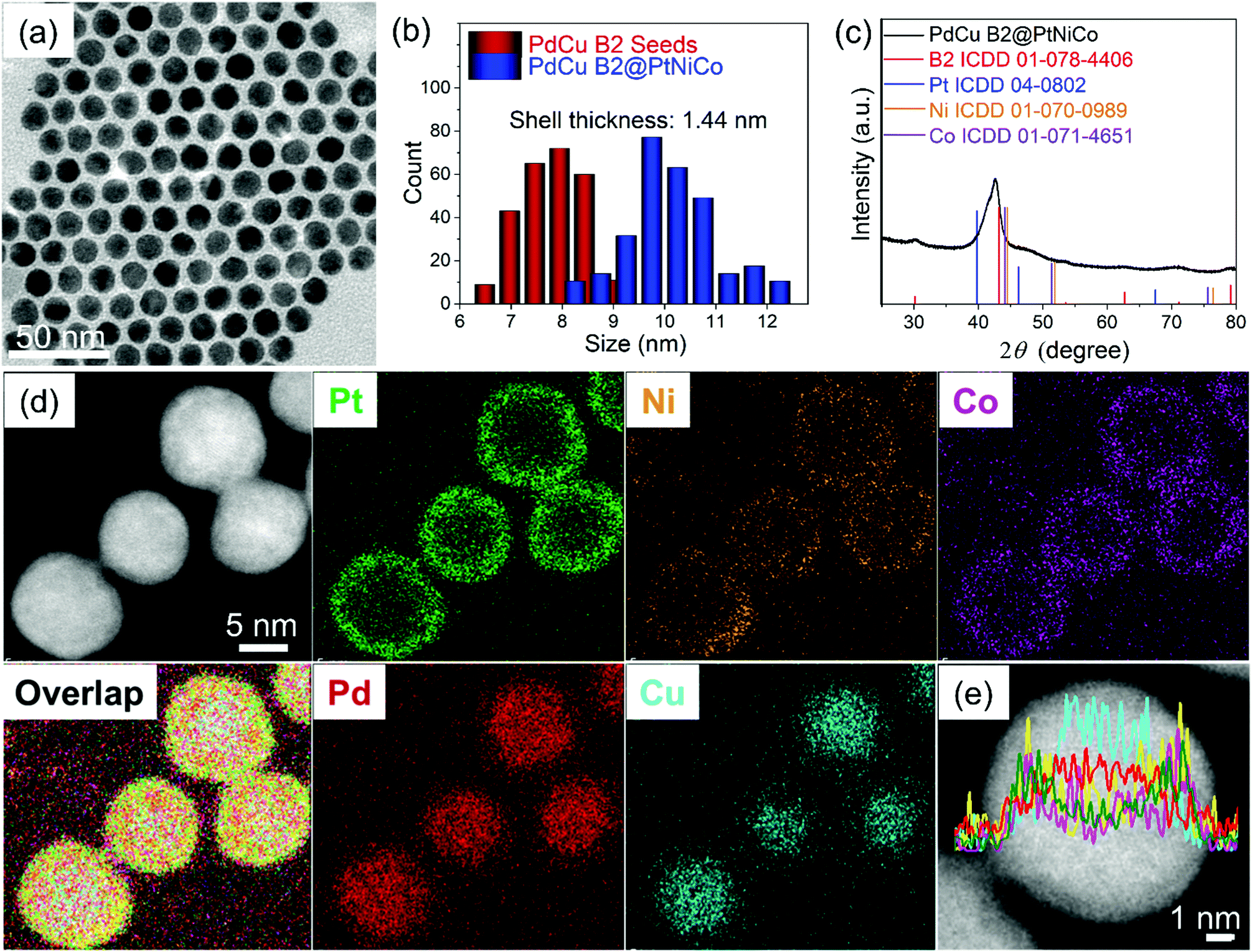 | ||
| Fig. 2 Morphological and compositional characterization of PdCu B2@PtNiCo NPs: (a) TEM bright-field image, (b) histogram of size distribution, (c) XRD pattern, (d) STEM-EDS elemental mapping images and (e) line scan profile. | ||
A probe spherical aberration (Cs)-corrected STEM high-angle annular dark field (HAADF) image was obtained along the PdCu B2 [100] zone axis to better understand the interface structure of the PdCu B2@PtNiCo NPs (Fig. 3). The power spectra obtained at core and shell of the HAADF image reveal the crystallographic orientation of the shell relative to the core. For the orientation relationship, when analyzed from the PdCu B2 [100] projection, one can observe that the (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1) facet of the shell was deposited on top of the (011) facet of the PdCu B2 and the projection of the shell is [110]. This analysis is supported by the angle-to-plane relationships also denoted in Fig. 3a. While for the interfacial orientation relationship, the analysis indicates that the (1) and (12) facets of the shell are in parallel with the (011) and (01) facets of the PdCu B2 core, respectively. The d-spacing for the (1) facet of the shell and (011) facet of the PdCu B2 core are measured as 0.236 and 0.224 nm, respectively, and the mismatch in d-spacing is −5.2%.20 By contrast, the mismatch at the interface is measured as 0.7%, where the d-spacing for the (12) facet of the shell and (01) facet of the PdCu B2 core are measured as 0.218 and 0.224 nm, respectively. Therefore, the interface between core and shell is coherent, and the shell is under tensile strain. Taken together, the deposition of the FCC PtNiCo shell proceeds in a manner that minimizes interfacial energy, just as it was observed with the deposition of PtM shells on PdCu B2 seeds.19
1) facet of the shell was deposited on top of the (011) facet of the PdCu B2 and the projection of the shell is [110]. This analysis is supported by the angle-to-plane relationships also denoted in Fig. 3a. While for the interfacial orientation relationship, the analysis indicates that the (1) and (12) facets of the shell are in parallel with the (011) and (01) facets of the PdCu B2 core, respectively. The d-spacing for the (1) facet of the shell and (011) facet of the PdCu B2 core are measured as 0.236 and 0.224 nm, respectively, and the mismatch in d-spacing is −5.2%.20 By contrast, the mismatch at the interface is measured as 0.7%, where the d-spacing for the (12) facet of the shell and (01) facet of the PdCu B2 core are measured as 0.218 and 0.224 nm, respectively. Therefore, the interface between core and shell is coherent, and the shell is under tensile strain. Taken together, the deposition of the FCC PtNiCo shell proceeds in a manner that minimizes interfacial energy, just as it was observed with the deposition of PtM shells on PdCu B2 seeds.19
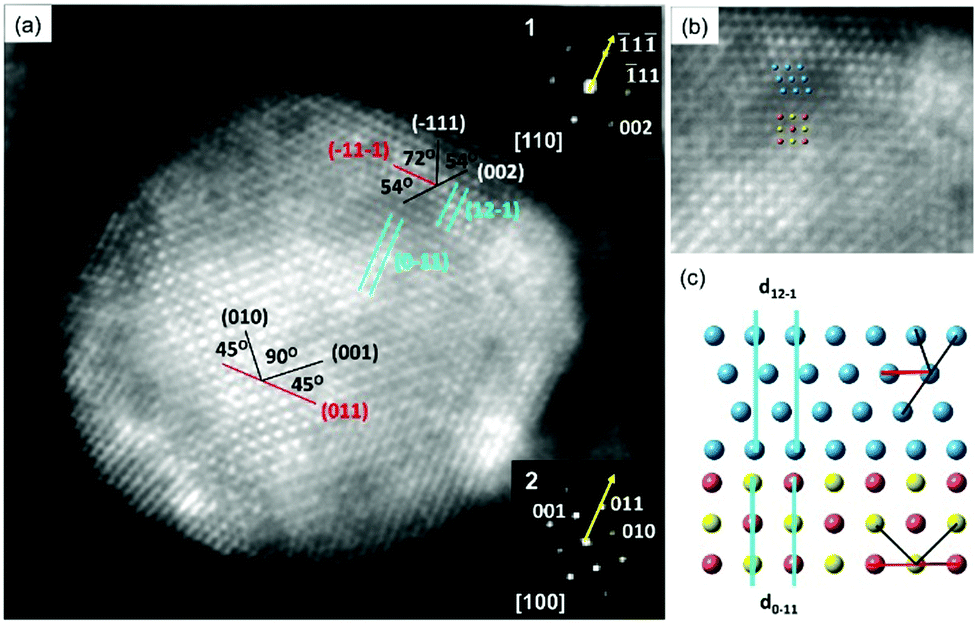 | ||
| Fig. 3 (a) An aberration-corrected STEM-HAADF image of PdCu B2@PtNiCo NPs along the PdCu B2 [100] zone axis and PtNiCo FCC [110] zone axis; the orientations were confirmed by the corresponding FFT in inset (1–2). The annotations indicate the angle-to-plane relationships used to determine the facets defining the core and shell interface. (b) Cropped atomic resolution images from (a). (c) The 2D atomic models show the core–shell interface, where the blue atoms are the repeating units for shell and the rest of the atoms in the bottom are the repeating units for the PdCu B2 core. | ||
To prevent NP coalescence during annealing, the PdCu B2@PtNiCo NPs were evenly dispersed on a carbon support (denoted as PdCu B2@PtNiCo/C; Fig. S3, ESI†). They were then heated at 600 °C for 10 h to obtain the PdCuPtNiCo HEAs/C (Fig. 4a). The size distribution of the PdCuPtNiCo HEAs/C is 10.6 ± 0.2 nm and shows that the monodispersity of the core@shell precursor NPs is largely transferred to the HEA NPs (Fig. S4, ESI†). X-ray photoelectron spectroscopy (XPS) reveals a surface atomic ratio for Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu:Pt:Ni:Co of 17.4:18.7:21.4:22.6:19.9, which is close to the bulk composition determined by EDS (Fig. S5, ESI†). Also, the binding energies of the major features for each metal match with metallic values (oxidation state 0). The XRD profile shows the disappearance of the intermetallic peaks after annealing (Fig. 4b). No peaks can be assigned to pure Pd, Cu, Pt, Ni, or Co, supporting the formation of a single-phase alloy. Based on Bragg's Law and the relationship between d-spacings and the unit cell parameter a for different crystal structure types – Fmm, Pmm, and Imm – the crystal structure can be assigned to the Fmm (FCC) structure (Table S2, ESI†). The STEM-HAADF image further supports the formation of single-phase and single-crystalline NPs (Fig. 4c). The STEM-EDS elemental mapping images (Fig. 4d) and line scan profile (Fig. 4e) show the complete mixing of the five metals within the individual NPs, indicating that annealing of multicomponent NPs is a viable pathway to HEA NPs. The homogeneous elemental distribution after annealing can be explained from the following views: (1) high interfacial strain leads to a lower barrier to diffusion, facilitating mixing;21 (2) thermodynamically, the increased entropy of mixing promotes formation of alloy structure;9 and (3) kinetically, the lattice distortion and low atomic diffusivity upon mixing can prevent NPs from phase separation upon cooling.22
:Cu:Pt:Ni:Co of 17.4:18.7:21.4:22.6:19.9, which is close to the bulk composition determined by EDS (Fig. S5, ESI†). Also, the binding energies of the major features for each metal match with metallic values (oxidation state 0). The XRD profile shows the disappearance of the intermetallic peaks after annealing (Fig. 4b). No peaks can be assigned to pure Pd, Cu, Pt, Ni, or Co, supporting the formation of a single-phase alloy. Based on Bragg's Law and the relationship between d-spacings and the unit cell parameter a for different crystal structure types – Fmm, Pmm, and Imm – the crystal structure can be assigned to the Fmm (FCC) structure (Table S2, ESI†). The STEM-HAADF image further supports the formation of single-phase and single-crystalline NPs (Fig. 4c). The STEM-EDS elemental mapping images (Fig. 4d) and line scan profile (Fig. 4e) show the complete mixing of the five metals within the individual NPs, indicating that annealing of multicomponent NPs is a viable pathway to HEA NPs. The homogeneous elemental distribution after annealing can be explained from the following views: (1) high interfacial strain leads to a lower barrier to diffusion, facilitating mixing;21 (2) thermodynamically, the increased entropy of mixing promotes formation of alloy structure;9 and (3) kinetically, the lattice distortion and low atomic diffusivity upon mixing can prevent NPs from phase separation upon cooling.22
 | ||
| Fig. 4 Morphological and compositional characterization of PdCuPtNiCo HEAs/C: (a) TEM bright-field image, (b) XRD pattern, (c) STEM-HAADF image, (d) STEM-EDS elemental mapping images and (e) line scan profile. | ||
With the successful demonstration of HEA NP formation from core@shell NPs, we next sought to verify their utility as electrocatalysts for the ORR in comparison to the PdCu B2@PtNiCo/C and commercial Pt/C. The NPs were dropcasted onto a glassy carbon electrode and prepared for evaluation by standard methods (see details in ESI†). Initial evaluations of their electrocatalytic activities toward the ORR were carried out in an O2-saturated 0.1 M KOH, where we note that the development of durable catalysts for the ORR in alkaline media is important given the tendency for reduced anion adsorption and better kinetics for the ORR compared to in acidic media.23,24Fig. 5a shows the ORR polarization curves from the catalysts, where the current densities were normalized to the geometrical area of the electrode. The PdCuPtNiCo HEAs/C exhibit a more positive onset potential (Eonset) (1.06 V) than those of the PdCu B2@PtNiCo/C (1.03 V) and commercial Pt/C (1.02 V). The enhancement of ORR activity with PdCuPtNiCo HEAs/C was also indicated by the positive half-wave potential (E1/2) (0.83 V) relative to PdCu B2@PtNiCo/C (0.79 V) and commercial Pt/C (0.81 V), which manifests that oxygen is more efficiently reduced on the surface of PdCuPtNiCo HEAs/C (Fig. S6, ESI†). The mass activities were normalized by the mass of Pt on the electrode, with the activity decreasing as PdCuPtNiCo HEAs/C > PdCu B2@PtNiCo/C > Pt/C (Fig. 5b). The kinetic current density (j) was calculated by the Koutecky–Levich (K–L) equation from Fig. 5a.25 The j of PdCuPtNiCo HEAs/C is greater than those of the PdCu B2@PtNiCo/C and Pt black in the potential region of 0.75 to 0.95 V, which indicates that PdCuPtNiCo HEAs/C has a larger kinetic current density, with better kinetic behavior than the core@shell counterpart or Pt black (Fig. 5c).26 Rotating ring-disk electrode (RRDE) experiments (Fig. S7, ESI†) further reveal selectivity in the ORR process for PdCuPtNiCo HEAs/C, with a low ratio of peroxide (1.10–1.85%) and higher electron transfer number (3.95–3.97) than those of the commercial Pt/C (1.53–4.07%; 3.89–3.96). The RRDE results indicate that the high efficiency for the ORR occurred on the PdCuPtNiCo HEAs/C through the four-electron transfer pathway.
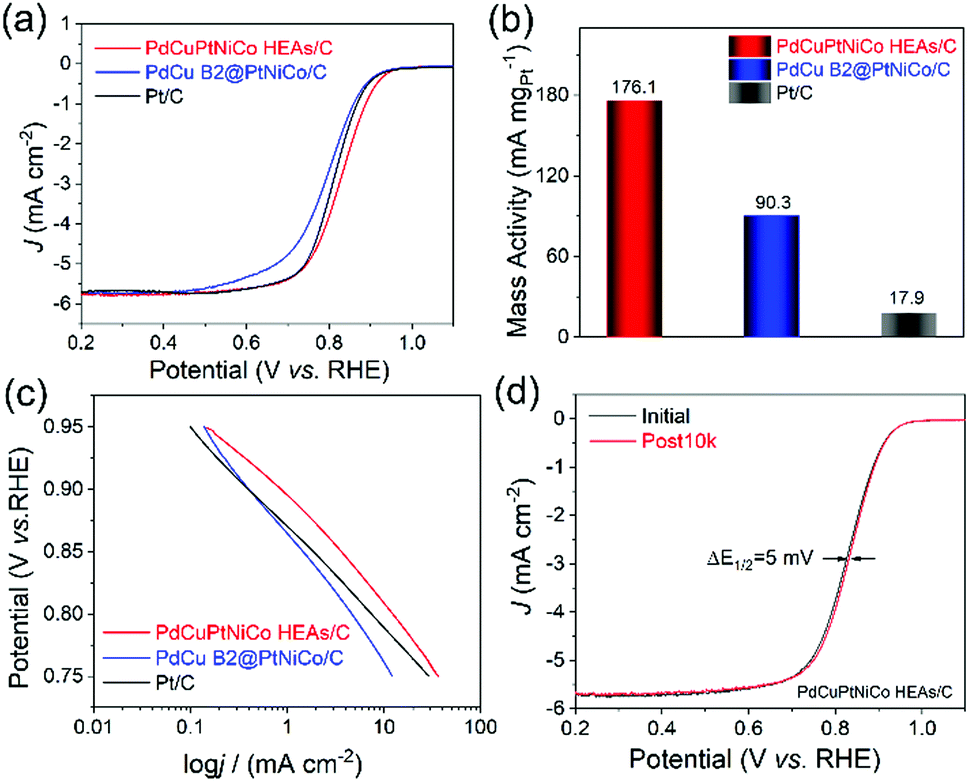 | ||
| Fig. 5 Evaluation of ORR performance of PdCuPtNiCo HEAs/C, PdCu B2@PtNiCo/C, and Pt/C. (a) LSV curves recorded in O2-saturated 0.1 M KOH solution with a rotation rate of 1600 rpm, (b) Mass activity at 0.9 V, and (c) Tafel plots. (d) ORR polarization curves of the PdCuPtNiCo HEAs/C before and after 10000 cycles. | ||
We also conducted ORR electrocatalysis tests in 0.1 M HClO4 solution (Fig. S8, ESI†). Unfortunately, our results do not exceed the results of commercial Pt/C. The performance difference in acidic and alkaline media is possibly attributed to the different ORR mechanisms proceeded in different electrolytes. Additionally, the charge transfer resistance (Rct) may vary at different electrolytes, leading to different reaction kinetics and varied ORR performance. The electrochemically active surface areas (ECSAs) of PdCuPtNiCo HEAs/C, PdCu B2@PtNiCo/C, and commercial Pt/C were calculated and the results show that the ECSA of the PdCuPtNiCo HEAs/C (79.4 m2 gPt−1 for acid medium; 79.9 m2 gPt−1 for alkaline medium) was larger than PdCu B2@PtNiCo/C (39.7 m2 gPt−1 for acid medium; 43.8 m2 gPt−1 for alkaline medium) and commercial Pt/C (76.7 m2 gPt−1 for acid medium; 55.3 m2 gPt−1 for alkaline medium) (Fig. S9, ESI†).
The stability tests for the catalysts in alkaline media show a surprising 5 mV increase in E1/2 for PdCuPtNiCo HEAs/C after 10000 cycles (Fig. 5d; 0.828 V vs. 0.833 V). In contrast, the E1/2 of PdCu B2@PtNiCo/C and commercial Pt/C display negative shifts of 6 mV and 8 mV under the same condition (Fig. S10, ESI†). TEM, STEM-EDS mapping, and line scan profile analysis were used to analyze the PdCuPtNiCo HEAs/C after stability testing to identify the origin of the increase in E1/2 (Fig. S11, ESI†). The spherical morphology and NP monodispersity of PdCuPtNiCo HEAs/C are well maintained, without obvious aggregation after testing. However, a Pt shell is evident after the accelerated durability test (ADT), where Pd, Cu, Ni, and Co are mainly located inside of the NPs. Leaching of non-noble metals from HEA NPs has been reported previously during the hydrogen evolution reaction and methanol oxidation reaction in alkaline medium.27 Here, such leaching appears to be limited by formation of the Pt shell.8 This observation is surprising given the low diffusivity of elements in HEAs; however, this effect does not appear to impact the activity or durability of NPs, suggesting that there is good adhesion between the Pt-rich skin and the HEA core. In the case of PdCu B2@PtNiCo/C, no aggregation and morphology changes after the ADT test are observed and the core@shell structure is maintained (Fig. S12, ESI†). TEM analysis of Pt/C before and after ADT test shows that there is aggregation (Fig. S13, ESI†). Chronoamperometric (i–t curve) tests performed in O2-saturated 0.1 M KOH solution at 0.7 V further verify the stability of PdCuPtNiCo HEAs/C, PdCu B2@PtNiCo/C, and Pt/C (Fig. S14, ESI†). After running for 30000 s, the PdCuPtNiCo HEAs/C exhibit higher retention of the initial current density compared with PdCu B2@PtNiCo/C and Pt/C. The superb activity for the ORR likely arises from the well-dispersed Pt/Pd content and the unique electronic effects of HEAs, although the formation of a Pt-rich skin also suggests that strain may be a contributing factor as well. Studies are ongoing to prepare additional materials for catalytic comparison in order to understand the origin of such enhancements and to identify the active sites for catalysis.28–30
Conclusions
Here, we report a new and potentially generalizable route to HEA NPs as monodisperse samples. This strategy is based on the synthesis of multimetallic core@shell NPs through SMCR and their subsequent conversion to HEA NPs by annealing. SMCR allows for multiple metals to be added into individual NPs in nearly equal amounts while achieving monodisperse samples that can be preserved through the annealing pathway when dispersed on a support. The well-dispersed HEA nanomaterials showed high performance and durability in catalyzing the ORR in alkaline media, where the enhanced ORR activity of PdCuPtNiCo HEAs/C can be attributed to the highly dispersed Pt/Pd content and unique features of HEAs.7,8 Given the flexibility of SMCR to achieve a multitude of multimetallic core@shell NPs, we envision that many different 5-element or higher-order alloys can be prepared simply by changing the compositions of the core and shell materials. Such experiments are on-going. In this way, the predictable synthesis of monodisperse HEA NPs for a diversity of applications has been enabled by this demonstration.Authors Contributions
Y. C. initiated synthesis and annealing experiments and electrocatalysis work. She also took lead in manuscript writing. X. Z. assisted in HRTEM and XRD analysis. S. L. A. B. collected the HRTEM images of samples at the Electron Microscopy Center at Indiana University (IU). I. S. validated the synthetic method. H. M. A. conducted ORR stability testing. K. C. collected and analyzed XPS data. L. X., Y. T., and S. E. S. supervised the project and student training, while providing research resources and support for manuscript preparation (including review and editing). Y. C. and S. E. S. are responsible for the project concept. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This manuscript is dedicated to Dr Chia-Kuang (Frank) Tsung, whose talent and friendship was taken away by the COVID-19 pandemic. Y. C., X. Z., S. L. A. B., I. S., H. M. A., K. C., and S. E. S. acknowledge financial support from IU and Y. C., S. L. A. B., I. S., H. M. A., K. C., and S. E. S. acknowledge support from U.S. DOE BES Award DE-SC0018961, both of which were used toward material synthesis, characterization, and catalytic evaluation. Access to the powder diffractometer was provided by NSF CRIF CHE-1048613. S. L. A. B. thanks U.S. NSF DGE-1342962 for a Graduate Research Fellowship. The atomic resolution HAADF STEM imaging was carried out in the Materials Research Laboratory Central Research Facilities, University of Illinois. We also want to thank the IU Electron Microscopy Center and Nanoscale Characterization Facility for access to the necessary instrumentation. Y. C. and Y. T. acknowledge financial support from the National Natural Science Foundation of China (No. 21875112), National and Local Joint Engineering Research Center of Biomedical Functional Materials and Priority Academic Program Development of Jiangsu Higher Education Institutions. Y. C. thanks the China Scholarship Council (Grant No. 201806860028) for the award of a fellowship, which supported her stay at IU.Notes and references
- X. Chang, M. Zeng, K. Liu and L. Fu, Adv. Mater., 2020, 32, 1907226 CrossRef CAS.
- H. Fang, J. Yang, M. Wen and Q. Wu, Adv. Mater., 2018, 30, 1705698 CrossRef.
- S. E. Skrabalak, Science, 2018, 359, 1467 CrossRef CAS.
- Y. Yao, Z. Liu, P. Xie, Z. Huang, T. Li, D. Morris, Z. Finfrock, J. Zhou, M. Jiao, J. Gao, Y. Mao, J. Miao, P. Zhang, R. Shahbazian-Yassar, C. Wang, G. Wang and L. Hu, Sci. Adv., 2020, 6, eaaz0510 CrossRef CAS.
- T. Löffler, A. Savan, H. Meyer, M. Meischein, V. Strotkötter, A. Ludwig and W. Schuhmann, Angew. Chem., Int. Ed., 2020, 59, 5844–5850 CrossRef.
- W. Zhang, P. K. Liaw and Y. J. S. C. M. Zhang, Sci. China Mater., 2018, 61, 2–22 CrossRef CAS.
- H.-J. Qiu, G. Fang, Y. Wen, P. Liu, G. Xie, X. Liu and S. Sun, J. Mater. Chem. A, 2019, 7, 6499–6506 RSC.
- S. Li, X. Tang, H. Jia, H. Li, G. Xie, X. Liu, X. Lin and H.-J. Qiu, J. Catal., 2020, 383, 164–171 CrossRef CAS.
- J.-W. Yeh, S.-K. Chen, S.-J. Lin, J.-Y. Gan, T.-S. Chin, T.-T. Shun, C.-H. Tsau and S.-Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- Z. Jin, J. Lv, H. Jia, W. Liu, H. Li, Z. Chen, X. Lin, G. Xie, X. Liu, S. Sun and H.-J. Qiu, Small, 2019, 15, 1904180 CrossRef CAS.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS.
- S. G. Kwon, G. Krylova, P. J. Phillips, R. F. Klie, S. Chattopadhyay, T. Shibata, E. E. Bunel, Y. Liu, V. B. Prakapenka, B. Lee and E. V. Shevchenko, Nat. Mater., 2015, 14, 215–223 CrossRef CAS.
- M. Bondesgaard, N. L. N. Broge, A. Mamakhel, M. Bremholm and B. B. Iversen, Adv. Funct. Mater., 2019, 29, 1905933 CrossRef CAS.
- M. Vara, L. T. Roling, X. Wang, A. O. Elnabawy, Z. D. Hood, M. Chi, M. Mavrikakis and Y. Xia, ACS Nano, 2017, 11, 4571–4581 CrossRef CAS.
- C. S. Bonifacio, S. Carenco, C. H. Wu, S. D. House, H. Bluhm and J. C. Yang, Chem. Mater., 2015, 27, 6960–6968 CrossRef CAS.
- J. T. L. Gamler, H. M. Ashberry, X. Sang, R. R. Unocic and S. E. Skrabalak, ACS Appl. Nano Mater., 2019, 2, 4538–4546 CrossRef CAS.
- C. Wang, D. P. Chen, X. Sang, R. R. Unocic and S. E. Skrabalak, ACS Nano, 2016, 10, 6345–6353 CrossRef CAS.
- N. Ortiz, R. G. Weiner and S. E. Skrabalak, ACS Nano, 2014, 8, 12461–12467 CrossRef CAS.
- C. Wang, X. Sang, J. T. L. Gamler, D. P. Chen, R. R. Unocic and S. E. Skrabalak, Nano Lett., 2017, 17, 5526–5532 CrossRef CAS.
- J. T. L. Gamler, A. Leonardi, H. M. Ashberry, N. N. Daanen, Y. Losovyj, R. R. Unocic, M. Engel and S. E. Skrabalak, ACS Nano, 2019, 13, 4008–4017 CrossRef CAS.
- B. P. Williams, A. P. Young, I. Andoni, Y. Han, W.-S. Lo, M. Golden, J. Yang, L.-M. Lyu, C.-H. Kuo, J. W. Evans, W. Huang and C.-K. Tsung, Angew. Chem., Int. Ed., 2020, 59, 10574–10580 CrossRef CAS.
- Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang, Mater. Today, 2016, 19, 349–362 CrossRef CAS.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- Y. Chen, Z. Li, Y. Zhu, D. Sun, X. Liu, L. Xu and Y. Tang, Adv. Mater., 2019, 31, 1806312 CrossRef.
- N. Zhang, Y. Feng, X. Zhu, S. Guo, J. Guo and X. Huang, Adv. Mater., 2017, 29, 1603774 CrossRef.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Nat. Commun., 2020, 11, 5437 CrossRef CAS.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3, 834 CrossRef CAS.
- V. M. Schmidt, R. Ianniello, E. Pastor and S. Gonzalez, J. Phys. Chem., 1996, 100, 17901 CrossRef CAS.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00656d |
| This journal is © The Royal Society of Chemistry 2021 |