
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantifying the effect of PEG architecture on nanoparticle ligand availability using DNA-PAINT†
Teodora
Andrian
 a,
Silvia
Pujals
*a and
Lorenzo
Albertazzi
*ab
a,
Silvia
Pujals
*a and
Lorenzo
Albertazzi
*ab
aInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac 15-21, 08028 Barcelona, Spain. E-mail: l.albertazzi@tue.nl; lalbertazzi@ibecbarcelona.eu; spujals@ibecbarcelona.eu
bDepartment of Biomedical Engineering, Institute for Complex Molecular Systems (ICMS), Eindhoven University of Technology, 5612AZ Eindhoven, The Netherlands
First published on 1st November 2021
Abstract
The importance of PEG architecture on nanoparticle (NP) functionality is known but still difficult to investigate, especially at a single particle level. Here, we apply DNA Point Accumulation for Imaging in Nanoscale Topography (DNA-PAINT), a super-resolution microscopy (SRM) technique, to study the surface functionality in poly(lactide-co-glycolide)–poly(ethylene glycol) (PLGA–PEG) NPs with different PEG structures. We demonstrated how the length of the PEG spacer can influence the accessibility of surface chemical functionality, highlighting the importance of SRM techniques to support the rational design of functionalized NPs.
Introduction
Nanomedicine aims to improve clinical outcomes and reduce the adverse side effects caused by the lack of selectivity for target tissues of small molecular drugs.1,2 A popular strategy to achieve this aim is the tethering of functional moieties on the surface of nanoparticles (NPs), which improves the interaction between the drug delivery system and the target cells.3,4 The grafting of targeting ligands to the surface of polymeric NPs is commonly implemented by using PEG as a spacer.5 The hydrophilic nature of PEG also endows the formulation with “stealth” properties, reducing fouling by plasma proteins6 and improving circulation times.7 However, the architecture of PEG chains has been shown to influence the association of nanoparticles to their target receptors8 and in turn their cellular uptake.9,10 Notably, covering the surface of NPs with identical length PEG chains can reduce the ligand free motion and hinder the accessibility towards target receptors, whilst ‘cocktail’ PEGylation – where ligand-free shorter PEG chains and ligand-tethered longer PEG chains are covering the NP surface – can increase ligand mobility and improve its accessibility to target receptors, as well as maintaining stealth properties.8–11Despite the acknowledgement of this phenomenon, the effect of PEG architecture on ligand accessibility has mainly been studied with ensemble assays based on average population results. In a pioneering study using transmission electron microscopy (TEM) and antibody fragments conjugated to gold nanoparticles, the authors were able to map the accessible epitopes on the surface of NPs at a single-particle level.12 Although TEM offers excellent resolution, quantification is limited by the density of ligands that can be quantified, and low throughput information that makes data analysis cumbersome.
Super-resolution microscopy (SRM) based on single-molecule localization (SMLM) techniques have demonstrated their superiority in the quantification of functional ligands,13–15 due to their nanoscale resolution (10–20 nm), molecular specificity and single-molecule sensitivity. DNA Point Accumulation for Imaging in Nanoscale Topography (DNA-PAINT) is a type of SMLM technique which offers various advantages over other microscopy techniques, such as greater multiplexing possibility,16,17 lower photobleaching rates and accurate molecule counting for a variety of functionalization densities.15,18 Consequently, DNA-PAINT has been used to map functional ligands on the surface of polystyrene,13,19 gold15 and PLGA–PEG NPs,20 exposing ligand distribution and heterogeneity on the NP surface. The exact number of functional ligands can be quantified using quantitative PAINT (qPAINT),13,19 a technique initially developed for the quantification of docking strands in DNA origami.21
In this work we use qPAINT to quantify the number of functional ligands on the surface of PLGA–PEG NPs and to study the role of spacer PEG length and target group content on their targeting ability. Finally, we study the ligand availability (%) by comparing the number of functional ligands quantified using qPAINT with the conjugation efficiency (CE%) calculated using ensemble measurements. First, we formulated the different NP formulations via the nanoprecipitation method, by mixing PLGA, PLGA–PEG5k–maleimide and PLGA–PEGx polymers, varying the amount of target group (maleimide) content and spacer PEG length in the PLGA–PEGx polymer (where x denotes molecular weight of 1k or 5k). Then, we conjugated the NPs to our model ligands, thiol-oligonucleotides (docking strands), and quantitatively studied the number of available ligands and distribution using DNA-PAINT and qPAINT for the different formulations, highlighting marked heterogeneity within the formulations. Finally, we demonstrated at a single particle and molecular level how PEG architecture can influence ligand number and availability.
Results and discussion
A brief outline of the DNA-PAINT and qPAINT quantification protocol is given in Fig. 1. NPs were first prepared manually via the nanoprecipitation method22 by mixing of PLGA–PEG5k, PLGA and PLGA–PEG5k–maleimide polymers with increasing maleimide content (10–100%) (1). Then they were conjugated with an excess of thiolated oligonucleotides (docking strands) used as a DNA-PAINT probe and as a model for a biological ligand via a thiol–maleimide reaction23 (2). The hydrodynamic diameter, TEM diameter and polydispersity index were generally consistent between formulations, except for 100% maleimide content for which we noted an increase in these parameters, probably due to reduced stability. For full protocol see ESI Experimental section† and for NPs characterization using DLS and ZP Table S1 and TEM Fig. S1.† We studied the availability of the conjugated functional ligands to the complementary imager strands using DNA-PAINT imaging3 (3). During imaging, DNA hybridization mediates the transient binding and unbinding of the complementary imager strands to the docking strands. Notably, if the functional ligand is not properly exposed in the imager strands, it will not be available for detection and thus the localizations identified reveal the number of available functional ligands. Finally, whilst the number of localizations gives an estimation of the total number of ligands, the exact number can be quantified using qPAINT (4), which uses kinetic information based on the dark time between binding events to quantify available ligands. Specific parameters used are the mean dark time between the binding events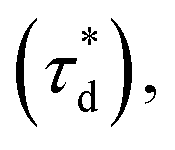 the second-order association rate constant (kON) between the complementary strands, and the known concentration of imager strands (Ci) through the equation
the second-order association rate constant (kON) between the complementary strands, and the known concentration of imager strands (Ci) through the equation 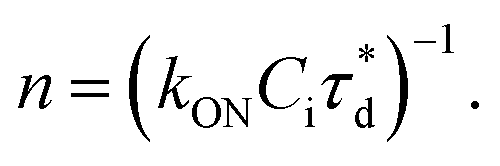 21 For details on DNA-PAINT imaging and qPAINT analysis see ESI Experimental section.†
21 For details on DNA-PAINT imaging and qPAINT analysis see ESI Experimental section.†
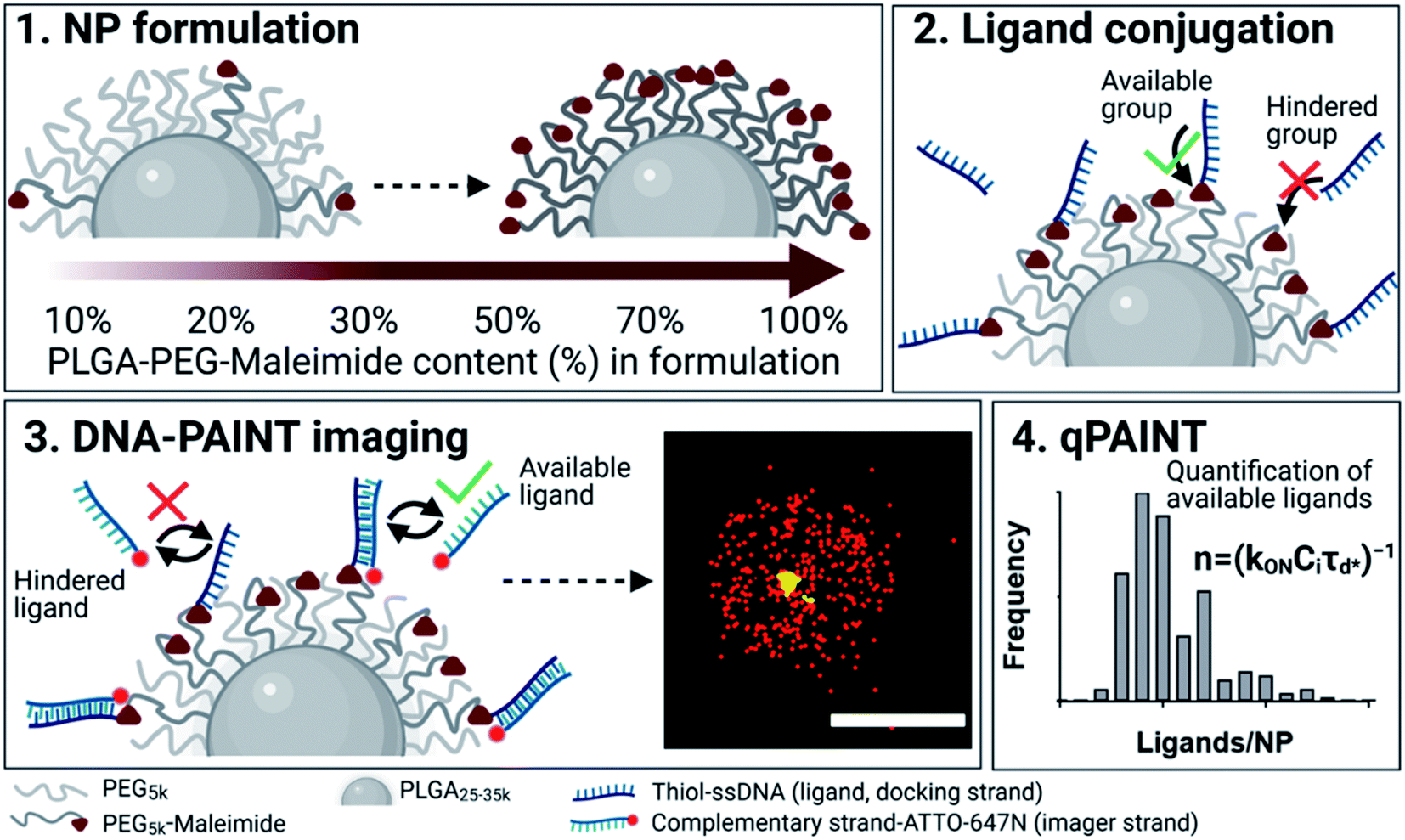 | ||
Fig. 1 Outline of DNA-PAINT and qPAINT protocol. (1) PLGA–PEG nanoparticles (NPs) are first formulated using varying maleimide contents (10–100%). (2) NPs are functionalized by conjugation with a thiol-single stranded DNA functional ligand (docking strand) via maleimide–thiol conjugation. Only some of the maleimide groups are available for conjugation with the functional ligand. (3) During DNA-PAINT imaging, complementary imager strands attached to ATTO-647N dye transiently bind and unbind to the available ligands on the NP surface, leading to the acquisition of a super-resolved image, whilst the hindered ligands are undetected. In red are the localizations representative of available ligands, and in yellow an encapsulated dye (DiI) used as a reference marker and drift corrector. Scale bar 100 nm. (4) qPAINT achieves quantification of the exact number of available ligands (n) by using the mean dark time between the binding events 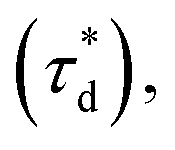 the second-order association rate constant (kON) between the complementary strands, and the known concentration of imager strands (Ci) through the equation the second-order association rate constant (kON) between the complementary strands, and the known concentration of imager strands (Ci) through the equation 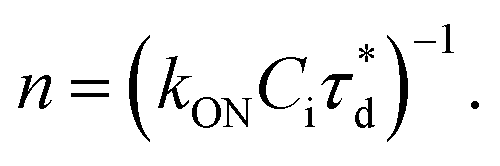 | ||
First, we show the DNA-PAINT and qPAINT results on the quantification of the number of available ligands per NP on 6 different PLGA–PEG NP formulations with increasing maleimide content (10–100%) conjugated to an excess of functional ligand. Please note a constant time was kept between NP formulation and imaging for all formulations. Fig. 2a shows a DNA-PAINT image of localizations (red) clusters representative of surface ligand number. In Fig. 2b we observe images of representative NPs from each formulation, illustrating a visual increase in number of localizations with maleimide content. This demonstrates that DNA-PAINT can discriminate formulations with different contents of surface functionalization. We then analyzed the x, y, t coordinates of the localizations using a previously described mean-shift clustering algorithm in Matlab13,19 and plotted the data of localizations/NP in frequency distribution graphs. The distributions show a clear increase in localizations/NP with maleimide content but also an increase in the distribution width (i.e. heterogeneity), which is expected with greater target group contents. This emphasizes that the average number of ligands is not and should not be used as a representation of the whole NP population. Despite the ligand conjugation and imager hybridization processes both being stochastic in nature, we observe non-symmetrical (i.e. non-Poissonian) localization distributions. We have recently attributed these results to an entanglement between ligand and size distribution using correlative microscopy.20 In addition, we observed that for NPs with greater maleimide content the distributions tail towards higher localization numbers. As seen in our previously published correlative results, the sub-populations of NPs at the extreme right of the localization distributions generally represent NPs with larger than expected diameters.
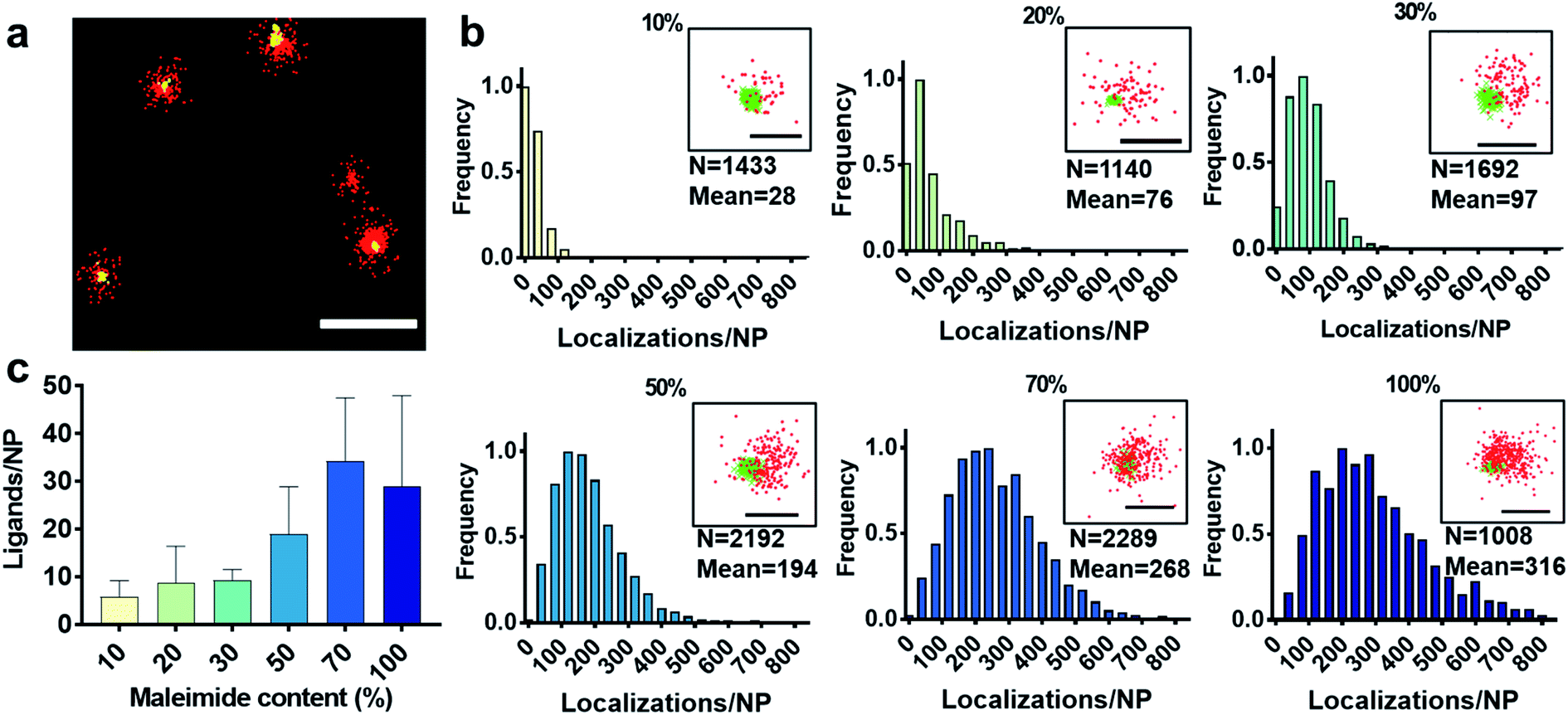 | ||
| Fig. 2 (a) Representative DNA-PAINT image of ligand-conjugated PLGA–PEG (PEG5k) 50% maleimide NPs displaying the number of localizations (red) per NP (scale bar = 200 nm). The yellow signal is DiI dye encapsulated in NP, used for mechanical drift correction and as a reference in NP identification. (b) Normalized frequency histograms of DNA-PAINT localizations for PLGA–PEG–maleimide (long PEG5k) NP at various maleimide contents (10–100%), the number of NP analysed (N) and the mean number of localizations. Close-up images of representative NP for each formulation (scale bar = 100 nm), whereby DNA-PAINT localizations are seen in red and DiI in green. (c) The number of available ligands/NP per formulation as retrieved by qPAINT. The data were analysed using Matlab software. | ||
We carried out two control experiments for our data, the first by using a non-complementary imager aimed to demonstrate that the DNA-hybridization between the docking and imager strand is specific, and the second control by imaging non-functional NPs using the correct imager aimed to demonstrate low non-specific attachment of imager strands to NPs. For both controls, the results show a much lower number of localizations, confirming the specificity of the method (Fig. S2†). Notably, DNA-PAINT offers a relatively high-throughput analysis, with 200–500 NPs analyzed per field of view, a clear advantage when compared to other single-particle microscopy techniques such as TEM. We then counted the exact number of available ligands on the NP surface using the previously established method qPAINT.13,19,21Fig. 2c illustrates a general increase in the quantified number of ligands per NP with maleimide content up to 70%, followed by a saturation in the number of ligands at 100% maleimide (for ligand distributions see Fig. S3†).
Next, we selected 3 formulations with a maleimide content typically used in literature (10%, 20% and 30%)24 and analyzed the ligand conjugation efficiency (CE%) using spectrophotometry to be between 20 and 30% (Table S2†) and confirmed the results by studying the CE% of a smaller molecule L-cysteine, with results between 40 and 50% (Table S3†). For information on how CE% was calculated please see ESI Experimental section.† Although the hydrophobic PLGA chains are assumed to form the core, and the hydrophilic PEG chains the outer layer, we expect that not all maleimide groups will be available for conjugation on the NP surface, since due to the miscibility of PEG and PLGA, some of the PEG–maleimide chains will be embedded in the NP core.24,25 It has been shown that on average, only 50% of the maleimide groups are available for ligand conjugation,24 values which closely agree with our CE% and L-cysteine assay results.
Then, based on the estimated number of theoretical maleimide groups per NP and the CE%, we calculated the average number of estimated ligands per NP for each formulation and compared these values with our qPAINT results. The experimental numbers were significantly lower than the estimated numbers e.g. 6 vs. 77 and 9 vs. 293 for 10% and 30% maleimide formulations, respectively. For results and calculations see Table S2 and ESI Experimental section.† In addition to the issue of maleimide availability, it has also been reported that with increasing molecular weight (e.g. at 5 kDa), the PEG chain becomes more flexible,26 leading to conjugated ligands to be entangled in the PEG chains,27 a phenomenon which could explain the low numbers of quantified ligands. This prompted us to analyze further the role of PEG length on the targeting ability of NPs.
PEG (typically 5 kDa)6 is a common linker used for the tethering of target groups (e.g. maleimide) on the NP surface, and used as a spacer to improve formulation stability and to decrease the adsorption of serum proteins.6,28 Despite this, it has been argued that the effect of PEG length on NP targeting ability must also be seriously considered.8,10–12,27,29
Particularly, it has been shown that PEGylation of NPs with identical spacer and ligand-tethered chain lengths at higher molecular weights can lead to a reduced number of accessible tethered ligands on the NP surface,12,29 as well as reduced target receptor accessibility.8,10,11 This phenomenon is thought to be due to hampered ligand free motion,8,10 as well as an increase in the flexibility of the PEG chain with increasing molecular weight26 which can lead to entangling of surface ligands into the PEG chains.27 We used DNA-PAINT and qPAINT to quantify at a single particle and molecular level the effect of spacer PEG chain length and on ligand availability. To this end, we tested the effect of substituting the 5 kDa PEG (PEG5k) spacer chain in the PLGA–PEG5k polymer with a shorter 1 kDa PEG (PEG1k) chain whilst maintaining the length of the PLGA and PLGA–PEG5k–maleimide strands unchanged, at 3 different maleimide concentrations (10–30%). Whilst the polydispersity index and TEM diameter were similar to the PEG5k formulations, we noted an increase in the hydrodynamic radius. It has been suggested that longer PEG chains such as 5 kDa can entangle among each other via van der Waals forces and hydrogen bonding, leading to a reduction in the size of the outer layer and thus hydrodynamic radius, as compared to shorter PEG chains.10,30 For formulation see ESI Experimental section† and for characterization using DLS and ZP Table S4 and TEM Fig. S4.†
With this approach we wanted to promote the sticking out of the PEG brushes bearing the maleimide groups for an improved conjugation efficiency (CE%) and ligand availability to imager strands (graphically shown in Fig. 3a). Using spectrophotometry, we found on average a 2-fold increase in CE% at all maleimide contents using the shorter PEG1k chains (Table 1). Using qPAINT, we observed on average a 7-fold increase at both 10% and 20% maleimide and a 3-fold increase at 30% maleimide contents in quantified ligands using the shorter PEG1k chains (Fig. 3b and Table 1). For localization distributions and negative controls see Fig. S5 and for ligand distributions see Fig. S6.† These results indicate that by varying surface PEG chain lengths we can improve the exposure of target groups (e.g. maleimide) which in turn improves conjugation efficiency, and we can enhance the exposure of conjugated functional ligands on the surface of NPs, which is expected to improve target cellular receptor recognition.
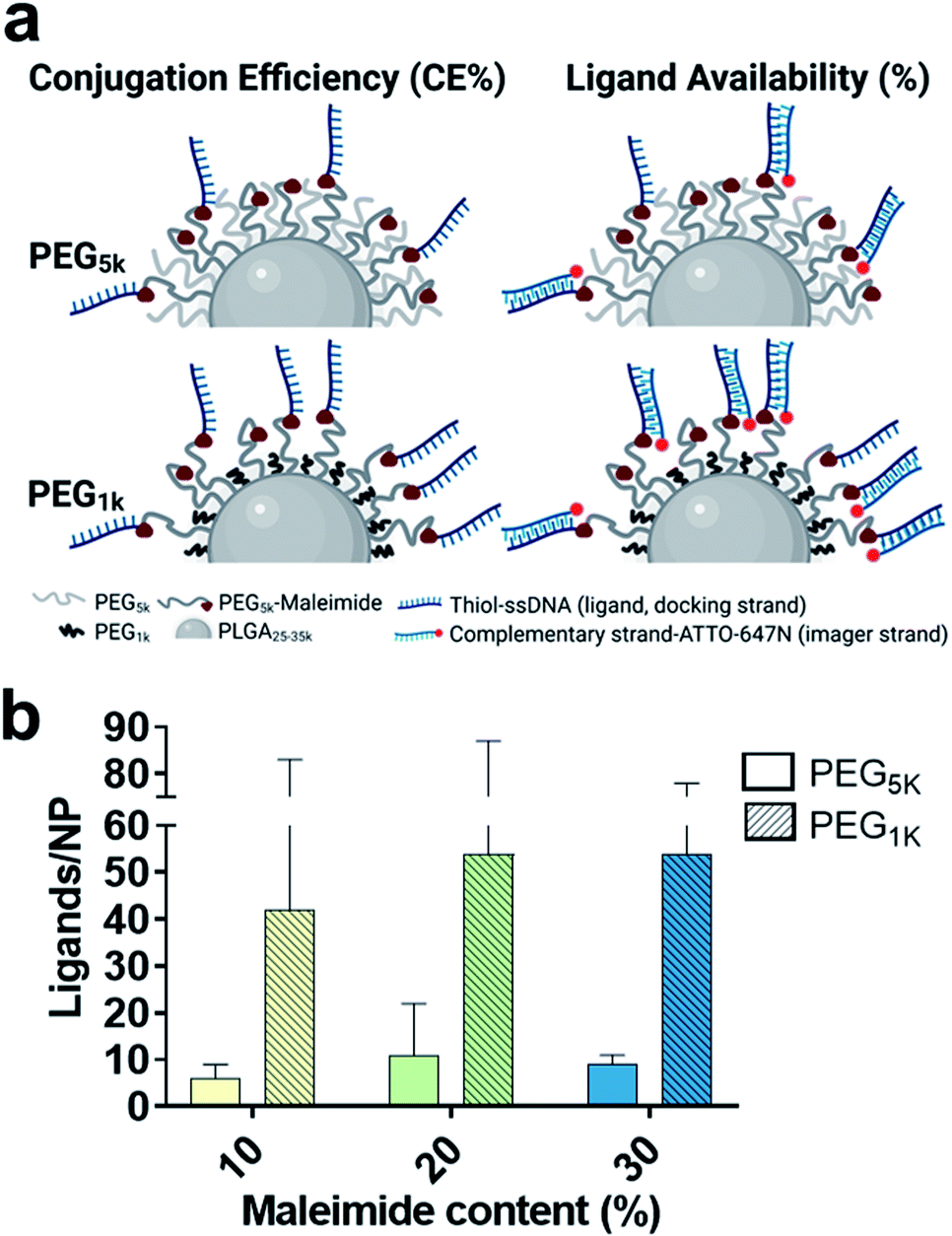 | ||
| Fig. 3 (a) Schematic representation of the influence of spacer PEG chain length on the conjugation efficiency (CE%) and ligand availability (%) of PLGA–PEG NPs. Shorter PEG chains (PEG1k) are expected to achieve better CE% and imager availability due to improved maleimide and ligand free motion, and reduced flexibility and entanglement of PEG chains. (b) Quantification of ligand number/NP by qPAINT with increasing maleimide content (10–30%) and varying spacer PEG chain length (PEG5k – blank, PEG1k – lined). Standard deviation bars are given for each formulation. | ||
| Maleimide content (%) | Spacer PEG chain length (kDa) | |||||
|---|---|---|---|---|---|---|
| Conjugation efficiency (%) | Quantified ligands (qPAINT) | Ligand availability (%) | ||||
| 5 | 1 | 5 | 1 | 5 | 1 | |
| a PLGA–PEG NPs with increasing maleimide content (10–30%) and varying spacer PEG chain length (PEG5k and PEG1k). The conjugation efficiency (%) was calculated using spectrophotometry, the number of accessible ligands by qPAINT and the ligand availability (%) by considering the number of available ligands out of the theoretical expected number of ligands. For calculations of CE% and ligand availability see ESI Experimental section and Table S2 in ESI. | ||||||
| 10 | 25 | 38 | 6 | 42 | 8 | 21 |
| 20 | 21 | 35 | 8 | 54 | 6 | 15 |
| 30 | 31 | 70 | 31 | 79 | 2 | 5 |
Lastly, to account for any changes between the formulations in diameter, theoretical maleimide groups/NP and CE%, we also compared the results in ligand availability (%) (Table 1). We calculated a 3-fold average increase in ligand availability using the lower PEG1k chain. Notably, for both PEG5k and PEG1k we found that ligand availability (%) is greater at lower maleimide contents. We expect this to be because at lower target group contents there are generally less conjugated ligands on the surface of the NPs, and therefore a reduced hindering effect, leading to an increase in ligand availability.
Conclusions
In summary, we demonstrated at a single-particle and single-molecule-level that ligand number and availability in polymeric NP could be greatly impacted by PEG architecture, with regular long PEG chain architecture causing entanglement of tethered groups and ligands within the polymeric chains. Still, although modification of PEG chain length generally improved ligand availability (maximum of 21% at 10% maleimide content), the overall picture is that the standard formulation method to conjugate functional ligands to polymeric NPs leads to NP with a low number of available ligands, and in hindsight a poor biological performance. Due to the unique single-particle and single-molecule properties and high-throughput capabilities of SMLM techniques, we strongly believe these results highlight their potential to be used in the routine design, quality control and optimization of nanomaterials with improved biological efficacy.Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by a Marie Skłodowska-Curie Horizon 2020 (713673), “la Caixa” Foundation (ID 100010434, code LCF/BQ/DI18/11660039) fellowship. S. P. and L. A. acknowledge the financial support by the Spanish Ministry of Science and Innovation (PID2019-109450RB-I00/AEI/10.13039/501100011033), European Research Council/Horizon 2020 (ERC-StG-757397), “la Caixa” Foundation (ID 100010434), and by the Generalitat de Catalunya (through the CERCA program and 2017 SGR 01536). L. A. thanks the support of NWO through the VIDI Grant 192.028. The authors thank Lidia Delgado Valderrama and Maria Yolanda Muela Castro from the Electron Cryomicroscopy Unit from CCiTUB. Figure schematics were created using BioRender.com.References
- T. M. Allen, Drug Delivery Systems: Entering the Mainstream, Science, 2004, 303(5665), 1818–1822, DOI:10.1126/science.1095833.
- C. L. Ventola, Progress in Nanomedicine: Approved and Investigational Nanodrugs, P T, 2017, 42(12), 742–755 Search PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Active Targeting Schemes for Nanoparticle Systems in Cancer Therapeutics, Adv. Drug Delivery Rev., 2008, 60(15), 1615–1626, DOI:10.1016/j.addr.2008.08.005..
- A. Friedman, S. Claypool and R. Liu, The Smart Targeting of Nanoparticles, Curr. Pharm. Des., 2013, 19(35), 6315–6329, DOI:10.2174/13816128113199990375.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery, Adv. Drug Delivery Rev., 2016, 99, 28–51, DOI:10.1016/j.addr.2015.09.012.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Müller, ‘Stealth’ Corona-Core Nanoparticles Surface Modified by Polyethylene Glycol (PEG): Influences of the Corona (PEG Chain Length and Surface Density) and of the Core Composition on Phagocytic Uptake and Plasma Protein Adsorption, Colloids Surf., B, 2000, 18(3–4), 301–313, DOI:10.1016/S0927-7765(99)00156-3.
- A. L. Klibanov, K. Maruyama, V. P. Torchilin and L. Huang, Amphipathic Polyethylene glycols Effectively Prolong the Circulation Time of Liposomes, FEBS Lett., 1990, 268(1), 235–237, DOI:10.1016/0014-5793(90)81016-H.
- T. Ishii, K. Miyata, Y. Anraku, M. Naito, Y. Yi, T. Jinbo, S. Takae, Y. Fukusato, M. Hori, K. Osada and K. Kataoka, Enhanced Target Recognition of Nanoparticles by Cocktail PEGylation with Chains of Varying Lengths, Chem. Commun., 2016, 52(7), 1517–1519, 10.1039/C5CC06661A.
- K. Miyata, Smart Polymeric Nanocarriers for Small Nucleic Acid Delivery, Drug Discov Ther., 2016, 10(5), 236–247, DOI:10.5582/ddt.2016.01061.
- C. P. Gomes, V. Leiro, C. D. F. Lopes, A. P. Spencer and A. P. Pêgo, Fine Tuning Neuronal Targeting of Nanoparticles by Adjusting the Ligand Grafting Density and Combining PEG Spacers of Different Length, Acta Biomater., 2018, 78, 247–259, DOI:10.1016/j.actbio.2018.08.005.
- S. M. Figueroa, D. Fleischmann, S. Beck and A. Goepferich, The Effect of Ligand Mobility on the Cellular Interaction of Multivalent Nanoparticles, Macromol. Biosci., 2020, 20(4), 1900427, DOI:10.1002/mabi.201900427.
- L. M. Herda, D. R. Hristov, M. C. Lo Giudice, E. Polo and K. A. Dawson, Mapping of Molecular Structure of the Nanoscale Surface in Bionanoparticles, J. Am. Chem. Soc., 2017, 139(1), 111–114, DOI:10.1021/jacs.6b12297.
- P. Delcanale, B. Miret-Ontiveros, M. Arista-Romero, S. Pujals and L. Albertazzi, Nanoscale Mapping Functional Sites on Nanoparticles by Points Accumulation for Imaging in Nanoscale Topography (PAINT), ACS Nano, 2018, 12(8), 7629–7637, DOI:10.1021/acsnano.7b09063.
- R. A. J. Post, D. van der Zwaag, G. Bet, S. P. W. Wijnands, L. Albertazzi, E. W. Meijer and R. W. van der Hofstad, A Stochastic View on Surface Inhomogeneity of Nanoparticles, Nat. Commun., 2019, 10(1), 1663, DOI:10.1038/s41467-019-09595-y.
- M. Horáček, D. J. Engels and P. Zijlstra, Dynamic Single-Molecule Counting for the Quantification and Optimization of Nanoparticle Functionalization Protocols, Nanoscale, 2020, 12(6), 4128–4136, 10.1039/C9NR10218C.
- S. S. Agasti, Y. Wang, F. Schueder, A. Sukumar, R. Jungmann and P. Yin, DNA-Barcoded Labeling Probes for Highly Multiplexed Exchange-PAINT Imaging, Chem. Sci., 2017, 8(4), 3080–3091, 10.1039/C6SC05420J.
- O. K. Wade, J. B. Woehrstein, P. C. Nickels, S. Strauss, F. Stehr, J. Stein, F. Schueder, M. T. Strauss, M. Ganji, J. Schnitzbauer, H. Grabmayr, P. Yin, P. Schwille and R. Jungmann, 124-Color Super-Resolution Imaging by Engineering DNA-PAINT Blinking Kinetics, Nano Lett., 2019, 19(4), 2641–2646, DOI:10.1021/acs.nanolett.9b00508.
- R. Jungmann, C. Steinhauer, M. Scheible, A. Kuzyk, P. Tinnefeld and F. C. Simmel, Single-Molecule Kinetics and Super-Resolution Microscopy by Fluorescence Imaging of Transient Binding on DNA Origami, Nano Lett., 2010, 10(11), 4756–4761, DOI:10.1021/nl103427w.
- P. Delcanale and L. Albertazzi, DNA-PAINT Super-Resolution Imaging Data of Surface Exposed Active Sites on Particles, Data Brief, 2020, 30, 105468, DOI:10.1016/j.dib.2020.105468.
- T. Andrian, P. Delcanale, S. Pujals and L. Albertazzi, Correlating Super-Resolution Microscopy and Transmission Electron Microscopy Reveals Multiparametric Heterogeneity in Nanoparticles, Nano Lett., 2021, 21(12), 5360–5368, DOI:10.1021/acs.nanolett.1c01666.
- R. Jungmann, M. S. Avendaño, M. Dai, J. B. Woehrstein, S. S. Agasti, Z. Feiger, A. Rodal and P. Yin, Quantitative Super-Resolution Imaging with QPAINT, Nat. Methods, 2016, 13(5), 439–442, DOI:10.1038/nmeth.3804.
- J. M. Barichello, M. Morishita, K. Takayama and T. Nagai, Encapsulation of Hydrophilic and Lipophilic Drugs in PLGA Nanoparticles by the Nanoprecipitation Method, Drug Dev. Ind. Pharm., 1999, 25(4), 471–476, DOI:10.1081/DDC-100102197.
- D. Papahadjopoulos and F. J. Martin, Irreversible Coupling of Immunoglobulin Fragments to Preformed Vesicles, J. Biol. Chem., 1982, 257(1), 286–288 CrossRef.
- L. Martínez-Jothar, S. Doulkeridou, R. M. Schiffelers, J. Sastre Torano, S. Oliveira, C. F. van Nostrum and W. E. Hennink, Insights into Maleimide-Thiol Conjugation Chemistry: Conditions for Efficient Surface Functionalization of Nanoparticles for Receptor Targeting, J. Controlled Release, 2018, 282, 101–109, DOI:10.1016/j.jconrel.2018.03.002.
- E. Vega, M. A. Egea, A. C. Calpena, M Espina and M. L. García, Role of Hydroxypropyl-β-Cyclodextrin on Freeze-Dried and Gamma-Irradiated PLGA and PLGA–PEG Diblock Copolymer Nanospheres for Ophthalmic Flurbiprofen Delivery, Int. J. Nanomedicine, 2012, 7, 1357–1371, DOI:10.2147/IJN.S28481.
- K. Abe, K. Higashi, K. Watabe, A. Kobayashi, W. Limwikrant, K. Yamamoto and K. Moribe, Effects of the PEG Molecular Weight of a PEG-Lipid and Cholesterol on PEG Chain Flexibility on Liposome Surfaces, Colloids Surf., A, 2015, 474, 63–70, DOI:10.1016/j.colsurfa.2015.03.006.
- R. Hennig, K. Pollinger, A. Veser, M. Breunig and A. Goepferich, Nanoparticle Multivalency Counterbalances the Ligand Affinity Loss upon PEGylation, J. Controlled Release, 2014, 194, 20–27, DOI:10.1016/j.jconrel.2014.07.062.
- Z. Amoozgar and Y. Yeo, Recent Advances in Stealth Coating of Nanoparticle Drug Delivery Systems: Stealth Coating of Nanoparticle Drug Delivery Systems, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2012, 4(2), 219–233, DOI:10.1002/wnan.1157.
- K. Abstiens, M. Gregoritza and A. M. Goepferich, Ligand Density and Linker Length are Critical Factors for Multivalent Nanoparticle–Receptor Interactions, ACS Appl. Mater. Interfaces, 2019, 11(1), 1311–1320, DOI:10.1021/acsami.8b18843.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Laganà, Effect of Polyethyleneglycol (PEG) Chain Length on the Bio–Nano-Interactions between PEGylated Lipid Nanoparticles and Biological Fluids: from Nanostructure to Uptake in Cancer Cells, Nanoscale, 2014, 6(5), 2782, 10.1039/c3nr05559k.
Footnote |
| † Electronic supplementary information (ESI) available: With detailed experimental procedures (NP formulation, conjugation, DNA-PAINT imaging, qPAINT analysis), and other supplementary information such as nanoparticle characterization, DNA-PAINT localization and qPAINT ligand histograms, DNA-PAINT negative controls, calculations (CE%, theoretical maleimide groups/NP, theoretical ligands/NP, ligand availability), cysteine assay and cysteine CE% results. See DOI: 10.1039/d1na00696g |
| This journal is © The Royal Society of Chemistry 2021 |