
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The size-dependent influence of palladium doping on the structures of cationic gold clusters†
Piero
Ferrari
 *a,
Laia
Delgado-Callico
b,
Olga V.
Lushchikova
c,
Gao-Lei
Hou‡
a,
Francesca
Baletto
b,
Joost M.
Bakker
c and
Ewald
Janssens
a
*a,
Laia
Delgado-Callico
b,
Olga V.
Lushchikova
c,
Gao-Lei
Hou‡
a,
Francesca
Baletto
b,
Joost M.
Bakker
c and
Ewald
Janssens
a
aQuantum Solid-State Physics, Department of Physics and Astronomy, KU Leuven, Leuven, Belgium. E-mail: piero.ferrari@kuleuven.be
bDepartment of Physics, King's College London, London, UK
cRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Nijmegen, The Netherlands
First published on 21st September 2021
Abstract
The physicochemical properties of small metal clusters strongly depend on their precise geometry. Determining such geometries, however, is challenging, particularly for clusters formed by multiple elements. In this work, we combine infrared multiple photon dissociation spectroscopy and density functional theory calculations to investigate the lowest-energy structures of Pd doped gold clusters, PdAun−1+ (n ≤ 10). The high-quality experimental spectra allow for an unambiguous determination of the structures adopted by the clusters. Our results show that the Pd–Au interaction is so large that the structures of PdAun−1+ and Aun+ are very different. Pd doping induces a 2D to 3D transition at much smaller cluster sizes than for pure Aun+ clusters. PdAun−1+ clusters are three-dimensional from n = 4, whereas for Aun+ this transition only takes place at n = 7. Despite the strong Au–Pd interaction, the Aun−1+ cluster geometries remain recognizable in PdAun−1+ up to n = 7. This is particularly clear for PdAu6+. In PdAu8+ and PdAu9+, Pd triggers major rearrangements of the Au clusters, which adopt pyramidal shapes. For PdAu4+ we find a geometry that was not considered in previous studies, and the geometry found for PdAu8+ does not correspond to the lowest-energy structure predicted by DFT, suggesting kinetic trapping during formation. This work demonstrates that even with the continuous improvement of computational methods, unambiguous assignment of cluster geometries still requires a synergistic approach, combining experiment and computational modelling.
Introduction
The physical and chemical properties of small metal clusters depend largely on the geometries adopted by these particles. For example, it has been predicted that different isomers of Aunq (n ≤ 11, q = 0, ±1) clusters adsorb and dissociate H2 differently, with the lowest-energy structures being not necessarily more reactive towards hydrogen.1 Moreover, the shape of the cluster and its electronic level sequence are strongly entangled, as it is particularly clear for the known planar magic clusters2 and oblate species like Ag15+.3 Furthermore, the reactivity of Aln− clusters has been shown to be size-dependent; Al12− is very reactive towards methanol and water, while Al13− is inert. Al13− adopts an icosahedral geometry, with an isotropic distribution of the total −1e charge. Instead, Al12− can either donate charge to or accept it from the molecules, increasing binding energies and reducing reaction barriers.4 Therefore, understanding the physical and chemical properties of a cluster requires precise knowledge about the adopted geometry.Determining the geometry of a cluster, however, is challenging even if the system is composed of only a few atoms. For instance, there has been an intensive debate about the geometries adopted by small Aun+ clusters, with contradictory claims about the planar or three-dimensional structure of Au8+, that was only recently solved.5 Even for a cluster as small as Au4+, evidence of a higher energy isomer present in molecular beams was shown not long ago.6 These challenges are further exacerbated in bimetallic species, with an increasing number of possible configurations, making it difficult to locate with high certainty the global minimum on the potential energy surface, based purely on theory.7 Moreover, even if the global minimum is identified (for the specific theoretical method used), there is no guarantee that this is the isomeric structure in the experiment, as formation conditions may favour other isomers.8 Hence, assigning a cluster geometry requires the combination of dedicated experiments with precise quantum chemical calculations.6,9–11
The geometries of gold clusters have fascinated the scientific community for decades. Remarkable structures have been identified, like the tetrahedral (pyramidal) shape of neutral Au20,12 and the hollow cages of anionic gold clusters.13 Moreover, Au clusters are known for remaining planar up to relatively large sizes; the largest planar neutral gold cluster, for example, is Au11.5,14,15 For cationic Aun+, this transition size is n = 8, with three-dimensional and planar Au8+ isomers coexisting in molecular beams.5 These unique structures play a decisive role in defining properties such as relative stabilities,16,17 reactivities,18,19 de-excitation mechanisms,20,21 and optical responses.22,23
To the best of our knowledge, experimental determination of the geometries adopted by doped cationic gold clusters has so far only been achieved for YAun−1+ (n ≤ 7)24 and the small AgAu2,3+ clusters.25 In the first case, Y doping induces a significant rearrangement of the gold cluster geometries, while in the second case, Ag acts as a substitutional dopant, where the symmetry of the cluster remains unchanged. Other doped Aun+ clusters have been studied by theoretical means, predicting a dopant-dependent structural influence. Density functional theory (DFT) calculations on MAun−1+ (M = Ti, Fe) clusters indicate substitutional doping in the n ≤ 8 size range.26 Similarly, calculations of MAu5+ (M = Sc, Ti, Cr, and Fe)27 and AgAun−1+ (n ≤ 18)28 predict substitutional doping. Instead, for YAun−1+ (n ≤ 15) clusters there are significant rearrangements of Aun+ upon doping,29 just as for BeAun−1+ (n ≤ 9).30
Pd doping significantly alters the properties of Au clusters. For example, it changes the size-to-size stability pattern of Aun+, where clusters with an odd number of atoms (even number of itinerant electrons) are experimentally found to be more stable.17 In PdAun−1+, instead, n ≤ 8 clusters are experimentally found to be more stable for odd n values (even total number of atoms), while even n Pd doped cationic gold clusters are more stable for n > 8.31 This change in the stability pattern with size is attributed to Au always delocalizing its 6s electron in PdAun−1+, whereas Pd can delocalize a 4d-electron depending on cluster size and cluster structure. Pd doping can also affect the cluster reactivity, illustrated by the increased CO and O2 binding energies on the doped clusters, compared to bare Aun+.32,33 For CO adsorption, the increase in binding energy becomes more pronounced from n = 7, as determined by a combination of mass spectrometry and statistical modelling.32 Furthermore, Pd doping was shown to quench optical absorption cross sections in the visible range.34,35 Finally, Pd is nowadays considered as a suitable dopant element in monolayer protected Au clusters, with diverse size-selected species synthesized in the laboratory.36,37
In this work, we unambiguously determine the geometry that PdAun−1+ (n ≤ 10) clusters adopt in a molecular beam, by combining infrared multiple photon dissociation (IRMPD) spectroscopy experiments with extensive density functional theory calculations. Our results show that Pd induces a transition from planar to three-dimensional structures on the Aun+ clusters, with PdAun−1+ adopting three-dimensional configurations from PdAu3+. The exact influence of Pd doping, however, is size-specific. Moreover, our work illustrates that dedicated experiments must be combined with accurate theoretical modelling for unambiguous assignment of cluster geometries.
Methods
Experiment
A molecular beam of Pd doped Au clusters is produced by laser ablation of bulk pure metal targets in a dual-target dual-laser source, using He gas for condensation.38 Complexes of cationic clusters with Ar are formed by adding 2% of Ar to the He carrier gas while the source is kept at 200 K. A 2 mm diameter skimmer initially shapes the cluster beam, followed by a 0.45 mm slit aperture. The cluster size distribution is irradiated by the laser light of the free-electron laser FELICE, adapted for intracavity experiments,39 and subsequently probed by a perpendicularly extracted reflectron time-of-flight mass spectrometer. The high pulse energy and large interaction volume reachable at FELICE allows for recording infrared spectra of high signal-to-noise ratio.IR spectra are obtained by calculating the IRMPD yield, defined as the natural logarithmic ratio ln(I0/I(ν)) of cluster–Ar complex intensities in mass spectra without (I0) and with (I(ν)) the IR irradiation at laser frequency ν.5 The PdAun−1+ clusters have high dissociation energies and are therefore difficult to fragment with the infrared light. Thus, Ar atoms are used as messenger species to record depletion spectra, since Ar has relatively low adsorption energies in PdAun−1+Arm and the Ar loss channel can be used as a probe for resonant infrared absorption. This approach has been successfully followed in the past, allowing, in combination with computed infrared spectra of different isomers, the structural identification of diverse clusters.40–42 Unavoidably, the drawback of this method is that the recorded IR spectra are those of PdAun−1+Arm, whose metal framework could have a slightly different geometry from the bare PdAun−1+ clusters. This possibility will be considered case by case. In the following, the infrared spectra of PdAun−1+Arm clusters are discussed for n = 3–10, except for n = 6. For this size, infrared spectra could only be recorded for PdAu5+Ar7 (see Fig. S12 in the ESI†); such a large number of attached Ar atoms does not allow a proper analysis of the geometry adopted by the bare cluster.
Calculations
DFT calculations are performed with the NWChem 6.8 software package,43 using the LC-ωPBEh functional in combination with the def2-TZVPP basis set. All electrons are included in the calculations for Ar and def2-ECP pseudopotentials are used for Au and Pd (19 and 18 valence electrons are included explicitly, respectively). Relativistic effects are accounted for by the pseudopotentials. Harmonic vibrational frequencies are calculated after a tight geometry optimization of each isomer, and the simulated infrared spectra are constructed by assuming Gaussian functions around each calculated frequency, with a full width at half maximum of 5 cm−1. To select the use of LC-ωPBEh, the IR spectrum of the PdAu2+Ar6 complex was used as benchmark, given the undoubtable triangular shape adopted by the metal PdAu2+ framework.The IRMPD spectrum in Fig. 1 has three intense bands, at 113, 147, and 203 cm−1, corresponding to (mostly) the Au–Au stretch, the Au2–Pd stretch and the PdAu2 breathing modes, respectively. The measurement is compared to computed spectra using three functionals: PBE (GGA), PBE0 (hybrid), and LC-ωPBEh (long-range separated hybrid). A more extensive benchmarking using additional functionals is presented in the ESI (Fig. S1†). The best agreement is found by using the LC-ωPBEh functional, which does not need a scaling factor, as commonly applied in IRMPD studies,12,24 to accurately reproduce the positions of the three bands. There is only a slight red shift for the 147 cm−1 band. A quantification of the agreement is presented later. A small frequency scaling factor (1.04) is required to account for anharmonicity for increasing cluster sizes (n ≥ 4). This scaling factor was determined for pure Aun+·Arm clusters (using the same experimental conditions).5 Given the results of Fig. 1, the LC-ωPBEh functional is chosen for further calculations. This functional has also been used to calculate properties of Pd and Ag doped Au clusters,25,31,32,44,45 and correctly predicted the vibrational modes of Au+·Arm and Pd+·Arm complexes.46 An extensive isomer global search of PdAun−1+ isomers was performed in ref. 31. Therefore, in the current work we consider the first six lowest-energy isomers identified in ref. 31 (except for PdAu4+; see the Results section for details) and we add the number of m Ar atoms as measured in the molecular beam. The Ar atoms are initially positioned on all possible atop coordination sites, followed by geometry optimization. Other coordination sites were considered but these were always found much higher in energy, as seen previously for Aun+.47
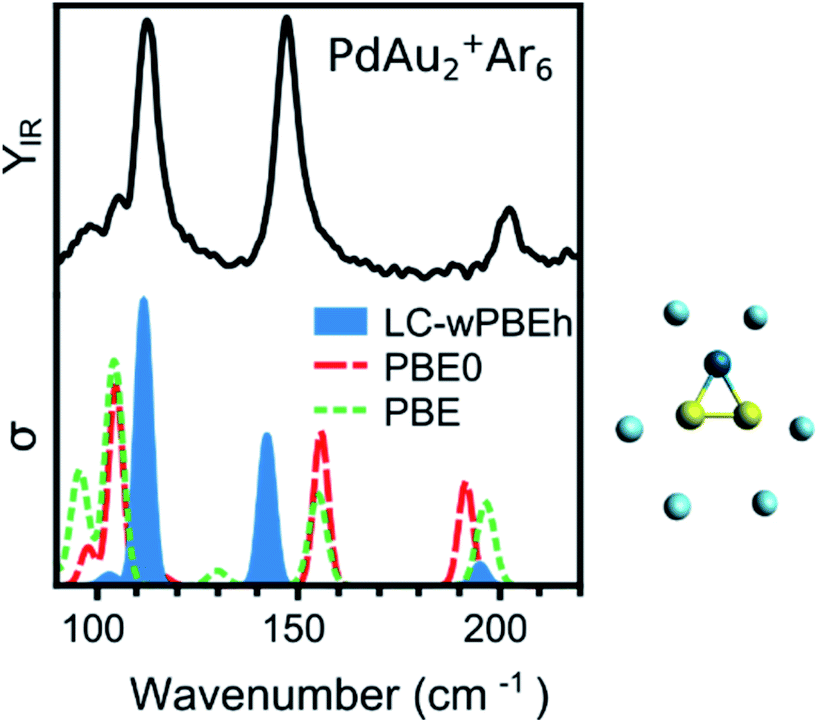 | ||
| Fig. 1 Experimental (top) and calculated (bottom) IR spectra of PdAu2+Ar6. Three functionals have been considered for the calculations (PBE, PBE0, and LC-ωPBEh), with LC-ωPBEh providing the best agreement. The cluster structure is depicted on the right of the figure. Gold, palladium, and argon atoms are represented by yellow, dark blue, and light blue spheres, respectively. | ||
Results
The IR spectra of PdAu3+Arm (m = 4–6) are shown in the left panel of Fig. 2. The spectrum of PdAu3+Ar4 has two clear bands at 102 and 147 cm−1, in addition to a weaker one at 123 cm−1, and possible low-intensity bands around 180–210 cm−1. The interaction of Ar with PdAu3+ is relatively strong (calculated adsorption energy around 0.2 eV), even when four argon atoms are attached to the cluster (see Table S1 in the ESI†). Still, this energy is much smaller than the 2.17 eV corresponding to the fragmentation energy of the bare PdAu3+ cluster.31 Nevertheless, Ar participates in the vibrational modes of the complex and thus, it cannot be considered as a spectator atom that does not disturb the IR spectrum. This raises the pertinent question whether Ar affects the structure of the metal framework. To address this point, Fig. 2 also contains the measured spectra of PdAu3+Ar5 and PdAu3+Ar6, which show many similarities with that of PdAu3+Ar4. The spectral similarities strongly suggest that, for these complexes, extra Ar atoms do not significantly change the geometry of the metal cluster. | ||
| Fig. 2 (left) Experimental IR spectra of PdAu3+Arm (m = 4, 5, 6) clusters. (right) Simulated IR spectra of the complexes, considering the lowest-energy isomer (see Fig. 5 for details). | ||
The experimental data of PdAu3+Arm (m = 4–6) is compared with simulated IR spectra using the putative lowest-energy structure for the metal framework (isomer 1), which adopts a 3D pyramidal geometry (other isomers are discussed later). The calculated IR spectra for the three Ar complexes all agree very well with the corresponding experiments, predicting the three observed bands and fairly reproducing their relative intensities. Moreover, the slightly broader feature seen for PdAu3+·Ar5 near 150 cm−1 is reproduced by a double band in the calculations, due to a symmetry breaking. The agreement between the calculations and the experiment, each with the same geometry for the metallic framework, confirms that Ar does not significantly modifies the geometry of PdAu3+. Accordingly, we conclude that the structure of the bare PdAu3+ cluster, for which the IR spectrum was not recorded, is the one in isomer 1. While Ar does not modify the isomeric configuration adopted by PdAu3+, it slightly modifies the Au–Pd bond lengths in the cluster. In PdAu3+, these are 2.660 Å, whereas in PdAu3+Ar4 they are 2.634 Å. IR spectra of the bare and tagged species of PdAu3+ are compared in Fig. S2 of the ESI,† with a representation of the PdAu3+Ar4 vibrational modes in Fig. S3.†
A similar analysis on the effect of Ar attachment on the metallic framework structure is made for PdAu6+, as summarized in Fig. 3. In the left panel, the measured spectra for PdAu6+Ar2, PdAu6+Ar3, and PdAu6+Ar4 are depicted, with the corresponding simulated spectra using the geometry of the putative lowest-energy configuration for PdAu6+, on the right. The recorded spectra are highly similar, with bands around 110, 127, 178, and 194 cm−1, suggesting that Ar is not affecting the structure adopted by the metal framework. The computed spectra for isomer 1 (other isomers are discussed later), a triangular Au6 with Pd above the plane coordinated to four Au atoms, reproduces all observed bands. A minor difference is the band at 127 cm−1 for which the calculations provide a doublet feature, but the experimental data only has a shoulder. Despite this small difference, the calculations employing isomer 1 reproduce very well the experimental results, confirming also that for this cluster size the attachment of many Ar atoms does not affect the underlying geometry adopted by the metallic framework in PdAu6+.
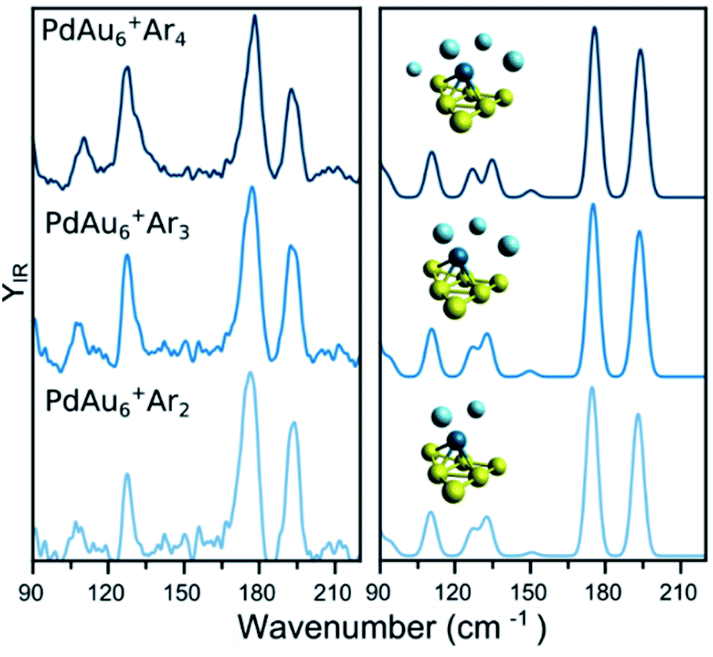 | ||
| Fig. 3 (left) Experimental IR spectra of PdAu6+Arm (m = 2, 3, 4) clusters. (right) Simulated IR spectra of the complexes, considering the lowest-energy isomer (see Fig. 5 for details). | ||
To assign the geometry of a specific PdAun−1+ cluster, the experimental data is compared with simulated IR spectra of different isomers of that size. As an example, Fig. 4 compares the experimental spectrum (bottom) with the simulated spectra (top) of two isomers of PdAu6+Ar2 (more isomers are discussed later). As already mentioned, the computed spectra for isomer 1 reproduce nicely the experiment. The computed spectra of isomer 4, where Pd substitutes a Au atom in Au7+,5 disagree with the experimental result, predicting bands at 97, 139, and 182 cm−1 that do not appear in the measurement. Comparing the results from both isomers, the geometry of isomer 1 is likely the one present in the molecular beam. The relative energy (ΔE) between the isomers also support this conclusion, as isomer 4 is found 0.63 eV higher.
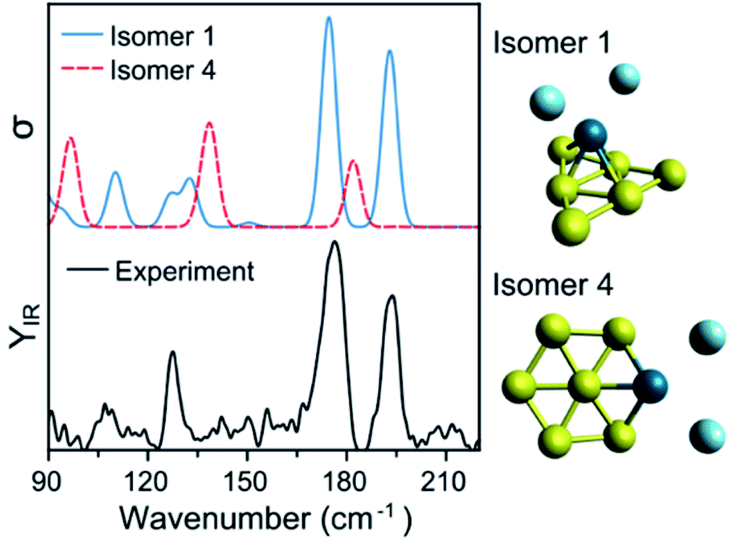 | ||
| Fig. 4 Experimental (bottom) and simulated (top) IR spectra of PdAu6+Ar2. Calculations for two isomers, with geometries depicted on the right of the figure. | ||
Such a visual comparison between the experimental and simulated IR spectra is illustrative, and has been proven useful for identifying cluster geometries.5,12 If many isomers are compared, a more quantitative comparison between experiment and calculations is advisable. For this reason, we directly contrast the simulated spectra with the experimental curve, using the Kullback–Leibler (DKL) divergence, which quantifies how much two curves differ from each other. DKL is zero for identical curves, and increases the more they diverge.48 Therefore, we can assign geometries by comparing DKL−1 values; a better agreement between experiment and calculation is reflected in a larger DKL−1 value. For this procedure, the computed vibrational modes of a specific isomer were used to construct a simulated IR spectrum, in which Gaussian functions were assumed centered at each vibrational frequency. These frequencies were scaled given the factor determined in ref. 5. The width of the Gaussian (5 cm−1) was determined by a prior visual inspection of the experimental and simulated IR spectra. The selected width was confirmed to provide the higher DKL−1 values. Using the benchmark analysis in Fig. 1 as an example, DKL−1 values of 0.08 (LC-ωPBEh), 0.05 (PBE0), and 0.04 (PBE) are calculated, indicating that LC-ωPBEh gives the best agreement. Another example of the procedure is presented in Fig. 4 for PdAu6+Ar2, with DKL−1 values of 0.14 and 0.04 obtained for isomers 1 and 4, respectively.
A summary of the structural assignment for the PdAun−1+ (n = 3–5, 7–10) clusters is shown in Fig. 5, with panel (a) showing the measured IR spectra of each complex. As mentioned, the spectrum of PdAu3+Ar4 has three bands at 102, 123, and 147 cm−1. For PdAu4+Ar4, two intense bands are observed, centered at 125 and 99 cm−1, with a side peak at 105 cm−1. The spectrum of PdAu6+Ar2, already discussed, consists of four clear bands, at 110, 127, 178, and 194 cm−1. For PdAu7+Ar4, four pronounced bands are seen, with maxima at 98, 110, 122 and 195 cm−1, in addition to some weaker features that could be hidden under the noise level in the 130 to 180 cm−1 spectral range. The IR spectrum of PdAu8+Ar3 has three clear bands at 126, 149, and 162 cm−1, with possible bands at 93, 106, 118, and 187 cm−1. Finally, the experimental IR spectrum of PdAu9+Ar1, with a signal-to-noise ratio a bit lower than for the other sizes, shows three clear bands at 126, 140, and 185 cm−1. For comparison, Fig. 5b presents the simulated IR spectra of the identified isomers, which are assigned as follow.
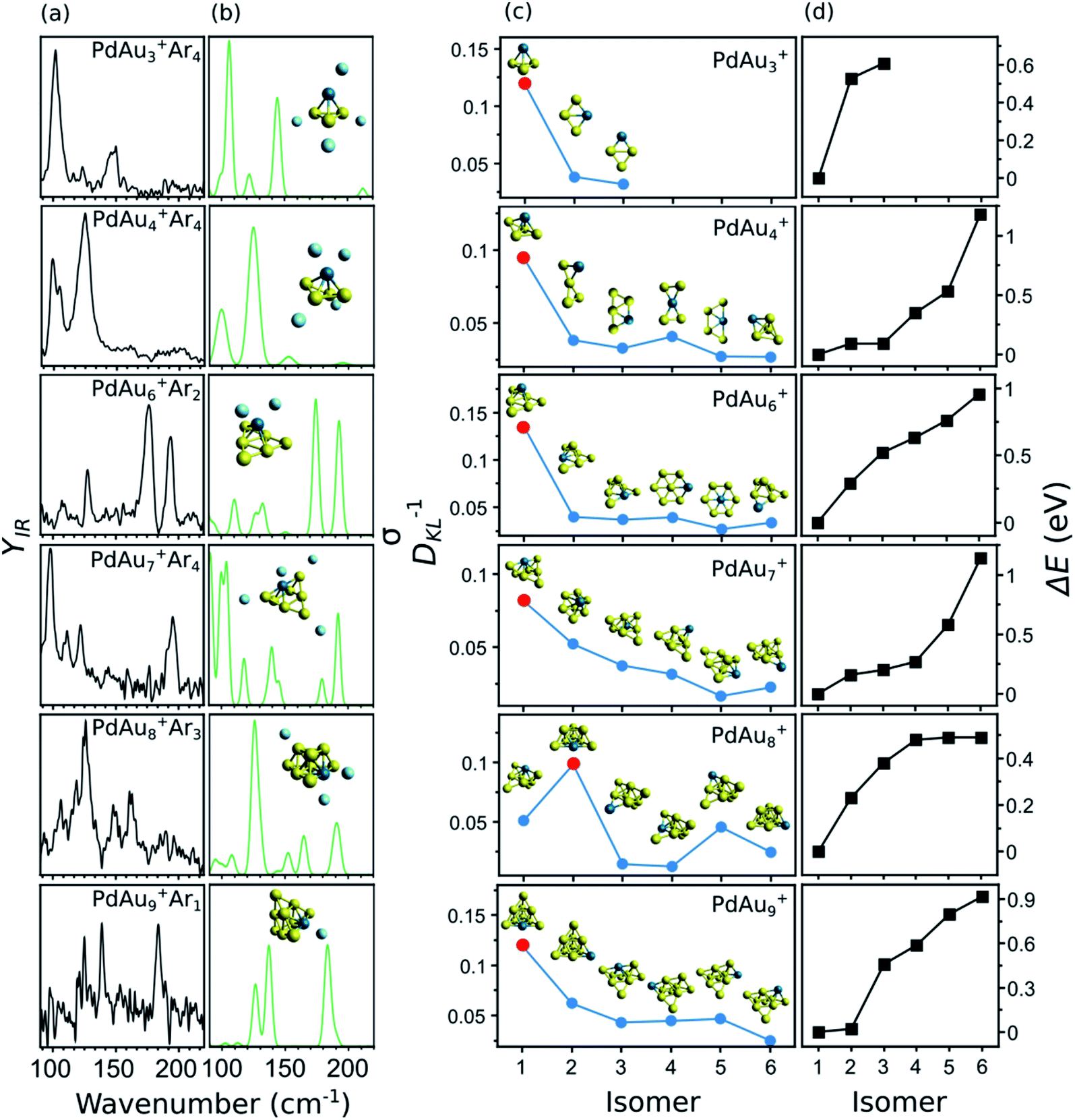 | ||
| Fig. 5 (a) Experimentally measured IR spectra of PdAun−1+Arm clusters. (b) Calculated IR spectra of the assigned isomers. The isomers are shown as insets. (c) Calculated values of DKL−1, comparing the measured IR spectrum of a cluster size with those simulated for each isomer. A higher DKL−1 value means a better correspondence between the experimental data and the calculations. The insets show the metallic framework of each isomer, although the simulated spectra used for calculating DKL−1 correspond to the complexes including Ar. The DKL−1 value of the assigned geometry is marked by a red dot. (d) Relative energy of each isomer with respect to the putative lowest-energy configuration (iso1). | ||
In panel (c) of Fig. 5, the blue circles show the DKL−1 values of six isomers (except for PdAu3+) for each PdAun−1+ (n = 4–5, 7–10) cluster. The relative energies (ΔE) of these isomers are given in panel (d), and are calculated with respect to isomer 1, which is the putative lowest-energy structure. As an inset in panel (c), the geometry of each considered isomer is depicted. As seen from the figure, for each cluster size, there is always one isomer that stands out for having a significantly higher DKL−1 value (highlighted in the figure by a red dot). This is the one we assign as the isomer present in the molecular beam. It corresponds to the putative lowest-energy structure in all cases, except for PdAu8+, for which the DKL−1 analysis points to isomer 2 as the geometry present in the experiment (0.23 eV above the computed lowest energy structure).
PdAu3+
Three isomers are considered for PdAu3+Ar4. In isomer 1, the metal atoms adopt a 3D pyramidal geometry, whereas isomers 2 and 3 form a rhombus, with Pd adopting different coordination sites. The DKL−1 analysis clearly assigns isomer 1 as the species present in the molecular beam, in agreement with the high relative energies of isomers 2 and 3, being 0.53 and 0.61 eV higher than isomer 1, respectively.PdAu4+
In previous DFT calculations, a twisted bow tie shaped structure (isomer 2) of PdAu4+ was predicted as the lowest in energy.31,32,34 In the current work, however, a bipyramid geometry was found 0.16 eV lower in energy (isomer 1). The simulated IR spectrum of isomer 1 agrees much better with the experimental data than the one of isomer 2 (DKL−1 values of 0.10 and 0.03, respectively). The other considered isomers yield equally low DKL−1 values, so they can be discarded as geometries present in the molecular beam. In a previous study, the visible/near-UV absorption spectrum of PdAu4+Ar1 was recorded and only isomer 2 was considered in the analysis.34 For this isomer, the experiment did not match the time-dependent DFT calculation of the optical spectrum. The ESI includes a calculation for isomer 1 of PdAu4+·Ar1 that agrees better with the experiment (Fig. S4†). This further supports the current structural assignment.PdAu6+
As was discussed in Fig. 3, the putative lowest-energy isomer of PdAu6+ (isomer 1) is formed by a triangular Au6 shape with Pd above it. Other isomers are considered, as detailed in Fig. 5c, with geometries similar to isomer 1, but with Pd adopting a different coordination (isomers 2, 3, and 6), or with the same planar shape as Au7+ and Pd substituting a Au atom (isomers 4 and 5). The DKL−1 analysis unambiguously favors isomer 1, with a DKL−1 value of 0.14, while the other isomers have values close to 0.03. Isomer 1 is also the lowest in energy, with isomer 2 having a relative energy of +0.29 eV.PdAu7+
The structural assignment is less obvious for PdAu7+. Isomer 1, where Pd adopts a symmetrical position substituting a Au atom in Au8+,5 correctly predicts the mode at 195 cm−1, although the agreement in the low-frequency range is not perfect (by comparing panels (a) and (b) in Fig. 5). Isomer 2, instead, does well below 150 cm−1, but the higher frequency modes are blue shifted with respect to the experiment (see Fig. S9 in the ESI†). The DKL−1 analysis suggests that isomer 1 (DKL−1 = 0.08) is the geometry present in the experiment, however, the DKL−1 value of isomer 2 (DKL−1 = 0.05) is not that different. The values for isomers 3 to 6 are around 0.03 and those isomers can thus be disregarded. Hence, isomer 1 is assigned in view of the better correspondence with the experiment and its lower relative energy, with perhaps some contribution from isomer 2.PdAu8+
The lowest-energy isomer of PdAu8+Ar3 (isomer 1) resembles the geometry assigned to Au9+ (formed by two Au6 triangles sharing three Au atoms),5 with the substitution of the most coordinated Au atom by the Pd dopant. The agreement between the experiment and the calculation for isomer 1 is low, with a DKL−1 value of 0.05. Instead, the calculated spectrum of isomer 2, a two-layer cluster with a PdAu5 base and a Au3 second layer, agrees much better (DKL−1 = 0.10). As shown in Fig. 5b, the calculation of isomer 2 reproduces all the experimental bands, except maybe the relative intensity of the 198 cm−1 band and the feature at 118 cm−1, although the latter simply can be noise. The other isomers are not considered because of their low DKL−1 values.This analysis provides strong evidence that isomer 2 is the isomer present in the molecular beam, even though it is computationally 0.23 eV less stable than isomer 1. Therefore, a study purely based on theory would incorrectly identify this cluster's geometry. We have three possible explanations for the assignment of a higher energy isomer: (1) Ar adsorption modifies the relative order of the isomeric structures. This explanation is unlikely since the calculated Ar binding energies differ by < 0.01 eV for isomer 1 and isomer 2 of PdAu8+Arm, whose relative energy differences are 0.19, 0.19, 0.19, and 0.23 eV for m = 0, 1, 2, and 3, respectively. Thus, irrespective of the number of Ar atoms isomer 1 is energetically preferred over isomer 2 by about 0.2 eV. (2) The calculated relative energies are not accurate. To explore this possibility, the relative energy between the isomers was also calculated using the double-hybrid B2PLYP functional. Double-hybrid DFT functionals are typically considered more accurate for total energy calculations, at the expense of very large computing times.49 See for example the correct prediction of dissociation energies of Aun+ clusters by the B2PLYP functional.50 This calculation yields an energy difference of 0.20 eV, thus essentially the same value as with LC-ωPBEh. This observation, however, does not rule out the possibility that DFT itself is not appropriate in this case, although given its success for the other sizes that explanation seems unlikely. (3) During the growth process, the PdAu8+Arm cluster is kinetically trapped in isomer 2. Kinetic trapping is certainly possible when clusters are formed by laser ablation and gas condensation, where the ablated plasma is rapidly cooled down by collisions with the carrier gas, in combination with a rapid supersonic expansion.51 In consequence, the produced size distributions are in many cases different from the expected thermodynamic equilibrium.52 Out of the three possibilities, option 3 seems most likely, considering that the 0.23 eV energy difference between the isomers is large enough as to not be ascribed to DFT uncertainty.53 Moreover, as detailed in the ESI (Fig. S5†), a scan of the potential energy surface of PdAu8+, along the reaction coordinate connecting isomers 1 and 2, shows a barrier of 0.18 eV with respect to isomer 2. This barrier could well trap the cluster in the geometry of isomer 2, given the experimental conditions.
PdAu9+
The two lower-energy isomers of PdAu9+Ar1 have a pyramidal structure with a different position of the Pd dopant. Four other isomers are considered, with the predicted shape of Au10+ (formed by adding a low-coordinated Au atom to Au9+) and a substitution of a Au atom by Pd, in different coordination sites. The energy difference between the first two isomers is very small (0.02 eV), but the simulated IR spectrum of isomer 1 is in much better agreement with the experiment, with a DKL−1 value of 0.12, in comparison with 0.06 for isomer 2. The calculation of isomer 1 predicts particularly well the bands at 126 and 140 cm−1, as is seen in Fig. 5b. These bands are not reproduced by the calculation of isomer 2 (Fig. S11 in the ESI†). All the other isomers agree poorly with the experiment and are found at higher relative energies. Accordingly, we assign isomer 1, which can be formed by adding a Au atom to the assigned isomer 2 of PdAu8+.Discussion
An overview of the identified PdAun−1+ (n = 3–5 and n = 7–10) clusters is shown in Fig. 6. The calculated lowest energy isomers of PdAun−1+ clusters with n = 2 (a trivial dimer) and n = 6 are extracted from ref. 31, and are added for completeness, even though the current study provided no experimental data for these sizes. For comparison, the figure also depicts the identified geometries for the pure Aun+ clusters, reproduced from ref. 5 and 31.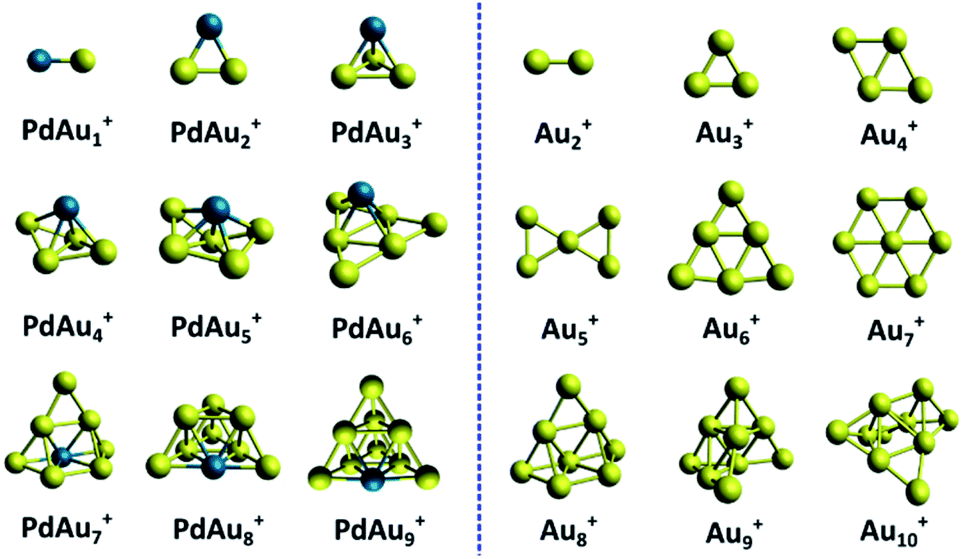 | ||
| Fig. 6 Comparison of the structures of PdAun−1+ and Aun+ clusters. The structures of Aun+ and PdAu5+ are reproduced from ref. 5 and 31. | ||
Comparing size-by-size, the geometries of the PdAun−1+ clusters are very different from those of Aun+, except for n = 8. This is very different from other doped Au clusters, such as AgAun−1+, where doping barely affects the cluster geometries.17,39,42 From PdAu3+, all Pd doped cationic gold clusters are unambiguously three-dimensional. This is in clear contrast with the pure Aun+ clusters, which are planar up to Au7+. Pd thus induces a drastic 2D to 3D transition at the smallest possible size, n = 4. We also note that the isomer identified for PdAu4+, which has not been considered earlier, can be formed by adding an additional Au atom to PdAu3+.
Previous studies have revealed that PdAu6+ is a particularly stable cluster, as a consequence of its closed-shell electronic configuration. To achieve such electronic configuration, the Pd dopant promotes one of its d electrons to the s shell (4d10 to 4d95s1), which subsequently delocalizes over the entire cluster. Such delocalization, however, was found to strongly depend on the geometry of PdAu6+, and was only predicted for some isomers.31 The current structural assignment confirms the analysis of ref. 31, proving that PdAu6+ can be described as a nearly planar Au6 triangle, with Pd adopting a μ4 configuration. Substituting one Au in Au7+ by Pd drastically changes the geometry (see Fig. 6).
In ref. 31, PdAu9+ was also identified as a species with enhanced stability, with a closed-shell electronic configuration. In this case, however, only the Au atoms delocalize electrons over the cluster volume (6s1), in contrast with the PdAu6+ cluster. Our analysis confirms the theoretical results from ref. 31, with PdAu9+ adopting a compact pyramidal geometry with high symmetry.
The Bader charges (calculated with the Multiwfn software package;54 see Section 9 in ESI†) reveal that the interaction of Pd with the gold framework is significant. Despite this, the Au framework in PdAu6+ remains similar to the geometry of Au6+. A similar observation can be made for the smaller n < 6 clusters. For example, PdAu3+ is formed by adding the Pd atom to the Au3+ triangle in a three-fold coordinated site. PdAu4+ adopts a three-dimensional structure with Pd at the fourfold coordinated site on the rhombus Au4+, which undergoes a minor out-of-plane distortion. In PdAu5+, the Pd atom is located above the central atom of the bow tie Au5+ structure. This bow tie geometry is distorted, but still recognizable in PdAu5+. For n = 9 and 10, instead, the Aun−1+ frameworks are not recognizable in PdAun−1+. This could indicate that the Au–Pd interaction is strong enough to induce an isomerization in larger Aun+ clusters (n > 7), but not for the smaller ones.
Conclusions
The structures of small Pd doped Au clusters, PdAun−1+ (n ≤ 10), were investigated by combining experimental infrared multiple photon dissociation spectroscopy with DFT calculations. The influence of Pd on the geometry of the pure Aun+ species was found to be size-dependent. PdAun−1+ clusters are three-dimensional from PdAu3+, in sharp contrast with pure gold clusters, which remain planar up to Au7+. Interestingly, up to n = 7, the geometry of Aun−1+ is recognizable in PdAun−1+. Instead, for n ≥ 9, the structures are drastically different. In PdAu7+, the Pd dopant substitutes a Au atom in Au8+, whereas for n = 9 and 10, the doped clusters adopt highly symmetric pyramidal shapes.Importantly, the geometry assigned to PdAu4+, which corresponds to the putative global minima of the cluster, was not considered in previous studies that assumed geometries of PdAun−1+ purely based on DFT calculations. Furthermore, the geometry determined for PdAu8+ does not correspond to the lowest-energy structure predicted by DFT, suggesting a kinetic trapping of a higher-energy isomer of this species during the formation process. In both cases, we show that theory alone cannot be used when determining the geometries of clusters, and that unambiguous assignment requires a joint experimental and computational effort.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the KU Leuven Research Council (project C14/18/073) and by the Research Foundation-Flanders (FWO), project G0A0519N. The research leading to this result has been supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. P. F. acknowledges the FWO for a postdoctoral grant. We gratefully acknowledge the support to the FELIX Laboratory by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO). The computational work has been performed under the Project HPC-EUROPA3 (INFRAIA-2016-1-730897), with the support of the EC Research Innovation Action under the H2020 Programme. We benefit of the computer resources and technical support provided by EPCC at The University of Edinburgh. LDC is supported by King's College London through the NMES Faculty Studentship Scheme. LDC and FB are grateful to their membership of the UK's HEC Materials Chemistry Consortium funded by EPSRC (EP/R029431). This work used ARCHER2 UK National Supercomputing Service (http://www.archer2.ac.uk). They further acknowledge the UK Materials and Molecular Modelling Hub for computational resources, MMM Hub, which is partially funded by EPSRC (EP/P020194 and EP/T022213). FB thanks the financial support offered by the Royal Society (No. RG 120207) and the technical support offered by the NMES Faculty at the King's College London to maintain local HPC facilities.References
- M. Gao, A. Lyalin, M. Takagi, S. Maeda and T. Taketsugu, J. Phys. Chem. C, 2015, 119, 11120–11130 CrossRef CAS
.
- E. Janssens, H. Tanaka, S. Neukermans, R. Silverans and P. Lievens, New J. Phys., 2003, 5, 46.1–46.10 CrossRef
- A. C. Reber, G. U. Gamboa and S. V. Khanna, J. Phys.: Conf. Ser., 2013, 438, 012002 CrossRef CAS
- A. C. Reber and S. N. Khanna, Acc. Chem. Res., 2017, 50, 255–263 CrossRef CAS PubMed
- P. Ferrari, G.-L. Hou, O. Lushchikova, F. Calvo, J. M. Bakker and E. Janssens, Phys. Chem. Chem. Phys., 2020, 22, 11572–11577 RSC
- M. S. W. Forstel and O. Dopfer, Angew. Chem., Int. Ed., 2019, 58, 3356–3360 CrossRef PubMed
- R. Ferrando, A. Fortunelli and R. L. Johnston, Phys. Chem. Chem. Phys., 2008, 10, 640–649 RSC
- P. Grammatikopoulos, S. Steinhauer, J. Vernieres, V. Singh and M. Sowwan, Adv. Phys.: X, 2016, 1, 81–100 CAS
- A. Fielicke, A. Kirilyuk, C. Ratsch, J. Behler, M. Scheffler, G. von Helden and G. Meijer, Phys. Rev. Lett., 2004, 93, 023401 CrossRef PubMed
- S. Gilb, P. Weis, F. Furche, R. Ahlrichs and M. M. Kappes, J. Chem. Phys., 2002, 116(10), 4094 CrossRef CAS
- J. Li, X. Li, G.-J. Zhai and L.-S. Wang, Science, 2003, 299, 864–867 CrossRef CAS PubMed
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed
- S. Bulusu, X. Li, L. Wang and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8326–8330 CrossRef CAS PubMed
- B. R. Goldsmith, J. Florian, J.-X. Liu, P. Gruene, J. T. Lyon, D. M. Rayner, A. Fielicke, M. Scheffler and L. M. Ghiringhelli, Phys. Rev. Mater., 2019, 3, 016002 CrossRef CAS
- H. Häkkinen, B. Yoon, U. Landman, X. Li, H.-J. Zhai and L.-S. Wang, J. Phys. Chem. A, 2003, 107, 6168–6175 CrossRef
- H. Häkkinen, Adv. Phys.: X, 2016, 1, 467–491 Search PubMed
- M. Vogel, K. Hansen, A. Herlert and L. Schweikhard, Phys. Rev. Lett., 2001, 87, 013401 CrossRef CAS PubMed
- E. Janssens, H. T. Le and P. Lievens, Chem.–Eur. J., 2015, 21, 15256–15262 CrossRef CAS PubMed
- M. Neumaier, F. Weigend, O. Hampe and M. M. Kappes, J. Chem. Phys., 2005, 122, 104702 CrossRef PubMed
- M. Vogel, K. Hansen, A. Herlert and L. Schweikhard, Eur. Phys. J. D, 2001, 16, 73–76 CrossRef CAS
- K. Hansen, P. Ferrari, E. Janssens and P. Lievens, Phys. Rev. A, 2017, 96, 022511 CrossRef
- A. N. Gloess, H. Schneider, J. M. Weber and M. M. Kappes, J. Chem. Phys., 2008, 128, 114312 CrossRef PubMed
- A. Shayeghi, R. L. Johnston and R. Schafer, Phys. Chem. Chem. Phys., 2013, 15, 19715 RSC
- L. Lin, P. Claes, T. Höltzl, E. Janssens, T. Wende, R. Bergmann, G. Santambrogio, G. Meijer, K. R. Asmis, M. T. Nguyen and P. Lievens, Phys. Chem. Chem. Phys., 2010, 12, 13907–13913 RSC
- A. Shayeghi, R. Schäfer, D. M. Rayner, R. L. Johnston and A. Fielicke, J. Chem. Phys., 2015, 143, 024310 CrossRef CAS PubMed
- M. Begona Torres, E. M. Fernandez and L. C. Balbas, J. Phys. Chem. A, 2008, 112, 6678–6689 CrossRef PubMed
- E. Janssens, H. Tanaka, S. Neukermans, R. E. Silverans and P. Lievens, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 085402 CrossRef
- P. Ferrari, L. Delgado-Callico, P. Lievens, F. Baletto and E. Janssens, J. Chem. Phys., 2020, 153, 244304 CrossRef CAS PubMed
- J. Barabás, P. Ferrari, V. Kaydashev, J. Vanbuel, E. Janssens and T. Höltzl, RSC Adv., 2021, 11, 29186–29195 RSC
- P. Shao, X.-Y. Kuang, Y.-R. Zhao, Y.-F. Li and S.-J. Wang, J. Mol. Model., 2012, 18, 3553–3562 CrossRef CAS PubMed
- P. Ferrari, H. A. Hussein, C. J. Heard, J. Vanbuel, P. Lievens, R. L. Johnston and E. Janssens, Phys. Rev. A, 2018, 97, 052508 CrossRef CAS
- H. A. Abdulhussein, P. Ferrari, J. Vanbuel, C. Heard, A. Fielicke, P. Lievens, E. Janssens and R. L. Johnston, Nanoscale, 2019, 11, 16130–16141 RSC
- S. M. Lang, A. Frank, I. Fleischer and T. M. Bernhardt, Eur. Phys. J. D, 2013, 67, 1–7 CrossRef
- V. Kaydashev, P. Ferrari, C. Heard, E. Janssens, R. L. Johnston and P. Lievens, Part. Part. Syst. Charact., 2016, 33, 364–372 CrossRef CAS
- Z. Wang, J. Jia and H.-S. Wu, Mater. Chem. Phys., 2015, 160, 105–110 CrossRef CAS
- M. A. Tofanelli, T. W. Ni, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2016, 55, 999–1001 CrossRef CAS PubMed
- A. K. Kacprzak, L. Lehtovaara, K. Akola, O. Lopez-Acevedo and H. Häkkinen, Phys. Chem. Chem. Phys., 2009, 11, 7123–7129 RSC
-
P. Ferrari, J. Vanbuel, Y. L. T. Li, E. Janssens and P. Lievens, Modifications of gas aggregation sources: the double laser ablation source approach, in Gas Aggregation Synthesis of Nanoparticles, Wiley-VCH, Weinheim, 2017, pp. 57–78 Search PubMed
- J. M. Bakker, V. J. F. Lapoutre, B. Redlich, J. Oomens, B. G. Sartakov, A. Fielicke, G. von Helden, G. Meijer and A. F. G. van der Meer, J. Chem. Phys., 2010, 132, 074305 CrossRef PubMed
- X. Li, P. Claes, M. Haertelt, P. Lievens, E. Janssens and A. Fielicke, Phys. Chem. Chem. Phys., 2016, 18, 6291–6300 RSC
- O. V. Lushchikova, D. M. M. Huitema, P. López-Tarifa, L. Visscher, Z. Jamshidi and J. M. Bakker, J. Phys. Chem. Lett., 2019, 10, 2151–2155 CrossRef CAS PubMed
- M. Fagiani, X. Song, P. Petkov, S. Debnath, S. Gewinner, W. Schöllkopf, T. Heine, A. Fielicke and K. Asmis, Angew. Chem., Int. Ed., 2017, 56, 501–504 CrossRef CAS PubMed
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 1181, 1477 CrossRef
- A. Shayeghi, C. J. Heard, R. L. Johnston and R. Schäfer, J. Chem. Phys., 2014, 140, 054312 CrossRef CAS PubMed
- A. Shayeghi, R. L. Johnston, D. M. Rayner, R. Schfer and A. Fielicke, Angew. Chem., Int. Ed., 2015, 54, 10675–10680 CrossRef CAS PubMed
- L. Delgado-Callico, P. Ferrari, J. M. Bakker, F. Baletto and E. Janssens, Theor. Chem. Acc., 2021, 140, 38 Search PubMed
- P. Ferrari and E. Janssens, Molecules, 2021, 26, 4082 Search PubMed
- L. Delgado-Callico, K. Rossi, R. Pinto-Miles, P. Salzbrenner and F. Baletto, Nanoscale, 2021, 13, 1172–1180 RSC
- L. Goerigk and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 576–600 CAS
- P. Ferrari and K. Hansen, Phys. Chem. Chem. Phys., 2021, 23, 14830–14835 RSC
- M. A. Duncan, Rev. Sci. Instrum., 2012, 83, 041101 CrossRef PubMed
- S. Neukermans, X. Wang, N. Veldeman, E. Janssens, R. Silverans and P. Lievens, Int. J. Mass Spectrom., 2006, 252, 145–150 CrossRef CAS
- Y. Wang and D. L. McDowell, Mech. Adv. Mater. Struct., 2020, 1–40 Search PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00587a |
| ‡ Current address: School of Physics, Xi'an Jiaotong University, Xi'an 710049, Shaanxi, China. |
| This journal is © The Royal Society of Chemistry 2021 |