
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of self-assembled nanostructures for intracellular drug delivery from diphenylalanine analogues with rigid or flexible chemical linkers†
Amutha
Arul
 a,
Priya
Rana
a,
Kiran
Das
b,
Ieshita
Pan
c,
Debasish
Mandal
d,
Adele
Stewart
e,
Biswanath
Maity
*b,
Soumyajit
Ghosh
*a and
Priyadip
Das
*a
a,
Priya
Rana
a,
Kiran
Das
b,
Ieshita
Pan
c,
Debasish
Mandal
d,
Adele
Stewart
e,
Biswanath
Maity
*b,
Soumyajit
Ghosh
*a and
Priyadip
Das
*a
aDepartment of Chemistry, SRM Institute of Science and Technology, SRM Nagar, Potheri, Kattankulathur, Tamil Nadu-603203, India. E-mail: priyadipcsmcri@gmail.com; priyadip@srmist.edu.in; soumyajitghosh89@gmail.com
bCentre of Biomedical Research, Sanjay Gandhi Post-Graduate Institute of Medical Sciences (SGPGI) Campus, Raebareli Road, Lucknow, Uttar Pradesh 226014, India. E-mail: bmaity28@gmail.com
cDepartment of Biotechnology, Saveetha School of Engineering, Saveetha Institute of Medical and Technical Sciences, Saveetha University, Tamil Nadu 602105, India
dSchool of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Bhadson Rd, Adarsh Nagar, Prem Nagar, Patiala, Punjab 147004, India
eDepartment of Biomedical Science, Charles E. Schmidt College of Medicine, Florida Atlantic University, Jupiter, FL 33458, USA
First published on 30th August 2021
Abstract
Self-assembly of molecular building blocks is a simple and useful approach to generate supramolecular structures with varied morphologies and functions. By studying the chemical properties of the building blocks and tuning the parameters of their self-assembly process, the resultant supramolecular assemblies can be optimized for the required downstream applications. To this end, in the present study we have designed and synthesized three different molecular building blocks composed of two diphenylalanine (FF) units connected to each other through three different linkers: ethylenediamine, succinic acid, or terephthalaldehyde. Under identical conditions, all the three building blocks self-assemble into supramolecular architectures with distinct morphologies. However, by varying the polarity of the self-assembly medium, the nature of the non-covalent interactions changes in such a way as to generate additional self-assembled structures unique to each building block. Utilizing microscopic and spectroscopic techniques, we characterized the morphological variety generated by each building block/linker combination. These data represent the first report analysing the diversity of nanostructures that can be generated from identical dipeptide-based molecular backbones simply by varying the chemical linker. We also demonstrate that the spherical assemblies and nanorod structures fabricated from these dipeptide/linker pairs can act as drug delivery systems. More specifically, the spherical assembly generated by two FF dipeptides linked via ethylenediamine and nanorods fabricated from terephthalaldehyde linked FF dipeptides were able to encapsulate the cancer chemotherapeutic agent doxorubicin (DOX) and chaperone the drug into cells. Thus, these supramolecular assemblies represent a new platform for the development of efficient and effective intracellular drug delivery systems.
Introduction
The fabrication of functional nano and microstructures with diverse morphologies is an area of active research in nanotechnology development. One “bottom-up” approach, referred to as self-assembly, involves the interaction of simple building blocks in a coordinated manner to form large and more complex supramolecular assemblies.1 The self-assembly of these core components can be directed by combining various non-covalent interactions including electrostatic interactions, hydrogen bonds, aromatic stacking interactions, hydrophobic interactions, non-specific van der Waals forces, and dipole–dipole interactions.2 While reasonably weak individually, the coordinated combination of these non-covalent molecular forces drives the process of self-organization allowing simple subunits to form ordered nanostructures and macroscopic objects with nano-scale order.3In nature, complex functional structures are generated from comparatively simple biological building blocks such as nucleic acids, lipids and amino acids by the process of bio-molecular self-assembly.4,5 Of the possible biological subunits available, peptides, specifically short peptides, provide unmatched biocompatibility, chemical diversity, biological recognition abilities and facile synthesis.6 While cyclic peptides, dendritic peptides, amphiphilic peptides, surfactant-like oligopeptides, copolypeptides, and aromatic dipeptides may be used, the construction of functional supramolecular architectures based on the use of short aromatic dipeptides is of particular interest due to their resemblance to very short amyloid-forming peptides and propensity for π-stacking interactions.5b,7 Families of self-assembled, well-ordered nanostructures generated from aromatic dipeptides have been previously shown to possess optimal thermal and chemical stability and significant rigidity at the nanoscale level.8
Among these aromatic short peptides, diphenylalanine (L-Phe-L-Phe, FF), a core recognition motif of Alzheimer's β-amyloid polypeptide, has been widely studied due to its structural simplicity, versatile functions and wide applications in biology and nanotechnology.5a,9 A variety of functional nanostructures such as spheres, nanotubes, nanofibrils, nanowires, hydrogels, and ordered molecular chains can be self-assembled by using FF as a building block9c,10 and find potential applications in diverse areas.11,12 Interestingly, the acid labile tert-butyl dicarbonate group (BOC) protected analogue BOC-Phe-Phe-OH (BOC-FF) can self-assemble into a tubular structure in aqueous medium and spherical assembly with metallic stiffness in the presence of ethanol,13 a process which can be mimicked via introduction of a thiol group into the diphenylalanine dipeptide.7b Similarly, varying the cationic dipeptide (H-Phe-Phe-NH2) concentration allows for a reversible transformation from a nanotube to a vesicle-like structure with an intermediate “necklace” morphology,11f,14 while a structural transformation from an organogel to flower-like microcrystals can be driven by the inclusion of ethanol as a co-solvent.9d FF nanotubes are also able to generate vertical arrays in the form of a “nano-forest” or align horizontally when modified with magnetic nanoparticles in the presence of an externally applied magnetic field.9a Thus, several parameters modify the self-assembly of FF based structures suggesting that FF-based supramolecular assemblies could be directed and optimized.15–17
While several protein assembly strategies exist, variation of the length and rigidity of connecting linkers between assembled units, which determines molecular flexibility and the rate of surface stacking, has proven challenging.18 Computational analyses predict that symmetrical protein assemblies require short or rigid linkers between proteins.19 In the case of protein polymers, relatively long linkers between monomeric building blocks enhance intramolecular assembly as Jung et al. demonstrated by incorporating various flexible and rigid peptide linkers into high valent GFP oligomers. In theory, oligomers with both flexible and rigid linkers could be fabricated to allow more versatile linker rigidity controls.18 Indeed, Das and Reches recently demonstrated that two hetero tripeptide units (BOC-Phe-Gly-Phe) connected through the flexible linker ethylenediamine exhibited solvent induced self-assembly and formed nanostructures with different shapes and sizes.20
However, there are no reports studying the impact of both flexible and rigid chemical linkers on the self-assembling ability of the FF building blocks, which display the unique capacity for self-assembly into a diverse array of nanostructures. Herein, we report the design and synthesis of three building blocks in which two identical FF units were connected through three different linkers: ethylenediamine (flexible linker), succinic acid (flexible linker) and terephthalaldehyde (rigid linker). We then demonstrate their self-assembling ability in different solvents identifying the morphological diversity of the resultant supramolecular assemblies and function as drug delivery vehicles in vitro with the ultimate aim of providing novel platforms for the development of nanostructures for biomedical applications.
Results and discussion
We report the synthesis of three molecules PA1 (BOC-FF-EA-FF-BOC), PA2 (OMe-FF-SA-FF-OMe) and PA3 (OMe-FF-TA-FF-OMe) (FF = diphenylalanine, EA = ethylenediamine, SA = succinic acid and TA = terephthalaldehyde). PA1 and PA2 comprise two repeating BOC-FF or OMe-FF units connected through ethylenediamine and succinic acid, respectively, using DCC and HOBT as coupling reagents. In PA3 two OMe-FF building blocks are linked through terephthalaldehyde by a simple condensation reaction in methanol at RT (Fig. 1, the synthesis procedure is provided in the ESI Experimental section as Schemes S1–S4†). All the synthesized products were isolated and characterized by standard analytical techniques (Fig. S1–S9†). We choose FF, the smallest self-assembled aromatic dipeptide moiety, as it represents the core recognition motif of Alzheimer's β-amyloid polypeptides, and, thus, has biological relevance in determining the amyloidal state of proteins as a basic structural motif. The incorporated linker ethylenediamine (EA) and succinic acid (SA) bridged two aromatic dipeptide units and acted as a flexible linker that should provide structural freedom during the self-assembly process.21 On the other hand, in PA3, the bridging unit terephthalaldehyde (TA) can be considered a rigid linker and will restrict the structural flexibility during the self-assembly process. | ||
| Fig. 1 Schematic representation of the synthesis of PA1, PA2 and PA3. | ||
We determined the critical aggregation concentration (CAC) for these newly synthesized molecules (PA1, PA2 and PA3) with the use of a tensiometer by measuring the surface tension in a wide range of concentrations. In the beginning, the surface tension dropped down considerably with increasing concentration before arriving at a plateau. The CAC values obtained from the intersection between the regression straight line of the linearly dependent region and the straight line passing through the plateau were 1.58 mg mL−1 for PA1, 1.20 mg mL−1 for PA2, and 1.31 mg mL−1 for PA3 (Fig. S10†). Thermogravimetric analysis was also performed to estimate the thermal stability of these newly synthesised peptides (PA1, PA2 and PA3) (Fig. S11†). This analysis showed that PA1, PA2 and PA3 were thermally stable up to 204 °C, 220 °C and 295 °C, respectively.
Next, we studied the self-assembly property of all synthesized compounds. To trigger the self-assembly process, each compound was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFP) to an initial concentration of 100 mg mL−1 followed by further dilution with polar solvents to achieve the desired concentration (2 mg mL−1). The polar solvents allowed the peptides to self-assemble into ordered assemblies. Field Emission Scanning Electron Microscopy (FE-SEM) and High-Resolution Transmission Electron Microscopy (HR-TEM) analysis showed that PA1 (ethylenediamine linker) self-assembled into a spherical assembly with nanomeric dimensions in 50% aqueous ethanol (Fig. 2A–D). Dynamic light scattering (DLS) analysis revealed that the average diameter of the spherical structure is 969.65 ± 17.66 nm (Fig. S12†). PA2 (succinic acid linker), conversely, self-assembled into rectangular plates with varying dimensions. These plates appear to be thin as well as symmetrical with a defined prominent border when analysed by SEM (Fig. 2E and F). The TEM analysis of the self-assembled structures formed by PA2 further confirmed the thin plate-like morphology (Fig. 2G and H). A similar morphology was previously obtained by the self-assembly of (4-phenyl-Phe)-(4-phenyl-Phe).12a In PA1, the terminal amine groups at both ends are blocked by tert-butoxycarbonyl (BOC) groups. In PA2, there are terminal ester groups (–CO2Me) at the two ends of the molecule. We suspect that the delicate balance between linker-dependent molecular flexibility and the geometrically restricted orientation of the building block originates from the π–π interaction controlling the self-assembly process of PA1 and PA2. This morphological difference between the self-assembled structures of PA1 and PA2 arises from the difference in the hydrophobicity of their terminal moieties. It is well known that hydrophobic interactions control the self-assembly process in water,22 play a significant role in the self-assembly process of aromatic homo dipeptides, and yield ordered nanostructures.11a,13 Therefore, the difference in hydrophobicity between PA1 and PA2 driven by the hydrophobic Boc group directs the self-assembly process and influences the size, shape and morphology of the self-assembled structures of PA1 and PA2.
 | ||
| Fig. 2 Microscopic analysis of the self-assembled structures formed by PA1, PA2 and PA3. Representative FE-SEM micrographs of the self-assembled structures formed by PA1 (A, B), PA2 (E, F), and PA3 (I, J) in 50% aqueous ethanol. Representative HR-TEM micrographs of the self-assembled structures formed by PA1 (C, D), PA2 (G, H), and PA3 (K, L) in 50% aqueous ethanol. | ||
The microscopic characterization (FESEM and TEM) analysis clearly confirmed the nanorod shaped morphology of terephthalaldehyde-linked PA3 in 50% aqueous ethanol (Fig. 2I–L). The combination of geometrically restricted π–π interactions between the aromatic moieties together with the planar nature of the amide bond and restricted flexibility of the linker controls the self-assembly process of the PA3 building block. Highly ordered molecular arrangements with high surface area/density as well as a hollow nanospace (for nanotubes) are required to achieve the cylindrical (nanorods/nanotubes) morphology from self-assembled organic building blocks.23 The restricted molecular flexibility of PA3 affords the ability to form highly ordered multi-layered molecular arrangements facilitated by π–π stacking interactions. The energetically more favourable lamellar molecular arrangements allow the structure to scroll up to form nanorods. This two dimensional layer closure process will consequently minimize the surface energy of the assembly and may result in nanorods breaking into pieces of smaller length or the occasional bundled assembly.24 FESEM and TEM micrographs estimated the diameter of the PA3-generated nanorods as ranging from ∼50 to 200 nm confirming the multi-layered nature of the nanorod formed by PA3 in 50% aqueous ethanol.
To get better insight into the secondary conformation as well as molecular configuration of the different self-assembled structures formed by PA1, PA2 and PA3, we have performed Fourier transform infrared (FT-IR) analysis and deconvoluted each spectrum in the amide-I region with a Gaussian function. The FT-IR spectra of the spherical structures self-assembled by PA1 displayed two dominant peaks at 1621 cm−1 and 1650 cm−1, and a high field minor peak at 1685 cm−1 (Fig. 3A). The peaks at 1621 cm−1 and 1685 cm−1 indicate an anti-parallel β-sheet conformation25 and the shoulder peak at 1650 cm−1 suggests a significantly disordered or random structure.26 The thin plate-like structures formed by PA2 exhibited two bands, one at 1660 cm−1, which correlates with the α-helix conformation,14 and another minor high field peak at ∼1695 cm−1, likely marker bands for a β-turn configuration (Fig. 3B).27 The FT-IR spectra for the self-assembled PA3 nanorods contained two distinctive bands with absorption maxima at 1659 cm−1, which is consistent with an α-helix conformation, and another shoulder band at 1676 cm−1, correlating with a β-turn conformation (Fig. 3C).27,28 Smaller linear peptides with 3–6 amino acid residues generally do not exhibit an α-helix conformation due to size restrictions. We suspect that the formation of a well-ordered supramolecular assembly by the aggregation of monomeric building blocks leads to antiparallel pairing of two monomeric units and the unanticipated α-helical conformation due to further aggregation of the initially assembled structures.29 Indeed, Reches and Gazit previously reported an α-helical secondary conformation generated from self-assembled nanostructures formed by different aromatic homodipeptides and charged termini capped diphenylalanine peptides.12 Furthermore, the presence of intense and sharp peaks corresponding to –N–Hamide (str) vibrations (at 3320 cm−1 for PA1 and 3283 cm−1 for PA2) (Fig. S13A and B†) also indicates the obvious formation of a H-bonded network of amides in the self-assembled states of PA1 and PA2. Such an intense peak of N–Hamide (str) is not detected for the nanorod structure formed by PA3 (Fig. S13C†). Based on these data, we propose that the amide bond formed by the conjugation of linkers (EA & SA) with FF building blocks triggered hydrogen bonded network formation during the self-assembly process. This is not possible for PA3 due to the formation of an imine bond by the terephthalaldehyde connective linker between two dipeptide building blocks.
 | ||
| Fig. 3 Deconvoluted FT-IR spectra of self-assembled PA1 (A), PA2 (B), and PA3 (C) in 50% aqueous ethanol. The dashed line indicates the original FTIR spectra and the solid line indicates the deconvoluted curves with a Gaussian function. Powder X-ray diffraction pattern of the self-assembled structure formed by (D) PA1, (E) PA2 and (F) PA3 in aqueous ethanol medium. | ||
Besides the FT-IR analysis, the nature of molecular assembly of these self-assembled structures was further clarified from the powder X-ray diffraction (PXRD) pattern of the dried mass of the self-assembled structures obtained from PA1, PA2 and PA3. In the wide region, diffraction peaks at 21.2° (with a d spacing value of 4.2 Å) for PA1 (Fig. 3D), 20.1° and 23.4° (with respective d spacing values of 4.4 Å and 3.7 Å) for PA2 (Fig. 3E), and 20.1° and 23.5° (with respective d spacing values of 4.4 Å and 3.7 Å) for PA3 (Fig. 3F) were observed, which suggest the presence of H-bonding and π–π stacking interactions among the building blocks.24 Additionally, as shown in Fig. 3F, diffraction peaks were identified for the nanorod assembly obtained from PA3 at 7.0° followed by other peaks in the wide angle region in a periodical order at 14.1°, 22.2° and 27.8°. The corresponding d-spacing values of these peaks obtained from Bragg's equation were 12.5 Å, 6.3 Å, 4.0 Å and 3.2 Å respectively. The ratios of 1/2, 1/3 and 1/4 are consistent with the lamellar arrangement of PA3 during the self-assembly.
To gain further insight into the self-assembly process of these peptide based molecules, we have studied the self-assembly of PA1, PA2 and PA3 in a polar solvent with maximum water content (100% aqueous medium). Recently, Ganesh et al. demonstrated that the hydrophobic substituent at the N or C terminus and aromatic π–π interactions play a significant role in determining the shape and morphology of the self-assembled nanostructures.10dPA1 in 100% water medium self-assembled into interlinked necklace-like spherical particles (Fig. 4A and B). With an enhanced H-bonding network, PA1 exhibited a rapid self-aggregation tendency and formed interlinked spherical units. HR-TEM images corroborated the morphology observed in SEM (Fig. 4C). In our previous report, we generated a polypeptide in which two identical tripeptide units (Phe-Gly-Phe) linked by an ethylenediamine linker exhibited solvent induced self-assembly. In 50% aqueous ethanol it self-assembled into nanospheres, and, in more polar 100% aqueous medium, formed a 3D network of aggregated spheres.20 The terminal hydrophobic Boc group in PA1 was expected to influence the molecular assembly process due to the enhanced hydrophobic forces with increasing water content. In 100% water PA2 self-assembled into nanobelt-like supramolecular structures (Fig. 4D–F), a different morphology from the self-assembled nanosheet obtained in 50% aqueous ethanol. The phenyl rings of the diphenylalanine facilitate lateral assembly to produce 2D nanosheet structures via interdigitation.30 The C-terminal methyl ester group of FF in PA2 promotes the molecular organization because of enhanced hydrophobic forces with increasing polarity of the solvent medium. In 100% aqueous medium the self-assembly process will help to minimize the solvophobic interaction from water as well as the surface energy of the assembly giving rise to the nanobelt-like morphology with smaller dimensions compared to the nanosheet generated in less polar 50% aqueous ethanol.
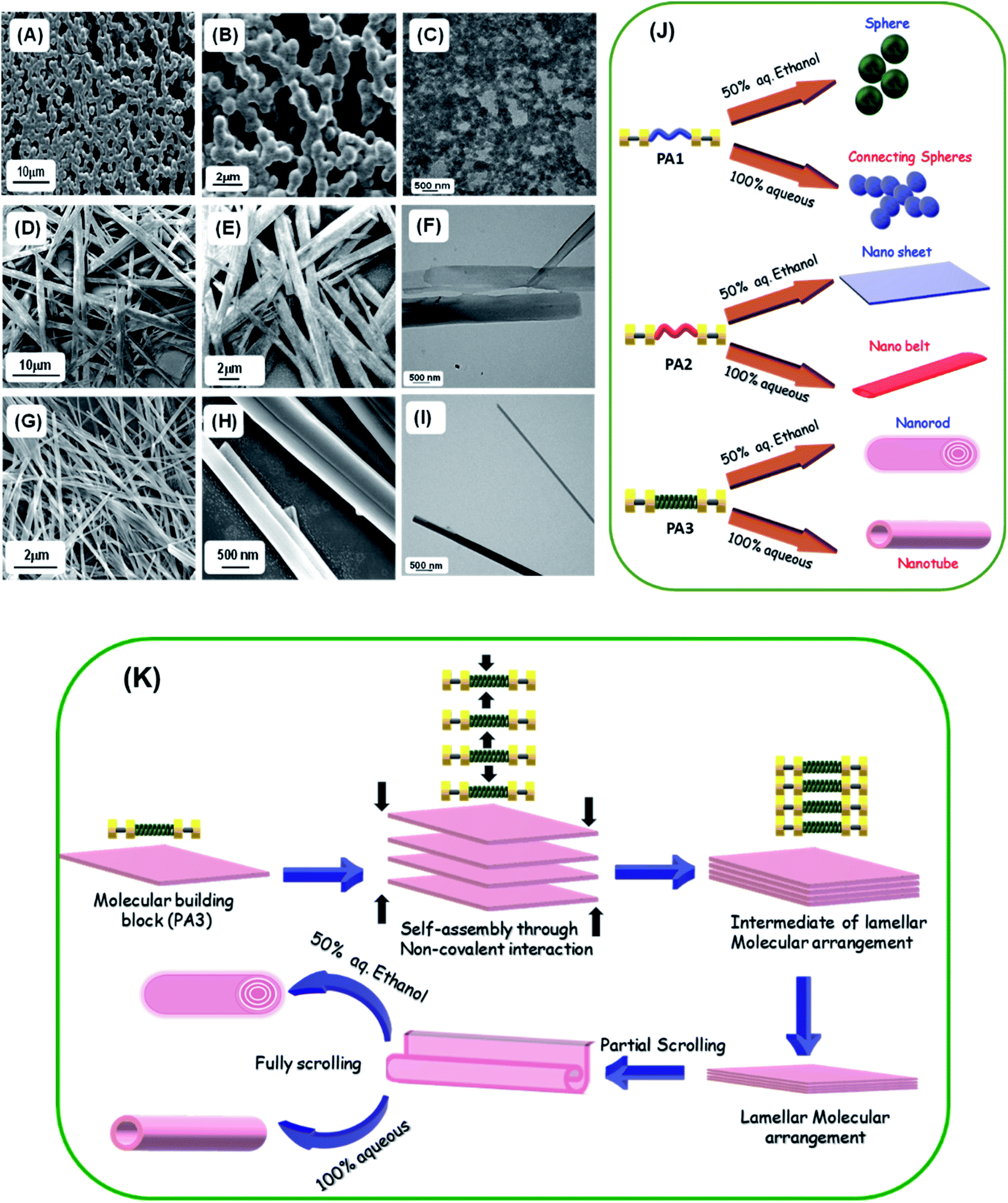 | ||
| Fig. 4 Microscopic analysis of the self-assembled structures formed by PA1, PA2 and PA3. Representative FE-SEM micrographs of the self-assembled structures formed by PA1 (A, B), PA2 (D, E), and PA3 (G, H) in 100% aqueous medium. Representative HR-TEM micrographs of the self-assembled structures formed by PA1 (C), PA2 (F), and PA3 (I) in 100% aqueous medium. (J) Pictorial representation of the different self-assembled structures with various morphologies formed by the peptides (PA1, PA2 and PA3) in different solvent media. Various supramolecular structures could be formed by the alternative organization of the monomeric building blocks. (K) A schematic illustration of the formation of nanorods and tubular structure by the self-assembly of PA3 in different solvent media through an intermediate lamellar molecular arrangement followed by layer closure or the scroll-up process. | ||
On the other hand, PA3 in 100% aqueous medium self-assembled into tubular structures (Fig. 4G–I). Non-covalent intermolecular interactions play a crucial role in directing the self-assembly process. Polar solvents with an enhanced H-bonding capacity may preferentially modify the nature of non-covalent interactions when compared to less polar or nonpolar solvents and direct the growth of the aggregation process of the building blocks to yield supramolecular assemblies with distinct morphology. In a solvent system with higher polarity, the stronger solvation effect contributes to the initial formation of solvent–building block solvated dimers by strong hydrogen bonding or electrostatic interactions.9d,31 This process is exacerbated by further aggregations of the solvated building blocks. However, in solvent systems with lower polarity, the relatively weaker solvation effect facilitates the initial formation of building block dimers followed by further assembly into supramolecular structures. Therefore, we may assume that the self-assembled layers generated from the initially obtained dimer within a polar solvent undergo a layer closure process, which could lead to tubular structures. This scroll-up process operates in a preferred direction, which can minimize the surface energy and form a thermodynamically stable nanotube. Similarly, XRD analysis of the dried mass obtained from self-assembled PA3 in 100% aqueous medium exhibited diffraction peaks at 7.3° followed by other peaks in the wide angle region in a periodical order at 14.7°, 22.2° and 28.1° (Fig. S14†). The corresponding d-spacing values of these identified peaks obtained from Bragg's equation were 12.1 Å, 6.0 Å, 4.0 Å and 3.1 Å respectively; the ratios of 1/2, 1/3 and 1/4 support the formation of a tubular structure through lamellar molecular arrangement followed by the scroll-up process. Fig. 4K represents the fabrication of nanorods and tubular structure by the self-assembly of PA3 in different solvent media through an intermediate lamellar molecular arrangement followed by layer closure or the scroll-up process.
These results clearly suggest that the precise organization of these building blocks could lead to various supramolecular structures due to the geometrically restricted interactions of the aromatic moieties and their complex hydrophobic and electrostatic nature (Fig. 4J). These can significantly impact the organization of the assembly formed by an ultra-short peptide.
We have also performed an in silico investigation to predict the most favourable conformer of the monomer for the supramolecular assembly. It was found that in the case of PA1 and PA2 with flexible ethylenediamine and succinic acid, respectively, there exist several interactions (Fig. 5A and B) in between the dipeptide FF moieties of both sides of the connecting linker. However, for PA3 with the non-flexible linker (terephthalaldehyde), the dipeptide FF units are far away (Fig. 5C). This arrangement may prefer intermolecular π–π stacking interactions followed by lamellar molecular arrangement. The optimization of the dimer of each of the systems starting from a minimum energy structure was also carried out to characterize the possible non-covalent interaction patterns between the monomeric building blocks in their self-assembled state. Non-covalent interactions were predominantly responsible for dimer stability. The computed stabilization energy was highest for PA3 dimers (∼41 kcal mol−1) and lowest for PA1 dimers (∼30 kcal mol−1). This is consistent with the structural analysis, which showed that building blocks with flexible linkers (PA1 and PA2) are more closed and have less interacting ability with other molecules, whereas, for PA3 with a non-flexible linker, dimers are more open and able to intermingle with surrounding molecules. The exact nature of these intermolecular non-covalent interactions influences the supramolecular arrangements of each dipeptide, though the precise origin of specific self-assembled structures remains to be determined. Further computational investigations on the nature of the bulk system of these building blocks are ongoing to provide insight into the molecular determinants of each self-assembled structure. The corresponding optimized dimer structures have been presented at the right column of Fig. 5.
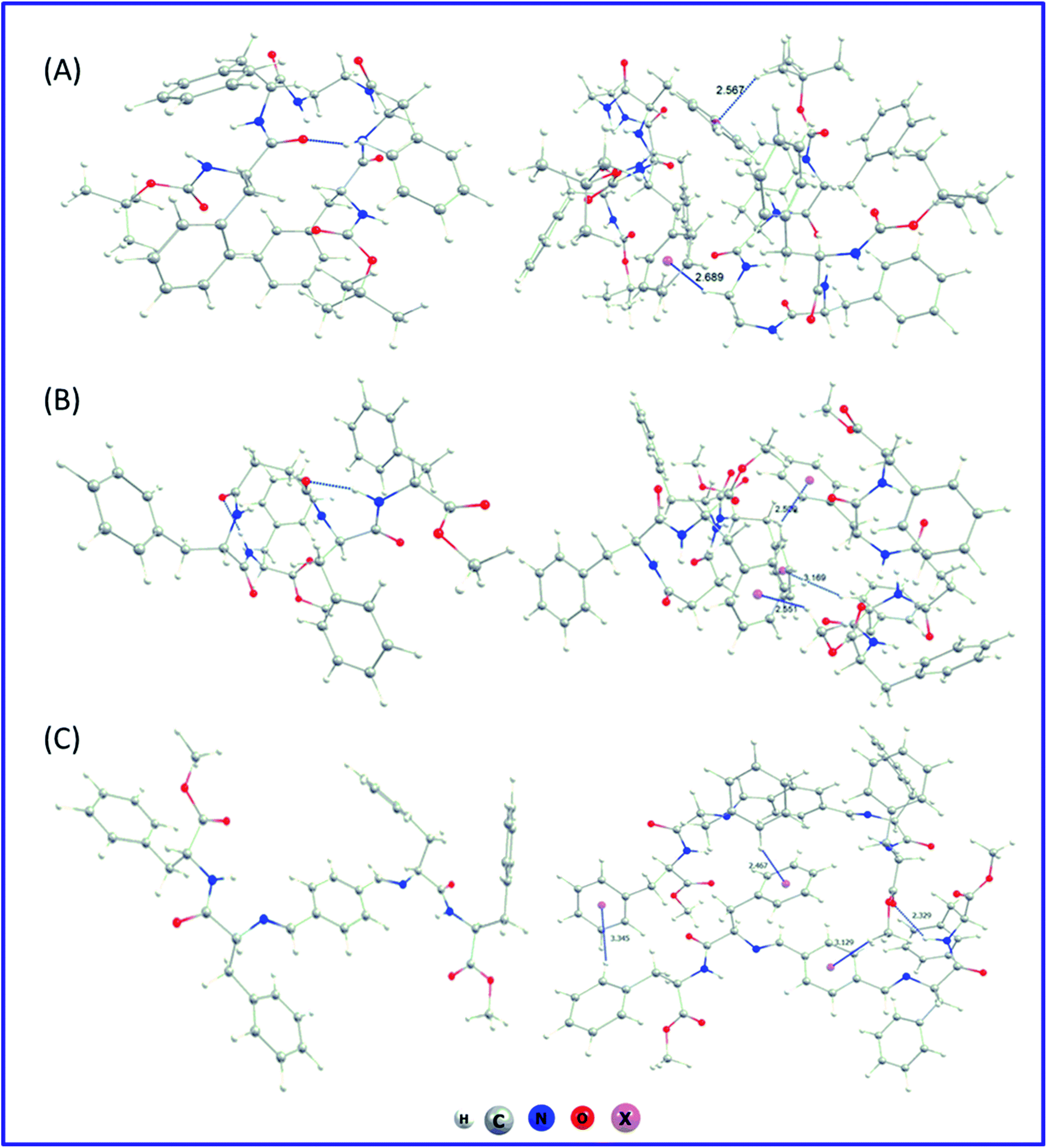 | ||
| Fig. 5 The best possible conformers (left-column) and optimized geometry of the dimers for (A) PA1, (B) PA2 and (C) PA3 (right-column) computed at the wB97XD/6-31G(d) level of theory. The blue dotted line shows the presence of non-covalent interactions. The dummy atom x is shown to focus the centre of the aromatic ring. | ||
We recorded the UV-Vis absorption spectra of PA1, PA2 and PA3. The UV-Vis absorption spectra of PA1 exhibit absorption peaks at 212 nm and 305 nm (Fig. 6A). PA2 showed absorption peaks at 250 nm, 258 nm and 264 nm (Fig. 6B), which could be attributed or preliminary assigned to the n–π*/π–π* and spin allowed intramolecular charge transfer. For PA3, the absorption maxima appeared at 344 nm along with a shoulder peak at 286 nm (Fig. 6C). Both peaks could be attributed to intramolecular charge transfer bands while a weaker n–π* transition was submerged under a strong π–π* transition and appeared as a shoulder peak at 286 nm.32
 | ||
| Fig. 6 UV-Vis absorption spectra of (A) PA1 (B) PA2 and (C) PA3 in 50% aqueous ethanol. Emission spectra of the peptides (monomer and self-assembled in 50% aqueous ethanol medium, 2 mg mL−1): (D) PA1, (E) PA2 and (F) PA3. Protease stability analysis of (G) PA1 (2 mg mL−1, λMon = 394 nm), (H) PA2 (2 mg mL−1, λMon = 360 nm) and (I) PA3 (2 mg mL−1, λMon = 437 nm) using different concentrations of Pronase from Streptomyces griseus. | ||
For preliminary understanding of the self-assembly process, we also recorded the emission spectra of these building blocks (PA1, PA2 and PA3) in both the monomeric and self-assembled state. The emission spectral analysis showed that, for PA1 in the monomeric state, the emission maxima appeared at 368 nm, while in the case of the self-assembled state it was red shifted (∼26 nm) and appeared at 394 nm (Fig. 6D). PA2 in the monomeric state exhibited a strong emission peak at 284 nm, but after self-assembly two red shifted emission maxima appeared at 294 nm and 360 nm (Fig. 6E). For PA3, the emission spectra of the monomeric state showed a characteristic emission peak at 374 nm. In the self-assembled state, the emission maxima were red shifted (∼63 nm) and appeared at 437 nm (Fig. 6F). It is already well known that self-assembly can drive the formation of fluorescent supramolecular architectures.10c However, the fluorescence is often suppressed due to the development of unfavourable excimers and exciplexes when the fluorophores are aggregated and condensed, a process termed Aggregation Caused Quenching (ACQ).33 Aggregation induced emission (AIE) by organic building blocks has also been described wherein subunits are non-fluorescent in the monomeric state but emit strong emission upon self-assembly in a polar solvent with red shifted emission maxima.34 The emission spectral analysis clearly revealed that in a polar solvent for all the three newly synthesized building blocks (PA1, PA2 and PA3) the emission maxima in the self-assembled state experienced a bathochromic shift when compared to the monomeric state. For PA3 in the self-assembled state, the emission maxima experienced a red shift of ∼63 nm when compared with the monomeric state and appeared at 437 nm, which can be attributed to π–π* stacking interactions of the self-assembled nanostructure. The self-assembled nanorods were further examined using a fluorescence microscope. As shown in Fig. 7A and B a characteristic blue fluorescence signal was observed for the nanorods obtained by the self-assembly of PA3 in aqueous ethanol.
 | ||
| Fig. 7 Fluorescence microscopic images of self-assembled nanorods (A, B) obtained from PA3. Fluorescence microscopic images of doxorubicin (DOX)-encapsulated spherical assemblies generated from PA1 (C, D) and nanorods from PA3 (E, F) showing the absolute morphological nature with high aspect ratios and with the characteristic fluorescence suggesting the presence of DOX across the self-assembled structures at the microscale. The plot of % of drug release from the drug encapsulated self-assembled spherical structures obtained from peptide PA1 (G) and nanorods obtained from peptide PA3 (H) with time; λMon = 590 nm, λExt = 490 nm. | ||
We also investigated the protease stability of these supramolecular structures formed by the self-assembly of PA1, PA2 and PA3 using Pronase from Streptomyces griseus (i.e. a mixture of endo- and exopeptidase capable of hydrolysing standard peptide bonds). Solutions of self-assembled structures (spheres from PA1, nanosheets from PA2 and nanorods from PA3) in PBS buffer (pH = 7.2) were incubated in the absence and presence of Pronase at 25 °C and fluorescence measurements performed at 1 hour intervals for 12 hours. Different Pronase concentrations (0.1 to 5.0 mg mL−1) were tested. The fluorescence measurement analysis revealed a steady decrease in the emission intensity with increasing time (Fig. 6G–I). This is due to the diminution in the concentration of the self-assembled peptides owing to the hydrolysis of the peptide bond by Pronase.35 High Pronase concentration was required to trigger a considerable fall in the emission intensity. A ∼50% decrease in emission intensity was observed at 5 mg mL−1 and after 12 hours (Fig. 6G–I).
We next sought to investigate the potential utility of our novel supramolecular architectures by assessing the potential of PA1 nanospheres and PA3 nanorods as drug delivery platforms. For this purpose, we self-assembled PA1 and PA3 in the presence of a therapeutically important anticancer chemotherapeutic drug, doxorubicin (DOX), in 50% aqueous ethanol generating DOX-PA1 and DOX-PA3, in which the emissive drug was encapsulated within the self-assembled structures. DOX is particularly useful for assessing the efficiency of intracellular drug delivery due to its intrinsic fluorescence. Fig. 7C, D and E, F clearly display the encapsulation of DOX into self-assembled structures formed by PA1 and PA3. The calculated encapsulation efficiencies (EEs) were 46.58% for the spherical assembly (PA1) and 51.71% for nanorods (PA3), while the loading capacities (LCs) were 12.65% (for the spherical assembly) and 14.0% (for nanorods). The drug release capability of these DOX encapsulated self-assembled structures (DOX-PA1 and DOX-PA3) was then evaluated by monitoring the steady state fluorescence. DOX-PA1 and DOX-PA3 were dispersed in saline buffer at RT and relocated to a dialysis bag (MWCO 3 kDa). The emission intensity of the aliquots was monitored over 70 h for DOX-PA1 and 80 h for DOX-PA3. We observed a steady/gradual increase in the emission intensity with time, clearly demonstrating an increase in the concentration of the DOX in the buffer medium (outside the dialysis bag) due to the release of encapsulated drug molecules. The emission intensity was recorded after 1 h followed by 2 h, 3 h and 4 h time intervals. A steady increase in the emission intensity up to 48 h for DOX-PA1 and 60 h for DOX-PA3 was observed after which there was no significant change in emission intensity, suggesting that the drug release process was completed and reached equilibrium. In order to confirm this hypothesis, the dialysis process continued up to 70 h for DOX-PA1 and 80 h for DOX-PA3 at which point a plateau was reached (Fig. 7G and H). Furthermore, DLS analysis revealed that after DOX encapsulation the average diameter of the spherical structures obtained from PA1 was 1033.23 ± 32.81 nm (Fig. S15†). Thus, there was no considerable alteration in the size of the self-assembled structures before and after DOX encapsulation. The rectangular nanosheets and nanobelts obtained from PA2 were not employed for the drug encapsulation and release study due to their larger dimensions (shape and size).
Hydrophobic peptides, which have a polar residue carrying little net charge or hydrophobic amino acid, are vital for cellular uptake. Herein, we have designed and synthesized peptides with the hydrophobic diphenylalanine moiety (N or C terminus protected), which should allow for translocation across cell membranes and promote cellular uptake. Therefore, these hydrophobic self-assembled nanoaggregates may function as a drug delivery platform.
The cytotoxicity of the self-assembled structures generated from PA1, PA2 and PA3 was tested in the normal human kidney cell line HEK293 by conventional MTT assays. Cell proliferation was estimated after 24 h of incubation following a standard protocol (details in the Experimental section†). After 24 h of incubation with PA1 spheres, PA2 nanosheets, or PA3 nanorods, a significant loss in cell viability was only observed at 100 μM (for PA1 spheres), a concentration well above what would typically be employed in vivo. Thus, at concentrations up to 75 μM, these compounds appear to be biocompatible (Fig. 8A and S16†). The non-cytotoxic nature as well as their propensity for drug encapsulation and release gives rise to the possibility that the PA1, PA2, and PA3 based self-assembled structures could be used for intracellular drug delivery. Based on the morphology, we employed the spherical assembly (from PA1) and nanorods (from PA3) due to their appropriate shape and size used for drug encapsulation and the drug release study. In this context, we next examined the cell diffusion capability of these supramolecular assemblies. HEK293 cells take up DOX-PA1 and DOX-PA3 within 6 h of the incubation as indicated by the red fluorescence in Fig. 8F and G. The DOX-encapsulated nanoparticles showed uniform distribution within cells and were able to release the incorporated drug molecule in a controlled manner resulting in an intracellular localization of the fluorescence intensities with uneven distribution, as shown by the merged image (Fig. 8H and I). These data provide a proof of concept that PA1 and PA3-based self-assembled structures might represent a novel platform for the development of drug delivery vehicles.
 | ||
| Fig. 8 (A) Cell viability was estimated by MTT assay. HEK293 cells were cultured in the presence of 0–100 μM of PA1 and PA3 at 37 °C for 24 h and after that the viability was assessed with a standard protocol. Fluorescence microscopy images of HEK293 cells treated for 6 h with DOX-PA1 and DOX-PA3. Bright field images (B, C) represent the morphology of the cells, nuclei stained with blue colour DAPI (D, E) and DOX incorporation in cells was captured in the red filter (F, G). The merged image is shown on the right (H, I). | ||
Conclusion
In summary, we have described the synthesis and self-assembly of three newly designed molecular building blocks, in which two diphenylalanine (the core recognition element of the β-amyloid polypeptide) units were connected through different linkers: ethylenediamine (PA1), succinic acid (PA2) and terephthalaldehyde (PA3). These dipeptide-based building blocks could be fabricated into different supramolecular architectures with various morphologies. Morphological transformation was also observed by altering the solvent polarity due to changes in non-covalent interactions. The mechanisms underlying the unique morphology of each self-assembled structure were investigated using various microscopic and spectroscopic techniques and their proteolytic stability was demonstrated by exposing the peptide-based structures to exogenous protease. Our results clearly demonstrate that the precise organization of these building blocks can generate unique supramolecular structures due to the geometrically restricted interactions of the aromatic moieties and their complex hydrophobic and electrostatic nature. Furthermore, the spherical assembly and the nanorods fabricated from PA1 and PA3 could encapsulate the cancer chemotherapeutic agent DOX and delivering the drug into the cell highlighting their potential as drug delivery vehicles.Materials and methods
All chemicals and solvents are commercially available and were used as received without further purification. Amino acid phenylalanine and N,N′-dicyclohexyl-carbodiimide, di-tert-butyl dicarbonate, 1-hydroxybenzotriazole, trimethylchlorosilane, and triethyl amine were purchased from Sisco Research Laboratories (SRL) Pvt. Ltd. Potassium hydrogen sulfate was purchased from Loba Chemie Pvt. Ltd. Trifluoro acetic acid was purchased from Finar Ltd. Terephthalaldehyde, ethylenediamine and succinic acid were purchased from Sigma Aldrich. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFP) was purchased from Tokyo Chemical Industry Co. Ltd. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Sigma (St Louis, MO, USA) and other regular laboratory chemicals were from Sisco Research Laboratory and Loba Chemicals (Mumbai, MH, India). DMEM and FBS were from Himedia Laboratories (Mumbai, MH, India).Synthesis of PA1, PA2 and PA3
The detailed protocol utilized to synthesize PA1, PA2 and PA3 is available in the ESI Experimental section (Schemes S1–S4 and Fig. S1–S9†).Self-assembly of PA1, PA2 and PA3
A fresh stock solution of peptides was prepared by dissolving the lyophilized forms of PA1, PA2 and PA3 in HFP to a concentration of 100 mg mL−1. We then blended these peptide solutions in several different proportions and diluted them with 50% aqueous ethanol and 100% distilled water to get the desired concentrations of these peptide based building blocks for self-assembly. The polarized solvent allowed the molecules to self-assemble.Field emission scanning electron microscopy (FE-SEM)
A 10 μL drop of a self-assembled solution of PA1, PA2 and PA3 in different solvent media was placed on a glass cover slip and allowed to dry at RT. The substrates were then coated with gold using a Leica EM ACE200 2–3 nm gold coater. SEM analysis was performed using a field emission scanning electron microscope (FE-SEM, JEOL JSM-7100F) operating at 18 kV.Transmission electron microscopy (TEM)
A 10 μL drop of a self-assembled solution of PA1, PA2 and PA3 was placed on a 200-mesh copper grid, covered by carbon-stabilized Formvars film. After 1 min, excess fluid was removed from the grid. The samples were analyzed using a transmission electron microscope, JEOL-JEM-2100 Plus (high resolution scintillator) operating at 200 kV.Microanalysis
C, H, N analysis was performed using a Vario Micro Cube (Elementar) instrument.Fourier transform infrared spectroscopy (FT-IR)
Fourier transform infrared spectra were recorded using an IRTracer-100 FT-IR spectrometer (Shimadzu) with a Deuterated Lanthanum α-Alanine doped TriGlycine Sulphate (DLaTGS) detector. These peptide-based building block self-assembly solutions were deposited on a CaF2 window and dried under vacuum. The peptide deposits were resuspended with D2O and subsequently dried to form thin films. The re-suspension procedure was repeated twice to ensure maximal hydrogen-to-deuterium exchange. The measurements were taken using 4 cm−1 resolutions and an average of 1000 scans. The transmittance minimal values were determined using the Lab solutions IR analysis program (IR Tracer).UV-Vis spectroscopy
UV-Vis absorption spectra of the synthesized building blocks were recorded using a UV/Vis spectrophotometer (Agilent, Cary 5000, Double beam UV-Vis absorption spectrometer).Fluorescence spectroscopy
Fluorescence measurements were performed at RT using a fluorescence spectrometer (Fluorolog, HORIBA). The emission spectra of the synthesized molecular building blocks were recorded using suitable excitation wavelengths. For the drug encapsulation and drug release study, the emission spectra were collected from 500 to 750 nm with an excitation wavelength of 490 nm. Both the excitation and emission slit widths were set to 2.0 nm. The fluorescence intensity at 590 nm was used for quantitative analysis.Drug encapsulation and drug release of PA1 and PA3 self-assembled structures
The incorporation of doxorubicin (DOX) was conducted during the self-assembly of PA1 and PA3. DOX at a concentration of 10−1 mol L−1 (dissolved in ethanol and water in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) was added to PA1 and PA3 (dissolved in HFP at 100 mg mL−1 concentration) at a desired dilution (2 mg mL−1, effective concentration) and the mixture incubated overnight. This mixture underwent spontaneous encapsulation of the drug molecule within the PA1-based spheres and PA3-based nanorods. Following conjugation, samples were prepared by drop casting (25 μL) of the conjugate mixture (drug encapsulated self-assembly) on a glass coverslip and dried in air. The remaining solvent was left to dry overnight at RT under vacuum. Then assemblies were washed carefully with ultrapure distilled water several times to remove the unbound or free DOX and dried properly at RT. The distinct morphologies of fluorescent DOX incorporated PA1 based spherical assemblies and PA3 based nanorods were characterized by fluorescence microscopy. The drug encapsulated spheres and nanorods were prepared by the above-mentioned protocol and kept for precipitation overnight. The solvent was decanted and the drug encapsulated self-assembled structures were re-dispersed in PBS (10 mM NaCl, pH = 7.4, 150 mM). After that, this suspension (2 mL) was transferred into a dialysis bag (MWCO 3 kDa), and the bag was dipped in 30 mL of PBS buffer at RT. The emission intensity of the buffer outside the dialysis bag was measured at different time intervals of 70 h (for spherical structures from PA1) and 80 h (nanorods from PA3). The volume of the solution was kept constant by adding 1 mL of the original PBS buffer solution after each measurement. The emission intensities were measured at RT using a fluorescence spectrometer. The emission spectra were recorded from 500 nm to 750 nm for the % drug release vs. time plot (λExt = 490 nm and λMon = 590 nm).
:1 ratio) was added to PA1 and PA3 (dissolved in HFP at 100 mg mL−1 concentration) at a desired dilution (2 mg mL−1, effective concentration) and the mixture incubated overnight. This mixture underwent spontaneous encapsulation of the drug molecule within the PA1-based spheres and PA3-based nanorods. Following conjugation, samples were prepared by drop casting (25 μL) of the conjugate mixture (drug encapsulated self-assembly) on a glass coverslip and dried in air. The remaining solvent was left to dry overnight at RT under vacuum. Then assemblies were washed carefully with ultrapure distilled water several times to remove the unbound or free DOX and dried properly at RT. The distinct morphologies of fluorescent DOX incorporated PA1 based spherical assemblies and PA3 based nanorods were characterized by fluorescence microscopy. The drug encapsulated spheres and nanorods were prepared by the above-mentioned protocol and kept for precipitation overnight. The solvent was decanted and the drug encapsulated self-assembled structures were re-dispersed in PBS (10 mM NaCl, pH = 7.4, 150 mM). After that, this suspension (2 mL) was transferred into a dialysis bag (MWCO 3 kDa), and the bag was dipped in 30 mL of PBS buffer at RT. The emission intensity of the buffer outside the dialysis bag was measured at different time intervals of 70 h (for spherical structures from PA1) and 80 h (nanorods from PA3). The volume of the solution was kept constant by adding 1 mL of the original PBS buffer solution after each measurement. The emission intensities were measured at RT using a fluorescence spectrometer. The emission spectra were recorded from 500 nm to 750 nm for the % drug release vs. time plot (λExt = 490 nm and λMon = 590 nm).
Drug encapsulation efficiency (EE) and loading capacity (LC)
DOX-PA1 and DOX-PA3 based assemblies were prepared by following the above-mentioned procedure and left to precipitate overnight. The aqueous medium was decanted and the emission intensity at the desired wavelength was measured. The drug encapsulation efficiency (EE), which is correlated with the concentration of the drug that was not incorporated or the free entrapped drug can be expressed by eqn (1).36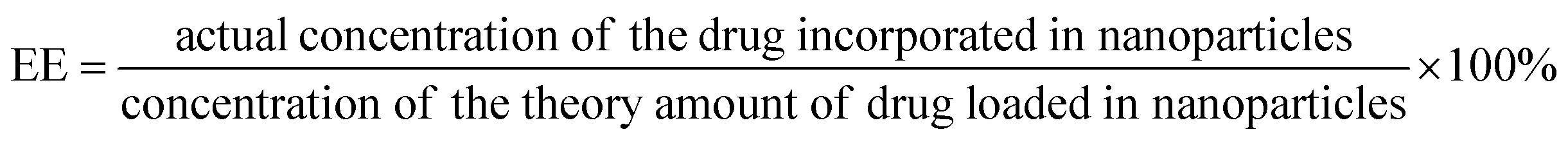 | (1) |
 | (2) |
 | (3) |
As the concentration of the drug is directly proportional to the emission intensity (eqn (2)), the emission of the drug incorporated in nanoparticles is equal to the total emission of the drug initially used subtracted by the emission intensity of the drug that was not incorporated, EE can be calculated using eqn (3):
The loading capacity (w/w % LC) can be calculated using the following expression:

The amount of the entrapped drug by the PA1 based spherical assembly is
| 0.543 × EE = 0.543 × 46.58% = 0.253 mg. |
| LC = (0.253/2) × 100% = 12.65% |
The amount of the entrapped drug by the PA3 based nanorods is
| 0.543 × EE = 0.543 × 51.71% = 0.280 mg. |
| LC = (0.280/2) × 100% = 14.0% |
Dynamic light scattering (DLS) analysis
Dynamic light scattering (DLS) analysis of the spherical assemblies formed by PA1 was performed using a Nano-zetasizer (Horiba SZ-100) and these measurements were performed at RT, 25 °C.X-ray diffraction (XRD) analysis
The PXRD pattern of the samples was recorded by using a PANalytical X'Pert Pro Powder X-ray diffractometer. Data collection was carried out at room temperature using Cu Kα radiation (1.5406 Å; 40 kV, 30 mA) as the X-ray source in 2θ continuous scan mode (Bragg–Brentano geometry) in the range of 5–30° at a scan rate of 1° min−1 and a time per step of 0.5 s.Thermogravimetric analysis (TGA)
Thermal analysis was carried out using a NETZSCH STA 449F3 TGA thermal analyzer with a heating rate of 10 °C min−1 in an N2 atmosphere.Cell culture
Normal kidney (HEK293) cells were procured from the National Centre for Cell Science (NCCS), Pune, India. The cells were cultured in DMEM media, supplemented with 10% FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. The cells were grown at 37 °C and 5% CO2.MTT assay
The MTT assay was performed to assess the effect of the test moieties on cellular proliferation. Cells were seeded at a density of 5 × 104 per well in 48 well plates and cultured with DMEM supplemented with 10% FCS with the media replaced after 24 h. Control cells at 70% confluence were treated with either vehicle (DMSO, 0.1%) or test compounds at various concentrations (2.5–100 μM) for 36 h. The MTT solution was prepared at a concentration of 1 mg mL−1 in medium without phenol red and 200 μL of MTT solution was added into each well after removing the previous culture medium. Cells were then incubated for at least 2 h at 37 °C with MTT solution and the reaction was stopped with 200 μL of DMSO for solubilization of the formazan crystals. The optical density of the wells was determined using a plate reader (Biotek Instrument) at a wavelength of 550 nm.Fluorescence imaging
For this experiment, we used HEK293 cells plated on sterile cover slips in 6 well plates with DMEM containing 10% FCS. After 24 h, the medium was removed and test compounds containing DMEM medium were added. After 6 h, the cells were washed three times with chilled PBS, fixed in 4% paraformaldehyde at RT for 10 min and mounted using Vectashield mounting media (Invitrogen). Cells were then analyzed by fluorescence microscopy (Optika B-100FL HBO, Italy) for morphological analysis and labelled fluorescence.Statistical analysis
Data were analyzed by one-way ANOVA. Statistical analyses were performed using Origin 6.1 software. Results were considered significantly different at p < 0.05. Values are expressed as means ± SEM.Computational details
We studied the assembly of PA1, PA2 and PA3. As these building blocks are quite large (more than 100 atoms), they likely have several possible conformations. First, to find out the best possible conformer we rigorously scanned possible conformations for each of the three molecules. An unconstrained molecular dynamics conformational search was performed using the Verlet velocity algorithm and NVE thermostat with other implemented parameters in Gabedit V.2.5.0 (A. R. Allouche, Gabedit, http://gabedit.sourceforge.net/). The minimum energy geometries were gleaned with the PM6 semi-empirical method using the program MOPAC 2009 (J. J. P. Stewart, MOPAC2009, Stewart Computational Chemistry, version 9.259W, http://openmopac.net/). For further authentication of the conformational analysis, the 10 lowest minimum structures were considered and optimized using density functional theory. The geometries of all the molecular species involved in this study are fully optimized at the wB97XD/6-31G(d) level of theory. wB97XD is a hybrid meta-GGA function37 containing Grimme's D2 empirical dispersion terms38 and the long-range corrections.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
P. D. acknowledges Council of Scientific and Industrial Research (CSIR), India for research funding (File No. 01(3077)/21/EMR-II) and the support of the Interdisciplinary Institute of Indian System of Medicine, SRM IST and Nano Research Centre (NRC), SRM IST for several characterization studies. S. G. thanks Science and Engineering Research Board, Department of Science and Technology, Government of India for Core Research Grant (File No. CRG/2020/000885). B. M. thanks the financial support from Indian Council of Medical Research (ICMR, No. 5/13/52/2015-NCD-III) and DST-SERB (EMR/2016/006873) for conducting the research.Notes and references
- (a) L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674–1684 CrossRef CAS PubMed; (b) A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS PubMed; (c) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (d) X. J. Zhao and S. G. Zhang, Chem. Soc. Rev., 2006, 35, 1105–1110 RSC.
- (a) M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed; (b) A. Aggeli, M. Bell, N. Boden, L. M. Carrick and A. E. Strong, Angew. Chem., Int. Ed., 2003, 42, 5603–5606 CrossRef CAS PubMed; (c) G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed; (d) M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef PubMed.
- (a) Q. He, L. Duan, W. Qi, K. W. Wang, Y. Cui, X. H. Yan and J. B. Li, Adv. Mater., 2008, 20, 2933–2937 CrossRef CAS; (b) E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC; (c) R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC; (d) S. G. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- (a) S. G. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed; (b) J. Li, Z. Chen, X. Wang, G. Brezesinski and H. Mohwald, Angew. Chem., Int. Ed., 2000, 39, 3059–3062 CrossRef CAS.
- (a) M. Yemini, M. Reches, J. Rishpon and E. Gazit, Nano Lett., 2005, 5, 183–186 CrossRef CAS PubMed; (b) V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. E. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS; (c) P. Das, I. Pan, E. Cohen and M. Reches, J. Mater. Chem. B, 2018, 6, 8228–8237 RSC; (d) E. P. Holowka, D. J. Prochain and T. J. Deming, J. Am. Chem. Soc., 2005, 127, 12423–12428 CrossRef CAS PubMed; (e) S. Ghosh, M. Reches, E. Gazit and S. Verma, Angew. Chem., Int. Ed., 2007, 46, 2002–2004 CrossRef CAS PubMed; (f) H. Zhang, J. Fei, X. Yan, A. Wang and J. Li, Adv. Funct. Mater., 2015, 25, 1193–1204 CrossRef CAS.
- (a) L. Adler-Abramovicha and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC; (b) E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC; (c) K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935–3953 RSC.
- (a) Z. M. Yang, K. M. Xu, L. Wang, H. W. Gu, H. Wei, M. J. Zhang and B. Xu, Chem. Commun., 2005, 4414–4416 RSC; (b) M. Reches and E. Gazit, Nano Lett., 2004, 4, 581–585 CrossRef CAS; (c) Z. A. C. Schnepp, R. Gonzalez-McQuire and S. Mann, Adv. Mater., 2006, 18, 1869–1872 CrossRef CAS.
- (a) N. Kol, L. Adler-Abramovich, D. Barlam, R. Z. Shneck, E. Gazit and I. Rousso, Nano Lett., 2005, 5, 1343–1346 CrossRef CAS PubMed; (b) S. Sivagnanam, A. Arul, S. Ghosh, A. Dey, S. Ghorai and P. Das, Mater. Chem. Front., 2019, 3, 2110–2119 RSC; (c) L. Adler-Abramovich, M. Reches, V. L. Sedman, S. Allen, S. J. Tendler and E. Gazit, Langmuir, 2006, 22, 1313–1320 CrossRef CAS PubMed; (d) I. Azuri, L. Adler-Abramovich, E. Gazit, O. Hod and L. Kronik, J. Am. Chem. Soc., 2013, 136, 963–969 CrossRef PubMed.
- (a) M. Reches and E. Gazit, Nat. Nanotechnol., 2006, 1, 195–200 CrossRef CAS PubMed; (b) T. H. Han, W. J. Lee, D. H. Lee, J. E. Kim, E. Y. Choi and S. O. Kim, Adv. Mater., 2010, 22, 2060–2064 CrossRef CAS PubMed; (c) X. Yan, P. Zhua and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC; (d) P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chem.–Eur. J., 2010, 16, 3176–3183 CrossRef CAS PubMed.
- (a) C. Guo, Y. Luo, R. H. Zhou and G. H. Wei, ACS Nano, 2012, 6, 3907–3918 CrossRef CAS PubMed; (b) X. Yan, Y. Cui, Q. He, K. W. Wang and J. Li, Chem. Mater., 2008, 20, 1522–1526 CrossRef CAS; (c) N. Na, X. Mu, Q. Liu, J. Wen, F. Wang and J. Ouyang, Chem. Commun., 2013, 49, 10076–10078 RSC; (d) D. Datta, O. Tiwari and K. N. Ganesh, Nanoscale, 2018, 10, 3212–3224 RSC.
- (a) A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS; (b) M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed; (c) O. Carny, D. E. Shalev and E. Gazit, Nano Lett., 2006, 6, 1594–1597 CrossRef CAS PubMed; (d) X. Yan, Y. Cui, W. Qi, Y. Su, Y. Yang, Q. He and J. Li, Small, 2008, 4, 1687–1693 CrossRef CAS PubMed; (e) M. Yemini, M. Reches, E. Gazit and J. Rishpon, Anal. Chem., 2005, 77, 5155–5159 CrossRef CAS PubMed; (f) X. Yan, Q. He, K. Wang, L. Duan, Y. Cui and J. Li, Angew. Chem., Int. Ed., 2007, 46, 2431–2434 CrossRef CAS PubMed; (g) M. Lingenfelder, G. Tomba, G. Costantini, L. C. Ciacchi, A. De Vita and K. Kern, Angew. Chem., Int. Ed., 2007, 46, 4492–4495 CrossRef CAS PubMed.
- (a) M. Reches and E. Gazit, Phys. Biol., 2006, 3, S10–S19 CrossRef CAS PubMed; (b) M. Reches and E. Gazit, Isr. J. Chem., 2005, 45, 363–371 CrossRef CAS.
- L. Adler-Abramovich, N. Kol, I. Yanai, D. Barlam, R. Z. Shneck, E. Gazit and I. Rousso, Angew. Chem., Int. Ed., 2010, 49, 9939–9942 CrossRef CAS PubMed.
- X. Yan, Y. Cui, Q. He, K. Wang, J. Li, W. Mu, B. Wang and Z. Ou-Yang, Chem.–Eur. J., 2008, 14, 5974–5980 CrossRef CAS PubMed.
- P. Kumaraswamy, R. Lakshmanan, S. Sethuraman and U. M. Krishnan, Soft Matter, 2011, 7, 2744–2754 RSC.
- S. Yuran, Y. Razvag and M. Reches, ACS Nano, 2012, 6, 9559–9566 CrossRef CAS PubMed.
- E. Mayans, G. Ballano, J. Casanovas, A. Diaz, M. M. Perez-Madrigal, F. Estrany, J. Puiggali, C. Cativiela and C. Aleman, Chem.–Eur. J., 2015, 21, 16895–16905 CrossRef CAS PubMed.
- H. Choi, H. Park, K. Son, H. M. Kim and Y. Jung, Chem. Sci., 2019, 10, 10428–10435 RSC.
- (a) N. P. King and Y. T. Lai, Curr. Opin. Struct. Biol., 2013, 23, 632–638 CrossRef CAS PubMed; (b) Y. T. Lai, E. Reading, G. L. Hura, K. L. Tsai, A. Laganowsky, F. J. Asturias, J. A. Tainer, C. V. Robinson and T. O. Yeates, Nat. Chem., 2014, 6, 1065–1071 CrossRef CAS PubMed.
- P. Das and M. Reches, Nanoscale, 2016, 8, 9527–9536 RSC.
- M. Slyngborg and P. Fojan, Phys. Chem. Chem. Phys., 2015, 17, 30023–30036 RSC.
- W. J. M. FrederixPim, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- (a) Y. Zhou and T. Shimizu, Chem. Mater., 2008, 20, 625–633 CrossRef CAS; (b) S. I. Stupp, V. L. Bonheur, K. Walker, L. S. Li, K. E. Huggins, M. Keser and A. Amstuts, Science, 1997, 276, 384–389 CrossRef CAS PubMed; (c) X. Gao and H. Matsui, Adv. Mater., 2005, 17, 2037–2050 CrossRef CAS PubMed.
- A. Maity, A. Dey, M. Gangopadhyay and A. Das, Nanoscale, 2018, 10, 1464–1473 RSC.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS PubMed.
- (a) D. M. Byler and H. Susi, Biopolymers, 1986, 25, 469–487 CrossRef CAS PubMed; (b) L. M. Gordon, P. W. Mobley, R. Pilpa, M. A. Sherman and A. J. Waring, Biochim. Biophys. Acta, Biomembr., 2002, 1559, 96–120 CrossRef CAS.
- M. Gupta, A. Bagaria, A. Mishra, P. Mathur, A. Basu, S. Ramakumar and V. S. Chauhan, Adv. Mater., 2007, 19, 858–861 CrossRef CAS.
- K. A. Feeney, N. Wellner, S. M. Gilbert, N. G. Halford, A. S. Tatham, P. R. Shewry and P. S. Belton, Biopolymers, 2003, 72, 123–131 CrossRef CAS PubMed.
- C. A. E. Hauser, R. Deng, A. Mishra, Y. Loo, U. Khoe, F. Zhuang, D. W. Cheong, A. Accardo, M. B. Sullivan, C. Riekel, J. Y. Ying and U. A. Hauser, Proc. Natl. Acad. Sci. U. S. A., 2011, 25, 1361–1366 CrossRef PubMed.
- T. Govindaraju, M. Pandeeshwar, K. Jayaramulu, G. Jaipuria and H. S. Atreya, Supramol. Chem., 2011, 23, 487–492 CrossRef CAS.
- J. Kim, T. H. Han, Y. I. Kim, J. S. Park, J. Choi, D. G. Churchill, S. O. Kim and H. Ihee, Adv. Mater., 2010, 22, 583–587 CrossRef CAS PubMed.
- (a) P. Das, A. Ghosh, M. K. Kesharwani, V. Ramu, B. Ganguly and A. Das, Eur. J. Inorg. Chem., 2011, 20, 3050–3058 CrossRef; (b) A. Trummal, L. Lipping, I. Kaljurand, I. A. Koppel and I. Leito, J. Phys. Chem. A, 2016, 120, 3663–3669 CrossRef CAS PubMed.
- (a) M. Belletete, J. Bouchard, M. Leclerc and G. Durocher, Macromolecules, 2005, 38, 880–887 CrossRef CAS; (b) W. Z. Yuan, P. Lu, S. M. Chen, J. W. Y. Lam, Z. M. Wang, Y. Liu, H. S. Kwok, Y. G. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- R. Bucci, P. Das, F. Iannuzzi, M. Feligioni, R. Gandolfi, M. L. Gelmi, M. Reches and S. Pellegrino, Org. Biomol. Chem., 2017, 15, 6773–6779 RSC.
- (a) Z. Zhang and S. S. Feng, Biomaterials, 2006, 27, 4025–4033 CrossRef CAS PubMed; (b) B. Chu, L. Zhang, Y. Qu, X. Chen, J. Peng, Y. Huang and Z. Qian, Sci. Rep., 2016, 6, 34069–34078 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00510c |
| This journal is © The Royal Society of Chemistry 2021 |