
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of morphology variations in colloidal CuGaS2 nanorods
Logan
Keating
 and
Moonsub
Shim
*
and
Moonsub
Shim
*
Department of Materials Science and Engineering, Frederick Seitz Materials Research Laboratory, University of Illinois, Urbana, Illinois 61801, USA. E-mail: mshim@illinois.edu
First published on 4th August 2021
Abstract
Cu2−xS nanocrystals can serve as templates and intermediates in the synthesis of a wide range of nanocrystals through seeded growth, cation exchange, and/or catalytic growth. This versatility can facilitate and accelerate the search for environmentally benign nanocrystals of high performance with variable shapes, sizes, and composition. However, expanding the compositional space via Cu2−xS nanocrystals while achieving necessary uniformity requires an improved understanding of the growth mechanisms. Herein we address several unusual and previously unexplained aspects of the growth of CuGaS2 nanorods from Cu2−xS seeds as an example. In particular, we address the origin of the diverse morphologies which manifest from a relatively homogeneous starting mixture. We find that CuGaS2 nanorods start as Cu2−xS/CuGaS2 Janus particles, the majority of which have a {10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 2}/{102} interface that helps to minimize lattice strain. We propose a mechanism that involves concurrent seed growth and cation exchange (CSC), where epitaxial growth of the Cu2−xS seed, rather than the anticipated catalytic or seeded growth of CuGaS2, occurs along with cation exchange that converts growing Cu2−xS to CuGaS2. This mechanism can explain the incorporation of the large number of anions needed to account for the order-of-magnitude volume increase upon CuGaS2 rod growth (which cannot be accounted for by the commonly assumed catalytic growth mechanism) and variations in morphology, including the pervasive tapering and growth direction change. Insights from the CSC growth mechanism also help to explain a previously puzzling phenomenon of regioselective nucleation of CuInSe2 on kinked CuGaS2 nanorods.
2}/{102} interface that helps to minimize lattice strain. We propose a mechanism that involves concurrent seed growth and cation exchange (CSC), where epitaxial growth of the Cu2−xS seed, rather than the anticipated catalytic or seeded growth of CuGaS2, occurs along with cation exchange that converts growing Cu2−xS to CuGaS2. This mechanism can explain the incorporation of the large number of anions needed to account for the order-of-magnitude volume increase upon CuGaS2 rod growth (which cannot be accounted for by the commonly assumed catalytic growth mechanism) and variations in morphology, including the pervasive tapering and growth direction change. Insights from the CSC growth mechanism also help to explain a previously puzzling phenomenon of regioselective nucleation of CuInSe2 on kinked CuGaS2 nanorods.
Introduction
Colloidal semiconductor nanocrystals are materials of broad interest, finding applications in multiple areas including sensors,1–3 luminescent solar concentrators,4,5 displays,6–8 and biomedical imaging.9,10 Anisotropic shapes and heterostructures provide additional benefits, such as polarized light emission,11–13 improved and/or directional conductivity, and the potential for multi-functional devices.14 For example, double-heterojunction nanorods have enabled simultaneous light emission and detection capabilities.14 However, high quality colloidal quantum dots and their anisotropic analogs are currently dominated by Cd-based II–VI materials, which face usage restrictions in consumer products.15 Alternative materials which do not face these restrictions are being widely pursued.Among Cd-free compositions being actively sought, materials synthesized through Cu2−xS templates or intermediates are promising. Cu2−xS nanocrystals are thought to undergo a combination of catalytic growth and cation exchange in addition to acting as seeds for heteroepitaxy to provide a wide range of compositions and morphologies.16 The high cation conductivity of Cu2−xS at temperatures greater than 105 °C (ref. 17 and 18) means that there are a multitude of different materials which can be accessed from a relatively simple starting material.19,20 In addition to the variety of compositions accessible, many different morphologies have also been observed for the same material. For example, restricting to CuGaS2, there are reports of at least 6 different variations on the common dot, platelet, and rod shapes, starting from Cu2−xS seeds.21–26 This combination of compositional and morphological diversity suggests that the Cu2−xS system may be ideal for tailoring nanocrystals for desired applications. However, a better understanding of how this diversity arises is needed for developing strategies to control morphology and composition, a prerequisite for most applications.
Even within the nanorod (NR) shape, CuGaS2 exhibits variations in morphology. These variations arise despite early growth periods exhibiting relatively uniform NRs (Fig. 1a). Nevertheless, there are common features, in particular, kinks and tapering ends, that might give insights into the growth mechanism. These morphological variations are also one of the hallmarks of NRs grown from Cu2−xS seeds, and has been observed in other related systems.27 As an example, tapering has been observed in the CuInS2 system,28,29 and kinking has been observed in several other systems which are thought to employ solid catalyst particles.30,31 These reports indicate that kinking and tapering are not unique to the Cu2−xS/CuGaS2 system. Hence, addressing how kinks and tapering arise may lead to the understanding necessary to achieve uniformity in shape and size of a wide range of materials synthesized through Cu2−xS seed-mediated routes.
 | ||
| Fig. 1 Transmission electron microscopy (TEM) images of CuGaS2 NR growth from aliquots taken at 0 s, 30 s, 1 min, 2 min, and 5 min. Darker contrast corresponds to Cu2−xS and lighter contrast to CuGaS2. From 0 s to 1 min (a–c), the morphology remains relatively uniform, with spherical Janus particles transitioning to rods as growth proceeds. From 1 min to 2 min (c and d), the predominant morphology is still “straight” NRs, though there is some indication of growth direction differentiation and tapering toward the Cu2−xS tip. By 5 min (e), the majority of NRs exhibit some form of a “kink” with pronounced tapering. | ||
One often-suggested mechanism of Cu2−xS seed-mediated growth of various materials, including CuGaS2 NRs, is the catalytic growth where Cu2−xS particles act as catalysts and supersaturation of precursors in these particles leads to precipitation of the second desired phase.32 Intermediate structures such as those seen in Fig. 1b–d readily invoke a mechanism similar to the well-known Vapor–Liquid–Solid (VLS) nanowire growth.33 In solution synthesis, the analogous mechanism is the Solution–Liquid–Solid (SLS) growth.34,35 Both of these growth mechanisms involve relatively low-melting metal catalyst particles. While Cu2−xS has a high melting point and remains solid at typical reaction temperatures,36 the cation sub-lattice is essentially in the liquid phase.17 Hence, an analog of SLS growth, sometimes referred to as Solution–Solid–Solid (SSS) growth, has been introduced.32 In this mechanism, the precursors are dissolved in solid, rather than liquid, catalyst particles with highly mobile cations. While it is reasonable that the additional cations can be readily incorporated (and replaced, as in cation exchange), there is no obvious means of incorporating additional anions. Yet an order of magnitude or larger volume increase from the initial Cu2−xS particles (Fig. 1a) to the final NRs (Fig. 1e) requires incorporation of a very large number of anions that is not accounted for. Alternative mechanisms proposed may include explanation of this anion sub-lattice extension but fail to explain frequently observed features such as kinks and tapering.
In this work, we systematically characterize and address the various morphologies observed in a typical CuGaS2 NR synthesis which proceeds from in situ generated Cu2−xS seeds. While NRs can initially grow in three different crystallographic directions, nearly all of them eventually return to the 〈0001〉 growth direction and exhibit tapering which occurs at the end of the growth. For convenience, crystallographic directions and planes given here and throughout refer to indices for the hexagonal anion sublattice for both Cu2−xS and CuGaS2. Based on our observations, we propose a growth mechanism where the Cu2−xS seeds grow epitaxially (that is, the growth of the Cu2−xS seed itself as opposed to the seeded heteroepitaxy of CuGaS2 on Cu2−xS seed) while cation exchange converts part of the growing seed to CuGaS2. This growth of Cu2−xS seeds rather than catalytic or SSS growth can easily account for the incorporation of a large number of anions needed to account for the volume change upon rod growth. As precursors deplete, cation exchange dominates over seed growth and the Cu2−xS tips become smaller and smaller until they are completely converted to CuGaS2, leading to the pervasive tapering. The three different initial growth directions lead to three primary NR morphologies, straight and two types of kinked (∼155° and 90°). Straight NRs grow along the 〈0001〉 direction. The two types of kinked NRs start to grow in the 〈102〉 or 〈100〉 direction but both return to the 〈0001〉 growth direction, which indicates that the kinking may be due to certain surface terminations being kinetically accessible at high precursor concentrations. Different surface terminations present in these kinked NRs help to explain the regioselective nucleation of CuInSe2 on CuGaS2 NRs.
Experimental section
Materials
Copper(I) acetate (CuOAc) (97%), gallium acetylacetonate (Ga(acac)3) (99.99%), 1-dodecanethiol (1-DDT) (98%), trioctylphosphine (TOP) (90%), indium(III) acetate (In(OAc)3) (99.99%), selenium (99.99%), and octadecene (90%) were obtained from Sigma Aldrich. All chemicals were used as received.Synthesis of CuGaS2 and CuGaS2/CuInSe2 nanorods
The CuGaS2 NRs were synthesized according to a previously reported procedure with minor modifications. To a 25 mL three-necked round bottom flask, CuOAc (24.5 mg, 0.2 mmol), Ga(acac)3 (73.4 mg, 0.2 mmol), and 1-DDT (5 mL) were added. The solution was stirred magnetically, and degassed under vacuum for approximately 15 s, at which point the flask was purged with Ar. The solution was then heated to 240 °C at a rate of 30 °C min−1. The yellow turbid solution became clear after which it turned to a yellow-orange suspension. Inclusion of a slight excess of CuOAc causes the solution to flash black at the thermolysis temperature of 1-DDT. This event was used as t = 0 for the reaction times indicated. The reaction was quenched via airjet to room temperature. A small amount of ethanol was added to the crude reaction mixture, and the solution was centrifuged at 2000 rpm until the nanorods aggregated and the solution was close to clear. The solvent was decanted, and the pellet was resuspended in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 chloroform:ethanol mixture, and centrifuged again until a pellet was formed. This process was repeated until the product had been centrifuged a total of 3 times. The resulting cleaned pellet was then resuspended in chloroform.
:2 chloroform:ethanol mixture, and centrifuged again until a pellet was formed. This process was repeated until the product had been centrifuged a total of 3 times. The resulting cleaned pellet was then resuspended in chloroform.
CuGaS2/CuInSe2 NRs were synthesized from purified CuGaS2 NRs according to a previously reported procedure.26 The purification of the NR heterostructures was also performed according to the same report.
Characterization
Samples for TEM were drop cast from purified solution on ultra-thin carbon grids from EMS Diasum (CF300-UL). High-resolution transmission electron microscopy (HRTEM) was performed on a JEOL 2011 LaB6. High-resolution scanning transmission electron microscopy (HRSTEM) measurements were carried out on a Thermo-Fisher Titan Z or a JEOL 2200FS.Results and discussion
A complete understanding of the mechanism which leads to the observed CuGaS2 morphologies requires consideration of many aspects of CuGaS2 synthesis, including the role of Cu2−xS seeds. We begin with a discussion on the synthetic conditions employed for CuGaS2 synthesis, followed by an exploration of the individual steps in NR growth, namely Janus particle formation, elongation, and tapering. We then consider the origin of the growth direction change. The effects of different growth directions, which lead to different surface terminations, are then discussed within the context of regioselective heterostructure nucleation.Synthesis of CuGaS2 NRs
The synthesis of CuGaS2 NRs is accomplished by thermolyzing 1-DDT in the presence of Cu and Ga precursors, where 1-DDT is the ligand, the solvent, and the anion precursor. Addition of 1-DDT to the initial reactants results in a yellow turbid solution which clears as it is heated and momentarily turns black around 235–240 °C, similar to the thermolysis temperature reported by other groups.37 We note that the degas time affects the growth rate of the NRs with longer degas times resulting in a slower growth rate. Additionally, the heating rate and the degree of temperature overshoot affect the growth rate with higher temperatures resulting in faster growth, as expected. Once thermolysis occurs, the solution then begins to turn yellow-orange as the growth of CuGaS2 NRs proceeds. Fig. 1 shows a time series of TEM micrographs, starting from the initial Cu2−xS nucleation, and proceeding to CuGaS2 NRs which are ∼100 nm in length. Ultraviolet-visible spectroscopy (UV-Vis), photoluminescence spectroscopy (PL), and X-ray diffraction (XRD) of ∼100 nm CuGaS2 NRs produced via this procedure have been reported in a previous work.26At the beginning of the synthesis Cu2−xS nanocrystals nucleate and grow but quickly convert to what appears to be a relatively monodisperse collection of Cu2−xS/CuGaS2 Janus particles (Fig. 1a). Rods of CuGaS2 then begin to extrude from these Janus particles (Fig. 1b). Continued growth leads to 3 distinct morphologies (Fig. 1c–e), all with some degree of tapering in the end where Cu2−xS is located and eventually lost (Fig. 1e). We refer to these morphologies as “straight”, “155°-kinked”, and “90°-bent”, with examples of 155°-kinked and 90°-bent NRs shown in Fig. 2a and b. All CuGaS2 NRs we have observed exhibit tapering (Fig. 2c). Straight NRs have their rod axis along 〈0001〉 direction. 155°-kinked NRs are characterized by two sections with distinct axes, which intersect to form a ∼155° angle. We note that while we identify the NRs as 155°-kinked, there is a distribution of observed angles. The 90°-bent rods have a broad flat section at the “head” of the rod, which is usually attached to a straight tapering NR. All of the final NRs appear to be single crystalline throughout. As shown in the FFT inset of Fig. 2a, the wurtzite-like phase is the dominant crystal phase observed and the NRs are single crystal in nature. Some NRs occasionally display stacking faults, however these domains are limited, and do not correlate to the observed morphology (i.e., the rods are wurtzite-like throughout rather than having two segments of different crystal phases with a change at the site of the kink). The diameter difference between the “head” and “tail” for a ∼100 nm long rod is typically 2.5 ± 1.4 nm. Tapering is observed to some extent in nearly all NRs after ∼2.5 min of growth and is pronounced in all NRs after 10 min.
 | ||
| Fig. 2 HRTEM images along with schematics of 155° (a) and 90° (b) “kinked” rods. The FFT in the inset of (a) demonstrates crystallinity. The same crystal phase is maintained before and after the kink in both cases. (c) HRTEM image and schematic of tapering, which is observed on all NRs, including straight NRs, after sufficient growth has occurred. Scale bars are 5 nm in all images. | ||
Cu2−xS/CuGaS2 Janus particle formation
The first step in CuGaS2 NR growth is the nucleation of a Cu2−xS seed, which is immediately followed by partial cation exchange to form a Cu2−xS/CuGaS2 Janus particle. When lattice fringes are visible throughout the entire particle in HRTEM, the darker contrast sections correspond to the expected lattice parameters for Cu2−xS (∼6.8 Å), and the lighter sections correspond to the expected parameters for wurtzite-like CuGaS2 (∼6.2 Å) as shown in Fig. 3a. | ||
| Fig. 3 (a) HRTEM image of a partially grown Cu2−xS/CuGaS2 NR showing the difference in lattice parameter. The 6.2 Å lattice parameter is consistent with the (0001) of CuGaS2, while the 6.8 Å lattice parameter is consistent with Cu2−xS (scale bar is 5 nm). (b) Illustrated HRTEM of an early growth Cu2−xS/CuGaS2 Janus particle showing how the interfacial angle between the two phases is defined with respect to (0001) plane (scale bar is 5 nm). (c) Detailed statistics of Cu2−xS/CuGaS2 interfacial angle for the {102} case, showing a distribution of values. | ||
Initial investigation of Janus particle formation revealed that there is a preferred crystallographic orientation of the interface between the Cu2−xS and CuGaS2 components. Fig. 3b shows an example particle, along with a guide to the eye indicating the measurement methodology. Statistical analysis of 150 particles with minimal elongation indicates that ∼83% of particles have an interface which is oriented between the {100} and {0001} planes. More detailed measurement of the interface angle was performed on these particles and the results are shown in Fig. 3c. The average of the most frequently observed angle between the Cu2−xS/CuGaS2 interface and the (0001) plane was 139 ± 10°, as shown in Fig. 3c. This angle is close to the ∼134° angle between the {102} and the {0001} expected for pure CuGaS2 and roxbyite or chalcocite phases of Cu2−xS. We note that the distribution skews towards higher angles and that it is difficult to determine this angle precisely for these small particles. Therefore, we assign the most frequently observed interface to be {102}/{102}. A recent work by the Schaak group has shown that the lattice strain between pseudo-roxbyite Cu2−xS and wurtzite-like CuGaS2 is minimized if the two anion lattices adopt an orientation close to {102}/{102}.19 Then, the observed preferential nucleation orientation may be attributed to strain minimization between the Cu2−xS and CuGaS2 lattice.
Elongation and tapering mechanism
As the reaction proceeds, elongation of the Cu2−xS/CuGaS2 Janus particles into NRs is observed. Typically, the particles grow from nearly spherical to ∼100 nm in length within 5 min, which gives a growth rate of roughly 0.3 nm s−1. Nanorods grown from Cu2−xS intermediates are sometimes thought to elongate through the SSS growth mechanism.32 This mechanism is a variation on the well-explored VLS33,38 and SLS34,39,40 mechanisms of growth, in which precursors dissolve into a liquid metal droplet (e.g., Au or Bi) and increase in concentration until supersaturation is reached. The solute phase then precipitates out, forming a NR with a diameter comparable to or smaller than the metal droplet (due to catalyst swelling). In the case of Cu2−xS, the liquid phase in which the precursor dissolves is the cation lattice (critical temperature ∼105 °C in the bulk17,18) while the anion lattice is assumed to be rigid. While there is not an accepted estimate for the cation liquification temperature of Cu2−xS at the nanometer size scale, Zheng et al. demonstrated facile phase transitions in high chalcocite Cu2S NRs under electron irradiation within the TEM with an estimated depression of 40 °C compared to the bulk.41 Several variations on the SSS mechanism for Cu2−xS have been reviewed previously,16 however how the anion lattice extends is left unexplained in the case where Cu2−xS plays the role of the “catalyst”. Additional cations and anions must both be incorporated for growth but with a solid anion lattice, the SSS growth mechanism can only account for cation incorporation. Hence, how the extension of the anion lattice occurs will play a critical role in our understanding of the growth mechanism and the resulting variations in morphologies. In our search of the superionic-conductor-based nanocrystal literature, among 66 reports, only 3 attempted to explore a potential mechanism for anion lattice extension and subsequent growth.42–44 Therefore, we first consider possible mechanisms based on the current literature to determine whether of not they can rationalize how CuGaS2 NRs of observed morphologies arise from Cu2−xS seeds.From the initial Janus particles and the final all-CuGaS2 products, we know that cation exchange must occur at or near the beginning and at the end of the NR growth. What is unclear is how CuGaS2 rods elongate in between. First, we consider the often-assumed “catalytic” growth or, more specifically, SSS growth, an analog of VLS or SLS, where the catalyst particle is not consumed. This mode of growth cannot account for the tapering observed unless we consider cation exchange gradually decreasing the catalyst particle size. However, this mechanism requires diffusion of Cu+, Ga3+ and S2− in the Cu2−xS lattice. While it might be feasible for small cations to be mobile, it is unlikely that the much larger anions would be able to diffuse sufficiently. Furthermore, the anion lattice must remain rigid, otherwise the entire particle would become liquid. While melting point depression in nanocrystals is well known, inspection of the Cu–Ga–S phase diagram indicates that a melting depression would have to be on the order of ∼1000 °C for the particle to liquify under the given reaction conditions.45In situ heating experiments on a related system, Ag2S/ZnS nanorods, support the conclusion that the particles remain solid well within the reaction temperatures being discussed.46 Hence, the catalytic SSS mechanism, even if we include cation exchange, cannot account for anion incorporation needed for the large volume change during the elongation into rod structures. Therefore, we rule out the SSS mechanism but reemphasize that cation exchange is necessary for the initial Janus particle formation and the final conversion of Cu2−xS at the tip, leading to tapering.
We also consider the possibility of diffusion-controlled growth.22,42 In this mechanism, the superionic conductor core is surrounded by an amorphous shell composed of the anion and the secondary cation precursors. As out-diffusion of the primary Cu+ cations occurs, nanoparticles elongate. While this mechanism does explain the observed tapering, we note that the Cu2−xS core is actually observed on the tapered end, as opposed to being embedded in the head as this model would predict. This mechanism also does not adequately address the issue of anisotropic growth. While diffusion within the particle may be anisotropic, if the surrounding shell is amorphous, then the diffusion should be isotropic, leading to a more pronounced tear drop shape which is not observed in our system. Furthermore, it is also unclear how kinked and bent structures, which are frequently observed in our and others' work are formed if cation diffusion within the NR is the primary mechanism for elongation.
Another commonly considered mechanism involving Cu2−xS and related materials is seeded growth. CuGaS2 might grow epitaxially, localized on the CuGaS2 seed part of the Janus particle with eventual conversion of remaining Cu2−xS via cation exchange at the end of the reaction. We consider this mechanism unlikely since the tapering tip of the final product has a much smaller diameter than the initial Janus particle. It is the larger diameter end of the final rod that is closer to the initial Janus particle diameter. If the Cu2−xS half of the Janus particle was converted at the end of the reaction, there should not be any tapering with diameter decreasing below the initial Janus particle diameter. While epitaxial ripening is possible and can be observed at long growth times (>1 h), any apparent tapering would result from the NR diameter increasing rather than decreasing from the initial Janus particle diameter. The observed final product has a diameter that decreases from the initial seed particle diameter.
Similar to catalytic SSS growth, we can also consider seeded growth of CuGaS2 along with cation exchange. In this case, Cu2−xS within the initial Janus particle must remain unreacted until the end, when cation exchange converts the original Cu2−xS to CuGaS2. However, there is no obvious reason why cation exchange would only occur at the beginning and the end of the reaction where precursor concentrations are at their extremes. If the cation exchange reaction were to occur concurrently with seeded growth, the Cu2−xS portion of the initial Janus particle would continuously decrease in size but the resulting CuGaS2 would retain the diameter of the original Cu2−xS component. The Cu+ ions exchanged out from the Cu2−xS lattice could diffuse out into the solution or diffuse on the surface of the growing particle and add to the CuGaS2 growth but that, again, would not alter the size and shape of the Cu2−xS region of the initial Janus particle as it converts to CuGaS2. Hence, this mechanism of seeded growth of CuGaS2 with concurrent cation exchange cannot account for the observed tapering to a much smaller diameter than the starting seed diameter.
Then, we consider epitaxial growth the of Cu2−xS seed rather than CuGaS2 with concurrent cation exchange. We refer to this growth as concurrent seed growth and cation exchange (CSC). In this case, Cu+ and S2− add directly to the Cu2−xS surface, which is accompanied by cation exchange that extends the CuGaS2 lattice as shown in Scheme 1. That is, Cu2−xS grows epitaxially on one end and cation exchange occurs on the opposite side of Cu2−xS near the interface with CuGaS2. The anisotropic growth of Cu2−xS may be attributed to a preferential inflow of Ga3+ at the interfacial region, which outcompetes Cu+ inflow. Given this situation and since homoepitaxy of Cu2−xS may be easier than that of CuGaS2, anisotropic growth of Cu2−xS can be anticipated. Once rod growth begins, poorer ligand coverage on the hemispherical Cu2−xS surface at the growing tip would further enhance anisotropic growth. As precursors deplete away, Cu2−xS growth slows and cation exchange starts to dominate, leading to a smaller and smaller Cu2−xS tip, which eventually converts completely to CuGaS2 as tapering occurs. This mechanism appears to be most plausible, as it accounts for the correlation between diameters of Cu2−xS and the elongating CuGaS2 as well as how the very large number of anions are incorporated.
 | ||
| Scheme 1 Elongation of Cu2−xS nanoparticles into CuGaS2 NRs via the concurrent seed growth and cation exchange (CSC) mechanism. First, a seed Cu2−xS particle is formed and rapidly converted into a Cu2−xS/CuGaS2 Janus particle via cation exchange. Cation exchange continues concurrent to anisotropic epitaxial growth of the Cu2−xS seed. This process continues until the rate of cation exchange begins to outpace the rate of Cu2−xS epitaxial growth due to depleting Cu+ concentration. At this point, the Cu2−xS tip continuously decreases in size and leads to tapering of the NR. Eventually the seed is completely converted to CuGaS2via cation exchange. | ||
Variations in growth direction
We now consider how different morphologies could arise within the CSC growth mechanism. In each of the three morphologies, 90°-bent, 155°-kinked, and straight NRs, the surface termination of a given growth direction plays a critical role in determining the stability and the longevity of a given growth direction.0} plane. As these NRs elongate, the growth direction always quickly changes from 〈100〉 to 〈0001〉. We have not observed a NR within this system where the 〈100〉 vector is the long axis of the NR. This change in growth direction results in a NR which appears to have a 90° angle between the “head” and “body” of the rod, as shown in Fig. 2b. These NRs are still single crystalline throughout, and do not display any other crystal phases other than the occasional stacking faults mentioned previously. Hence, the initial {100} interface of the Janus particle and high Cu+ concentration allows brief growth of Cu2−xS along 〈100〉 direction. However, this growth direction imposes energetically unfavorable high index planes for surface terminations and quickly forces the growth of Cu2−xS to be along 〈0001〉 direction, leading to the 90°-bent morphology.
2〉 and 〈0001〉 of 152°. The mismatch between this expected value and the observed kink angle may be due in part to the rods undergoing continuous tapering (as in Fig. 4c) and the fact that the change in direction is unlikely to be abrupt, which would give rise to a distribution of angles, along with the uncertainty in measuring angles. However, we note that the zone axis of the kinked rods observed in HRTEM images is usually 〈20〉. That means the observed angle is actually the angle between the projections of 〈102〉 and 〈0001〉 directions on the {20} plane. In this case the expected kink angle is 155°, in excellent agreement with the experimentally measured angle.
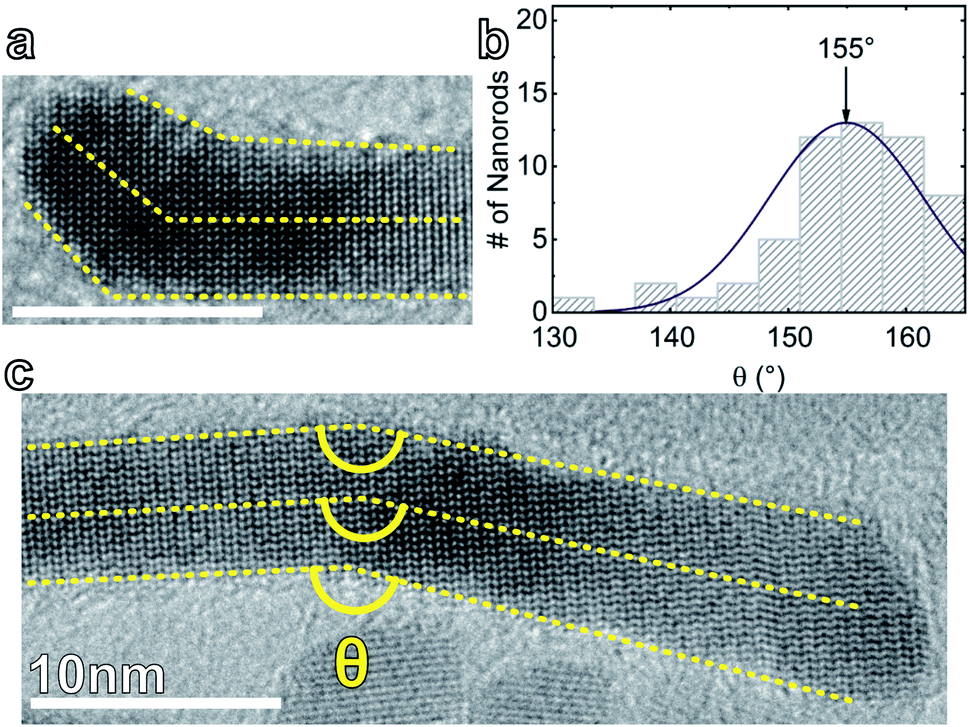 | ||
| Fig. 4 (a) Example HRTEM of a NR with a short section of 〈102〉 growth, which is immediately followed by 〈0001〉 growth. At lower magnifications, this rod might have been categorized as “straight”. Scale bar is 10 nm. (b) Distribution of measured kink angles from CuGaS2 NRs. (c) Annotated HRTEM of a kinked NR showing the methodology for measuring the kink angle. | ||
On the other hand, there is a non-negligible discrepancy between the measured initial internal nucleation angle (139 ± 10°) and the kink angle (155 ± 7°). The expected angle between the vector normal to the (102) plane, which we estimate as 〈101〉, and the identified growth vector, 〈102〉, is 16°, which is the noted discrepancy. This difference in angle means that there is a slight deviation in the growth direction from that expected of the initial Janus particle interface. That is, one often expects the growth to be perpendicular to the heterointerface formed. However, growth along direction perpendicular to the initial Cu2−xS/CuGaS2 interface would lead to highly unfavorable surface termination with high index planes. Therefore, we expect and observe growth at a slightly different direction along 〈102〉 that would give rise to low-index facets.
As the NR continues to elongate, the thermodynamic driving force begins to favor Cu2−xS growth along 〈0001〉 direction, leading to a change in growth direction and therefore, the kink. Growth along 〈0001〉 giving rise to a more stable NR orientation than along 〈102〉 may be expected given that the 〈102〉 growth direction would require multiple polar side facets whereas the 〈0001〉 growth direction would likely lead to non-polar {100} or {20} facets. Furthermore, both 90°-bent and 155°-kinked NRs arise because the final growth is along 〈0001〉, supporting the assertion that 〈0001〉-oriented NRs are energetically the most stable. Then, within the CSC mechanism, growth along 〈102〉 is feasible at high Cu+ concentrations where fast growth of Cu2−xS occurs but depleting Cu+ precursor concentration and therefore slowing Cu2−xS growth rate would lead to a change in the growth direction to the energetically favored 〈0001〉 direction, resulting in kinked NRs.
2〉 direction and change orientation to 〈0001〉 resulting in a ∼155°-kinked rods. An example of such a kinked rod with short 〈102〉 growth segment is shown in Fig. 4a. These short-growth ∼155°-kinked rods initially led to an underestimation of the kinked NR population. When it became clear that the kinking of NRs was more frequent than initially thought, we revisited the initial statistics and found that the population of purely straight NRs was actually the minority. Consistent with a previous report,19 we expect the strain within the initial Janus particle to be minimized when the heterointerface is oriented {102}/{102}. Then the predominance of this interface initiates most of the rod growth to be along 〈102〉, leading to the kinked rather than the straight NRs to be prevalent.
0〉 growth direction is seen in the case of the 90°-bent NRs and is only observed for very short sections of NRs and represents a small population (Fig. 5a). This behavior may be explained by the large surface energy difference between the terminating facets for 〈100〉 vs. 〈0001〉 growth direction. The 〈102〉 growth direction is observed in the 155°-kinked NRs, and can be observed for significant stretches of NRs. The 〈0001〉 growth direction is observed in the straight NRs, as well as in the 90°-bent and 155°-kinked NRs (i.e. all NRs eventually change to the 〈0001〉 growth direction). Given that NRs with 〈0001〉 long axis are the most stable, we postulate that the 〈100〉 and 〈102〉 growth directions are kinetically accessible in the early stages of growth due to the high concentration of precursors. As growth proceeds, the concentration of precursors decreases, and the thermodynamically stable product becomes dominant, 〈0001〉 growth direction in this case.
 | ||
| Fig. 5 (a) Categorical statistics which show a correlation between the distribution of initial nucleation angles (given as growth vectors) and the observed final morphology. Particles which exhibit very short kinked segments, or where the orientation of the {0001} could not be reliably assigned, were placed in the ambiguous category. (b) HRSTEM of Cu2−xS/CuGaS2 NR with two crystallographic planes making up the interface, presumably occurring at the initial stage of growth direction change. We note that we label the vector ∼ normal to the (102) plane instead of the plane for easier comparison with the kinked growth vector. (c) HRSTEM of Cu2−xS/CuGaS2 NR immediately after growth direction change. Note that the two different particles in (b) and (c) are from the same aliquot, indicating that these morphologies coexist under normal reaction conditions. | ||
Fig. 5b shows a NR at an early reaction time with its growth direction transitioning from 〈102〉 to 〈0001〉. Fig. 5c shows a NR which has just completed the transition and is now growing in the 〈0001〉 direction. These images suggest that as the Cu2−xS growth direction changes to the more thermodynamically favored 〈0001〉 direction, there is a corresponding shift in the interface through a combination of {102} and {0001} planes.
We note that a modulation of growth direction via reaction conditions has also been reported in the VLS literature. In particular, precursor pressure has been shown to induce kinking in VLS NRs.47,48 At very high growth rates kinks have been shown to arise spontaneously, indicating that multiple surface terminations become accessible at high reactant concentrations,49 consistent with the above outlined reason for the occurrence of kinks and bends in our CuGaS2 NRs. Preliminary results on intentional concentration variations indicate that this may indeed be the case in the Cu2−xS/CuGaS2 system. However, further investigation is needed to suppress other complicating factors such as separate homogeneous nucleation that can also occur with a sudden increase in precursor concentration.
Regioselective heterostructure nucleation
The different termination facets arising from changes in the growth direction of kinked NRs can help to explain an interesting and potentially useful phenomenon. While pursuing epitaxial growth of CuInE2 (E = S, Se) on CuGaS2 NRs, we frequently observed regioselective heterostructure growth. We define regioselectivity as growth which is consistently localized on a particular facet or area of a nanorod (e.g. tips vs. side facets). For sections of 〈0001〉 growth, we observed CuInE2 island growth on all side facets (Fig. 6a). However, selective CuInE2 heteroepitaxy, where one side of the NR was bare of CuInE2 was often observed for 〈102〉 growth sections (Fig. 6b). One possible explanation for this phenomenon in a known polymorphic system, such as CuGaS2 would be that the 〈102〉 sections are a different crystal structure than the 〈0001〉 sections (e.g., chalcopyrite vs. wurtzite-like). However, as shown here and in our prior investigation by HRTEM, SAED, and XRD, the CuGaS2 NRs are wurtzite-like throughout.26 We therefore expect the surface termination of the CuGaS2 NR to be the primary determining factor in the nucleation of CuInE2.
The expected surface termination for a section of CuGaS2 rod with 〈0001〉 growth direction is {100} with an overall hexagonal shape. While we acknowledge that the {20} surface termination is possible we expect the {100} termination based on the observed kink angle. Assuming that the 〈102〉 sections of the rod maintain hexagonal cross-section, the surface termination should be a mixture of {2![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 01} and {101}, with two {101} facets and four {201} facets (shown schematically in Fig. 6c). We note that the {100} facets are charge neutral, while the {101} facets can be net positive or negative, depending on the surface termination. We denote cation-terminated facets as (101) and anion-terminated facets as (01), adopting the convention for the {0001} and {000} facets of wurtzite. Cation or anion termination of surfaces has been shown previously to significantly affect the growth rate of nanocrystals.50 We therefore postulate that the regioselective nature of CuInE2 heterostructure growth is due to the difference in reactivity between the {101} cation- and anion-terminated surfaces. Similar to Kim et al. we expect the (01) anion-terminated surfaces to be more reactive, and therefore more conducive to heterostructure growth, while the (101) cation-terminated surfaces are less reactive.50 Hence, regioselective nucleation of CuInSe2 is preferential only on the segments of CuGaS2 NRs where the growth direction is 〈102〉, thus explaining the observed difference in facet-selectivity of heteroepitaxy shown in Fig. 6c.
01} and {101}, with two {101} facets and four {201} facets (shown schematically in Fig. 6c). We note that the {100} facets are charge neutral, while the {101} facets can be net positive or negative, depending on the surface termination. We denote cation-terminated facets as (101) and anion-terminated facets as (01), adopting the convention for the {0001} and {000} facets of wurtzite. Cation or anion termination of surfaces has been shown previously to significantly affect the growth rate of nanocrystals.50 We therefore postulate that the regioselective nature of CuInE2 heterostructure growth is due to the difference in reactivity between the {101} cation- and anion-terminated surfaces. Similar to Kim et al. we expect the (01) anion-terminated surfaces to be more reactive, and therefore more conducive to heterostructure growth, while the (101) cation-terminated surfaces are less reactive.50 Hence, regioselective nucleation of CuInSe2 is preferential only on the segments of CuGaS2 NRs where the growth direction is 〈102〉, thus explaining the observed difference in facet-selectivity of heteroepitaxy shown in Fig. 6c.
 | ||
| Fig. 6 (a) Low magnification STEM of CuGaS2/CuInSe2 NRs. (b) STEM of a CuGaS2/CuInSe2 NR heterostructure which shows an absence of CuInSe2 nuclei on a section of the CuGaS2 NR (rectangle outlined by dotted line) prior to the kink. (c) 3D-model of the underlying CuGaS2 NR with facets labeled and color-coded. The yellow spheres represent the anion lattice, while the blue spheres represent the cation lattice. The cation-only (101) plane and anion-only (01) plane are visible, along with the non-polar {100} facets. (d) Statistical distribution of CuInSe2 growth on CuGaS2 NRs showing a strong facet dependence in the case of heteroepitaxy on 〈102〉 oriented NR segments. | ||
Conclusions
We have investigated CuGaS2 NR growth with the goal of understanding the origin of the morphological diversity which emerges from a relatively monodisperse system. We have categorized the various observed morphologies as 155°-kinked, 90°-bent and straight. The kink angles are average values as there is a distribution, which arises from variations in tapering and growth direction change rates. The abundances of the three morphologies correlate strongly with those of the three heterointerfaces observed in the initial Cu2−xS/CuGaS2 Janus particles. All three morphologies exhibit tapering. In addition to these observations, the mechanism of S2− addition (which increases in number by more than an order of magnitude) is a critical component for understanding the growth of NRs from this system. In our proposed CSC mechanism, the Cu2−xS part of the initial Janus particle is the component that actually grows epitaxially (seed growth) and the concurrent cation exchange at the heterointerface converts the opposite end of the growing Cu2−xS to CuGaS2. This mechanism can account for or is consistent with all observed features of CuGaS2 NR growth. In particular:(1) The anion sub-lattice expansion can be accounted for by Cu2−xS seed, rather than CuGaS2, growth.
(2) The prevalent tapering of the NRs occurs as the diameter of the Cu2−xS particle decreases due to cation exchange dominating over Cu2−xS growth with depleting Cu+ precursor concentration.
(3) Kinks/bends can occur through Cu2−xS growth direction change, driven by the thermodynamically more stable 〈0001〉 rod long-axis orientation.
(4) The dominance of the 155°-kinked morphology is due to the preference of the lattice-strain-minimizing {102}/{102} interface in the initial Cu2−xS/CuGaS2 Janus particles that forms through cation exchange.
These results have allowed us to reveal that the 155°-kinked NR can induce regioselective nucleation of CuInE2 on its cation-terminated (101) surfaces within the 〈102〉 growth direction segment. Insights gained through our proposed CSC mechanism should help to devise strategies for developing a wide variety of anisotropic nanocrystals and their heterostructures with uniform size and shape.
Abbreviations
| CSC | Concurrent seed growth and cation exchange |
| TEM | Transmission electron microscopy |
| NRs | Nanorods |
| VLS | Vapor–liquid–solid |
| SLS | Solution–liquid–solid |
| SSS | Solution–solid–solid |
| CuOAc | Copper(I) acetate |
| Ga(acac)3 | Gallium acetylacetonate |
| 1-DDT | 1-Dodecanethiol |
| TOP | Trioctylphosphine |
| In(OAc)3 | Indium(III) acetate |
| HRTEM | High-resolution transmission electron microscopy |
| HRSTEM | High-resolution scanning transmission electron microscopy |
| UV-Vis | Ultraviolet-visible spectroscopy |
| PL | Photoluminescence spectroscopy |
| XRD | X-ray diffraction |
| EDS | Energy-dispersive X-ray spectroscopy |
| DCG | Diffusion-controlled growth |
Funding sources
This work was supported by the NSF (Grant 1825356).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the generous financial support of the NSF. We thank Nuri Oh for preliminary studies that have led to this work. Research was carried out in part in the Materials Research Laboratory Central Research Facilities, University of Illinois. We have made significant use of VESTA as a tool to visualize crystal structures, lattice vectors and planes, and wish to thank Koichi Momma for his generosity in making this program publicly available. We also owe a great debt to F. C. Frank for his excellent paper on four-dimensional vectors,51 which was an immense help in preparing this manuscript.References
- C. Livache, B. Martinez, N. Goubet, J. Ramade and E. Lhuillier, Front. Chem., 2018, 6, 1–11 CrossRef PubMed.
- A. Jagtap, B. Martinez, N. Goubet, A. Chu, C. Livache, C. Gréboval, J. Ramade, D. Amelot, P. Trousset, A. Triboulin, S. Ithurria, M. G. Silly, B. Dubertret and E. Lhuillier, ACS Photonics, 2018, 5, 4569–4576 CrossRef CAS.
- X. Tang, M. M. Ackerman, M. Chen and P. Guyot-Sionnest, Nat. Photonics, 2019, 13, 277–282 CrossRef CAS.
- Y. You, X. Tong, W. Wang, J. Sun, P. Yu, H. Ji, X. Niu and Z. M. Wang, Adv. Sci., 2019, 6, 1801967 CrossRef PubMed.
- L. R. Bradshaw, K. E. Knowles, S. McDowall and D. R. Gamelin, Nano Lett., 2015, 15, 1315–1323 CrossRef CAS PubMed.
- J. Yang, M. K. Choi, U. J. Yang, S. Y. Kim, Y. S. Kim, J. H. Kim, D. H. Kim and T. Hyeon, Nano Lett., 2021, 21 DOI:10.1021/acs.nanolett.0c03939.
- Y. Jiang, S. Y. Cho and M. Shim, J. Mater. Chem. C, 2018, 6, 2618–2634 RSC.
- M. K. Choi, J. Yang, T. Hyeon and D. H. Kim, npj Flexible Electron., 2018, 2, 1–14 CrossRef.
- Z. Yue, F. Lisdat, W. J. Parak, S. G. Hickey, L. Tu, N. Sabir, D. Dorfs and N. C. Bigall, ACS Appl. Mater. Interfaces, 2013, 5, 2800–2814 CrossRef CAS PubMed.
- F. Ma, C. Chen Li and C. Yang Zhang, J. Mater. Chem. B, 2018, 6, 6173–6190 RSC.
- A. Rizzo, C. Nobile, M. Mazzeo, M. De Giorgi, A. Fiore, L. Carbone, R. Cingolani, L. Manna and G. Gigli, ACS Nano, 2009, 3, 1506–1512 CrossRef CAS PubMed.
- J. Hu, L. Li, W. Yang, L. Manna, L. Wang and A. P. Alivisatos, Science, 2001, 292, 2060–2063 CrossRef CAS PubMed.
- J. Schneider, W. Zhang, A. K. Srivastava, V. G. Chigrinov, H. S. Kwok and A. L. Rogach, Nano Lett., 2017, 17, 3133–3138 CrossRef CAS PubMed.
- N. Oh, B. H. Kim, S.-Y. Cho, S. Nam, S. P. Rogers, Y. Jiang, J. C. Flanagan, Y. Zhai, J.-H. Kim, J. Lee, Y. Yu, Y. K. Cho, G. Hur, J. Zhang, P. Trefonas, J. A. Rogers and M. Shim, Science, 2017, 355, 616–619 CrossRef CAS PubMed.
- M. J. Anc, N. L. Pickett, N. C. Gresty, J. A. Harris and K. C. Mishra, ECS J. Solid State Sci. Technol., 2013, 2, R3071–R3082 CrossRef CAS.
- J. Kolny-Olesiak, CrystEngComm, 2014, 16, 9381–9390 RSC.
- L. W. Wang, Phys. Rev. Lett., 2012, 108, 2–6 Search PubMed.
- K. Wakamura, Solid State Ionics, 2000, 130, 305–312 CrossRef CAS.
- B. C. Steimle, J. L. Fenton and R. E. Schaak, Science, 2020, 367(6476), 418–424 CrossRef CAS PubMed.
- C. R. McCormick and R. E. Schaak, J. Am. Chem. Soc., 2021, 143(2), 1017–1023 CrossRef CAS PubMed.
- M. Zhao, F. Huang, H. Lin, J. Zhou, J. Xu, Q. Wu and Y. Wang, Nanoscale, 2016, 8, 16670–16676 RSC.
- S. Das Adhikari, A. Dutta, G. Prusty, P. Sahu and N. Pradhan, Chem. Mater., 2017, 29, 5384–5393 CrossRef.
- S. H. Chang, B. C. Chiu, T. L. Gao, S. Lou Jheng and H. Y. Tuan, CrystEngComm, 2014, 16, 3323–3330 RSC.
- M. D. Regulacio, C. Ye, S. H. Lim, Y. Zheng, Q.-H. H. Xu and M.-Y. Y. Han, CrystEngComm, 2013, 15, 5214–5217 RSC.
- Z. Liu, Q. Hao, R. Tang, L. Wang and K. Tang, Nanoscale Res. Lett., 2013, 2–7 Search PubMed.
- N. Oh, L. P. Keating, G. A. Drake and M. Shim, Chem. Mater., 2019, 31, 1973–1980 CrossRef CAS.
- M. Kruszynska, H. Borchert, J. Parisi and J. Kolny-Olesiak, J. Am. Chem. Soc., 2010, 132, 15976–15986 CrossRef CAS PubMed.
- S. H. Choi, E. G. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 2520–2521 CrossRef CAS PubMed.
- S. T. Connor, C. M. Hsu, B. D. Weil, S. Aloni and Y. Cui, J. Am. Chem. Soc., 2009, 131, 4962–4966 CrossRef CAS PubMed.
- X. Yang, B. Zhou, C. Liu, Y. Sui, G. Xiao, Y. Wei, X. Wang and B. Zou, Nano Res., 2017, 10, 2311–2320 CrossRef CAS.
- X. Geng, H. Liu, L. Zhai, Z. Xiong, J. Hu, C. Zou, Y. Dong, Y. Yang and S. Huang, J. Alloys Compd., 2014, 617, 961–967 CrossRef CAS.
- J. Wang, K. Chen, M. Gong, B. Xu and Q. Yang, Nano Lett., 2013, 13, 3996–4000 CrossRef CAS PubMed.
- R. S. Wagner and W. C. Ellis, Appl. Phys. Lett., 1964, 4, 89–90 CrossRef CAS.
- T. J. Trentler, K. M. Hickman, S. C. Goel, A. M. Viano, P. C. Gibbons and W. E. Buhro, Science, 1995, 270, 1791–1794 CrossRef CAS.
- F. Wang, A. Dong and W. E. Buhro, Chem. Rev., 2016, 116, 10888–10933 CrossRef CAS PubMed.
- D. J. Chakrabarti and D. E. Laughlin, Bull. Alloy Phase Diagrams, 1983, 4, 254 CrossRef.
- R. Xie, M. Rutherford and X. Peng, J. Am. Chem. Soc., 2009, 131, 5691–5697 CrossRef CAS PubMed.
- V. G. Dubrovskii, Cryst. Growth Des., 2017, 17, 2544–2548 CrossRef CAS.
- W. E. Buhro, K. M. Hickman and T. J. Trentler, Adv. Mater., 1996, 8, 685–688 CrossRef CAS.
- F. Wang, A. Dong, J. Sun, R. Tang, H. Yu and W. E. Buhro, Inorg. Chem., 2006, 45, 7511–7521 CrossRef CAS PubMed.
- H. Zheng, B. J. Rivest, T. A. Miller, B. Sadtler, A. Lindenberg, M. F. Toney, L. Wang, C. Kisielowski and A. P. Alivisatos, Science, 2011, 333, 206–209 CrossRef CAS PubMed.
- G. Prusty, A. K. Guria, B. K. Patra and N. Pradhan, J. Phys. Chem. Lett., 2015, 6, 2421–2426 CrossRef CAS PubMed.
- S. Noor Mohammad, J. Chem. Phys., 2009, 131(22), 224702 CrossRef CAS PubMed.
- Y. Vahidshad, M. Nawaz Tahir, A. Iraji Zad, S. M. Mirkazemi, R. Ghasemzadeh, H. Huesmann and W. Tremel, J. Phys. Chem. C, 2014, 118, 24670–24679 CrossRef CAS.
- M. Kokta, J. R. Carruthers, M. Grasso, H. M. Kasper and B. Tell, J. Electron. Mater., 1976, 5, 69–89 CrossRef CAS.
- J. H. Kim, J. G. Kim, J. Song, T. S. Bae, K. H. Kim, Y. S. Lee, Y. Pang, K. H. Oh and H. S. Chung, Appl. Surf. Sci., 2018, 436, 556–561 CrossRef CAS.
- B. Tian, P. Xie, T. J. Kempa, D. C. Bell and C. M. Lieber, Nat. Nanotechnol., 2009, 4, 824–829 CrossRef CAS PubMed.
- C. M. L. Bozhi Tian, T. Cohen-Karni, Q. Qing, X. Duan, P. Xie, B. Tian, T. Cohen-Karni, Q. Qing, X. Duan, P. Xie and C. M. Lieber, Science, 2010, 329, 830 CrossRef PubMed.
- P. Madras, E. Dailey and J. Drucker, Nano Lett., 2009, 9, 3826–3830 CrossRef CAS PubMed.
- D. Kim, Y. K. Lee, D. Lee, W. D. Kim, W. K. Bae and D. C. Lee, ACS Nano, 2017, 11, 12461–12472 CrossRef CAS PubMed.
- F. C. Frank, Acta Crystallogr., 1965, 18, 862–866 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |