
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlation between the TiO2 encapsulation layer on Pt and its electrochemical behavior†
Raquel Aymerich
Armengol
a,
Joohyun
Lim
 *b,
Marc
Ledendecker
c,
Katharina
Hengge
a and
Christina
Scheu
*a
*b,
Marc
Ledendecker
c,
Katharina
Hengge
a and
Christina
Scheu
*a
aMax-Planck Institut für Eisenforschung GmbH, Max-Planck-Straße 1, 40237, Germany. E-mail: c.scheu@mpie.de
bDepartment of Chemistry, Kangwon National University, Chuncheon, Gangwon 24341, Republic of Korea. E-mail: jlim@kangwon.ac.kr
cDepartment of Technical Chemistry I, Technical University Darmstadt, Alarich-Weiss-Straße 8, 64287, Germany
First published on 15th July 2021
Abstract
Supported metal catalysts with partial encapsulation resulting from strong metal–support interactions show distinctive structural features which strongly affect their functionalities. Yet, challenges in systematic synthesis and in-depth characterization for such systems limit the present understanding of structure–property relationships. Herein, the synthesis and characterization of two Pt/TiO2 models are conducted by a simple change of the synthesis order, while keeping all other parameters constant. They differ in containing either bare or encapsulated Pt nanoparticles. The presence of an extremely thin and inhomogeneous TiO2 layer is clearly demonstrated on 2–3 nm sized Pt nanoparticles by combination of imaging, energy dispersive X-ray spectroscopy and electron energy loss spectroscopy performed in a transmission electron microscope. The two Pt/TiO2 systems exhibit differences in morphology and local structure which can be correlated with their electrochemical activity and stability using cyclic voltammetry experiments. Beyond enhanced particle stability, we report an increase in H+ intercalation on titania and reduced Pt activity due to partial encapsulation by TiO2. Finally, the growth of an encapsulation layer as a result of cyclic voltammetry measurements is discussed. These results shed light on the in-depth structure–property relationship of catalysts with strong metal–support interactions which leads to enhanced functional materials for electrochromic devices and energy applications.
1. Introduction
The properties of supported catalysts depend on the local structure and interaction between the catalyst and the support. A particular example is materials showing strong metal–support interactions (SMSI). The electronic and structural modifications of SMSI yield high stability against agglomeration as well as enhanced activity and selectivity.1–3SMSI can generally be achieved through annealing of metal catalysts supported by reducible metal oxides such as TiO2, WO3 or Nb2O5 in a reducing atmosphere.4,5 During the annealing process, atoms of the metal oxide support can diffuse and cover the metal catalyst forming an encapsulation layer. Thin and/or partial encapsulation of the catalyst by the support material was described to be responsible for extraordinary stability, activity and selectivity in various metal/metal oxide systems.6,7 Yet, impermeable and thick support layers over the catalysts can also lead to a decrease in performance by blocking active sites.8,9 Therefore, probing the existence and specific role of the encapsulation layer of SMSI materials is crucial to understanding their performance in catalytic reactions. Although the existence of thin encapsulation layers on metal catalysts was recently proved by electron microscopy,10–14 structure–property relationships were rarely investigated, particularly in the case of electrochemical properties.15–17 The lack of systematic studies is related to both challenges in the synthesis and characterization of comparable model materials with and without an encapsulation layer. Moreover, the high temperature and reducing atmosphere employed to obtain SMSI materials often cause sintering and perfect encapsulation of the catalyst instead of thin and/or inhomogeneous encapsulation layers.18–20
Among various supported metal systems, Pt/TiO2 is recognized as a promising material for electrochromic devices21 and as an anode in fuel cells where it is investigated as a substitute for platinum nanoparticles supported on carbon (Pt/C) due to its superior corrosion stability.22,23
Furthermore, a very recent study reported that encapsulated Pt on TiO2 can act as a selective catalyst for the hydrogen oxidation reaction.16 On the one hand, the selective H+ permeability through the TiO2 encapsulation layer prevents the oxygen reduction reaction on Pt. On the other hand, selectivity can also be driven by exploiting the conductivity dependence of the material. This depends on the mixed valences of Ti and the reversible formation of conductive HxTiO2 or insulating TiO2 under H2 and O2 atmospheres, respectively.24
In the present study, we aim to determine the effect of SMSI encapsulation layers on the electrochemical behavior of Pt/TiO2 nanomaterials. For this purpose, we synthesize and analyze Pt nanoparticles on TiO2 nanowires with and without a TiO2 encapsulation layer and compared their performance and stability. A combination of (scanning) transmission electron microscopy ((S)TEM), energy-dispersive X-ray spectroscopy (EDS) and electron energy loss spectroscopy (EELS) techniques is used to probe the presence of an extremely thin encapsulation layer. The structure of the materials is correlated to the electrochemical response and stability by cyclic voltammetry (CV). Our results determine the precise structure of a supported metal catalyst material and reveal how an extremely thin encapsulation layer affects its electrochemical behavior. Such an encapsulation layer can form directly during synthesis or during the CV experiments. The findings are essential for understanding structure–property relationships for electrocatalysts showing SMSI.
2. Synthesis of Pt/TiO2
Pt nanoparticles on TiO2 nanowires were synthesized by two different chemical routes (Fig. 1). In both cases, TiO2 nanowires were grown on a fluorine-doped tin oxide (FTO) electrode through a hydrothermal method and etched to a hollow structure with a high specific surface area.25,26 Divalent tin cations (Sn2+) were adsorbed to the surface oxyanions and hydroxyl groups, which served as nucleation points for the reduction of the Pt precursor to Pt nanoparticles.27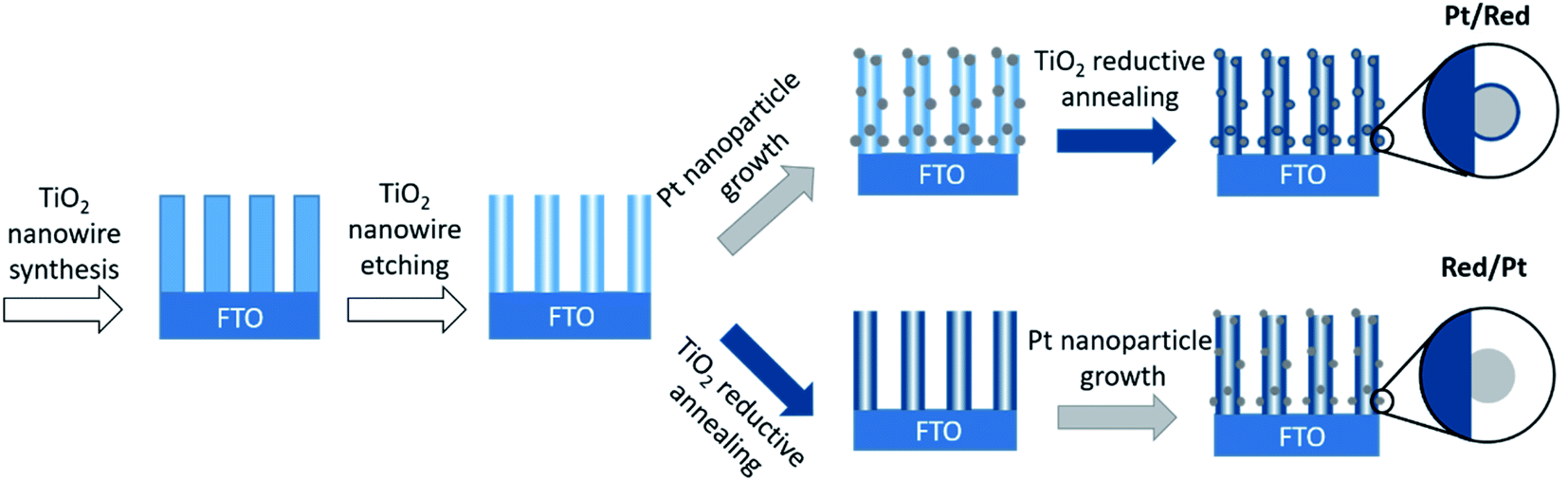 | ||
| Fig. 1 Schematic drawings of the synthesis of two Pt/TiO2 systems. Note that only the order of the synthesis steps was changed. | ||
To obtain the first Pt/TiO2 model with a TiO2 encapsulation layer on Pt nanoparticles (denoted as Pt/Red), Pt nanoparticles were grown on the surface of hollow TiO2 nanowires and the entire Pt/TiO2 composite was subsequently annealed in the presence of sodium borohydride as a reducing agent under a nitrogen atmosphere. The reduction process induces the formation of a TiO2 encapsulation layer on Pt nanoparticles4 and increases the conductivity of the titania support by introducing oxygen vacancies and trace impurities.28,29 To obtain the second Pt/TiO2 model without an encapsulation layer (denoted as Red/Pt), hollow TiO2 nanowires were reduced before the growth of Pt nanoparticles. This different reaction order allows us to synthesize two distinct Pt/TiO2 materials to study the TiO2 encapsulation layer on Pt, while fixing most other variables (Fig. 1).
The size, morphology, and crystal structure of Pt/TiO2 were verified by electron microscopy (Fig. 2). Scanning electron microscopy (SEM) shows TiO2 nanowires grown on a FTO electrode in the top-view (Fig. 2a), while the high angle annular dark-field (HAADF)-STEM image in Fig. 2b displays a single detached nanowire composed of smaller nanobundles in the side-view. After etching the inner part of TiO2 nanowires, only the external nanobundles remain (Fig. 2c and d). This yields hollow TiO2 nanowires with a larger surface area to accommodate a larger amount of Pt nanoparticles.
 | ||
| Fig. 2 Top-view SEM and side-view HAADF-STEM micrographs of the as-synthesized TiO2 nanowire array (a and b) before and (c and d) after etching. Side-view TEM and HAADF-STEM micrographs for a single nanobundle of (e and f) Pt/Red and (g and h) Red/Pt. Please note that (f) and (h) were taken at different regions compared to (e) and (g). | ||
TEM and HAADF-STEM images display Pt nanoparticles in black and white, respectively, for both Pt/Red (Fig. 2e and f) and Red/Pt (Fig. 2g and h). Pt nanoparticles are homogeneously distributed over the TiO2 surface (see ESI movie1† for the 3D reconstructed volume from a single nanobundle of Pt/Red using electron tomography). The high magnification STEM image in Fig. 2f reveals the (111) lattice planes of Pt nanoparticles and the (110) lattice planes of rutile TiO2.
3. Particle size and catalyst loading
A larger average size of Pt particles in Pt/Red (2.4 ± 0.6 nm) than that of Red/Pt (1.6 ± 0.4 nm) was found (Fig. S1†). The encapsulation layer in Pt/Red is barely visible in HRTEM images (Fig. 3a). From the obtained micrographs, one can conclude that the average thickness of the encapsulation layer is below 1–2 (111)Pt atomic layers. We believe that this layer is not from any contaminations including carbon based on our careful sample preparation which included plasma cleaning. | ||
| Fig. 3 (a) HRTEM image of Pt nanoparticles of Pt/Red with a visible TiO2 encapsulation layer (indicated with white arrows). In the other Pt surface parts the layer is <5 Å which would correspond to 2 (111)Pt atomic layers. HAADF-STEM image and EDS maps of (b–d) Pt/Red and (f–h) Red/Pt. Ti and Pt are denoted in red and yellow colors, respectively. White arrows indicate the areas where Pt is exposed out of a single TiO2 nanobundle. (e) HAADF-STEM image of Pt/Red with the white square indicating a Pt nanoparticle on the surface above the vacuum where EELS analysis in (i) was performed. (i) Intensity distribution of the EELS Ti L2,3-edge from region marked in (e). | ||
The presence of such a thin TiO2 layer seems to efficiently prevent the growth of Pt nanoparticles at high temperature30 since the average size of Pt nanoparticles in Pt/Red is not distinguishable from the particle size before the reductive annealing (2.5 ± 0.4 nm, Fig. S1†). The stability of nanoparticles at high temperatures is beneficial for catalysis, yet it is rarely reported even for SMSI systems.18,19
Inductively coupled plasma mass spectroscopy (ICP-MS) determined a higher Pt concentration per electrode area for Pt/Red (2.85 ± 0.08 μg cm−2) than that for Red/Pt (0.37 ± 0.01 μg cm−2).
We hypothesize that the difference in the size of the Pt nanoparticles and Pt loading is related to a different number of surface adsorption sites on the TiO2 substrates. In the synthetic approach employed to deposit Pt nanoparticles,27 Sn2+ cations are adsorbed onto oxyanions and hydroxyl groups of TiO2 by electrostatic interactions. The Sn2+ cations reduce Pt4+ and the yielded Pt0 nanoparticles remain adsorbed at the surface sites while Sn4+ cations are dissolved in the solution. The Pt nanoparticles are able to grow at these sites. However, in Red/Pt the oxyanions and hydroxyl groups are partially removed during heat treatment before deposition of the Pt. Thus, fewer growth sites for Pt are available in the Red/Pt system, resulting in lower Pt loading and smaller nanoparticles than those of Pt/Red.
4. Spectroscopy analysis of the encapsulation layer
To further characterize the TiO2 encapsulation layer on Pt nanoparticles, both Pt/TiO2 systems were analyzed using spectroscopy techniques (Fig. 3). EDS maps obtained in STEM mode display Ti (red color) distributed around the Pt nanoparticles (yellow color) of Pt/Red (Fig. 3b–d). This Ti corresponds to the TiO2 encapsulation layer grown during the reductive annealing of Pt on the TiO2 support. In contrast, the EDS of Red/Pt does not reveal a Ti layer covering the Pt nanoparticles (Fig. 3f–h) and no visible encapsulation layer was found by HRTEM analyses (Fig. S2†).To corroborate our results with shorter electron beam exposure and higher spatial resolution, EELS was conducted in STEM mode. Representative spectra were measured on encapsulated Pt nanoparticles lying on the surface of TiO2 nanobundles to prevent contributions from the preexisting TiO2 (see Fig. 3e). Fig. 3i displays the intensity distribution of the Ti L2,3-edge over the Pt/Red Pt nanoparticle indicated in Fig. 3e. The visualized Ti signal implies the formation of a TiO2 encapsulation layer surrounding the Pt nanoparticle on Pt/Red, while no Ti coverage was found for Red/Pt (Fig. S3†). The Ti signal intensity is higher at the Pt/TiO2 interface and spreads inhomogeneously throughout the Pt nanoparticle, while fading completely at the edge of the nanoparticle (see Fig. S4†). This suggests a partial TiO2 encapsulation on the Pt nanoparticles. An “incomplete” encapsulation is desirable for highly stable and selective catalysts without losing activity during application.
5. Electrochemical behavior
The electrochemical behavior of the two Pt/TiO2 systems was compared using CV by scanning from 0 to 1.2 VRHE at a scan rate of 200 mV s−1 in an oxygen saturated 0.1 M perchloric acid (HClO4) electrolyte. The results after stabilization of the electrodes (see Fig. S5†) can be found in Fig. 4a. The majority of the electrochemical response of the electrodes in the range 0–0.2 VRHE arises from the hollow TiO2 nanowires. Nevertheless, contributions of Pt can also be observed in both samples despite the low Pt loading.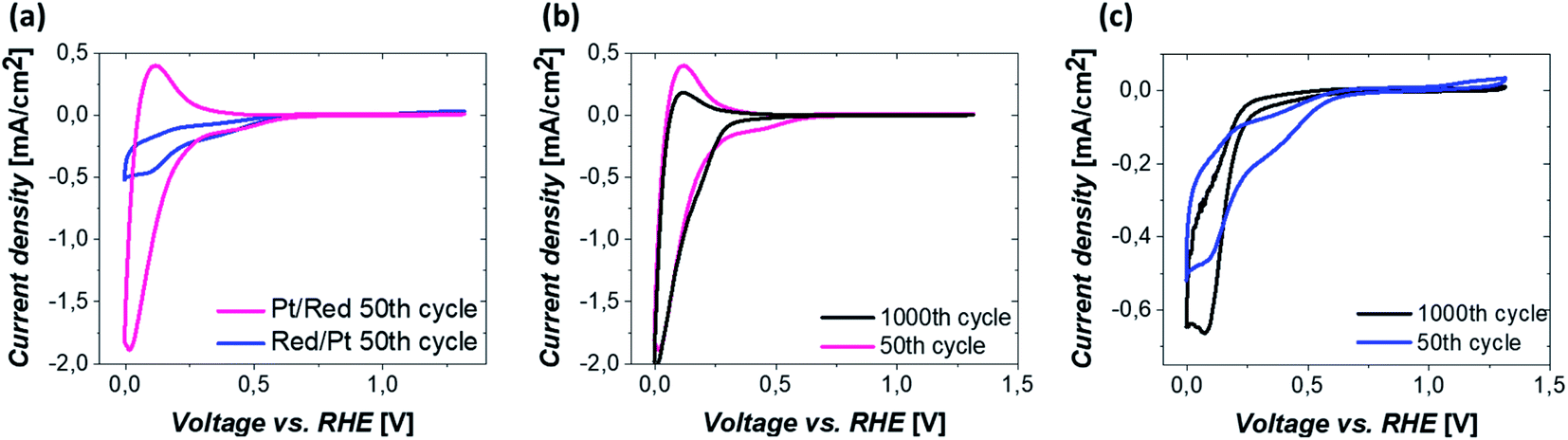 | ||
| Fig. 4 (a) CVs after 50 CV cycles of Pt/Red and Red/Pt. Comparison of CVs after 50 and 1000 CV cycles of (b) Pt/Red and (c) Red/Pt. All measurements were conducted under an oxygen atmosphere in 0.1 M HClO4 electrolyte at a scan rate of 200 mV s−1. | ||
The major CV feature of Pt/Red after 50 cycles (Pt/Red50CV) is the broad anodic peak at ca. 0.1 VRHE and its corresponding cathodic peak at ca. 0.0 VRHE. These peaks can be attributed to both hydrogen underpotential deposition (HUPD) on Pt and H+ intercalation on TiO2 leading to HxTiO2:31–33
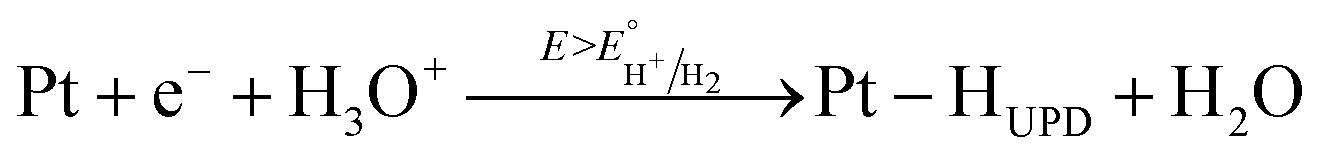 | (1) |
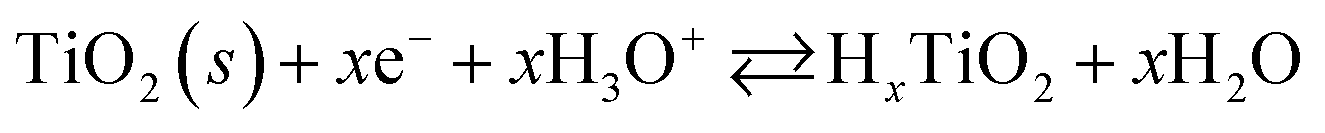 | (2) |
The overlap between HUPD and H+ intercalation hinders a detailed understanding of the exact contribution of each reaction to the overall observed charge and limits a straightforward evaluation of the electrochemically active surface area.34 Yet, the single broad peak in the range of 0 and 0.1 VRHE implies that the H+ intercalation in TiO2 is a major contributor for Pt/Red50cv. In contrast, the 50th cycle of Red/Pt (named Red/Pt50cv) shows a markedly different peak shape and current density compared to Pt/Red50cv with one cathodic maximum at 0.1 VRHE and one anodic maximum at 0.2 VRHE. Such a difference suggests a relatively low H+ intercalation to HUPD contribution compared to Pt/Red.
The reason behind the enhanced contribution of H+ intercalation in the encapsulated Pt/Red system compared to Red/Pt is hydrogen spillover. This consists in the migration of adsorbed H on Pt towards TiO2, aiding the formation of the HxTiO2 phase.35–38 Furthermore, hydrogen spillover is particularly boosted in materials with SMSI due to the presence of a larger metal–metal oxide interface15 which explains the increased H+ intercalation in Pt/Red.
The two Pt/TiO2 systems exhibit a cathodic peak at ca. 0.4 VRHE after the 50th cycle. This feature can be attributed to the presence of Pt (HUPD or Pt reduction, which can take place in a broad potential range due to the H+ intercalation on TiO2 (ref. 39)). The oxygen reduction reaction (ORR) also takes place in both systems33 but is more remarkable in Red/Pt50CV, visibly shifting the H+ intercalation and HUPD peaks towards negative current densities. Moreover, only Red/Pt50CV shows an additional peak at 1.2 VRHE which can be assigned to the onset of Pt oxidation. This suggests a higher accessibility of Pt-related reactions for Red/Pt than for Pt/Red despite its lower Pt content as determined by ICP-MS. Clearly, Pt/Red possesses a lower number of exposed Pt active sites due to the partial TiO2 encapsulation of Pt nanoparticles.
Our results imply that the electrochemical behavior of Pt/TiO2 can be finely controlled by extremely thin encapsulation layers on the platinum surface. Because of the enhanced H+ intercalation, Pt/Red can be a promising material in electrochromic applications such as smart displays.40,41 Furthermore, the encapsulating layer can be used for catalytic reactions with higher selectivity for fuel cell applications.16,42
To investigate the electrochemical stability of both Pt/TiO2 systems, 1000 CV measurements between 0 and 1.2 VRHE were conducted under oxygen saturation conditions and compared with the 50th cycle. The anodic peak of Pt/Red after 1000 cycles at 0.1 VRHE reduces in intensity, while the corresponding cathodic peak slightly increases (Pt/Red1000CV, Fig. 4b). This might be the consequence of a growth in thickness and Pt surface coverage of the TiO2 encapsulation layer, which further enhances the HxTiO2 phase formation. In contrast, for 1000 times cycled Red/Pt (Red/Pt1000CV, Fig. 4c), the shape of the CV peaks attributed to HUPD and H+ intercalation changed and the anodic peak at 0.2 V almost disappeared. This behavior could be explained by the formation of an encapsulation layer also for Red/Pt during cycling even at room temperature.17 In such a case, due to the gradual encapsulation, the contribution of H+ intercalation in Red/Pt increases yielding a broader peak in the range of 0–0.25 VRHE for Red/Pt1000CV which resembles that of Pt/Red50CV. During cycling, the Pt oxidation peak at 1.2 VRHE disappears, as well as the peak at 0.4 VRHE. These changes indicate the loss of Pt active sites either as a consequence of catalyst degradation or due to the growth of TiO2 over Pt nanoparticles during electrochemical redox cycling.17
To confirm the CV-driven encapsulation of Pt nanoparticles and further structural changes in the employed potential region, the local structure of the two Pt/TiO2 systems was analyzed after 1000 CV cycles (Fig. 5). HRTEM analyses (Fig. 5a) and EDS maps (Fig. 5b–d) confirmed the presence of encapsulated Pt nanoparticles on Pt/Red1000CV. Notably, EDS analyses for Red/Pt1000CV also show a low Ti signal at the Pt nanoparticles (Fig. 5g and j). EELS analyses confirmed the existence of a TiO2 layer covering the Pt nanoparticles after cycling for both samples. HAADF images and the intensity distribution of the Ti L2,3-edges of selected particles are shown in Fig. 5e and f for Pt/Red1000CV and in Fig. 5j and k for Red/Pt1000CV, respectively. The presence of a Ti signal observed by EDS and EELS in Red/Pt1000CV, which was not observed for the initial material, strongly supports the growth of an encapsulation layer as a result of electrochemical cycling. A Ti L2,3-edge signal is, however, not present for all Pt nanoparticles in the Red/Pt system, suggesting the presence of both encapsulated and uncovered Pt nanoparticles after 1000 CV cycles. This is consistent with the observed voltammogram for Red/Pt1000CV (Fig. 4c), which shows intermediate behavior between encapsulated (Pt/Red50CV, Fig. 4b) and encapsulation-free models (Red/Pt50CV, Fig. 4c).
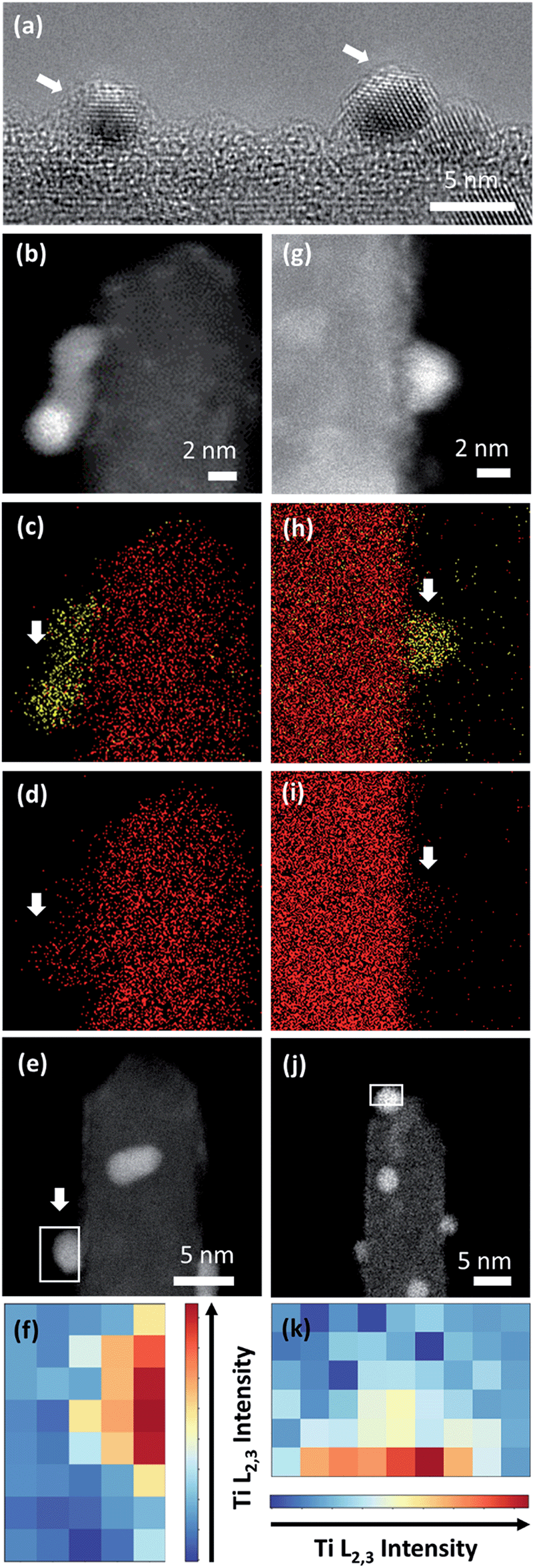 | ||
| Fig. 5 (a) HRTEM image of Pt nanoparticles of Pt/Red after 1000 CV cycles with a visible TiO2 encapsulation layer (indicated with white arrows). HAADF-STEM image and EDS maps of (b–d) Pt/Red and (h–i) Red/Pt after 1000 CV cycles. Ti and Pt are denoted with red and yellow colors, respectively. White arrows indicate the areas where Pt is exposed to vacuum and attached to a single TiO2 nanobundle. HAADF-STEM image after 1000 CV cycles of Pt/Red (e) and Red/Pt (j) indicating the area where EELS analysis was performed. (f and k) Intensity distribution of EELS Ti L2,3 core loss on a Pt nanoparticle after 1000 CV cycles for Pt/Red and Red/Pt, respectively. | ||
Pt nanoparticle degradation might also contribute to the electrochemical changes observed prior to and after electrochemical cycling. Fig. 5b and g show Pt nanoparticles for the Pt/Red1000CV and Red/Pt1000CV, respectively. The average nanoparticle size of Red/Pt increased from 1.6 ± 0.4 to 3.3 ± 0.7 nm (relative size increase of 100%) with cycling (Fig. S1c and e†). For Red/Pt, the encapsulation of a few Pt nanoparticles by TiO2 does not prevent an average growth in the relative particle size during cycling. In the case of Pt/Red, the nanoparticles grew from 2.4 ± 0.6 to 3.0 ± 0.6 nm (relative size increase of ∼25%) (Fig. S1b and d†). This demonstrates that for Pt/Red, the presence of an encapsulation layer clearly increases the stability and prevents agglomeration. Our results highlight the protective role of the initial thin encapsulation layer against particle aggregation during the employed electrochemical degradation cycles.19,43–45
Finally, the change in the oxidation state of Ti was investigated. TiO2 was initially partially reduced to increase the conductivity of the Pt/TiO2 material system through the reduction step.46 Therefore, the stability of the oxidation state of Ti during electrochemical cycles could also modify the properties of Pt/TiO2 materials. EELS mapping was conducted on TiO2 nanobundles prior to and after 1000 CV cycles and the averaged spectra over the whole area are shown in Fig. 6 for both Pt/Red and Red/Pt. Both spectra show Ti L2,3 white lines and similar energy losses for the uncycled samples (see Fig. S6†). However, for Pt/Red, the peaks show lower splitting and are shifted towards lower energy losses by ca. 0.4 eV after 1000 cycles (Fig. 6a). This indicates a slight reduction of the TiO2 support upon electrochemical cycling. Such a reduction is a consequence of the enhanced H+ intercalation in Pt/Red which is not fully reversible.16 In contrast, Red/Pt displays similar energy loss of the Ti L2,3 edge before and after electrochemical cycling (Fig. 6b), indicating a higher stability of the TiO2 support with no or only slight reduction. This can be attributed either to lower H+ intercalation and/or higher reversibility of such a reaction for Red/Pt.
 | ||
| Fig. 6 EELS spectrum showing Ti-L2,3 white lines from a nanobundle of (a) Pt/Red and (b) Red/Pt samples before and after 1000 CV cycles. | ||
In both samples, the lack of re-oxidation of the partially reduced TiO2 after electrochemical cycling underlines its high stability, a promising characteristic for future application of Pt/TiO2 materials.
6. Conclusions
Two Pt/TiO2 systems consisting of Pt nanoparticles on partially reduced TiO2 nanowires were synthesized with and without partial TiO2 encapsulation of the Pt surface. The presence of an inhomogeneous TiO2 encapsulation layer thinner than 1–2 (111)Pt layers was confirmed on Pt nanoparticles (<3 nm) by a combination of HRTEM, EDS and EELS. The encapsulated nanoparticles possess higher stability against agglomeration at high temperatures and in electrochemical measurements. We could also correlate the partial encapsulation of Pt with an enhanced and partially irreversible H+ intercalation, explained by enhanced hydrogen spillover on the encapsulated Pt/TiO2, as well as reduced Pt activity. Moreover, our results also demonstrate the formation of nanoparticle encapsulation caused by electrochemical cycling, thus modifying the long-term electrochemical behavior and application of the materials.7. Experimental section
7.1 Materials
Titanium(IV) butoxide (Ti(OC4H9)4), hydrochloric acid (HCl, 37 wt%), hydrogen hexachloroplatinate solution (H2PtCl6, ∼8 wt% in H2O), sodium formate (HCOONa), tin(II) chloride dihydrate (SnCl2·2H2O), sodium borohydride (NaBH4) and fluorine-doped tin oxide glass (F:SnO2, FTO) were purchased from Sigma Aldrich. All reagents were used without further purification.7.2 Synthesis
Growth of Pt nanoparticles. Two TiO2 electrodes (for Pt/Red: without reduction, for Red/Pt: after reduction) were placed inside a beaker. Deionized water (10 ml), SnCl2·2H2O (20 mg) and HCl (20 μl) were added in this order. The beaker was sonicated (ca. 10 s) in an ultrasound bath and stirred for 10 min. The electrodes were then rinsed with HCl solution (0.2 M) and soaked in H2PtCl6 solution (10 ml, 0.2 mM). Pt nanoparticles were grown by dropwise addition of HCOONa solution (5 ml, 4 mM) and stirring for 5 h.27
Reductive annealing. Two (Pt/)TiO2 electrodes were introduced in a crucible over NaBH4, with the glass side contacting the chemical powder. The crucible was heated under nitrogen flow in a tubular furnace at 400 °C for 2 h.47 After cooling down to room temperature, the electrodes were removed from the furnace and rinsed with deionized water to eliminate excess NaBH4.
7.3 Characterization
Conflicts of interest
There are no conflicts to declare.Acknowledgements
RAA is grateful for the financial support from the International Max Planck Research School for Interface Controlled Materials for Energy Conversion (IMPRS-SurMat). JL acknowledges the financial support from the Alexander von Humboldt Foundation. ML acknowledges the Federal Ministry of Education and Research (BMBF) in the framework of NanoMatFutur (SynKat) for financial support (project number 03XP0265). This study was supported by 2020 Research Grant from Kangwon National University. Andrea Mingers is acknowledged for performing the ICP-MS measurements and Alena Folger for her contribution to the initial stages of the project. We also want to thank Olga Kasian for valuable discussions.References
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2(11), 955 CrossRef CAS.
- J. Zhang and J. W. Medlin, Surf. Sci. Rep., 2018, 73(4), 117 CrossRef CAS.
- N. Cheng, L. Zhang, K. Doyle-Davis and X. Sun, Electrochem. Energy Rev., 2019, 2, 539 CrossRef.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100(1), 170 CrossRef CAS.
- K. Fujiwara, K. Okuyama and S. E. Pratsinis, Environ. Sci.: Nano, 2017, 4(11), 2076 RSC.
- B.-J. Hsieh, M.-C. Tsai, C.-J. Pan, W.-N. Su, J. Rick, H.-L. Chou, J.-F. Lee and B.-J. Hwang, Electrochim. Acta, 2017, 224, 452 CrossRef CAS.
- S. Kattel, B. Yan, J. G. Chen and P. Liu, J. Catal., 2016, 343, 115 CrossRef CAS.
- I. Jiménez-Morales, S. Cavaliere, D. Jones and J. Rozière, Phys. Chem. Chem. Phys., 2018, 20(13), 8765 RSC.
- F. Liu, J. Ftouni, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2019, 11(8), 2079 CrossRef CAS.
- X. Y. Shi, W. Zhang, C. Zhang, W. T. Zheng, H. Chen and J. G. Qi, J. Microsc., 2016, 262(3), 203 CrossRef CAS PubMed.
- M. G. Willinger, W. Zhang, O. Bondarchuk, S. Shaikhutdinov, H.-J. Freund and R. Schlögl, Angew. Chem., Int. Ed., 2014, 53(23), 5998 CrossRef CAS PubMed.
- S. Bernal, J. J. Calvino, M. A. Cauqui, J. M. Gatica, C. López Cartes, J. A. Pérez Omil and J. M. Pintado, Catal. Today, 2003, 77(4), 385 CrossRef CAS.
- S. Zhang, P. N. Plessow, J. J. Willis, S. Dai, M. Xu, G. W. Graham, M. Cargnello, F. Abild-Pedersen and X. Pan, Nano Lett., 2016, 16(7), 4528 CrossRef CAS PubMed.
- J. Liu, ChemCatChem, 2011, 3(6), 934 CrossRef CAS.
- C.-J. Pan, M.-C. Tsai, W.-N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S.-Y. Cheng and B.-J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154 CrossRef CAS.
- B. M. Stühmeier, S. Selve, M. U. M. Patel, T. N. Geppert, H. A. Gasteiger and H. A. El-Sayed, ACS Appl. Energy Mater., 2019, 2(8), 5534 CrossRef.
- E. Hornberger, A. Bergmann, H. Schmies, S. Kühl, G. Wang, J. Drnec, D. J. S. Sandbeck, V. Ramani, S. Cherevko, K. J. J. Mayrhofer and P. Strasser, ACS Catal., 2018, 8(10), 9675 CrossRef CAS.
- J. Zhang, H. Wang, L. Wang, S. Ali, C. Wang, L. Wang, X. Meng, B. Li and D. S. SuF.-S. Xiao, J. Am. Chem. Soc., 2019, 141(7), 2975 CrossRef CAS PubMed.
- L. Wang, J. Zhang, Y. Zhu, S. Xu, C. Wang, C. Bian, X. Meng and F.-S. Xiao, ACS Catal., 2017, 7(11), 7461 CrossRef CAS.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9(2), 120 CrossRef CAS PubMed.
- K.-W. Park, Inorg. Chem., 2005, 44(9), 3190 CrossRef CAS PubMed.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2(1), 18 CrossRef CAS PubMed.
- N. R. Elezovic, V. R. Radmilovic and N. V. Krstajic, RSC Adv., 2016, 6(8), 6788 RSC.
- S.-M. Jung, S.-W. Yun, J.-H. Kim, S.-H. You, J. Park, S. Lee, S. H. Chang, S. C. Chae, S. H. Joo, Y. Jung, J. Lee, J. Son, J. Snyder, V. Stamenkovic, N. M. Markovic and Y.-T. Kim, Nat. Catal., 2020, 3, 639 CrossRef CAS.
- L. Pan, H. Huang, C. K. Lim, Q. Y. Hong, M. S. Tse and O. K. Tan, RSC Adv., 2013, 3(11), 3566 RSC.
- B. Liu and E. S. Aydil, J. Am. Chem. Soc., 2009, 131(11), 3985 CrossRef CAS PubMed.
- L.-S. Zhong, J.-S. Hu, Z.-M. Cui, L.-J. Wan and W.-G. Song, Chem. Mater., 2007, 19(18), 4557 CrossRef CAS.
- J. Lim, S.-H. Kim, R. Aymerich Armengol, O. Kasian, P.-P. Choi, L. T. Stephenson, B. Gault and C. Scheu, Angew. Chem., Int. Ed., 2020, 59(14), 5651 CrossRef CAS PubMed.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44(7), 1861 RSC.
- M. Rahmati, M.-S. Safdari, T. H. Fletcher, M. D. Argyle and C. H. Bartholomew, Chem. Rev., 2020, 120(10), 4455 CrossRef CAS PubMed.
- G. Jerkiewicz, Electrocatalysis, 2010, 1(4), 179 CrossRef CAS.
- Y.-S. Kim, S. Kriegel, K. D. Harris, C. Costentin, B. Limoges and V. Balland, J. Phys. Chem. C, 2017, 121(19), 10325 CrossRef CAS.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C.-j. Liu, J. Mater. Chem. A, 2017, 5(5), 1808 RSC.
- T. Binninger, E. Fabbri, R. Kötz and T. J. Schmidt, J. Electrochem. Soc., 2014, 161(3), H121 CrossRef CAS.
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95(3), 759 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112(5), 2714 CrossRef CAS PubMed.
- M. Choi, S. Yook and H. Kim, ChemCatChem, 2015, 7(7), 1048 CrossRef CAS.
- M. Xiong, Z. Gao and Y. Qin, ACS Catal., 2021, 11(5), 3159 CrossRef CAS.
- S. Proch, K. Kodama, S. Yoshino, N. Takahashi, N. Kato and Y. Morimoto, Electrocatalysis, 2016, 7(5), 362 CrossRef CAS.
- R. R. Kharade, S. S. Mali, S. P. Patil, K. R. Patil, M. G. Gang, P. S. Patil, J. H. Kim and P. N. Bhosale, Electrochim. Acta, 2013, 102, 358 CrossRef CAS.
- W. Wu, M. Wang, J. Ma, Y. Cao and Y. Deng, Adv. Electron. Mater., 2018, 4(8), 1800185 CrossRef.
- J. Durst, A. Orfanidi, P. J. Rheinländer, F. Hasché, C. Eickes, P. Suchsland, M. Binder and H. A. Gasteiger, ECS Trans., 2015, 69(17), 67 CrossRef CAS.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329(5994), 933 CrossRef CAS PubMed.
- Q. Wan, S. Hu, J. Dai, C. Chen and W.-X. Li, J. Phys. Chem. C, 2019, 123(17), 11020 CrossRef CAS.
- S.-Y. Huang, P. Ganesan, S. Park and B. N. Popov, J. Am. Chem. Soc., 2009, 131(39), 13898 CrossRef CAS PubMed.
- A. Folger, J. Kalb, L. Schmidt-Mende and C. Scheu, Nanomaterials, 2017, 7, 289 CrossRef PubMed.
- H. Tan, Z. Zhao, M. Niu, C. Mao, D. Cao, D. Cheng, P. Feng and Z. Sun, Nanoscale, 2014, 6(17), 10216 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00423a |
| This journal is © The Royal Society of Chemistry 2021 |