
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCelery-derived porous carbon materials for superior performance supercapacitors†
Sirui
Liu‡
a,
Yaping
Xu‡
a,
Jinggao
Wu
c and
Jing
Huang
 *ab
*ab
aState Key Laboratory of Silkworm Genome Biology, Key Laboratory of Sericultural Biology and Genetic Breeding, Ministry of Agriculture and Rural Affairs, College of Biotechnology, Southwest University, Chongqing 400715, PR China. E-mail: hj41012@163.com
bInstitute for Clean Energy & Advanced Materials, Faculty of Materials and Energy, Southwest University, Chongqing 400715, P. R. China
cKey Laboratory of Rare Earth Optoelectronic Materials & Devices, College of Chemistry and Materials Engineering, Huaihua University, Huaihua 418000, PR China
First published on 31st July 2021
Abstract
Supercapacitors are of paramount importance for next-generation applications, demonstrating high energy output and an ultra-long cycle life, and utilizing green and sustainable materials. Herein, we utilize celery, a common biomass from vegetables, by a facile low-cost pyrolysis and activation method for use in high-voltage, high-energy, and high-power supercapacitors. The as-synthesized hierarchically porous carbon materials with a high surface area of 1612 m2 g−1 and a large quantity of nitrogen and phosphorus heteroatoms exhibit a high specific capacitance of 1002.80 F g−1 at 1 A g−1 and excellent cycling stability of 95.6% even after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles (10 A g−1) in aqueous electrolytes. Moreover, the assembled symmetric cell delivers a high energy density of 32.7 W h kg−1 at 1200 W kg−1 and an ultra-high stability (loss of 4.8% after 10000 cycles). Therefore, the outstanding electrochemical performance of the materials will be of use in the development of high-performance, green supercapacitors for advanced energy storage systems.
000 cycles (10 A g−1) in aqueous electrolytes. Moreover, the assembled symmetric cell delivers a high energy density of 32.7 W h kg−1 at 1200 W kg−1 and an ultra-high stability (loss of 4.8% after 10000 cycles). Therefore, the outstanding electrochemical performance of the materials will be of use in the development of high-performance, green supercapacitors for advanced energy storage systems.
1. Introduction
Nowadays, the energy crisis and environmental deterioration are growing global issues which seriously hinder the progress of human society.1,2 Therefore, accelerating the innovation and development of alternative energy and storage devices with high energy conversion efficiency and green features is an urgent need.3 At present, energy storage devices are playing important roles in solving problems in energy reserves and supply.4 Common energy storage devices are traditional capacitors, rechargeable batteries and supercapacitors.5,6 Supercapacitors are new energy storage devices between traditional capacitors and rechargeable batteries. Compared with storage batteries and traditional capacitors, supercapacitors have the characteristics of high power density, great cycling stability, low maintenance cost, long life, fast charging and discharging, safety and greenness.7,8 As a result, supercapacitors are now widely used in electronic devices, hybrid electric vehicles, pulsing techniques and so on.9,10However, low energy density is a common problem in most supercapacitors.11 In order to resolve this problem, researchers promote the performance of supercapacitors by constantly improving the properties of electrode materials and electrolyte.12 With respect to electrode materials, usual electrode materials are (i) carbons, (ii) metal oxides and (iii) conducting polymers.13,14 Compared with metal oxides and conducting polymers, carbon materials have large specific surface areas, meso-/micro-porosity, non-toxicity, high chemical stability and good electrical conductivity for accelerating the diffusion of electrolyte ions,15,16 which makes the study of carbon-based materials of much interest. Common carbon-based materials include biomass, carbon fibers, carbon black, carbon aerogels, carbon nanotubes, graphene and so on.17 For carbon fibers, graphene, carbon aerogels, carbon nanotubes, etc., they can't be used for commercialization owing to their high cost, non-renewable carbon precursors and complex synthesis process.18–20 Meanwhile, for biomass-derived carbon, it could just make up for these shortcomings. Biomass has a natural structure with natural multi-stage pores, which makes the synthesis of biomass-based carbon easier and safer, cheaper, and greener than that of other carbon materials. In addition, it is also abundant and renewable.21–23 Based on the above facts, it could be deduced that biomass is a good precursor of electrode materials applied to supercapacitors.24 Currently, a variety of biomass have been used as precursors of carbon materials for supercapacitors, such as bamboo, hair, wheat, bagasse, orange peel, silk, pig bone, etc.11,21,25 Although most biomass-based carbons have good electrochemical performance, they still have drawbacks such as limited regional distribution and difficulties in their production, collection, and transport, which could restrict their further industrialization.25–28
Celery is a kind of cellulose-rich, hollow and porous plant, widely planted in China. This herein motivates us to develop a facile, non-templating and readily scalable strategy for the efficient synthesis of porous carbon through pre-treatment via carbonization of celery and subsequent activation with KOH as the agent (Scheme 1). In this study, we have investigated the effects of the proportion of KOH/precursors and carbonization conditions on the pore structures of the biocarbons and have also systematically evaluated the electrochemical performances of the biocarbons.
 | ||
| Scheme 1 Synthesis of porous carbon from celery. | ||
2. Experimental section
2.1 Materials and methods
:1.5 were thoroughly ground in an agate mortar. After grinding, the same pyrolysis process was carried out in the tube furnace according to the preparation of CC. Finally, the obtained carbon was named as CAC-4. For comparison, the different mass ratios of CC and KOH (2:1, 3:2, 1:1, and 1:2) were also investigated in the above process. The samples were denoted as CAC-1, CAC-2, CAC-3 and CAC-5. In addition, CAC-6, CAC-7 and CAC-8 were prepared at 600, 700 and 900 °C following a similar procedure to that of CAC-3. In the whole process, all carbonized samples were neutralized with diluted HCl and washed with deionized water, and then dried at 80 °C for 12 h.
2.2 Characterization
The XRD patterns of all samples were recorded using powder X-ray diffraction (Shimadzu XRD-7000). The surface morphology and structure of the samples were observed using scanning electron microscopy (FESEM, JSM-7800F) and transmission electron microscopy (TEM, JEOL 2100). Nitrogen sorption isotherms were obtained using an Autosorb-1 (Quantachrome Instruments). The specific surface area was calculated by the modified Brunauer–Emmett–Teller (BET) method. The pore size distributions and the pore volume were analyzed from the adsorption branch isotherms by the density functional theory (DFT) method. Moreover, the total pore volume (Vtot) was estimated from the amount adsorbed at a relative pressure P/P0 of 0.990. The micropore volume (Vmic) and micropore surface area (Smic) were determined using t-plot theory. Raman spectra were acquired with a Jobin-Yvon HR 800 spectrometer. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo Fisher Scientific (Escalab 250xi, USA). Fourier transform infrared (FT-IR) spectra were recorded on a Thermo Scientific Nicolet iS 50 spectrometer.2.3 Electrochemical measurements
For the two-electrode system, a homogeneous slurry of the electroactive material, polytetrafluoroethylene (PTFE), and acetylene black with a weight ratio of 80:10:10 in ethanol was formed and pasted onto the nickel foam current collector (1 cm × 1 cm), and then vacuum dried at 80 °C for 12 h. The loading of the active material for each working electrode was measured to be ∼3 mg cm−2. And then, a glass-fiber filter paper (Waterman, GF/B) as a separator and 1 M KOH aqueous solution as the electrolyte were used to assemble a test cell. For the three-electrode system, the working electrode was manufactured via the dispersion of active carbon in the mixture of Nafion and ethanol (1:20) dropped on the glassy carbon electrode, accompanied by platinum foil Hg/HgO as the counter and reference electrodes, respectively. Electrochemical characterization was carried out on an electrochemical workstation (Shanghai Chenhua Instrument Co. Ltd, China).
For the two-electrode system, the gravimetric specific capacitance of a single electrode is calculated using the equation
| Csp = 2I × Δt/m × ΔV | (1) |
For the three-electrode system, the gravimetric specific capacitance can be calculated using the following equation:
| Csp = I × Δt/m × ΔV | (2) |
The energy density E (W h kg−1) and the power density P (W kg−1) are calculated using the following equations:
| E = CspV2/(2 × 3.6) | (3) |
| P = 3600E/Δt | (4) |
3. Results and discussion
3.1 Materials characterization
The porous structure of pristine celery derived porous carbon is analyzed by field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM). On account of the FESEM image of CAC-4 (Fig. 1a), it is indicated that huge numbers of pores are randomly distributed over their surfaces. This abundant pore formation originates from the evolution of gases such as H2 and CO2 owing to carbon lamellae upon decomposition of KOH at high temperatures. The TEM images (Fig. 1b) reveal the presence of randomly distributed, worm-like, irregularly shaped pores, mainly located in the region of the micropores. The TEM images also illustrate the absence of any graphitic domains, indicating the amorphous nature of CAC-4.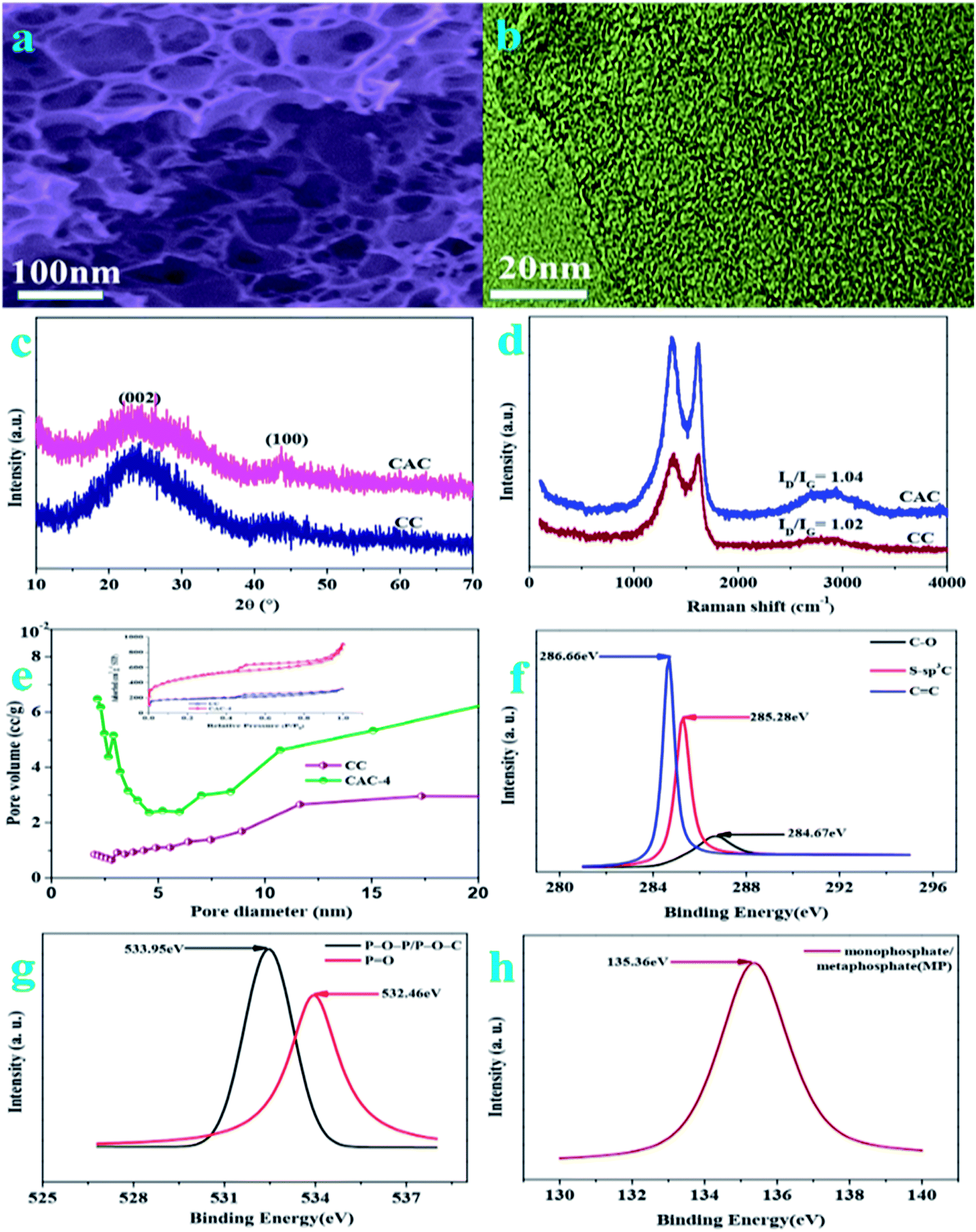 | ||
| Fig. 1 (a) FESEM image of CAC-4; (b) TEM image of CAC-4; (c) XRD patterns of CC and CAC-4; (d) Raman spectra of CC and CAC-4; (e) N2 adsorption/desorption isotherms and the pore size distributions of CC and CAC-4; (f) high resolution C 1s of CAC-4; (g) high resolution O 2p of PSAC-3; (h) high resolution P 2p of CAC-4. | ||
On account of FT-IR in the 500–4000 cm−1 region (Fig. S1†), the chemical nature of the as-synthesized samples could be characterized. The strong peak at 3427 cm−1 is typically ascribed to the O–H stretching vibration (hydroxyl groups and chemisorbed water) and/or N–H symmetric stretching vibration.1 The bands at 2978 cm−1 are due to the C–H stretching vibrations. The bands at 1655 cm−1 are due to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (COOH functional groups). The spectra at 1439 cm−1 and 1325 cm−1 are attributed to the deformation vibrations of C–H and O–H, respectively. The signal located at 1049 cm−1 is attributed to C–O–C (symmetric angular deformation of ethers).3 Based on the FT-IR analysis, the existence of oxygen and phosphorus species in the as-synthesized porous ACs could be confirmed.
O (COOH functional groups). The spectra at 1439 cm−1 and 1325 cm−1 are attributed to the deformation vibrations of C–H and O–H, respectively. The signal located at 1049 cm−1 is attributed to C–O–C (symmetric angular deformation of ethers).3 Based on the FT-IR analysis, the existence of oxygen and phosphorus species in the as-synthesized porous ACs could be confirmed.
The X-ray diffraction (XRD) patterns (Fig. 1c) of the as-synthesized samples demonstrate two characteristic peaks located at 2θ of ∼26.4° and ∼43.7°, owing to the (002) and (100) planes of graphitic carbon layers, respectively.29 The broad diffraction peak of carbon is due to the abundant micropores in the samples, confirming the amorphous carbon structure. According to the spectra of CAC-4 and CC, the activation of KOH could lead to increasing porosity as the spectra of CAC-4 show a broader peak.30 The Raman spectra of CAC-4 in Fig. 1d indicate two peaks located at about ∼1360 cm−1 and ∼1618 cm−1, attributed to D (disordered carbon) and G (graphitic carbon).31 The G band is ascribed to graphitic in-plane stretching from sp2 carbon and the D band corresponds to defects and disorders in the carbon structure owing to disordered carbon atoms with dangling bonds. By virtue of the activation of KOH, the ID/IG ratio obviously increases from 1.02 for CC to 1.04 for CAC-4, confirming that the graphitization intensity gradually becomes weaker, which agrees well with the XRD analysis and TEM images.
The porosity parameters of these carbon samples are further characterized by N2 adsorption/desorption isothermal analysis (Fig. 1e), and the relevant data are listed in Table 1. The isotherm of CC (inset: Fig. 1e) is type I, indicating the microporous nature of the carbon with a surface area of ∼658 m2 g−1 and a pore volume of 0.69 cm−3 g−1. CAC-4 also shows a type I isotherm owing to strong adsorption on micropores, followed by an H4 hysteresis by virtue of the presence of mesopores. The BET surface area of CAC-4 is calculated to be ∼1613 m2 g−1 with a pore volume of 1.034 cm−3 g−1. In light of the DFT pore size distribution curve, most pores are smaller mesopores less than 10 nm in size, with major pores ranging from 2 to 8 nm. The average pore sizes of CAC-4 and CC are 3.296 nm and 3.303 nm, respectively. The activation of KOH could lead to increasing the textural properties and further maximizing the ion adsorption sites, owing to generating a huge amount of gas (CO and CO2) from the activation agent (KOH).32 In view of the effect of the activation temperature, the BET surface area, pore volume and average pore size of CAC-6 (600 °C) and CAC-8 (900 °C) are 728.388 m2 g−1, 0.463 cm−3 g−1, and 2.934 nm, and 1611.417 m2 g−1, 1.051 cm−3 g−1, and 2.778 nm, respectively. With the increase of the activation temperature from 600 °C to 800 °C, the textural properties similarly increase. Meanwhile, with the activation temperature further increasing to 900 °C, the pores may collapse, which unfavorably contributes to the decrease of the textural properties. Therefore, CAC-4 could effectively be conducive to fast ion movement and accommodation of large solvated ions, further resulting in high capacitance and providing quick electrolyte diffusion into deeper pores.33
| Sample | S BET | V tot | S mic | S me | S ma | V mic | V me | V ma |
|---|---|---|---|---|---|---|---|---|
| a S BET: BET surface area. b V tot: total volume. c S mic: micropore surface area. d S me: mesopore surface area. e S ma: macropore surface area. f V mic: micropore volume. g V me: mesopore volume. h V ma: macropore volume. | ||||||||
| CC | 657.70 | 0.69 | 175.34 | 467.36 | 14.996 | 0.16 | 0.45 | 0.08 |
| CAC-1 | 1271.6 | 0.836 | 163.36 | 865.62 | 242.61 | 0.21 | 0.583 | 0.043 |
| CAC-2 | 1096.4 | 0.799 | 149.06 | 669.38 | 277.95 | 0.125 | 0.386 | 0.288 |
| CAC-3 | 1630.1 | 1.155 | 363.62 | 1068.5 | 197.99 | 0.362 | 0.646 | 0.147 |
| CAC-4 | 1612.6 | 1.034 | 116.32 | 1023.2 | 473.17 | 0.321 | 0.587 | 0.126 |
| CAC-5 | 1587.8 | 1.467 | 105.72 | 937.26 | 544.81 | 0.285 | 0.786 | 0.396 |
| CAC-6 | 728.39 | 0.463 | 96.42 | 561.68 | 70.288 | 0.145 | 0.236 | 0.082 |
| CAC-7 | 986.15 | 0.78 | 123.64 | 625.73 | 236.78 | 0.175 | 0.473 | 0.132 |
| CAC-8 | 1611.4 | 1.051 | 107.42 | 1016.5 | 487.48 | 0.352 | 0.603 | 0.096 |
The surface functionalities and incorporation of O and P heteroatoms in CAC are investigated by XPS, which could also contribute to high pseudocapacitance. To understand the chemical nature of dopants in CAC-4, the C 1s, O 1s, and P 2p spectra are deconvoluted. The high resolution C 1s (Fig. 1f) spectra have been resolved into three peaks centered at around 284.67, 285.28, 286.0, and 286.66 eV, which originate from C–C, S–sp3C, and CC, respectively. The O 2p (Fig. 1g) could also be resolved into two peaks, P–O–P/P–O–C (533.95 eV) and PO (532.46 eV), both of which are beneficial to electrochemical redox activity and wettability.34 The high-resolution P 2p region (Fig. 1h) reveals the presence of monophosphate/metaphosphate (MP) (135.36 eV).35 The P-doped configuration of CAC-4 could inhibit the formation of electrophilic oxygen species but also greatly stabilizes the electrochemical interface of the graphene electrode. These results agree well with the analysis of its FT-IR spectrum. The presence of heteroatoms increases the polarity of the pores and thereby facilitates easy contact between the pores and electrolyte. In general, the co-doping effect of heteroatoms could greatly modify the chemical and surface nature of the porous carbon and improve the ion and electron transfer kinetics of carbon, which could contribute to a superior performance.36
3.2 Electrochemical behaviors of the electrode
As described in Fig. 2a, the CV curves for CAC-4 retain typical rectangular shapes at different scan rates, even at a high scan rate of 200 mV s−1, demonstrating its excellent capacitive behavior and rate performance.37 No redox peaks/humps due to the pseudocapacitive interaction of ions with functional groups are observed. Moreover, the slight changes of the curve shapes demonstrate rapid migration at high scanning rates with an increase of the scanning rate.38 At the same time, CAC-4 encloses the larger area than CC on the basis of the CV curves (Fig. S2a†) owing to the chemical activation. On account of the effect of ratios of precursor/KOH, CV curves of CAC samples (Fig. 2b) at a scan rate of 100 mV s−1 show that CAC-4 with a ratio of 1:1.5 exhibits the largest area of the loop, suggesting the highest capacitance.39 As for the temperature performance, the CV curve of CAC-4 (Fig. S2b†) presents the largest encircled area, demonstrating the superior performance at a temperature of 800 °C. Above all, CAC-4 displays the largest CV curve area due to its broader PSD and higher mesoporosity that contribute to facile and fast electrolyte ion transport and shorten the diffusion distances from the electrolyte to the inner micropore surface.40
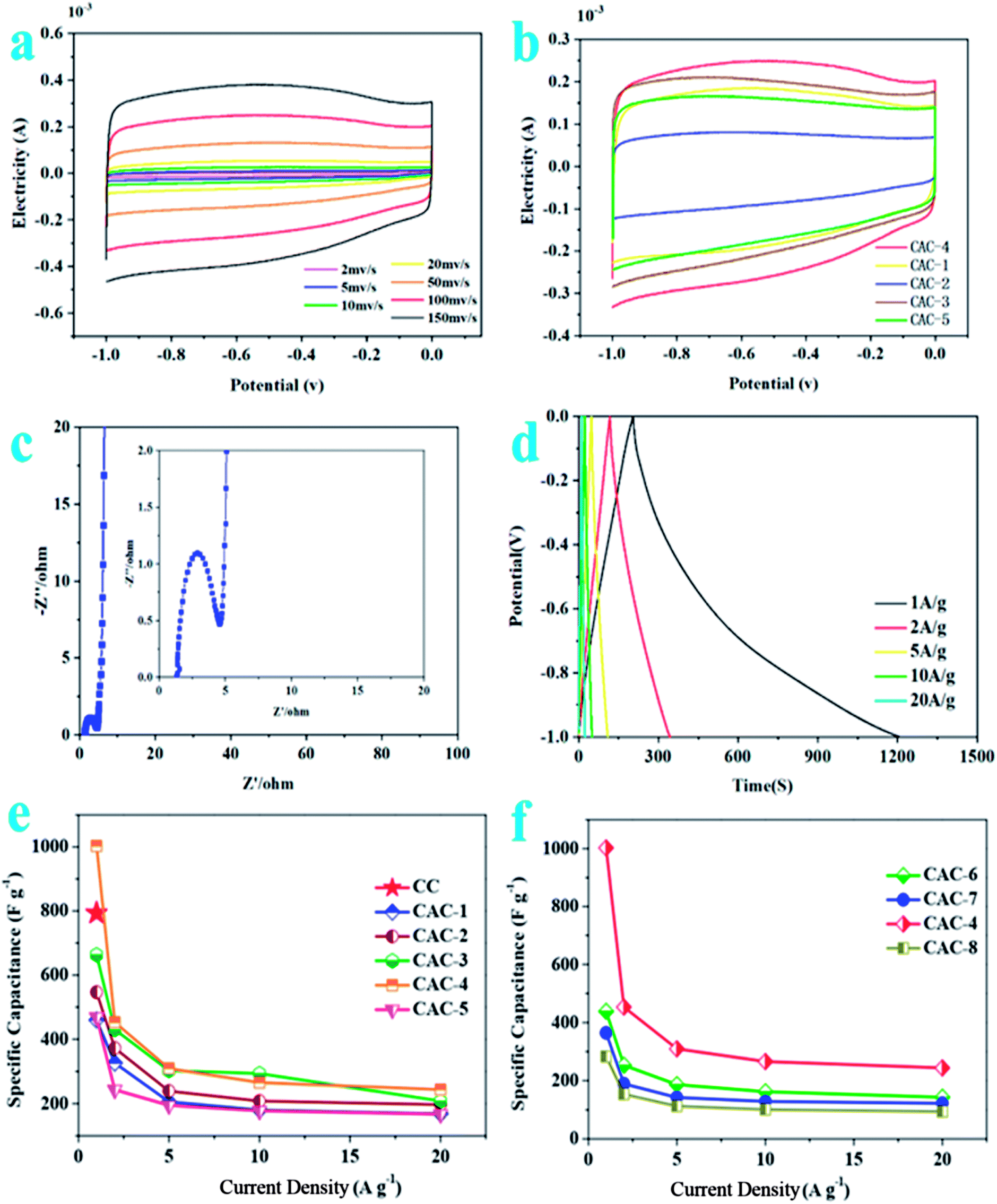 | ||
| Fig. 2 (a) CV curves of CAC-4 at different scan rates. (b) CV curves of CAC samples prepared under different ratios of precursor/KOH at 100 mV s−1. (c) Nyquist plots of CAC-4. (d) Galvanostatic charge/discharge curves of CAC-4 at different current densities. (e) Capacitances of CAC samples prepared under different ratios of precursor/KOH at different current densities. (f) Capacitances of CAC samples prepared under different temperatures at different current densities. | ||
The Nyquist plots (Fig. 2c) display an initial solution resistance, a semi-circle in the high-frequency region that can be attributed to charge transfer resistance originating from electronic and ionic conductivity of the carbon electrode, and a Warburg tail at low frequency indicating a capacitive-type storage. This semicircle is associated with the Faraday process of the charge transfer at the electrode/electrolyte interface.41 The small semicircle of CAC-4 indicates the good ionic conductivity owing to the high degree of graphitization that leads to high electrical conductivity of electrode materials. CAC-4 also exhibits a low intercept (1.29 Ω) on the real axis owing to the surface wettability derived from the coexistence of P and O. The steep linear curve of CAC-4 in the low-frequency region is closer to a vertical line, demonstrating that the porous carbon can improve the capacitive behavior effectively.42 The electrode materials have low ionic resistance inside the porous structure that facilitates the fast diffusion of electrolyte ions into the porous network. The electrode materials have good conductivity and low charge transfer resistance that favor fast charge transfer between the electrolytes and electrode materials.43
The GCD curves (Fig. 2d) are approximately linear and quasi-symmetrical, and almost without a voltage drop (IR), indicating a good capacitive behavior, and high coulombic efficiency and conductivity.44 This also demonstrates both EDLC and pseudocapacitive performance, which is in accordance with the CV results. Based on the discharge curves, CAC-4 delivers a specific capacitance of 1002.80 F g−1 at 1 A g−1, which is much higher than that of CC (∼223.1 F g−1). A capacitance of 244 F g−1 can still be retained at 20 A g−1, revealing the good energy storage output capacity at high current density. The capacitances are also higher than those of many other carbonaceous capacitive materials derived from biomass.45–48 The negligible value of the internal resistance (IR) drop implied a low internal resistance due to the superior electrical conductivity and well-developed hierarchical porosity. The good conductivity of hierarchical porous carbon can improve the electrical properties of the composite electrodes. The specific capacitances decrease gradually with the increase of discharging current density possibly owing to inadequate time for electrolyte ion diffusion throughout the pores. The lowered capacitance at higher current densities is also ascribed to the insufficient electrolyte ion diffusion kinetics across the micropores due to the narrow pore size at higher operating current densities, and the reduced accumulation amount of electrolyte ions onto the porous electrode interface leads to the decrement in specific capacitance.49 As the ratio of KOH/CC is increased, the specific capacitance increases until reaching an optimum value, beyond which it drastically decreases again. According to Fig. 2e, it is obvious that the specific capacitances of the CAC samples increase with the decrease of the ratio of CC to KOH (2:1 vs. 3:2 vs. 1:1 vs. 2:3; 459.67 vs. 546.43 vs. 663.03 vs. 1002.80 F g−1 at 1 A g−1; CAC-1 vs. CAC-2 vs. CAC-3 vs. CAC-4). However, the specific capacitances for the CAC samples also progressively decrease with the decrease of the ratio of CC/KOH (2:3 vs. 1:2, 1002.80 vs. 468.73 F g−1, CAC-4 vs. CAC-5). In addition, the specific capacitance of CAC-4 at 800 °C (Fig. 2f) obviously exceeds those of CAC-6 (at 600 °C) and CAC-7 (at 700 °C) as well as CAC-8 (at 900 °C) (800 vs. 600 vs. 700 vs. 900 °C, 1002.80 vs. 438.47 vs. 364.50 vs. 283.10 F g−1 at 1 A g−1; CAC-4 vs. CAC-6 vs. CAC-7 vs. CAC-8), due to the hierarchically porous 3D structure, which allows efficient infiltration of the electrolyte, the direct channels allowing fast transport of ions and the highly conductive 3D carbon frameworks (CFs) ensuring efficient current collection.50
As shown in Fig. S2c,† the specific capacitance decreases slowly at the first 1000 cycles and maintains at about 95.6% of the initial specific capacitance with a slight fluctuation over 10000 cycles, indicating a good cycling stability.51 This benefits from the unique microporous and mesoporous ratios, stable porous biocarbon structures, high oxygen content, lower impedance, and the specific surface area of the CAC-4 biocarbon itself (Fig. 3).
 | ||
| Fig. 3 Schematic diagram of the formation of the supercapacitor. | ||
3.3 Supercapacitor performance
The electrochemical performance of CAC-4 based supercapacitors is evaluated in a fully assembled two-electrode cell with 1 M KOH electrolyte. Shown in Fig. 4a, the CV curves in a symmetrical two electrode configuration at different scan rates ranging from 2 to 200 mV s−1 exhibit an approximately rectangular shape with slight faradaic humps, indicating the combination of double-layer capacitive and pseudocapacitive behavior. Even at a high scan rate of 200 mV s−1, the CV profiles still retain a rectangular shape without obvious distortion, demonstrating its excellent capacitive behavior and rate performance. The specific capacitance of the supercapacitor device is greatly dependent on efficient utilization of pores for double layer formation by ions.52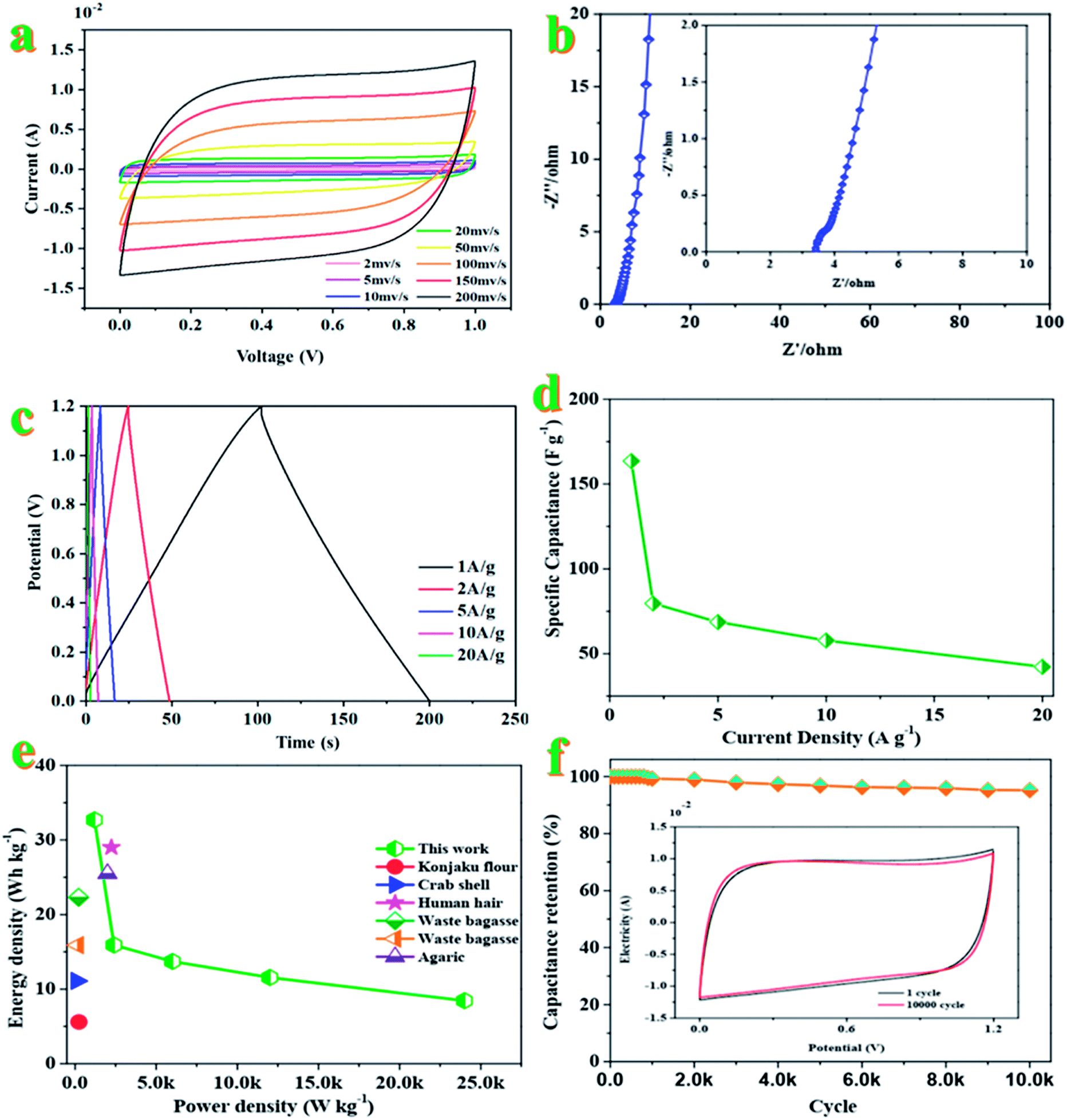 | ||
| Fig. 4 (a) CV curves of the CAC//CAC symmetrical supercapacitor at different scan rates in the voltage window of 0–1.0 V. (b) Nyquist plots of the CAC//CAC symmetrical supercapacitor. (c) Galvanostatic charge/discharge curves of the CAC//CAC symmetrical supercapacitor at different current densities. (d) Specific capacitances of the as-assembled CAC symmetrical supercapacitor based on the total mass of the active materials of the two electrodes at different current densities. (e) Ragone plots of the CAC symmetrical supercapacitor and other previously reported carbon-based symmetric supercapacitors. (f) Cycle performance of the CAC symmetrical supercapacitor at a current density of 10 A g−1 (inset photograph of CV curves for recycling). | ||
The Nyquist plots of the device (Fig. 4b) indicate a small impedance of 3.32 Ω with a vertical line in the low-frequency region, demonstrating fast and efficient ionic transport in the KOH electrolyte.53 As for Fig. 4c, the approximate symmetry and the linear GCD curves over the whole voltage window of 0–1.2 V confirm the reversible charge–discharge process, consistent with the CV results mentioned above. Based on the GCD curves, the supercapacitor displays a specific capacitance of 163.5 F g−1 at 1 A g−1. Generally, the excellent rate capacity of the device is closely associated with the low polarization effects of the electrode, which is governed by the high electron conductivity of the electrodes and rapid electrolyte diffusion.54Fig. 4d compares the capacitance of the device in a range of current density from 1 to 20 A g−1, which brings about the change of capacitance from ∼164 to ∼42 F g−1. The good capacitance values could be attributed to the interconnected honeycomb-like structure combining the developed hierarchical porosity that facilitated rapid electrolyte transfer and the enhanced degree of graphitization that imparted good electrical conductivity. The specific capacitance is decreased with an increase of the current density, which may be attributed to the slow diffusion and the migration of protons through the electrodes.55 The lowered capacitance at higher current densities is also attributed to the insufficient electrolyte ion diffusion kinetics across the micropores owing to the narrow pore size at higher operating current densities; the reduced accumulation amount of electrolyte ions onto the porous electrode interface results in the decrement in specific capacitance.56 The presence of dual heteroatoms such as O and P greatly reduces such resistance and increases the pore utilization to achieve a high capacitance.57 The Ragone plot of the device in KOH electrolyte calculated from discharge curves at different current densities is displayed in Fig. 4e. This assembled two-electrode supercapacitor shows a maximum energy density of 32.7 W h kg−1 with a power density of 1200 W kg−1 at 1 A g−1. Even at a power density of 24 kW kg−1, the energy density still reached 8.4 W h kg−1 at a current density of 20 A g−1, which is higher than those of commercially available supercapacitors (3–5 W h kg−1) and other previously reported biomass-derived heteroatom-doped carbonaceous symmetric supercapacitors.58–62 It is well known that the energy density was suppressed with the increase of current density since the limited pores on the surface were accessed by electrolyte ions for fast discharging at high current density, while almost all pores could be utilized at a low current density. These results indicate that heteroatom doped carbon materials could reach high capacitance in aqueous solution. The nanoporous carbon-based SC can deliver a great mass of energy and power densities simultaneously, owing to its ultrafast electron/ion diffusion kinetics during the electrochemical process. The superior energy–power output behavior effectively bridges the performance gap between batteries and capacitors.63
The specific capacitance decreased slowly at the initial stage and then tended to be stable at 95.2% of the initial capacitance after 10000 cycles (Fig. 4f). Only 4.8% of the initial capacitance is lost after 10000 cycles, indicating the high electrochemical stability. The high cycling stability can also be confirmed by the CVs before and after 1000 charging–discharging cycles (inset in Fig. 4f). Both curves are almost fully overlapped except a faint decay in plateau current for the latter, further evidencing the excellent cycling stability of the supercapacitor.64 The superior SC performance (high specific capacitance and high capacity retention) of the nanoporous carbon material could be attributed to the high specific surface area with ultrahigh pore volume and a high degree of graphitization, leading to providing more charge storage sites as well as improving the diffusion kinetics of electrolyte ions at high current rates.65,66
4. Conclusions
In summary, a porous carbon doped with oxygen and phosphorus is derived from a sustainable, low-cost, and environmentally abundant celery biomass precursor by a one-step pyrolysis and activation process. The as-obtained carbon materials have abundant micropores with a super-high surface area of 1612.639 m2 g−1 and heteroatoms of N and P, which contributes to a high specific capacitance of 1002.80 F g−1 at 1 A g−1. The symmetric supercapacitor displays a remarkable specific energy of 32.7 W h kg−1 at 1.2 kW kg−1 and excellent cycling stability with 95.2% retention after 10000 cycles. Therefore, this work provides a facile strategy to synthesize a porous carbon with excellent electrochemical performance, which would be a promising material for supercapacitors, adsorbents, and catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Faculty of Materials and Energy and Institute for Clean Energy & Advanced Materials, Southwest University and Chongqing Key Laboratory for Advanced Materials and Technologies of Clean Electrical Power Sources. This work was sponsored by the Natural Science Foundation of Chongqing, China (cstc2020jcyj-msxmX0019).References
- S. Y. Gao, X. G. Li, L. Y. Li and X. J. Wei, Nano Energy, 2017, 33, 334 CrossRef CAS.
- A. M. Elshahawy, C. Guan, X. Li, H. Zhang, Y. T. Hu, H. J. Wu, S. J. Pennycook and J. Wang, Nano Energy, 2017, 39, 162 CrossRef CAS.
- Y. N. Gong, D. L. Li, C. Z. Luo, Q. Fu and C. X. Pan, Green Chem., 2017, 19, 4132 RSC.
- D. D. Shan, J. Yang, W. Liu, J. Yan and Z. J. Fan, J. Mater. Chem. A, 2016, 4, 13589 RSC.
- J. Wang, P. Nie, B. Ding, S. Y. Dong, X. D. Hao, H. Dou and X. G. Zhang, J. Mater. Chem. A, 2017, 5, 2411 RSC.
- Y. L. Shao, M. F. E. Kady, J. Y. Sun, Y. G. Li, Q. H. Zhang, M. F. Zhu, H. Z. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233 CrossRef CAS PubMed.
- S. J. Song, F. W. Ma, G. Wu, D. Ma, W. D. Geng and J. F. Wan, J. Mater. Chem. A, 2015, 3, 18154 RSC.
- S. A. Dong, X. J. He, H. F. Zhang, X. Y. Xie, M. X. Yu, C. Yu, N. Xiao and J. S. Qiu, J. Mater. Chem. A, 2018, 6, 15954 RSC.
- F. Zhang, T. F. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. S. Chen, Energy Environ. Sci., 2013, 6, 1623 RSC.
- M. Biswal, A. Banerjee, M. Deo and S. Ogale, Energy Environ. Sci., 2013, 6, 1249 RSC.
- Y. S. Yun, S. Y. Cho, J. Y. Shim, B. H. Kim, S. J. Chang, S. J. Baek, Y. S. Huh, Y. Tak, Y. W. Park, S. Park and H. J. Jin, Adv. Mater., 2013, 25, 1993 CrossRef CAS PubMed.
- C. Zhong, Y. D. Deng, W. B. Hu, J. L. Qiao, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2015, 44, 7484 RSC.
- Y. G. Wang, Y. F. Song and Y. Y. Xia, Chem. Soc. Rev., 2016, 45, 5925 RSC.
- Y. Huang, H. F. Li, Z. F. Wang, M. S. Zhu, Z. X. Pei, Q. Xue, Y. Huang and C. Y. Zhi, Nano Energy, 2016, 22, 422 CrossRef CAS.
- C. Zequine, C. K. Ranaweera, Z. Wang, S. Singh, P. Tripathi, O. N. Srivastava, B. K. Gupta, K. Ramasamy, P. K. Kahol, P. R. Dvornic and R. K. Gupta, Sci. Rep., 2016, 6, 31704 CrossRef CAS PubMed.
- W. Q. Tian, Q. M. Gao, Y. L. Tan, K. Yang, L. H. Zhu, C. X. Yang and H. Zhang, J. Mater. Chem. A, 2015, 3, 5656 RSC.
- Z. Y. Peng, Y. B. Zou, S. Q. Xu, W. B. Zhong and W. T. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 22190 CrossRef CAS PubMed.
- R. Thangavel, A. G. Kannan, R. Ponraj, V. Thangavel, D. W. Kim and Y. S. Lee, J. Mater. Chem. A, 2018, 6, 17751 RSC.
- K. Xie, X. T. Qin, X. Z. Wang, Y. N. Wang, H. S. Tao, Q. Wu, L. J. Yang and Z. Hu, Adv. Mater., 2012, 24, 347 CrossRef CAS PubMed.
- Y. Y. Zhang, J. J. He, Z. Gao and X. D. Li, Nano Energy, 2019, 65, 104045 CrossRef CAS.
- S. C. Wei, H. Zhang, Y. Q. Huang, W. K. Wang, Y. Z. Xia and Z. B. Yu, Energy Environ. Sci., 2011, 4, 736 RSC.
- S. Y. Lu, M. Jin, Y. Zhang, Y. B. Niu, J. C. Gao and C. M. Li, Adv. Energy Mater., 2017, 9, 1702545 Search PubMed.
- J. Niu, R. Shao, J. J. Liang, M. L. Dou, Z. L. Li, Y. Q. Huang and F. Wang, Nano Energy, 2017, 36, 322 CrossRef CAS.
- Z. Li, Z. W. Xu, X. H. i Tan, H. L. Wang, C. M. B. Holt, T. Stephenson, B. C. Olsen and D. Mitlin, Energy Environ. Sci., 2013, 6, 871 RSC.
- W. J. Qian, F. X. Sun, Y. H. Xu, L. H. Qiu, C. H. Liu, S. D. Wang and F. Yan, Energy Environ. Sci., 2014, 7, 379 RSC.
- M. H. Yu, Z. F. Wang, H. Z. Zhang, P. P. Zhang, T. Zhang, X. H. Lu and X. L. Feng, Nano Energy, 2019, 65, 103987 CrossRef CAS.
- K. L. Hong, L. Qie, R. Zeng, Z. Q. Yi, W. Zhang, D. Wang, W. Yin, C. Wu, Q. J. Fan, W. X. Zhang and Y. H. Huang, J. Mater. Chem. A, 2014, 2, 12733 RSC.
- X. L. Wu, L. L. Jiang, C. L. Long and Z. J. Fan, Nano Energy, 2015, 13, 527 CrossRef CAS.
- R. R. Cessa, H. Grebel, Z. Jiang, C. Fukuda, H. Pita, T. S. Chowdhury, Z. Dong and Y. Wan, Environ. Prog. Sustainable Energy, 2018, 37, 155 CrossRef.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605366 Search PubMed.
- Y. C. Liu, B. B. Huang, X. X. Lin and Z. L. Xie, J. Mater. Chem. A, 2017, 25, 13009 RSC.
- Z. N. Yu, L. Tetard, L. Zhai and J. Thomas, Energy Environ. Sci., 2015, 3, 702 RSC.
- B. B. Chang, W. W. Shi, S. C. Han, Y. N. Zhou, Y. X. Liu, S. R. Zhang and B. C. Yang, Chem. Eng. J., 2018, 350, 585 CrossRef CAS.
- M. L. Sánchez, A. Primo and H. García, Angew. Chem., Int. Ed., 2013, 52, 11813 CrossRef PubMed.
- R. Huang, J. Wang, B. Zhang, K. H. Wu, Y. Zhang and D. S. Su, Catal. Sci. Technol., 2018, 8, 1522 RSC.
- Y. N. Zhou, J. Li, X. P. Gao, W. Chu, G. P. Gao and L. W. Wang, J. Mater. Chem. A, 2021, 9, 9979 RSC.
- T. L. Mei, C. Wang, M. Liao, J. X. Li, L. Y. Wang, C. Q. Tang, X. M. Sun, B. J. Wang and H. S. Peng, J. Mater. Chem. A, 2021, 9, 10104 RSC.
- H. Zheng, Q. P. Cao, M. N. Zhu, D. Xu, H. Y. Guo, Y. Li and J. H. Zhou, J. Mater. Chem. A, 2021, 9, 10120 RSC.
- W. Y. Liang and I. Zhitomirsky, J. Mater. Chem. A, 2021, 9, 10335 RSC.
- Y. He, L. Xie, S. X. Ding, Y. J. Long, X. Y. Zhou, Q. Hu and D. M. Lin, Dalton Trans., 2021, 50, 4923 RSC.
- K. Ghosh and M. Pumera, Nanoscale, 2021, 13, 5744 RSC.
- W. Kim, H. J. Lee, S. J. Yoo, C. K. Trinh, Z. Ahmad and J. S. Lee, Nanoscale, 2021, 13, 5868 RSC.
- J. Zhuge, F. Rouhani, F. Bigdeli, X. M. Gao, H. Kaviani, H. J. Li, W. Wang, M. L. Hu, K. G. Liu and A. Morsali, Dalton Trans., 2021, 50, 2606 RSC.
- J. Li, L. Yu, Y. P. Li, G. R. Wang, L. P. Zhao, B. Peng, S. Y. Zeng, L. Shi and G. Q. Zhang, Nanoscale, 2021, 13, 692 RSC.
- A. Noori, M. F. E. Kady, M. S. Rahmanifar, R. B. Kaner and M. F. Mousavi, Chem. Soc. Rev., 2019, 48, 1272 RSC.
- F. Wang, X. Wu, X. Yuan, Z. Liu, Y. Zhang, L. Fu, Y. Zhu, Q. Zhou, Y. Wu and W. Huang, Chem. Soc. Rev., 2017, 46, 6816 RSC.
- M. Yuan, X. Guo, Y. Liu and H. Pang, J. Mater. Chem. A, 2019, 7, 22123 RSC.
- W. J. Liu, H. Jiang and H. Q. Yu, Energy Environ. Sci., 2019, 12, 1751 RSC.
- Y. Zhang, X. Liu, S. Wang, L. Li and S. Dou, Adv. Energy Mater., 2017, 7, 1700592 CrossRef.
- Z. D. Hao, Q. Zhao, J. D. Tang, Q. Q. Zhang, J. B. Liu, Y. H. Jin and H. Wang, Mater. Horiz., 2021, 8, 12 RSC.
- M. Z. Iqbal, J. Khan, H. T. A. Awan, M. Alzaid, A. M. Afzal and S. Aftab, Dalton Trans., 2020, 49, 16715 RSC.
- Y. Zhou, H. L. Qi, J. Y. Yang, Z. Bo, F. Huang, M. S. Islam, X. Y. Lu, L. M. Dai, R. Amal, C. H. Wang and Z. J. Han, Energy Environ. Sci., 2021, 14, 1854 RSC.
- R. Mo, F. Li, X. Tan, P. Xu, R. Tao, G. Shen, X. Lu, F. Liu, L. Shen, B. Xu, Q. Xiao, X. Wang, C. Wang, J. Li, G. Wang and Y. Lu, Nat. Commun., 2019, 10, 1474 CrossRef PubMed.
- X. Feng, Y. Bai, M. Q. Liu, Y. Li, H. Y. Yang, X. R. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036 RSC.
- C. C. Wang, D. T. Wu, Y. Qin and Y. Kong, Chem. Commun., 2021, 57, 4019 RSC.
- B. S. Soram, J. Y. Dai, I. S. Thangjam, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2020, 8, 24040 RSC.
- L. Kang, M. Y. Zhang, J. Zhang, S. D. Liu, N. Zhang, W. J. Yao, Y. Ye, C. Luo, Z. W. i Gong, C. L. Wang, X. F. Zhou, X. Wu and S. C. Jun, J. Mater. Chem. A, 2020, 8, 24053 RSC.
- Y. Liu, Z. Xiao, Y. Liu and L. Z. Fan, J. Mater. Chem. A, 2018, 6, 160 RSC.
- Y. Zhang, S. Liu, X. Zheng, X. Wang, Y. Xu, H. Tang, F. Kang, Q. H. Yang and J. Luo, Adv. Funct. Mater., 2017, 27, 1604687 CrossRef.
- M. Zohair, K. Moyer, J. E. Rathert, C. Meng, J. Waugh and C. L. Pint, ACS Nano, 2020, 14, 2308 CrossRef CAS PubMed.
- B. Zhu, B. Liu, C. Qu, H. Zhang, W. Guo, Z. Liang, F. Chen and R. Zou, J. Mater. Chem. A, 2018, 6, 1523 RSC.
- R. Thangavel, A. G. Kannan, R. Ponraj, V. Thangavel, D. W. Kim and Y. S. Lee, J. Mater. Chem. A, 2018, 6, 17751 RSC.
- R. Velmurugan, P. Alagammai, M. Ulaganathan and B. Subramanian, J. Mater. Chem. A, 2020, 8, 24148 RSC.
- Y. L. Zhang, H. X. Chao, H. Y. Liu, X. S. Wang, W. Xing, H. Hu and M. B. Wu, Chem. Commun., 2020, 56, 12777 RSC.
- S. Kumar, S. Telpande, V. Manikandan, P. Kumar and A. Misra, Nanoscale, 2020, 12, 19438 RSC.
- K. S. Kumar, N. Choudhary, D. Pandey, Y. Ding, L. Hurtado, H. S. Chung, L. Tetard, Y. Jung and J. Thomas, J. Mater. Chem. A, 2020, 8, 12699 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00342a |
| ‡ Equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2021 |