
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of amyloid beta in small extracellular vesicles via Raman spectroscopy
Meruyert
Imanbekova
a,
Sorina
Suarasan
 a,
Tatu
Rojalin
b,
Rachel R.
Mizenko
b,
Silvia
Hilt
c,
Meghna
Mathur
d,
Paula
Lepine
d,
Michael
Nicouleau
d,
Nguyen-Vi
Mohamed
d,
Thomas M.
Durcan
d,
Randy P.
Carney
b,
John C.
Voss
c and
Sebastian
Wachsmann-Hogiu
*a
a,
Tatu
Rojalin
b,
Rachel R.
Mizenko
b,
Silvia
Hilt
c,
Meghna
Mathur
d,
Paula
Lepine
d,
Michael
Nicouleau
d,
Nguyen-Vi
Mohamed
d,
Thomas M.
Durcan
d,
Randy P.
Carney
b,
John C.
Voss
c and
Sebastian
Wachsmann-Hogiu
*a
aDepartment of Bioengineering, McGill University, Montreal, QC H3A 0E9, Canada. E-mail: sebastian.wachsmannhogiu@mcgill.ca
bDepartment of Biomedical Engineering, University of California, Davis, CA 95616, USA
cDepartment of Biochemistry & Molecular Medicine, University of California, Davis, CA 95616, USA
dThe Early Drug Discovery Unit (EDDU), Montreal Neurological Institute and Hospital, McGill University, Montreal, QC H3A 2B4, Canada
First published on 7th June 2021
Abstract
One of the hallmarks of Alzheimer's disease (AD) pathogenesis is believed to be the production and deposition of amyloid-beta (Aβ) peptide into extracellular plaques. Existing research indicates that extracellular vesicles (EVs) can carry Aβ associated with AD. However, characterization of the EVs-associated Aβ and its conformational variants has yet to be realized. Raman spectroscopy is a label-free and non-destructive method that is able to assess the biochemical composition of EVs. This study reports for the first time the Raman spectroscopic fingerprint of the Aβ present in the molecular cargo of small extracellular vesicles (sEVs). Raman spectra were measured from sEVs isolated from Alzheimer's disease cell culture model, where secretion of Aβ is regulated by tetracycline promoter, and from midbrain organoids. The averaged spectra of each sEV group showed considerable variation as a reflection of the biochemical content of sEVs. Spectral analysis identified more intense Raman peaks at 1650 cm−1 and 2930 cm−1 attributable to the Aβ peptide incorporated in sEVs produced by the Alzheimer's cell culture model. Subsequent analysis of the spectra by principal component analysis differentiated the sEVs of the Alzheimer's disease cell culture model from the control groups of sEVs. Moreover, the results indicate that Aβ associated with secreted sEVs has a α-helical secondary structure and the size of a monomer or small oligomer. Furthermore, by analyzing the lipid content of sEVs we identified altered fatty acid chain lengths in sEVs that carry Aβ that may affect the fluidity of the EV membrane. Overall, our findings provide evidence supporting the use of Raman spectroscopy for the identification and characterization of sEVs associated with potential biomarkers of neurological disorders such as toxic proteins.
1. Introduction
Alzheimer's Disease is the most common form of dementia and has an overwhelming impact on patients' lives and their families. The formation of Aβ senile plaques and tau tangles are the hallmark of AD. Aβ is a 36–43 amino acid peptide that is derived from proteolysis of amyloid precursor protein (APP). Understanding the role of Aβ in the molecular pathways that lead to pathological changes in the brain of patients with AD is a long-standing goal in the AD research field. While the mechanisms of age-related accumulation of Aβ in the AD patients' brain remains unclear, it has been hypothesized that the alterations in the metabolism of APP could be related to AD progression. The non-amyloidogenic pathway, which prevents the formation of the toxic Aβ forms, proceeds from the proteolysis of APP on the cell surface by α-secretase followed by γ-secretase. On the other hand, the amyloidogenic pathway includes cleavage of APP by β-secretase generating 99 amino acid C-terminal fragment that is then cut by γ-secretase, leading to generation of the neurotoxic Aβ40 and Aβ42 peptides.1,2 The Aβ42 peptide is shown to be more hydrophobic and prone to form fibrils compared to Aβ40 peptide and is found to be highly prevalent in senile plaques.3 Moreover, several studies showed that intracellular Aβ42 can be located in multivesicular bodies of neurons and further enveloped into small extracellular vesicles – exosomes.4,5Exosomes are nanometer-sized small extracellular vesicles (sEVs) derived from the endocytic pathway and released from the cells upon diffusion of cytosolic multivesicular bodies with the plasma membrane. Exosomes have been detected in different fluids of the human body including serum, plasma, saliva, breast milk, amniotic fluid, semen, and urine.6 Their molecular cargo reflects the state of the releasing cells and contains membrane proteins, endosome-associated proteins, cytosolic proteins, lipids, and nucleic acids. Functions of sEVs in normal physiology and in a variety of pathological processes are under extensive study. They are known to facilitate intercellular communication between neighboring cells or distant cells and to play a role in cardiovascular diseases,7 cancer,8 metabolic9 and neurological disorders,10,11 and autoimmune diseases.12,13 Due to the lack of explicit consensus in the field of extracellular vesicles on the appropriate nomenclature, and to adhere to the MISEV 2018 guidelines14 we chose to use the term “small extracellular vesicles” or “sEVs” for the purpose of this study to refer to EVs in the approximate size range of 50–200 nm. In more broader contexts we used a collective term “extracellular vesicle” or “EVs”.
With relevance to neurodegenerative diseases, it has been proposed that the generation and progression of many neurodegenerative disorders are associated with exosome-mediated transport of misfolded proteins15–17 and specific RNA species in exosomes.18–20 Furthermore, recent clinical studies showed elevated levels of AD-associated proteins, tau, and Aβ, in exosomes isolated from plasma, serum, and cerebrospinal fluid (CSF) of AD patients.21–25 These findings stimulate further exploration of the sEVs as potential biomarkers of neurodegenerative diseases.
Extracellular vesicles are characterized by a wide variety of methods. Morphological features of EVs are described by nanoparticle tracking analysis (NTA),26 electron microscopy (EM),27 and atomic force microscopy techniques (AFM).28 Their molecular cargo is characterized mainly by flow cytometry, western blot, immunoprecipitation, and immunohistochemistry methods29 as well as by mass spectrometry, and quantitative polymerase chain reaction. In addition, there are several emerging techniques that complement traditional methods for EVs characterization by their ability to reveal new information about the EVs molecular cargo or to characterize the composition of individual EVs. These methods include fluorescence-based techniques,30,31 atomic force microscopy,32 surface plasmon resonance (SPR),33 Raman spectroscopy,34,35 and electrochemical sensing methods.36 Among these novel approaches, Raman spectroscopy enables sensitive label-free detection and analysis of EVs protein content.
Raman spectroscopy is an optical method where a laser beam is used to irradiate a sample resulting in inelastic scattering of photons. The difference in energy of these photons corresponds to the chemical bonds that are present in the sample.37 Due to its label-free and non-destructive nature with high chemical specificity, Raman spectroscopy has great application potential in the characterization of extracellular vesicles. Several studies have been published in the past decade using Raman spectroscopy as a tool to analyze the biochemical content of EVs. The pioneering work reporting the first Raman spectrum of sEVs was published in 2009.38 Later studies demonstrated the use of Raman spectroscopy for characterization of single extracellular vesicles,39 as well as clusters of EVs trapped in the laser focus.40 In addition, recent studies have indicated that Raman spectroscopy can be used for tissue characterization by analyzing the spectral signature of cancer EVs for prostate cancer diagnosis,41,42 as well as tissue-specific EVs derived from mesenchymal stromal cells43 and peripheral blood mononuclear cells.44 Furthermore, urinary EVs from diabetic patients and hyperglycemic endothelial cells45 have been successfully characterized by Raman spectroscopy. Immune-capture based single EV Raman spectroscopy46 has also been reported as a promising approach.
In the research field of neurological disorders, Raman spectroscopy has been used to investigate structural features and changes of toxic proteins such as Aβ,47–49 α-synuclein,50,51 and tau52 by analyzing the amide bands in the protein spectrum that is particularly sensitive to the protein's conformational state and environment. Moreover, differences in the Raman fingerprint of blood samples of patients compared to a healthy control have been reported for a variety of neurological conditions such as AD,53 Parkinson's disease (PD),54,55 dementia with Lewy bodies,56 and Huntington disease.57 Recent reports have demonstrated the ability of Raman spectroscopy to accurately distinguish PD58 and Amyotrophic Lateral Sclerosis (ALS)59 patients from healthy control group based on their EVs profile. Our group previously demonstrated the application of laser tweezers Raman spectroscopy for exosomes heterogeneity analysis39 and surface-enhanced Raman spectroscopy for biochemical analysis of EVs.60–64
However, to our knowledge, specific Raman studies indicating Aβ association within sEVs have not been reported. Here we report for the first time the use of Raman spectroscopy for the identification and characterization of Aβ associated with sEVs, as well as the structural and dynamical effects of Aβ on the membrane of sEVs.
2. Materials and methods
2.1 Aβ1–42 pure protein preparation
Aβ1–42 protein samples were prepared by resuspension of the stock Aβ1–42 protein (stock number: A9810, Sigma-Aldrich, USA) in DMSO to a final concentration of 10−6 M and vortexed prior usage.2.2 Cell culture models
In this work, we used sEVs derived from the MC65 cell culture model and midbrain organoids, as described next.MC65 cells were cultured in a 75 ml flask in Dulbecco's Modified Eagle medium supplemented with 4.5 mg ml−1D-glucose, non-essential amino acids, 1 mM sodium pyruvate, and 10% (v/v) heat-inactivated fetal bovine serum (FBS), supplemented with 0.1 mg ml−1 tetracycline, 50 IU ml−1 penicillin, and 50 g ml−1 streptomycin. In order to prevent the addition of nonspecific FBS EVs, we cultured the cells with an EV-depleted FBS (Life Technologies®). This ensures that the resulting sEVs in the cell culture medium supernatant only originate from the plated cells. The MC65 cells were cultured in the presence of TC for 24 h and the growth media was then collected MC65(TC+) for further isolation of sEVs. Expression of APPC99 in MC65 cells was induced by removing TC from the cell culture medium and cells cultured for another 16 h. At this point, the cell culture media MC65(TC−) was harvested and centrifuged at 2000g for 30 min to remove any cells and debris.
2.3 Isolation of sEVs from cell culture media
sEVs were isolated by differential ultracentrifugation with two rounds of spinning. First, we employed a low-speed centrifugation of the sEVs containing media to remove the cell portions, cell debris, apoptotic bodies or large biopolymers, and microvesicles. For this, 34 ml of the cell culture media from MC65 cells and midbrain organoids were centrifuged at 300g for 10 min, followed by 2000g for 10 min centrifugation and a final step centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min. All low-speed centrifugations (300–10000g) were performed using a Beckman Coulter Microfuge 20R centrifuge with a FA361.5 Biosafe rotor. The second round is a high-speed centrifugation which has the following steps: 120000g for 90 min, collected supernatant was discarded, and the pellet was dispersed in ultrapure water and centrifugated one more time at 120000g for 90 min to pellet the sEVs. UC was performed using Beckman Optima TLX Ultracentrifuge with an SW 28 swinging bucket rotor. The resulting pellets were finally resuspended in up to 100 μl of ultrapure water and stored at −80 °C until use. The samples were aliquoted (50 μl) to reduce freeze-thaw cycling which may otherwise damage the sEVs. In this way, only one freeze-thaw cycle is used, which has been shown previously to not have a significant effect on the integrity of sEVs.67,68 Moreover, dispersion and aliquoting of the resulting pellet allows characterization of the same isolated sEVs sample by complementary characterization methods to meet MISEV guidelines (e.g., electron microscopy, SP-IRIS, NTA, etc.).
000g for 30 min. All low-speed centrifugations (300–10000g) were performed using a Beckman Coulter Microfuge 20R centrifuge with a FA361.5 Biosafe rotor. The second round is a high-speed centrifugation which has the following steps: 120000g for 90 min, collected supernatant was discarded, and the pellet was dispersed in ultrapure water and centrifugated one more time at 120000g for 90 min to pellet the sEVs. UC was performed using Beckman Optima TLX Ultracentrifuge with an SW 28 swinging bucket rotor. The resulting pellets were finally resuspended in up to 100 μl of ultrapure water and stored at −80 °C until use. The samples were aliquoted (50 μl) to reduce freeze-thaw cycling which may otherwise damage the sEVs. In this way, only one freeze-thaw cycle is used, which has been shown previously to not have a significant effect on the integrity of sEVs.67,68 Moreover, dispersion and aliquoting of the resulting pellet allows characterization of the same isolated sEVs sample by complementary characterization methods to meet MISEV guidelines (e.g., electron microscopy, SP-IRIS, NTA, etc.).
2.4 sEVs characterization
:1 Solution A and blocking solution. Antibodies were diluted 1:600 in a blocking mixture. After the final mix, 750 μl of the solution was taken out of each well and 250 μl of antibody mixture was added. Chips were then incubated at room temperature for 1 h. After incubation, 500 μl of Solution A was added to each well. 750 μl of the solution was then immediately taken out and replaced by 750 μl of new Solution A. This was shaken at 500 rpm for 3 min followed by removing 750 μl of solution from each well. 750 μl of Solution B was then added to each well and the plate was shaken at 500 rpm for 3 min followed by removing 750 μl of solution. This was repeated 3 times. 750 μl of MilliQ water was then added to each well and shaken at 500 rpm for 3 min for a total of 5 shaking steps after antibody incubation. Each chip was washed in two successive dishes of MilliQ water, taking care to avoid drying of the chip between dishes. In the final dish, the chip was tipped at a 45-degree angle and slowly pulled out of the water. These were then dried on absorbent paper and added to the chuck. Chips were scanned by SP-IRIS and all three fluorescent channels. Data were analyzed with fluorescence cut-offs of 600, 400, and 400 arbitrary units for the blue, green, and red channels, which were chosen by limiting the number of particles on the negative control MIgG spot to less than 10 for all chips.
2.5 Raman spectroscopy setup and data acquisition
A WITec Confocal Raman microscopy system (WITec Alpha300R) with a 633 nm HeNe laser, maximum power of 5 mW at the sample, coupled into a microscope equipped with a 50× objective (NA 0.8, WD 0.58 mm, theoretical laser focal spot diameter ∼1 μm), a spectrometer (UHTS400 NIR, 400 mm focal length, with a 300 grooves per mm grating corresponding to a spectral resolution of <6.8 rel. cm−1/pixel at 633 nm), and a CCD camera was used for these experiments. The acquisition time for sEVs characterization was 60 s. The spectra were collected after air-drying 5 μl of isolated sEVs solutions on a glass cover slip from multiple points within the droplet fingerprint focusing on small aggregates of sEVs and in the rim area of the droplet. This approach allows size-based separation of sEVs from possible contaminants such as large EVs or protein aggregates, via convection currents that drive smaller particles to the outside of the ring. This is not possible if the spectra are measured from pellets where the EVs are clumped together, making the separation of larger aggregates (including protein aggregates) from the actual sEVs more challenging. Moreover, measuring Raman spectra of sEVs in liquid pellets presents difficulties due to their intrinsic Brownian motion, which will cause particles to move in and out of the laser beam. In addition, the momentum of the photons in the laser beam may push particles out of the focal region and, if not controlled properly, may make the measurements less accurate.2.6 Data pre-processing and statistical analysis
The statistical analysis and data processing were performed using WITec Project Five build-in software (ImageLab) and OriginPro (OriginLab, Northampton, MA). Prior analysis the quality of Raman spectra was assessed, and data pre-processing was performed in order to minimize insignificant variability. Pre-processing of the data included correction of baseline by subtraction of the spectral background from glass, cosmic rays, and other background deviations. Next, in order to enhance the spectral quality, we reduced the noise by applying Savitzky–Golay smoothing and then the data were normalized. Principal component analysis (PCA) was performed using OriginPro PCA for spectroscopy app. PCA was performed on a range of 900–1800 cm−1, 1540–1800 cm−1, and 2800–3100 cm−1. The variance-covariance matrix was utilized for further analysis and the reduction of initially complex data was achieved by PCA. Next, to build the PCA score plot we used the first two principal components (PCs). The optimal number of PCs to describe major features of the spectra was chosen based on the size of the corresponding eigenvalues of the PCA scree plot. The eigenvalues of the PCs after the first two were significantly smaller suggesting that the rest of the PCs may not have much interpretive values and add relatively little to the information already retained by first two PCs. Peak deconvolution was achieved by using the OriginPro built-in Multiple Peak Fit tool. The peak positions were chosen based on existing literature and further deconvolved using Voigt peak shape function.3. Results
The workflow of the study is represented in Fig. 1, which describes schematically the steps followed to isolate, characterize, and analyze the sEVs. Specifically, in this study, we employed three different sEVs groups, isolated from two types of cell cultures: 2D MC65 neuroblastoma cell line and 3D midbrain organoids. The MC65 cell line is an in vitro AD model that provides a neuronal source of sEVs containing Aβ. We believe that it is important to fully investigate and understand the signatures of sEVs associated Aβ in simulated conditions before examining human samples. The study of an in vitro model of AD allows the investigation of possible roles Aβ protein has in neurons,69–71 and subsequently in neuronal sEVs, and may provide valuable insights into the pathogenesis of AD. Future work building on this data will apply Raman-based detection of AD in clinical settings. sEVs isolated from 3D midbrain organoids serve as an additional negative control in this study and represent healthy brain neurons. As described in the Methods section, sEVs isolation was achieved by first centrifuging the cell culture media several times at low speed to remove the remaining cell fragments, debris, and microvesicles, followed by two cycles of high-speed centrifugation. We expect that, in accordance with previous reports, the remaining pellet contains the small sEVs of interest. We will further denote the sEVs isolated from untreated and tetracycline treated MC65 cells line as TC− sEVs and TC+ sEVs, respectively. The sEVs isolated from organoids culture media are labeled as osEVs. The sEVs were characterized by established methodologies such as NTA and TEM and were further studied by Raman spectroscopy to reveal their biochemical content. Subsequently, the recorded spectra were analyzed by PCA to identify the Aβ content of each sEVs group.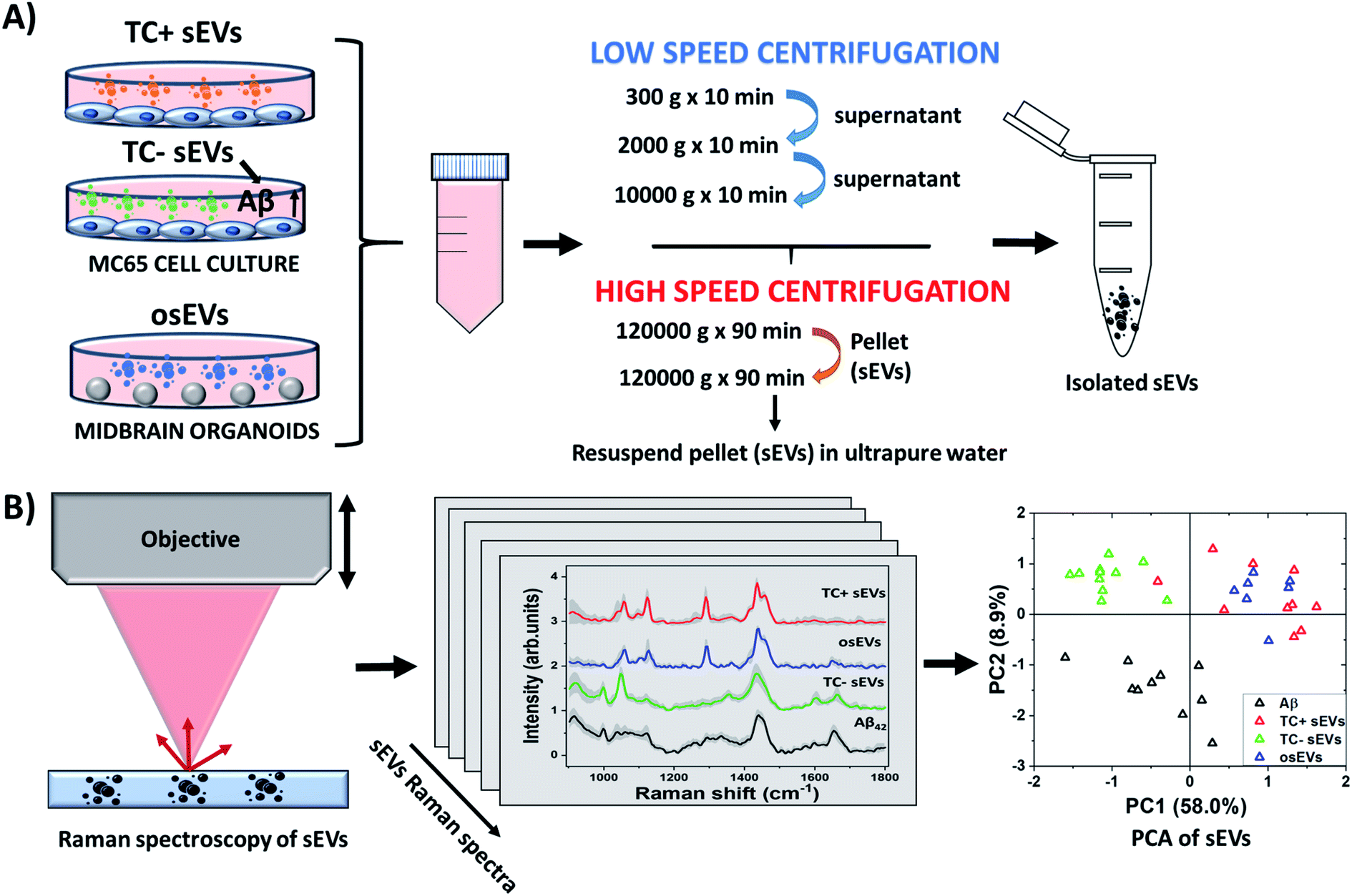 | ||
| Fig. 1 Schematic illustration of sEVs isolation and characterization by Raman spectroscopy. (A) sEVs were isolated by differential ultracentrifugation from the MC65 AD cell culture model, which is under tetracycline promoter regulation, and midbrain organoids developed from PBMCs of healthy donors. (B) The biochemical content of the isolated sEVs was characterized by Raman spectroscopy. The collected spectra were further analyzed by PCA. | ||
3.1 sEVs characterization by NTA and TEM
First, we characterized the size and concentration of the isolated sEVs via NTA. Fig. 2A shows the size distribution plots for all analyzed sEVs groups. The mean concentration of TC− sEVs, as measured by NTA, 6.5 × 109 EVs ml−1, was higher than the mean concentration of TC+ and osEVs samples, which was 4.2 × 109 EVs ml−1 and 4.5 × 107 EVs ml−1, respectively. Additionally, the mean particle size as measured by NTA was 157.3 nm ± 3.8 nm, 164.1 nm ± 11.2 nm, and 293.5 ± 2.7 nm for TC− sEVs, TC+ sEVs, and osEVs, respectively. One can see that the mean particle size of TC− sEVs was comparable with the one that is recorded for TC+ sEVs. On the other hand, we observed a slightly larger particle size for osEVs. TEM images, presented in Fig. 2B confirm this result, showing an increased size for organoids sEVs. Moreover, TEM images revealed the sEVs cup-shaped morphology, which is a typical experimental artefact related to deflation of EV structure during the sample preparation.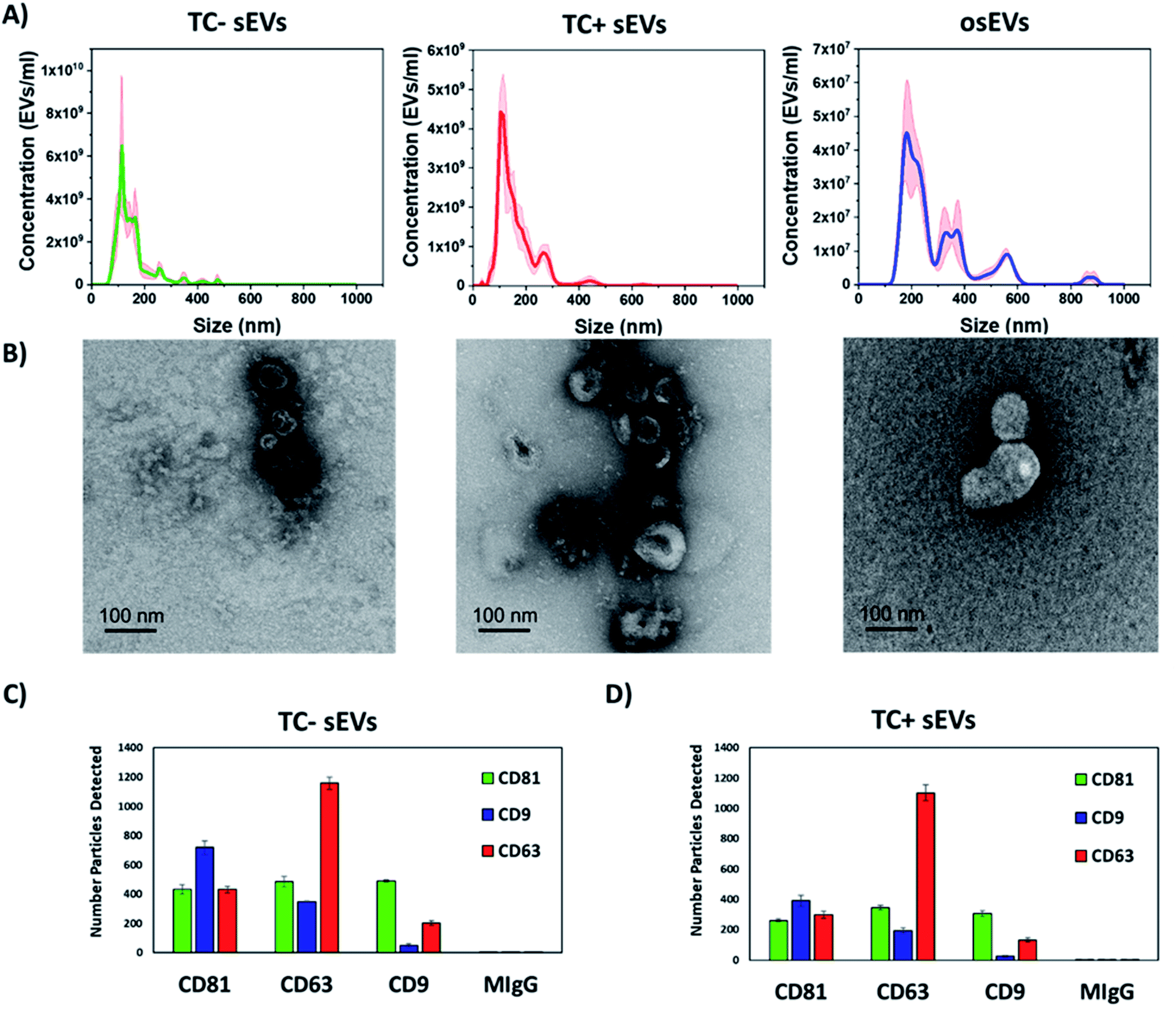 | ||
| Fig. 2 Characterization of sEVs by transmission electron microscopy, nanoparticle tracking analysis, and SP-IRIS. (A) Graphs show the concentration of sEVs as a function of particle size for TC− sEVs, TC+ sEVs, and osEVs. Shaded areas represent error bars. (B) Electron micrographs of TC− sEVs, TC+ sEVs, and osEVs showing the cup-shaped morphology. Scale bar is 100 nm. Expression of typical EVs surface proteins CD81, CD63, and CD9 as well as the negative control MIgG in the (C) TC− sEVs and (D) TC+ sEVs samples. | ||
To confirm that sEVs were enriched during ultracentrifugation, expression of sEVs associated tetraspanins, CD9, CD63, and CD81, were tested by immuno-capture and immuno-fluorescence, using the SP-IRIS method implemented into the ExoView R100 instrument. This equipment utilizes a micropatterned chip with an array of spatially distinct antibody spots. During incubation, sEVs are captured by these antibodies and subsequently labeled with fluorescent detection antibodies. By directly imaging these antibody arrays, up to four co-expressed surface proteins (capture antibody and three fluorescent detection channels) can be detected on a single sEV.
For both sEVs populations, all three tetraspanins were expressed with both capture and fluorescence detection of each tetraspanin. Furthermore, the tetraspanin profile of each sEVs population was very similar with most CD9 positive sEVs detected on the CD81 capture spot, the most CD63 positive sEVs detected on the CD63 capture spot, and similar amounts of CD81 positive sEVs captured on each spot. These results show that the co-expression of these tetraspanins is highly consistent between these sEVs populations.
In addition, we note that the resuspension of sEVs in ultrapure water did not notably change the characteristics of analyzed sEVs. Their size, morphology, and surface protein expression (Fig. 2) is comparable to the ones reported for sEVs resuspended in PBS or commercially available EVs resuspension buffers that maintain osmotic pressure. We believe that the ability of EVs to withstand the isotonic solution pressure can be explained by the higher rigidity of the EVs lipid bilayer that is enriched with cholesterol, sphingomyelin, and gangliosides compared to the membranes of their cells of origin.72,73 Moreover, we experimentally determined that the composition of the resuspension buffer did not majorly impact the physical or chemical nature of sEVs (data not shown). To do this, we isolated sEVs by differential centrifugation from cell culture media and resuspended them using either 0.1% filtered PBS or ultrapure water, both as the final buffer as well as during intermediate steps of processing. Then, we characterized sEVs by NTA, resistive pulse sensing (RPS), and SP-IRIS methods. The results of concentration and size distribution analysis did not show a major difference between the two groups of sEVs. We found that the sEVs resuspended in water had a similar concentration (8.8 × 1011 particles per ml) compared to sEVs resuspended in PBS (2.4 × 1011 particles per ml), indicating similar yield for particles. Finally, we determined that both sEVs groups had similar CD9, CD63, and CD81 tetraspanins profiles, which further suggests that the chemical nature of sEVs remains generally similar regardless of the choice of resuspension buffer.
3.2 Raman spectroscopy analysis of sEVs isolated from MC65 (TC−/+) cells and midbrain organoids cell culture media
The MC65 AD cell culture model used in this study overexpresses 99-amino acid carboxyl-terminal fragments (βCTF) of APP under tetracycline promoter regulation. This model is designed to mimic the pathological pathway of APP that leads to amyloidogenesis. This pathway involves cleavage of mature APP by β- and γ-secretases, where β-secretase cleaves the amino terminus of Aβ, and membrane-associated βCTF. Further, βCTF is cleaved by γ-secretase resulting in the release of Aβ40 or Aβ42 peptides and APP intracellular domains (AICDs).65 As βCTF undergoes endocytosis, it can be trafficked to endosomal compartments such as multivesicular bodies (MVBs) and possibly enveloped in sEVs or exosomes.74 It has been shown previously that APP CTFs are overabundant in cerebrospinal fluid (CSF) of AD patients and suggested to be potential diagnostic biomarkers of AD.75 The control samples of sEVs are isolated from the same cell culture model in the presence of tetracycline (TC+) and midbrain organoids sEVs (osEVs). The midbrain organoids were developed from PBMCs of healthy individuals and were used in this study because they are biochemically and biophysically more similar to tissues due to their ability to mimic cell-matrix and cell–cell interactions. Therefore, they are representative of healthy brain neurons.For the Raman spectroscopy analysis, the isolated sEVs were resuspended in ultrapure water and placed on a clean glass microscope slide to allow air-drying. Spectra were mostly recorded from the small aggregates of sEVs and from the edge of the dried sample, where sEVs accumulate preferentially due to the “coffee-ring effect”. This effect is observed upon the evaporation of water from droplet samples that contain small-sized particles. Explicitly, in a sample with a heterogeneous particle size distribution, the smallest particles flow radially toward the contact line during the drying process. The angle between the surface of the drying EVs sample and the microscope slide decreases progressively during water evaporation which limits the size of the particles that can approach the edge of the droplet. Therefore, after drying, the particles will be separated based on their size due to convective currents inside the droplet, as reported by Jeong et al.76 As the droplet dries, the smaller (lighter) particles such as sEVs are deposited and concentrated at the outer edge of the dried sample, and the bigger (heavier) particles, such as large EVs or protein aggregates that could be co-isolated during differential ultracentrifugation, are concentrated closer to the center region. By positioning the laser spot in the ring and adjacent to the ring area we ensure that we measure particle sizes in the typical sEVs range according to MISEV 2018 nomenclature and as measured here by NTA and TEM (Fig. 2). This is particularly important for the acquisition of reliable Raman spectra. In our case, we were able to record high-quality spectra with 633 nm continuum laser excitation at relatively low power of few mW and acquisition times on the order of one minute.
Next, we analyzed the collected Raman spectra from TC− sEVs (n = 11), TC+ sEVs (n = 10), and osEVs (n = 7) samples and compared them with spectra recorded from pure Aβ42 protein (n = 10). The Raman spectra of the “fingerprint region” 900–1800 cm−1 from all sEVs groups and Aβ42 pure protein represent a complex set of peaks with shared features among all sEVs samples and some variations (Fig. 3A). The Raman peak assignments are given in Table 1. All sEVs groups shared the same peak positions at 1123 cm−1 and 1290 cm−1 assigned to C–N vibration and amide III α-helix protein structure, respectively. Peaks at 1436 cm−1 and 1453 cm−1 are assigned to the lipid content, specifically to the CH2 and CH3 deformation in lipids and triglycerides. Additionally, Aβ42 pure protein spectra presented two distinct peaks at 1000 cm−1 and 1600 cm−1, assigned to the breathing of the benzene ring and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration corresponding to phenylalanine, respectively. These peaks can be also observed in the TC− sEVs spectra, suggesting that these sEVs could potentially carry Aβ protein. Amide I region was also located at similar positions for TC− sEVs, osEVs, and Aβ42 pure protein covering the area 1650–1668 cm−1. In the osEVs spectra, these peaks can be observed, while they are missing in the TC+ sEVs spectra.
C vibration corresponding to phenylalanine, respectively. These peaks can be also observed in the TC− sEVs spectra, suggesting that these sEVs could potentially carry Aβ protein. Amide I region was also located at similar positions for TC− sEVs, osEVs, and Aβ42 pure protein covering the area 1650–1668 cm−1. In the osEVs spectra, these peaks can be observed, while they are missing in the TC+ sEVs spectra.
 | ||
| Fig. 3 Characterization and analysis of the “fingerprint region” 900–1800 cm−1 of TC− sEVs, TC+ sEVs, osEVs, and Aβ42 pure protein. (A) Average Raman spectra of fingerprint region 900–1800 cm−1 of TC− sEVs, TC+ sEVs, osEVs, and Aβ42 pure protein. Spectra are offset for clarity. Shaded areas represent ±1 standard deviation. (B) The score plot of the first two principal components for each sEVs group. Colors represent each sEVs group as shown in the legend. | ||
| Position (cm−1) | Peak assignment | Reference |
|---|---|---|
| 1000 | Phenylalanine ring breathing of protein | Hernández et al., 201379 |
| 1123 | C–N of proteins | Teh et al., 200980 |
| 1290 | Amide III | Bandekar et al., 199281 |
| 1436 | Fatty acids, triglycerides, CH2 or CH3 deformations | Verma et al., 197782 |
| 1453 | Proteins, CH, CH2 or CH3 deformations of long chain fatty acids, phospholipids | Notingher et al., 200983 |
| 1600 | CC of phenylalanine |
Hernandez et al., 201379 |
| 1650–1660 | Unsaturated fatty acids cis form, amide I α-helix | Bandekar et al., 199281 |
| 1667–1668 | Amide I β-sheet | Bandekar et al., 199281 |
| 2845 | CH2 symmetrical stretching of fatty acids, triglycerides | Song et al., 202084 |
| 2878 | CH2 asymmetric stretching (lipids) | Song et al., 202084 |
| 2900 | CH2 asymmetric stretching (lipids) | Song et al., 202084 |
| 2930 | CH3 symmetric stretching of proteins/lipids (cholesterol) | Rygula et al., 201385 |
| 2960 | CH3 symmetric stretching of proteins | Lis et al., 197686 |
| 3060 | Proteins aromatic CH stretching mode/nucleic acids | Rygula et al., 201385 |
Then, we performed PCA of the collected data and the results are shown in Fig. 3B. The first two principal components represented 58.0% and 8.9% variability of the total variance, respectively. It is important to note that these scores may be influenced by both spectrum intensity and spectrum shape.77,78 The samples are spread along the PC1 axis with TC− sEVs located on the negative side, while TC+ sEVs and osEVs are distributed loosely on the positive side of the axis. Aβ42 pure protein spectra form an elongated cluster between PC1 and PC2. By this, it is clear that the different sample groups can be distinguished from each other based on their Raman spectra, which also serves as a valuable starting point for further analyses.
Raman spectroscopy can effectively determine the secondary structure of proteins.81 The peaks centered at 1667–1668 cm−1 assigned to CO and a small contribution of C–N stretch corresponds to β-sheet protein conformation. The peaks located at 1650–1660 cm−1 region arising from the coupling of C–N stretching vibration and N–H bending vibrations correspond to an α-helix structure. Therefore, our attention was further focused on the amide I region which is mostly affected by the secondary structure of the proteins. The α-helix rich structure might originate from Aβ peptide bonded to a plasma membrane. In an attempt to identify specific peaks within the amide I region, we performed peak deconvolution analysis. Fig. 4 depicts the deconvolution of the amide I region in TC− sEVs and Aβ42 pure protein spectra. For this, the most intense peaks at 1600 cm−1 and 1650 cm−1 in the spectra were centered, fixed, and fitted until high values of R2 were obtained. The recorded Raman spectra are shown as solid lines and deconvolved peaks are marked as dashed lines.
 | ||
| Fig. 4 Analysis of the Raman spectra of the amide I region 1540–1800 cm−1. (A) Deconvolution of amide I region of averaged spectra of TC− sEVs and (B) Aβ42 pure protein indicated the presence of two peaks at 1600 cm−1 (labeled as peak 1) and 1650 cm−1 in TC− sEVs spectra and 1663 cm−1 for Aβ42 pure protein spectra (labeled as peak 2). Deconvolved spectra are shown as dotted lines. (C) The score plot of the first two principal components for each sEVs group. Colors represent each sEVs group as shown in the legend. Colored regions are to provide visual aids. (D) Comparison of the PC1 and PC2 loadings with Aβ42 pure protein spectrum and with average spectra of each sEVs group. Dotted lines represent zero-axes of the PCA loadings. Shaded areas represent ±1 standard deviation. Spectra are offset for clarity. | ||
The amide I region deconvolution clearly identifies the presence of a peak at 1650 cm−1 in the spectra of TC− sEVs (Fig. 4A) that corresponds to an α-helical conformation of the protein. The peak at 1663 cm−1 in the spectra of Aβ42 pure protein is assigned to an α-helical conformation with a potential contribution from “disordered structures” (Fig. 4B). On the other hand, the amide I region of TC+ sEVs and osEVs is too weak to provide a reliable fit and to obtain information. In addition, the presence of only α-helical structure of the proteins in the spectra of TC− sEVs confirms that the collected spectra represent the proteins within sEVs and not insoluble protein or peptide deposits, which typically adopt an enriched β-sheet conformation.87
The deconvolution of the Aβ42 pure protein spectra identified strong peaks centered at 1600 cm−1 and 1663 cm−1. The broad peaks in the amide I region of TC− sEVs spectra indicated the presence of mainly monomeric form or small oligomers of Aβ. A previous NMR study characterized Aβ associated with a phospholipid bilayer-mimicking environment as a monomeric amphipathic α-helix conformer.88 Next, the Raman spectra region between 1540–1800 cm−1 that includes the amide I region was analyzed by PCA. Fig. 4C depicts the score plot of the first two principal components that cumulatively represent 48.1% variability of the total variance. In order to highlight different cluster regions, shaded ellipse areas are shown in the plot. One can observe that TC− sEVs and Aβ42 pure protein are closely clustered in the positive side of the PC1 axis. In contrast, TC+ sEVs and osEVs spectra are dispersed along the PC1–PC2 plane. Next, the analysis of PC loadings showed the contribution of the individual wavenumber to PC1 and PC2 (Fig. 4D). While the PC1 loading resembles the spectra of Aβ42 pure protein, the biochemical meaning of the second PC is more difficult to interpret. Taken together, these data revealed that the major secondary structure of the proteins and potentially Aβ within the analyzed sEVs is typical to the α-helix form of proteins.
Additionally, Raman spectra in the “high-wavenumber region” can provide valuable information about the biochemical composition of the sEVs. Fig. 5 compares the experimentally recorded and deconvolved Raman bands that were obtained under the same experimental conditions as for the amide I region. Two major peaks were present within all sEVs spectra in this region at 2845 cm−1 and 2878 cm−1. These peaks are characteristic vibrational features of lipids and correspond to symmetrical and asymmetrical CH2 vibrations, respectively. The analysis of Aβ42 pure protein did not show the presence of the 2845 cm−1 peak and only showed weak intensity of the 2878 cm−1 peak. Specifically, these peaks can be attributed to the presence of long acyl chain lipids such as fatty acids and ceramides. In addition, there was a small contribution of cholesterol to these peaks. On the other hand, the characteristic peak of the proteins is located at 2930 cm−1. Fig. 5A and B depicts stronger intensities at 2930 cm−1 in deconvolved peaks of the TC− sEVs and Aβ42 pure protein compared to TC+ sEVs (Fig. 5C) and osEVs (Fig. 5D).
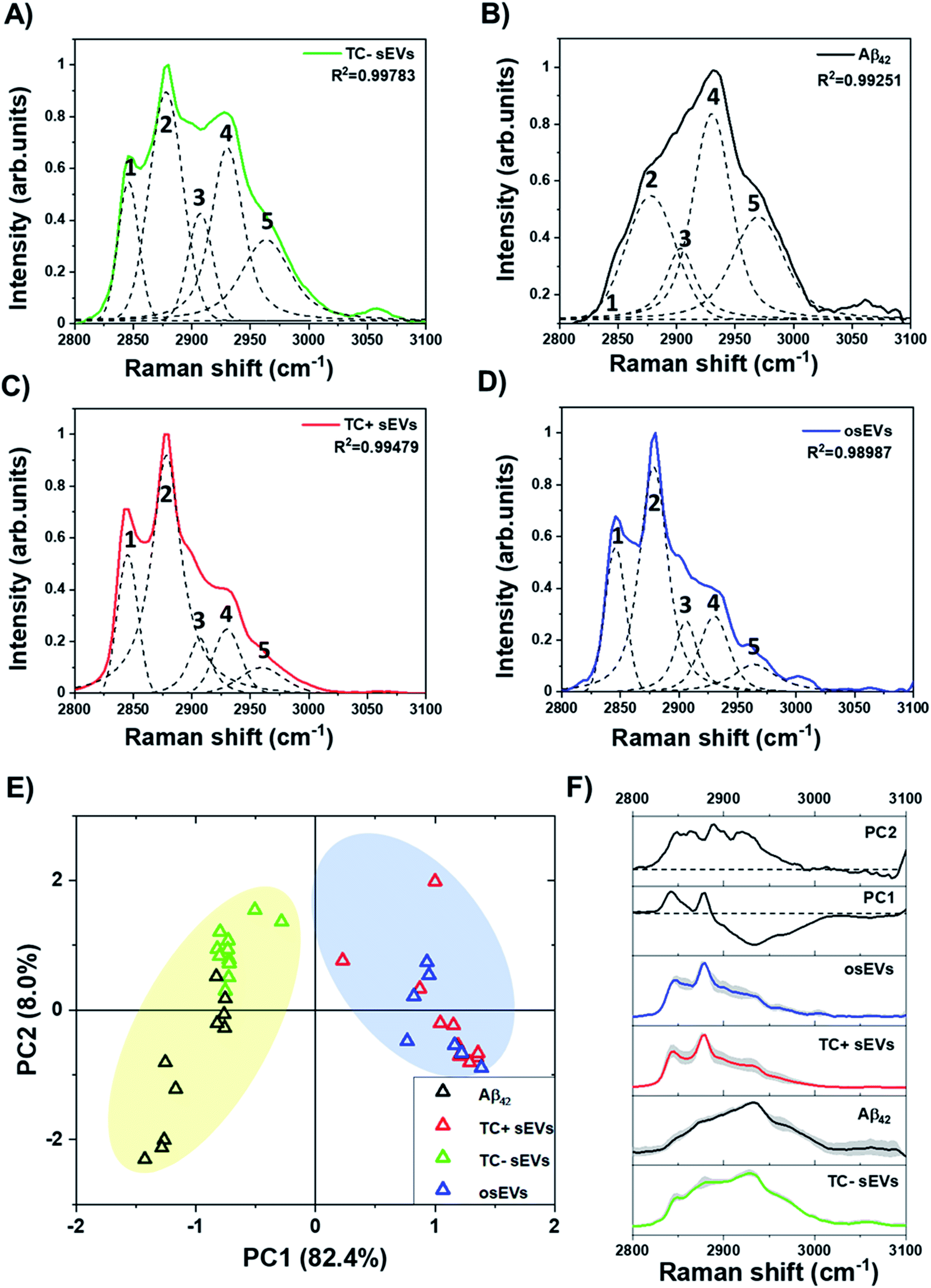 | ||
| Fig. 5 Analysis of the “high-wavenumber region” 2800–3100 cm−1 of normalized Raman spectra. Raman spectra and peak deconvolution for (A) TC− sEVs, (B) Aβ42 pure protein, (C) TC+ sEVs, and (D) osEVs. Dotted lines represent five peaks that were analyzed in this region, labeled with numbers as follows: 1 – 2845 cm−1, 2 – 2878 cm−1, 3 – 2900 cm−1, 4 – 2930 cm−1, and 5 – 2960 cm−1. (E) The PCA score plot of the first two principal components. Colors represent each sEVs group as shown in the legend. Colored regions are to provide visual aids. (F) Comparison of the PC1 and PC2 loadings and average spectra of sEVs groups. Shaded areas represent ±1 standard deviation. Dotted lines represent zero-axes of the PCA loadings. Spectra are offset for clarity. | ||
It has been shown previously that the ratio of Raman intensities at 2930 cm−1 and 2845 cm−1 (I2930/I2845 cm−1) reflects the ratio of the protein and lipid content.89,90Table 2 shows the calculated Raman intensity ratio for all analyzed sEVs groups. The intensity values are calculated for the area under the curve for each peak.
| sEVs group | I 2930 cm−1 | I 2845 cm−1 | Ratio I2930/I2845 cm−1 |
|---|---|---|---|
| TC− sEVs | 29.16947 | 11.97269 | 2.43633 |
| TC+ sEVs | 8.97397 | 12.17685 | 0.73696 |
| osEVs | 11.55123 | 12.63436 | 0.91427 |
One can see that TC− sEVs had a higher I2930/I2845 cm−1 intensity ratio compared to the TC+ sEVs and osEVs, indicating a higher concentration of proteins within TC− sEVs. These results indicate that Aβ protein could be present in the TC− sEVs and could be at higher concentrations than in TC+ sEVs and osEVs. To complement these findings, we performed PCA of the peaks in the “high-wavenumber region” between 2800–3100 cm−1 of Raman spectra of all sEVs groups and Aβ42 pure protein. Fig. 5E represents the score plot in the PC1–PC2 plane where the first PC was responsible for 82.4% of the variability and PC2 carried 8.0% of variability of the total variance. It can be clearly seen that TC− sEVs and Aβ42 pure protein spectra were clustered on the negative side of the PC1 axis while the TC+ sEVs and osEVs were clustered on the positive side of the PC1 axis. Fig. 5F shows the loading plots of PC1 and PC2 and the average spectra of the sEVs analytes and Aβ42 pure protein control protein. The loading spectrum for PC1 had several peaks at both positive and negative sides where the most significant wavenumbers are 2930 cm−1, 2845 cm−1, and 2878 cm−1 and resembled the spectra of Aβ42 pure protein and sEVs groups spectra. In contrast, the chemical meaning of the second principal component was not clear from the shape of the loading. Finally, PCA was able to successfully cluster similar spectra and segregate different ones.
Next, we evaluated the effect of Aβ on sEVs lipid membrane composition and structure. For this, we used the ratio of Raman peaks at 2845 cm−1 (CH2 sym.) to 2878 cm−1 (CH2 asym.) that describe an estimated lipid fluidity or degree of unsaturation91,92 (Table 3). The higher the I2845/I2878 cm−1 ratio is, the more unsaturated lipids are present, and the higher is the fluidity of the EV membrane. The results show that all three groups of sEVs had the same degree of saturation. Furthermore, in order to analyze the structure of lipids, we calculated the ratio of Raman peak intensities at 2845 cm−1 (C–H stretch of CH2) to 2930 cm−1 (C–H stretch of CH3) that has been shown to correlate with the number of C atoms in the fatty acid chain.92,93 We observed a slight change in the lipids structure, with the higher prevalence of unsaturated lipids with a longer chain in the TC+ sEVs and osEVs, and prevalence of lipids with a shorter chain length in TC− sEVs. This observation together with previously published reports indicates the effect of Aβ association to EV membrane fluidity by changing the structure of EV membrane lipids.94
| sEVs group | I 2878 cm−1 | Degree of unsaturation (CH2 sym. − CH2 asym.) (ratio I2845/I2878 cm−1) | Chain length prediction (CH2/CH3) (ratio I2845/I2930 cm−1) |
|---|---|---|---|
| TC− sEVs | 32.06708 | 0.37336 | 0.41045 |
| TC+ sEVs | 45.10705 | 0.26995 | 1.35690 |
| osEVs | 34.37304 | 0.36756 | 1.09376 |
4. Discussion
Alzheimer's disease is a neurodegenerative disease that remains challenging to diagnose in early stages. This prognostic uncertainty of existing diagnostic methods in combination with high costs and invasiveness of current diagnostic procedures further emphasizes the importance of developing sensitive and accurate alternative tests for early AD diagnosis. The overall goal of the study was to explore the use of sEVs as carriers of toxic proteins. We used Raman spectroscopy to characterize sEVs associated with Aβ protein as potential biomarkers for AD diagnosis. First, we demonstrated a clearly different biochemical profile of Aβ associated sEVs compared to the control sEVs groups. In particular, intense peaks at 1650 cm−1 and 2930 cm−1 and their similarities with the spectra of pure Aβ protein indicate the presence of the Aβ protein in TC− sEVs. On the contrary, less intense, or lacking bands at these positions in TC+ sEVs and osEVs confirm the hypothesis that these peaks are associated with Aβ protein. The observed differences in the PCA results in the amide I and “high-wavenumber regions” of the spectra can be explained by additional contributions from other proteins in sEVs cargo in the amide I region, as well as a lower overall signal-to-noise ratio in this region compared to the “high-wavenumber region”.In order to evaluate a priori the ability of Raman spectroscopy to detect Aβ in our sEVs we performed an estimate of the number of Aβ molecules in our laser spot. First, we calculated the number of Aβ molecules per sEV based on published data.22 Fiandaca et al.22 reported the Aβ42 concentration (pg ml−1) in total exosomes solution and the number of exosomes per ml. To determine the mass of Aβ42 per sEV, we divided the Aβ concentration (expressed in pg ml−1) by the number of exosomes per ml. Next, we converted the mass of Aβ to the number of molecules per sEV by first converting the mass of Aβ to moles using the molar mass and further converting to the number of molecules using the Avogadro's number. We applied this procedure to the values reported in the aforementioned study. The reported concentration of Aβ42 is 18.5 pg ml−1 in exosomes (2.78 × 109 particles per ml) isolated from plasma of AD patients (n = 3) and 0.83 pg ml−1 in exosomes (3.49 × 109 particles per ml) extracted from age matching healthy individuals (n = 3). We used the values of the exosomal Aβ42 protein concentration extracted from AD patients' plasma to estimate the concertation of the protein in our TC− sEVs. The concentration of Aβ42 protein obtained from the analysis of healthy controls was used to calculate the protein concentration in TC+ sEVs and osEVs. Beginning with the number of Aβ molecules per one sEV in TC−/+ sEVs and osEVs solutions, we calculated approximately 885 Aβ molecules per sEV, 31.5 molecules per sEV, and 30 molecules per sEV, respectively. The calculated values indicate a higher load of Aβ in TC− sEVs. Next, knowing the number of Aβ molecules per sEV we can calculate the expected number of Aβ molecules in our laser spot by assuming that the laser spot is a cylinder with 0.5 μm radius and 2 μm height and the sample is composed of concentrated sEVs filling the laser beam. Then, we calculated the estimated volume of one sEV of each group based on the mean size of sEVs analyzed by NTA. Subsequently, we calculated the number of sEVs of each group in the laser spot described above. The estimated number of Aβ molecules in the laser beam spot is 6.8 × 105 Aβ molecules for TC− sEVs, 2.1 × 102 Aβ molecules for TC+ sEV, and 1.7 × 102 Aβ molecules for osEVs. These estimates are supported by the differences of Raman intensities, where a linear relationship is expected to the number of molecules in the analyte. It is important to note that the 2930 cm−1 peak corresponds to overall protein concentration within analyzed sEVs. However, the main difference between TC− MC65 cells and TC+ MC65 cells is the presence of tetracycline and overexpression of Aβ in TC− MC65 cells. This indicates that the isolated sEVs will have mainly the same molecular composition and the major variability is the presence of Aβ in TC− sEVs as detected by Raman spectroscopy. In addition, it is important to mention that PCA can be used for further semi-quantitative analysis of Aβ in future studies of disease diagnosis using human clinical samples.
Next, the deconvolution of the amide I region of TC− sEVs showed that Aβ associated with sEVs is in an α-helical conformational form and in the size of a monomer or a small oligomer. These findings may shine light on a potential mechanism of propagation of neurodegeneration by sEVs carrying toxic oligomers. There is no consensus in the field regarding the structure of the toxic oligomers. The process of transformation of the monomers into toxic oligomers has been shown to be structure dependent. Specifically, it has been noted that toxic oligomers, as well as Aβ fibrils, have a β-sheet enriched secondary structure that provides a high adherence site for further fibrillation.95–97 Conversely, a number of studies showed that early oligomers of Aβ and α-synuclein have an α-helical secondary structure and are prompted by helix–helix interactions.51,98 This knowledge and our results further suggest that sEVs may be involved in toxic oligomers spread within the neurons in CNS.
In addition, we observed differences in the lipid structures of sEVs. The lipids with longer fatty acid chains are prevalent in control sEVs groups, TC+ sEVs, and osEVs. On the other hand, TC− sEVs have shorter fatty acid chain lengths. Since the main difference between TC− sEVs and TC+ sEVs is the presence of the Aβ protein, we suggest that the association of Aβ protein with plasma membrane alters plasma membrane fluidity. The plasma membrane fluidity depends on several factors, such as degree of fatty acids saturation, length of fatty acid tail, cholesterol content, and temperature. Specifically, the lengths of fatty acids tails affect the membrane rigidity by creating intermolecular interactions between phospholipid tails. In the case of TC− sEVs we observe a two-fold reduction of the chain length and as a result, a potential increase in membrane fluidity. However, the cause of this phenomenon remains to be explored. One possible explanation for the increased EV membrane fluidity is the formation of transmembrane oligomeric pore structures that are proposed to occur with the peptide's interaction with the EV plasma membrane. In addition, the length of the fatty acid chain shortens with an increase in temperature. However, this parameter should not affect our results since sEVs from all three groups were analyzed under the same experimental conditions.
Overall, our results confirm that Aβ protein is present in sEVs and can be detected via Raman spectroscopy. Moreover, our study uncovered the role of Aβ protein in the plasma membrane fluidity, paving the way for other studies on this topic. Future studies using clinical samples of AD patients will be necessary to demonstrate the potential of sEVs for early AD diagnosis. Further, studies of the sEVs derived from AD patients and healthy controls via Raman spectroscopy will possibly indicate spectral biomarkers that may correlate to the development of AD. The analysis of molecular conformation of sEVs associated Aβ protein is particularly important in understanding the role of sEVs in the propagation of neurodegeneration as it has been previously proposed in the literature. Potential pathologies underlying AD other than misfolded proteins and their conformers can be explored via Raman spectroscopy in sEVs from clinical samples. For instance, a comparison of the metal ions contents in EVs that has been shown to correlate with aggregation of Aβ protein and deposition of plaques. Moreover, another area of great interest is exploring lipidomic changes that may contribute to the disease development and may potentially be detected in EVs molecular content via Raman spectroscopy. The main drawback of the technique that limits its translation to clinic is the relatively low intensity of the Raman signal. Nonetheless, this limitation can potentially be addressed by technologies aimed at enhancing Raman signals such as plasmonic nanomaterials in surface enhanced Raman spectroscopy, or coherent Raman techniques.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
This work was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), Discovery Grant RGPIN-2018-05675 (to S. W. -H.); M. I. acknowledges the Bolashak International Scholarship for PhD studies; T. R. acknowledges The Sigrid Juselius Foundation, Helsinki, Finland; R. M. and R. P. C. acknowledge funding support from the NIH (1R01CA241666); TMD acknowledges funding support from the Van Berkom foundation, the CQDM and the McGill Healthy Brains for Healthy Lives. We also acknowledge Dr Carol Chen and Narges Abdian for prior work establishing and profiling the AIW002-02 iPSC used for MBO generation.References
- R. J. O'Brien and P. C. Wong, Annu. Rev. Neurosci., 2011, 34, 185–204 Search PubMed.
- T. A. Bayer, O. Wirths, K. Majtényi, T. Hartmann, G. Multhaup, K. Beyreuther and C. Czech, Brain Pathol., 2001, 11, 1–11 Search PubMed.
- S. G. Younkin, J. Physiol., 1998, 92, 289–292 Search PubMed.
- L. Rajendran, M. Honsho, T. R. Zahn, P. Keller, K. D. Geiger, P. Verkade and K. Simons, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11172–11177 Search PubMed.
- T. Zheng, J. Pu, Y. Chen, Y. Mao, Z. Guo, H. Pan, L. Zhang, H. Zhang, B. Sun and B. Zhang, Front. Aging Neurosci., 2017, 9, 12 Search PubMed.
- C. Théry, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 Search PubMed.
- F. Jansen, G. Nickenig and N. Werner, Circ. Res., 2017, 120, 1649–1657 Search PubMed.
- R. Kalluri, J. Clin. Invest., 2016, 126, 1208–1215 Search PubMed.
- M.-J. Lee, D.-H. Park and J.-H. Kang, Ann. Pediatr. Endocrinol. Metab., 2016, 21, 119 Search PubMed.
- D. M. Pegtel, L. Peferoen and S. Amor, Philos. Trans. R. Soc., B, 2014, 369, 20130516 Search PubMed.
- C. Caruso Bavisotto, F. Scalia, A. Marino Gammazza, D. Carlisi, F. Bucchieri, E. Conway de Macario, A. J. L. Macario, F. Cappello and C. Campanella, Int. J. Mol. Sci., 2019, 20, 434 Search PubMed.
- A. Anel, A. Gallego-Lleyda, D. de Miguel, J. Naval and L. Martínez-Lostao, Cells, 2019, 8, 154 Search PubMed.
- L. Tan, H. Wu, Y. Liu, M. Zhao, D. Li and Q. Lu, Autoimmunity, 2016, 49, 357–365 Search PubMed.
- C. Théry, et.al. , J. Extracell. Vesicles, 2018, 7, 1535750 Search PubMed.
- S. A. Bellingham, B. Guo, B. Coleman and A. F. Hill, Front. Physiol., 2012, 3, 124 Search PubMed.
- L. J. Vella, R. A. Sharples, R. M. Nisbet, R. Cappai and A. F. Hill, Eur. Biophys. J., 2008, 37, 323–332 Search PubMed.
- M. Perrotte, M. Haddad, A. Le Page, E. H. Frost, T. Fulöp and C. Ramassamy, Neurobiol. Aging, 2020, 86, 102–111 Search PubMed.
- D. J. Cha, D. Mengel, M. Mustapic, W. Liu, D. J. Selkoe, D. Kapogiannis, D. Galasko, R. A. Rissman, D. A. Bennett and D. M. Walsh, Front. Neurosci., 2019, 13, 1208 Search PubMed.
- S. A. Bellingham, B. M. Coleman and A. F. Hill, Nucleic Acids Res., 2012, 40, 10937–10949 Search PubMed.
- A. F. Hill, J. Neurosci., 2019, 39, 9269–9273 Search PubMed.
- M. Sardar Sinha, A. Ansell-Schultz, L. Civitelli, C. Hildesjö, M. Larsson, L. Lannfelt, M. Ingelsson and M. Hallbeck, Acta Neuropathol., 2018, 136, 41–56 Search PubMed.
- M. S. Fiandaca, D. Kapogiannis, M. Mapstone, A. Boxer, E. Eitan, J. B. Schwartz, E. L. Abner, R. C. Petersen, H. J. Federoff and B. L. Miller, J. Alzheimer’s Dis., 2015, 11, 600-7.e1 Search PubMed.
- D. Kapogiannis, M. Mustapic, M. D. Shardell, S. T. Berkowitz, T. C. Diehl, R. D. Spangler, J. Tran, M. P. Lazaropoulos, S. Chawla, S. Gulyani, E. Eitan, Y. An, C. W. Huang, E. S. Oh, C. G. Lyketsos, S. M. Resnick, E. J. Goetzl and L. Ferrucci, JAMA Neurol., 2019, 76, 1340–1351 Search PubMed.
- P. Spitzer, L.-M. Mulzer, T. J. Oberstein, L. E. Munoz, P. Lewczuk, J. Kornhuber, M. Herrmann and J. M. Maler, Sci. Rep., 2019, 9, 7089 Search PubMed.
- C. Z. J. Lim, Y. Zhang, Y. Chen, H. Zhao, M. C. Stephenson, N. R. Y. Ho, Y. Chen, J. Chung, A. Reilhac, T. P. Loh, C. L. H. Chen and H. Shao, Nat. Commun., 2019, 10, 1144 Search PubMed.
- D. Bachurski, M. Schuldner, P.-H. Nguyen, A. Malz, K. S. Reiners, P. C. Grenzi, F. Babatz, A. C. Schauss, H. P. Hansen, M. Hallek and E. Pogge von Strandmann, J. Extracell. Vesicles, 2019, 8, 1596016 Search PubMed.
- R. Linares, S. Tan, C. Gounou and A. R. Brisson, in Exosomes and Microvesicles, Springer, 2017, pp. 43–54 Search PubMed.
- S. Sharma, M. LeClaire and J. K. Gimzewski, Nanotechnology, 2018, 29, 132001 Search PubMed.
- C. Gardiner, D. D. Vizio, S. Sahoo, C. Théry, K. W. Witwer, M. Wauben and A. F. Hill, J. Extracell. Vesicles, 2016, 5, 32945 Search PubMed.
- M. S. Panagopoulou, A. W. Wark, D. J. S. Birch and C. D. Gregory, J. Extracell. Vesicles, 2020, 9, 1710020 Search PubMed.
- M. P. Bebelman, P. Bun, S. Huveneers, G. van Niel, D. M. Pegtel and F. J. Verweij, Nat. Protoc., 2020, 15, 102–121 Search PubMed.
- S. Y. Kim, D. Khanal, B. Kalionis and W. Chrzanowski, Nat. Protoc., 2019, 14, 576–593 Search PubMed.
- T. Rojalin, B. Phong, H. J. Koster and R. P. Carney, 2019, 7.
- A. Gualerzi, S. A. A. Kooijmans, S. Niada, S. Picciolini, A. T. Brini, G. Camussi and M. Bedoni, J. Extracell. Vesicles, 2019, 8, 1568780 Search PubMed.
- G. Devitt, K. Howard, A. Mudher and S. Mahajan, ACS Chem. Neurosci., 2018, 9, 404–420 Search PubMed.
- Y. An, T. Jin, Y. Zhu, F. Zhang and P. He, Biosens. Bioelectron., 2019, 142, 111503 Search PubMed.
- N. Colthup, Introduction to infrared and Raman spectroscopy, Elsevier, 2012 Search PubMed.
- F. Lavialle, S. Deshayes, F. Gonnet, E. Larquet, S. G. Kruglik, N. Boisset, R. Daniel, A. Alfsen and I. Tatischeff, Int. J. Pharm., 2009, 380, 206–215 Search PubMed.
- Z. J. Smith, C. Lee, T. Rojalin, R. P. Carney, S. Hazari, A. Knudson, K. Lam, H. Saari, E. L. Ibañez, T. Viitala, T. Laaksonen, M. Yliperttula and S. Wachsmann-Hogiu, J. Extracell. Vesicles, 2015, 4, 28533 Search PubMed.
- I. Tatischeff, E. Larquet, J. M. Falcón-Pérez, P.-Y. Turpin and S. G. Kruglik, J. Extracell. Vesicles, 2012, 1, 19179 Search PubMed.
- C. Krafft, K. Wilhelm, A. Eremin, S. Nestel, N. von Bubnoff, W. Schultze-Seemann, J. Popp and I. Nazarenko, Nanotechnology, Biology and Medicine, 2017, 13, 835–841 Search PubMed.
- W. Lee, A. Nanou, L. Rikkert, F. A. W. Coumans, C. Otto, L. W. M. M. Terstappen and H. L. Offerhaus, Anal. Chem., 2018, 90, 11290–11296 Search PubMed.
- A. Gualerzi, S. Niada, C. Giannasi, S. Picciolini, C. Morasso, R. Vanna, V. Rossella, M. Masserini, M. Bedoni and F. Ciceri, Sci. Rep., 2017, 7, 1–11 Search PubMed.
- H. Zhang, A. C. Silva, W. Zhang, H. Rutigliano and A. Zhou, PloS One, 2020, 15, e0235214 Search PubMed.
- M. Roman, A. Kamińska, A. Drożdż, M. Platt, M. Kuźniewski, M. T. Małecki, W. M. Kwiatek, C. Paluszkiewicz and E. Ł. Stępień, Nanotechnology, Biology and Medicine, 2019, 17, 137–149 Search PubMed.
- P. Beekman, A. Enciso-Martinez, H. S. Rho, S. P. Pujari, A. Lenferink, H. Zuilhof, L. W. M. M. Terstappen, C. Otto and S. Le Gac, Lab Chip, 2019, 19, 2526–2536 Search PubMed.
- D. Kurouski, R. P. Van Duyne and I. K. Lednev, Analyst, 2015, 140, 4967–4980 Search PubMed.
- D. Röhr, B. D. C. Boon, M. Schuler, K. Kremer, J. J. M. Hoozemans, F. H. Bouwman, S. F. El-Mashtoly, A. Nabers, F. Großerueschkamp, A. J. M. Rozemuller and K. Gerwert, Acta Neuropathol. Commun., 2020, 8, 222 Search PubMed.
- M. Ji, M. Arbel, L. Zhang, C. W. Freudiger, S. S. Hou, D. Lin, X. Yang, B. J. Bacskai and X. S. Xie, Sci. Adv., 2018, 4, eaat7715 Search PubMed.
- N. C. Maiti, M. M. Apetri, M. G. Zagorski, P. R. Carey and V. E. Anderson, J. Am. Chem. Soc., 2004, 126, 2399–2408 Search PubMed.
- M. M. Apetri, N. C. Maiti, M. G. Zagorski, P. R. Carey and V. E. Anderson, J. Mol. Biol., 2006, 355, 63–71 Search PubMed.
- G. Ramachandran, E. A. Milán-Garcés, J. B. Udgaonkar and M. Puranik, Biochemistry, 2014, 53, 6550–6565 Search PubMed.
- E. Ryzhikova, O. Kazakov, L. Halamkova, D. Celmins, P. Malone, E. Molho, E. A. Zimmerman and I. K. Lednev, J. Biophot., 2015, 8, 584–596 Search PubMed.
- C. W. Ong, Z. X. Shen, Y. He, T. Lee and S. H. Tang, J. Raman Spectrosc., 1999, 30, 91–96 Search PubMed.
- N. Mammadova, C. M. Summers, R. D. Kokemuller, Q. He, S. Ding, T. Baron, C. Yu, R. J. Valentine, D. S. Sakaguchi and A. G. Kanthasamy, Neurobiol. Dis., 2019, 121, 1–16 Search PubMed.
- M. Paraskevaidi, C. L. M. Morais, D. E. Halliwell, D. M. A. Mann, D. Allsop, P. L. Martin-Hirsch and F. L. Martin, ACS Chem. Neurosci., 2018, 9, 2786–2794 Search PubMed.
- A. Huefner, W.-L. Kuan, S. L. Mason, S. Mahajan and R. A. Barker, Chem. Sci., 2020, 11, 525–533 Search PubMed.
- A. Gualerzi, S. Picciolini, C. Carlomagno, F. Terenzi, S. Ramat, S. Sorbi and M. Bedoni, Nanotechnology, Biology and Medicine, 2019, 22, 102097 Search PubMed.
- C. F. Morasso, D. Sproviero, M. C. Mimmi, M. Giannini, S. Gagliardi, R. Vanna, L. Diamanti, S. Bernuzzi, F. Piccotti and M. Truffi, Nanotechnology, Biology and Medicine, 2020, 102249 Search PubMed.
- C. Lee, R. P. Carney, S. Hazari, Z. J. Smith, A. Knudson, C. S. Robertson, K. S. Lam and S. Wachsmann-Hogiu, Nanoscale, 2015, 7, 9290–9297 Search PubMed.
- S. Suarasan, J. Liu, M. Imanbekova, T. Rojalin, S. Hilt, J. C. Voss and S. Wachsmann-Hogiu, J. Mater. Chem. B, 2020, 8, 8845–8852 Search PubMed.
- M. Avella-Oliver, R. Puchades, S. Wachsmann-Hogiu and A. Maquieira, Sens. Actuators, B, 2017, 252, 657–662 Search PubMed.
- M. Kahraman, P. Daggumati, O. Kurtulus, E. Seker and S. Wachsmann-Hogiu, Sci. Rep., 2013, 3, 3396 Search PubMed.
- T. Rojalin, H. J. Koster, J. Liu, R. R. Mizenko, D. Tran, S. Wachsmann-Hogiu and R. P. Carney, ACS Sens., 2020, 5, 2820–2833 Search PubMed.
- B. L. Sopher, K.-i. Fukuchi, A. C. Smith, K. A. Leppig, C. E. Furlong and G. M. Martin, Mol. Brain Res., 1994, 26, 207–217 Search PubMed.
- N.-V. Mohamed, M. Mathur, R. da Silva, L. Beitel, E. Fon and T. Durcan, MNI Open Res., 2019, 3, 1 Search PubMed.
- Y. Yuana, A. N. Böing, A. E. Grootemaat, E. van der Pol, C. M. Hau, P. Cizmar, E. Buhr, A. Sturk and R. Nieuwland, J. Extracell. Vesicles, 2015, 4, 29260 Search PubMed.
- Y. Cheng, Q. Zeng, Q. Han and W. Xia, Protein Cell, 2019, 10, 295–299 Search PubMed.
- E. Valera, R. Dargusch, P. A. Maher and D. Schubert, J. Neurosci., 2013, 33, 10512–10525 Search PubMed.
- A. Currais, O. Quehenberger, A. M Armando, D. Daugherty, P. Maher and D. Schubert, npj Aging Mech. Dis., 2016, 2, 16012 Search PubMed.
- P. F. Copenhaver, T. S. Anekonda, D. Musashe, K. M. Robinson, J. M. Ramaker, T. L. Swanson, T. L. Wadsworth, D. Kretzschmar, R. L. Woltjer and J. F. Quinn, Dis. Models Mech., 2011, 4, 634–648 Search PubMed.
- K. Laulagnier, C. Motta, S. Hamdi, S. Roy, F. Fauvelle, J.-F. Pageaux, T. Kobayashi, J.-P. Salles, B. Perret, C. Bonnerot and M. Record, Biochem. J., 2004, 380, 161–171 Search PubMed.
- I. Parolini, C. Federici, C. Raggi, L. Lugini, S. Palleschi, A. De Milito, C. Coscia, E. Iessi, M. Logozzi, A. Molinari, M. Colone, M. Tatti, M. Sargiacomo and S. Fais, J. Biol. Chem., 2009, 284, 34211–34222 Search PubMed.
- L. Rajendran, M. Honsho, T. R. Zahn, P. Keller, K. D. Geiger, P. Verkade and K. Simons, Proc. Natl. Acad. Sci., 2006, 103, 11172–11177 Search PubMed.
- M.-S. García-Ayllón, I. Lopez-Font, C. P. Boix, J. Fortea, R. Sánchez-Valle, A. Lleó, J.-L. Molinuevo, H. Zetterberg, K. Blennow and J. Sáez-Valero, Sci. Rep., 2017, 7, 2477 Search PubMed.
- H. Jeong, H. Shin, J. Yi, Y. Park, J. Lee, Y. Gianchandani and J. Park, Lab Chip, 2019, 19, 3326–3336 Search PubMed.
- S. Zou, M. Hou, J. Li, L. Ma and Z. Zhang, Sci. Rep., 2017, 7, 6186 Search PubMed.
- R. Yasukuni, R. Gillibert, M. N. Triba, R. Grinyte, V. Pavlov and M. Lamy de la Chapelle, Nanophotonics, 2019, 8, 1477–1483 Search PubMed.
- B. Hernández, F. Pflüger, S. G. Kruglik and M. Ghomi, J. Raman Spectrosc., 2013, 44, 827–833 Search PubMed.
- S. K. Teh, W. Zheng, D. P. Lau and Z. Huang, Analyst, 2009, 134, 1232–1239 Search PubMed.
- J. Bandekar, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1992, 1120, 123–143 Search PubMed.
- S. P. Verma and D. F. H. Wallach, Biochim. Biophys. Acta, Lipids Lipid Metab., 1977, 486, 217–227 Search PubMed.
- I. Notingher, S. Verrier, S. Haque, J. M. Polak and L. L. Hench, Biopolymers, 2003, 72, 230–240 Search PubMed.
- D. Song, T. Chen, S. Wang, S. Chen, H. Li, F. Yu, J. Zhang and Z. Zhang, Analyst, 2020, 145, 626–635 Search PubMed.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyk and M. Baranska, J. Raman Spectrosc., 2013, 44, 1061–1076 Search PubMed.
- L. J. Lis, S. C. Goheen, J. W. Kauffman and D. F. Shriver, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1976, 443, 331–338 Search PubMed.
- B. Shivu, S. Seshadri, J. Li, K. A. Oberg, V. N. Uversky and A. L. Fink, Biochemistry, 2013, 52, 5176–5183 Search PubMed.
- M. Coles, W. Bicknell, A. A. Watson, D. P. Fairlie and D. J. Craik, Biochemistry, 1998, 37, 11064–11077 Search PubMed.
- M. Ji, D. A. Orringer, C. W. Freudiger, S. Ramkissoon, X. Liu, D. Lau, A. J. Golby, I. Norton, M. Hayashi and N. Y. R. Agar, Sci. Transl. Med., 2013, 5, 201ra119 Search PubMed.
- I. Anna, P. Bartosz, P. Lech and A. Halina, Oncotarget, 2017, 8, 85290 Search PubMed.
- K. Larsson and R. P. Rand, Biochim. Biophys. Acta, Lipids Lipid Metab., 1973, 326, 245–255 Search PubMed.
- J. Kiskis, H. Fink, L. Nyberg, J. Thyr, J.-Y. Li and A. Enejder, Sci. Rep., 2015, 5, 13489 Search PubMed.
- L. E. Jamieson, A. Li, K. Faulds and D. Graham, R. Soc. Open Sci., 2018, 5, 181483 Search PubMed.
- W. Gibson Wood, G. P. Eckert, U. Igbavboa and W. E. Müller, Biochim. Biophys. Acta, Biomembr., 2003, 1610, 281–290 Search PubMed.
- J. C. Stroud, C. Liu, P. K. Teng and D. Eisenberg, 2012, 109, 7717–7722.
- L. Yu, R. Edalji, J. E. Harlan, T. F. Holzman, A. P. Lopez, B. Labkovsky, H. Hillen, S. Barghorn, U. Ebert, P. L. Richardson, L. Miesbauer, L. Solomon, D. Bartley, K. Walter, R. W. Johnson, P. J. Hajduk and E. T. Olejniczak, Biochemistry, 2009, 48, 1870–1877 Search PubMed.
- E. Cerf, R. Sarroukh, S. Tamamizu-Kato, L. Breydo, S. Derclaye, Y. F. Dufrêne, V. Narayanaswami, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biochem. J., 2009, 421, 415–423 Search PubMed.
- A. Abedini and D. P. Raleigh, Phys. Biol., 2009, 6, 015005 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |