
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermomechanically controlled fluorescence anisotropy in thin films of InP/ZnS quantum dots†
Sylwia
Parzyszek
 ,
Damian
Pociecha
,
Joanna Maria
Wolska
* and
Wiktor
Lewandowski
*
,
Damian
Pociecha
,
Joanna Maria
Wolska
* and
Wiktor
Lewandowski
*
Faculty of Chemistry, University of Warsaw, Pasteura 1 st., 02-093 Warsaw, Poland. E-mail: jokos@chem.uw.edu.pl; wlewandowski@chem.uw.edu.pl
First published on 9th August 2021
Abstract
Macroscopic scale sources of polarized light play a fundamental role in designing light-emitting devices. In this communication we report the formation of nano- and macro-scale ordered, layered assemblies of InP/ZnS quantum dots (QDs) exhibiting fluorescence anisotropy (FA), as well as thermo- and mechano-responsive properties. The long-range organization of small, quasi-isotropic nanoparticles was achieved by introducing liquid crystal molecules to the surface of QDs, without the need to use an organic matrix. Melting/crystallization of the ligand at 95 deg. C translated to a reversible reconfiguration of QDs thin film between 2D layered and body-centered cubic structures, characteristic for a temperature range below and above the melting point, respectively. The low-temperature, layered structure exhibited mechano-responsiveness which was key to introduce and control the sample alignment. Interestingly, transverse and parallel alignment modes of QDs layers were achieved, depending on the temperature of mechanical shearing. As prepared QD samples exhibited fluorescence anisotropy strongly correlated to the macroscopic orientation of the layers. Correlated small-angle X-ray diffraction (SAXRD) and fluorescence spectroscopy studies confirmed the mm-scale alignment of the thin films of QDs. Such films may be advantageous for developing efficient, densely packed, and uniform macro-scale FA sources.
Introduction
Materials exhibiting controlled fluorescence anisotropy (FA) are highly demanded for the rapidly developing lighting and display technologies,1–6 fueling materials research towards the realization of efficient and cost-effective FA sources.7–9 Quantum dots (QDs, down-converting semiconductor nanocrystals) are considered particularly appealing FA sources since they exhibit long-term stability, high quantum efficiency, and high color purity, superior to organic materials. Further, the emission wavelength (QDs band-gap) can be tuned by adjusting nanocrystals' composition and size through colloidal synthesis.10 For example, rod-like,11 nanoplatelets,12 tetrapods,13 dot-in-rod,14 or dot-in-plate15 were shown to exhibit FA in dispersion, at the single-particle level. However, many practical applications rely on device-scale, tuneable polarized luminescence from thin films of QDs, which requires the formation of anisotropic, configurable assemblies with long-range order ranging from nm to mm scales.16Over the last two decades several tools and methods to obtain ordered, thin films of QDs (often referred to as superlattices, SL) were developed.17–20 In the case of anisotropic nanocrystals their unidirectional alignment in SL is required to coherently enhance the intrinsic directional properties of individual QDs and achieve macroscopically anisotropic fluorescence.21 Such orientationally ordered assemblies can be achieved by exploiting evaporation/destabilization of dispersions, assembling QDs at the interfaces, by using a template- and external factors-assisted assembly or other methods.22–24 Conversely, in the case of quasi-isotropic QDs, the formation of non-close packed, anisotropic thin films is required to explore their anisotropic, collective interactions and thus induce FA.25,26 An elegant way to achieve structurally anisotropic thin films is by selective adsorption of ligands on different crystallographic facets of quasi-isotropic QDs, which results in an anisotropic organic shell of the nanocrystals, and translates to the anisotropic distribution of QDs in space.27–29 However, besides few sparse examples,30,31 most anisotropic and quasi-isotropic QDs SLs are static, with restricted tunability of structure and optical characteristics of the materials.
In this context, hybrid materials comprising QDs and liquid crystals (QDs/LC) are particularly appealing since they benefit from the soft, ordered, and responsive character of mesogens, allowing to drive the organization/orientation of assemblies of QDs and enabling the formation of macroscopically (device-scale) ordered systems. For example, LC film doped with CdSe/CdS dot-in-rod QDs can serve as a backlight source for a display, absorbing unpolarized light and illuminating display with reemitted polarized light. Matching the polarized luminescence direction with the LC cell polarizer, by controlling the orientation of rod-like QDs, can translate to lowering the energy losses.1,32 Moreover, QDs/LC nanocomposites allow for electric field driven, fast-rate switching of the conformation of the LC matrix. Along this line, it has been proven that nematic LC doped with cuboidal CsPbBr3 perovskite QDs can exhibit electrically controlled, continuously tuneable FA,33 while doping semiconductor nanocrystals to a chiral nematic host can lead to composites exhibiting circularly polarized luminescence with electrically controlled dissymmetry factor.34 These QDs/LC nanocomposites allow for ordered QDs thin-film tunability, long-range order, and exhibit directional optical properties, making them interesting for applications as low-cost on-demand single photon sources or energy-efficient displays.16,34–37 However, one of the key parameters determining optical quality of these devices is compatibility between the ligand shell of quantum dots and the LC matrix.16,38,39 Several groups have recently demonstrated that precise control of the architecture of the ligand shell can lead to enhanced dispersion or tuneable organization of QDs in the LC matrix,36 however further research on QDs/LC matrix interactions is required to fully capitalize on the potential of these devices, e.g. through increasing the QDs concentration to maximize the optical response.40,41 For example, partial covering of QDs (or other types of NPs) with liquid crystalline organic ligands, chemically compatible with the LC matrix, has been recently shown to allow overcoming the colloidal instability of QDs dispersions in the LC host16,42 and increase the uniformity of the films, but, even this way, it is usually impossible to achieve doping higher than 5 wt% and further research on this topic is required.16,38,39,43–46
An alternative way to maximize the QDs concentration in an anisotropic material is to use LC-coated QDs in the neat state, not doped to a LC matrix; in such a case the distances between QDs are defined solely by the size of organic ligands. This way, fluorescence anisotropy could be potentially achieved for isotropic nanocrystals, driven by their collective, anisotropic interactions, not relying on the anisotropy of LC matrix or QDs shape. Thus, for LC-coated QDs there is no need for optimization of QDs/LC matrix interactions or synthetizing anisotropic nanocrystals. The driving force for the assembly is that LC ligands exhibit steric and electromagnetic interactions leading to their bundling and imposing the anisotropic shape of the organic coating. Further, the temperature-dependent ligand–ligand interactions allow for achieving a morphable character of the organic shell, leading to structural transitions in a densely packed thin film of QDs. For example, the formation of long-range ordered thin films with P213 cubic symmetry was evidenced for quasi-spherical CdSe/ZnSe QDs coated with short alkyl thiols and dendrimeric LC ligands after prolonged heat-annealing.47 Reversible structural transitions of QDs assembly have been recently reported by some of us. Quasi-spherical PbS/CdS QDs covered with a mixed shell comprising alkyl and LC ligands were shown to spontaneously form SLs; depending on ligands ratio in the organic coating, these QDs exhibited lamellar to body-centered cubic or body-centered tetragonal to face-centered cubic phase transitions.31 Preliminary optical investigations confirmed that symmetry of the SL influenced the position of the emission band maxima, further confirming that collective interactions of QDs can be modulated. Unfortunately, no device-scale aligned samples were prepared thus it was not possible to detect directional optical properties of the LC-coated QDs thin films.
It is also worth mentioning that ‘smart’, thermomechanically responsive materials42,48 can be achieved using the LC-ligands-based approach. For example, some of us have reported on metallic NPs covered with dimeric LC ligands, which exhibited various modes of SL alignment.43 It can be thus expected that also QDs covered with similar LC ligands could lead to hybrid composites exhibiting mechano-tuneable, anisotropic structures.
The stability of quantum dot thin films is another important aspect in the context of their practical applications.50 The relatively high molecular weight of liquid crystalline ligands usually ensures the high durability of LC coated nanoparticles against mechanical and thermal workup.41,43,49 However, in contrast to polymeric ligand shells offering a similar increase of stability, LC ligands form anisotropic shells at low temperatures, leading to a superior optoelectronic coupling between nanocrystals.
Herein we report a macroscopic fluorescence anisotropy of red-emitting InP/ZnS QDs coated partially with dimeric LC ligands. These ligands ensure the assemble of QDs into superlattices with temperature-dependent structure (in the neat state). A body-centered cubic structure is formed at elevated temperatures, above the melting point of ligands, while an anisotropic, lamellar structure is formed at low temperature. Importantly, control of the orientation of the assembly is possible by mechanical shearing at different temperatures, leading to mm-scale films with uniformly anisotropic structure exhibiting macroscopically anisotropic emission.
Results and discussion
We used a modified Tessier's method to prepare InP/ZnS QDs, which yielded oleylamine covered, 2.8 nm size quasi-isotropic (tetrahedral) QDs (see Experimental section and ESI Note 1†).51 To obtain QDs covered with a mixture of LC and 1-dodecanethiol (DDT) ligands, a ligand–exchange reaction was performed in two steps (Fig. 1a). First, oleylamine coated InP/ZnS QDs (Fig. 1b and S1–S3†) were mixed with an excess of DDT. DDT-coated QDs are usually considered to have lower stability than oleylamine coated QDs, but we solved this problem by storing QDs in dispersions with an excess amount of free DDT molecules and purification of these QDs directly before further use.52 The obtained DDT-coated QDs exhibited an emission band centered at ∼620 nm in a toluene dispersion, similar to oleylamine coted QDs of the same size in the original report on their synthesis.51 Then, the second ligand exchange procedure was performed to introduce LC ligand (compound L, Fig. 1c, ESI Note 2†) to the surface of QDs, yielding the final material QDs/L. The L ligand design was dictated by two criteria. Firstly, we aimed at producing lamellar assemblies of QDs, since this symmetry can lead to high anisotropy of collective interactions of nanoparticles. Such properties were achieved for hybrid NP systems comprising linear or branched ligands.53 Secondly, we aimed at mechanothermal control of sample alignment.50 Thus, the L ligand was designed to comprise two rigid parts connected by an 11-carbon long flexible spacer via ether and ester linkages. Two flexible alkoxy chains were attached at the distant ends of the aromatic units, endowing L with LC properties and allowing attachment to the surface of nanoparticles via a terminal thiol moiety.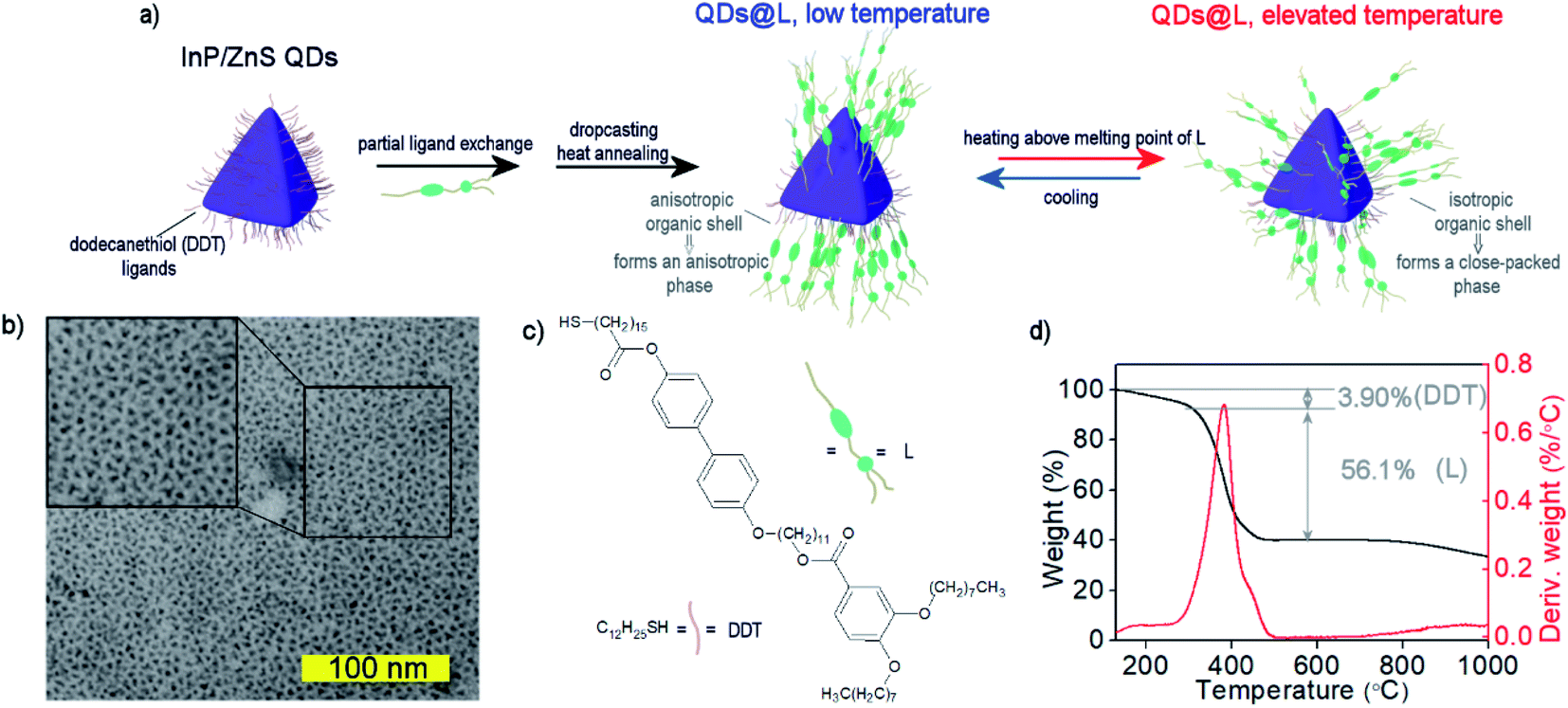 | ||
| Fig. 1 Structural characterization of InP/ZnS QDs. (a) Scheme showing functionalization of QDs with liquid crystal ligand (L showed in panel (c)) and temperature-dependent ligands' distribution around QDs. (b) TEM image of InP/ZnS QDs. The inset presents magnification of 100×100 nm square. (c) The chemical formulas of organic ligands. (d) Thermogravimetric analysis of QDs/LC hybrid material revealed that it contains 40% of InP/ZnS QDs, 3.90% DDT, and 56.1% LC. | ||
To determine the composition of the organic shell of QDs/L we performed thermogravimetric analysis (TG, Fig. 1d) in an inert atmosphere. It revealed that ∼40% of the nanoparticle mass comes from the inorganic core, which translates to ∼40% of the volume of inorganic material in the tested materials. Two separate events of mass losses were evidenced, which can be ascribed to the size/type of the ligand. The mass losses were 3.90% for 120–250 °C, and 56.1% for 250–510 °C, corresponding to detachment of DDT and L ligands, respectively, and evidencing a threefold molar excess of L over DDT in the organic shell.
Partial exchange of DDT ligands with mesogenic L molecules did not considerably affect the fluorescent properties of QDs. The emission maximum remained at 615 nm, and PL quantum yield was 80%, as determined using rhodamine 101 in ethanol as a reference.
Due to the mixed organic coating, we expected the temperature-responsive shape of the organic shell. To test this hypothesis the temperature-dependent, small-angle X-ray diffraction (SAXRD) experiments were performed. For this purpose toluene dispersion of QDs/L was drop-casted on Kapton foil and dried to remove the solvent. The sample was thermally annealed by heating up to 140 °C and slowly cooled down to 30 °C. Then, diffraction patterns were collected on heating to 140 °C in 3 K steps (Fig. 2a), which revealed a clear phase transition at 95 °C. Below the transition, diffraction patterns can be ascribed to lamellar structure (Lam) with the layer periodicity of 10.5 nm, and mean in-plane interparticle distance of 7.2 nm (Fig. S4a†). Notably, this means that surface-to-surface distance between QDs along a direction normal to the layer is almost doubled, in comparison to in-plane QDs surface-to-surface distance (Table S1†). High-temperature phase diffractograms can be indexed assuming a body-centered cubic (bcc) structure, with the unit cell parameter a = 10.0 nm (Fig. S4b and S5†). Based on the recorded diffractograms we could calculate volume per single QD (Table S1†), this parameter was found to be ∼7% larger in the higher temperature bcc phase. The effect is most probably due to the increase of organic coating volume caused by growing entropic demands of the flexible ligands, which is typical for thermoresponsive liquid crystalline nanoparticles.54 It is worth noting that there were no qualitative differences between subsequent batches of QDs covered with L, or after exposing the materials to air for a few days, evidencing high reproducibility and reasonable durability of the self-assembly properties of QDs (Fig. S6†).
 | ||
| Fig. 2 X-ray analysis of the long-range order in QDs/L thin films. (a) Small-angle X-ray diffraction (SAXRD) temperature map obtained for an unaligned (multidomain) sample of QDs/L hybrid material, it exhibits lamellar structure at low temperatures that rearrange to bcc on heating. (b and c) show the azimuthal distribution of the intensity of the diffraction signal (001) related to lamellar structure for samples sheared at 40 °C and 80 °C, respectively. Note that the layer orientation is rotated by 90 deg. in samples sheared at different temperatures. (d) SAXRD pattern of an aligned sample of QDs/L sheared at 40 °C. Arrow indicates the shearing direction. (e) SAXRD pattern of an aligned sample of QDs/L sheared at 80 °C. Arrow indicates the shearing direction. (f and g) Schematically represent the procedure of macroscopic orientation of QDs/L material by the mechanical shearing. | ||
The flexibility of the QDs mixed organic coating built of DDT and LC molecules define the final structure of QDs/L assemblies in thin films and facilitate macroscale domains orientation.55 We expected that the proposed ligand architecture should allow for anisotropic orientation of InP/ZnS QDs, leading to optical anisotropy. To obtain macroscopic-scale aligned samples the mechanical stress alignment procedure was applied.56 The QDs/L were drop-casted on the Kapton substrate and after thermal annealing, the mechanical shearing was performed at two temperatures in the range of lamellar phase stability (40 and 80 °C, samples QDs/L/40 and QDs/L/80, respectively). It should be noted that the applied procedure did not affect the observed structure formed by hybrid QDs, in both cases signals characteristic for the lamellar phase were detected however, two different alignment modes, transverse and parallel, were observed depending on the shearing temperature. The SAXRD diffractograms for samples QDs/L/40 and QDs/L/80 (Fig. 2d, e and S7†) evidenced different orientations of the QDs layers with respect to the shearing direction; the layer normal was parallel to the shearing direction for QDs/L/40, while perpendicular for QDs/L/80. It is worth noting that qualitatively better alignment was achieved for sample sheared at the higher temperature, as evidenced by the narrower distribution of diffracted signal intensity vs. azimuthal angle (Fig. 2b and c). The better orientation for QDs/L/80 results probably from the lower viscosity of the material at elevated temperature.
The same alignment procedure was applied to prepare samples for FA measurements, however in this case hybrid QD material was deposited on glass substrates. Using SAXRD nanography (Fig. S7†) it was proved that uniform alignment was achieved on a macro scale – diffractograms taken from various spots on the sample showed the same orientation of the QD layers.
The emission of the QDs/L thin films were first measured for a non-aligned sample (Fig. S8†) evidencing a relatively narrow (FWHM = 62 nm) emission band centered at 623 nm. Thus the initial temperature annealing caused a drop of PL intensity by ca. 22%, however the following heating–cooling cycles did not affect the intensity of PL, nor did storing the material under an inert atmosphere over a period of a week, although we expect limited durability of these materials without protective encapsulation. The FA measurements were performed for QDs/L/40 and QDs/L/80 samples on a glass substrate (Fig. 3a, c) using a fluorometer setup with two rotating polarizers, and emission intensity was measured with polarizers either in parallel (I‖) or perpendicular (I⊥) alignment. Normalized fluorescence spectra for QDs/L/40 and QDs/L/80 are presented in Fig. 3b, d. Notably, samples sheared at different temperatures exhibit the opposite sign of anisotropy of fluorescence, which results from the opposite orientation of QD layers to the shearing direction in both samples. It should be stressed that the sign of anisotropy changes when the samples are rotated by 90 deg (Fig. S9 and S10†), apparently, in all cases the higher intensity of emitted light is observed when the exit polarizer is parallel to the QDs layers, proving that the fluorescence is partially polarized along the layers. The anisotropy of fluorescence intensity for QDs/L/80 is considerably higher than for QDs/L/40, such difference may arise from the more uniform alignment of sample sheared at the higher temperature. The FA for both sheared samples is significantly higher than the one measured in the control experiment (Fig. S11†).
 | ||
| Fig. 3 FA measurements of macro-scale aligned QDs/L thin films exhibiting orthogonal lamellar structure. (a and c) show QDs/L/40 and QDs/L/80 samples, prepared on glass substrates, under UV irradiation, images correspond to 5 mm × 5 mm area. (b and d) Fluorescence anisotropy was detected for QDs/L/40 and QDs/L/80 respectively. | ||
Experimental section
Fabrication of InP/ZnS QDs stabilized with 1-dodecanethiol
A seed-mediated Tessier et al. procedure was used to prepare InP/ZnS QDs with slight modifications as described in ESI Note 1.†51 A fraction containing 2.8 nm diameter tetrahedral QDs, capped with oleylamine were obtained by size-focusing precipitation with growing amounts of acetone. These QDs were modified with a monolayer of dodecanethiol ligands using a ligand exchange reaction. A three-fold molar excess of dodecanethiol (in respect of the estimated amount of oleylamine) was added to InP/ZnS capped with DDT dispersion in toluene and left overnight in a glass vial, ensuring magnetic stirring. After this time, the reaction mixture was precipitated several times with acetone, to remove the majority of unbound ligands, and dispersed in toluene. For prolonged storage, it is critical that DDT-coated InP/ZnS QDs should be stored at room temperature in dispersions containing an excess of free DDT molecules.Fabrication of QDs/L
For modifying QDs' surface with L ligand, a ligand exchange reaction was carried out using DDT-coated QDs. Right before the ligand exchange, the DDT-coated QDs were purified from the unbound ligands by several precipitations with acetone. Successful purification of DDT-coated QDs was confirmed with the thin layer chromatography technique. For ligand exchange, the purified QDs (0.5 ml, 10 mg ml−1) were mixed with a toluene solution of liquid crystal (L, 2.0 ml, 10 mg ml−1) and left on stirring (400 rpm) for 1 h. The final QDs/L were achieved by purifying the ligand exchange mixture, by precipitation/dispersion protocol using acetone as the antisolvent. Thin-layer chromatography was used to confirm the purity of the final material.Conclusions
We proposed and successfully realized a facilitated approach towards anisotropic, densely packed thin films of quasi-isotropic QDs exhibiting anisotropic fluorescence at the macroscopic scale. For this purpose, InP/ZnS QDs were modified with liquid crystalline ligands which ensured self-assembly of QDs into long-range ordered structures. The reversible structural transformation of the films, between lamellar and bcc structures, was thermally induced due to a morphable character of the organic shell. Mechanical shearing of the material allowed us to obtain unidirectionally aligned lamellar superlattices of QDs/L. Orthogonally oriented lamellar superlattices were obtained simply by varying the temperature at which the alignment procedure was applied. For such macroscopically oriented samples we observed anisotropy of fluorescence in the visible range, originating from the anisotropic spatial distribution of QDs. Our LC-based approach enables the formation of mechanically and thermally durable, uniform thin films of QD exhibiting FA with high, 40% wt loading of inorganic QDs cores and variable macroscopic scale alignment, providing a simple and feasible basis for more effective, configurable sources of polarized light. Further research could be directed towards enhancing durability of the LC-coated QDs materials and exploring the possibility to induce electrically or optically responsiveness.Author contributions
WL – conceptualization, funding acquisition, supervision; SP – investigation, visualization; SP, WL – methodology, writing – original draft; SP, WL, JW, DP – formal analysis, writing – review & editing.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This research was funded the REINFORCE project (Agreement No. First TEAM2016-2/15) carried out within the First Team program of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund.References
- A. K. Srivastava, W. Zhang, J. Schneider, A. L. Rogach, V. G. Chigrinov and H.-S. Kwok, Adv. Mater., 2017, 29, 1701091 CrossRef PubMed.
- J. Kim, J. Peretti, K. Lahlil, J. P. Boilot and T. Gacoin, Adv. Mater., 2013, 25, 3295–3300 CrossRef CAS PubMed.
- Y. Wang, J. Shi, J. Chen, W. Zhu and E. Baranoff, J. Mater. Chem. C, 2015, 3, 7993–8005 RSC.
- L. Tao, K. Lan, C.-L. Zhong, Y.-J. Zhou, P. Wang, F. Fan, Z. Shen and H.-L. Xie, J. Mater. Chem. C, 2020, 8, 16561–16568 RSC.
- D. Yan, R. Gao, M. Wei, S. Li, J. Lu, D. G. Evans and X. Duan, J. Mater. Chem. C, 2013, 1, 997–1004 RSC.
- D. Yan, W. Jones, G. Fan, M. Wei and D. G. Evans, J. Mater. Chem. C, 2013, 1, 4138–4145 RSC.
- L. Zhang, J. H. Teng, S. J. Chua and E. A. Fitzgerald, Appl. Phys. Lett., 2009, 95, 261110 CrossRef.
- M. P. Aldred, P. Vlachos, A. E. A. Contoret, S. R. Farrar, W. Chung-Tsoi, B. Mansoor, K. L. Woon, R. Hudson, S. M. Kelly and M. O'Neill, J. Mater. Chem., 2005, 15, 3208–3213 RSC.
- E. Matioli, S. Brinkley, K. M. Kelchner, Y. L. Hu, S. Nakamura, S. DenBaars, J. Speck and C. Weisbuch, Light: Sci. Appl., 2012, 1, 22 CrossRef.
- C. Z. N. Peidong Yang, L. Dou and P. Yang, Nat. Rev. Mater., 2017, 2, 17070 CrossRef.
- J. Hu, L. S. Li, W. Yang, L. Manna, L. W. Wang and A. P. Alivisatos, Science, 2001, 292, 2060–2063 CrossRef CAS PubMed.
- C. Huo, C. F. Fong, M. R. Amara, Y. Huang, B. Chen, H. Zhang, L. Guo, H. Li, W. Huang, C. Diederichs and Q. Xiong, Nano Lett., 2020, 20, 3673–3680 CrossRef CAS PubMed.
- S. Asokan, K. M. Krueger, V. L. Colvin and M. S. Wong, Small, 2007, 3, 1164–1169 CrossRef CAS PubMed.
- F. Pisanello, L. Martiradonna, P. Spinicelli, A. Fiore, J. P. Hermier, L. Manna, R. Cingolani, E. Giacobino, M. De Vittorio and A. Bramati, Superlattices Microstruct., 2010, 47, 165–169 CrossRef CAS.
- E. Cassette, B. Mahler, J. M. Guigner, G. Patriarche, B. Dubertret and T. Pons, ACS Nano, 2012, 6, 6741–6750 CrossRef CAS PubMed.
- W. Zhang, M. F. Prodanov, J. Schneider, S. K. Gupta, T. Dudka, V. V. Vashchenko, A. L. Rogach and A. K. Srivastava, Adv. Funct. Mater., 2019, 29, 1805094 CrossRef.
- A. V. Baranov, E. V. Ushakova, V. V. Golubkov, A. P. Litvin, P. S. Parfenov, A. V. Fedorov and K. Berwick, Langmuir, 2015, 31, 506–513 CrossRef CAS PubMed.
- R. Li, K. Bian, T. Hanrath, W. A. Bassett and Z. Wang, J. Am. Chem. Soc., 2014, 136, 46 Search PubMed.
- Y. Nagaoka, H. Zhu, D. Eggert and O. Chen, Science, 2018, 362, 1396–1400 CrossRef CAS PubMed.
- J. Park, S. Jeong, J. Bang, B. Kim, H. Doh, S. Cho, S. Hwang and S. Kim, Chem. Mater., 2016, 28, 5329–5335 CrossRef CAS.
- W. D. Kim, D. Kim, D. E. Yoon, H. Lee, J. Lim, W. K. Bae and D. C. Lee, Chem. Mater., 2019, 31, 3066–3082 CrossRef CAS.
- K. Deng, Z. Luo, L. Tan and Z. Quan, Chem. Soc. Rev., 2020, 49, 6002–6038 RSC.
- P. D. Cunningham, J. B. Souza, I. Fedin, C. She, B. Lee and D. V. Talapin, ACS Nano, 2016, 10, 5769–5781 CrossRef CAS PubMed.
- S. E. Seo, M. Girard, M. Olvera de la Cruz and C. A. Mirkin, Nat. Commun., 2018, 9, 4558 CrossRef PubMed.
- M. Rey, A. D. Law, D. M. A. Buzza and N. Vogel, J. Am. Chem. Soc., 2017, 139, 17464–17473 CrossRef CAS PubMed.
- P. Akcora, H. Liu, S. K. Kumar, J. Moll, Y. Li, B. C. Benicewicz, L. S. Schadler, D. Acehan, A. Z. Panagiotopoulos, V. Pryamitsyn, V. Ganesan, J. Ilavsky, P. Thiyagarajan, R. H. Colby and J. F. Douglas, Nat. Mater., 2009, 8, 354–359 CrossRef CAS PubMed.
- C. Giansante, Acc. Chem. Res., 2020, 53, 1458–1467 CrossRef CAS PubMed.
- D. Kim and D. C. Lee, J. Phys. Chem. Lett., 2020, 11, 2647–2657 CrossRef CAS PubMed.
- H. Lee, D. E. Yoon, S. Koh, M. S. Kang, J. Lim and D. C. Lee, Chem. Sci., 2020, 11, 2318–2329 RSC.
- S. W. Winslow, D. M. Smilgies, J. W. Swan and W. A. Tisdale, J. Phys. Chem. C, 2020, 124, 13456–13466 CrossRef CAS.
- D. Grzelak, S. Parzyszek, P. Moroz, P. Szustakiewicz, M. Zamkov and W. Lewandowski, Chem. Mater., 2019, 31, 7855–7863 CrossRef CAS.
- T. Du, J. Schneider, A. K. Srivastava, A. S. Susha, V. G. Chigrinov, H. S. Kwok and A. L. Rogach, ACS Nano, 2015, 9, 11049–11055 CrossRef CAS PubMed.
- P. Satapathy, P. K. Santra, A. Haque, C. V. Yelamaggad, S. Das and S. K. Prasad, Adv. Opt. Mater., 2019, 7, 1–9 Search PubMed.
- A. Bobrovsky, K. Mochalov, V. Oleinikov, A. Sukhanova, A. Prudnikau, M. Artemyev, V. Shibaev and I. Nabiev, Adv. Mater., 2012, 24, 6216–6222 CrossRef CAS PubMed.
- L.-J. Chen, J.-D. Lin, S.-Y. Huang, T.-S. Mo and C.-R. Lee, Adv. Opt. Mater., 2013, 1, 637–643 CrossRef.
- A. L. Rodarte, F. Cisneros, J. E. Hein, S. Ghosh and L. S. Hirst, Photonics, 2015, 2, 855–864 CrossRef CAS.
- R. Kumar and K. K. Raina, Liq. Cryst., 2016, 43, 994–1001 CrossRef CAS.
- T. Dudka, W. Zhang, J. Schneider, S. K. Gupta, M. F. Prodanov, V. V. Vashchenko, A. K. Srivastava and A. L. Rogach, Adv. Mater. Technol., 2019, 4, 1900695 CrossRef CAS.
- M. Bugakov, N. Boiko, P. Samokhvalov, X. Zhu, M. Möller and V. Shibaev, J. Mater. Chem. C, 2019, 7, 4326–4331 RSC.
- G. Kocakülah, G. Önsal, K. Goksen, İ. Ercan and O. Köysal, Phys. B, 2018, 550, 47–59 CrossRef.
- Y. Shen and I. Dierking, Appl. Sci., 2019, 9, 2512 CrossRef CAS.
- H. Yu, C. Welch, W. Qu, C. J. Schubert, F. Liu, G. Siligardi and G. H. Mehl, Mater. Horiz., 2020, 7, 3021–3027 RSC.
- M. F. Prodanov, N. V. Pogorelova, A. P. Kryshtal, A. S. Klymchenko, Y. Mely, V. P. Semynozhenko, A. I. Krivoshey, Y. A. Reznikov, S. N. Yarmolenko, J. W. Goodby and V. V. Vashchenko, Langmuir, 2013, 29, 9301–9309 CrossRef CAS PubMed.
- L. S. Hirst, S. T. Riahinasab and C. N. Melton, SPIE Newsroom, 2017 DOI:10.1117/2.1201702.006865.
- A. L. Rodarte, Z. S. Nuno, B. H. Cao, R. J. Pandolfi, M. T. Quint, S. Ghosh, J. E. Hein and L. S. Hirst, ChemPhysChem, 2014, 15, 1413–1421 CrossRef CAS PubMed.
- A. L. Rodarte, B. H. Cao, H. Panesar, R. J. Pandolfi, M. Quint, L. Edwards, S. Ghosh, J. E. Hein and L. S. Hirst, Soft Matter, 2015, 11, 1701–1707 RSC.
- M. Matsubara, W. Stevenson, J. Yabuki, X. Zeng, H. Dong, K. Kojima, S. F. Chichibu, K. Tamada, A. Muramatsu, G. Ungar and K. Kanie, Chem, 2017, 2, 860–876 CAS.
- C. C. Hewa-Rahinduwage, X. Geng, K. L. Silva, X. Niu, L. Zhang, S. L. Brock and L. Luo, J. Am. Chem. Soc., 2020, 142, 12207–12215 CrossRef CAS PubMed.
- W. Lewandowski, T. Łojewska, P. Szustakiewicz, J. Mieczkowski and D. Pociecha, Nanoscale, 2016, 8, 2656–2663 RSC.
- J. M. Wolska, D. Pociecha, J. Mieczkowski and E. Górecka, Chem. Commun., 2014, 50, 7975–7978 RSC.
- M. D. Tessier, D. Dupont, K. De Nolf, J. De Roo and Z. Hens, Chem. Mater., 2015, 27, 4893–4898 CrossRef CAS.
- C. Radhakrishnan, M. K. F. Lo, C. M. Knobler, M. A. Garcia-Garibay and H. G. Monbouquette, Langmuir, 2011, 27, 2099–2103 CrossRef CAS PubMed.
- W. Lewandowski, M. Wójcik and E. Górecka, ChemPhysChem, 2014, 15, 1283–1295 CrossRef CAS PubMed.
- M. Bagiński, E. Tomczyk, A. Vetter, R. N. S. Suryadharma, C. Rockstuhl and W. Lewandowski, Chem. Mater., 2018, 30, 8201–8210 CrossRef.
- H. K. Bisoyi and S. Kumar, Chem. Soc. Rev., 2011, 306–319 RSC.
- D. Rogez, H. Brandt, H. Finkelmann and P. Martinoty, Macromol. Chem. Phys., 2006, 207, 735–745 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic recipes, additional TEM, EDS, SAXRD and light emission data. See DOI: 10.1039/d1na00290b |
| This journal is © The Royal Society of Chemistry 2021 |