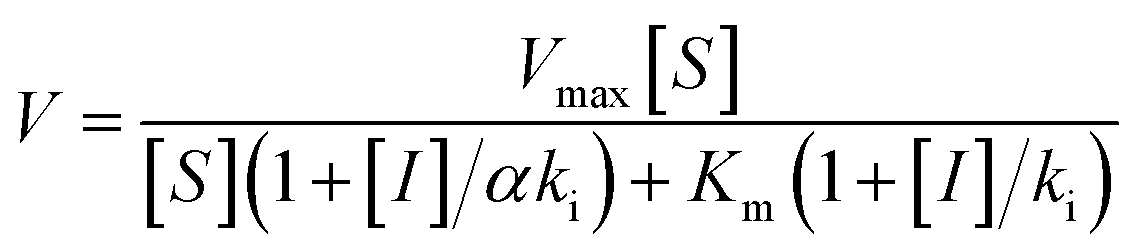DOI:
10.1039/D1NA00226K
(Paper)
Nanoscale Adv., 2021,
3, 5102-5110
One-pot bottom-up synthesis of a 2D graphene derivative: application in biomolecular recognition and nanozyme activity†
Received
25th March 2021
, Accepted 20th July 2021
First published on 21st July 2021
Abstract
The synthesis of two-dimensional (2D) nanosheets such as graphene and its derivatives through a bottom-up approach has many advantages such as growth control and functionalization, but it is always challenging to get the desired material. Herein, we have reported the synthesis of water soluble 2D-nanosheets through a bottom-up approach from 2,4,6-tribromo-3-hydroxybenzoic acid via a self-coupling pathway and characterized them using several techniques. AFM and TEM analyses reveal that the synthesized material has a layered structure with a thickness of ∼1.2 nm. Also, the prepared nanosheets are amorphous in nature with high negative charge (−38 ± 2.5 mV). The flexible nature of 2D-nanosheets and their functionality can be used in many related applications. Therefore, we have utilized the synthesized 2D-nanosheets in biomolecular recognition studies. It was found that the enzymatic activity of α-chymotrypsin can be controlled reversibly in the presence of the synthesized 2D-nanosheets. The kinetic study revealed that the nanosheet surface selectively binds to the active sites of the enzyme through a competitive pathway. Furthermore, we explored the nanozyme activity of the material in a peroxidase-like activity assay of two bio-active molecules: Nicotinamide Adenine Dinucleotide Phosphate (NADH) and dopamine. The results suggest that the prepared material efficiently catalyzed the oxidation of NADH to biological cofactor NAD+ and dopamine to aminochrome in the presence of H2O2. These synthesized graphene-like 2D-nanosheets with functional groups can be further tuned with other functionalities, which can open a new window for other related applications.
Introduction
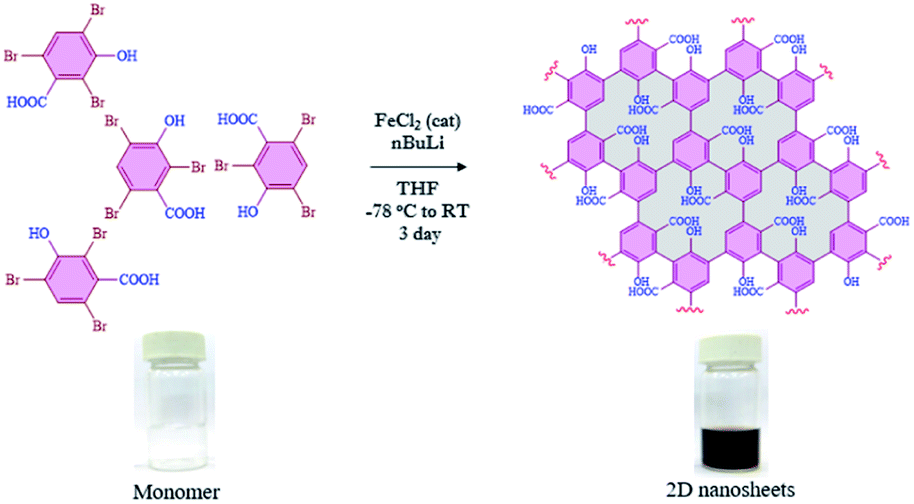
Since the discovery of graphene in 2004,1,2 other two-dimensional (2D) nanomaterials such as g-C3N4,3,4 porous carbon,5,6 transition-metal dichalcogenides7,8 and black phosphorus9,10 have attracted a lot of attention in a variety of applications such as catalytic activity,11–13 energy storage,14,15 adsorption16–18 and molecular recognition.19–21 Furthermore, those materials are also considered in several biological applications including bio-imaging,22 drug delivery,23 sensing,24,25 nanozyme activity,26,27 transduction,28 DNA detection,29 immobilization of enzymes,30etc. The evolution of 2D-nanosheets from molecules to materials (bottom-up approach) has also been employed in broad applications, due to their controlled functionalities and tunable physical and chemical properties.31,32 Moreover, we have also found that the functionalized 2D-nanosheets exhibit many advantages compared to unfunctionalized 2D-nanosheets such as stability, and tunable physical and optical properties.33–35 Graphene or related 2D nanomaterials have been implemented in various optoelectronic applications such as broadband optical response, strong and tunable light–matter interactions, device fabrication, etc.36,37 The synthesis of 2D nanosheets such as graphene nanoribbons,38–40 metal organic frameworks (MOFs),41–43 covalent organic frameworks (COFs),44–46 self-assembled 2D-nanosheets,47,48 dendrimers49,50 and squarely designed small organic molecular51 structures via a bottom-up approach was reported earlier using various methodologies. In all those reports, the main focus was related to their structure, morphology and functional groups.52–55 In all of those reported studies either multistep synthesis or multi component reactions were involved which limits the scalability and leads to the presence of possible impurities. In most of the reports the synthesized graphene-like derivatives are nano-ribbon-like structures due to 1D growth of monomer building blocks such as diphenyl-10,10′-dibromo-9,9′-bianthracene (DP-DBBA),38 2,7,11,16-tetrabromotetrabenzo phenazine (TBTBP),56 and poly(para-dibenzo-coronenylene) (PPDBC)57 which formed porous hetero atom doped ribbon like structures. However, the preparation of extended 2D nanosheets using a bottom-up approach has rarely been reported.58 In this regard we have chosen 2,4,6-tribromo-3-hydroxybenzoic acid (TBHBA) as the novel building block and iron-catalyzed homocoupling of aryl bromides as a mild reaction condition59 for the preparation of 2D graphene-like nanosheets. To the best of our knowledge, the synthesis of 2D-nanosheets from a single molecule through self-coupling by treating with mild reagents in one-pot has never been reported. In this regard, we report the one pot synthesis of water soluble 2D nanosheets through a bottom-up approach from TBHBA in the presence of n-butyl lithium (n-BuLi), and FeCl2 as a catalyst at −78 °C. The synthesized material was characterized by several techniques and we observed the formation of single layered 2D-nanosheets having ∼1.2 nm thickness. This synthesized material was found to have a flexible nature, large specific surface area with a controlled functionality and hydrophobic/hydrophilic ratio (Fig. 1).
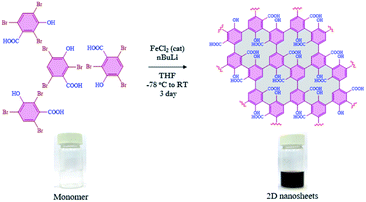 |
| | Fig. 1 Reaction scheme for the bottom-up synthesis of 2D nanosheets from TBHBA via a covalent coupling reaction in the presence of n-BuLi and FeCl2 as a catalyst from −78 °C to room temperature. The change in color of the solution indicates the formation of nanomaterials with extended conjugation. | |
To investigate the applicability of the synthesized 2D-nanosheets, we have performed a biomolecular recognition study. Herein, we have considered a model proteolytic enzyme, α-chymotrypsin (α-ChT), which has a well-defined structure with hydrophobic patches and positively charged residues around the active site.60 It has been reported that the development of an artificial inhibitor of α-ChT is highly relevant to many therapeutic applications, as the deficiency of proteolytic inhibitors causes neurodegenerative diseases including Alzheimer's,61 thromboembolism,62 emphysema,63etc. As α-ChT has positively charged residues with hydrophobic pockets at the active site, negatively charged materials with hydrophobic residues can bind to the active site of α-ChT and can modulate the enzymatic activity. It has been reported that α-ChT can be specifically recognized by negatively charged nanomaterials with hydrophobic residues.64,65 As our synthesized 2D-nanosheets have both of those properties (anionic functionality and hydrophobic backbone), we expect the possible inhibition of α-ChT in the presence of 2D-nanosheets. Hence, we have measured the enzymatic activity of α-ChT in the presence of 2D-nanosheets and we observed highly efficient enzymatic inhibition. The inhibition efficiency is comparable to that of the best artificial inhibitor reported so far. We have also observed that the inhibition is reversible in nature without any effect on the protein's secondary structure. This implies the suitability of this system as an artificial protein receptor. Based on our structural analysis, the synthesized nanosheets contain several oxygenated functionalities which may effectively take part in various redox reactions. Hence we have explored the peroxidase-like activity of the as-synthesized nanosheets in the presence of H2O2 as in the case of graphene oxide.66 Peroxidase enzymes such as glucose oxidase (GOx) and horseradish peroxidase (HRP) are very critical enzymes used for the oxidation of glucose67 and degradation of H2O2 (ref. 68) respectively. Gox and HRP are responsible for diverse biological processes such as cell wall synthesis and degradation, signalling oxidative stress, removal of xenobiotics, etc. Apart from that, peroxidase activity has a wide range of practical applications ranging from detection of blood glucose to waste water treatment. In this regard, nanomaterial-based enzymes (nanozymes) played a crucial role due to the low cost of production, stability and a versatile range of working conditions.69–71 The nanozyme activity always attracts additional advantages in many biological and catalytic applications. To determine the efficacy of this material as a nanozyme we have considered two bio-active molecules: NADH and dopamine. Redox reactions involved with NADH and dopamine are highly relevant in various biological processes. The results suggest that the prepared 2D-material efficiently catalyzed the oxidation of NADH to biological cofactor NAD+ and dopamine to aminochrome in the presence of H2O2 as reported for similar materials.72,73 Those two applications support the formation of a graphene oxide-like 2D nanomaterial.
Results and discussion
Synthesis and characterization
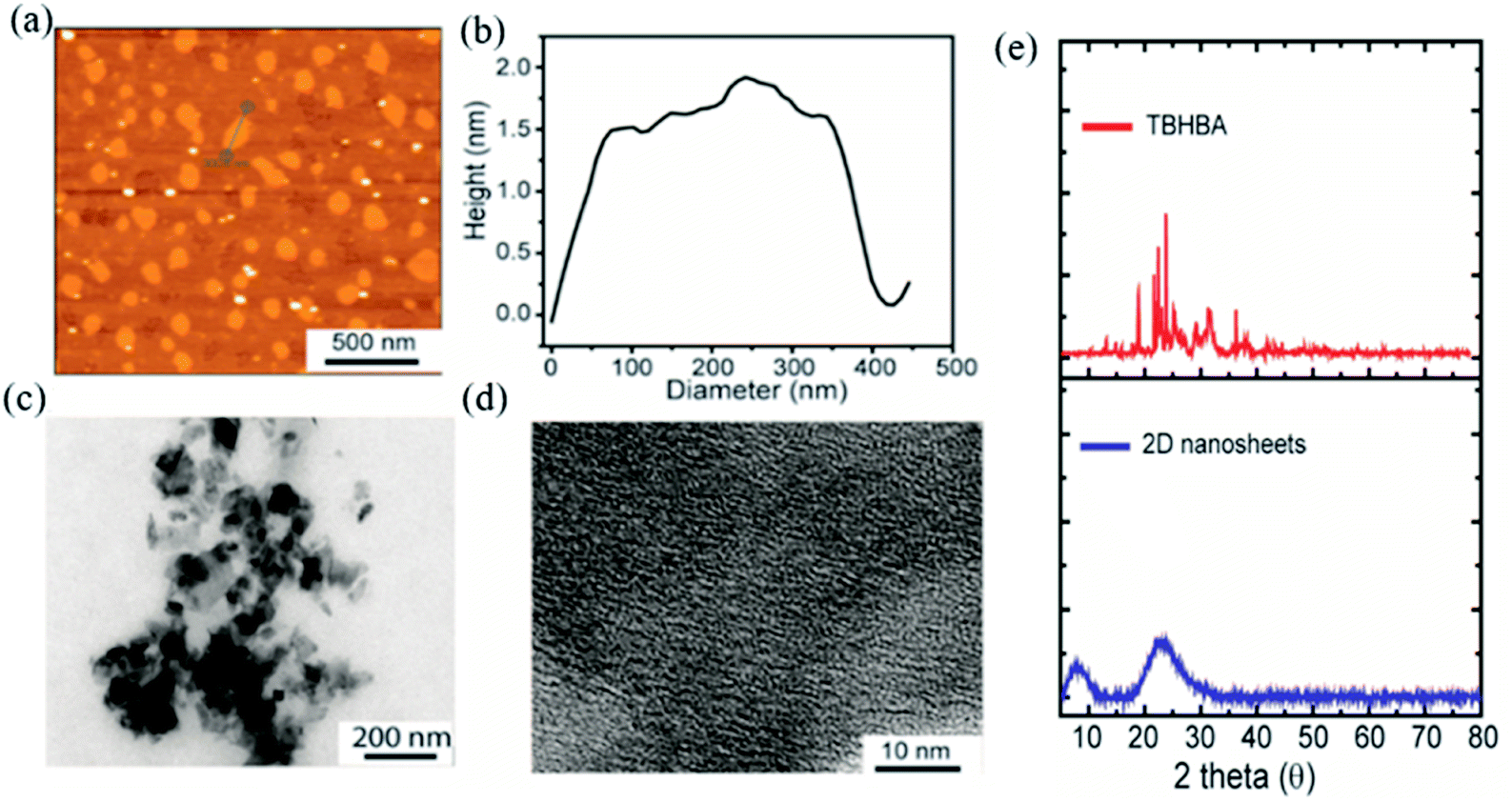
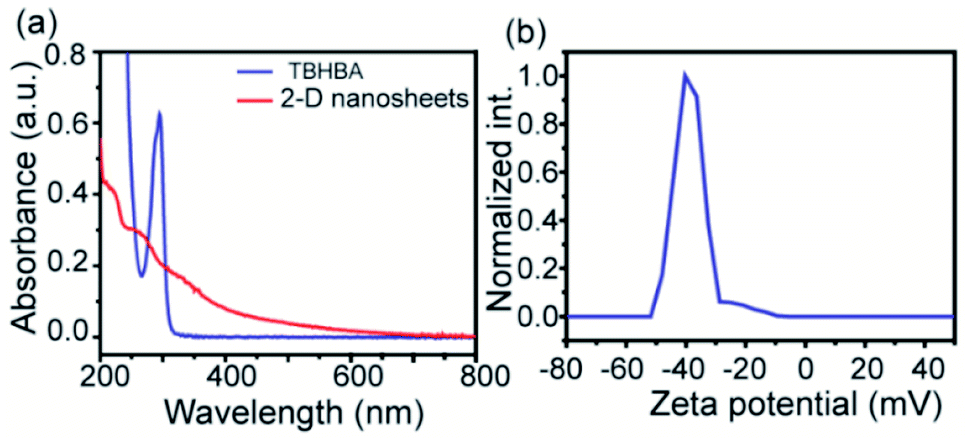
The evolution of functionalized carbon-based nanomaterials from small molecules has attracted great interest in various applications due to control over size, functionality and properties. Here we have synthesized 2D carbon based nanosheets using TBHBA as the starting source in the presence of n-butyl lithium (n-BuLi) and ferrous chloride as a catalyst along with triethylamine as an additive in dry tetrahydrofuran (THF) solvent. By a self-coupling reaction, water soluble negatively charged (carboxylate and hydroxyl groups) 2D nanosheets have been prepared through a bottom-up approach. We had observed that during the progress of the reaction, the color of the reaction mixture changed from pale yellow to dark brown. This suggests that self-polymerization has taken place. The reaction mixture was quenched by a few drops of 1 N HCl solution and then organic solvent was evaporated. The obtained black solid was redispersed in distilled water and centrifuged at 4000 rpm to remove the insoluble residues. Finally, the prepared material was purified through dialysis in 5 mM sodium phosphate buffer. After the purification, the synthesized material was characterized by various microscopic and spectroscopic techniques. The atomic force microscopy (AFM) study revealed that the morphology of the material is layered in structure with ∼1.2 nm thickness (Fig. 2a and b). The average lateral diameter of this layered material is 200–400 nm. The synthesized material suggests the formation of a single layered material similar to a well-known 2D material: graphene oxide (GO). After plotting the size as well as thickness distribution of the synthesized 2D material we found that the lateral diameter varied from 50 to 350 nm but the thickness was very consistent ∼1.5 nm which indicates the formation of a 2D nanomaterial without any aggregation (Fig. S1†). Similarly, the morphology has also been confirmed by the Transmission Electron Microscopy (TEM) technique (Fig. 2c). Like AFM analysis, it also suggests a layered structure and the HRTEM image indicate its amorphous nature like GO (Fig. 2d). The synthesized 2D-nanosheets have also been characterized through the powder X-ray diffraction technique and compared with starting materials (Fig. 2e). We observed a broad hump at 8° and 23.6° which supports the amorphous nature of the synthesized material compared to the starting material as we observed in TEM analysis. Among the spectral analyses, the UV-Vis Spectrum reveals that the synthesized 2D nanosheets have three significant absorption peaks at 218 nm, 270 nm and 350 nm originating from hydroxyl (–OH), carboxyl (–COOH) and the conjugated aromatic moiety respectively. On the other hand, the starting source TBHBA shows one sharp peak at 295 nm due to the n to π* electronic transition (Fig. 3a). This study also suggests that the self-coupling reaction has taken place and formed 2D-nanosheets with extended conjugation and functionality. To examine the charge of the 2D-nanosheets we measured the zeta potential (z) of the solution, and we found that the charge of the solution is −35 ± 1.8 mV (Fig. 3b). This strongly suggests the formation of nanosheets containing free carboxylic acid and hydroxyl groups present in the material as shown in Fig. 1.
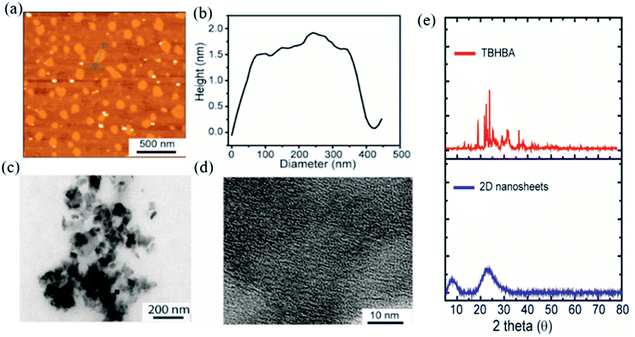 |
| | Fig. 2 Characterization of the synthesized 2D-nanosheets. (a) AFM image indicates the formation of 2D-nanosheets without any visible aggregation. (b) Height profile diagram indicates the formation of a single layered material. (c and d) TEM and HRTEM analyses indicate the formation of a 2D-material with amorphous nature. (e) Powder X-ray diffraction (p-XRD) pattern of TBHBA (red line) and synthesized 2D nanosheets (blue line). | |
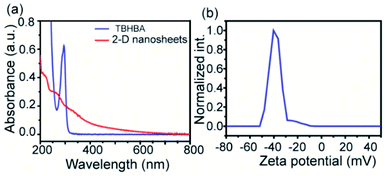 |
| | Fig. 3 UV-vis spectra of TBHBA and synthesized 2D-nanosheets (a) and zeta potential plot of the synthesized 2D-nanosheets (b). | |
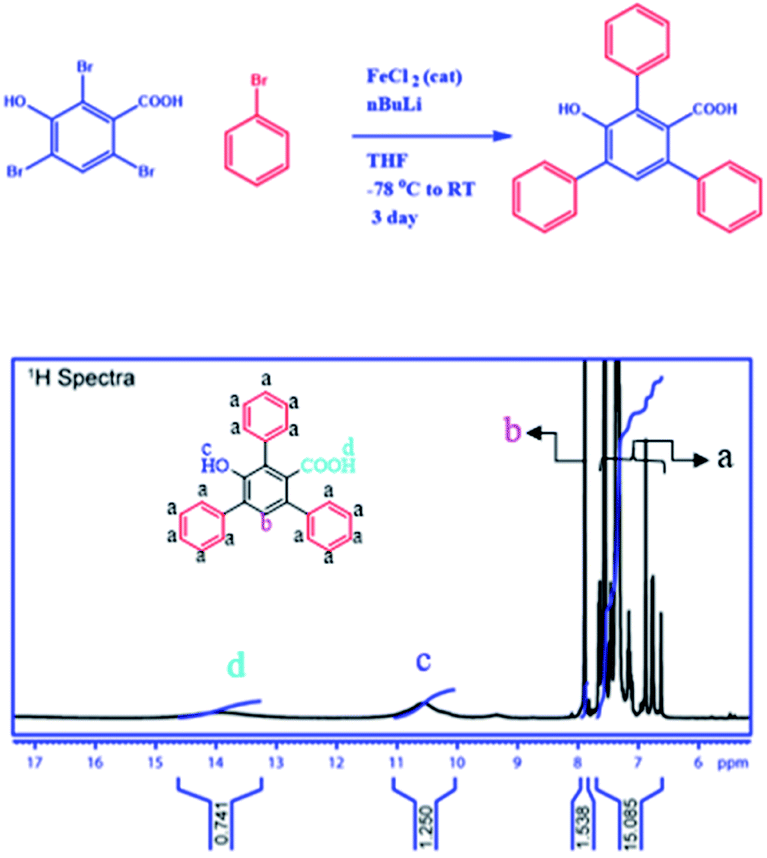
To establish the backbone structure of the synthesized 2D-nanosheets we have carried out the NMR and IR analyses. To determine the structure, we have synthesized monomers under the same conditions and analyzed them through 1H and 13C NMR (Fig. 4 and S2–3†). According to the NMR analysis the monomer contains both the hydroxyl and carboxyl functionalities with conjugated benzene rings. This is the building block for the 2D-nanosheet.
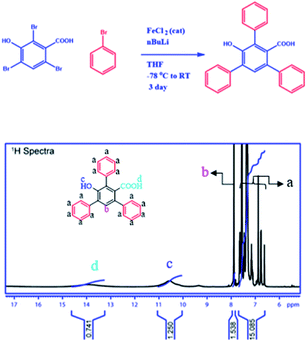 |
| | Fig. 4 Reaction scheme for the monomer from TBHBA via the covalent coupling reaction in the presence of n-BuLi and FeCl2 as a catalyst from −78 °C to room temperature. 1H NMR spectra of the monomer in deuterated DMSO-D6 solvent. | |
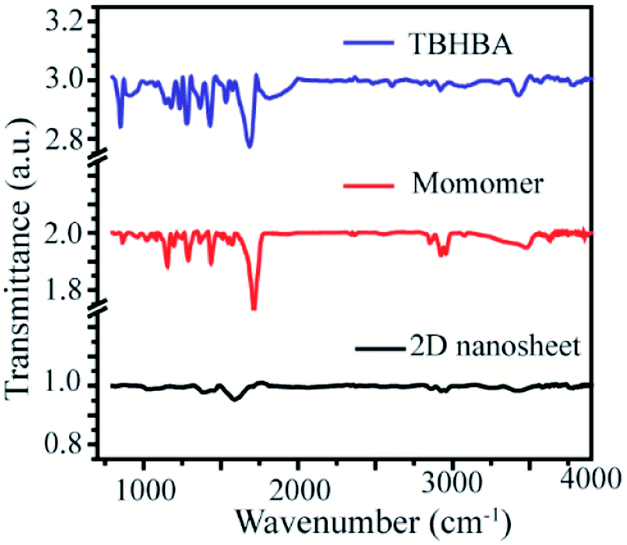
Moreover, to confirm this, we have also performed Fourier Transformed Infrared Spectroscopy (FT-IR) which revealed that the significant stretching frequency for hydroxyl and carbonyl groups was present at 3418 cm−1 and 1595 cm−1 respectively, whereas the hydroxy and carbonyl stretching frequencies of the starting material appeared red shifted at 3428 cm−1 and 1684 cm−1 respectively. We also found that the significant intensity of C–H stretching and wagging frequency appears at 2920–2965 cm−1 and 1284 cm−1 respectively, as well as the disappearance of the C–Br bond from the monomer spectra was observed. We have also observed the presence of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching frequency at 1400 cm−1 in all the spectra (Fig. 5). This result suggests that the synthesized material has an extended conjugated 2D structure.
C stretching frequency at 1400 cm−1 in all the spectra (Fig. 5). This result suggests that the synthesized material has an extended conjugated 2D structure.
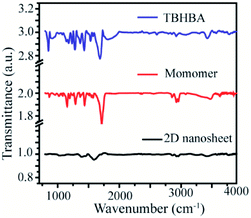 |
| | Fig. 5 Fourier transform Infrared (FT-IR) spectra of TBHBA (top), the monomer (middle) and synthesized 2D nanosheets (bottom). | |
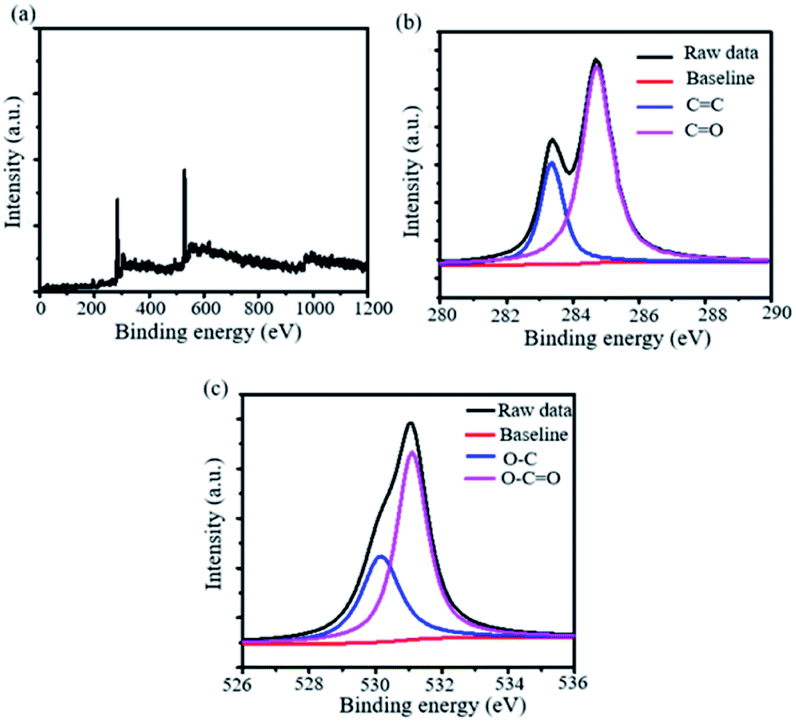
To investigate the elemental composition of the synthesized 2D-nanosheets, we have utilized X-ray photoelectron spectroscopy (XPS). The XPS analysis revealed that two sharp distinguished peaks were appearing in the spectrum. This suggests that these peaks are from the 1s spectra of carbon and oxygen at 284.4 and 532.6 eV respectively (Fig. 6a). Also, we have deconvoluted the 1s spectra of both the elements and it suggests the presence of sp2 C associated with the benzene ring (CC) and carboxylic acid (CO) (Fig. 6b). Similarly, we observed the presence of C–O and O–CO bonds in the case of 1s spectra of oxygen (Fig. 6c). We have also observed that the C–Br bond disappeared from the wide XPS spectra. This suggests that the C–C bond has formed which causes extended conjugation.
 |
| | Fig. 6 X-ray photoelectron spectra (XPS) of the synthesized 2D nanomaterial. (a) Wide, (b) C 1s and (b) O 1s XPS spectra of the synthesized material. | |
Enzymatic inhibition: the synthesized 2D nanosheet has been characterized and it was found to be negatively charged and flexible in nature. This synthesized material can be used for the protein surface recognition study, specifically for positively charged proteins with aromatic/hydrophobic residues. In this regard we have considered a positively charged protein α-chymotrypsin (α-ChT) as mentioned in the introduction section.
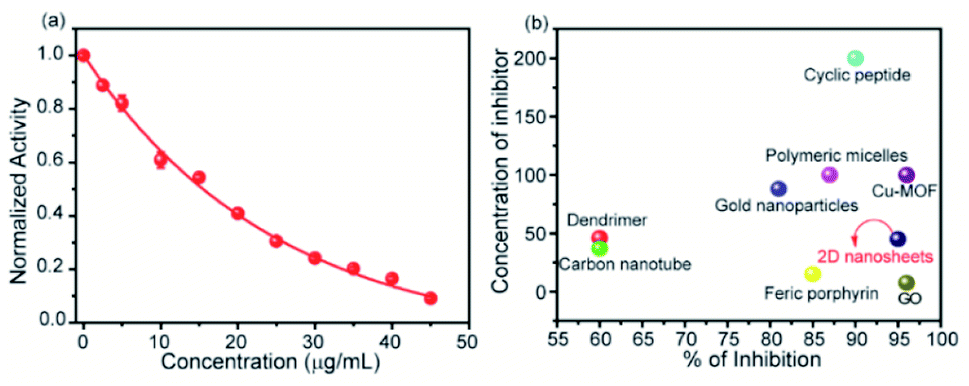
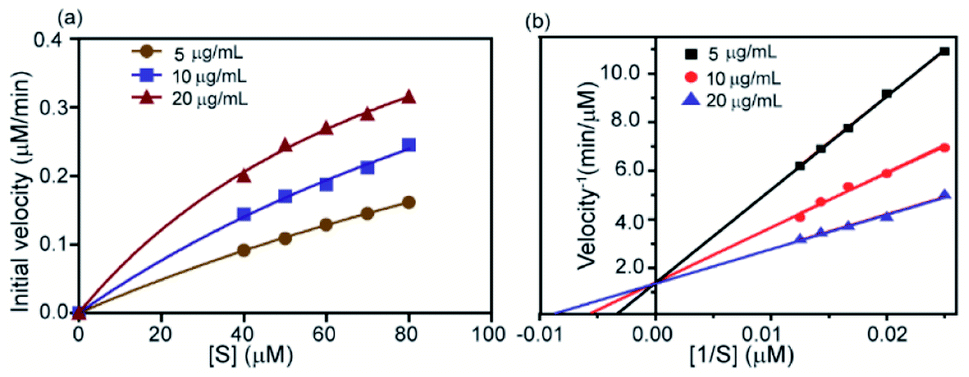
To measure the degree of inhibition efficiency of negatively charged 2D nanosheets the enzymatic activity was measured in the presence of chromogenic substrate N-succinyl L-phenylalanine p-nitroanilide (SPNA) (Fig. 7, S4†). The enzymatic activity study was carried out by preincubating α-ChT (3.2 μM) in the presence of synthesized 2D nanosheets at various concentrations ranging from 0 to 45 μg mL−1 (whereas the stock solution was 100 μg mL−1). The activity of α-ChT reduces to 94% at 40 μg mL−1. As reported earlier, not only the presence of negative charge but also π-conjugated hydrophobic pockets (aromatic moiety) played a crucial role in binding with the positively charged hydrophobic patches of α-ChT around its active sites.60 We have performed an enzymatic study at different pHs ranging from 4 to 10 and we found that at extremely low and high pH the enzymatic activity has been reduced. This indicates, either under those conditions effective interaction does not happen, or protein gets denatured. Hence, we have studied the effect of pH on the secondary structure of α-ChT by a circular dichroism study. As expected, we have observed denaturation of protein at extremely high and low pH. So the optimized pH conditions for the enzymatic study is 7.4, physiological condition (Fig. S5†). Moreover we have also tried to carry out this interaction in 100% serum but due to the opaqueness we were not able to monitor the enzymatic activity. So we have tried some diluted buffer conditions and we found that up to 8% serum solution the efficiency of enzymatic inhibition was not affected. This indicates that the presence of serum proteins will not affect the 2D nanosheet and α-ChT interaction. At higher concentrations (16%) we still see equally effective inhibition but with some inconsistency (Fig. S6†). This difficulty could be overcome by using other substrates such as NIR or fluorescence based substrates. Further we have also performed the activity study of α-ChT with time and it showed that the complete complexation is not instant, rather takes time up to 165 min for complete complexation. (Fig. S7†). Compared to other reported artificial inhibitors of α-ChT under a similar category, such as carbon nanotubes, gold nanoparticles, micelles, etc., our synthesized 2D nanosheets exhibit a much better inhibition effect (Table S1†). The inhibition efficacy is only comparable with that of graphene oxide (GO) which is known as the best-known artificial inhibitor for α-ChT60 (Fig. 7b). This result also suggests the structural similarity of our synthesized material with GO. To establish the mode of inhibition, rate constant (Ki), velocity (Vmax) and Michaelis–Menten constant (Km), a kinetic study was carried out. In this study, at first, we fixed the concentration of α-ChT and the synthesized 2D nanosheets and varied the concentration of the substrate (SPNA) ranging from 0 to 80 μM. The data were fitted by nonlinear regression using GraphPad software based on the general equation for the velocity (V) of an enzyme reaction in the presence of an inhibitor (Ii).
 |
| | Fig. 7 Enzymatic activity of α-ChT as a function of the concentration of 2D nanosheets in 5 mM sodium phosphate buffer at pH 7.4 (a), and inhibition efficiency of the synthesized material compared to other materials (b). | |
This is a well-known equation in the literature which can be split into three specific forms describing competitive, non-competitive and uncompetitive inhibition mechanisms depending on the slope value (α). Specifically, α = 1 implies that the inhibitor does not affect the binding of the substrate to the enzyme, which is non-competitive inhibition, when α ≫ 1 binding of the inhibitor prevents the interaction of the substrate which is competitive inhibition and when α ≪ 1 inhibitor binding increases enzymatic substrate binding, and the equation describes mostly uncompetitive inhibition. For fitting the data (Fig. 8a) we have considered synthesized 2D nanosheet [I] concentrations as a static value and obtained the values of Vmax and Km as the best-fit values for the 7.01 × 10−4 and 39.81 ± 7.1 respectively. Here we obtained α = 1.4, indicating that the synthesized 2D nanosheets inhibit α-ChT through a competitive pathway. The mode of inhibition can also be illustrated using a Lineweaver–Burk plot (Fig. 8b). Here we have observed that different concentrations of synthesized 2D nanosheets (inhibitor) produce lines with the same y-axis intercept, indicating competitive inhibition. From this data set a Ki of 2.41 mg mL−1 was observed.
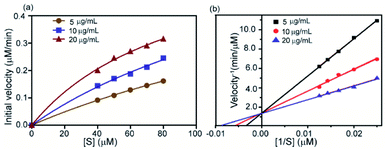 |
| | Fig. 8 Kinetics study of enzyme inhibition by the complex formed between α-ChT and synthesized 2D nanosheets. The enzyme velocity as a function of substrate concentration (SPNA) with the inhibitor (a) and Lineweaver–Burk plot (b) in sodium phosphate buffer 5 mM pH 7.4. | |
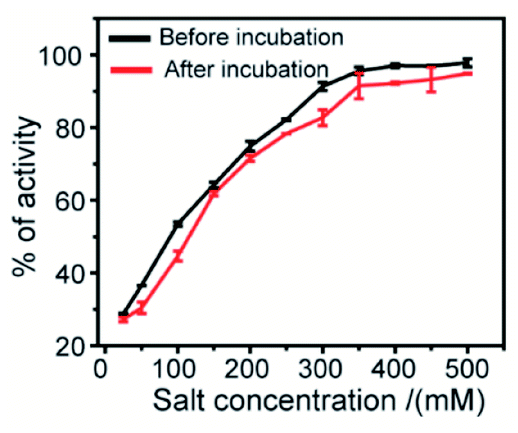
The effect of ionic strength can be determined by measuring the enzymatic activity in the presence of various salt concentrations. This study describes the nature of interaction between the synthesized 2D nanosheets (inhibitor) and guest molecules (enzyme). To estimate the degree of reversibility, we have performed two parallel experiments. In the first experiment, 3.2 μM α-ChT was preincubated with 50% inhibitory concentration of receptor (synthesized 2D nanosheets, 20 μg mL−1) for 30 min and then various concentrations of salt solutions (NaCl) were added to reach the final concentration from 0 to 500 mM. In the second experiment, we incubated the 3.2 μM α-ChT and mixed it with the synthesized 2D nanosheets in the presence of salt solution at the same concentrations as before without the 30 min preincubation. The activity of α-ChT could be restored while increasing salt concentrations. It suggests that the extent of binding can be monitored by the hydrolysis of the SPNA substrate. It was observed that in both cases the α-ChT between synthesized 2D nanosheets and α-ChT is electrostatic in nature and can be reversed (Fig. 9). Also, control experiments were carried out under identical conditions without the addition of α-ChT and the synthesized 2D nanosheets. It was reported in earlier studies that the interaction of the inhibitor with the biomolecules can be reversed by increasing the ionic strength of the medium, since the electrostatic forces can be attenuated by the presence of competitive ions.74,75
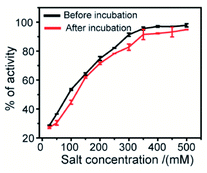 |
| | Fig. 9 Salt study assay of the complex between enzyme α-ChT and synthesized 2D nanosheets in 5 mM sodium phosphate buffer at pH 7.4. | |
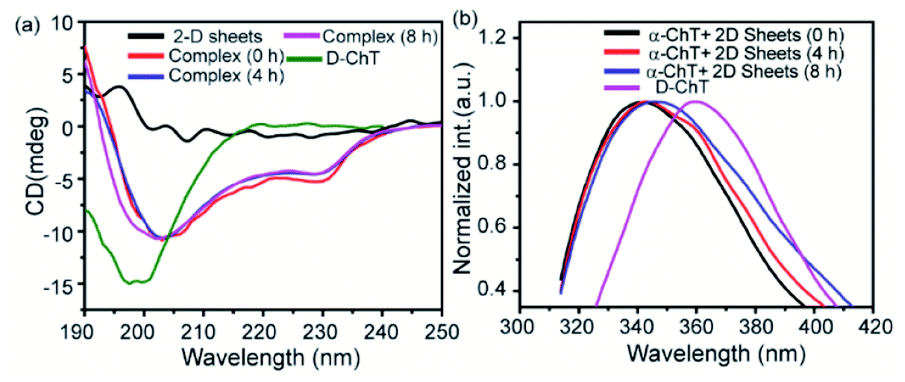
To compare the secondary structure of native α-ChT and the complex formed (i.e., between α-ChT and synthesized 2D nanosheets), The Circular Dichroism (CD) and Fluorescence Spectroscopy techniques were utilized. Firstly, we have performed the CD experiment which provides detailed information related to the secondary structure of protein (i.e., combination of helix, sheet and random coil). Here we found that the enzyme α-ChT shows two characteristic minima at 232 nm and 202 nm for the alpha helix and random coil respectively and was compared with the complex form. The result shows that the degree of denaturation of α-ChT upon binding with synthesized 2D nanosheets was insignificant (Fig. 10a). This suggests that the synthesized 2D nanosheet does not affect the secondary structure of α-ChT. On the other hand, we have also investigated the secondary structure of the enzyme through the fluorescence spectroscopy technique. This study revealed that the native α-ChT characteristic emission peak is at 340 nm and after structural denaturation the peak was red shifted to 364 nm because of the exposure of the tryptophan (Trp) residues to the aqueous environment. Our study suggests that upon the addition of the synthesized 2D nanosheet to the α-ChT solution (3.2 μg mL−1) the secondary structure has not changed (Fig. 10b Table S2†). Herein we have also observed that the characteristic emission peak shows a minimum red shift which indicates that the degree of denaturation of protein is very low (12%) and is comparable to that of native α-ChT over time. In previous reports it was suggested that α-ChT denaturation could be caused by a more hydrophobic environment or strong electrostatic interactions and this denaturation process can be prevented at a suitable ratio of hydrophobic and hydrophilic moieties.
 |
| | Fig. 10 Secondary structure of the enzyme in the presence of synthesized 2D nanosheets at different time intervals: circular dichroism (a) and fluorescence spectroscopy (b). | |
Nanozyme activity.
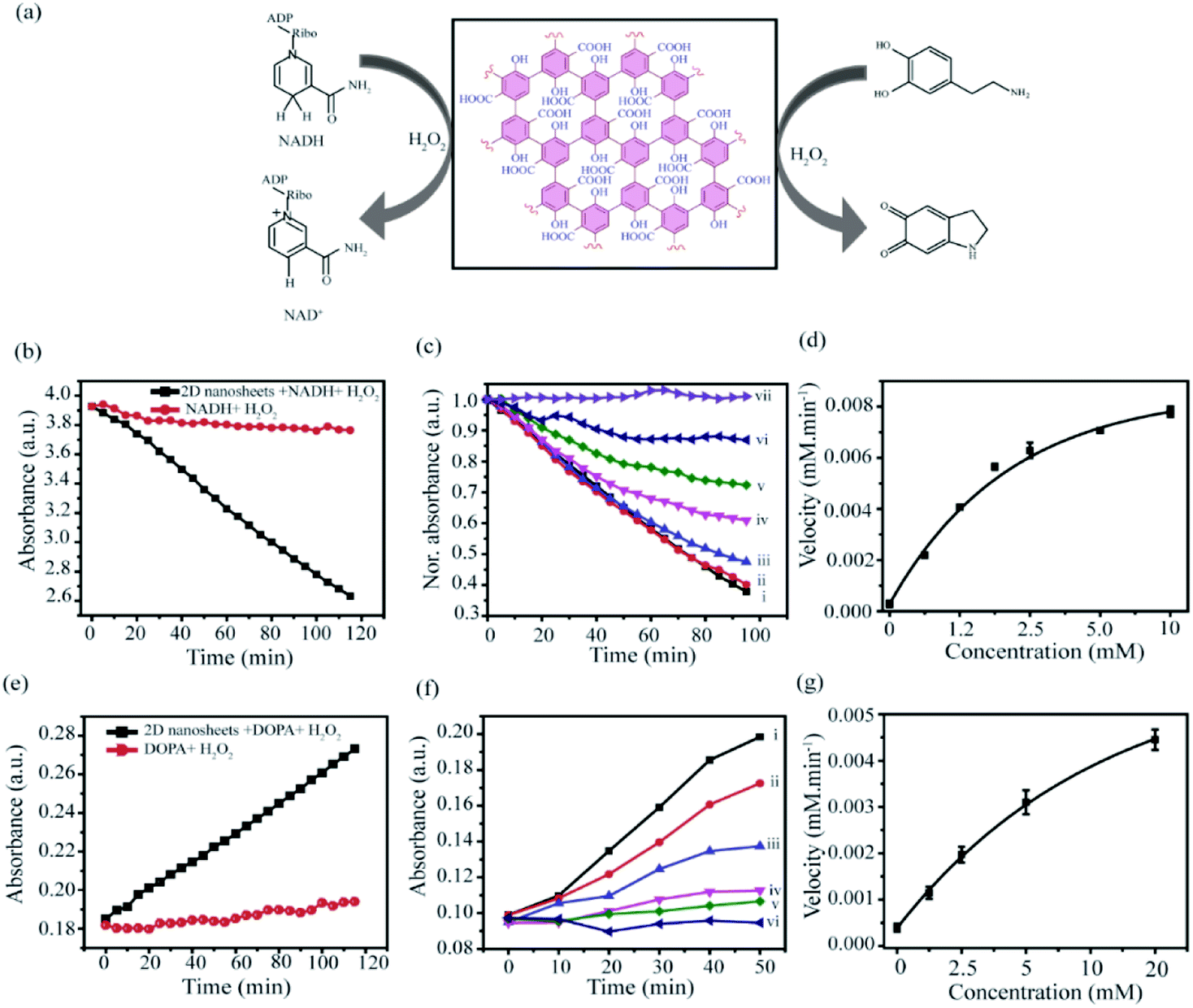
Derivatives of graphene and graphene-like materials are well known in nanozyme applications. Various oxygenated functionalities such as hydroxyl, epoxide, carboxylate, etc. play a crucial role in those phenomena.76 As we have established the presence of those functionalities in our synthesized nanosheets, so we have explored the potential of this material as a nanozyme. So far only graphene derivatives from bulk materials (top-down method) have been used for nanozyme applications. Qu et al. first observed the peroxidase-like activity of graphene oxide and applied in glucose detection.66 After that, several modified graphene-based systems were applied as peroxidase nanozymes in various applications such as Willner et al. reported Cu2+ mediated nanographene in enzyme mimetic activity.66,77,78 However, graphene derivatives prepared by the bottom-up approach have not been explored in peroxidase-like activity so far. Therefore, we have investigated whether the prepared materials can exhibit enzyme-like properties. In this regard we have considered two well-known assays: (1) NADH and (2) dopamine to evaluate the nanozyme activity of 2D-nanosheet (Fig. 11a). The NADH peroxidase-like activity measured in the presence of H2O2 which has specific absorbance at 340 nm. In the presence of the nanozyme, NADH is oxidized to form biological cofactor NAD+, and the absorbance intensity at 340 nm decreases gradually over the period of time (Fig. 11b). The activity assay suggests that these 2D nanosheets can efficiently mimic the NADH peroxidase-like activity when tested at different concentrations of 2D-nanosheets ranging from 0 to 80 μg mL−1 and it was observed that with increasing concentrations the rate of oxidation of NADH increases (Fig. S8a, c†) linearly. This indicates that the catalytic activity can efficiently mimic the NADH peroxidase-like activity in the presence of H2O2. Similarly, under the same conditions the control experiment was carried out where very low oxidation of NADH was observed in the presence of only H2O2 (Fig. 11b). The kinetic study reveals that the rate of oxidation of NADH to NAD+ increases with increasing concentration of NADH (0–10 mM) (insight) and the rate is 1.546 × 10−4 mM min−1 (Fig. 11c and d). Moreover, we have also examined the catalytic oxidation of dopamine using H2O2 in the presence of 2D nanosheets. The absorbance of the oxidized product, aminochrome, was measured at 480 nm (Fig. 11d). We observed that the material catalyzed the oxidation of dopamine to aminochrome in the presence of H2O2. Similarly, we have also performed a control study and it clearly suggested that in the absence of 2D-nanosheets the reaction does not happen significantly (Fig. 11d). We have also regulated time-dependent absorbance of the oxidized product aminochrome with different concentrations of synthesized materials. Similar to NADH we have also observed a rate enhancement with increasing concentrations of 2D-nanosheets (Fig. S8b, d†). Moreover, kinetic studies suggest that with increasing the concentration of dopamine from 0 to 20 mM the absorbance of oxidized product aminochrome increases. From this study we have calculated the rate of oxidation, which is 5.716 × 10−5 mM min−1 (insight) (Fig. 11e). Based on the above two experiments, we can assume that our synthesized 2D-nanosheets behave like natural enzymes and are comparable with many other reported oxidases like nanozymes.
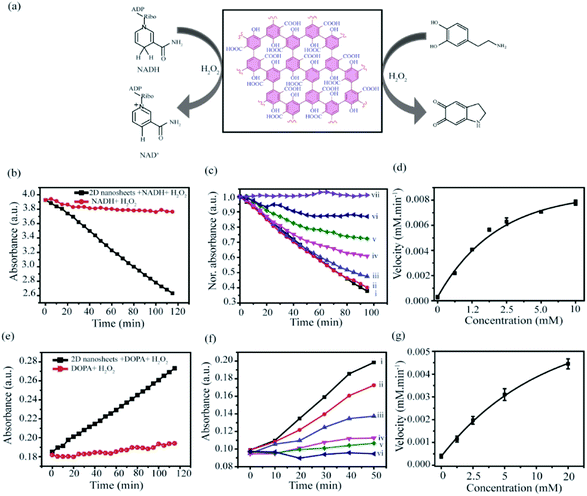 |
| | Fig. 11 (a) Scheme of the nanozyme activity of NADH and dopamine in the presence of 2D nanosheets. (b–d) NADH activity assay in the presence of 5 mM H2O2 (b) and the kinetic studies of NADH, measured at 340 nm at different concentrations of NADH (i) 10, (ii) 5, (iii) 2.5, (iv) 1.25, (v) 0.625, (vi) 0.312 to (vii) 0 mM (c and d). (e–g) Dopamine activity assay in the presence of H2O2 (e) and the kinetic assay mesured at various concentration of dopamine (f and g). The concentration of dopamine are, (i) 20, (ii) 10, (iii) 5, (iv) 2.5, (v) 1.25, to (vi) 0 mM. All studies have been done in 5 mM sodium phosphate buffer solution, pH 7.4. | |
Conclusion
In summary, we have reported a new bottom-up approach for the synthesis of a graphene-like 2D material with controlled functionalities. According to this approach, 2,4,6-tribromo-3-hydroxybenzoic acid was self-coupled in the presence of nBuLi and FeCl2 under mild conditions. The synthesized material has been characterized by several techniques and we have established that the synthesized material is a single layered 2D nanosheet having certain functional groups (carboxylic and hydroxy) which makes it water soluble and negatively charged. Owing to these properties, we have applied the synthesized 2D nanosheets in molecular recognition studies. We have found that the synthesized 2D nanosheet efficiently binds to the enzyme surface and inhibits the enzymatic activity without any effect on its secondary structure. The interaction is mainly electrostatic in nature and can be reversed in the presence of competitive ions. Furthermore, we have also examined the nanozyme activity of the synthesized 2D-nanosheets. They exhibit excellent peroxidase-like activity from NADH to biological cofactor NAD+ and dopamine to aminochrome in the presence of H2O2. Even though here we have demonstrated biological applications, but similar to previous reports a 2D material based layered film can be prepared and various electronic properties can be measured. This reported method can provide an easy access of preparation of 2D-nanosheets with a controlled ratio of functionalities which can be further tuned by post modification. This may open a new window for other related applications of graphene derivatives in biology, sensing, electronic field, etc.
Conflicts of interest
The author declares no competing financial interest.
Acknowledgements
The authors would like to thank DST-SERB (CVD/2020/000855) for financial support. SP thanks IISc for a doctoral fellowship.
References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS.
- X. D. Zhang, X. Xie, H. Wang, J. J. Zhang, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS.
- W. Xing, W. Tu, Z. Han, Y. Hu, Q. Meng and G. Chen, ACS Energy Lett., 2018, 3, 514–519 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- A. Stein, Z. Y. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- K. F. Mak and J. Shan, Nat. Photonics, 2016, 10, 216–226 CrossRef CAS.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- F. Xia, H. Wang and Y. Jia, Nat. Commun., 2014, 5, 4458 CrossRef CAS PubMed.
- F. R. Fortea-Perez, M. Mon, J. Ferrando-Soria, M. Boronat, A. Leyva-Perez, A. Corma, J. M. Herrera, D. Osadchii, J. Gascon, D. Armentano and E. Pardo, Nat. Mater., 2017, 16, 760–766 CrossRef CAS PubMed.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- Y. R. Girish, S. Pandit, S. Pandit and M. De, Chem.–Asian J., 2017, 12, 2393–2398 CrossRef CAS.
- J. Q. Lv, Y. X. Tan, J. F. Xie, R. Yang, M. X. Yu, S. S. Sun, M. D. Li, D. Q. Yuan and Y. B. Wang, Angew. Chem., Int. Ed., 2018, 57, 12716–12720 CrossRef CAS PubMed.
- Y. Li, Y. X. Xu, W. P. Yang, W. X. Shen, H. G. Xue and H. Pang, Small, 2018, 14, 1704435 CrossRef PubMed.
- S. Hermes, M. K. Schroter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS PubMed.
- G. Q. Lin, H. M. Ding, D. Q. Yuan, B. S. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS PubMed.
- S. Pandit and M. De, ChemistrySelect, 2017, 2, 10004–10009 CrossRef CAS.
- M. Xu, S. Yuan, X.-Y. Chen, Y.-J. Chang, G. Day, Z.-Y. Gu and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 8312–8319 CrossRef CAS PubMed.
- S. Pandit and M. De, J. Phys. Chem. C, 2017, 121, 600–608 CrossRef CAS.
- S. Pandit and M. De, ACS Appl. Nano Mater., 2020, 3, 3829–3838 CrossRef CAS.
- G. Das, F. Benyettou, S. K. Sharma, T. Prakasam, F. Gandara, V. A. de la Pena-O'Shea, N. Saleh, R. Pasricha, R. Jagannathan, M. A. Olson and A. Trabolsi, Chem. Sci., 2018, 9, 8461 RSC.
- G. Y. Zhang, X. L. Li, Q. B. Liao, Y. F. Liu, K. Xi, W. Y. Huang and X. D. Jia, Nat. Commun., 2018, 9, 2785 CrossRef PubMed.
- J. Q. Dong, X. Li, S. B. Peh, Y. D. Yuan, Y. X. Wang, D. X. Ji, S. J. Peng, G. L. Liu, S. M. Ying, D. Q. Yuan, J. W. Jiang, S. Ramakrishna and D. Zhao, Chem. Mater., 2019, 31, 146–160 CrossRef CAS.
- S. Jhulki, A. M. Evans, X. L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam, S. Barlow, J. L. Bredas, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 783–791 CrossRef CAS PubMed.
- P. P. Sun, J. Hai, S. H. Sun, S. Y. Lu, S. Liu, H. W. Liu, F. J. Chen and B. D. Wang, Nanoscale, 2020, 12, 825–831 RSC.
- D. Sun, X. Pang, Y. Cheng, J. Ming, S. J. Xiang, C. Zhang, P. Lv, C. C. Chu, X. L. Chen, G. Liu and N. F. Zheng, ACS Nano, 2020, 14, 2063–2076 CrossRef CAS PubMed.
- S. A. A. Razavi, M. Y. Masoomi and A. Morsali, Inorg. Chem., 2017, 56, 9646–9652 CrossRef CAS PubMed.
- H. T. Zhang, J. W. Zhang, G. Huang, Z. Y. Du and H. L. Jiang, Chem. Commun., 2014, 50, 12069–12072 RSC.
- Y. X. Pang, H. Li, J. Farmakes, F. Xiao, B. C. Chen, S. Q. Ma and Z. Y. Yang, J. Am. Chem. Soc., 2018, 140, 16032–16036 CrossRef PubMed.
- R. Ma and T. Sasaki, Acc. Chem. Res., 2015, 48, 136–143 CrossRef CAS.
- J. Q. Dong, K. Zhang, X. Li, Y. H. Qian, H. Zhu, D. Q. Yuan, Q. H. Xu, J. W. Jiang and D. Zhao, Nat. Commun., 2017, 8, 1142 CrossRef.
- J. H. Deng, Y. Q. Wen, J. Willman, W. J. Liu, Y. N. Gong, D. C. Zhong, T. B. Lu and H. C. Zhou, Inorg. Chem., 2019, 58, 11020–11027 CrossRef CAS PubMed.
- G. J. Guan and M. Y. Han, Adv. Sci., 2019, 6, 1901837 CrossRef CAS PubMed.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS.
- T. Tan, X. T. Jiang, C. Wang, B.
C. Yao and H. Zhang, Adv. Sci., 2020, 7, 2000058 CrossRef CAS PubMed.
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Boggild, S. Borini, F. H. L. Koppens, V. Palermo, N. Pugno, J. A. Garrido, R. Sordan, A. Bianco, L. Ballerini, M. Prato, E. Lidorikis, J. Kivioja, C. Marinelli, T. Ryhanen, A. Morpurgo, J. N. Coleman, V. Nicolosi, L. Colombo, A. Fert, M. Garcia-Hernandez, A. Bachtold, G. F. Schneider, F. Guinea, C. Dekker, M. Barbone, Z. P. Sun, C. Galiotis, A. N. Grigorenko, G. Konstantatos, A. Kis, M. Katsnelson, L. Vandersypen, A. Loiseau, V. Morandi, D. Neumaier, E. Treossi, V. Pellegrini, M. Polini, A. Tredicucci, G. M. Williams, B. H. Hong, J. H. Ahn, J. M. Kim, H. Zirath, B. J. van Wees, H. van der Zant, L. Occhipinti, A. Di Matteo, I. A. Kinloch, T. Seyller, E. Quesnel, X. L. Feng, K. Teo, N. Rupesinghe, P. Hakonen, S. R. T. Neil, Q. Tannock, T. Lofwander and J. Kinaret, Nanoscale, 2015, 7, 4598–4810 RSC.
- C. Moreno, M. Vilas-Varela, B. Kretz, A. Garcia-Lekue, M. V. Costache, M. Paradinas, M. Panighel, G. Ceballos, S. O. Valenzuela, D. Pena and A. Mugarza, Science, 2018, 360, 199–203 CrossRef CAS PubMed.
- J. M. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. L. Feng, K. Mullen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS.
- W. L. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137–9144 CrossRef CAS.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- O. M. Yaghi and H. L. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed.
- Y. Y. Lin, M. R. Thomas, A. Gelmi, V. Leonardo, E. T. Pashuck, S. A. Maynard, Y. Wang and M. M. Stevens, J. Am. Chem. Soc., 2017, 139, 13592–13595 CrossRef CAS PubMed.
- A. de Leon, B. J. Rodier, Q. M. Luo, C. M. Hemmingsen, P. R. Wei, K. Abbasi, R. Advincula and E. B. Pentzer, ACS Nano, 2017, 11, 7485–7493 CrossRef CAS PubMed.
- S. C. Zimmerman, F. Zeng, D. E. C. Reichert and S. V. Kolotuchin, Science, 1996, 271, 1095 CrossRef CAS PubMed.
- U. Boas and P. M. H. Heegaard, Chem. Soc. Rev., 2004, 33, 43–63 RSC.
- B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- Y. W. Peng, Y. Huang, Y. H. Zhu, B. Chen, L. Y. Wang, Z. C. Lai, Z. C. Zhang, M. T. Zhao, C. L. Tan, N. L. Yang, F. W. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698–8704 CrossRef CAS PubMed.
- S. L. Qiu, M. Xue and G. S. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- W. C. Mai, Y. Zuo, X. C. Zhang, K. Y. Leng, R. L. Liu, L. Y. Chen, X. D. Lin, Y. H. Lin, R. W. Fu and D. C. Wu, Chem. Commun., 2019, 55, 10241–10244 RSC.
- R. M. Crooks, M. Q. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS PubMed.
- R. Pawlak, X. S. Liu, S. Ninova, P. D'Astolfo, C. Drechsel, S. Sangtarash, R. Haner, S. Decurtins, H. Sadeghi, C. J. Lambert, U. Aschauer, S. X. Liu and E. Meyer, J. Am. Chem. Soc., 2020, 142, 12568–12573 CrossRef CAS.
- D. Beyer, S. Y. Wang, C. A. Pignedoli, J. Melidonie, B. K. Yuan, C. Li, J. Wilhelm, P. Ruffieux, R. Berger, K. Mullen, R. Fasel and X. L. Feng, J. Am. Chem. Soc., 2019, 141, 2843–2846 CrossRef CAS PubMed.
- P. W. Fritz and A. Coskun, Chem-Us, 2020, 6, 1046–1048 CrossRef CAS.
- D. Toummini, F. Ouazzani and M. Taillefer, Org. Lett., 2013, 15, 4690–4693 CrossRef CAS PubMed.
- M. De, S. S. Chou and V. P. Dravid, J. Am. Chem. Soc., 2011, 133, 17524–17527 CrossRef CAS.
- M. Clauberg and J. G. Joshi, P. Natl. Acad. Sci. U. S. A, 1993, 90, 1009–1012 CrossRef CAS PubMed.
- P. E. Stein and R. W. Carrell, Nat. Struct. Biol., 1995, 2, 96–113 CrossRef CAS PubMed.
- U. I. Ekeowa, B. Gooptu, D. Belorgey, P. Hagglof, S. Karlsson-Li, E. Miranda, J. Perez, I. MacLeod, H. Kroger, S. J. Marciniak, D. C. Crowther and D. A. Lomas, Clin. Sci., 2009, 116, 837–850 CrossRef CAS PubMed.
- N. O. Fischer, C. M. McIntosh, J. M. Simard and V. M. Rotello, P. Natl. Acad. Sci. U. S. A, 2002, 99, 5018–5023 CrossRef CAS PubMed.
- C. C. You, M. De, G. Han and V. M. Rotello, J. Am. Chem. Soc., 2005, 127, 12873–12881 CrossRef CAS PubMed.
- Y. J. Song, K. G. Qu, C. Zhao, J. S. Ren and X. G. Qu, Adv. Mater., 2010, 22, 2206–2210 CrossRef CAS PubMed.
- C. M. Wong, K. H. Wong and X. D. Chen, Appl. Microbiol. Biotechnol., 2008, 78, 927–938 CrossRef CAS PubMed.
- P. H. Ling, S. Cheng, N. Chen, C. H. Qian and F. Gao, ACS Appl. Mater. Interfaces, 2020, 12, 17185–17192 CrossRef CAS PubMed.
- J. J. X. Wu, X. Y. Wang, Q. Wang, Z. P. Lou, S. R. Li, Y. Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- S. Ghosh, P. Roy, N. Karmodak, E. D. Jemmis and G. Mugesh, Angew. Chem., Int. Ed., 2018, 57, 4510–4515 CrossRef CAS.
- N. Singh, S. K. NaveenKumar, M. Geethika and G. Mugesh, Angew. Chem., Int. Ed., 2021, 60, 3121–3130 CrossRef CAS PubMed.
- Y. X. Wang, X. Zhang, Z. M. Luo, X. Huang, C. L. Tan, H. Li, B. Zheng, B. Li, Y. Huang, J. Yang, Y. Zong, Y. B. Ying and H. Zhang, Nanoscale, 2014, 6, 12340–12344 RSC.
- M. Gao, Z. H. Wang, H. H. Zheng, L. Wang, S. J. Xu, X. Liu, W. Li, Y. X. Pan, W. L. Wang, X. M. Cai, R. A. Wu, X. F. Gao and R. B. Li, Angew. Chem., Int. Ed., 2020, 59, 3618–3623 CrossRef CAS PubMed.
- B. S. Sandanaraj, D. R. Vutukuri, J. M. Simard, A. Klaikherd, R. Hong, V. M. Rotello and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 10693–10698 CrossRef CAS PubMed.
- A. Verma, J. M. Simard and V. M. Rotello, Langmuir, 2004, 20, 4178–4181 CrossRef CAS PubMed.
- D. Wang, X. L. Song, P. Li, X. J. J. Gao and X. F. Gao, J. Mater. Chem. B, 2020, 8, 9028–9034 RSC.
- Y. L. Dong, H. G. Zhang, Z. U. Rahman, L. Su, X. J. Chen, J. Hu and X. G. Chen, Nanoscale, 2012, 4, 3969–3976 RSC.
- S. Wang, R. Cazelles, W. C. Liao, M. Vazquez-Gonzalez, A. Zoabi, R. Abu-Reziq and I. Willner, Nano Lett., 2017, 17, 2043–2048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, enzyme activity and nanozyme activity of the synthesized material. See DOI: 10.1039/d1na00226k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*