
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand exchange reactions on thiolate-protected gold nanoclusters
Yanan
Wang
 and
Thomas
Bürgi
*
and
Thomas
Bürgi
*
Department of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, 1211 Geneva 4, Switzerland. E-mail: thomas.buergi@unige.ch
First published on 6th April 2021
Abstract
As a versatile post-synthesis modification method, ligand exchange reaction exhibits great potential to extend the space of accessible nanoclusters. In this review, we summarized this process for thiolate-protected gold nanoclusters. In order to better understand this reaction we will first provide the necessary background on the synthesis and structure of various gold clusters, such as Au25(SR)18, Au38(SR)24, and Au102(SR)44. The previous investigations illustrated that ligand exchange is enabled by the chemical properties and flexible gold–sulfur interface of nanoclusters. It is generally believed that ligand exchange follows a SN2-like mechanism, which is supported both by experiments and calculations. More interesting, several studies show that ligand exchange takes place at preferred sites, i.e. thiolate groups –SR, on the ligand shell of nanoclusters. With the help of ligand exchange reactions many functionalities could be imparted to gold nanoclusters including the introduced of chirality to achiral nanoclusters, size transformation and phase transfer of nanoclusters, and the addition of fluorescence or biological labels. Ligand exchange was also used to amplify the enantiomeric excess of an intrinsically chiral cluster. Ligand exchange reaction accelerates the prosperity of the nanocluster field, and also extends the diversity of precise nanoclusters.
 Yanan Wang | Yanan Wang received her Bachelor's degree in Applied Chemistry and Master's degree in bioengineering from China University of Petroleum (East China) in 2014 and 2017, respectively. Since 2017, she has been working in the group of Prof. Thomas Bürgi as a PhD candidate in the Department of Physical Chemistry of the University of Geneva. Currently, her research mainly focuses on the synthesis and characterization of chiral gold nanoclusters and their potential applications. |
 Thomas Bürgi | Thomas Bürgi studied chemistry and obtained hid PhD (1995) at the University of Berne (Switzerland). After a postdoc at MIT, he did his habilitation at ETH, Zürich. He became assistant professor at the University of Neuchâtel (Switzerland, 2005) and full professor at the University of Heidelberg (2008). In 2010 he moved to the University of Geneva, where he is professor of physical chemistry. His research focuses on fundamental aspects and applications of chiral metal clusters, plasmon-based metamaterials and the development of in situ spectroscopy. |
1. Introduction
During the past decade, the interest in monolayer-protected metal clusters has drastically increased. The progress in the field has been reported in review articles focussing for example on precise synthesis,1–5 metal doping,6,7 and applications of gold nanoclusters.8,9 In general, the chemical properties of nanomaterials largely depend on their surface properties. As a flexible surface chemistry method, ligand exchange reaction (LER) is therefore an important tool for the development of nanomaterials. For instance, Dinkel and co-workers summarized the findings obtained by in situ spectroscopic measurements to probe the ligand exchange at the surface of colloidal gold and silver nanoparticles. Specifically, the results obtained by the inherently surface sensitive technique second-harmonic scattering (SHS) was discussed.10 In addition, Xu and co-workers concentrated on exchange reactions on metal–organic frameworks (MOFs).11 More focussing on metal clusters, Zhu and co-worker published a review on transformations of precise nanoclusters induced by ligand exchange.12 They focused on the size conversion of gold and silver nanoclusters during ligand exchange reaction. However, a comprehensive summary focussing on the ligand exchange reaction of thiolate-protected gold clusters is still rare.13Recently, many new findings have emerged on the mechanism and peculiarities of the ligand exchange reactions on thiolate-protected clusters, including ligand exchange between clusters,14 ligand exchange including chiral thiols,15 and ligand exchange induced amplification of enantiomeric excess.16 These new findings are of great interest and further expanded the knowledge in the field of the ligand exchange reaction on thiolate-protected gold nanoclusters. Considering the recent findings in the field and its implications for applications an attempt to summarize to current knowledge seems appropriate.
The ligand exchange reaction is an important post-synthesis method for the modification and further functionalization of gold nanoclusters. In this review, we specifically address the ligand exchange reactions (LERs) on thiolate-protected gold nanoclusters. The main points to be addressed are illustrated in Scheme 1. We mainly focus on atomically precise gold nanoclusters with known crystal structure, such as Au25(SR)18, Au38(SR)24, and Au102(SR)44, which allows to obtain molecular level insight into the process. The clusters contain diverse surface motifs, e.g. monomeric and dimeric Au–sulfur staples and the distinct surface thiolates show different reactivity towards ligand exchange. It is also emerging from recent experiments and calculations that the surface region of these clusters, i.e. the Au–sulfur interface, is far from being static. Furthermore, not only free thiols in solution can exchange with thiols on the cluster, but thiols can exchange among the clusters without addition of free thiols. In addition, LERs also takes place between free thiols and supported clusters. Ligand exchange can lead to important modifications of the cluster properties. For example, LERs can induce chirality on achiral nanoclusters and also lead to the enhancement of enantiomeric excess of chiral clusters. LERs can promote size transfer of clusters, and phase transfer between organic and aqueous phase. In general, ligand exchange reaction is an important process that largely extends the diversity of precise nanoclusters and their potential for applications. In the following, after an introduction to monolayer-protected gold clusters, we summarize the current knowledge on LERs with special focus on the more recent findings.
 | ||
| Scheme 1 Schematic diagram of ligand exchange reactions on thiolate-protected gold nanoclusters. | ||
2. Precise monolayer-protected gold nanoclusters
2.1 Synthesis of precise gold nanoclusters
Metal nanoclusters are composed of tens to hundreds of atoms and bridge the gap between single atoms or molecules and nanoparticles.5,17 Different from the plasmon excitation in nanoparticles, which is a collective excitation of conduction electrons, metal nanoclusters are characterized by strong quantum confinement effect and molecular-like properties.2,18 Nowadays, metal nanoclusters include several sub-classes, such as thiolate-protected silver nanoclusters, phosphine-protected gold nanoclusters, thiolate-protected gold nanoclusters and so on. Among them, thiolate-protected gold nanoclusters are the most studied due to their relatively long history and unique properties.3,18,19 In the 1990s, triggered by the work of Brust and Schiffrin, severeal groups started the research on gold nanoclusters and obtained a series of ultrasmall species.20 Typically in these syntheses mixtures of different cluster sizes were obtained. At this size every atom counts and minor changes in composition can have large effects on electronic and optical properties of the nanoclusters. Therefore, the precise control of the composition and purity is critically important.3In order to obtain pure nanoclusters, Whetten and co-workers tried to separate cluster mixtures by polyacrylamide gel electrophoresis (PAGE)21 and solvent-extraction,22,23 however, it is challenging to obtain pure cluster samples in this way. Murray and co-workers prepared the Au25 cluster by a two-phase protocol24,25 or the conversion of Au11 to Au25,26 but mixtures of clusters of different size were obtained. After purification by solvent-extraction and other methods, clusters of different size were obtained with very low yield. Later, Tsukuda and co-workers applied PAGE,27 recycling size exclusion chromatograph28 and solvent-extraction29 to isolate the precise nanoclusters in a time-consuming strategy. After about ten years of struggling to obtain pure cluster samples in high yield, there was a strong need for efficient methods towards synthesis of precise gold nanoclusters.
The first breakthrough was reported for Au25, a prototypical gold cluster. In 2008 Zhu and co-workers reported a high yield synthesis of Au25(SCH2CH2Ph)18 by kinetic control with a two-phase method.30 Later on, by using THF as solvent, Wu and co-workers achieved a big improvement of the synthesis of Au25 nanoclusters using a one-pot method.31 Importantly, a “size-focusing” process was identified in the growth of Au25 nanoclusters, which accounts for the formation of atomically monodisperse Au25 nanoclusters from the one-pot reaction.
Similar to the success in the synthesis of monodisperse Au25, a facile solution synthetic method for obtaining monodisperse Au38(S–C12H25)24 was developed by exploring a two phase ligand exchange process in which glutathione-capped Aun clusters are utilized as the starting material.32 Here a LER was carried out at elevated temperature (e.g. 80 °C) to obtain high purity Au38(S–C12H25)24 clusters, still at relatively low yield. After that, Qian and co-workers modified the protocol by replacing methanol by acetone in the first step (synthesis of Aun(SG)m) and by using an excess of phenylethylthiol (PhC2H4SH) as the etching ligand for the ligand exchange reaction.33 Using these modifications they could increase the synthesis yield of Au38 to 25% (Au atom).33 This work illustrated that the formation of Au38(SC2H4Ph)24 resulted from the conversion from larger clusters in the thiol etching induced growth process in a remarkable “size-focusing” process. The above results revealed that size-focusing is a very efficient methodology for synthesizing atomically precise gold nanoclusters. The underlying principle can be illustrated as shown in Fig. 1.2 Initially several nanoclusters with different sizes and vastly different stability coexist in the reaction mixture. The most stable nanocluster within the initially controlled size distribution will be sieved out.2 After the success in obtaining Au25 and Au38 nanoclusters using the size focussing method, a series of Aun(SR)m nanoclusters were synthesized in high yield and on a large scale, using this methodology, such as Au23,34 Au64,35 Au99,36 Au144 (ref. 37) and Au333.38 As the size-focusing involves core-etching from larger clusters to smaller clusters, the size (distribution) of the initial thiolate-protected gold cluster is critical. In addition, the high yield product may still coexist with some by-products, making additional purification steps such as size exclusion chromatography (SEC) necessary. Knoppe and co-workers used SEC to separate Au38 and Au40 clusters, which differ by only two gold atoms.39 This example demonstrates the power of this separation technique.
 | ||
| Fig. 1 Schematic of the “size-focusing” process. Adapted with permission from ref. 2. Copyright 2010 American Chemical Society. | ||
The development of synthesis methods resulted in several monodisperse nanoclusters including Au25(SR)18, Au38(SR)24, Au99(SR)42,36 and Au144(SR)60.37 Such clusters have already become excellent candidates for applications in catalysis,36,40,41 as optical active materials,42,43 as chemical sensors,44,45 and for biological applications.46,47
The availability of numerous atomically precise thiolate-protected gold clusters allowed one to study the evolution of properties as a function of size. Compared with the bulk metal, which has continuous energy band structure, ultrasmall nanoclusters possess size-dependent electronic structure, a HOMO–LUMO gap and molecule-like properties. The HOMO–LUMO gap of Au25(SR)18 was found to be 1.3 eV,30 which is bigger than the value of 1.0 eV found for Au38(SR)24.33 This implies that the larger clusters have a smaller energy gap, as expected. Having many additional clusters at hand, the relationship between HOMO–LUMO gap and size of the nanoclusters became clearer. There is a general trend of shrinking gap with increasing size. Plots of the HOMO–LUMO gap Eg against n−1/3 or against ln(n) (n: number of gold atoms) revealed linear relationship and two groups of clusters.17 The two sets of clusters have difference structure and atomic packing, which will be addressed in the next subsection.
2.2 Structure of precise monolayer-protected gold nanoclusters
In order to understand the fundamental science of nanoclusters, it is necessary to determine their structures. X-ray single-crystal diffraction is the most straightforward and important technique for the structure determination of nanoclusters. Among the most important milestones in the research history of structural characterization of nanoclusters goes back to 2007, when Kornberg and co-workers published the first total X-ray structure of a thiolate-protected gold cluster, containing 102 gold atoms and 44 p-mercaptobenzoic acid (p-MBA) ligands.48 The structure of Au102(p-MBA)44 was unexpected and did not match the view emerging from the “cluster of clusters” growth mechanism proposed for cluster chemistry.49 As shown in Fig. 2A, the gold core has a decahedron structure and is surrounded by a gold thiolate layer characterized by “staple” motifs. In these structures, one gold atom is bound to two sulfur atoms. Similar structures were previously predicted theoretically as gold-thiolate rings.50 The Au102(p-MBA)44 cluster contains 19 short and two long motif SR(AuSR)x (x = 1, 2). In addition, the cluster is chiral because of the geometry of the equatorial atoms (Fig. 2B). The stability of the cluster structure has several origins. The cluster has 58 free electrons and thus has a closed electronic shell within a superatom model.51 The thiolate monolayer contributes as well to the stability of the cluster and within this layer additional stability stems from the π–π stacking between phenyl rings of the p-MBA molecules.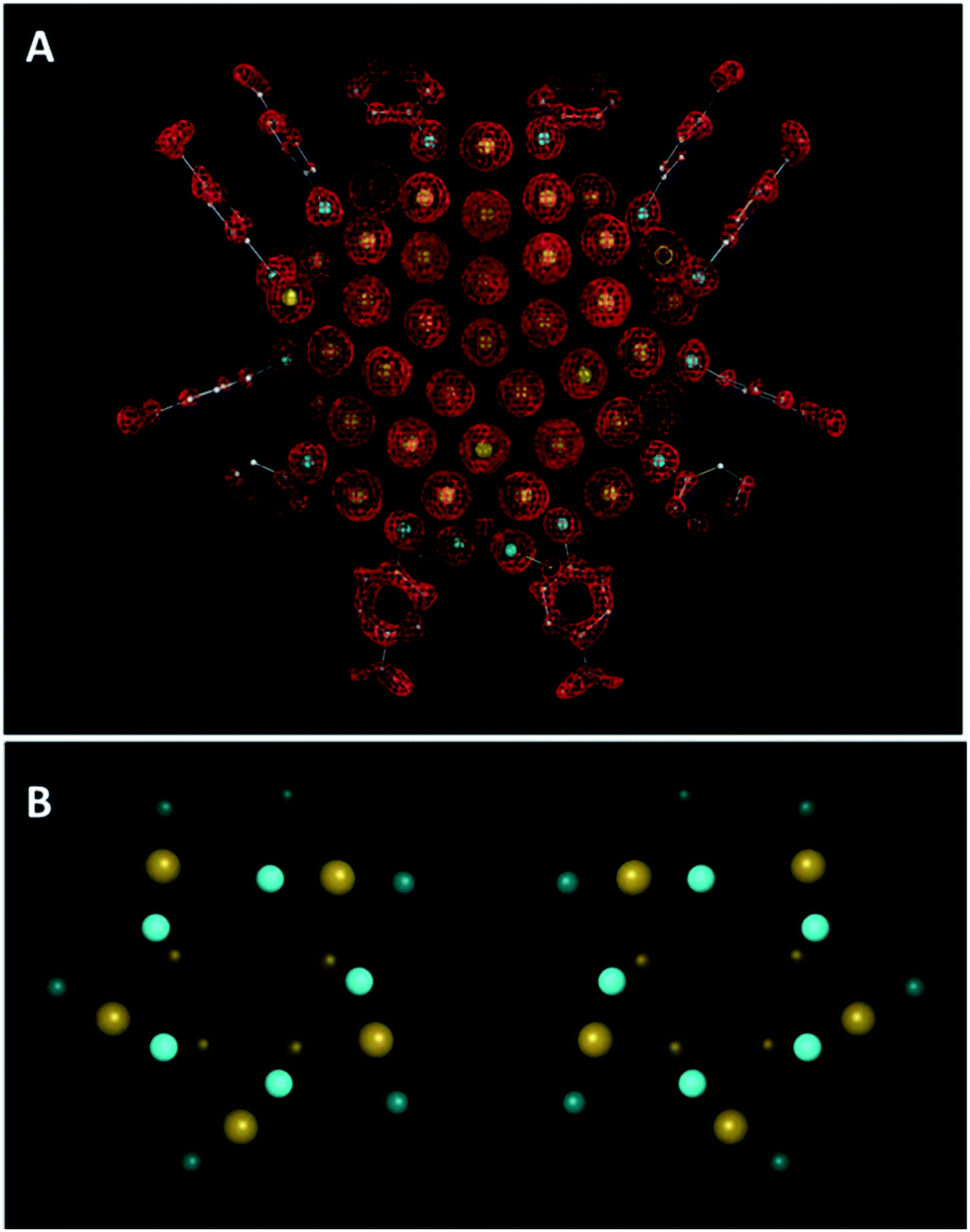 | ||
| Fig. 2 X-ray crystal structure of the Au102(p-MBA)44 crystal. (A) Electron density map and atomic structure (Au atoms depicted as yellow spheres, and p-MBA shown as framework and with small spheres [S in cyan, C in gray, and O in red]). (B) Top view of the two enantiomeric clusters. Colour scheme as in (A), only the sulfur atoms of p-MBA are shown. Adapted with permission from ref. 48. Copyright 2007 American Association for the Advancement of Science. | ||
Owing to the development of precise synthesis of nanoclusters and X-ray single-crystal diffraction, structure determination of gold nanoclusters ushered in a period of vigorous development after the first breakthrough of Au102(p-MBA)44.48 The experimental crystal structure of ultrasmall gold nanocluster Au25(SR)18 was reported separately by Jin's and Murray's groups.52,53 Grönbeck and co-workers also predicted the same Au25(SR)18 structure by DFT calculations.54 X-ray crystallographic analysis shows that the Au25 cluster has an Au13 icosahedral core (Fig. 3) surrounded by a monolayer containing 12 gold atoms and 18 thiolates (Fig. 3). The crystal structure reveals three types of gold atoms ((i) one central gold atom (ii) 12 gold atoms as the vertices of the icosahedron, and (iii) 12 gold atoms forming staple) and two types of SR groups (terminal SR groups which link to the gold core, and central SR groups which link to two staple gold atoms). The high symmetry structure shown above was obtained from the anion Au25(SR)18−, which is an 8 electron superatom with S2P6 electronic structure, but a similar structure was found for neutral Au25(SR)18.52
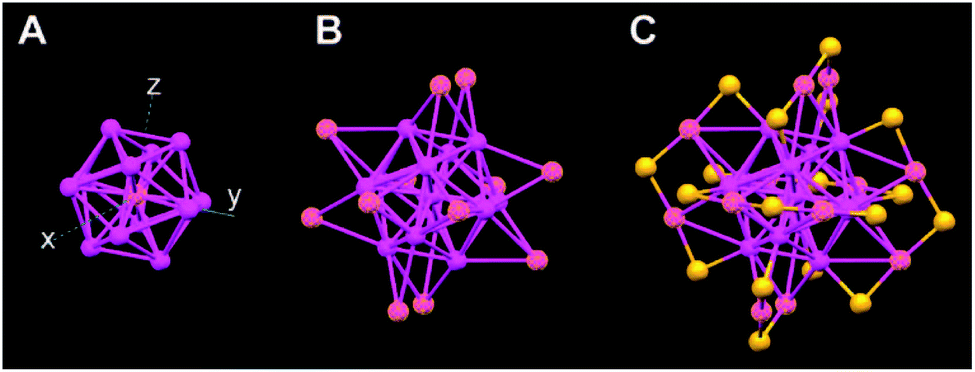 | ||
| Fig. 3 Crystal structure of the Au25(SR)18 cluster. (A) The icosahedral Au13 core; (B) the Au13 core plus the exterior 12 Au atoms; (C) whole Au25 cluster protected by 18 thiolate ligands (just S is shown for clarity, magenta, Au; yellow, S). Adapted with permission from ref. 52. Copyright 2008 American Chemical Society. | ||
Compared with Au25(SR)18, the crystal structure of Au38(SR)24 is slightly more complex.55 As illustrated in Fig. 4A, the Au38(SR)24 cluster has a face-fused biicosahedral Au23 core is covered by six dimeric (–SR–Au–SR–Au–SR–) and three monomeric staples (–SR–Au–SR–). A similar structure has been predicted by Zeng56 just slightly differing in the arrangement of the dimeric staples on the icosahedral Au13 unit. The monomeric staples were found for Au38(SR)24 but not for Au25(SR)18 which is just protected by six dimeric staples. This observation led to the prediction that the smaller size clusters need longer staples for the protection considering the large curvature for small gold core.57
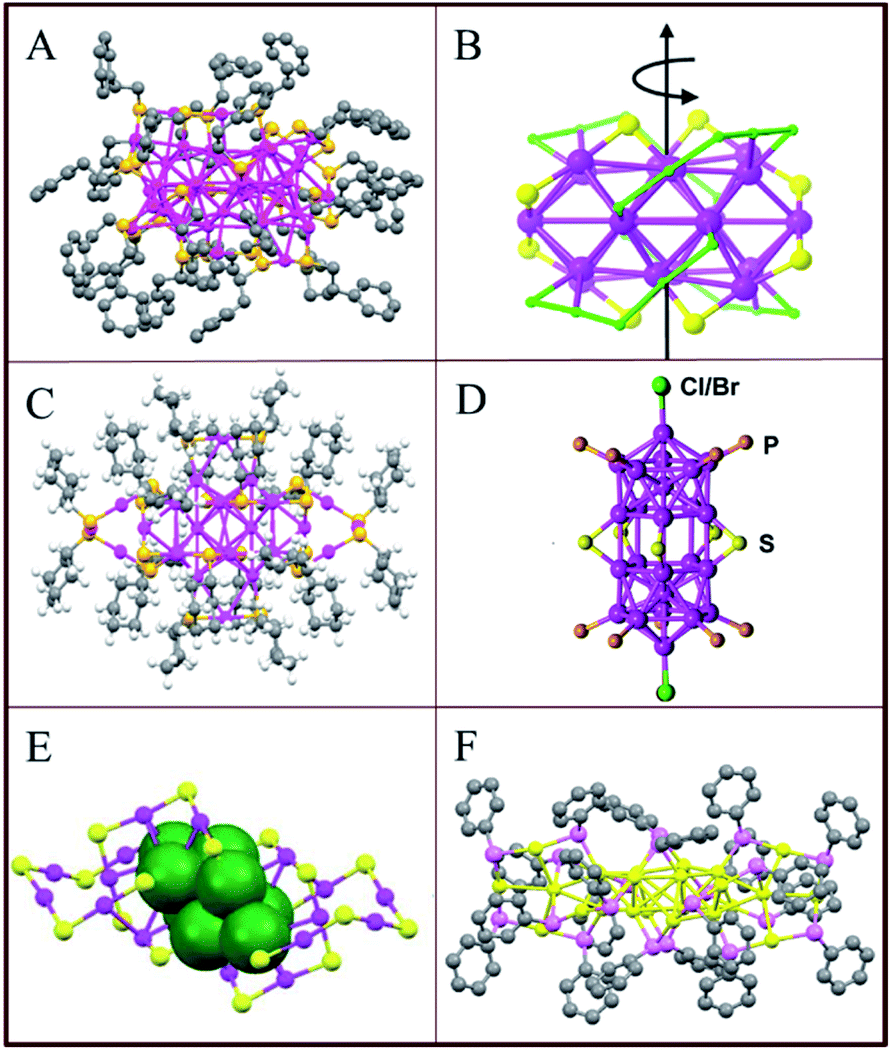 | ||
| Fig. 4 Crystal structures of (A) Au38(PET)24, adapted with permission from ref. 55. Copyright 2010 American Chemical Society. (B) Au28(TBBT)20, adapted with permission from ref. 58. Copyright 2013 American Chemical Society. (C) Au28(S–c-C6H11)20, adapted with permission from ref. 59. Copyright 2016 American Chemical Society. (D) [Au24(PPh3)10(SC2H4Ph)5X2]+, adapted with permission from ref. 60. Copyright 2012 American Chemical Society. (E) Au24(SCH2Ph-tBu)20, adapted with permission from ref. 61. Copyright 2014 Royal Society of Chemistry. (F) Au24(SeC6H5)20, adapted with permission from ref. 62. Copyright 2014 American Chemical Society. The ligands are partially omitted for clarity. | ||
Similar to the case of Au102(SR)44 and Au38(SR)24, the structure of Au28(TBBT)20 (where TBBT = 4-tert-butylbenzenethiolate) is also chiral (Fig. 4B).48,55,58 The cluster has an Au20 chiral kernel protected by four dimeric “staples” and eight bridging thiolates, and the chirality of the structure arises due to the rotational arrangement of the four dimeric staples and the arrangement of eight bridging thiolates. Later, Jin's group reported an isomer of Au28(TBBT)20 obtained after the ligand exchange with excess SH–c-C6H11 ligand. The obtained Au28(S–c-C6H11)20 has two trimeric staples arranged on the surface as shown in Fig. 4C.59 The above results revealed that, for a given number of gold atoms, the structure of a cluster may be modified by the ligand. This was also observed for Au24 clusters.60–62 Au24L20 clusters have different structures depending on the ligands (L): (i) Mixed ligand shell composed of phosphine and thiolate (Fig. 4D),60 (ii) thiolate (Fig. 4E)61 and (iii) selenolate-capped Au24 clusters (Fig. 4F).62 This confirms that the structure of gold clusters is affected by the protecting ligand on the surface.
Apart from the clusters we mentioned above, the crystal structure of some other clusters have also been reported including Au18(SR)14,63 Au20(TBBT)16,64 Au21(S–tBu)15,65 Au36(SR)24,66 Au92(SR)44,67 Au130(p-MBT)50,68 Au144(SCH2Ph)60.69 Among those clusters, Au144(SR)60 has attracted a lot of attention. The structure of Au144(SCH2Ph)60 was predicted by Olga Lopez-Acevedo and co-workers in 2009,70 but the experimental structure was published almost one decade later, in 2018, by Zhikun Wu and co-workers.69 The Au144(SCH2Ph)60 has a Au12 icosahedral inner core covered by two Au shells resulting in a Au114 core. 30 monomeric staple motifs are distributed on the surface of the Au114 core in a highly ordered pattern. Because of the arrangement of the 30 monomeric staples, the Au144(SCH2Ph)60 structure is chiral. With more and more crystal structures of gold clusters determined, the influence of size and structure on the HOMO–LUMO gap can be established. Atom packing plays an important role for Eg, as mentioned above, and the FCC (face-centered cubic) structures have much higher Eg energies than the icosahedral ones of comparable sizes.17
2.3 Flexibility of the gold–sulfur interface of gold nanoclusters
The formation of a gold–sulfur bond is the driving force for the anchoring of thiol ligands on either gold surfaces, nanoparticles or clusters,71 leading to the formation of self-assembled monolayers (SAM).72 From the available structural investigations it emerges that the staple-type binding motif is the preferred structure element of the gold–sulfur interface, as first evidenced by the crystal structure determination of Au102(SR)44.48 After analysis of various nanocluster structures such as Au25(SR)18, Au38(SR)24 and Au28(SR)20, the bridging motif (Fig. 5A), monomeric staple (Fig. 5B) and dimeric staple motif (Fig. 5C) are the most common surface structure elements. Actually the tetrameric staple motif also has been evidenced for Au24(SCH2Ph-tBu)20.61 Also other staple motifs are expected in order to adapt to the different curvatures of nanoclusters.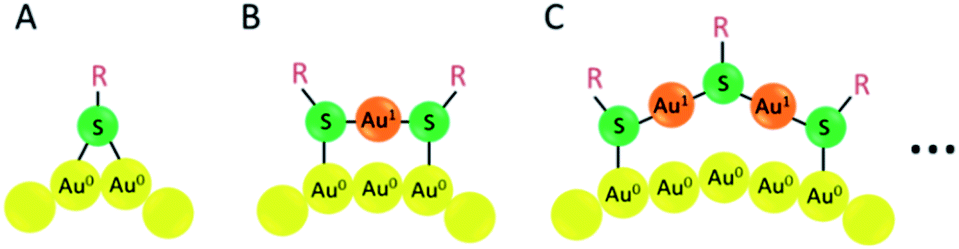 | ||
| Fig. 5 Schematic drawing of the bridging thiolate (A), monomeric Au–SR–Au staple motif (B) and dimeric Au–SR–Au–SR–Au staple motif. Yellow: Au (0) atom; orange: Au(I) atom; green: sulphur atom. | ||
The properties of gold–sulfur interface and its dynamics determine the further functionalization of thiolate-protected clusters and their applications. However, due to the experimental difficulties in probing the dynamic properties of the interface, this field was barren for a long time. Since the structure of the Au38(SR)24 cluster been solved it became a good candidate for studying the gold–sulfur interface.55 The cluster is intrinsically chiral due to the chiral arrangement of the dimeric staples at the poles of the cluster. Dolamic and co-workers reported the separation of the enantiomers of Au38(SCH2CH2Ph)24 (ref. 73) by chiral high-performance liquid chromatography. Using enantiopure Au38(SR)24 clusters Knoppe and co-workers showed that the cluster can racemize (Fig. 6, top).74 The reaction was started with A-Au38(SCH2CH2Ph)24 and C-Au38(SCH2CH2Ph)24 (A: anticlockwise, C: clockwise), respectively, and the evolution of the clusters was followed by circular dichroism spectroscopy. From this investigation, the racemization of the enantiopure Au38(SR)24 clusters takes place at modest temperatures without significant decomposition. Furthermore, from the temperature-dependent kinetic data, the activation barrier for the rearrangement within the thiolate–gold interface of the Au38(SR)24 clusters is about 28 kcal mol−1, which is lower than the gold–sulfur bond energy (about 50 kcal mol−1). The relatively low activation barrier indicated that the racemization process of Au38(SR)24 proceeds without complete Au–S bond breaking and evidenced the flexibility of the gold-thiolate interface. In addition, using density functional theory (DFT) computations, Häkkinen and co-workers proposed a mechanism for this inversion of the Au–S framework of Au38(SR)24 as shown in Fig. 6 (bottom).75 In their model, the racemization proceeds via a rotational reconstruction of the metal core without any Au–S bonds being broken.
 | ||
| Fig. 6 Schematic drawing of the racemization reaction of Au38(SR)24 cluster. (Top) In this scheme (view along the long axis of the cluster), the left-handed cluster (left, A-Au38(SR)24) is converted into the right-handed enantiomer (right, C-Au38(SR)24) and vice versa. The two enantiomers are separated by an activation barrier of ca. 28 kcal mol−1 as determined from experiments. Adapted with permission from ref. 74. Copyright 2012 American Chemical Society. (Bottom) Density functional theory computational simulation of the chiral inversion of Au38(SR)24 clusters through rotation of the three gold core atoms at the poles of the cluster. Adapted with permission from ref. 75. Copyright 2019 American Chemical Society. | ||
The flexibility of the gold–sulfur interface is affected by many factors, such as Pd doping, which is normally used for increasing the stability of the cluster and enhance its reactivity in catalytic and ligand exchange reactions. Barrabés and co-workers reported that the racemization process takes place at significantly lower temperature after doping the Au38(SR)24 cluster with two Pd atoms compared with the parent cluster.76 This case indicated that the Pd doping of the cluster renders the Au–S interface more flexible. In contrast, after introducing of a rigid dithiolate, 1,1′-binaphthyl-2,2′-dithiol (BINAS), into the ligand shell of the Au38(SR)24 cluster, the racemization drastically slows down.77 For example, the racemization of Au38(2-PET)22(BINAS)1 at 70 °C is about 27 times slower compared to the parent cluster, meaning that the introduction of the dithiol reduces the flexibility of the Au–S interface. Interestingly, the vibrational spectrum of the Au–S interface is also drastically influenced by introducing one BINAS dithiol into the ligand shell of the cluster, possibly related to the fact that the dithiol bridges two staples.78 Similarly, the racemization of the Au40(SR)24 cluster was reported to take place at higher temperature compared with Au38(SR)24 but with a similar activation barrier, further confirming the flexibility of gold–sulfur interface.79
The racemization described above is not the only dynamic process observed for the Au38(SR)24 cluster. In fact, ligands can migrate on the cluster surface between different symmetry unique sites. It has been reported that HPLC can be used to separate and isolate one specific regioisomer after ligand exchange between Au38(2-PET)24 clusters and enantiopure chiral [2.2]-paracyclophane-4-thiol 1 (PCP-4-SH).80 The isolated species is stable at room temperature, however, the adsorbed thiolate migrates between the different symmetry-unique sites at 80 °C as followed by HPLC.80 The mechanism underlying this observation is not yet clear: it might involve the exchange of two ligands between different sites on one cluster or the exchange of ligands between clusters (see below).
The chemical properties and flexibility of the gold–sulfur interface, as discussed above, form the basis for the post-synthesis modification and further functionalization of the gold nanoclusters. Among them, ligand exchange reaction is extraordinarily sparkly and we will now focus on this topic in the following sections.
3. Ligand exchange reactions
3.1 Mechanism of ligand exchange reactions
Depending on the flexibility and the reactivity of the gold–sulfur interface, the thiolates within the ligand shell can be displaced by other thiols in the solution, which is named place-exchange reactions, or more commonly ligand exchange reactions (LERs). For thiolate-protected clusters this reaction was first reported by Murry and co-workers.81–85 LERs were studied by 1H NMR spectroscopy, thermal analysis and IR spectroscopy.81,82,84,85 Results of these experiments revealed some factors which may affect the process. The chemical reactivity is mostly independent of the nanoparticle or cluster size,84 but the stability of the sample is a factor determining the fate of the cluster after the ligand exchange reactions.86 The rate and extent of ligand exchange reactions was found to increase with increasing positive electronic charge on the Au cluster core,82 but it decreases with increasing size of the entering ligand and the carbon chain length of the protecting monolayer.85 The ligand exchange reactions discussed in the following sections have been summarized in Table 1.| Section | Topic | Reaction | Reference |
|---|---|---|---|
| 3.1 | Ligand exchange with free ligand | Au102(p-MBA)44 + parabromobenzene thiol (p-BBT) → Au102(p-MBA)40(p-BBT)4 | 88 |
| Anti-influenza N9 neuraminidase NC10 antibody + Au25(SG)18 → ScFv-Au25(SG)18-complex | 87 | ||
| [Au25(SC2H4Ph)18]−+ HSCH2Ph-tBu → Au24(SCH2Ph-tBu)20 | 86 | ||
| Au24Cd(SC2H4Ph)18 + HSCH2Ph-tBu → Au24Cd(SCH2Ph-tBu)18 + Au24Cd(SCH2Ph-tBu)17(SC2H4Ph)1 | |||
| Intercluster ligand exchange | [Au25(SC10H21)18]− + [Au25(SC12H25)18]− → Au25(SC12H25)18−x(SC10H21)x | 89 | |
| Au25(SBut)18 + Au25(2-PET)18 → Au25(SBut)18−x(2-PET)x | 14 | ||
| Au38(2-PET)24−2x(R-BINAS)x + Au25(2-PET)18 → Au25(2-PET)16(R-BINAS)1 + Au38(2-PET)24−2x(R-BINAS)x | 16 | ||
| 3.2 | Site-selective ligand exchange | Au24Pd(SC2H4Ph)18 + C12H25SH → Au24Pd(SC2H4Ph)18−x(SC12H25)x | 92 |
| [Au25(SC2H4Ph)18]− + HSePh/HTePh → [Au25(SC2H4Ph)18−X(SePh/TePh)x | 99 | ||
| Au24Cd(SCH2Ph)18 + SCH2Ph-tBu → Au24Cd(SCH2Ph-tBu)17(SCH2Ph)1 | 86 | ||
| PdAu24(2-PET)18 + S-BINAS → PdAu24(2-PET)16(S-BINAS)1 | 108 | ||
| 3.3 | Au11(PPh3)7Cl3 (deposited on Al2O3) + L-glutathione(GSH) → Au11(PPh3)7(GS)xCl3−x | 109 | |
| 4.1 | Ligand exchange induced chirality | [Au25(PET)18−][TOA+] + R/S-BINAS → [Au25(PET)18−2X(R/S-BINAS)X−][TOA+] | 107 |
| [Au38(SR)24]/[Au40(SR)24] + R/S-BINAS → [Au38(SR)24−2X(R/S-BINAS)X]/[Au40(SR)24−2X(R/S-BINAS)X] | 106 | ||
| A-Au38(2-PET)24 + C-Au38(2-PET)22(R-BINAS)1 → C-Au38(2-PET)24 + A-Au38(2-PET)22(R-BINAS)1 | 16 | ||
| 4.2 | Ligand exchange induced size transformation | Au28(SPh-tBu)20 + HS–c-C6H11 ←→ Au28(S–c-C6H11)20 + HSPh-tBu | 59 |
| Au25(PET)18 + 4-tert-butylbenzenelthiol (TBBTH) → Au28(TBBT)20 | 58 | ||
| Au11(PPh3)7Cl3 + L-glutathione (GSH) → Au25(GS)18 | 26 and 110 | ||
| Au144(SC2H4Ph)60 + HSPh → Au99(SPh)42 | 128 | ||
| Au38(SC2H4Ph)24 + HSPh-tBu → Au36(SPh-tBu)24 | 120 | ||
| Au23(S–c-C6H11)16 + HTBBT → Au36(TBBT)24 | 127 | ||
| Au329(PET)84 + HSPh-tBu → Au279(SPh-tBu)84 | 129 | ||
| Au36(SPhX)24 (where X = –H or –tBu) + HS-tBu → Au30(S-tBu)18Au30(S-tBu)18 + HSPhX (where X = –H or -tBu) → Au36(SPhX)24 | 12 and 125 | ||
| Au44(TBBT)28 + 2,4-dimethylbenzenethiol (2,4-DMBTH) ←→ Au44(2,4-DMBT)26 + TBBTH | 130 and 131 | ||
| Au43(S–c-C6H11)25 + 2,4-DMBTH ←→ Au44(2,4-DMBT)26 + HS–c-C6H11 | |||
| Au43(S–c-C6H11)25 + TBBTH → Au44(TBBT)28 + HS–c-C6H11 | |||
| Au25(SBut)18 + R/S-phenylpropane-1-thiol (R/S-PPT) → Au28(R/S-PPT)21 | 15 | ||
| 4.3 | Ligand exchange induced phase transfer | Au24Ag20(2-Spy)4(PA)20Cl2 + mercaptosuccinic acid (MSA) → Au24Ag20-MSA | 138 |
| [Ag141Br12(S-Adm)40]3+ + Phenylacetylene(PAH) → [Ag141Br12−nCln(S-Adm)38PA2]3+ | 139 | ||
AuxAg44−x(PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)m(Ph3P)nClP + Tiopronin → AuxAg44−x(SR)wClz C)m(Ph3P)nClP + Tiopronin → AuxAg44−x(SR)wClz |
140 | ||
| 4.4 | Addition of fluorescence properties | AuNC@DDAB + dihydrolipoic acid (DHLA) → AuNC@DHLA | 141 |
| AuNAC@Ag (NAC = N-acetylcysteine) + L-glutathione (GSH) → AuNAC@AgSG | 147 |
Evidence from related investigations suggested an associative pathway for the mechanism of ligand exchange reactions, which has been corroborated by various experimental and computational studies.14,87,88 For example, Heinecke and co-workers reported the first crystal structure of Au102(p-MBA)40(p-BBT)4 (p-BBT = parabromobenzene thiol), which is formed by the ligand exchange reaction of Au102(p-MBA)44 with p-BBT as the incoming ligand.88 Available crystal structures of Au102(p-MBA)44 and Au102(p-MBA)40(p-BBT)4 were underpinned by a computational DFT study on energetics, reaction intermediates and pertinent transition states during the ligand exchange reactions, revealing microscopic details of this process. The associative ligand exchange mechanism, as emerging from a computational study, is illustrated in Fig. 7, where methane thiol is the incoming ligand. For associative ligand exchange, the more solvent exposed sulfur sites on Au102(p-MBA)44 are more reactive and “nucleophilic attacked” by the incoming thiol(ate), creating an intermediate that has both incoming and outgoing ligands simultaneously bound to the accessible gold atom (Fig. 7b). Then the intermediate structure changed to the well-known “hemiring” unit with one Au replaced by an H atom (Fig. 7c). Depending on the orientation of the residue, the observed bond length between the hydrogen of the incoming ligand and the sulfur of the outgoing ligand become shorter and finally the sulfur of the outgoing ligand is released from the gold core (Fig. 7d). To complete exchange, the hydrogen atom of the adsorbed methane thiol is transferred to the outgoing ligand, which is then released to the solution (Fig. 7e). The intermediate states b (gold bound to three sulfur atoms) and d (breaking of S–H bond) have maximum energy during the exchange process.
 | ||
| Fig. 7 Scheme of ligand exchange process with methane thiol and Au102(SR)44 clusters. Top-left panel: energy behavior during exchange process (corresponding configuration were depicted as a sketch in the bottom panel); top-right panel: hemiring-like intermediate c. Configurations close to b and d have been confirmed to be at the local energy maximum by structural relaxations to the intermediate and final states. Adapted with permission from ref. 88. Copyright 2012 American Chemical Society. | ||
A similar process may take place when labelling proteins with nanoclusters (as shown in Fig. 8). The initial reaction of this modification is a ligand exchange reactions of the anti-influenza N9 neuraminidase NC10 antibody against a glutathione ligand on a gold cluster as studied by means of ab initio QM/MM calculations.87 Compared to the reaction with free thiol, the example displayed here concerns a large protein and may have two substitution modes as illustrated in Fig. 8. In the calculations ligand exchange of the side and apex glutathione ligands were considered, and showed that the intermediate from the side ligand is more stable. This investigation indicates that the essential features of the ligand exchange reaction are independent of the nature of the thiolate ligands and the presence of the protein. But the presence of positive residues at the protein C-terminal tail is critical for forming attractive intermolecular interactions with the carboxylate groups of the SG ligands, facilitating the adsorption of the protein cysteine on the gold cluster surface.
 | ||
| Fig. 8 Mechanism of the ligand exchange reaction in the scFv⋯Au25(SG)18− complex computed by QM/MM calculations. (a) Substitution at the side SG. (b) Substitution at the apex SG. For clarity, only three SG ligands of the dimeric staples are shown, and other SG groups and gold atoms far from the recation centre are shown colorless or semitransparent. Part of the secondary structure of the protein is displayed, together with the nucleophilic cysteine. Water molecules are not shown. Adapted with permission from ref. 87. Copyright 2017 Royal Society of Chemistry. | ||
Because of the fast dynamics and complexity of the ligand exchange it is difficulty to disclose the microscopic details of the SN2-like process by experimental alone. However, with the assistance of computational method such as DFT calculations, some light could be shed on the SN2-like mechanism of initial ligand exchange. Interestingly, recently Wu and co-workers proposed an unimolecular nucleophilic substitution (SN1)-like mechanism (in addition to a SN2-like process),86 based on single crystal structure analysis of ligand exchanged clusters. The crystal structures showed that part of the sulfur atoms retained their configuration upon ligand exchange, which seems not possible for a SN2-like process. However, this argument only holds if the configuration at the sulfur atom is retained in solution, which was not addressed.
Apart from the possibility for exchange of thiols via free ligand, as discussed above, there is a second mechanism for thiol exchange, which takes place through intercluster collisions. In 2013, Yoshiki Niihori and co-workers observed the intercluster LERs between Au25(SC10H21)18 and Au25(SC12H25)18 when they studied the influence of Pd atom doping of thiolate-protected Au25 nanoclusters on ligand exchange reactivity.89 In that case the authors assumed that this exchange resulted from the detachment of ligand or gold–ligand species from the cluster.89 Later, Salassa and co-workers carried out the intercluster LERs between Au25(SBut)18 and Au25(2-PET)18, and experimentally showed that the process is fast. As shown in Fig. 9A, 15 min after mixing the two clusters at room temperature peaks from clusters with mixed ligand layer already appear in the mass spectrum.14 In addition, the interclusters ligand exchange seems to take place without release of thiol or thiol–gold complex into the solution, as illustrated in Fig. 9B. In this experiment Au25(SBut)18 was put inside a dialysis membrane and thus the two clusters were physically separated, whereas low molecular weight species could still penetrate and pass the membrane. The MALDI mass spectra showed that the clusters did not undergo any exchange of thiols.14
 | ||
| Fig. 9 (A) Positive-ion MALDI mass spectra of Au25(SBut)18 and Au25(2-PET)18 mixture collected at different time after mixing the two clusters; (B) experiment where Au25(SBut)18 was put inside a dialysis membrane whereas Au25(2-PET)18 was added outside the dialysis membrane. Positive-ion MALDI mass spectra of the solution inside/outside the dialysis membrane at t = 0 (a/b) and after 5 days (c/d). Adapted with permission from ref. 14. Copyright 2017 American Chemical Society. | ||
Recently, we confirmed that bidentate thiol ligands can also exchange between clusters by observing the mass peak belonging to Au25(2-PET)16(R-BINAS)1 (BINAS = 1,1′-binaphthyl-2,2′-dithiol) after mixing Au38(2-PET)24−2x(R-BINAS)x (x corresponds to the number of BINAS ligands, which was about 2 on average in this experiment) and Au25(2-PET)18 clusters.16 Compared to the exchange of monothiols higher temperatures are needed for the process to take place, 70 °C in this case. However, compared with the free thiol exchange, the mechanism of intercluster ligand exchange is still rather unexplored and further investigations are needed.
Over the past decades, since the first report of ligand exchange reactions for gold nanoclusters, this reaction has been widely used to introduce new ligands to the parent nanoclusters,77,88,90–92 and to add functionalities or chemical properties to clusters as a post-modification method.87,93 This methodology strongly extends the possibilities to modify surface properties of gold nanoclusters, normally without changing the metal core.93 Understanding the mechanism of the ligand exchange reaction may help us engineer the surface chemistry of nanomaterials in order to build multifunctional nano-platforms.
3.2 Site-selective ligand exchange reactions
Several reports show that during the ligand exchange reaction, many sites on the ligand shell exchange slowly or not at all, whereas others are comparatively reactive.85,94,95 These observations revealed that the ligand shell is heterogeneous and offers a diversity of ligand binding sites and the exchange reaction occurs preferentially at selected ones.83 Before going into detail, it is essential to discuss the diversity sulfur groups in the ligand shell which represent the reactive site.Since the dynamic exchange process were mostly investigated with Au25(SR)18 and Au38(SR)24,80,96 which are easily prepared and relatively stable, we focus some more on their crystal structure as illustrated in Fig. 10. The Au25(SR)18 cluster (Fig. 10 left structure) has an icosahedral Au13 core surrounded by six SR-Au-SR-Au-SR staple units as we mentioned before.77,89,92,97–99 Because of the symmetry of Au25(SR)18, six dimeric staples share the same chemical environment, but the SR groups in one staple can be divided into central –SR group, which is bound to the two gold atoms in the staple, and terminal –SR groups, which are linked to the gold core.100 In contrast, Au38(SR)24 consists of a biicosahedral Au23 core (Fig. 10 right structure) and is covered not only by six dimeric units but also by three monomeric units. The structure of Au38(SR)24 is elongated, with the three monomeric staples at the equator and the six dimeric staples at the two poles.101 The dimeric staples can be divided into central –SR group and terminal –SR groups. However, the two terminal –SR groups in the dimeric staples of Au38(SR)24 have different chemical environment.80,102 Overall the 24 thiolate ligands are divided into four groups of symmetry unique sites with different chemical environment, which can give rise to preferential exchange sites during LERs.88,92
 | ||
| Fig. 10 Crystal structures of Au25(SR)18 and Au38(SR)24 clusters. Some terminal –SR (orange circle) and central –SR (red circle) are marked in the scheme. Au25(SR)18 nanocluster has three possible ligand substitution sites marked as A, B, C, and Au38(SR)24 has eight possible ligand substitution sites marked as A-H. Bond distances also are given in Å. Color code: Au core = gold, Au staple = orange, S = green. Adapted with permission from ref. 100 and ref. 102. Copyright 2015 &2016 American Chemical Society. | ||
From recent experimental and computational studies, it became clear that LERs between clusters and free monothioles in solution start preferentially at the terminal SR groups of Au25(SR)18 and Au38(SR)24 clusters,92,100,102via an associative SN2-like mechanism.88 This tendency was experimentally demonstrated by Yoshiki Niihori and co-workers.92 In this case the isomer distributions of the product after ligand exchange between Au24Pd(SC2H4Ph)18 and C12H25SH were determined by high resolution high-performance liquid chromatography. The quantitative evaluation of the expected coordination isomers and the products obtained by the reactions, showed that the exchange reaction starts at the thiolate which is bound to the core site. This also holds for exchange with other chalcogenate ligands such as HSePh or - HTePh.99 Niihori and co-workers also found that Pd doping of the Au25(SR)18 cluster drastically increased the ligand exchange rate. Pd doping reduces the number of valance electrons of the metal core, which facilitates the attack by the incoming ligand. Furthermore Pd doping induces the distortion of the cluster geometry.89
In addition, the site preference of LERs can be investigated by computational methods. Aikens and co-workers employed density functional theory (DFT) to examine the ligand exchange on model Au25(SH)18− and Au38(SH)24 clusters with an incoming methanethiol.100,102 They calculated the intermediates and transition states, and predicted the barrier heights and reaction energies for this ligand exchange process. In Fig. 10, the different possible ligand substitution sites are marked on the crystal structure of Au25(SR)18 (Fig. 10 left) and Au38(SR)24 (Fig. 10 right) nanoclusters. The former cluster offers three different sites, whereas the latter has eight. The energies of the intermediates, transition state and products corresponding to ligand exchange at different sites determined by DFT calculation are summarized at Table 2. The results for Au25 indicated that the most favourable ligand exchange process is at site B, and the site C has highest energy barrier, which resulted in ligand exchange at the central SR group. This tendency was consistent with the one for Au38(SR)24 as is evident from the calculated energies listed at Table 2. LERs at site C had higher energy barriers compared with other sites, meaning that the reaction between the staple gold atom and the sulfur atom of the central-SR units will proceed at a slower rate. Here, we should mention that each sulfur atom is participating in two bonds, for example Au25 A&B, Au38 C&D, and Au38 E&F, which represent different reactive sites, however, those two sites will lead to the same product. Above predictions from computational studies on Au25(SH)18− and Au38(SR)24 indicated that the exchange is preferred at the terminal SR groups, which is congruous with the experimental results.88,92 However, this might not always be the case as shown by Zhu and co-workers. After the ligand exchange reaction between Au24Cd(SCH2Ph)18 and SCH2Ph-tBu Au24Cd(SCH2Ph-tBu)17(SCH2Ph)1 has been obtained.86 The integrated area of the peaks extracted from 1H NMR spectra of this species illustrated that the remaining parent ligand SCH2Ph was equally distributed among the terminal and central sulfur atoms, in contrast with the results discussed above. This result may be caused by several reasons, such as steric hindrance within the ligand shell or the inevitable migration of the ligand on the cluster surface at 80 °C. In addition, Pengo and co-workers also showed by NMR spectroscopy that the distribution of incoming ligand on the central and terminal sites depends on the properties of the ligand.98
| TS1 | Int | TS2 | Products | |
|---|---|---|---|---|
| a Energies are relative to the original cluster and free ligand. | ||||
| Au25 A | 0.56 | 0.19 | 0.76 | −0.01 |
| Au25 B | 0.62 | 0.29 | 0.65 | 0.05 |
| Au25 C | 0.78 | 0.35 | 1.15 | −0.04 |
| Au38 A | 0.68 | 0.22 | 0.80 | −0.06 |
| Au38 B | 0.61 | 0.29 | 0.76 | −0.07 |
| Au38 C | 0.85 | 0.21 | 0.85 | −0.05 |
| Au38 D | 0.59 | 0.34 | 0.89 | −0.11 |
| Au38 E | 0.59 | 0.21 | 0.80 | −0.04 |
| Au38 F | 0.75 | 0.06 | 0.56 | −0.04 |
| Au38 G | 0.54 | 0.21 | 0.65 | −0.05 |
| Au38 H | 0.91 | 0.15 | 0.77 | −0.05 |
Apart from the monothiolate ligands, nanoclusters are also reactive towards dithiol ligands. Amala Dass and co-workers systematic studied LERs with aliphatic dithiol ligands of various chain length, HS-(CH2)n-SH.104 They documented that C3 and C4 prefer interstaple coupling, and C5 and C6 are good candidates for intrastaple binding, whereas the length of C2 ligand is not enough for the bidentate binding. Aromatic dithiols have also been used for the LERs. However, they are sterically more demanding and more rigid compared to aliphatic thiols. One example is 1,1′-binaphthalene-2,2′-dithiol (BINAS), which is a chiral rigid dithiol ligand (Fig. 11 left), used to introduce chirality to achiral clusters.77,105 Several studies provide insight into the LERs on BINAS with Au25 and Au38 clusters,77,103,105–108 and from the experimental and computational results it emerges that the bidentate ligand connects two neighboring staples by interstaple coupling.103,105,108 Sels and co-workers reported the isolation of different exchange products and isomers of clusters containing one or two BINAS adsorbed PdAu24(2-PET)18 clusters.108 The investigated structure of the PdAu24(SR)16(S-BINAS)1 cluster is shown in Fig. 11, which indicates the two different sulfur environments of the cluster and marks some bond distances.103 As displayed in Fig. 11, there are several possible binding sites for BINAS at the ligand shell, illustrated with curved lines. However, considering the distances between the two S-atoms in undistorted BINAS, 4.1 Å, the interstaple binding site between a Sapex and a Score sulfur atom has the appropriate distance, 4.05 Å. Based on X-ray absorption spectroscopy (XAS), the authors also showed that the BINAS interstaple binding mode does not perturb the cluster structure.103
 | ||
| Fig. 11 3D sketch of S-BINAS (left) and MAu24(SR)16(S-BINAS)1 (right), where M = Pt, Pd. Other ligands apart from BINAS are omitted for clarity. The two types of sulfur environments are indicated with Score (also called terminal) and Sapex (also called central). Color code: Au = yellow, S = green, C = beige, H = white, Pt and Pd = blue. Adapted with permission from ref. 103. Copyright 2017 American Chemical Society. | ||
Compared with the monothiolate ligand exchange it may be easier to isolate isomers of clusters with mixed ligand shell when using dithiolate ligands. A better understanding of the preferred reactive sites of the LERs will enable the rational design of mixed ligand shell clusters.
3.3 Comparison of ligand exchange reactions of clusters in solution and on supports
LERs is a process that takes advantage of the dynamic nature of the thiol-gold surface,72 and in most cases it is performed with clusters in solution. As a further development, Truttmann and co-workers recently reported the first successful ligand exchange reaction on supported (immobilized) Au11 nanoclusters.109 In this case, Au11(PPh3)7Cl3 clusters were deposited on Al2O3, and then the dropcasts sample was exposed to a solution of thiol ligand. In such a procedure cluster–ligand combinations are possible that are not accessible through normal LERs due to solvent incompatibility. Indeed, in the study mentioned above the hydrophilic thiol ligand, L-glutathione, and the hydrophobic 2-PET (2-phenylethanethiol) ligand were used. According to previous reports, when performing ligand exchange with Au11(PPh3)7Cl3 and GSH in solution, the Au11 clusters grow to form Au25.26,110 Here, in contrast, no cluster growth has been observed as indicated from MALDI-MS after ligand exchange reaction on supported clusters. As evidenced by IR (PM-IRRAS, ATR-IR) and NMR spectroscopy, partial ligand exchange took place and the halide ligands of the parent nanocluster were primarily exchanged with thiolates.In contrast to the ligand exchange in solution, the reaction on the supported cluster did not induce a change of the core size of the nanoclusters. However, due to the hindered accessibility of the ligand on the support side of the cluster, the number of exchanged ligands was relatively low compared to typical solution phase ligand exchange reactions. In addition, similar to the functionalization in solution, fluorescence could be introduced to supported clusters by reacting them with fluorescein probe, which also provides new insight for use of the clusters in biological applications.109
4. Functionalization of gold nanoclusters induced by LERs
4.1 Chirality induced by LERs
Chirality is a geometric property of objects, which widely exists in nature from molecules over proteins111,112 to even larger structures. During the past decade, with the use of X-ray single-crystal diffraction, it has emerged that chirality is a ubiquitous property for gold nanoclusters. Because of the potential applications in sensing, catalysis, molecular recognition and so on, chiral gold nanoclusters have attracted a lot of interest.113–115The reported chiral gold nanoclusters can be categorized into three types: (i) chiral Au–S framework with achiral ligands, (ii) achiral Au–S framework with chiral ligands,114,115 and (iii) a combination of the two (chiral Au–S framework and chiral ligand). The type I chiral nanoclusters are sometimes also called intrinsically chiral nanoclusters, and some examples are Au20(SR)16, Au28(SR)20, Au38(SR)24, Au102(SR)44 and Au133(SR)52, in which all the different R groups are achiral. The chirality of type II nanoclusters is due to the ligands SR. Fundamentally, such chiral clusters can be prepared by direct synthesis using a chiral thiol.116 However, resulting from the solubility and steric effect of different ligands, direct synthesis of nanoclusters with some ligands was unsuccessful.93 Alternatively, ligand exchange is a good method to incorporate chirality or other functionality onto the gold nanoclusters.
By using ligand exchange on achiral Aum(SR)n with chiral thiols (SR*), a series of chiral Aum(SR)n−x(SR*)x clusters with chiroptical activity have become available.15,107,117,119 For example, Si and co-workers reported the ligand exchange on [Au25(PET)18−][TOA+] (in the following named as Au25) with R/S-BINAS.107 As illustrated in Fig. 12, the CD (left panel) and UV-Vis absorption (right panel) spectra of Au25 clusters were recorded before (a) and after exchange with R/S-BINAS (b/c). As expected, the Au25 cluster is optically inactive before ligand exchange. Once the cluster exchange with BINAS, intense bands at 305, 352, and 390 nm are observed in the CD spectra with opposite sign for clusters covered by the two enantiomers of BINAS. The CD signals refer to the chiral ligand but the spectra are different compared with the one of free BINAS. Furthermore, Fig. 12 reveals that the absorption spectrum of the cluster became less defined after exchange with BINAS. Apart from Au25 clusters, similar exchange with R/S-BINAS also takes place on Au38 and Au40 clusters.106 In addition, as we mentioned before, the introduction of BINAS ligand on Au38 clusters stabilized the structure against inversion (enantiomerization), probably due to the reduced flexibility of the gold–sulfur interface.77 Also, different from the LERs with monothiols, exchange with dithiol ligand reduces the number of isomers. Due to the reduction of the reaction rate after the first exchange, it is also relatively easy to obtain species with just one ligand exchanged.77,105
 | ||
| Fig. 12 (A) CD spectra of Au25 clusters before exchange with R/S-BINAS (a), after exchange with R-BINAS (b), and after exchange with S-BINAS (c); (B) UV-vis absorption spectra of the corresponding samples. Adapted with permission from ref. 107. Copyright 2009 American Chemical Society. | ||
Compared with the direct synthesis, the ligand exchange of nanoclusters may lead to a heterogeneous system with a distribution of the number of exchanged ligands when the exchange is incomplete. In order to obtain completely exchanged nanoclusters, large excess of free incoming ligand with respect to the clusters and/or repetition of the exchange reaction are necessary, with purification of the intermediate cluster after each step.
The optical activity is usually considerably stronger for the type I chiral clusters, i.e. clusters with intrinsic chirality in their Au–S framework, compared to type II chiral clusters, i.e. achiral Au–S framework with chiral ligands. The absorption spectrum of a given cluster is roughly ligand independent. In contrast, when the chirality of the nanoclusters is due to the ligand, the chiroptical properties will be ligand-dependent,107 which offer various possibilities to influence the optical response. Fig. 13 shows circular dichroism (CD) spectra of Au25 and Au38 clusters stabilized with three different chiral R* groups. All these chiral Au25(SR*)18 nanoclusters reveal the typical UV/Vis absorption spectrum of Au25, but their CD signals show differences depending on the R* groups (Fig. 13 left panel).117 A similar phenomenon was also observed for Au38(SR*)24 nanoclusters with different chiral thiolate ligands (Fig. 13 right panel).118 This ligand-dependnce provides a possibility to engineer the optical characteristics of a given gold nanocluster.
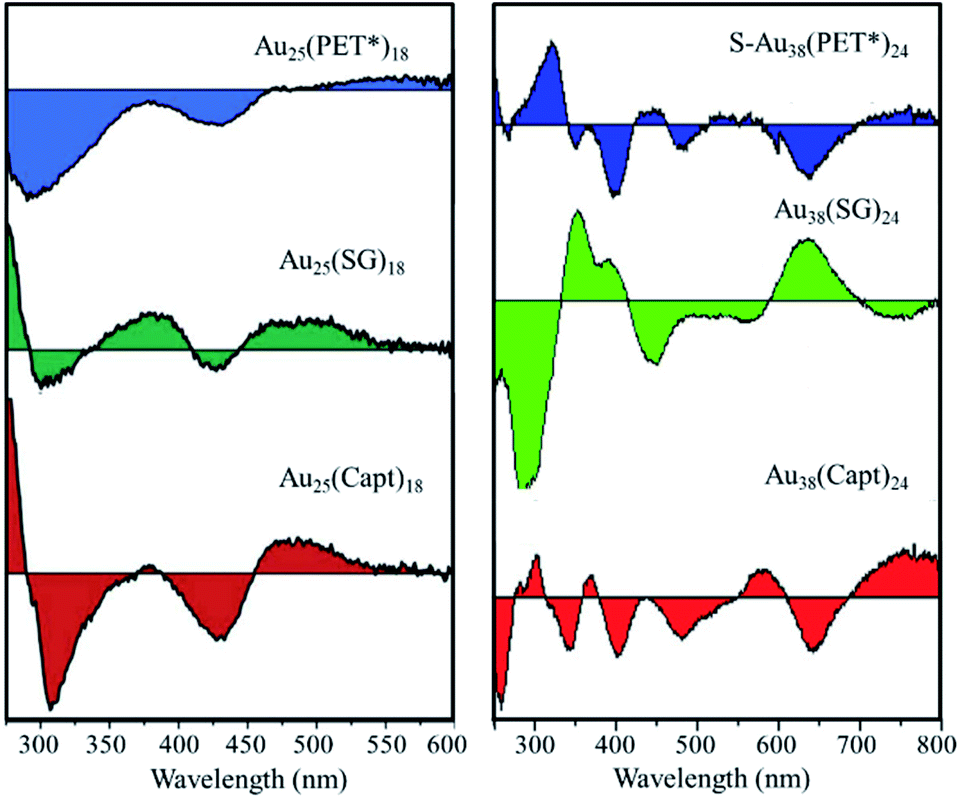 | ||
| Fig. 13 Effect of the chiral ligand type on the CD spectra of Au25(SR*)18 and Au38(SR*)24. PET* = S-phenylpropane-1-thiol, SG = glutathione, Capt = captopril. Left panel: adapted with permission from ref. 117. Copyright 2012 Royal Society of Chemistry. Right panel: adapted with permission from ref. 118. Copyright 2014 Wiley-VCH. | ||
In addition, the chiral ligand R-BINAS was also used to amplify the enantiomeric excess of Au38(SR)24 at 70 °C.16 In a mixture containing the two enantiomers of the Au38 cluster and the corresponding clusters containing one R-BINAS in their ligand shell (diastereomers), the fraction of anticlockwise (A) clusters increases with time at the expense of clockwise (C) clusters (Fig. 14B). For the experiment shown in Fig. 14B the initial sample was prepared with high fraction of C-Au38. The sample was then heated to 70 °C and the HPL chromatograms were recorded as a function of time. The fractions of different species were determined from the HPL chromatograph and plotted as function as time (Fig. 14B), showing the increase of A-Au38. The proposed mechanism of the process is shown in Fig. 14A. This dynamic inversion is due to the diastereoselective intercluster exchange of R-BINAS between chiral clusters and the fast racemization of Au38(2-PET)24. This example shows that the dynamic nature of these clusters can be used as a benefit. Overall, the ligand exchange reaction with chiral ligands will continue to add new impetus to the field of chiral nanomaterials and its applications.
 | ||
| Fig. 14 Amplification of the enantiomeric excess after introducing chiral R-BINAS to the Au38 nanoclusters. (A) Scheme for the dynamic inversion system, (B) evolution of different clusters species under 70 °C as a function of time. The nomenclature of the four different species assigned as follow: (A-38, 24, R-0) corresponded to A-Au38(2-PET)24; Adapted with permission from ref. 16. Copyright 2020 Nature Research. | ||
4.2 LERs induced size transformation of clusters
LERs is a process that takes advantage of the dynamic nature of the thiol-gold interface,72 and in most cases it does not cause any change in size or structure of the cluster, only replacing one capping ligand by another one.94 During the past decade, due to the development of precise synthesis methods and structure determination by X-ray single-crystal diffraction numerous new nanoclusters were reported. LERs became an important new methodology for controlling the size and structure of nanoclusters, and the process was named ligand-exchange-induced size/structure transformation (LEIST for short), which was first proposed by the Jin's group.120Up to now, most of the reported LEIST work focuses on PPh3-stabilized121,122 and thiolate-protected Au nanoclusters.123–125 However, the driving forces for LEIST is quite different for these two kinds of Au nanoclusters.12 First, for LEIST of PPh3-stabilized Au nanoclusters, exchanging the phosphine ligands by thiolate ligands changes the surface bonding completely (from Au–P to Au–S). The different coordination modes, Au–P vs. Au–S, lead to different surface motifs and drives the structural transformation of the cluster.12,121 Second, for LEIST of thiolate-protected Au nanoclusters, the new thiolates would not significantly alter the gold–ligand coordination; however, due to the distinctly different physical–chemical properties (e.g., size, rigidity, bulkiness, interactions etc.) of different thiolate ligands, the original structures may not be the most stable for the ligand-exchanged nanoclusters, and thus the structural transformation takes place.12 In the following we mainly focus on the LEIST on thiolate-protected Au nanoclusters. Depending on the size or structural change of the thiolate-protected Au nanoclusters resulting from the LEIST, this process been classified into three groups.
(I) Transformation between structural isomers without size change. For example, Au28(SR)20 nanocluster reversibly changes its structure upon ligand exchange between R = c-C6H11 and R = Ph-tBu at elevated temperatures (e.g., 80 °C) as shown in Fig. 15.59 The structures of these two nanoclusters are remarkable different. Au28(SPhtBu)20 contains a FCC Au20 kernel capped by four Au2(SR)3 staple motifs and eight bridging thiolates, whereas Au28(S–c-C6H11)20 adopts a more lose structure with a FCC Au20 kernel plus two Au1(SR)2, two Au3(SR)4, and eight bridging SR staple-like structures. DFT calculations revealed that the origin of reversible isomerization lay in the thiolate ligand's carbon tail structure, which was found to dictate the isomer's stability.59 In LEIST the isomerization of clusters is rare.
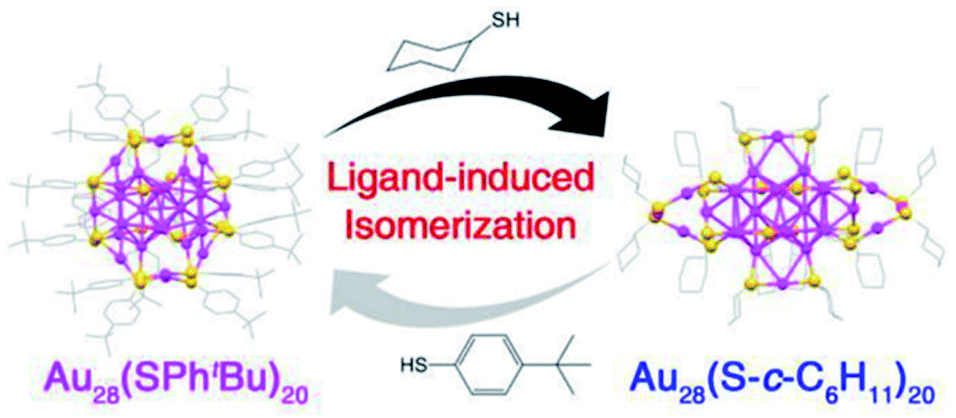 | ||
| Fig. 15 Ligand-exchange induced transformation between Au28(SPh-tBu)20 and Au28(S–c-C6H11)20 nanoclusters. Color codes: purple sphere, Au; yellow sphere, S; gray sphere, C. For clarity, all H atoms are omitted. Adapted with permission from ref. 59. Copyright 2016 American Chemistry Society. | ||
(II) Transformations from a smaller to a larger nanocluster.26,58,61,110,125–127 An interesting example is the transformation of Au25(PET)18 into Au28(TBBT)20.58 Au25(PET)18 is probably the most studied thiolate-protected nanocluster, due to its prototypical character. At 80 °C and in large excess of TBBT the Au25(PET)18 cluster is transformed into Au28(TBBT)20. It is worth to mention that the resulting Au28(TBBT)20 cluster is chiral, and the origin of chirality is primarily rooted in the rotating arrangement of the four dimeric staples as well as the arrangement of the bridging thiolates as we mentioned before. Moreover, the pair of enantiomers of Au28(TBBT)20 can be separated by chiral-HPLC.58 Another example is the transformation of Au11 to Au25 upon ligand exchange with GSH.26,110
(III) Transformations from a larger to a smaller nanocluster.12,64,120,123–125,128,129 For instance, highly stable Au144(SC2H4Ph)60 reacted with thiophenol, HSPh, to form a different 99 atom cluster species Au99(SPh)42.128 In addition, Zeng and co-worker reported that, by LEIST, the very stable and widely investigated Au38(SC2H4Ph)24 cluster was transformed to Au36(SPh-tBu)24 as shown in Fig. 16A.120 To unravel details of the intriguing one-size-to-another size transformation, Zeng carried out time-dependent mass spectrometry and optical spectroscopy analyses (Fig. 16B) and found a remarkable disproportionation in the transformation of rod-like biicosahedral Au38(SC2H4Ph)24 to tetrahedral Au36(SPh-tBu)24. From the evolution of the mass spectra and corresponding UV curves, the reaction pathway can be roughly divided into four stages (Fig. 16C): (i) (0–5 min) ligand exchange reaction occurs between PET and TBBT; (ii) (10–15 min) ligand exchange reaction continues leading to TBBT-triggered structural distortion of Au38 with an optical feature at 550 nm (Fig. 16D); (iii) (20–60 min) critical stage for the disproportionation of Au38 to Au36 and Au40, and (iv) size conversion of Au40 to Au36 evidenced by the decrease and complete disappearance of the Au40 peak in time-dependent mass spectra. Recently, other LEIST, such as transformations from Au23(S–c-C6H11)16 to Au36(TBBT)24 and from Au329(PET)84 to Au279(SPh-tBu)84 also revealed similar pathways but without disproportionation. In these cases the conversion through ligand exchange followed by the size focusing ultimately lead to size growth.127,129
 | ||
| Fig. 16 (A) Ligand-exchange induced transformation from Au38(SC2H4Ph)24 to Au36(SPhtBu)24. (B) Time-dependent ESI-MS and UV-vis spectra of the transformation. (C) Reaction pathway for the transformation. (D) Kinetics (monitored by absorbance at 550 nm) for the transformation. Adapted with permission from ref. 120. Copyright 2013 American Chemical Society. | ||
Apart from the ireversible transformation mentioned above, Amala Dass and co-workers presented the first reversible interconversion between two nanomolecules Au36(SPhX)24, (where X = –H or -tBu) and Au30(S-tBu)18 as illustrated in Fig. 17.12,125 In this case, the gold core converted between bicuboctahedral Au20 and 4-fused cuboctahedron Au24, and the staple arrangement of these two cluster surfacers was also different. More interesting, the reversible conversion easily takes place under the same thermochemical conditions with different thiol ligands. Later, Wu and co-workers achieved the interconversion among Au44(TBBT)28, Au44(2,4-DMBT)26, and Au43(S–c-C6H11)25 nanoclusters by the ligand exchange,130,131 and they also investigated the thermostability of these nanoclusters. As monitored by time-dependent optical absorption, the Au44(2,4-DMBT)26 cluster was less thermostable than Au44(TBBT)28, and much more stable than Au43(S–c-C6H11)25 at 80 °C.130 The interconversion not only offer many possibilities to obtain new nanoclusters but also leads to valuable insight into the inherent influence of the ligand on the composition and atomic structure of thiolate-protected gold clusters.
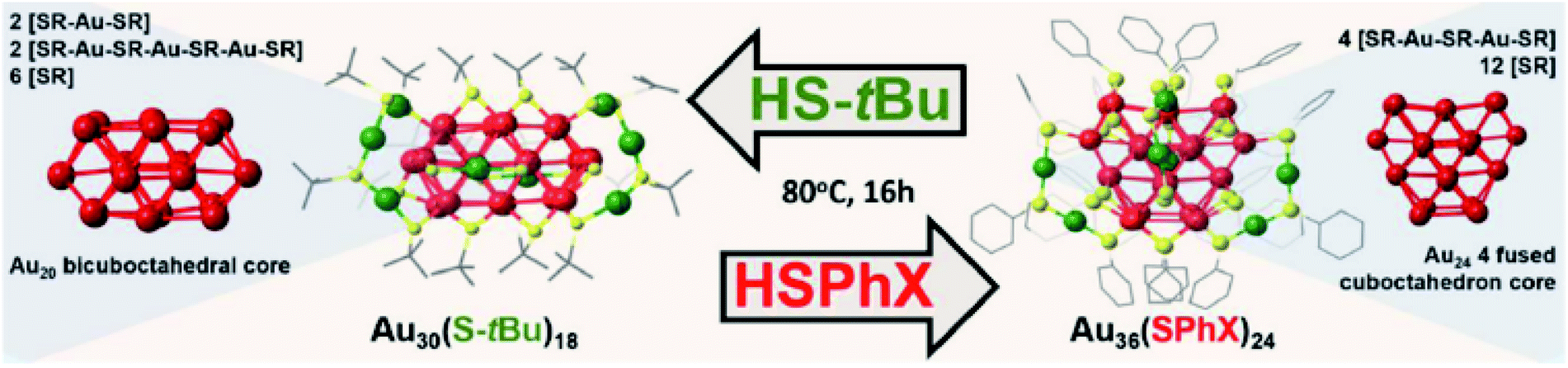 | ||
| Fig. 17 Schematic of the conditions for the interconversion between Au30(S-tBu)18 and Au36(SPhX)24. Reaction take place under the same thermochemical conditions with different thiol ligands (thiophenol and tert-butyl). Left panel: crystal structure of Au30(S-tBu)18, right panel: crystal structure of Au36(SPhX)24. Color code: Au core atom: red, Au staple atom: green, sulphur: yellow, carbon: gray. Hydrogen atoms has been eliminated for clarity. Adapted with permission from ref. 125. Copyright 2017 American Chemistry Society. | ||
Focussing on the experimental conditions of the LEIST, mentioned above, it seems that the conditions necessary for LEIST to take place are large excess of incoming ligand, normally more than 100 times compared to the endogenous ligand, and elevated temperatures.124 In addition, it was argued that the incoming thiol must be significantly different from the endogenous thiol for the transformation to happen.61 However, we reported a new transformation of Au25(SR)18 into Au28(SR)21 by LER with a chiral ligand (R- or S-phenylpropane-1-thiol) at mild conditions, i.e. at room temperature and with low thiol excess (with incoming to outgoing ligand molar ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).15 In this case, the van der Waals interactions within the ligand shell and the “bulkiness” of the methyl are the main factors driving this process. In summary, LEIST provides new and exciting avenues to explore novel atom-precise nanoclusters and opens a new strategy to investigate the size growth of nanoclusters.
:1).15 In this case, the van der Waals interactions within the ligand shell and the “bulkiness” of the methyl are the main factors driving this process. In summary, LEIST provides new and exciting avenues to explore novel atom-precise nanoclusters and opens a new strategy to investigate the size growth of nanoclusters.
4.3 LERs induced organic/aqueous phase transfer of clusters
There are many types of ligands that can be used to stabilize nanoparticles or nanoclusters, as shown in Fig. 18.132 Depending on the solubility of the surface ligand, one can distinguish hydrophobic and hydrophilic clusters. However, for some (biological) applications, the hydrophobic clusters need to be dissolved in aqueous solutions. Also hydrophilic clusters sometimes need to made accessible to reactions in organic solvent. In this situation, phase transfer will be the first option to relocate the clusters to the desired phase. | ||
| Fig. 18 A nanoparticle (nanocluster) stabilized with different hydrophobic (top panel) and hydrophilic (bottom panel) ligand molecules (skeletal drawings or space filling models were used to represent the ligands). Adapted with permission from ref. 132. Copyright 2010 Royal Society. | ||
In general, for phase transfer processes, reagents such as tetraoctylammonium bromide,133,134 (−)-1R,2S–N-dodecyl-N-methylephedrinium bromide135 and others have been successfully used. These chemical reagents form an additional molecular layer on the clusters and thus change their surface properties. This strategy was mainly used for the phase transfer from aqueous to organic phase.133–137 In addition to this strategy, ligand exchange has also been utilized as an important strategy for the phase transfer. For instance, when hydrophobic Au11(PPh3)7Cl3 reacted with GSH ligand in solution, the Au11 clusters grew to form water-soluble Au25 nanoclusters,26,110 which is a good evidence for ligand exchange. In addition, Zheng and co-workers reported the crystal structure of intermetallic nanocluster Au24Ag20(2-SPy)4(PA)20Cl2 and also investigated the phase transfer process after performing ligand exchange with mercaptosuccinic acid (MSA),138 leading to transfer of the Au24Ag20 cluster from DCM to water phase. Other reported cases of phase transfer induced by ligand exchange reactions, include ligands such as (phenylacetylene) PAH139 and Tiopronin.140 The latter ligand also introduced cancer therapy functions to the cluster.
It emerges from the cases mentioned above, that the phase transfer from aqueous to organic phase relies on phase transfer reagents or ligand modification, and in many cases the core size of the cluster is maintained. However, the inverse process, transfer from organic to aqueous phase, mostly relies on the ligand exchange reactions. Furthermore, the phosphine-stabilized clusters are more prone to be transferred to another phase after reaction with thiol ligands. For the choice of the ligand used for the phase transfer, three considerations have to be made: (i) the affinity to the metal core of new ligand compared with the original one; (ii) the solubility of the new ligand in the target solvent, and (iii) the capability of the ligand to maintain the core size of the cluster after phase transfer.
4.4 Florescence induced by LERs
Fluorescent gold nanoclusters have been widely used for biological applications such as cell identification, to study interaction, differentiation, and tracking.109,142–145 Compared with the more traditional QDs, fluorescent gold nanoclusters have decent quantum yield, excellent biocompatibility, good photostability, and lower cytotoxicity. The clusters had negligible influence on the cell viability at the considered dose.146 However, because of the synthesis method and the solubility of ligands, the diversity of the fluorescent gold nanoclusters has been limited.LER is a very efficient strategy for preparing fluorescent gold nanoclusters. As one of the most intuitive cases, the non-fluorescent gold nanoclusters (AuNC@DDAB) can be converted to brightly red emitting nanoclusters (AuNC@DHLA) through an elegant ligand exchange reaction with dihydrolipoic acid (DHLA) as shown in Fig. 19.141 In this system, the DDAB-stabilized gold nanoparticles (AuNP@DDAB) are etched by the addition of Au precursors (HAuCl4 or AuCl3) to smaller nanoclusters (AuNC@DDAB), and the organic soluble and hydrophobic AuNC@DDAB become water soluble upon ligand exchange with dihydrolipoic acid (AuNC@DHLA). The AuNP@DDAB solution shows red color due to surface plasmon absorption, which is absent to AuNC@DDAB and AuNC@DHLA. More interesting, after ligand exchange with DHLA, the AuNC@DHLA solution shows strong red photoluminescence as shown in the Fig. 19. Here, the phase transfer and addition of fluorophore has been achieved together by the LERs.
 | ||
| Fig. 19 Strategy to fabricate hydrophilic fluorescent Au nanoclusters. Size distribution of three different Au nanoclusters extracted from 100 particles. Pictures of particle solutions under daylight and UV excitation illustrated respectively. Adapted with permission from ref. 141. Copyright 2009 American Chemical Society. | ||
In addition, Xu et al. reported ligand exchange by GSH and N-acetylcysteine (NAC) on AuNAC@Ag. The products showed maximum 20-fold fluorescence enhancement.147 During this ligand exchange process, silver ions and GSH have synergistic effects and the PL enhancement was found to be proportional to the concentration of GSH. This fluorescence enhancement was also used for selective imaging of intracellular glutathione.147 Besides dissolved clusters, the fluorescence can also be introduced to supported nanoclusters by using ligand exchange on the immobilized sample, as we mentioned above.109
5 Conclusions
In conclusion, ligand exchange reaction plays an important role in the field of nanoclusters. As one of the post-synthesis methods for the modification and functionalization of the nanoclusters, the process is enabled by the flexibility of the gold–sulfur interface. Experimental and computational investigations show that LERs between clusters and free monothiols in solution starts preferably at the terminal SR groups of the staple motifs, which are linked to the gold core, via an associative SN2-like mechanism. The process takes place in solution phase but also on immobilized Au nanoclusters. Understanding of the preferred reactive sites of the process will help us engineer precise clusters with mixed ligand shell. Compared with the monothiolate ligand exchange, dithiolate ligands may offer an easier way to obtain precise clusters with mixed ligand shell. Intercluster ligand exchange is also an important property of thiolate-protected metal clusters and has to be considered whenever clusters with different ligands are in the same solution. Despite the considerable effort made, the microscopic details of intercluster ligand exchange remain obscure.Ligand exchange reactions are in general easy to perform and offer a great potential to introduce or amplify properties of the clusters, such as introduction of chirality to achiral nanoclusters and the amplification of optical activity and enantiomeric excess, size transformation of the cluster, phase transfer, and the addition of fluorescent groups. Thus, ligand exchange reactions will continue to play an important role in the future and a better understanding of the process will further increase the potential of this method.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TB acknowledges the generous support of the Swiss National Science Foundation (grant 200020_192232) and the University of Geneva. YW thanks the China Scholarship Council fellowship (201706450070).References
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- X.-R. Song, N. Goswami, H.-H. Yang and J. Xie, Analyst, 2016, 141, 3126–3140 RSC.
- S. Maity, D. Bain and A. Patra, Nanoscale, 2019, 11, 22685–22723 RSC.
- R. Dinkel, W. Peukert and B. Braunschweig, J. Phys.: Condens. Matter, 2017, 29, 133002 CrossRef PubMed.
- M.-M. Xu, Q. Chen, L.-H. Xie and J.-R. Li, Coord. Chem. Rev., 2020, 421, 213421 CrossRef CAS.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- Y. Niihori, S. Hossain, B. Kumar, L. V. Nair, W. Kurashige and Y. Negishi, APL Mater., 2017, 5, 053201 CrossRef.
- G. Salassa, A. Sels, F. Mancin and T. Bürgi, ACS Nano, 2017, 11, 12609–12614 CrossRef CAS PubMed.
- Y. Wang, B. Nieto-Ortega and T. Bürgi, Chem. Commun., 2019, 55, 14914–14917 RSC.
- Y. Wang, B. Nieto-Ortega and T. Bürgi, Nat. Commun., 2020, 11, 4562 CrossRef CAS PubMed.
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- T. Tsukuda, Bull. Chem. Soc. Jpn., 2012, 85, 151–168 CrossRef CAS.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802, 10.1039/C39940000801.
- T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2000, 104, 2630–2641 CrossRef CAS.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- T. G. Schaaff, M. N. Shafigullin, J. T. Khoury, I. Vezmar, R. L. Whetten, W. G. Cullen, P. N. First, C. Gutiérrez-Wing, J. Ascensio and M. J. Jose-Yacamán, J. Phys. Chem. B, 1997, 101, 7885–7891 CrossRef CAS.
- R. L. Donkers, D. Lee and R. W. Murray, Langmuir, 2004, 20, 1945–1952 CrossRef CAS.
- J. B. Tracy, M. C. Crowe, J. F. Parker, O. Hampe, C. A. Fields-Zinna, A. Dass and R. W. Murray, J. Am. Chem. Soc., 2007, 129, 16209–16215 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- H. Tsunoyama, P. Nickut, Y. Negishi, K. Al-Shamery, Y. Matsumoto and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 4153–4158 CrossRef CAS.
- N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu and T. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610 CrossRef CAS PubMed.
- M. Zhu, E. Lanni, N. Garg, M. E. Bier and R. Jin, J. Am. Chem. Soc., 2008, 130, 1138–1139 CrossRef CAS PubMed.
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC.
- H. Qian, M. Zhu, U. N. Andersen and R. Jin, J. Phys. Chem. A, 2009, 113, 4281–4284 CrossRef CAS PubMed.
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS PubMed.
- A. Das, T. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 18264–18267 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, G. Li and R. Jin, Chem. Mater., 2014, 26, 2635–2641 CrossRef CAS.
- G. Li, C. Zeng and R. Jin, J. Am. Chem. Soc., 2014, 136, 3673–3679 CrossRef CAS PubMed.
- H. Qian and R. Jin, Nano Lett., 2009, 9, 4083–4087 CrossRef CAS PubMed.
- H. Qian, Y. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 696–700 CrossRef CAS PubMed.
- S. Knoppe, J. Boudon, I. Dolamic, A. Dass and T. Bürgi, Anal. Chem., 2011, 83, 5056–5061 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 238 CrossRef CAS PubMed.
- M. Zhou, T. Higaki, Y. Li, C. Zeng, Q. Li, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2019, 141, 19754–19764 CrossRef CAS PubMed.
- A. Sels, R. Azoulay, W. J. Buma, M. A. J. Koenis, V. P. Nicu and T. Bürgi, J. Phys. Chem. C, 2019, 123, 22586–22594 CrossRef CAS.
- Z. Wu, M. Wang, J. Yang, X. Zheng, W. Cai, G. Meng, H. Qian, H. Wang and R. Jin, Small, 2012, 8, 2028–2035 CrossRef CAS PubMed.
- T. Shu, J. Wang, L. Su and X. Zhang, Anal. Chem., 2016, 88, 11193–11198 CrossRef CAS PubMed.
- S. K. Katla, J. Zhang, E. Castro, R. A. Bernal and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 75–82 CrossRef CAS PubMed.
- D. Yang, G. Yang, S. Gai, F. He, G. An, Y. Dai, R. Lv and P. Yang, Nanoscale, 2015, 7, 19568–19578 RSC.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- B. K. Teo and H. Zhang, Coord. Chem. Rev., 1995, 143, 611–636 CrossRef CAS.
- H. Häkkinen, M. Walter and H. Grönbeck, J. Phys. Chem. B, 2006, 110, 9927–9931 CrossRef PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- Y. Pei, Y. Gao and X. C. Zeng, J. Am. Chem. Soc., 2008, 130, 7830–7832 CrossRef CAS PubMed.
- D.-e. Jiang, W. Chen, R. L. Whetten and Z. Chen, J. Phys. Chem. C, 2009, 113, 16983–16987 CrossRef CAS.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-e. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS PubMed.
- A. Das, T. Li, K. Nobusada, Q. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2012, 134, 20286–20289 CrossRef CAS PubMed.
- A. Das, T. Li, G. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, Nanoscale, 2014, 6, 6458–6462 RSC.
- Y. Song, S. Wang, J. Zhang, X. Kang, S. Chen, P. Li, H. Sheng and M. Zhu, J. Am. Chem. Soc., 2014, 136, 2963–2965 CrossRef CAS PubMed.
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145–3149 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2014, 136, 11922–11925 CrossRef CAS PubMed.
- L. Xiong, S. Yang, X. Sun, J. Chai, B. Rao, L. Yi, M. Zhu and Y. Pei, J. Phys. Chem. C, 2018, 122, 14898–14907 CrossRef CAS.
- C. Zeng, H. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon, R. N. Barnett, R. L. Whetten, U. Landman and R. Jin, Angew. Chem., Int. Ed., 2012, 51, 13114–13118 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 8710–8713 CrossRef CAS PubMed.
- Y. Chen, C. Zeng, C. Liu, K. Kirschbaum, C. Gayathri, R. R. Gil, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2015, 137, 10076–10079 CrossRef CAS PubMed.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed.
- O. Lopez-Acevedo, J. Akola, R. L. Whetten, H. Grönbeck and H. Häkkinen, J. Phys. Chem. C, 2009, 113, 5035–5038 CrossRef CAS.
- E. Pensa, E. Cortés, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benítez, A. A. Rubert and R. C. Salvarezza, Acc. Chem. Res., 2012, 45, 1183–1192 CrossRef CAS PubMed.
- T. Bürgi, Nanoscale, 2015, 7, 15553–15567 RSC.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- S. Knoppe, I. Dolamic and T. Bürgi, J. Am. Chem. Soc., 2012, 134, 13114–13120 CrossRef CAS PubMed.
- S. Malola and H. Häkkinen, J. Am. Chem. Soc., 2019, 141, 6006–6012 CrossRef CAS PubMed.
- N. Barrabés, B. Zhang and T. Bürgi, J. Am. Chem. Soc., 2014, 136, 14361–14364 CrossRef PubMed.
- S. Knoppe and T. Bürgi, Phys. Chem. Chem. Phys., 2013, 15, 15816–15820 RSC.
- B. Varnholt, P. Oulevey, S. Luber, C. Kumara, A. Dass and T. Bürgi, J. Phys. Chem. C, 2014, 118, 9604–9611 CrossRef CAS.
- B. Varnholt, I. Dolamic, S. Knoppe and T. Bürgi, Nanoscale, 2013, 5, 9568–9571 RSC.
- L. Beqa, D. Deschamps, S. Perrio, A.-C. Gaumont, S. Knoppe and T. Bürgi, J. Phys. Chem. C, 2013, 117, 21619–21625 CrossRef CAS.
- A. C. Templeton, M. J. Hostetler, C. T. Kraft and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 1906–1911 CrossRef CAS.
- Y. Song and R. W. Murray, J. Am. Chem. Soc., 2002, 124, 7096–7102 CrossRef CAS PubMed.
- R. L. Donkers, Y. Song and R. W. Murray, Langmuir, 2004, 20, 4703–4707 CrossRef CAS PubMed.
- R. Guo, Y. Song, G. Wang and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 2752–2757 CrossRef CAS PubMed.
- M. J. Hostetler, A. C. Templeton and R. W. Murray, Langmuir, 1999, 15, 3782–3789 CrossRef CAS.
- N. Yan, N. Xia and Z. Wu, Small, 2020, 2000609 CrossRef PubMed.
- V. Rojas-Cervellera, L. Raich, J. Akola and C. Rovira, Nanoscale, 2017, 9, 3121–3127 RSC.
- C. L. Heinecke, T. W. Ni, S. Malola, V. Mäkinen, O. A. Wong, H. Häkkinen and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 13316–13322 CrossRef CAS PubMed.
- Y. Niihori, W. Kurashige, M. Matsuzaki and Y. Negishi, Nanoscale, 2013, 5, 508–512 RSC.
- M. Kluenker, M. Mondeshki, M. N. Tahir and W. Tremel, Langmuir, 2018, 34, 1700–1710 CrossRef CAS PubMed.
- T. W. Ni, M. A. Tofanelli, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2014, 53, 6500–6502 CrossRef CAS PubMed.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 CrossRef CAS PubMed.
- E. S. Shibu, M. A. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 CrossRef CAS.
- G. H. Woehrle, L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 2005, 127, 2172–2183 CrossRef CAS PubMed.
- L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 1997, 119, 12384–12385 CrossRef CAS.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 RSC.
- C. A. Fields-Zinna, J. F. Parker and R. W. Murray, J. Am. Chem. Soc., 2010, 132, 17193–17198 CrossRef CAS PubMed.
- P. Pengo, C. Bazzo, M. Boccalon and L. Pasquato, Chem. Commun., 2015, 51, 3204–3207 RSC.
- S. Hossain, W. Kurashige, S. Wakayama, B. Kumar, L. V. Nair, Y. Niihori and Y. Negishi, J. Phys. Chem. C, 2016, 120, 25861–25869 CrossRef CAS.
- A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2015, 119, 20179–20187 CrossRef CAS.
- S. Knoppe, R. Azoulay, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2012, 134, 20302–20305 CrossRef CAS PubMed.
- A. Fernando and C. M. Aikens, J. Phys. Chem. C, 2016, 120, 14948–14961 CrossRef CAS.
- A. Sels, G. Salassa, S. Pollitt, C. Guglieri, G. Rupprechter, N. Barrabés and T. Bürgi, J. Phys. Chem. C, 2017, 121, 10919–10926 CrossRef CAS.
- V. R. Jupally, R. Kota, E. V. Dornshuld, D. L. Mattern, G. S. Tschumper, D.-e. Jiang and A. Dass, J. Am. Chem. Soc., 2011, 133, 20258–20266 CrossRef CAS PubMed.
- B. Molina, A. Sánchez-Castillo, S. Knoppe, I. L. Garzón, T. Bürgi and A. Tlahuice-Flores, Nanoscale, 2013, 5, 10956–10962 RSC.
- S. Knoppe, A. C. Dharmaratne, E. Schreiner, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2010, 132, 16783–16789 CrossRef CAS PubMed.
- S. Si, C. Gautier, J. Boudon, R. Taras, S. Gladiali and T. Bürgi, J. Phys. Chem. C, 2009, 113, 12966–12969 CrossRef CAS.
- A. Sels, N. Barrabés, S. Knoppe and T. Bürgi, Nanoscale, 2016, 8, 11130–11135 RSC.
- V. Truttmann, C. Herzig, I. Illes, A. Limbeck, E. Pittenauer, M. Stöger-Pollach, G. Allmaier, T. Bürgi, N. Barrabés and G. Rupprechter, Nanoscale, 2020, 12, 12809–12816 RSC.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS PubMed.
- S. Mason, Trends Pharmacol. Sci., 1986, 7, 20–23 CrossRef CAS.
- J. L. Bada, Nature, 1995, 374, 594–595 CrossRef CAS PubMed.
- C. Gautier and T. Bürgi, ChemPhysChem, 2009, 10, 483–492 CrossRef CAS PubMed.
- H. Yao, Prog. Nat. Sci.: Mater. Int., 2016, 26, 428–439 CrossRef CAS.
- C. Zeng and R. Jin, Chem. - Asian J., 2017, 12, 1839–1850 CrossRef CAS PubMed.
- M. Zhu, H. Qian, X. Meng, S. Jin, Z. Wu and R. Jin, Nano Lett., 2011, 11, 3963–3969 CrossRef CAS PubMed.
- S. Kumar and R. Jin, Nanoscale, 2012, 4, 4222–4227 RSC.
- Q. Xu, S. Kumar, S. Jin, H. Qian, M. Zhu and R. Jin, Small, 2014, 10, 1008–1014 CrossRef CAS PubMed.
- S. Knoppe, N. Kothalawala, V. R. Jupally, A. Dass and T. Bürgi, Chem. Commun., 2012, 48, 4630–4632 RSC.
- C. Zeng, C. Liu, Y. Pei and R. Jin, ACS Nano, 2013, 7, 6138–6145 CrossRef CAS PubMed.
- M.-B. Li, S.-K. Tian, Z. Wu and R. Jin, Chem. Mater., 2016, 28, 1022–1025 CrossRef CAS.
- S. Wang, H. Abroshan, C. Liu, T.-Y. Luo, M. Zhu, H. J. Kim, N. L. Rosi and R. Jin, Nat. Commun., 2017, 8, 848 CrossRef.
- P. R. Nimmala, S. Theivendran, G. Barcaro, L. Sementa, C. Kumara, V. R. Jupally, E. Apra, M. Stener, A. Fortunelli and A. Dass, J. Phys. Chem. Lett., 2015, 6, 2134–2139 CrossRef CAS.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS PubMed.
- A. Dass, T. C. Jones, S. Theivendran, L. Sementa and A. Fortunelli, J. Phys. Chem. C, 2017, 121, 14914–14919 CrossRef CAS.
- Z. Gan, J. Chen, J. Wang, C. Wang, M.-B. Li, C. Yao, S. Zhuang, A. Xu, L. Li and Z. Wu, Nat. Commun., 2017, 8, 14739 CrossRef CAS PubMed.
- M. P. Maman, A. S. Nair, H. Cheraparambil, B. Pathak and S. Mandal, J. Phys. Chem. Lett., 2020, 11, 1781–1788 CrossRef CAS PubMed.
- P. R. Nimmala and A. Dass, J. Am. Chem. Soc., 2014, 136, 17016–17023 CrossRef CAS PubMed.
- S. K. Eswaramoorthy, N. A. Sakthivel and A. Dass, J. Phys. Chem. C, 2019, 123, 9634–9639 CrossRef CAS.
- H. Dong, L. Liao and Z. Wu, J. Phys. Chem. Lett., 2017, 8, 5338–5343 CrossRef CAS PubMed.
- L. Liao, S. Zhuang, C. Yao, N. Yan, J. Chen, C. Wang, N. Xia, X. Liu, M.-B. Li, L. Li, X. Bao and Z. Wu, J. Am. Chem. Soc., 2016, 138, 10425–10428 CrossRef CAS PubMed.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- M. A. Habeeb Muhammed and T. Pradeep, J. Cluster Sci., 2009, 20, 365–373 CrossRef CAS.
- J. W. Padelford, T. Wang and G. Wang, ChemElectroChem, 2016, 3, 1201–1205 CrossRef CAS.
- S. Knoppe, O. A. Wong, S. Malola, H. Häkkinen, T. Bürgi, T. Verbiest and C. J. Ackerson, J. Am. Chem. Soc., 2014, 136, 4129–4132 CrossRef CAS PubMed.
- H. Yao and M. Iwatsu, Langmuir, 2016, 32, 3284–3293 CrossRef CAS PubMed.
- H. Yao and S. Tsubota, Chem. Phys., 2017, 493, 149–156 CrossRef CAS.
- Y. Wang, H. Su, C. Xu, G. Li, L. Gell, S. Lin, Z. Tang, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2015, 137, 4324–4327 CrossRef CAS PubMed.
- L. Ren, P. Yuan, H. Su, S. Malola, S. Lin, Z. Tang, B. K. Teo, H. Häkkinen, L. Zheng and N. Zheng, J. Am. Chem. Soc., 2017, 139, 13288–13291 CrossRef CAS PubMed.
- X. Zan, Q. Li, Y. Pan, D. J. Morris, P. Zhang, P. Li, H. Yu and M. Zhu, ACS Appl. Nano Mater., 2018, 1, 6773–6781 CrossRef CAS.
- C.-A. J. Lin, T.-Y. Yang, C.-H. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J.-L. Shen, H.-H. Wang, H.-I. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395–401 CrossRef CAS PubMed.
- Y. Bai, T. Shu, L. Su and X. Zhang, Crystals, 2020, 10, 357 CrossRef.
- Y. Guo, H. T. N. N. Amunyela, Y. Cheng, Y. Xie, H. Yu, W. Yao, H.-W. Li and H. Qian, Food Chem., 2021, 335, 127657 CrossRef CAS PubMed.
- Y. Zheng, L. Lai, W. Liu, H. Jiang and X. Wang, Adv. Colloid Interface Sci., 2017, 242, 1–16 CrossRef CAS PubMed.
- L. Shang, F. Stockmar, N. Azadfar and G. U. Nienhaus, Angew. Chem., Int. Ed., 2013, 52, 11154–11157 CrossRef CAS PubMed.
- Y. Wang, L. Hu, L. Li and J.-J. Zhu, Journal of Analysis and Testing, 2017, 1, 13 CrossRef.
- X. Hu, Y. Zheng, J. Zhou, D. Fang, H. Jiang and X. Wang, Chem. Mater., 2018, 30, 1947–1955 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |