
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile photochemical strategy for the synthesis of high-performance amorphous MoS2 nanoparticles†
Haifeng
Sun
a,
Manlin
Yang
a,
Shan
Pu
a,
Lichen
Gou
b,
Caizhi
Lv
 b,
Juan
He
b,
Xiandeng
Hou
*ab and
Kailai
Xu
*a
b,
Juan
He
b,
Xiandeng
Hou
*ab and
Kailai
Xu
*a
aKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: houxd@scu.edu.cn; xukailai@scu.edu.cn
bAnalytical & Testing Center, Sichuan University, Chengdu, Sichuan 610064, China
First published on 26th March 2021
Abstract
It is difficult to avoid the formation of polysulfides by traditional chemical methods, and the synthesis of high purity amorphous MoS2 nanomaterials under ambient conditions is still a challenging task. Here we present a new and facile photochemical strategy for the synthesis of amorphous MoS2 nanomaterials, which is achieved by irradiating a mixed solution containing ammonium molybdate, formic acid and sodium sulfide simply with a Xe lamp for 3 min. The mechanism study reveals that the key step in this synthesis is the photolysis of formic acid to produce free radicals which can rapidly reduce Mo6+ to Mo4+, which then combines with S2− to form MoS2 and inhibits the formation of S–S2− by preventing S2− from participating in the reduction reaction. In addition, the results of a series of experiments indicate that the as-prepared amorphous MoS2 features a small particle size, uniform morphology and relatively large specific surface area, and shows excellent performance in the removal of inorganic heavy metal ions (mercury, lead and cadmium ions) and organic pollutants (rhodamine B and tetracycline), catalase catalysis and a lithium battery anode, showing its great potential and broad application prospects in the fields of environmental remediation, clean energy and green catalysis.
Introduction
Molybdenum disulfide (MoS2) nanomaterial has a wide scope of application in various fields including photocatalysis,1–3 electrochemical catalysis,4–7 electronic devices,8,9 optoelectronic devices,10,11 sensors,12,13 and environmental pollutant removal,14–17 owing to its specific physico-chemical properties, such as relatively large surface area, strong adsorption capacity, adjustable band gap, abundant edge sites and good chemical stability.18 Compared with crystals, short-range ordered and long-range disordered amorphous materials can offer large active surface areas with more defects and reaction sites. Up to now, however, amorphous MoS2 nanomaterial has not attracted enough attention and its potential application has not been fully explored.19 The current methods for synthesizing amorphous nanomaterials usually need to take advantage of low temperature, rapid reaction, competitive coordination/adsorption, and lattice distortion to prevent or hinder the transformation of the material from amorphous to crystalline.20 It should be noted that pure amorphous MoS2 cannot be obtained by directly mixing a molybdenum source, acid and sulfur source under ambient conditions. Since the generated elemental sulfur during the reaction will complex with S2− in the solution to form S–S2−, it is difficult to obtain a single product MoS2, but mostly MoSx or MoS3 is produced.21–23 Therefore, it is necessary to develop a new synthesis strategy to inhibit the formation of S–S2− by preventing S2− from participating in the reduction reaction so as to synthesize stable amorphous MoS2 nanomaterial in high yield at ambient temperature and pressure.As is well known, photochemistry can provide a reaction path that is difficult to complete in classical thermochemical reactions or shortens the reaction process to make the reaction gentler.24,25 Photons as a “traceless and green reagent” make the photochemical process greener and more sustainable.26 For the past few years, photo-redox catalysis has made tremendous progress and widely been used in many aspects such as polycyclic addition and functionalization of organic substrates,27 noble metal catalysis,28–30 semiconductor synthesis,31,32 carbon dioxide reduction,33,34 polymer synthesis35–37 and drug synthesis.38–41 As far as we know, there has been no report on the photochemical synthesis of MoS2. On the other hand, it has been reported that Mo6+ in aqueous solution can be reduced to Mo(CO)6 through photolysis of formic acid.42 Therefore, we believe that the application of photochemistry has great potential in the synthesis of MoS2 nanomaterial with a new morphology and characteristics.
Herein, we propose a novel method of using light from a Xe lamp as an energy source and a traceless catalyst to quickly and simply synthesize amorphous MoS2 nanomaterials under ambient conditions. A molybdenum source, formic acid and a sulfur source were directly mixed and then reacted under full-spectrum xenon lamp illumination (for details, please refer to the experimental synthesis part and Fig. S1 in the ESI†).
The key step of the proposed method is the photolysis of formic acid to produce free radicals, which could rapidly reduce Mo6+ to Mo4+, which then combines with S2− to form MoS2. Because there is no need for high temperature, high pressure and any catalyst substance, the proposed synthetic method is economically feasible and environmentally friendly. More importantly, a kind of amorphous MoS2 nanomaterial with smaller and more uniform particle size can be readily obtained, owing to the photocatalysis strategy.
Results and discussion
In the first step of synthesis, the mixed solution of (NH4)6Mo7O24 and HCOOH is colorless and transparent. After dropping in Na2S solution, the solution mixture became bright orange-brown but was still clear and transparent (as shown in Scheme 1 and Fig. S2†). Upon exposure to the light from a Xe lamp, the solution turned dark brown with poor transparency, and a large amount of precipitation appeared after merely 3 min. After centrifugation, washing and vacuum drying, the product (the black solid powder as shown in Fig. S2†) was characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM) and scanning probe microscopy (SPM) (Fig. 1a–c and S3†). The results indicated that the synthesized product has a uniform particle size of about 15 nm, and both lattice patterns and sharp characteristic peaks, in the TEM and X-ray diffraction (XRD) characterization (Fig. 1d), respectively, were not observed, indicating that the synthesized MoS2 is amorphous. There have been many synthetic methods via direct addition of sodium sulfide as a sulfur source, but the resultant products are generally MoSx.21 In order to prove that our product was MoS2 instead of MoSx, Raman, XPS, and EDS characterization were conducted, respectively. The Raman results (Fig. 1e) showed that distinct J1, J2, J3, E2g and A1g peaks appeared, and the position of each peak except for J2 roughly corresponded to the theoretical calculation value of 1T′-MoS2 (Table S1†).41 J2, the mode which tends to shorten the distance between the two zigzag chains, was 10 cm−1 lower than the theoretical value. This redshift may be caused by the amorphous state of MoS2, which is not bound by a crystal lattice and has easier atomic movement. After testing for 1T′-MoS2, a higher energy (shorter wavelength) laser was used to obtain the Raman spectra, and the Raman standard peaks E2g and A1g of 2H-MoS2 were found. Obviously, under the irradiation of the higher laser energy, the 1T′ phase changes to the 2H phase. Then, the amorphous MoS2 was investigated by XPS (Fig. 1f), with the 226.5 eV peak attributed to the S2S binding energy of MoS2, and the Mo 3d peaks of 1T′-MoS2 were located at 229.3 eV (3d5/2) and 232.6 eV (3d3/2), respectively. It shows that molybdenum is tetravalent. The energy-dispersive X-ray spectroscopy (EDS) test (Fig. S4†) shows Mo and S peaks, with an atomic ratio of Mo to S of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, consistent with the composition of MoS2. Therefore, MoS2 was successfully synthesized by this method.
:2, consistent with the composition of MoS2. Therefore, MoS2 was successfully synthesized by this method.
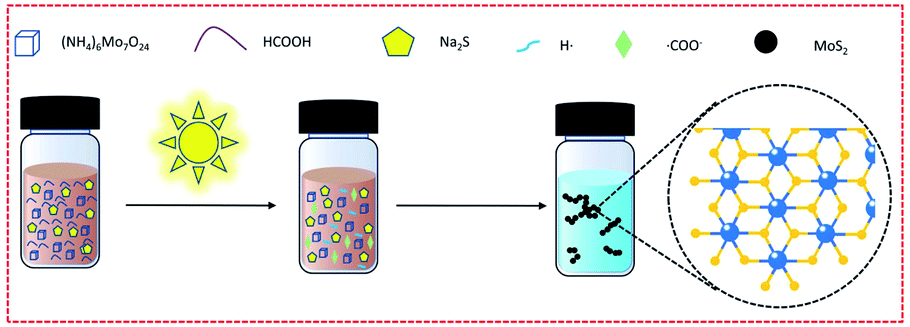 | ||
| Scheme 1 Schematic diagram of the preparation reaction. | ||
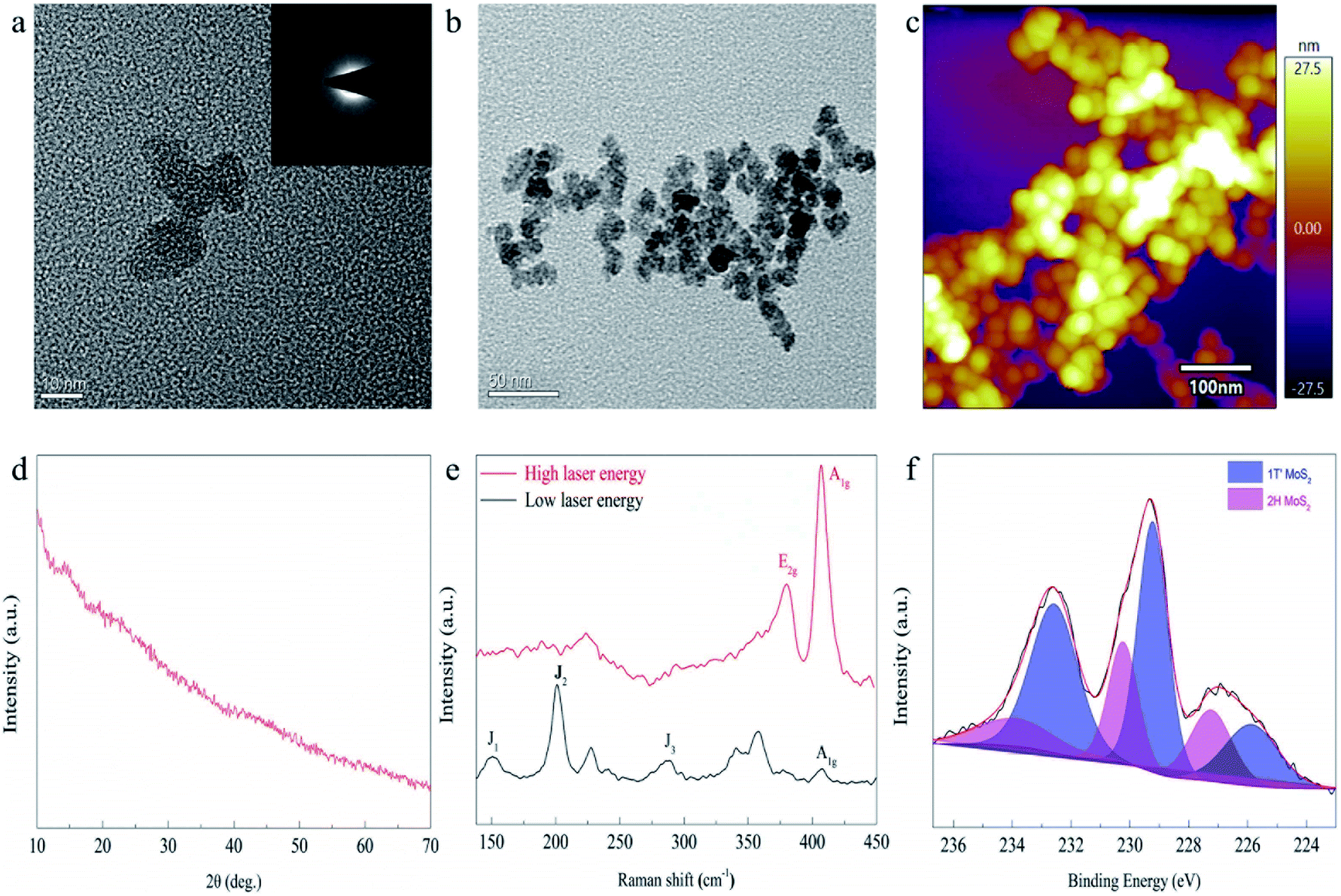 | ||
| Fig. 1 Morphology and structural phase characterization of MoS2 prepared through photocatalysis at ambient temperature and pressure. (a and b) TEM characterization, (c) SPM image, (d) XRD characterization, (e) Raman image of the as-prepared MoS2 and (f) high-resolution XPS spectrum of Mo 3d. | ||
For this unconventional synthesis, it is necessary to explore the mechanism of formation of MoS2. It has been reported that formic acid will generate free radicals in the presence of UV light, which can help Mo6+ reduction to Mo0.42 To verify the existence of free radicals, electron paramagnetic resonance (EPR) spectra (Fig. 2a) were collected, which show that only formic acid (neither hydrochloric acid nor sulfuric acid) cracks to generate free radicals (˙COO− and ˙OH−). After adding Na2S solution and (NH4)6Mo7O24·4H2O solution, no free radical signal was detected. The Raman spectra (Fig. 2b) show that the photocatalytic reaction of formic acid can generate 1T′-MoS2, but in the case of hydrochloric acid and sulfuric acid, MoSx was produced. From these phenomena, one can speculate that formic acid can absorb the energy of light and split to produce highly reactive free radicals. Mo6+ receives electrons from free radicals and reduces to Mo4+, and then collides with S2− in the solution to form Mo–S chemical bonds. In this process, since free radicals with stronger reduction capacity react rapidly with Mo6+, the S2− is free from the reduction reaction. As a result, elemental sulfur could not be generated and form S–S2− in the solution, eventually preventing the formation of molybdenum trisulfide. Moreover, because the light can irradiate the entire reaction solution during the synthesis process, a large number of MoS2 particles with a short-range ordered structure can be rapidly produced in each part of the reaction solution at the same time, with the lattice structure having no chance to grow, and thereby forming an amorphous structure.
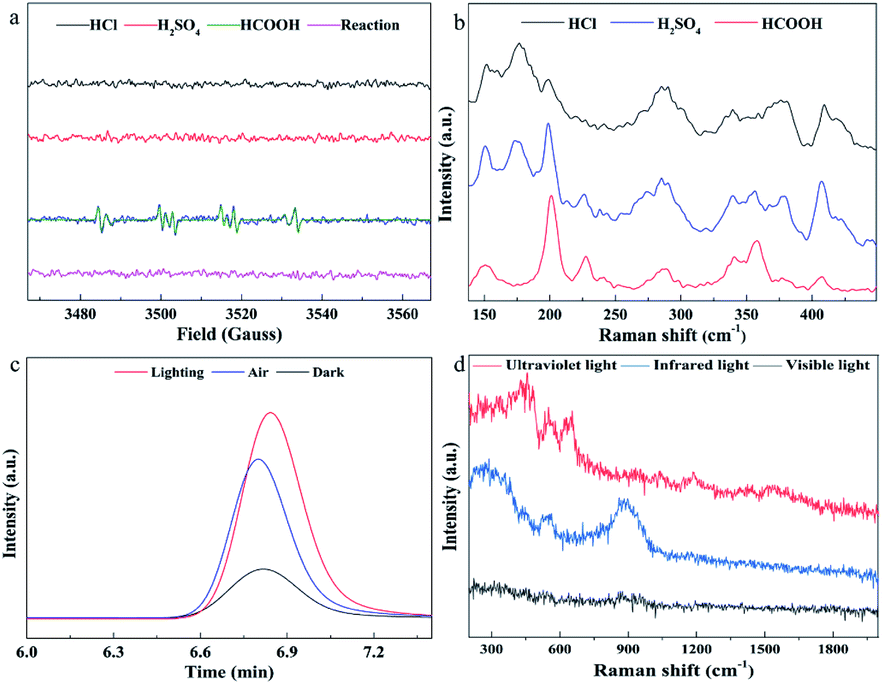 | ||
| Fig. 2 Mechanism study. (a) EPR spectra; (b) Raman characterization of the products after synthesis with hydrochloric acid, sulfuric acid and formic acid under Xe lamp irradiation, respectively; (c) gas chromatographic analysis of carbon dioxide; (d) Raman characterization of products synthesized under light irradiation in different wavelength regions. | ||
Finally, according to the above inference, the reaction formula can be written as follows:
| HCOOH → H+ + HCOO− |
| HCOO− → H˙ + ˙COO− |
| (Mo7O24)6− + 14˙COO− + 48H+ = 7Mo4+ + 14CO2 + 24H2O |
| (Mo7O24)6− + 14H˙ + 34H+ = 7Mo4+ + 24H2O |
| Mo4+ + 2S2− = MoS2(S) + 4H+ |
| (NH4)6Mo7O24 + 41HCOOH + 14Na2S = 7MoS2↓ + 7CO2↑ + 24H2O + 6HCOONH4 + 28HCOONa |
So as to further verify whether the inferred mechanism is correct, gas chromatography was used to detect the products in the gas phase. First, the reactants were added to a quartz bottle with headspace, and then nitrogen was used to purge the carbon dioxide, and the quartz bottle was quickly sealed. One group of quartz bottles were under xenon lamp illumination, and the other group was placed in a dark environment as a blank control experiment. 1 mL of gas above the solution was taken for gas chromatography measurements. Fig. 2c shows that a large amount of carbon dioxide is generated in the synthesis. Since in our reaction system carbon dioxide can only come from the decomposition and oxidation of formic acid, the experimental results are in good agreement with the speculated mechanism.
In order to investigate the effect of light in different wavelength regions on the reaction, corresponding filters were added to the xenon lamp and the synthesis was carried out under ultraviolet, visible and infrared light irradiation, respectively. When a large amount of precipitate appeared in the solution (3 min), the reaction was stopped, the precipitate was separated and Raman tests were performed (Fig. 2d). The Raman spectra show that all have no characteristic Raman spectral peaks of 1T′-MoS2 and 2H-MoS2, indicating that none of the samples were MoS2, meaning that MoS2 was not synthesized. Consequently, the full-spectrum light source is a necessary condition in our synthesis system. This may be due to the fact that the full-spectrum light source contains not only ultraviolet rays, which can decompose formic acid into free radicals, but also visible and infrared light, which provide sufficient photons as energy to maintain the reaction.
Because of the smaller and more uniform particle size and the formation of a lot of voids owing to the chain like entanglement (Fig. 1c), it can be speculated (Fig. 3a) that the as-synthesized amorphous MoS2 with a large surface area and more active sites would show excellent performance in adsorption of heavy metal ions, removal of organic pollutants, catalysis and batteries. First, the ability of this material to adsorb heavy metal ions was investigated. After optimizing the adsorption pH (Fig. S6d†) and adsorption time (Fig. S7†), the maximum adsorption capacity was measured, and the results are shown in Fig. 3b. The adsorption capacity for heavy metal ions increases with the increase of equilibrium concentration, and then reaches equilibrium. The isotherm data were fitted using the Langmuir isotherm model of eqn S(4),† and an excellent correlation coefficient (R2 > 0.99) was obtained. It shows that the adsorption process is mainly a monolayer adsorption mechanism. The maximum metal ion adsorption capacities were calculated from the Langmuir model and the values are summarized in Table S3.† Specifically, at pH 6, the maximum capacities are 169.2 mg g−1 (Cd2+), 269.2 mg g−1 (Pb2+) and 2789.2 mg g−1 (Hg2+), respectively. In order to illustrate the advantages of the synthesized amorphous MoS2 in mercury removal, we compared some mercury removal materials previously reported (Table S4†). The results show that the synthesized material has excellent mercury removal ability. In addition, we tested the removal of organic pollutants in water by the as-synthesized MoS2 (Fig. 3c and S9†) and found that the amorphous MoS2 can quickly remove tetracycline, up to 85%.
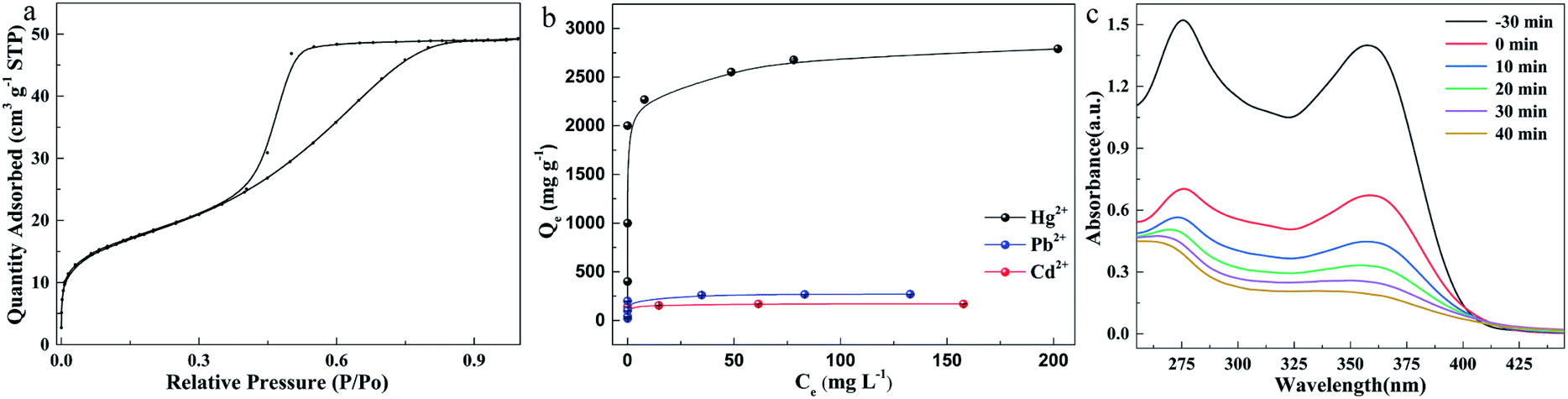 | ||
| Fig. 3 Material performance. (a) Liquid nitrogen adsorption curve; (b) maximum adsorption capacity for Hg2+, Cd2+ and Pb2+ at pH 6; (c) absorbance of tetracycline solution at different times during removal by MoS2. Experimental conditions: (a) MoS2 (m/v), 0.25 mg mL−1; temperature, 25 °C; Hg2+ residence time, 200 min; Cd2+ and Pb2+ residence time, 15 min; (b) MoS2, 10 mg; tetracycline, 25 mL, 50 mg L−1. | ||
o-Phenylenediamine (OPD) solution is a colorless substrate, which can appear as a yellow solution under the action of hydrogen peroxide and iron. As a catalase, MoS2 can promote the reaction and improve the sensitivity of Fe2+ detection (Fig. 4a and S13†). The linear calibration equation was found to be A = 0.212 + 0.33c with a correlation coefficient of 0.995. The linear range was from 0.1 to 1.4 μM with an LOD of 0.08 μM. The results show that the MoS2/OPD/H2O2 sensor for Fe2+ detection is much more sensitive than the OPD/H2O2 sensor, indicating that the sensitivity of Fe2+ sensors can be improved by using the synthesized MoS2.
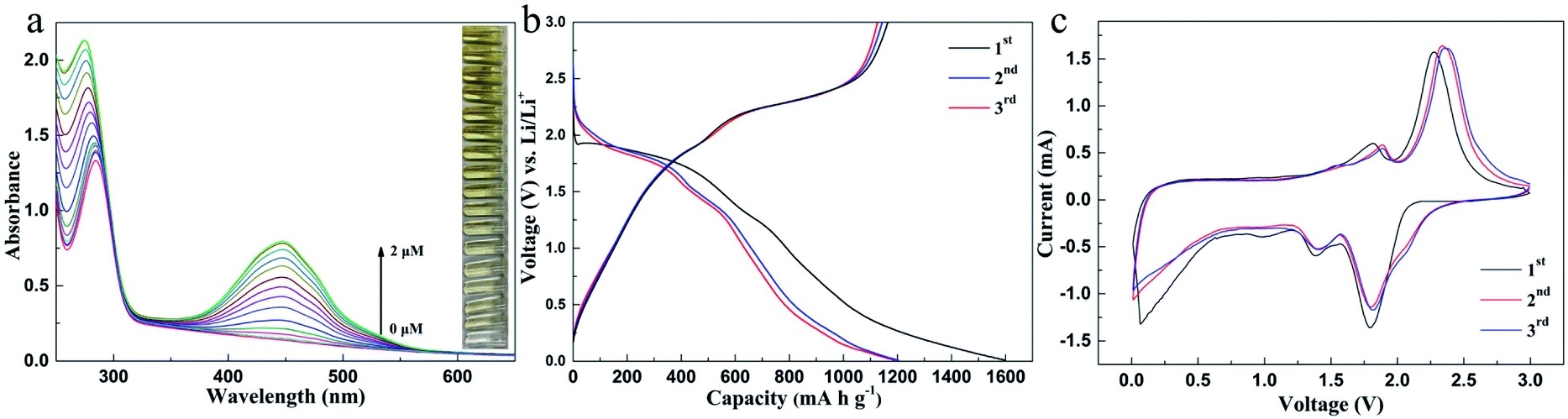 | ||
| Fig. 4 Material performance. (a) Determination of Fe2+ with MoS2 as the H2O2 enzyme; (b) charge–discharge voltage profiles of the MoS2 electrode at 200 mA g−1; (c) cycle curves at a scan rate of 0.2 mV s−1 between 0.01 and 3.0 V. Experimental conditions: (a) MoS2 0.1 mg mL−1, H2O2, 0.1 mM, and OPD 0.5 mM; Fe2+ (0, 0.001, 0.005, 0.01, 0.02, 0.05, 0.1, 0.2, 0.4, 0.6, 0.8, 1, 1.2, 1.4, 1.6, 1.8, and 2 μM). | ||
The synthesized MoS2 was also applied to the anode of a lithium battery, and the second and third cycle charge discharge voltage curves at 200 mA g−1 are shown in Fig. 4b. The initial charge and discharge capacities are 1202 and 1563 mA h g−1, respectively, and the initial coulombic efficiency of the first cycle is 77%. Fig. 4c shows the three initial cyclic voltammograms (CVS) of the MoS2 electrode in the potential window of 0.01–3.0 V (vs. Li+/Li) at a scanning rate of 0.2 mV s−1. The reduction peak at 1.01 V is attributed to the intercalation of lithium ions into the MoS2 lattice.
| MoS2 + xLi+ + xe− → LixMoS2 |
The oxidation peaks at 1.65 V and 2.30 V during the process of loose lithium removal are attributed to the reversible reaction of Mo and MoS2 and the formation of S by LiS2. A new peak appeared at 1.86 V during the subsequent lithium reaction, indicating the formation of LiS2. In fact, after the first cycle, MoS2 is transformed into the composite of Mo nanoparticles and S nanoparticles, and the second cycle curve coincides with the third cycle curve, which proves that the prepared MoS2 has good cycling stability. Although with unmodified MoS221 the energy loss is large during the cell cycle (Fig. S18†), the high charge discharge capacity proves that amorphous MoS2 has a number of active sites and large surface area, indicating its great potential as the negative electrode of a lithium battery.
Experimental section
Synthesis of MoS2
All chemicals were of analytical grade and used as received without further purification. First, 2 mL of 5 mM (NH4)6Mo7O24·4H2O and 2 mL of 50% v/v formic acid were added to a 10 mL quartz bottle, to which 2 mL of 10 mM sodium sulfide was then added. The sealed quartz bottle was placed 10 cm under a xenon lamp, and lit under the full spectrum for 3 min. The resultant product was collected by centrifugation (10000 rpm for 10 min), then washed at least three times with distilled water and absolute ethanol to remove ions and possible remnants, and vacuum dried at 40 °C for 3 h.
Materials characterization
The morphology was characterized by SEM, TEM and SPM. EDS and XPS were used for elemental analysis. EPR was applied to detect free radicals. Raman spectroscopy and XRD were used to detect the characteristic spectral peaks of the products. BET was used to determine the surface area and adsorption type of the product. CO2 was detected by high performance gas chromatography. The UV spectrum was obtained with a UV spectrophotometer. The electrochemical performance of the lithium battery was tested using an electrochemical workstation. See the ESI† for details.Mechanism exploration
60 μL of DMPO was added to the reaction solution prepared according to the “synthesis” step. For comparsion, 2 mL of sulfuric acid, hydrochloric acid, and formic acid were added to 4 mL of deionized water in 10 mL quartz bottle, respectively, and then 60 μL of DMPO was added to each solution. Then every quartz bottle was sealed and placed under a xenon lamp for 3 min. The solution after the reaction was taken out with a quartz capillary and put into an EPR instrument for detection.During the GC test, the sample injection temperature was set at 150 °C, the chromatographic column temperature at 100 °C, and the thermal conductivity detector temperature at 120 °C. The solution was added to a quartz bottle according to the “synthesis” procedure, purged with nitrogen gas, and sealed. One group was used as a control experiment without light irradiation, and the other group reacted normally. After the reaction, 1 mL of the gas in the headspace of the quartz bottle in the control group and the experimental group were taken and injected with a gas injection needle for GC measurements, respectively, and 1 mL of air was directly injected for comparison.
Removal of tetracycline
25 mL of 50 mg L−1 tetracycline was added into a customized reaction vessel with cooling circulating water. 10 mg of synthetic amorphous MoS2 was weighed and added to the prepared solution. First, the solution was stirred for 30 min in the dark, then a Xe lamp was turned on, and 1 mL of the solution was drawn every 10 min, which was then centrifuged and measured with a UV-vis spectrophotometer.Removal of rhodamine B
25 mL of 50 mg L−1 rhodamine B was added into a customized reaction vessel with cooling circulating water. 10 mg of synthetic amorphous MoS2 was weighed and added to the prepared solution. First, the solution was stirred for 30 min in the dark, then a Xe lamp was turned on, and 1 mL of the solution was drawn every 10 min, which was then centrifuged and measured with a UV-vis spectrophotometer.Colorimetric detection of Fe2+
20 μL of MoS2 nanosheets (0.1 mg mL−1), appropriate volume, 200 μL, of Fe2+ standard solution, and selected volume, 980 μL, of water were micropipetted directly into a cell. Then, 100 μL of OPD (10 mM) and 500 μL of acetate buffer (200 mM, pH = 4.5) were added. Finally, 200 μL of H2O2 (1 mM) was added to give a total volume of 2.00 mL. After the reaction period, the solution was put into an ultraviolet spectrophotometer for measurement, and the scanning wavelength range was 250–650 nm.Cell assembly and electrochemical measurements
MoS2 was fabricated by mixing the active materials with acetylene black (AB) and carboxymethyl cellulose (CMC), at a weight ratio of 70:20:10. The mixture was mixed with water to form a slurry. The slurry was uniformly pasted onto Cu foil with a blade. The electrodes were dried at 40 °C in a vacuum oven for 3 h. CR2025-type coin cells were assembled in a glove box for electrochemical characterization. A solution of 1 M LiPF6 at a 1:1:1 weight ratio of ethylene carbonate (EC), diethyl carbonate (DEC), and dimethyl carbonate (DMC) was used as the electrolyte. Lithium metal disks were used as the counter electrodes for electrochemical testing. A polypropylene (PP) membrane was used as the separator. The cells were charged and discharged at a current density of 0.2 A g−1 within the voltage range of 0.01–3.0 V for 50 cycles.
Conclusions
A facile photochemical synthesis strategy was proposed to produce high-performance amorphous MoS2 nanoparticles at ambient temperature and pressure. The synthesis technique based on the photolysis of formic acid could effectively prevent the formation of MoSx or MoS3 and thus successfully promote the yield of MoS2. Physical and chemical characterization suggests that the resultant amorphous MoS2 has the structure of 1T′-MoS2 with a small particle size, uniform morphology and relatively large specific surface area. Owing to its short-range ordered and long-range disordered structures with a significant number of defects and reaction sites, the as-prepared amorphous MoS2 shows remarkable performance in the removal of heavy metal ions (mercury, lead and cadmium ions), organic pollutants (rhodamine B and tetracycline), catalase catalysis and a lithium battery anode. This work opens up a new way for the synthesis of MoS2 nanomaterial and enhances or widens its promising applications in the fields of environmental remediation, clean energy, green catalysis and batteries.Author contributions
K. L. Xu, X. D. Hou and H. F. Sun conceived the idea. H. F. Sun contributed to all of the experimental work. M. Yang contributed to the experimental work. P. Shan contributed to the lithium battery experimental work related to material synthesis. L. C. Gou contributed to the experimental work related to Raman testing. C. Z. Lv contributed to the experimental work related to adsorption testing. J. He assisted with BET characterization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21876118) and the Ministry of Education through the 111 Project (Grant No. B17030). We would like to thank Dr Yunfei Tian, Dr Shanlin Wang and Dr Hanjiao Chen of the Analytical & Testing Center at Sichuan University for their help in XPS, TEM and EPR data collection. We would also like to thank Prof. Xiaodong Guo and Prof. Dan Xiao of the School of Chemical Engineering for their help in battery making and its performance test.Notes and references
- L. Wang, X. Liu, J. Luo, X. Duan, J. Crittenden, C. Liu, S. Zhang, Y. Pei, Y. Zeng and X. Duan, Angew. Chem., Int. Ed. Engl., 2017, 56, 7610–7614 CrossRef CAS PubMed.
- B. Han and Y. H. Hu, Energy Sci. Eng., 2016, 4, 285–304 CrossRef CAS.
- S. Min and G. Lu, J. Phys. Chem. C, 2012, 116, 25415–25424 CrossRef CAS.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- D. Wang, Z. Pan, Z. Wu, Z. Wang and Z. Liu, J. Power Sources, 2014, 264, 229–234 CrossRef CAS.
- X. Ren, W. Wang, R. Ge, S. Hao, F. Qu, G. Du, A. M. Asiri, Q. Wei, L. Chen and X. Sun, Chem. Commun., 2017, 53, 9000–9003 RSC.
- K. Lee, H. Y. Kim, M. Lotya, J. N. Coleman, G. T. Kim and G. S. Duesberg, Adv. Mater., 2011, 23, 4178–4182 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- W. Wang, X. Zeng, J. H. Warner, Z. Guo, Y. Hu, Y. Zeng, J. Lu, W. Jin, S. Wang, J. Lu, Y. Zeng and Y. Xiao, ACS Appl. Mater. Interfaces, 2020, 12, 33325–33335 CrossRef CAS PubMed.
- A. Midya, A. Ghorai, S. Mukherjee, R. Maiti and S. K. Ray, J. Mater. Chem. A, 2016, 4, 4534–4543 RSC.
- W. Zhang, P. Zhang, Z. Su and G. Wei, Nanoscale, 2015, 7, 18364–18378 RSC.
- Z. Ashkavand, E. Sadeghi, R. Parvizi and M. Zare, ACS Appl. Mater. Interfaces, 2020, 12, 34283–34296 CrossRef CAS PubMed.
- Z. Wang and B. Mi, Environ. Sci. Technol., 2017, 51, 8229–8244 CrossRef CAS PubMed.
- Z. Wang, A. Sim, J. J. Urban and B. Mi, Environ. Sci. Technol., 2018, 52, 9741–9748 CrossRef CAS PubMed.
- Q. Li, N. Zhang, Y. Yang, G. Wang and D. H. Ng, Langmuir, 2014, 30, 8965–8972 CrossRef CAS PubMed.
- K. Ai, C. Ruan, M. Shen and L. Lu, Adv. Funct. Mater., 2016, 26, 5542–5549 CrossRef CAS.
- I. Song, C. Park and H. C. Choi, RSC Adv., 2015, 5, 7495–7514 RSC.
- Z. Yang, J. Hao and S. P. Lau, J. Appl. Phys., 2020, 127, 220901 CrossRef CAS.
- H. Zhao, X. Chen, G. Wang, Y. Qiu and L. Guo, 2D Materials, 2019, 6, 032002 CrossRef CAS.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916 CrossRef CAS.
- H. Ye, L. Wang, S. Deng, X. Zeng, K. Nie, P. N. Duchesne, B. Wang, S. Liu, J. Zhou, F. Zhao, N. Han, P. Zhang, J. Zhong, X. Sun, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2017, 7, 1601602 CrossRef.
- A. Albini and M. Fagnoni, ChemSusChem, 2008, 1, 63–66 CrossRef CAS PubMed.
- A. Albini and M. Fagnoni, Green Chem., 2004, 6, 1 RSC.
- H. Norbert, Chem. Rev., 2008, 108, 1052–1103 CrossRef PubMed.
- D. Cambie, C. Bottecchia, N. J. Straathof, V. Hessel and T. Noel, Chem. Rev., 2016, 116, 10276–10341 CrossRef CAS PubMed.
- P. Fageria, S. Uppala, R. Nazir, S. Gangopadhyay, C. H. Chang, M. Basu and S. Pande, Langmuir, 2016, 32, 10054–10064 CrossRef CAS PubMed.
- D. Marie-Christine and A. Didier, Chem. Rev., 2004, 104, 293–346 CrossRef PubMed.
- L. M. Katherine, R. D. Matthew, W. Dashan and C. S. Juan, J. Am. Chem. Soc., 2006, 128, 15980–15981 CrossRef PubMed.
- Y. Cao, W. Geng, R. Shi, L. Shang, G. I. Waterhouse, L. Liu, L. Z. Wu, C. H. Tung, Y. Yin and T. Zhang, Angew. Chem., Int. Ed. Engl., 2016, 55, 14952–14957 CrossRef CAS PubMed.
- C. M. Gonzalez, W. C. Wu, J. B. Tracy and B. Martin, Chem. Commun., 2015, 51, 3087–3090 RSC.
- L. M. Xue, F. H. Zhang, H. J. Fan and X. F. Bai, Adv. Mater. Res., 2011, 183–185, 1842–1846 CAS.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- F. Kettling, B. Vonhoren, J. A. Krings, S. Saito and B. J. Ravoo, Chem. Commun., 2015, 51, 1027–1030 RSC.
- K. A. Ogawa, A. E. Goetz and A. J. Boydston, J. Am. Chem. Soc., 2015, 137, 1400–1403 CrossRef CAS PubMed.
- G. Zhang, I. Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- J. Xuan, X. D. Xia, T. T. Zeng, Z. J. Feng, J. R. Chen, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed. Engl., 2014, 53, 5653–5656 CrossRef CAS PubMed.
- H. Wang, Y. Ren, K. Wang, Y. Man, Y. Xiang, N. Li and B. Tang, Chem. Commun., 2017, 53, 9644–9647 RSC.
- J. B. McManus and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 2880–2883 CrossRef CAS PubMed.
- T. T. Zeng, J. Xuan, W. Ding, K. Wang, L. Q. Lu and W. J. Xiao, Org. Lett., 2015, 17, 4070–4073 CrossRef CAS PubMed.
- M. Calandra, Phys. Rev. B, 2013, 88, 245428 CrossRef.
- J. Hu, H. Chen, X. Hou and X. Jiang, Anal. Chem., 2019, 91, 5938–5944 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00165e |
| This journal is © The Royal Society of Chemistry 2021 |