
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvophobic-controlled synthesis of smart magneto-fluorescent nanostructures for real-time inspection of metallic fractures†
Fernando
Menegatti de Melo
 *ab,
João V.
Mattioni
a,
Fernando
Dias
b,
Ying
Fu
c and
Henrique E.
Toma
a
*ab,
João V.
Mattioni
a,
Fernando
Dias
b,
Ying
Fu
c and
Henrique E.
Toma
a
aSupramolecular Nanotech Laboratory, Institute of Chemistry, University of São Paulo, Avenida Professor Lineu Prestes, 748, 05508000, São Paulo, Brazil. E-mail: fernando.menegatti.melo@usp.br
bMetal-Chek do Brasil Indústria e Comércio, Research & Development Department, Rua das Indústrias, 135, 12926674, Bragança Paulista, Brazil
cSchool of Information Technology, Halmstad University, SE-30118 Halmstad, Sweden
First published on 28th April 2021
Abstract
The production of materials that contain more than one functional constituent, the so-called multifunctional materials, is quite relevant in advanced technology. By acting as building blocks, nanoparticles can be suitably explored for generating higher-order multifunctional structures. In this regard, herein, a special clustered magneto-fluorescent superstructure has been developed for non-destructive detection of flaws and shallow subsurface discontinuities in industrial ferromagnetic materials. The strategy consists of the solvophobic-controlled assembly of organic-based maghemite cores and water-based II–VI quantum dots, in the presence of hexadecyltrimethyl-ammonium bromide, CTAB, as a compatibilizer agent. This composite exhibited a high magnetic response (σmax = 66 emu g−1) and uniform size, in addition to tunable optical properties (QY = 78%). The strategy of utilizing nanoparticles as magneto-fluorescent nanoprobes to identify tiny slits represents a great advance, for improving the capability of precisely revealing the fracture boundary locations by visual real-time inspection. The nanoscale probes exhibit a low signal-to-noise ratio and a higher competitive performance in relation to the existing micrometric detection systems.
Introduction
Co-assembling two types of distinct nanoparticles with contrasting properties allows the production of new classes of hybrid nanoparticles, displaying combined features, but keeping the individual characteristics of the original building blocks. Despite the exciting perspectives of these materials, their development around magneto-fluorescent materials remains quite challenging, mostly because of quantum restrictions imposed by the FRET mechanism at the nanoscale, requiring research to deal with chemical compatibility, structural defects, and synthesis methodology. To fully accomplish the design of magneto-fluorescent nanoparticles, some important requisites should be fulfilled, e.g., a high magnetic response in the presence of an external magnetic field, controlled uniform sizes and an optimized fluorescence response.For this reason, efforts have been devoted to developing synthetic strategies for producing such multifunctional materials, encompassing for instance the heterostructure crystal growth,1–3 co-encapsulation into organic structures or inorganic materials,4–8 and template-based synthesis via either chemical bonding or physical attachment.9–11 For instance, six main prototypes can be identified in this area: (i) a magnetic core coated with a silica shell containing fluorescent components by incorporating dyes such as 7-(dimethylamino)-4-methylcoumarin-3-isothiocyanate, tetramethyl-rhodamine-5-isothio-cyanate or (1-methyl-4-(E)-2-(4-[methyl(2-sulfanylethyl)-amino]phenyl)-1-ethenyl)pyridinium iodide;12–14 (ii) polymer-coated magnetic nanoparticles functionalized with fluorescent species by the layer-by-layer technique using poly(allylamine hydrochloride) and poly(sodium 4-styrenesulfonate), by exploiting their electrostatic interactions with charged quantum dots;15–17 (iii) ionic aggregates consisting of a magnetic core and fluorescent ionic compounds using polyhedral silsesquioxane which can ionically interact with water-based magnetic nanoparticles, providing a strategic spacer to interact with fluorescent porphyrin, for applications in hyperthermia;18,19 (iv) fluorescent labelled lipid-based magnetic nanoparticles that use amphiphilic molecules which can be linked to various water-based species with the same charge, e.g., by coating hydrophobic magnetic nanoparticles with the polystyrene250-block-poly(acrylic acid13) block copolymer, which will interact with fluorescent species;20–22 (v) a magnetic core covalently bound to a fluorescent entity via a long molecular linker as a spacer, as obtained from the thermolysis of Fe(CO)5 and reduction of Pt(acac)2 by hexadecane-1,2 diol followed by the deposition of sulphur and cadmium at the surface of the nanoparticles;23 (vi) organized magnetic nanoparticle-quantum dots embedded in nanocomposites, using water-in-oil emulsion.24 All the above papers have interesting approaches. However, it should be noticed that they focus on biomedical applications such as bioimaging, bio- and chemo-sensing, cell tracking and sorting, bioseparation, drug delivery and therapy. Considering large scale production and performance, such systems can hardly be applied in magnetic particle inspection (MPI) baths, which are the subject of this paper. For that reason, we are suggesting another type of magneto-fluorescent nanoparticle synthesis that fulfils the above-mentioned requirements as a strong magnetic and fluorescence response.
Magnetic particle inspection (MPI) is the most important technique to identify structural defects in ferromagnetic materials in industry, e.g. aerospace. It is a real-time, non-destructive testing (NDT) for identifying metallic fractures in industrial ferromagnetic materials. Most common MPI commercial products are based on micrometric particles in association with expensive fluorescent organic dyes chemically mixed with isothiocyanate derivatives. Their functioning is based on induced magnetic attraction exerted by the fracture borders, thus exposing their precise location for visual fluorescence detection.25 In this regard, nanomaterials can detect the occurrence of smaller flaws which are not accessible by the micrometric MPI agents, without generating any background noise as we will comment later. By allowing the detection of smaller flaws, the use of QDs is particularly rewarding in MPI applications because they are highly luminescent and stable under harsh conditions.
In comparison with traditional dyes, they convey many advantages, including high brightness at a lower concentration and broad optical absorption characteristics since they use a lower amount of materials to produce a bright indicator working under a variety of exciting sources and displaying narrow band emission spectra associated with quantum size effects.26–31 Previously, we have demonstrated the possibility of exploring a side-chain interaction between two water-based distinct materials for MPI assays.22 Along this line, efforts were still necessary to improve the net performance, such as, maximizing their magnetic response by improving the iron oxide crystallinity and working on the quantum dot recovery to minimize the background noise during visual detection. So, herein, we carried out the engineering and production of a clustered multifunctional magneto-fluorescent superstructure, MghNP@QD (γ-Fe2O3@C17H35COO@CTAB@CdTe@C3H5O2S), as a new class of magneto-fluorescent nanoparticles, based on solvophobic-controlled synthesis. An organic-based multi-magnetic core of stearate-functionalized maghemite nanoparticles, MghNPs, was assembled with water-based 3-mercaptopropionate-functionalized cadmium telluride quantum dots, QDs, in association with the compatibilizer hexadecyltrimethyl-ammonium bromide, CTAB, as shown in Fig. 1. They exhibited a higher magnetic response compared with our previous work22 and a typical superparamagnetic behavior. In addition, we have obtained tunable and uniform sizes and optimized fluorescence features for real-time NDT with negligible background noise.
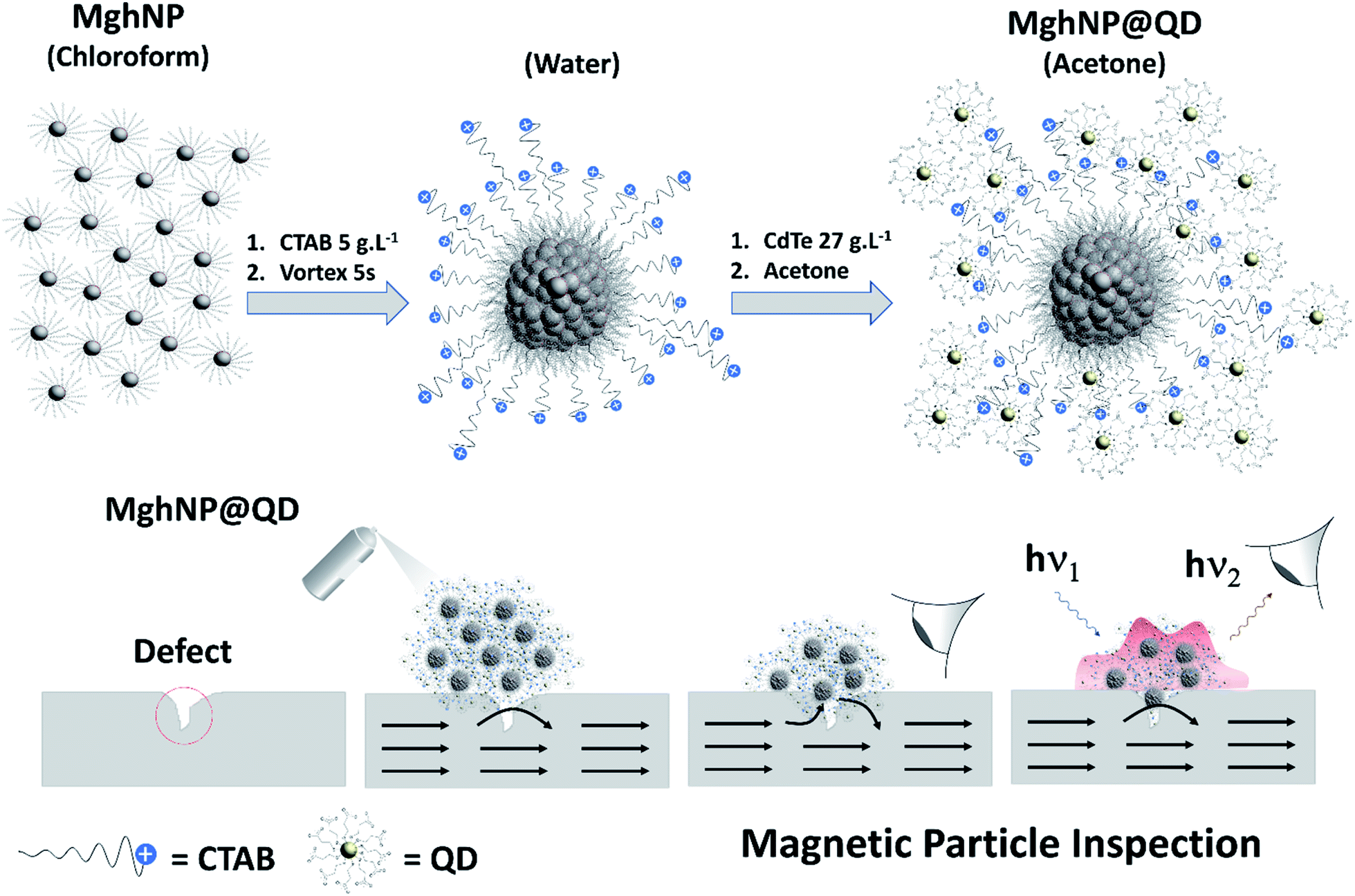 | ||
| Fig. 1 Schematic diagram showing the as-prepared clustered multifunctional magneto-fluorescent nanoparticles to be applied in real-time non-destructive detection of structural defects in industrial ferromagnetic materials. Gray spheres surrounded by stearate molecules (C17H35COO−) represent stearate-functionalized maghemite nanoparticles, MghNPs (γ-Fe2O3@C17H35COO). Blues spheres with positive charge represent CTAB molecules (C19H42BrN). Yellow spheres surrounded by 3-mercaptopropionate molecules (C3H5O2S−) represent the 3-mercaptopropionate-functionalized cadmium telluride quantum dots, QD (CdTe@C3H5O2S) (structural formulas are shown in the ESI†). | ||
Results and discussion
Synthesis and characterization of MghNPs
To produce a clustered multifunctional magneto-fluorescent superstructure, stearate-functionalized maghemite nanoparticles were exploited as the anchoring surface to 3-mercaptopropionate-functionalized cadmium telluride quantum dots, using CTAB as the mediator, as depicted in Fig. 1. Generally speaking, magnetic nanoparticles are widely used for several applications. However, their organic synthesis remains unclear, since the influence of the precursor on the shape of the nanoparticles has been scarcely investigated. For that reason, we have studied the thermal decomposition synthesis of hydrated (h) and dehydrated (d) iron(III) stearate, FeSt3, and iron(II) stearate, FeSt2, as a function of sodium oleate (NaOl)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oleic acid (HOl) ratio and sodium stearate (NaSt):stearate acid (HSt) ratio obtaining four different shapes of magnetic nanoparticles, Fig. 2 (Fig. S1–S4, ESI†).
:oleic acid (HOl) ratio and sodium stearate (NaSt):stearate acid (HSt) ratio obtaining four different shapes of magnetic nanoparticles, Fig. 2 (Fig. S1–S4, ESI†).
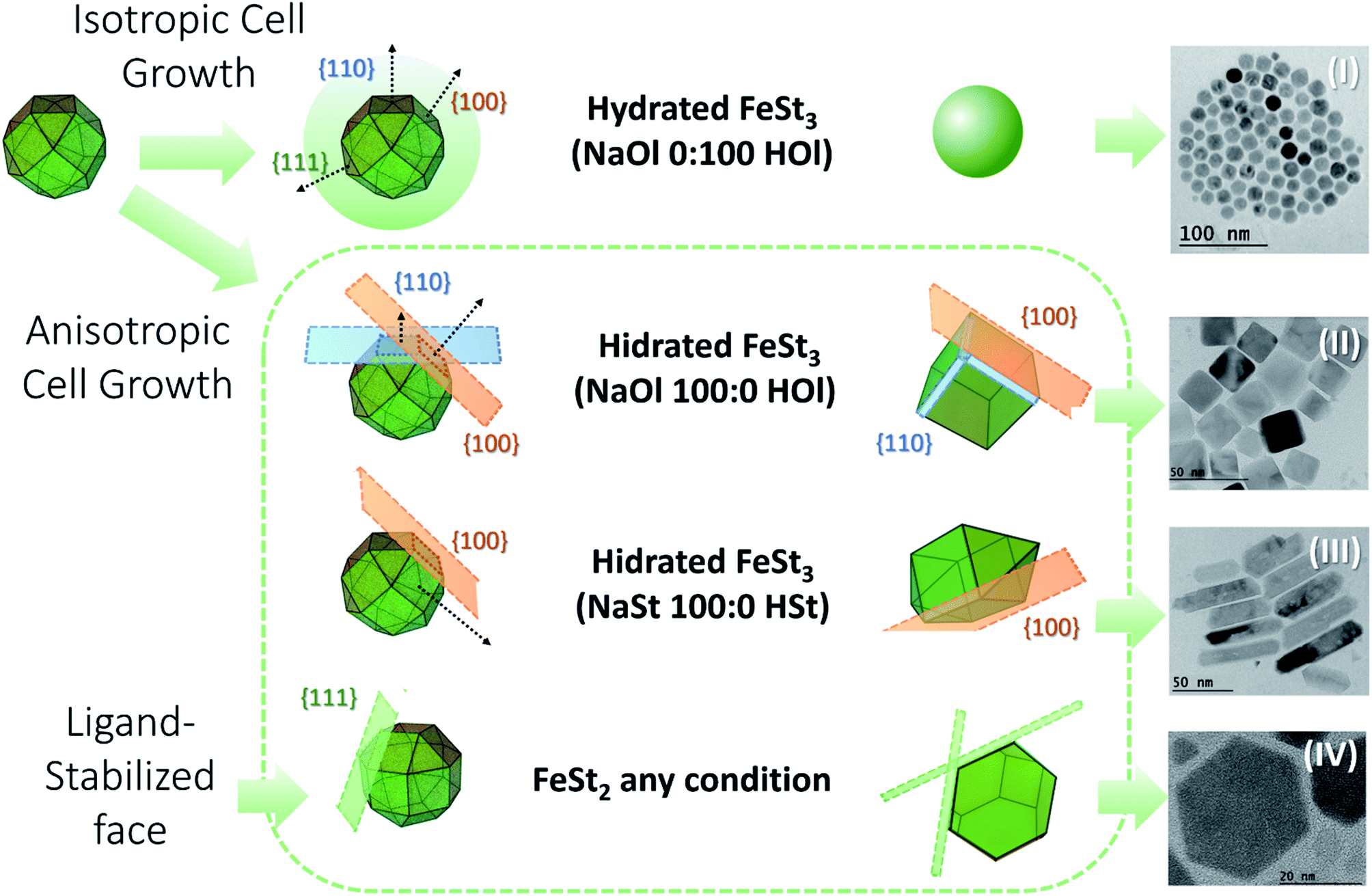 | ||
| Fig. 2 Summary of the thermal decomposition synthesis of hydrated and dehydrated in house iron(III) stearate, FeSt3 (C54H105FeO6), and iron(II) stearate, FeSt2 (C36H72FeO4), as a function of sodium oleate (NaOl, C18H33NaO2):oleic acid (HOl, C18H34O2) ratio and sodium stearate (NaSt, C18H35NaO2):stearate acid (HSt, C18H36O2) ratio. (I) Sphere-like nanoparticles. (II) Cube-like nanoparticles. (III) Rod nanoparticles. (IV) Plate nanoparticles (structural formulas are shown in the ESI†). | ||
As shown previously by Cotin, et al. 2018 (ref. 32) it was confirmed that NaOl triggered the nanoparticle shape from spheres to cubes and plates and NaSt drove the nanoparticle shape from spheres to plates. It was not so clear if the presence of water can play an important role in the structure of the nanoparticles, since most of them are cubes mixed with regular spheres. So, we decided to work with hydrated FeSt3, eliminating the time consuming step of dehydration, and facilitating the iron(III) precursor manipulation. In this way, briefly, in house iron(III) stearate, FeSt3, was solubilized in octadecene, heated to 120 °C in N2 for 60 minutes, and heated again to 200 °C for 60 minutes in the presence of 50:50 sodium oleate:oleic acid ratio. Then it was heated again up to 320 °C and kept for 60 minutes. The as-synthesized magnetite nanoparticles were purified by repetitive dispersion in chloroform and sedimentation with acetone, in order to form a stock colloidal solution (see the Experimental section). One should note that most commonly employed magnetic materials are based on magnetite.33–39 Despite the fact that this is an inverse spinel iron oxide with high loading magnetic content (Fe3O4), it is not a good candidate as a magneto-fluorescent building block since its Fe(II) → Fe(III) intervalence transitions superimposed to the O2− → Fe(III) charge-transfer band promotes its typical black colour. This feature makes it unsuitable for magneto-fluorescent materials because the optical absorption and quenching by the FRET mechanism become rather critical, decreasing the luminescence intensity of any entity at the iron oxide surface.
Another important detail to mention is the preference for the iron-stearate complex instead of the iron–oleate (C54H99FeO6) one. Despite the fact that the synthesis of the iron–oleate complex is well known in the literature,40,41 it forms a waxy solid material which is very difficult to purify and manipulate.
For the above-mentioned reasons, we synthesized the iron-stearate complex and promoted the assisted oxidation of the magnetite stock colloidal chloroform solution by air bubbling until complete evaporation of the solvent. In this way, another iron oxide phase, maghemite, was produced. Although maghemite has a slightly smaller (∼25%) magnetic moment as compared with magnetite, it is more stable in air and has the advantage of exhibiting a much lower optical absorption in the visible region. The oxidation of magnetite, MgNPs, to maghemite, MghNPs, was successfully achieved,42,43 as shown in Fig. 3, and in the following discussion. The large-area transmission electron microscopy (TEM) images in Fig. 3A and B and in Fig. S5, S6A and S6D (see ESI†) show that the resulting quasi-spherical nanoparticles, before and after the oxidation process, have an average diameter of 14.58 ± 1.62 nm and 11.97 ± 1.59 nm respectively, Fig. 3D. This is probably related to the leached Fe(II) that generated several vacancies and surface defects, slightly decreasing the average diameter of the particles. Raman spectra, Fig. 3C, were obtained as well. The magnetite crystal belongs to the cubic space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, and thus, five Raman active modes (one A1g band, one Eg and three T2g) are predicted. In the Raman spectra of MgNPs, only the Td sites occupied by Fe(III) ions contribute to Raman activity while Fe(II) is not directly involved. The most intense band of magnetite, MgNPs, is observed at 668 cm−1, assigned to the A1g vibrational mode. Other three phonon modes have been reported for magnetite at 194 cm−1 (T2g), 303 cm−1 (Eg) and 528 cm −1 (T2g), leading to weak bands usually masked by the poor signal/noise ratio in this region. Thus, after the oxidation process, the broad bands at 370, 460, 721 and 1404 cm−1 in the Raman spectra are consistent with the formation of maghemite, MghNPs. As a matter of fact, it is possible to see the νC–C stretching from the stearate ligand for both iron oxide phases, which was confirmed by FTIR spectroscopy in Fig. S7 (see ESI†) and some impurities directly involved with small ligand degradation denoted by *, Fig. 3C.
m, and thus, five Raman active modes (one A1g band, one Eg and three T2g) are predicted. In the Raman spectra of MgNPs, only the Td sites occupied by Fe(III) ions contribute to Raman activity while Fe(II) is not directly involved. The most intense band of magnetite, MgNPs, is observed at 668 cm−1, assigned to the A1g vibrational mode. Other three phonon modes have been reported for magnetite at 194 cm−1 (T2g), 303 cm−1 (Eg) and 528 cm −1 (T2g), leading to weak bands usually masked by the poor signal/noise ratio in this region. Thus, after the oxidation process, the broad bands at 370, 460, 721 and 1404 cm−1 in the Raman spectra are consistent with the formation of maghemite, MghNPs. As a matter of fact, it is possible to see the νC–C stretching from the stearate ligand for both iron oxide phases, which was confirmed by FTIR spectroscopy in Fig. S7 (see ESI†) and some impurities directly involved with small ligand degradation denoted by *, Fig. 3C.
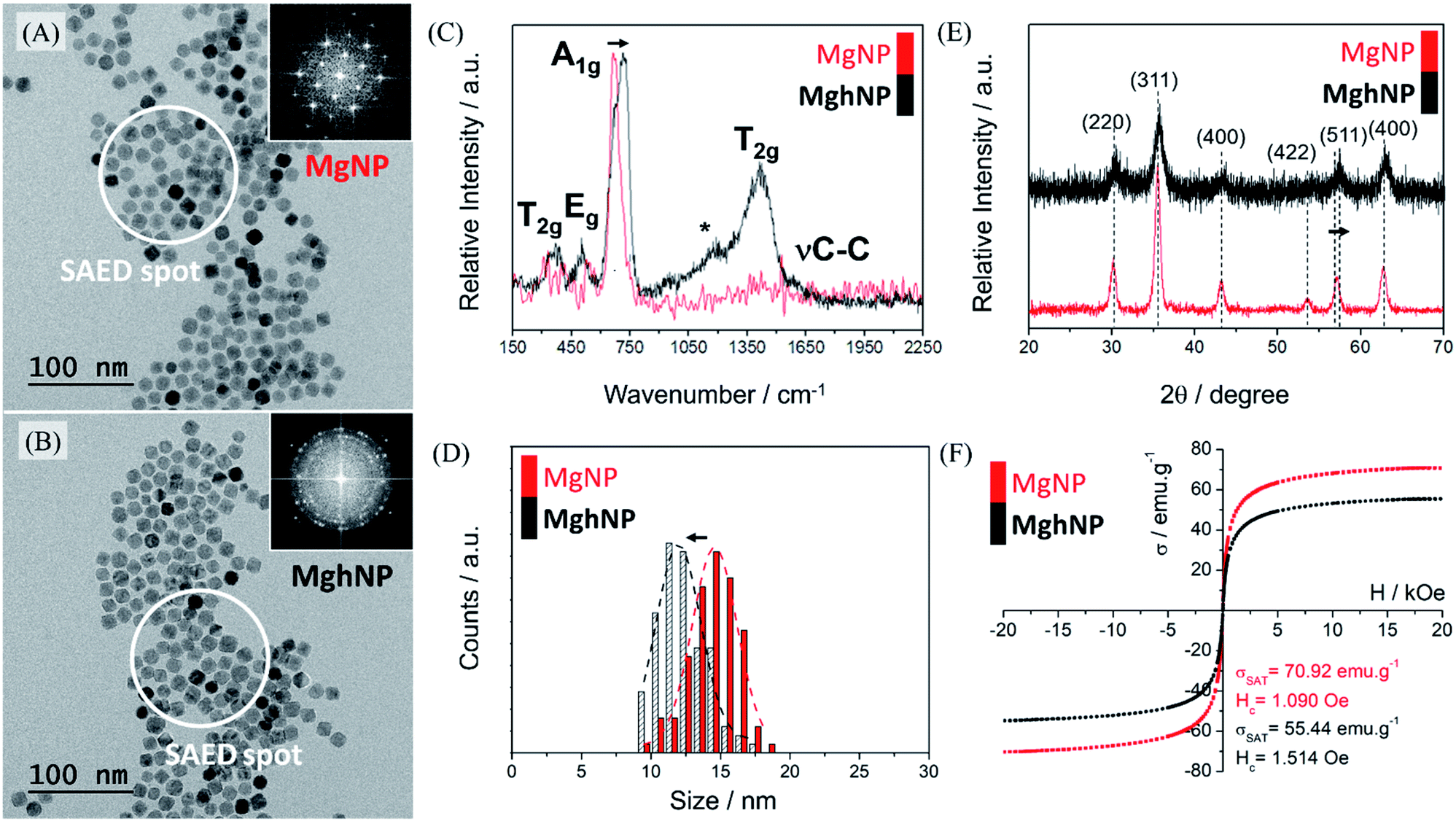 | ||
| Fig. 3 (A and B) HRTEM micrographs of MgNPs (magnetite, Fe3O4) and MghNPs (maghemite, γ-Fe2O3) respectively; insets show SAED measurement for both materials. (C) Raman spectra of MgNPs and MghNPs with active modes assigned. (D) Statistical average diameter of MgNPs and MghNPs (1000 particles). (E) Powder diffraction of MgNPs and MghNPs with diffraction planes assigned. (F) VSM measurements of MgNPs and MghNPs indicating the slight decrease of the magnetic response after the oxidation process as well as the slight increase of the coercivity field. | ||
The powder diffraction of MgNPs and MghNPs has indicated the spinel structure of both materials and the successful oxidation too by the (511) shifts, Fig. 3E.44 A Rietveld refinement confirmed the quasi-total phase transition, Fig. S8 (see ESI†). As easily shown by DRX and specific area electron diffraction, SAED, in Fig. 3A and B, inset, and in Fig. S6I and S6II (see ESI†), the crystallinity of MghNPs is not so good compared with MgNPs. However, this was expected because of the vacancy and defect formation at the surface. In addition, VSM measurements, Fig. 3F, showed the maintenance of superparamagnetic behavior, since the coercivity field is lower than 12.5 Oe,45 and a high loading magnetic content before and after the oxidation process.
Synthesis and characterization of QDs
Cadmium telluride quantum dots were synthesized as previously reported by our group.22 In general, the biggest problem of the synthesis in aqueous medium is the competition between the cadmium hydroxide formation and the Cd complex precursor stabilization. Both are directly linked with the medium pH. As we shown, the existence of a polymer as a precursor explains two aspects of the 3-mercaptopropionate-based quantum dot nucleation process.Firstly, the alkaline solution addition should be done slowly to prevent the Cd–S–Cd bond breaking and the formation of Cd(OH)2. In this case, the gradual MPA deprotonation leads to better stabilization of the polymer, which will be the precursor for the CdTe nucleus formation. The second point is related to the (CdTe)2 monomer concentration and stabilization. With the addition of telluride to this precursor, the telluride anions enter the Cd coordination sphere forming Cd2Te2(MPA)2 complexes, which are responsible for the supersaturation. A higher mercaptopropionic acid concentration leads to the stabilization of the Cd-MPA precursor and consequently more efficient formation and stabilization of monomers.
Accordingly, the supersaturation value rises with the ligand concentration leading to a burst nucleation. At the end of the nucleation step the higher MPA concentration at the nanocrystal surface hinders the growth step. Therefore, this shows that the quantum dot size, and consequently their optical properties, can be fine-tuned by changing the ligand concentration. In this way, by carefully controlling the variables, highly luminescent quantum dots can be successfully designed, and their properties are precisely controlled. By changing the Cd:3-MPA ratio we were able to synthesize QDs with emission wavelengths ranging from 514 to 650 nm. This control is essential to the efficiency of the hybrid MPI probe since we can choose the more suitable QDs to avoid energy transfer to the magnetic nanoparticles. Fig. 4 shows that the resulting spherical synthesized nanoparticles are regular, homogeneous and well-crystalline. Generally speaking, PL from ‘‘pure’’ quantum dots arises from the radiative band-edge recombination.
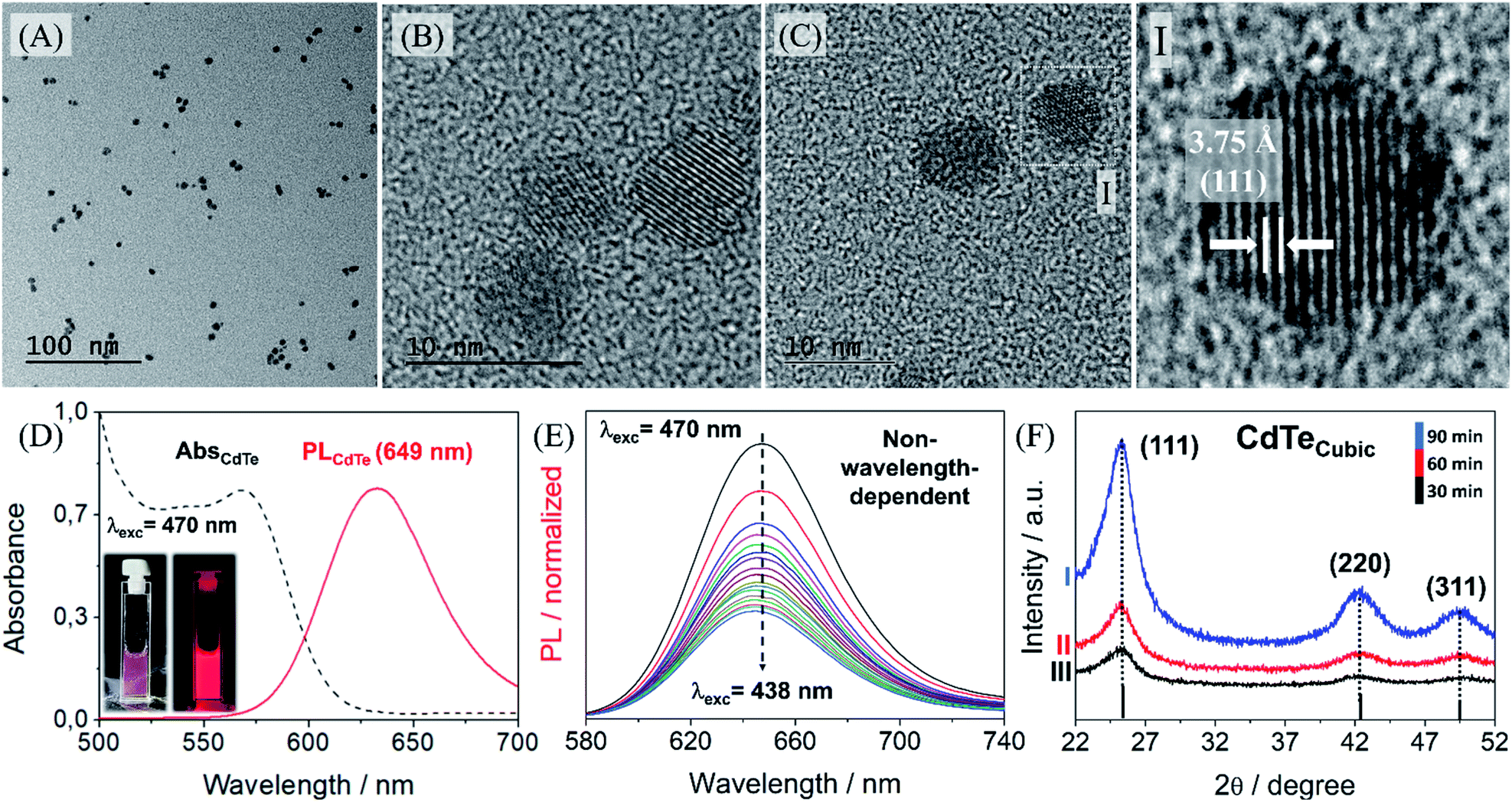 | ||
| Fig. 4 (A–C) TEM micrography of quasi-spherical cadmium telluride nanoparticles, QDs, highlighting the interplanar distance of (111) according to JCPDS 15-0770 (I). (D) Absorption and emission spectra of QDs; inset showing the QD suspension under visible excitation, left, and under 470 nm excitation, right. (E) Emission spectra under different excitation wavelengths, from 470 nm to 438 nm, showing a radiative band-edge recombination without trap states. (F) Power diffraction of the blende CdTe-cubic taken during different synthesis times. | ||
Upon absorption of a photon, an electron from the valence band is promoted to the conduction band leaving a hole behind, and the following recombination results in the emission of a photon, the so-called fluorescence. This is the main mechanism of PL in the case of defect-free and impurity-free quantum dots, in which no interstates within the bandgap exist. In these cases, the PL properties are entirely governed by the quantum confinement effects. Since the exciton confinement effects confer size-dependence, spectral heterogeneity may appear due to the distribution of particle sizes, with some influence of variation in the composition of surface-modifying groups. In our case the PL emission band is not so broad, as we can note in Fig. 4D, varying mainly from 630 nm to 680 nm. Another particularity of these in-house quantum dots is their excitation-independent emission, Fig. 4E. This means that the surface traps are minimal and allowed us to optimize their optical features by means of the precisely optimized RGB analysis, as previously reported22, Fig. S9 (see ESI†). This has allowed us to select a red-emitting QD, 649 nm, to maximize the reduction of the overlap between the emission spectra of QDs and the absorption spectra of maghemite nanoparticles. Besides, these QDs have a cubic structure containing three major peaks, 111, 220 and 311 that confirm their good crystallinity and purity, Fig. 4F. As we proceeded with the synthesis, FTIR showed efficient functionalization of the cadmium telluride quantum dots in Fig. S10 (see ESI†).
Engineering of MghNP@QD
Because of the unique physical properties, nanoparticles are often described as artificial atoms. The ability to assemble these artificial atoms into desired and higher-ordered architectures may open a new way to fabricate multifunction materials of interest for different applications. Previous approaches for controlling the size and shape of artificial atom assemblies include spray drying, DNA-induced assembly and solvophobic interaction. On the basis of this last example, it is common to use organic-based iron oxide nanoparticles as building blocks to produce water-based iron oxide nanoparticle-micelles. The formation of nanoparticle-micelles is always driven by hydrophobic van der Waals interaction between the hydrocarbon chain of the iron oxide nanoparticle ligand (e.g., oleic acid) and the hydrocarbon chain of some surfactants (e.g., dodecyltrimethyl-ammonium bromide, DTAB, C15H34BrN).46–48 As described, after the nanoparticle-micelles were introduced into polyvinylpyrrolidone, PVP (C6H9NO)n, in ethylene glycol (C2H6O2) solution, the van der Waals interactions between nanoparticles, ligands and surfactants are weakened, and then, nanoparticle-micelles decomposed due to the loss of DTAB molecules in the solution.49 As a result, a solvophobic interaction between nanoparticle ligands and PVP in ethylene glycol solution is induced, leading to nanoparticle aggregation and formation of superstructures.However, there are many problems with this approach for the present purposes. The first thing we must consider is related to the oleic acid as an iron oxide ligand as we have already mentioned. Its wax form is difficult to manipulate and purify. Besides, it is expensive and not environmentally friendly. That was the major reason why we decided to develop a new iron oxide synthesis with stearic acid. Iron stearate is much easier to work with and to purify, mainly because of its solid form. The second thing we have to think about is related to quantum dot synthesis. Hot injection approaches are not suitable for up-scaling, neither the use of unstable trioctylphosphines. For that reason, the water-based approach is much more convenient both from the environmental point of view and facility, since it has already been systematized previously.22 So the challenge is to make organic-based iron oxide nanoparticles compatible with water-based quantum dots, thus allowing the magnetic momentum of the core and the fluorescence features of the shell to be maximized. For these purposes, a good strategy is using a compatibilizer, e.g., hexadecyltrimethyl-ammonium bromide, CTAB, to attach both quantum dots and nanoparticles.
We have already used CTAB as a linker between water-based maghemite nanoparticles and water-based quantum dots, by means of a side chain interaction.22 However, it is necessary to maximize the total magnetic momentum of the nanoparticles. A possible solution would be controlling the aggregation process. As a matter of fact, one can put several iron oxide nanoparticles together, by means of solvophobic interactions using the electrostatic properties to attach quantum dots at the surface of such multi-magnetic cores and then, maximizing their magnetic response.
For our purposes, it is mandatory to keep the surfactant at the surface of the multi-core nanoparticles because of the negatively charged cadmium telluride quantum dot. In that way, losing CTAB molecules at the multi-magnetic core would not be convenient and that was the reason why we decided to optimize the uses of CTAB and not DTAB. Despite it has already been reported that CTAB nanoparticle-micelles remain nearly unchanged in ethylene glycol solution and no spherical nanoparticle assembly is formed,49 we have examined a large-area TEM micrograph and found that even using 20 g L−1 CTAB solution, certain agglomeration was produced, Fig. S11 (see ESI†).
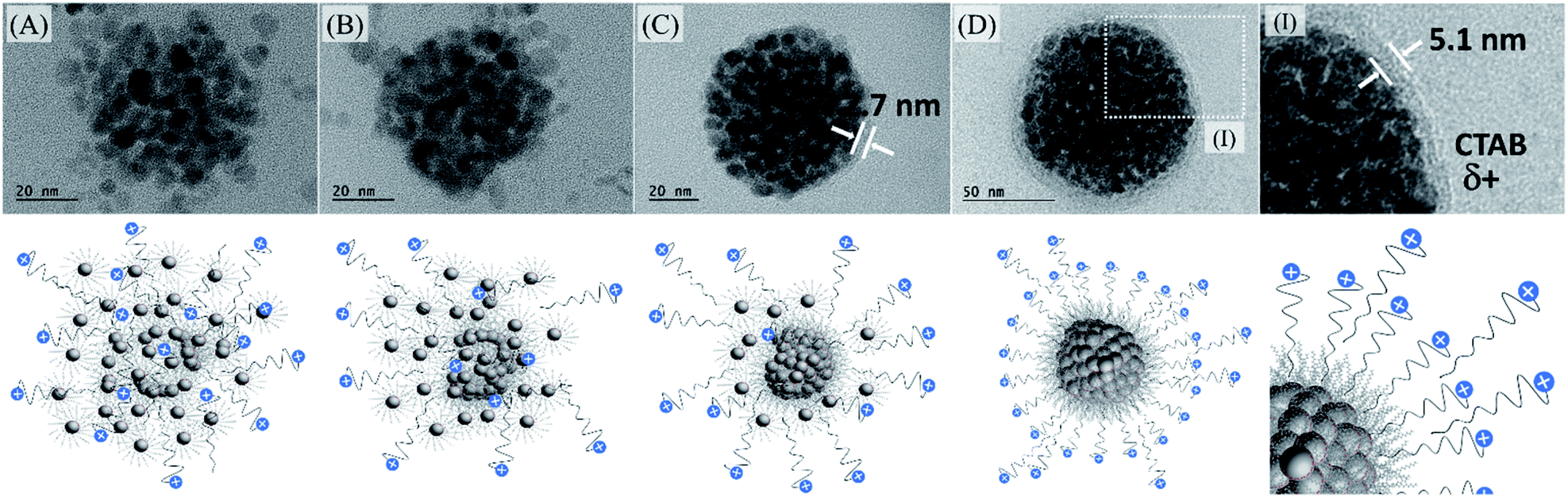
We also observed that at smaller surfactant concentration, some agglomeration occurred without the use of PVP in ethylene glycol. After iterative centrifugations, at 14000 rpm for 30 minutes, in the presence of acetone, it is possible to concentrate the multi-magnetic core encompassing the organic-based iron oxide nanoparticles surrounded by CTAB with +30 mV zeta potential, to be used in the next step. Probably, as shown in Fig. 5, it is related to CTAB availability to recover all individual iron oxide nanoparticles. As we increase the CTAB concentration, more CTAB molecules are available to stabilize individual nanoparticles. However, as we decrease the CTAB concentration, fewer CTAB molecules are available to stabilize individual nanoparticles and, in that case, the iron oxide nanoparticles tend to agglomerate and tend to be at the surface of them.
 | ||
| Fig. 5 (A–D) HRTEM micrographs of the multi-magnetic core of maghemite nanoparticles according to different CTAB solution concentrations, 15, 11, 8 and 5 g L−1 respectively. As the amount of CTAB molecules decreases, the agglomeration becomes favored. (I) Digital zoom in highlighting the CTAB coating. | ||
So, it is quite important to say that there is a specific organic-based magnetic nanoparticle:CTAB ratio used to obtain this type of system in an optimized way (under intellectual protection). For this reason, just working on different magnetic nanoparticle:CTAB ratios does not allow the system to be controlled and several nanoparticles can be outside the agglomerated ones producing irregular spheres or will be most individually stabilized, Fig. S12 (see ESI†). In that way, establishing the ideal condition for the multi-magnetic core formation, and iterative precipitation and redispersion, the best conjugation of QDs with MghNPs has been achieved, MghNP@QD. As shown in Fig. 5DI, CTAB provides a convenient linker due to its long aliphatic carbon chain which decreases extensively the FRET mechanism between quantum dots and stearate-functionalized maghemite.
It is important to say that the Förster critical distance, from 4 to 8 nm,11 is mandatory to prevent charge-transfer interactions. Another point is that CTAB is not completely organized at the surface. It can impose a non-regular coverage of QD, that is attracted to the positively charged multi-magnetic core, Fig. 6, before precipitating with acetone. However, for our purposes, this is not a problem and can be even convenient during the washing process in the real MPI test as we will show later. This new class of clustered multifunctional magneto-fluorescent superstructures (117.0 ± 4.8 nm, Fig. S13, see ESI†) was quite well characterized as we can see in Fig. 6. First, by means of energy-dispersive X-ray spectroscopy, it was possible to show several chemical differences between EDS spot X and EDS spot Y. In EDS spot X we found many elements that are directly involved with the chemical nature of organic-based maghemite nanoparticles, e.g. carbon from stearate recovery, and iron and oxygen from the maghemite core. Besides, it is possible to see sodium, copper and silicon probably from the grid and salts used in the synthesis. Weak signals assigned as sulphur and cadmium are related to quantum dot nanoparticles that are above the multi-magnetic core and were detected by their secondary electrons.
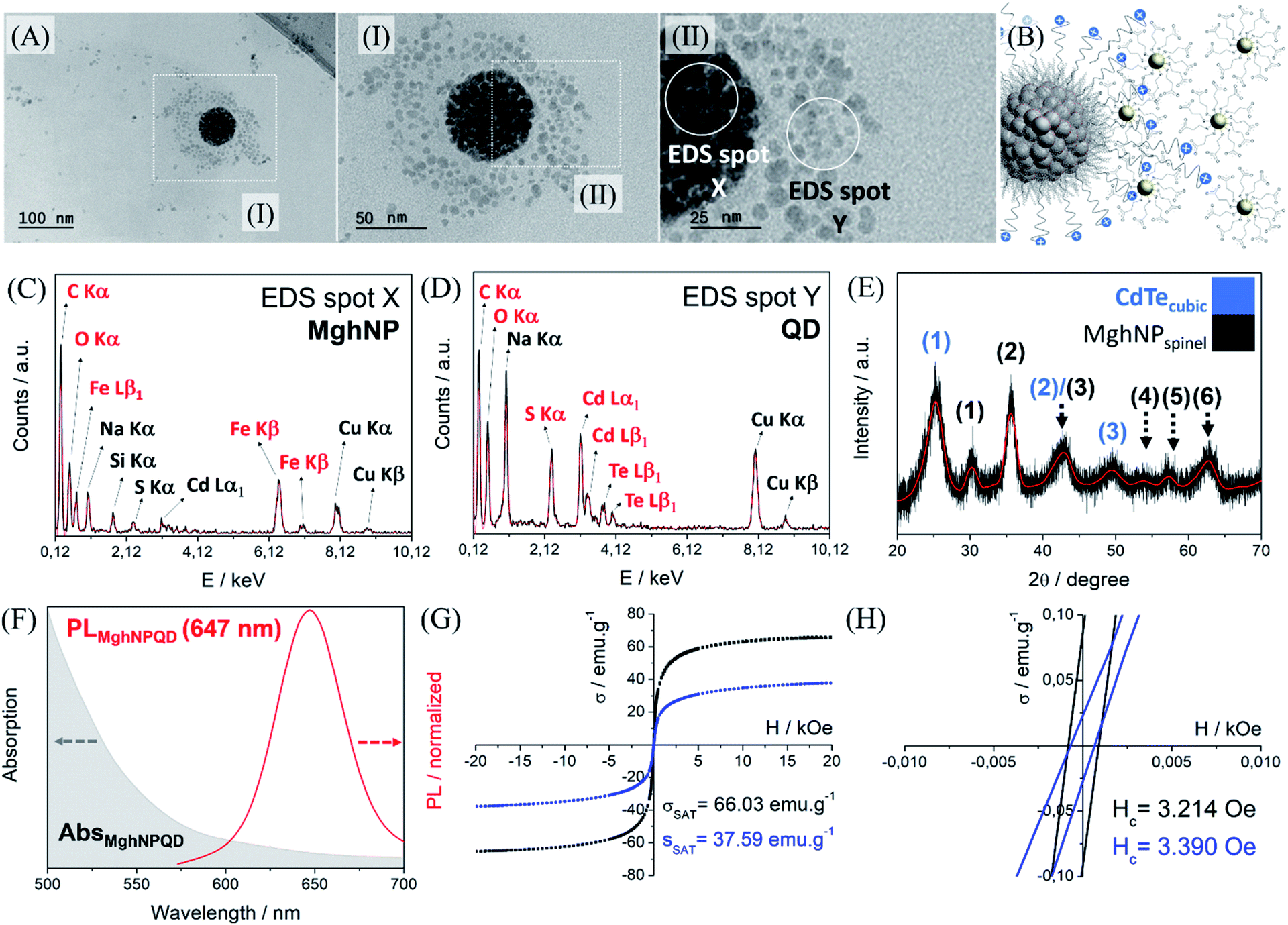 | ||
| Fig. 6 (A) TEM micrographs of the MghNP@QD. (I and II) Zoom in of MghNP@QD highlighting two different regions where EDS measurement was performed. (B) Schematic representation of the clustered multifunctional magneto-fluorescent superstructure, MghNP@QD. (C and D) EDS measurement took from MghNP@QD in two different regions as indicated before. In red, the elements directly coming from the analyzed nanoparticles. In black, the elements not directly coming from the analyzed nanoparticles. (E) Powder diffraction of MghNP@QD indicating the presence of both nanoparticles. (F) Absorption in the UV-vis region of MghNP@QD and its respective emission spectra. (G) VSM analysis of multi-magnetic core maghemite–micelle, in black, and of MghNP@QD, in blue. (H) Coercivity field of the multi-magnetic core maghemite-micelle, in black, and of MghNP@QD, in blue. | ||
In EDS spot Y we found many elements that are directly involved with the chemical nature of cadmium telluride nanoparticles, e.g. carbon, oxygen and sulfur from mercaptopropionate coverage, and cadmium and telluride from the inorganic core. Sodium and copper are present in the analysis most from the salt impurity and grid. Powder diffraction shows the presence of a cubic structure containing three major peaks, 111, 220 and 311, (1), (2) and (3), in blue, and the presence of a spinel structure from maghemite, in black, Fig. 6E. It is important to see that this clustered multifunctional magneto-fluorescent superstructure was designed according to some quantum restrictions. To minimize quenching mechanisms in MghNP@QD, it was decided to use red-emitting quantum dots to distance the maghemite absorption spectra from quantum dot emission spectra, Fig. 6F. These results show that even in the case of maghemite, a certain degree of FRET effect occurs because of the small overlap between the emission spectra of QDs and the absorption spectra of maghemite nanoparticles. By comparing with the quantum dot emission spectra, Fig. 4D, we observed a subtle blue shift. This means that quantum dots do not suffer significant interference from the photon absorber core.
Moreover, it is possible to note that the magnetic properties are preserved (see ESI,† superparamagnetism vs. monodomain), like the superparamagnetic behavior, even after the hybrid nanoprobe formation, Fig. 6G and H. By comparing with the magnetic response of individual organic-based maghemite nanoparticles, Fig. 3F, 55.44 emu g−1, it was found that the saturation magnetization of the multi-magnetic core of maghemite is higher, 66.03 emu g−1. This had already been expected since we are increasing the magnetic momentum of the core as we agglomerate them. For the clustered multifunctional magneto-fluorescent superstructure, the saturation magnetization is still high enough for our purposes. In this way, we believe that their full potential has been achieved, fulfilling the criteria of a high magnetic response in the presence of an external magnetic field, tunable and uniform sizes, long-term colloidal stability, and optimized fluorescence.
The clustered multifunctional magneto-fluorescent superstructure should be purified before its use since it is made by iterative precipitation with acetone and redispersion in water. At the end of this process, we used a magnetic gradient to separate the un-reacted multi-magnetic core (higher magnetic response) from the MghNP@QD (lower magnetic response). By comparing with our previously reported multifunctional material,22 there is a great improvement in the magnetic response and the quantum yield increased from 69% to 78% (see ESI†).
Real MPI test of MghNP@QD
The use of nanoparticles can greatly improve the spatial resolution of inspection baths, allowing us to detect the occurrence of sub-micrometric flaws. Another advantage is related to the lowest concentration necessary to identify the shallow subsurface (around 10−6 mol L−1) and the absence of a background signal, which is one of the problems of the conventional probes, Fig. S14 and S15 (see ESI†).50 In order to test the MghNP@QD as the MPI agent, we have used an official high coercivity magnetic stripe for the MPI bath, with inter stripe separation of 300 μm as shown in Fig. 7A. We dropped an acetone suspension containing MaghNP@QD on the card and waited a few seconds, until most of the acetone evaporated. The study of the magnetophoretic response of this system and the time taken by the particles to go to the magnetic strip is seen in Fig. S16 (see ESI†). Our agent has a superparamagnetic behavior, as we have shown in Fig. 6G and H, and this is another advantage compared with conventional ones, since it does not retain any magnetic information and can be easily washed away after removing the applied field. In this way, we are avoiding any background noise, Fig. 7V and S14 and S15 (see ESI†).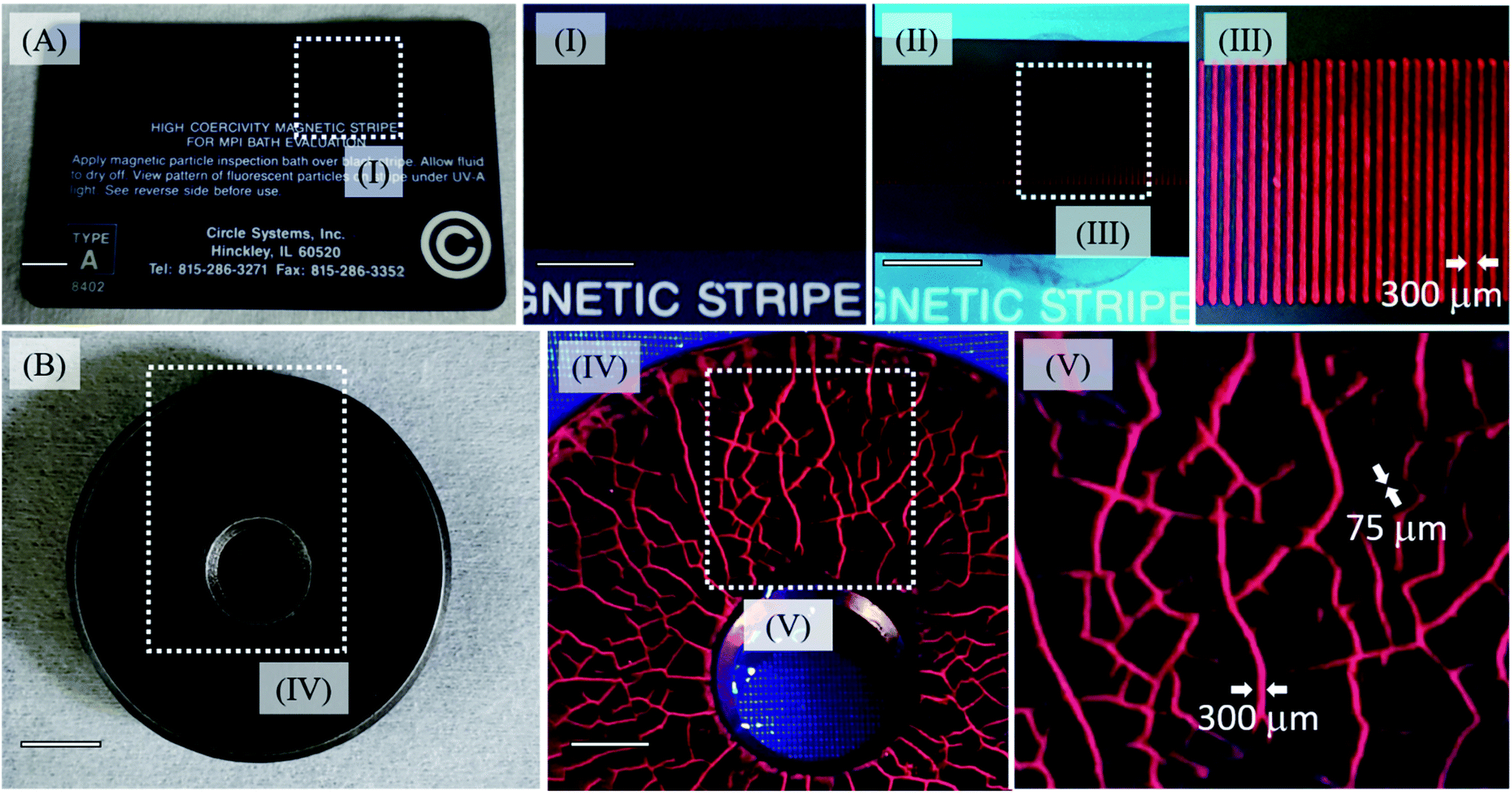 | ||
| Fig. 7 (A) Official high coercivity magnetic striped card for MPI bath evaluation (0.3 Tesla) highlighting magnetic lines before MghNP@QD uses (I) and after MghNP@QD uses (II). Under 470 nm excitation, no background noise was detected, (Fig. S15A†) and magnetic lines were clearly identified (III). (B) Real reference block (disc with coarse and fine cracks in the surface, produced by grinding and stress corrosion, see ESI† for more information) under visible light irradiation highlighting the shallow subsurface, in the presence of MghNP@QD under 470 nm excitation, clearly identified with no background noise (Fig. S15B†) and high resolution. | ||
Besides, because of the chemical nature of the MghNP@QD formation, the electrostatic force can break under water washing. It helps us to separate the magnetic content and fluorescent one to be discarded properly after the uses. For this reason, using acetone as a solvent to produce MghNP@QD becomes quite significant. Moreover, it is important to say that since the magneto-fluorescent samples were sealed in a vessel, the composite was kept suspended in acetone for 5 minutes. It is not a problem because before application as the NDT probe, the user must shake the aerosol to guarantee the resuspension of the nanocomposite as recommended by ASTM E1444/E1444M – 16e1 Standard Practice for Magnetic Particle Testing. Once we produce the final material, it is going to maintain its structure for a long time (more than 10 months) if we keep this composite in acetone and under 30 °C. This test corroborates the potential use of the magneto-fluorescence nanoparticles in water-bath MPI tests, as well as for capturing magnetic information, and processing magnetic codes and images for many industrial applications. To evaluate the real applicability of the new clustered multifunctional magneto-fluorescent superstructure synthesized in this work, we performed a standard test according to the NDT procedure, Fig. 7B, using a reference block (disc with coarse and fine cracks in the surface, produced by grinding and stress corrosion). The multifunctional material shows an excellent capability to recognize the flaws. In addition, the concentration of the fluorescent species in the new class of clustered multifunctional magneto-fluorescent superstructure is much lower in relation to the conventional MPI agents (e.g. typically around 1.0 g L−1). It is important to say that, although the (Cd, Te) elements in the quantum dots can be cytotoxic, in the nanoparticulated form this will depend on several factors, such as size, charge, concentration, outer capping material bioactivity and oxidation, photolytic response, and mechanical stability. For that reason, there are many aspects to investigate, including its possible bio-uses, such as the capability to activate multiprotein oligomers responsible for activation of inflammatory processes called inflammasome51 in a theranostic approach. In that way, we cannot say it will make its use unviable as a NDT probe.
Conclusions
In summary, a new class of clustered multifunctional magneto-fluorescent superstructures was achieved. The strategy consisted of the solvophobic-controlled assembly of organic-based maghemite cores and water-based II–VI quantum dots, in the presence of hexadecyltrimethyl-ammonium bromide as a compatibilizer agent. This nanoprobe exhibited a higher magnetic core response compared with the last nanoprobe published previously22 (from 47.9 emu g−1 to 63.0 emu g−1), tunable and uniform sizes, long-term structural stability in acetone compared with the previous paper,22 and optimized fluorescence for real-time non-destructive detection of flaws and shallow subsurface discontinuities in industrial ferromagnetic materials with no background signal. For the very first time, this specific nanocomposite reported here showed an excellent capability to recognize the defects, exposing the precise fracture border location in a real proof body by means of visual fluorescent detection with no background noise compared with the previous paper22 at a low concentration and with a high magnetic load, holding a highly competitive performance in relation to the existing detection probes on the market.Experimental section
MghNP synthesis
First, the iron–stearate complex was prepared by reacting iron chloride and sodium stearate in a 1:3 ratio respectively. In a typical synthesis of the iron–stearate complex, 10.8 g of iron chloride (FeCl3·6H2O, 40 mmol, Sigma-Aldrich) and 5.8 g of sodium stearate (C18H35NaO2, 120 mmol, Sigma-Aldrich) were dissolved in 400 mL of water. The resulting suspension was heated to 70 °C and kept at that temperature for 5 hours. When the reaction was completed, the iron–stearate complex was filtered and washed several times with a mixture of ethanol and water at a 2:3 ratio, respectively. After washing, the final complex was left in an oven for 48 h. Then, 900 mg (1 mmol) of the as-prepared iron–stearate was dissolved in 40 mL of octadecene (Sigma-Aldrich) in a three-neck round-bottom flask under mechanical stirring, 400 rpm, and heated for 60 minutes at 120 °C. After this, a mixture (50:50 ratio, % w/w) of sodium oleate:oleic acid (Sigma-Aldrich) was carefully added and the mixture was heated to 200 °C for 60 minutes. The system was heated again to 320 °C for 60 minutes. After cooling, the purification was carried out by successive cycles of precipitation and dispersion of the nanoparticles by using ethanol and chloroform respectively. Then, the organic-based magnetite nanoparticles, MgNPs, in 20 mL of chloroform, were kept under vigorous air bubbling (2 L min−1). The procedure was repeated five times to obtain organic-based maghemite nanoparticles, MghNPs.
QD synthesis
Briefly, in a 125 mL three-neck round-bottom flask, a desired amount of 3-mercaptopropionic acid (Sigma-Aldrich) was added to a 5 mmol L−1 cadmium acetate solution (Labsynth) in the 1:2.3 ratio (Cd:MPA) and the pH of that was adjusted to 8.9 using 1 mol L−1 NaOH (Labsynth). This solution was deoxygenated for 30 minutes and heated up to the water refluxing temperature. Besides, into a 50 mL Schlenk flask, tellurium 200 Mesh (6.1 g, 48 mmol), sodium (2.2 g, 96 mmol) and naphthalene (0.1 g, 0.8 mmol) were stirred in THF (20 mL) for 24 hours at 60 °C. After this time, the reaction medium was filtered, and the crude product was washed with THF (20 mL) and then dried under vacuum. After that, a 2.5 mmol L−1 sodium telluride solution (it can be substituted by commercial sodium tellurite, Na2TeO3) was prepared and added abruptly to the Cd(II)/ligand solution. The reaction was monitored in real-time and proceeded for 140 min under refluxing.
MghNP@QD synthesis
In a typical synthesis of the clustered multifunctional magneto-fluorescent superstructure, after obtaining MghNPs and QDs, 1 mL of chloroform solution containing 2.5–4.0 mg 12 nm MghNPs was injected into 1.0 mL CTAB aqueous solution (5.0–8.0 mg mL−1 in Nanopure water). The solution was thoroughly mixed by vortex for 5 s. After removing the chloroform from the mixture by blowing N2 at 40 °C, a dark-turbid solution of MghNP-micelle aqueous solution was obtained. Then, 1.0 mL of acetone was added, and the MghNP-micelle solution was centrifuged (14000 rpm, 30 min). This procedure was repeated twice. The supernatant was withdrawn and the solid was dispersed in 4 mL water. After that, the desired amount of QDs was added (400 μL to 4 mL, 25 g L−1) to the above-mentioned MghNP-micelle solution and gently shaken for 12 h. The resulting clear suspension was precipitated by using acetone and dried in a hood under room temperature.
Real-time kinetics study
The formation kinetics of QDs were monitored in real-time using MasterView® software as previously reported.22 Generally, a conventional webcam was used for collecting the transmitted and reflected light from the reaction system. The signal generated by the CCD camera was sent to the graphics processing unit of the computer. From these signals, the software reconstructs the image and stores the average data from the red, green and blue, RGB, channels, as a function of time in a predetermined spatial region. The data collection covered all the synthetic procedures, and the RGB spectral evolution was displayed in real-time. To minimize the QD excitation and the fluorescence interference in the RGB spectra, a light bulb with maximum emission between 600 and 800 nm was used as the illumination source. This way, only the scattered and transmission light is collected by the CCD and the RGB time evolution is related to changes in the QD suspension color. So, during the formation of nanoparticles, light is absorbed and scattered at distinct intensities and wavelengths, depending on the particles size and composition. Consequently, the RGB counts start to increase or decrease depending on the contents of scattering and absorbing materials in the three-neck flask, allowing us to obtain specific water-based quantum dots.22Real MPI test
By using a high coercivity magnetic stripe for MPI bath evaluation, type A, 8402, provided by Circle Systems and a Reference Block, provided by Metal-Chek do Brasil (Brazilian company specialized in NDT and MPI) the MghNP@QD colloidal solution (10−6 mol L−1) was dropped onto the surface and analyzed after acetone vaporization. The reference block was made of steel (Grade 90MnCrV8) hardened at 860 °C for 2 h and quenched in oil to give a surface hardness of 63 HRC to 70 HRC.Materials characterization
The fine particle morphology was analyzed by high resolution transmission electron microscopy (HRTEM) on a JEOL, model JEM 2100, operating with a LaB6 electron emitting filament, with a maximum acceleration voltage of 200 kV, using a drop casting technique (electron diffraction, SAED, was carried out using the same JEM 2100). Magnetic hysteresis curves were obtained using a vibrating sample magnetometer manufactured by EG&G Princeton Applied Research, model 4500, using magnetic fields of up to 20 kOe (saturation field). The electromagnet used was the Walker Scientific HR8 model. The Gauss meter used was Lake Shore-model450. The diffraction patterns were obtained on a Bruker D8 Discover facility. The measurement was obtained with a primary twin (0.6 mm; primary axial Soller) and a secondary twin (maximum aperture 5 mm; secondary axial Soller and nickel filter in 1D mode), with 0.05 as an increment and 1.5 as a step from 20 to 70 degrees and Rietveld refinement was performed using MAUD 2.91 software and CIF files were analyzed using VESTA 3.4.7 software. Electronic absorption spectra were recorded on a diode-array Hewellet-Packard 8453A spectrophotometer. Photoluminescent emission was recorded on Photon Technology equipment, using an InGaAs detector and FelixGX 1317 software. Infrared spectra were recorded on an ALPHA Bruker FTIR spectrometer (KBr pellets, 96 scans) and Raman spectra were recorded on a WITEC confocal microscope, with an excitation wavelength of 532 nm, 0.1 mW of laser power, and integration time of 2 s. Scanning fluorescence was carried out on a WITec Raman confocal microscope, using 488 nm excitation and 2.9 mW of laser power (170 points per line, 180 lines per image, scan width of 170 μm and scan height of 180 μm). Sample irradiation and collection of the backscattered radiation were performed using the same microscope objective (Olympus, 100×).Author contributions
Fernando Menegatti de Melo: conceptualization, data curation, formal analysis, investigation, project administration, writing – original draft and writing – review & editing; João Victor Mattioni: data curation, formal analysis, investigation; Fernando Dias: conceptualization, methodology, resources; Ying Fu: conceptualization, formal analysis, methodology, writing – original draft; Henrique Eisi Toma: conceptualization, funding acquisition, project administration, supervision, writing – original draft.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We greatly acknowledge the support from the São Paulo Research Foundation (FAPESP) (Grant 2018/21489-1; Grant 2019/14771-5), the National Council for Scientific and Technological Development (CNPq) (Grant 482383/2013-5), the Research Development Foundation (FUNDEP) (Grant SIBRATECnano – ciclo03/2017) and Metal-Chek do Brasil (MC) (Grant 5415/17.1.00778.46.8). The authors greatly acknowledge Alfredo Duarte who obtained several HRTEM images for us and Lucas Quartarolli for the helpful tips with Rietveld refinement. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.References
- W. H. Suh, Y. H. Suh and G. D. Stucky, Nano Today, 2009, 4, 27 CrossRef CAS.
- R. F. Gibson, Compos. Struct., 2010, 92, 2793 CrossRef.
- J. E. Lee, N. Lee, T. Kim, J. Kim and T. Hyeon, Acc. Chem. Res., 2011, 44, 893 CrossRef CAS PubMed.
- N. C. Bigall, W. J. Parak and D. Dorfs, Nano Today, 2012, 7, 282 CrossRef CAS.
- D. V. Talapin, E. V. Shevchenko, M. I. Bodnarchuk, X. Ye, J. Chen and C. B. Murray, Nature, 2009, 461, 964 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204 CrossRef CAS PubMed.
- T. Wang, J. Zhuang, J. Lynch, O. Chen, J. Wang, X. Wang, D. LaMontagne, H. Wu, Z. Wang and Y. C. Cao, Science, 2012, 338, 358 CrossRef CAS.
- F. X. Redl, K. S. Cho, C. B. Murray and S. O'Brien, Nature, 2003, 423, 968 CrossRef CAS PubMed.
- Y. S. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580 CrossRef CAS PubMed.
- Z. W. Quan and J. Y. Fang, Nano Today, 2010, 5, 390 CrossRef CAS.
- K. Chou and A. Dennis, Sensors, 2015, 15, 13288 CrossRef PubMed.
- Y. Lu, Y. Yin, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 183 CrossRef CAS.
- H. M. Johng, J. S. Yoo, T. J. Yoon, H. S. Shin, B. C. Lee, C. Lee, J. K. Lee and K. S. Soh, J. Evidence-Based Complementary Altern. Med., 2007, 4, 77 CrossRef.
- L. Levy, Y. Sahoo, K. S. Kim, E. J. Bergey and P. N. Prasad, Chem. Mater., 2002, 14, 3715 CrossRef CAS.
- X. Hong, J. Li, M. J. Wang, J. J. Xu, W. Guo, J. H. Li, Y. B. Bai and T. J. Li, Chem. Mater., 2004, 16, 4022 CrossRef CAS.
- M. Fang, P. S. Grant, M. J. McShane, G. B. Sukhorukov, V. O. Golub and Y. M. Lvov, Langmuir, 2002, 18, 6338 CrossRef CAS.
- Y. Okamoto, F. Kitagawa and K. Otsuka, Anal. Chem., 2007, 79, 3041 CrossRef CAS PubMed.
- F. J. Feher, K. D. Wyndham, D. Soulivong and F. Nguyen, J. Chem. Soc., Dalton Trans., 1999, 9, 1491 RSC.
- S. A. Corr, A. O'Byrne, Y. K. Gun'ko, S. Ghosh, D. F. Brougham, S. Mitchell, Y. Volkov and A. Prina-Mello, Chem. Commun., 2006, 43, 4474 RSC.
- C. Becker, M. Hodenius, G. Blendinger, A. Sechi, T. Hieronymus, D. Muller-Schulte, T. Schmitz-Rode and M. Zenke, J. Magn. Magn. Mater., 2007, 311, 234 CrossRef CAS.
- X. Z. Li, L. Wang, C. Zhou, T. T. Guan, J. Li and Y. H. Zhang, Clin. Chim. Acta, 2007, 378, 168 CrossRef CAS PubMed.
- F. M. Melo, D. Grasseschi, B. B. N. S. Brandão, Y. Fu and H. E. Tome, ACS Appl. Nano Mater., 2018, 1, 2858 CrossRef.
- Y. Sahoo, A. Goodarzi, M. T. Swihart, T. Y. Ohulchanskyy, N. Kaur, E. P. Furlani and P. N. Prasad, J. Phys. Chem. B, 2005, 109, 3879 CrossRef CAS PubMed.
- O. Chen, L. Riedemann, F. Etoc, H. Herrmann, M. Coppey, M. Barch, C. T. Farrar, J. Zhao, O. T. Bruns, H. Wei, P. Guo, J. Cui, R. Jensen, Y. Chen, D. K. Harris, J. M. Cordero, Z. Wang, A. Jasanoff, D. Fukumura, R. Reimer, M. Dahan, R. K. Jain and M. G. Bawendi, Nat. Commun., 2014, 5, 1 Search PubMed.
- C. E. Betz, Principles of Magnetic Particle Inspection, Magnaflux: Harwood Heights, IL, 1st edn, 1967 Search PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS PubMed.
- A. F. Morais, I. G. N. Silva, S. P. Sree, F. M. Melo, G. Brabants, H. F. Brito, J. A. Martens, H. E. Toma and C. E. A. Kirschhock, Chem. Commun., 2017, 53, 7341 RSC.
- T. Jamieson, R. Bakhshi, D. Petrova, R. Pocock, M. Imani and A. M. Seifalian, Biomaterials, 2007, 28, 4717 CrossRef CAS PubMed.
- A. Ekimov, J. Lumin., 1996, 70, 1 CrossRef CAS.
- R. He and H. Gu, Colloids Surf., A, 2006, 272, 111 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545 CrossRef CAS.
- G. Cotin, C. Kiefer, F. Perton, D. Ihiawakrim, C. B. Andujar, S. Moldovan, C. Lefevre, O. Ersen, B. Pichon, D. Mertz and S. B. Colin, Nanomaterials, 2018, 8, 881 CrossRef PubMed.
- F. M. Melo, N. Almeida, N. S. Uezu, C. Alberto, O. Ramirez, A. Domingues and H. E. Toma, J. Nanosci. Nanotechnol., 2017, 17, 1 CrossRef PubMed.
- F. M. Melo, A. T. Silveira, L. F. Quartarolli, F. F. Kaid, D. R. Cornejo and H. E. Toma, J. Magn. Magn. Mater., 2019, 487, 165324 CrossRef CAS.
- D. G. Silva, F. M. Melo, A. T. Silveira, B. C. Cruz, C. C. P. Prado, L. C. P. Vasconcelos, V. A. S. Lucas and H. E. Toma, J. Chem. Educ., 2016, 93, 1929 CrossRef.
- D. G. Silva, S. H. Toma, F. M. Melo, L. V. C. Carvalho, A. Magalhães, E. Sabadini, A. D. Santos, K. Araki and H. E. Toma, J. Magn. Magn. Mater., 2016, 397, 28 CrossRef.
- F. Y. Cheng, C. H. Su, Y. S. Yang, C. S. Yeh, C. Y. Tsai, C. L. Wu, M. T. Wu and D. B. Shieh, Biomaterials, 2005, 26, 729 CrossRef CAS PubMed.
- B. Feng, R. Y. Hong, L. S. Wang, L. Guo, H. Z. Li, J. Ding, Y. Zheng and D. G. Wei, Colloids Surf., A, 2008, 328, 52 CrossRef CAS.
- A. Jordan, R. Scholz, P. Wust, H. Schirra, S. Thomas, H. Schmidt and R. Felix, J. Magn. Magn. Mater., 1999, 194, 185 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS.
- F. X. Redl, C. T. Black, G. C. Papaefthymiou, R. L. Sandstrom, M. Yin, H. Zeng, C. B. Murray and S. P. O'Brien, J. Am. Chem. Soc., 2004, 126, 14583 CrossRef CAS PubMed.
- D. L. Faria, S. V. Silva and M. T. Oliveira, J. Raman Spectrosc., 1997, 28, 873 CrossRef.
- I. Chourpa, L. Douziech-Eyrolles, L. Ngaboni-Okassa, J. F. Fouquenet, S. Cohen-Jonathan, M. Soucé, H. Marchais and P. Dubois, Analyst, 2005, 130, 1395 RSC.
- W. Kim, C. Suh, S. Cho, K. Roh, H. Kwon, K. Song and I. Shon, Talanta, 2012, 94, 348 CrossRef CAS PubMed.
- D. L. Leslie-Pelecky and R. D. Rieke, Chem. Mater., 1996, 8, 1770 CrossRef CAS.
- H. Fan, E. Leve, J. Gabaldon, A. Wright, R. E. Haddad and C. J. Brinker, Adv. Mater., 2005, 17, 2587 CrossRef CAS.
- H. Xu, L. Cui, N. Tong and H. Gu, J. Am. Chem. Soc., 2006, 128, 15582 CrossRef CAS PubMed.
- A. K. Boal, F. Ilhan, J. E. DeRouchey, T. T. Albrecht, T. P. Russel and V. M. Rotello, Nature, 2000, 404, 746 CrossRef CAS PubMed.
- J. Zhuang, H. Wu, Y. Yang and C. Cao, Angew. Chem., 2008, 47, 2208 CrossRef CAS PubMed.
- P. J. Shull, Nondestructive Evaluation: Theory, Techniques, and Applications, CRC Press, 2002 Search PubMed.
- F. Liu, W. Ye, J. Wang, F. Song, Y. Cheng and B. Zhang, Int. J. Nanomed., 2017, 12, 5135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00149c |
| This journal is © The Royal Society of Chemistry 2021 |