
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the coupling between the components in a graphene–mesoporous germanium nanocomposite using high-pressure Raman spectroscopy†
Denis
Machon
 *abc,
Stéphanie
Sauze
ab,
Richard
Arès
ab and
Abderraouf
Boucherif
ab
*abc,
Stéphanie
Sauze
ab,
Richard
Arès
ab and
Abderraouf
Boucherif
ab
aInstitut Interdisciplinaire d'Innovation Technologique (3IT), Université de Sherbrooke, 3000 Boulevard Université, Sherbrooke, J1K OA5 Québec, Canada
bLaboratoire Nanotechnologies et Nanosystèmes (LN2), CNRS UMI-3463, Institut Interdisciplinaire d′Innovation Technologique (3IT), Université de Sherbrooke, 3000 Boulevard Université, Sherbrooke, J1K OA5 Québec, Canada
cUniv Lyon, Université Claude Bernard Lyon 1, CNRS UMR 5306, Institut Lumière Matière, F-69622 Villeurbanne, France. E-mail: denis.machon@univ-lyon1.fr
First published on 10th March 2021
Abstract
The nature of the interface between the components of a nanocomposite is a major determining factor in the resulting properties. Using a graphene–mesoporous germanium nanocomposite with a core–shell structure as a template for complex graphene-based nanocomposites, an approach to quantify the interactions between the graphene coating and the component materials is proposed. By monitoring the pressure-induced shift of the Raman G-peak, the degree of coupling between the components, a parameter that is critical in determining the properties of a nanocomposite, can be evaluated. In addition, pressure-induced transformations are a way to tune the physical and chemical properties of materials, and this method provides an opportunity for the controlled design of nanocomposites.
Introduction
With the advent of graphene synthesis, an avenue in nanocomposite science and technology has emerged with the opportunity to modify the surface chemistry of materials by coating the surface with graphene.1 This opened up the field of graphene-based nanocomposites with applications in energy and environmental areas, among others.2–5 For instance, practical applications of graphene-based materials in lithium batteries, supercapacitors and fuel cells are now foreseen.6,7 On the other hand, the high surface-to-volume ratio in mesoporous materials enhances properties related to surface effects such as reactivity and catalysis, Li insertion (batteries) and supercapacitance, among others.8,9 This is the case, for example, of porous silicon which has attracted enormous interest because of its inherent properties.10,11Therefore, the combination of these two nanomaterials, graphene and mesoporous semi-conductors, is highly promising. Such a nanocomposite was obtained with mesoporous silicon,12 and more recently with mesoporous germanium (MP-Ge).13 Such achievements allowed incorporating the remarkable properties of graphene for ample improvement of semiconductor-based devices, possibly beyond the limits set by their nature or fabrication processes. For instance, in the graphene–mesoporous silicon nanocomposite, the temperature stability of the materials has been drastically improved thanks to the graphene coating.14
Germanium has suitable characteristics which make it a good candidate for the direct growth of graphene: (i) Ge has catalytic activity that allows the decrease of the energy barrier for decomposition of the carbon precursor,15 (ii) carbon has a low solubility in the Ge substrate, even at its melting temperature, and (iii) the difference between the thermal expansion coefficients of Ge and carbon is small16,17 and this leads to the drastic decrease of the formation of wrinkles in the graphene layer.18 The combination of mesoporous Ge (MP-Ge) and graphene is therefore a template material for using the large specific surface area of mesoporous germanium and the properties of graphene in a semiconductor to design a versatile 3D material. Thus, a nanomaterial made of MP-Ge and graphene is expected to exhibit a decreased thermal conductivity associated with the MP-Ge and an improved electrical conductivity with graphene leading to potentially interesting thermoelectric properties.19,20
However, if the chemical and physical properties of a nanocomposite highly depend on the morphology of the different component materials, the nature of their interface is also a major determining factor of their resulting properties.21,22 In the case of the graphene–mesoporous germanium nanocomposite (Gr–MP-Ge), it has been shown that the carbon coating of mesoporous Ge does not create Ge–C covalent bonding,13 and the nature of the interaction between the components of the nanocomposite and its intensity remains to be characterized. Raman spectroscopy is a powerful tool not only as it provides a very rich fingerprint of graphene crystallinity or allows stacking order determination, but also as it provides access to understanding the adhesion property when it is combined with mechanical deformation.
Thus, determining the mechanical behaviour of graphene-based nanocomposites using Raman spectroscopy has been a subject of investigation,22–24 and this method has been proposed to characterize the interface of a nanocomposite through the stress-induced Raman peak position shift.25 However, in many cases, systems investigated are model systems with a planar configuration and, very often, with a well-crystallized graphene coating. However, the majority of graphene-based nanocomposite materials that are developed for applications are complex nanomaterials synthesized at moderate temperatures.26 In this sense, the nanocomposite investigated in this study is a template of this complexity.
The aim of this work is to propose a proof of concept for a new approach allowing the quantification of the degree of coupling between graphene and the other component of a complex nanocomposite. This cohesive interaction between the graphene and the other component can be determined based on pressure-induced shifts of the Raman G-peak of the graphene layers. Therefore, we developed a methodology to determine a universal (it does not depend on the nature of the component on which the graphene is deposited) phenomenological (valid for complex nanostructures) parameter that accounts for the interaction between the components.
In addition, pressure-induced transformations are a way to tune the physical and chemical properties of materials through phase transitions or decoupling of the components. This method provides an opportunity for the controlled design of nanocomposites.27
Experimental
The graphene–mesoporous germanium nanocomposite (Gr–MP-Ge) was synthesized and characterized following the process reported in ref. 13 (Fig. 1). In short, bipolar electrochemical etching and chemical vapor infiltration (CVI) were employed for the nanostructuration of the Ge substrate and the subsequent 3D nanographene coating of the porous structure, respectively. The CVI step corresponds to the infiltration of a mixture of hydrogen/acetylene in a furnace under a controlled atmosphere (argon). The temperature treatment may vary and this allows us to tune the graphene quality (domain size and disorder).13 In this work, the treatment temperature was 873 K. | ||
| Fig. 1 Cross-sectional SEM images of the graphene–mesoporous germanium (Gr–MP-Ge) nanocomposite after carbonization treatment at 873 K. | ||
High pressure was generated using a membrane diamond anvil cell (DAC) with low-fluorescence diamonds. Gr–MP-Ge samples were placed into a 125 μm chamber drilled in an indented stainless-steel gasket. Paraffin oil was used as the pressure-transmitting medium (PTM). With this PTM, hydrostaticity is ensured up to 4–5 GPa.28 The pressure was probed by the shift of the R1 fluorescence line of a small ruby chip.
The Raman experiment was carried out using a HORIBA LabRam HR Evolution Raman spectrometer operated with a 532 nm wavelength compatible with our high-pressure setup (diamond anvil cell – DAC), which can detect an inelastic signal down to about 6 cm−1. The laser power was set at 5 mW at the entrance of the DAC to avoid heating. The beam was focused on the sample using a 50× objective, with a beam diameter of ∼2 μm at the sample. The scattered light was collected in backscattering geometry using the same objective.
Results and discussion
Characterization at ambient pressure
Structural characterization of the Gr–MP-Ge has been carried out and reported in ref. 13. According to XPS measurements performed at different etching times to evaluate the presence of carbon at different depths, the coating covers the pore walls as indicated by the presence of carbon after different etching times.13 The coating is sp2 carbon (Fig. S1†), and no trace of sp3 is detected, excluding the presence of amorphous graphite,29 nor of graphene oxide. Fig. 2a shows the Raman spectra of the reference Ge wafer (before porosification, after porosification and after graphenization). The initial Ge wafer is characterized by the presence of the T2g mode located at ∼298 cm−1. The position of this peak and its width (full width at half maximum, FWHM = 3 cm−1) indicate a highly crystalline quality. After porosification, the T2g peak shows significant broadening as well as the appearance of a lower frequency peak. The resemblance to the vibrational density of states indicates the presence of a high density of defects leading to the breaking of the Raman spectroscopy selection rules.30 The thermal treatment during graphenization at T = 873 K induces defect annealing leading to the disappearance of the low-frequency band and a narrowing of the T2g mode. | ||
| Fig. 2 (a) Raman spectra of a Ge wafer (Ge), a mesoporous Ge (MP-Ge) sample (after porosification of the wafer), and a graphene–mesoporous germanium nanocomposite (Gr–MP-Ge). (b) Raman signature of the graphene deposition (D- and G-peaks). (c) Raman spectra of the region from 2500 cm−1 to 3100 cm−1 showing the 2D peak (see text for discussion). | ||
The presence of carbon as a graphite-like material is evidenced by the Raman signature in the range of 1000–1800 cm−1 as shown in Fig. 2b.31 The spectrum consists of the D- and G-peaks that are characteristic of sp2 carbon with the presence of disorder.31,32 The analysis of such spectra for graphite has been proposed in the literature,29 and it has been shown that the intensity ratio of the D-peak to the G-peak is correlated with the upshift of the G-peak position. This corresponds to stage 1 defined by Ferrari and Robertson.29 The underlying reason is the presence of nanodomains. According to ref. 33, when using a 532 nm laser excitation, one has
 | (1) |
The ratio I(D)/I(G) measured in our case (∼0.85) indicates that nanodomains with a typical size of ∼6 nm have grown on the mesoporous Ge substrate.
The signature of graphene is usually attested by the 2D peak shape and intensity. If the conclusion is straightforward for well crystallized graphene, it is more difficult to conclude for nanographene (graphene made of nanodomains) grown at low temperature (typically below 900 K). Fig. 2c shows the spectral region corresponding to the 2D peak. The signal shows broad features that are typical of the ones obtained for the low-temperature growth of nanographene as reported in the literature.34 It indicates some degree of disorder of the coating.35
When the treatment temperature following the infiltration of acetylene is increased (Fig. S2†), the Raman spectra show marked 2D and D + D′ indicating a lower degree of disorder but being compatible with the spectra obtained for nanographene.36
These results agree with gravimetric measurements (estimating between 2 and 3 layers) and are discussed below. The following results indicate that there is conformation and adhesion of the carbon coating on the substrate (meaning that the number of layers is strictly lower than 3 (ref. 37)) and prove that the number of layers is at most 2.
In summary, the graphene-based nanocomposite investigated in this study is made of mesoporous germanium covered by mono- or bi-layer nanographene. It is worth noting that because of the small bandgap of germanium, the penetration length of the laser with a wavelength of 532 nm is of the order of 20 nm. On the other hand, the laser spot size is of the order of 2–3 microns leading to a large area being probed during measurements. As the high corrugation starts from the surface, the coupling between the nanographene coating and the mesoporous germanium is monitored during the experiment thanks to the pressure-induced shift of the G-peak on a large scale as discussed below.
High-pressure experiments
Let us first discuss the pressure-induced effect on the mesoporous-Ge component of the nanocomposite (Fig. 3). As shown in Fig. 3a, the pressure-induced upshift and broadening of the T2g mode is observed until the phase transition to the β-tin (Ge-II) phase above ∼9.9 GPa. At 11.7 GPa, the spectrum is made of the two peaks of Ge-II in coexistence with the T2g mode of Ge-I.38 Ge-II is observed up to the highest pressure reached in this experiment i.e., 18.5 GPa. At first glance, this behaviour is very similar to that of the Ge wafer under pressure previously reported.30 However, closer examination of the spectral parameters obtained by fitting the T2g peak by a Lorentzian function shows some differences.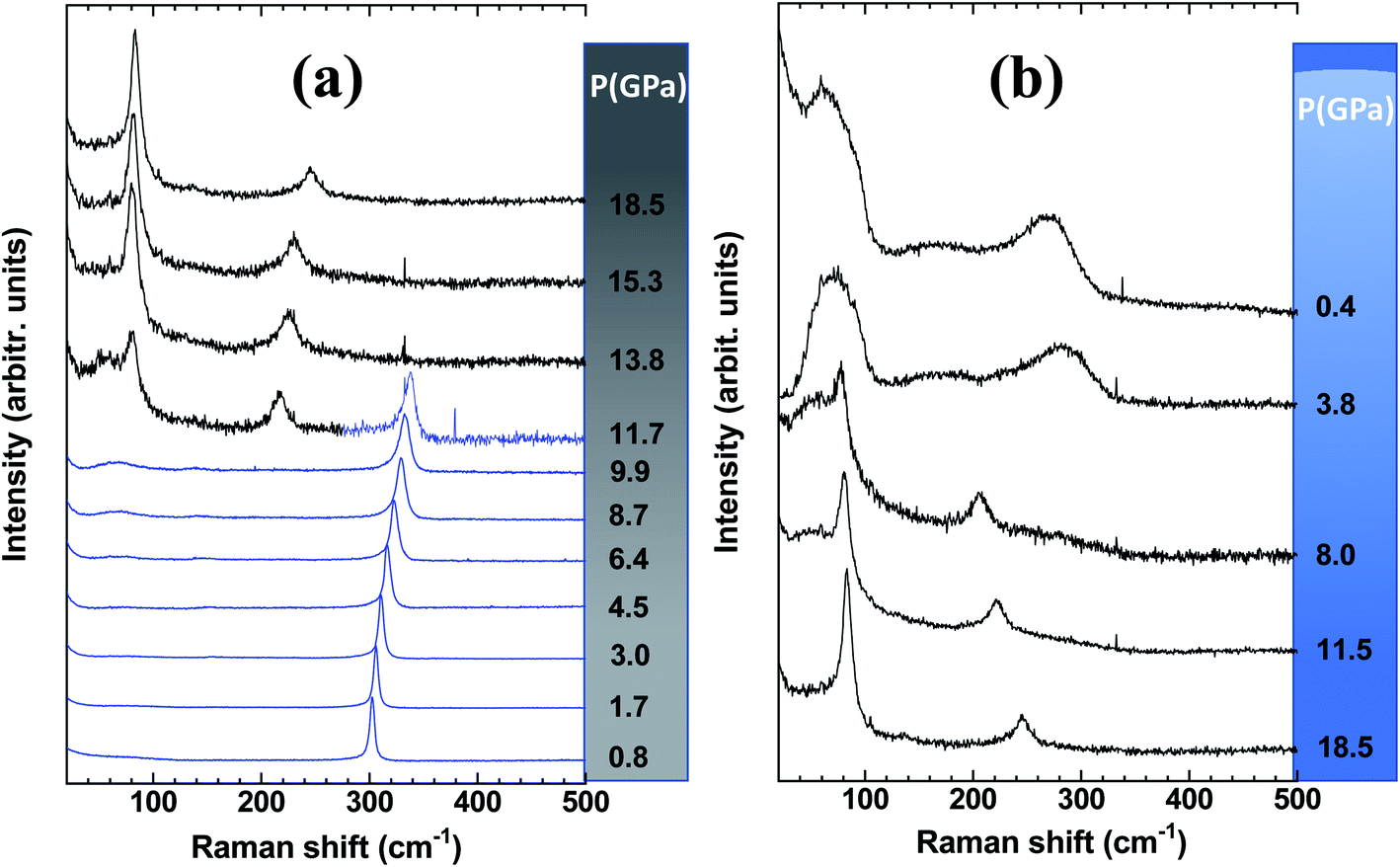 | ||
| Fig. 3 Spectral range of Raman spectra highlighting the MP-Ge component of a Gr–MP-Ge sample (a) during compression with the observation of two phases Ge-I (blue) and Ge-II (black) after the transition above 9.9 GPa and (b) on decompression with the observation of amorphization below 8.0 GPa. | ||
Fig. 4 shows the position of the T2g peak (Fig. 4a) and its width (Fig. 4b) as a function of pressure. As noted in ref. 30, the pressure-induced shift follows an almost linear variation across the pressure range where hydrostaticity is ensured (up to P ∼ 4 GPa with this PTM28). At higher pressure, the peak position shows a significant deviation from a linear dependence on pressure. This effect has already been observed for mesoporous Ge but also for bulk Ge under non-hydrostatic conditions.30 Adjusting the experimental data points with a quadratic relationship ν(P) = ν(P = 0) + AP + BP2 yields A = 4.1(1) and B = −0.08(1). This relationship is also found in silicon and has been detailed in ref. 39 and ref therein as resulting from a Taylor expansion of the exponential function ν(P) = ν(P0)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−γχ(P − P0)]. These values of A and B are very close to those obtained for bulk Ge compressed under similar conditions.30 This is an interesting observation as it points out that the carbon coating does not modify the pressure transmission to the Ge porous substrate and that the mechanical behaviour is mainly dominated by Ge.
exp[−γχ(P − P0)]. These values of A and B are very close to those obtained for bulk Ge compressed under similar conditions.30 This is an interesting observation as it points out that the carbon coating does not modify the pressure transmission to the Ge porous substrate and that the mechanical behaviour is mainly dominated by Ge.
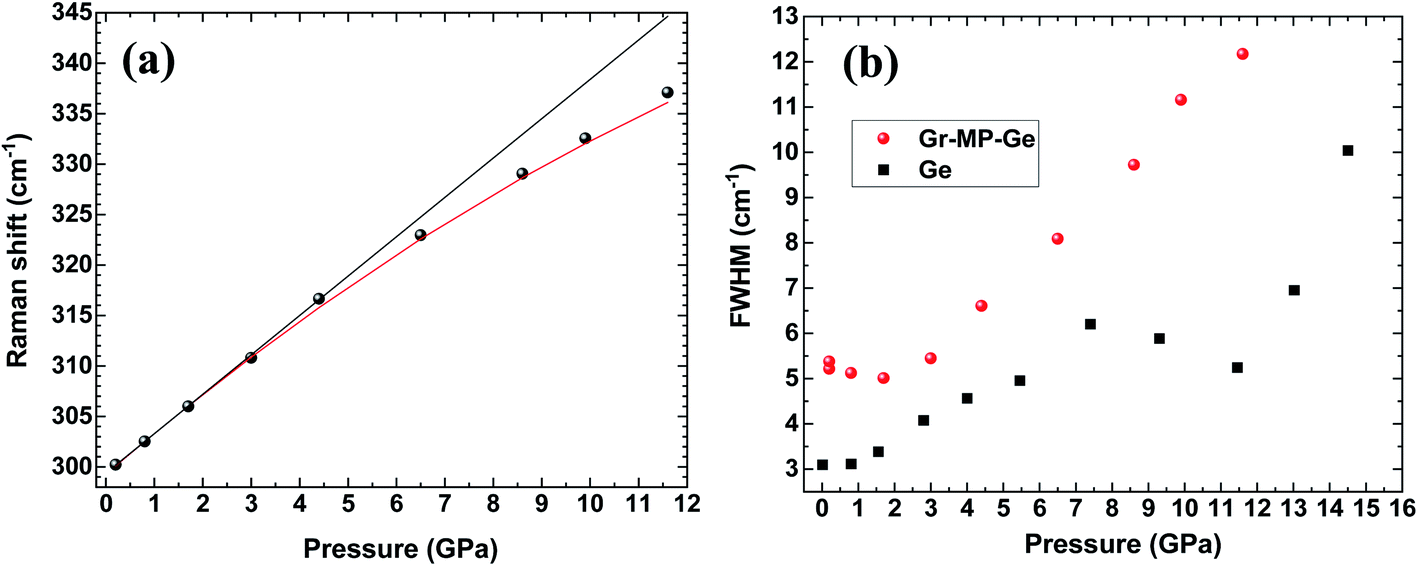 | ||
| Fig. 4 (a) Peak position and (b) full width at half maximum (FWHM) of the T2g mode in Gr–MP-Ge as a function of pressure (circles). The FWHM of the same mode measured on a Ge wafer is shown for comparison (square).30 In (a) the black line is a linear fit of the data up to 4.5 GPa (limit of hydrostaticity) and the red curve is a quadratic fit of the dataset. | ||
Considering the FWHM of the T2g peak with increasing pressure (Fig. 4b), it starts to increase between 3 and 4 GPa when the hydrostaticity provided by the pressure-transmitting medium is no longer ensured.28 At higher pressure, the peak width increases monotonically without any effect of the pressure-induced phase transition from Ge-I to Ge-II. The FWHM of the T2g peak is compared with that of the initial wafer.30 In the case of the latter, a decrease of this parameter was observed across the transition as a sign of a certain stress relaxation during the transformation.30,40 The absence of such an effect on the Gr–MP-Ge sample may be related to the heterogeneous nature of the sample that probably does not allow cooperative relaxation through the nucleation of Ge-II that is spatially distributed. Such an effect of heterogeneity on the phase transformation has already been underlined using the Ginzburg–Landau description of phase transitions.40,41
On decompression, Gr–MP-Ge exhibits a surprising behaviour compared to bulk Ge subjected to a similar pressure cycle (Fig. 3b). In bulk Ge, the pressure release results in different phases depending on the degree of hydrostaticity. Generally, the metastable ST12 phase (also named Ge-III) is favored by shear stresses.42 In our present experiments, the high-pressure crystalline Ge-II phase transforms on decompression into a low-density amorphous (LDA) state below ∼8.0 GPa. Pressure-induced amorphization is a phenomenon usually observed during the compression stage with possibly a subsequent polyamorphic transformation.43 This is, for example, the sequence observed in mesoporous germanium30 or in nanosized Ge.44 Examples of crystal-to-amorphous state transformation during decompression are scarcer.43 This phenomenon was discussed theoretically in 1988 (ref. 45) and can be summarized, as in Fig. 5. It shows free energy as a function of pressure in different cases. For the bulk sample, the transformation from phase Ge-I to Ge-II occurs when the associated G(P) functions cross (this defines the transition pressure). In the case of the mesoporous Ge (MP-Ge) component of the Gr–MP-Ge, the initial position of G is higher due to the interface and the energy contributions of the defects. However, contrary to the mesoporous Ge studied in ref. 30, our Gr–MP-Ge sample was annealed during graphene coating, and the surface and defect energy contributions are consequently lower. This is attested by the changes observed in the Raman spectra shown in Fig. 2a, where the T2g peak is narrower, and the low-frequency band is less intense after graphene deposition. This explained why the present sample undergoes a polymorphic transformation on compression rather than a pressure-induced amorphization (to the high-density amorphous (HDA) state) as observed in ref. 30).
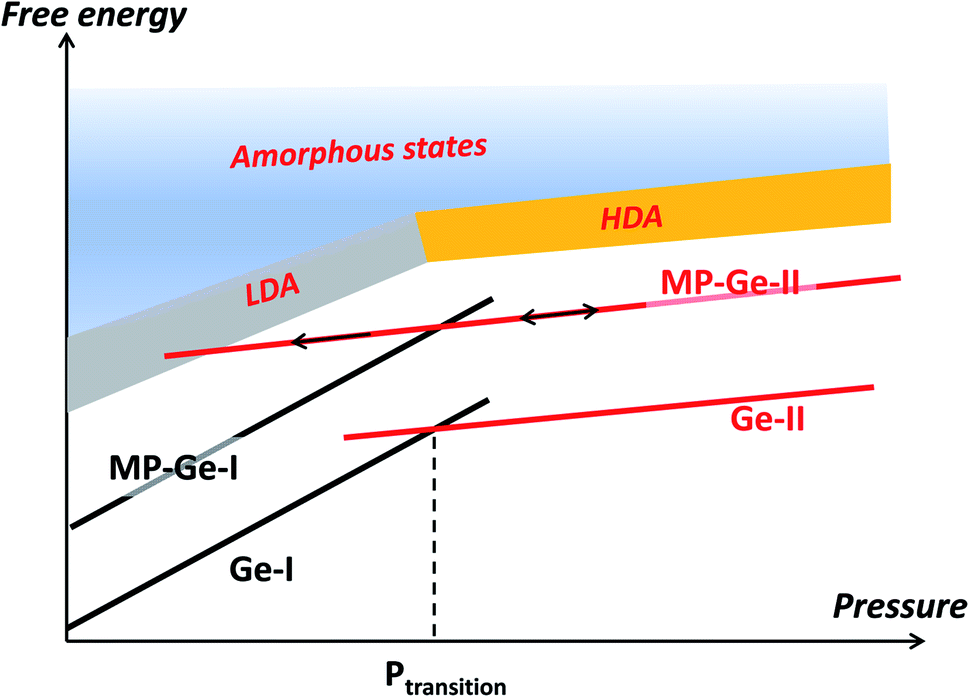 | ||
| Fig. 5 Free energy as a function of pressure. In the case of bulk germanium (Ge), the phase transformation occurs at the crossing of the respective free energies of Ge-I and Ge-II, defining the transition pressure. In the case of the mesoporous sample (MP-Ge), the interface induces an initial energy increase. Compression and subsequent decompression allow the system to enter the energy range where metastable states such as the low-density amorphous (LDA) state can be observed. | ||
However, during decompression in our experiment on Gr–MP-Ge, back-transformation into MP-Ge-I is hindered, certainly because the transition is highly reconstructive with a high kinetic barrier. When MP-Ge-II is decompressed, it does not back-transform to MP-Ge-I but remains in phase II until the stability range of the LDA state is attained and amorphization occurs. Several parameters (and their combination) can be at the origin of this amorphization process. First, the amorphization of Ge-II during decompression has already been observed at low temperature and, consequently, it is certainly related to phase transformation kinetics.46 Second, as for pressure induced-amorphization in porous Ge, the defect density and the heterogeneous structure are favorable to allow the system to enter the amorphous energy landscape due to the contribution of interface energy.30 Third, with the same idea of inhomogeneity, the effect of non-hydrostaticity has been shown to control the transition pathway on decompression, resulting in controlling the nucleation of metastable states.41
Let us now analyze the Raman signature of the sp2 carbon bonds (G-peak) with increasing and subsequently decreasing pressure (Fig. 6). The position of the G-peak during the pressure cycle is shown in Fig. 7. In the first pressure range, from 0.1 GPa to ∼7 GPa, the peak position shifts linearly with pressure by 5.8(1) cm−1 GPa−1. Above 8 GPa, the peak variation deviates from this linear trend. After a transitional range between ∼8 GPa and 13.5 GPa, a linear variation is found again but with a different linear pressure coefficient of 4.5(2) cm−1 GPa−1. The same slope is observed on the decompression path. This value of the slope for the G-peak position as a function of pressure is similar to that of free-standing graphite reported in the literature and lies between 4.2 and 4.7 cm−1 GPa−1.47,48 The change in the pressure-induced shift regime is concomitant with the pressure-induced phase transition of the germanium substrate i.e., the MP-Ge-I-to-MP-Ge-II transformation (Fig. 3).
 | ||
| Fig. 6 Raman spectra of the G-peak of the graphene-based coating (a) during compression and (b) during subsequent decompression. | ||
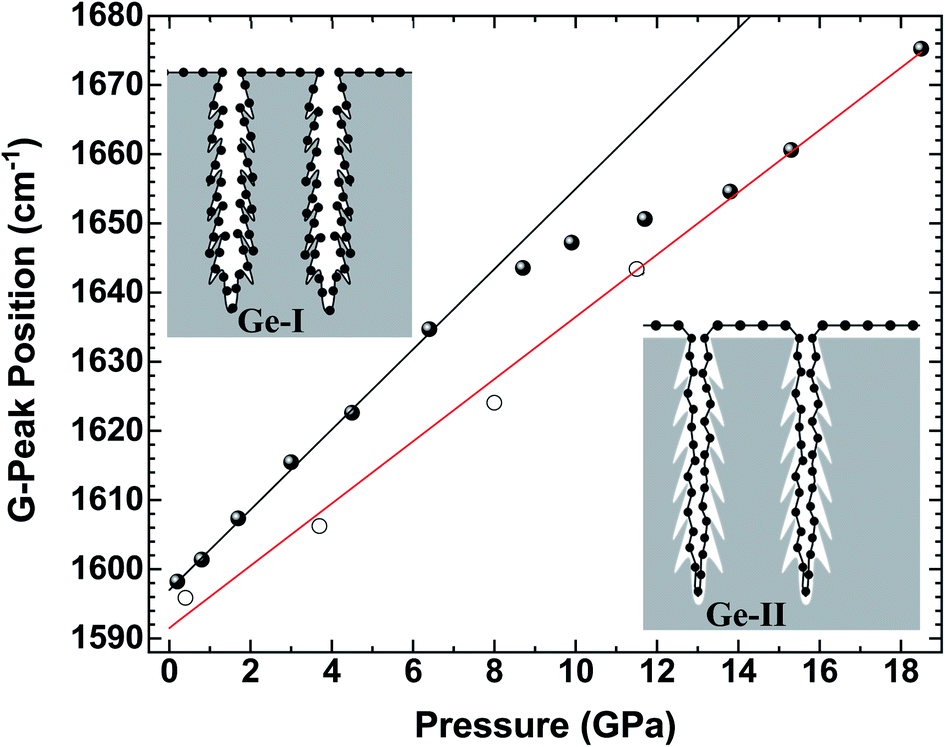 | ||
| Fig. 7 Peak position of the G-peak in Gr–MP-Ge as a function of pressure on compression (full circles) and on decompression (open circles). The lines are fits of the G-peak positions before and after the transformation of Ge. The slope of the black line is 5.8(1) cm−1 GPa−1, and that of the red line is 4.5(2) cm−1 GPa−1. | ||
The pressure-induced shift of the G-peak for the Gr–MP-Ge in the range of [0.1–8.0 GPa] is higher than for free standing graphite. This higher pressure-dependency is very similar to what is observed in compression experiments on graphene deposited on substrates.49,50 This is related to the adhesion interaction between graphene and the substrate. When the latter is significantly more compressible than carbon-based materials, its deformation drags the graphene, inducing an additional stress component which can be measured by Raman spectroscopy.50 As a matter of fact, the Raman shift of the G-peak is attributed to the variation in the length of the C–C bond. In high-pressure experiments on graphene deposited on various substrates, the main starting hypothesis which was proposed to explain the higher pressure-induced shift relied on the idea that the strain is transferred from the substrate to the graphene, and not the pressure itself.49
The deformation of the substrate is induced by the application of pressure. Therefore, the substrate linear deformation is εS = βSP, where βS is the substrate linear compressibility and P is the applied pressure. Substrate deformation is transmitted to the graphene component, totally or partially depending on the substrate, yielding εG = αεS with εG and εS being the deformations of graphene and the substrate, respectively. The parameter α then corresponds to the quantification of the transmission of the deformation related to the degree of adhesion between the 2D-system and its substrate. This relation leads to  , where βG is the graphene linear bulk modulus and σ is the equivalent stress associated with the transmitted deformation of the substrate. This point is important as it clearly appears that the stress on graphene is not equal to the applied pressure.
, where βG is the graphene linear bulk modulus and σ is the equivalent stress associated with the transmitted deformation of the substrate. This point is important as it clearly appears that the stress on graphene is not equal to the applied pressure.
The value of βG is taken as the in-plane linear bulk modulus of graphite, thus assuming that the reduction of dimensionality does not induce modifications of the C–C chemical bond and the associated mechanical properties. Thanks to X-ray diffraction experiments on graphite, it was determined that βG = 1250 GPa.47
By following the procedure proposed in ref. 50, the value of α can be determined using the linear bulk modulus of germanium ( , where B0 = 75 GPa is the bulk modulus of the cubic diamond Ge phase51). The low calculated value of α ≈ 3% indicates a rather low coupling between the carbon-layers and the mesoporous Ge structure. For comparison, this parameter is 20% in the case of a monolayer of exfoliated graphene deposited on a Si/SiO2 substrate in a planar geometry.50 Such a difference can certainly be explained by a difference in the surface properties, in particular a marked difference in substrate roughness that can lead to partial stress transmission. For instance, in the case of MoS2, the adhesion with the substrate is not complete, as MoS2 is more rigid and cannot conform totally to a not-perfectly flat substrate. In that case, two different pressure-induced behaviours related to a high and a low conformation of MoS2 to the substrate roughness, respectively, were observed, leading to different pressure-induced shifts.52 Here, well identified regions with different conformations are not observed because of (i) the broadness of the G-peak induced by the nanodomains and (ii) the size of the low and high conformed regions that must be much smaller with graphene as it is much more flexible than MoS2.
, where B0 = 75 GPa is the bulk modulus of the cubic diamond Ge phase51). The low calculated value of α ≈ 3% indicates a rather low coupling between the carbon-layers and the mesoporous Ge structure. For comparison, this parameter is 20% in the case of a monolayer of exfoliated graphene deposited on a Si/SiO2 substrate in a planar geometry.50 Such a difference can certainly be explained by a difference in the surface properties, in particular a marked difference in substrate roughness that can lead to partial stress transmission. For instance, in the case of MoS2, the adhesion with the substrate is not complete, as MoS2 is more rigid and cannot conform totally to a not-perfectly flat substrate. In that case, two different pressure-induced behaviours related to a high and a low conformation of MoS2 to the substrate roughness, respectively, were observed, leading to different pressure-induced shifts.52 Here, well identified regions with different conformations are not observed because of (i) the broadness of the G-peak induced by the nanodomains and (ii) the size of the low and high conformed regions that must be much smaller with graphene as it is much more flexible than MoS2.
Therefore, it is difficult to extract the fine evolution of such nanocomposites with a complex nanostructure, and this is why determining the phenomenological parameter α is highly informative. This quantity summarizes the graphene–substrate adhesion, friction, graphene bending and ripple formation, and substrate roughness and commensurability, regardless of the complexity of the nanostructure. In addition, this method is universal as it can be used for any graphene-based nanocomposite.
It is worth noting interesting information emerging from this result and related to the number of graphitic layers deposited during the synthesis of the Gr–MP-Ge. It has been shown that adhesion between n-layer graphene and a substrate may occur only for n ≤ 2. Above this value, the bending modulus of graphene is large enough to avoid the conformation of the n-layered system to the substrate topology.37 This result is in good agreement with the estimate of 2–3 layers in the Gr–MP-Ge obtained by gravimetric methods13 and with the Raman spectra shown in Fig. 2 and S2.†
Above 8 GPa, the variation of the G-peak position with pressure starts to decrease. This effect occurs concomitantly with the pressure-induced phase transformation from Ge-I to Ge-II, which is characterized by a volume reduction of ∼20%.53 Such drastic and sudden volume variation certainly leads to the decoupling of graphene and germanium components. In fact, after the transition, the pressure-induced shift recovers a lower pressure-dependency similar to that of free-standing graphite (Fig. 7). Therefore, the parameter α tends to zero. Consequently, because of this decoupling, the carbon layer must have very little effect on the amorphization mechanism of Ge during decompression as presented above, and this phenomenon is intrinsically related to germanium.
It is interesting to note that the regime change of the pressure-induced shift of the graphene coating occurred at a lower pressure than that observed in the clear Raman signature of the Ge high-pressure phase (8.0 GPa vs. 9.9 GPa). The Raman spectral features of graphene are highly sensitive to its environment making it an attractive sensor for phase transitions.54 Therefore, depositing graphene layers as a gauge can help to detect phase transitions such as polyamorphic transformations, isostructural phase transitions, etc.
Conclusions
A graphene–mesoporous germanium nanocomposite (Gr–MP-Ge) was studied under pressure using Raman spectroscopy. Two frequency regions have received special interest: (i) the region [20–500 cm−1] that allows the monitoring of the pressure-induced transformations of the germanium substrate and (ii) the region [1500–1850 cm−1] corresponding to the G-peak of the graphene-like coating.The pressure-induced variations of both Raman signatures were found to be related. First, the G-peak pressure-dependency is larger than that expected for graphene-based materials. This effect recalls what is observed in graphene deposited on substrates. This pressure-dependency has been explained on the basis of the additional stress component generated by substrate deformation. The pressure-induced phase transformation in the porous germanium substrate led to the decoupling of interactions in a domino effect, and the pressure-induced Raman shift returns to that of free-standing graphite. Therefore, pressure-induced variations of the Raman spectral features are a valuable tool for exploring the coupling between the components of a graphene-based nanocomposite. It allows quantifying the interactions between these components and observing the unbinding of the components. In addition, pressure cycling allows us to modify the nature of one component, here, from a crystal to an amorphous structure, providing the opportunity of tuning the associated physical properties such as thermal or electrical conductivities. This is an important step in the design of nanomaterials.27 Finally, this work proposes a unified protocol that allows characterizing and comparing the interactions between graphene and the other component in different graphene-based nanocomposites. The results of this study invite us to more systematically explore graphene-based (nano)composite materials under pressure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
LN2 is a joint International Research Laboratory (Unité Mixte Internationale UMI 3463) funded and co-operated in Canada by Université de Sherbrooke (UdeS) and in France by CNRS as well as Université de Lyon (UdL, especially including ECL, INSA Lyon, and CPE) and Université Grenoble Alpes (UGA). It is also associated with the French national nanofabrication network RENATECH and is supported by the Fonds de Recherche du Québec Nature et Technologie (FRQNT). The authors would like to thank the CECOMO and the PLECE platforms at Université Lyon 1 for the use of the spectrometer and DAC, respectively. This work was supported by the LABEX iMUST (ANR-10-LABX-0064) of the Université de Lyon, within the program “Investissements d’Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR).References
- I. A. Kinloch, J. Suhr, J. Lou, R. J. Young and P. M. Ajayan, Science, 2018, 362, 547–553 CrossRef CAS.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed.
- H. Chang and H. Wu, Energy Environ. Sci., 2013, 6, 3483 RSC.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2017, 90, 75 CrossRef CAS.
- N. Mahmood, C. Zhang, H. Yin and Y. Hou, J. Mater. Chem. A, 2014, 2, 15 RSC.
- L. Ji, P. Meduri, V. Agubra, X. Xiao and M. Alcoutlabi, Adv. Energy Mater., 2016, 6, 1502159 CrossRef.
- Y. Ye, C. Jo, I. Jeong and J. Lee, Nanoscale, 2013, 5, 4584 RSC.
- L. Wang, W. Ding and Y. Sun, Mater. Res. Bull., 2016, 83, 230 CrossRef CAS.
- Handbook of Porous Silicon, ed. L. Canham, Springer, 2015 Search PubMed.
- A. Roland, A. Dupuy, D. Machon, F. Cunin, N. Louvain, B. Fraisse, A. Boucherif and L. Monconduit, Electrochim. Acta, 2019, 323, 1347582 CrossRef.
- A. R. Boucherif, A. Boucherif, G. Kolhatkar, A. Ruediger and R. Arès, Small, 2017, 13, 1603269 CrossRef PubMed.
- S. Sauze, M. Reza Aziziyan, P. Brault, G. Kolhatkar, A. Ruediger, A. Korinek, D. Machon, R. Arès and A. Boucherif, Nanoscale, 2020, 12, 23984 RSC.
- G. Kolhatkar, A. Boucherif, A. R. Boucherif, A. Dupuy, L. G. Fréchette, R. Arès and A. Ruediger, Nanotechnology, 2018, 29, 145701 CrossRef PubMed.
- P. W. Loscutoff and S. F. Bent, Annu. Rev. Phys. Chem., 2006, 57, 467–495 CrossRef CAS PubMed.
- J. H. Lee, S. G. Kang, H. S. Jang, J. Y. Moon and D. Whang, Adv. Mater., 2019, 31, 1803469 CrossRef PubMed.
- R. I. Scace and G. A. Slack, J. Chem. Phys., 1959, 30, 1551 CrossRef CAS.
- W. Bao, F. Miao, Z. Chen, H. Zhang, W. Jang, C. Dames and C. N. Lau, Nature Nanotech, 2009, 4, 562–566 CrossRef CAS PubMed.
- P. Thiyagarajan, M.-W. Oh, J.-C. Yoon and J.-H. Jang, Appl. Phys. Lett., 2014, 105, 033905 CrossRef.
- J.-H. Lee and J. C. Grossman, Appl. Phys. Lett., 2009, 95, 013106 CrossRef.
- G. Anagnostopoulos, C. Androulidakis, E. N. Koukaras, G. Tsoukleri, I. Polyzos, J. Parthenios, K. Papagelis and C. Galiotis, ACS Appl. Mater. Interfaces, 2015, 7, 4216 CrossRef CAS PubMed.
- Z. Dai, L. Liu and Z. Zhang, Adv. Mater., 2019, 31, 1805417 CrossRef CAS PubMed.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2017, 90, 75 CrossRef CAS.
- G. Wang, L. Liu and Z. Zhang, Composites, Part A, 2021, 141, 106212 CrossRef CAS.
- G. Wang, Z. Dai, L. Liu, H. Hu, Q. Dai and Z. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 22554 CrossRef CAS.
- P. Samorì, I. A. Kinloch, X. Feng and V. Palermo, 2D Mater., 2015, 2, 030205 CrossRef.
- D. Machon, V. Pischedda, S. Le Floch and A. San-Miguel, J. Appl. Phys., 2018, 124, 160902 CrossRef.
- J. W. Otto, J. K. Vassiliou and G. Frommeyer, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 3253 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- A. Boucherif, S. Radescu, R. Arès, A. Mujicà, P. Mélinon and P. D. Machon, J. Phys. Chem. C, 2018, 122, 10929 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc., A, 2007, 362, 2271 CrossRef PubMed.
- M. J. Matthews, M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus and M. Endo, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, R6585 CrossRef CAS.
- M. H. Rümmeli, A. Bachmatiuk, A. Scott, F. Börrnert, J. H. Warner, V. Hoffman, J.-H. Lin, G. Cuniberti and B. Büchner, ACS Nano, 2010, 4, 4206 CrossRef PubMed.
- A. Merlen, J. Buijnsters and C. Pardanaud, Coatings, 2017, 7, 153 CrossRef.
- J.-B. Wu, M.-L. Lin, X. Cong, H.-N. Liua and P.-H. Tan, Chem. Soc. Rev., 2018, 47, 1822 RSC.
- J. Nicolle, D. Machon, P. Poncharal, O. Pierre-Louis and A. San-Miguel, Nano Lett., 2011, 11, 3564 CrossRef CAS PubMed.
- H. Olijnyk, Phys. Rev. Lett., 1992, 68, 2232 CrossRef CAS.
- G. Lucazeau, J. Raman Spectrosc., 2003, 34, 478 CrossRef CAS.
- X. Yan, D. Tan, X. Ren, W. Yang, D. He and H.-K. Mao, Appl. Phys. Lett., 2015, 106, 171902 CrossRef.
- D. Machon, L. Piot, D. Hapiuk, B. Masenelli, F. Demoisson, R. Piolet, M. Ariane, S. Mishra, S. Daniele and M. Hosni, et al. , Nano Lett., 2014, 14, 269 CrossRef CAS.
- B. Haberl, M. Guthrie, B. D. Malone, J. S. Smith, S. V. Sinogeikin, M. L. Cohen, J. S. Williams, G. Shen and J. E. Bradby, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 144111 CrossRef.
- D. Machon, F. Meersman, M. C. Wilding, M. Wilson and P. F. McMillan, Prog. Mater. Sci., 2014, 61, 216 CrossRef CAS.
- N. R. C. Corsini, Y. Zhang, W. R. Little, A. Karatutlu, O. Ersoy, P. D. Haynes, C. Molteni, N. D. M. Hine, I. Hernandez and J. Gonzalez, Nano Lett., 2015, 15, 7334 CrossRef CAS PubMed.
- P. Richet, Nature, 1988, 331, 56 CrossRef CAS.
- V. V. Brazkhin, A. G. Lyapin, S. V. Popova and R. N. Voloshin, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 7549 CrossRef PubMed.
- M. Hanfland, H. Beister and K. Syassen, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 12598 CrossRef CAS PubMed.
- A. F. Goncharov, I. N. Makarenko and S. M. Stishov, Sov. Phys. JETP, 1989, 69, 380 Search PubMed.
- C. Bousige, F. Balima, D. Machon, G. S. Pinheiro, A. Torres-Dias, J. Nicolle, D. Kalita, N. Bendiab, L. Marty, V. Bouchiat, G. Montagnac, A. G. Souza Filho, P. Poncharal and A. San-Miguel, Nanolett, 2017, 17, 21 CrossRef CAS PubMed.
- D. Machon, C. Bousige, R. Alencar, A. Torres-Dias, F. Balima, J. Nicolle, G. S. Pinheiro, A. G. Souza Filho and A. San-Miguel, J. Raman Spectrosc., 2018, 49, 121 CrossRef CAS.
- D. A. Young, Phase Diagrams of the Elements, Univ. of California Press, Berkley, 1991 Search PubMed.
- R. S. Alencar, K. D. A. Saboia, D. Machon, G. Montagnac, V. Meunier, O. P. Ferreira, A. San-Miguel and A. G. Souza Filho, Phys. Rev. Mater., 2017, 1, 024002 CrossRef.
- J. C. Jamieson, Science, 1963, 139, 762 CrossRef CAS.
- A. Forestier, F. Balima, C. Bousige, G. Pinheiro, R. Fulcrand, M. Kalbac, D. Machon and A. San-Miguel, J. Phys. Chem. C, 2020, 124, 11193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00123j |
| This journal is © The Royal Society of Chemistry 2021 |