
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyperthermia-mediated changes in the tumor immune microenvironment using iron oxide nanoparticles
Gil
Covarrubias†
ab,
Morgan E.
Lorkowski†
ab,
Haley M.
Sims†
a,
Georgia
Loutrianakis†
a,
Abdelrahman
Rahmy
a,
Anthony
Cha
a,
Eric
Abenojar
 c,
Sameera
Wickramasinghe
c,
Taylor J.
Moon
a,
Anna Cristina S.
Samia
c and
Efstathios
Karathanasis
*ab
c,
Sameera
Wickramasinghe
c,
Taylor J.
Moon
a,
Anna Cristina S.
Samia
c and
Efstathios
Karathanasis
*ab
aDepartment of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio, USA. E-mail: stathis@case.edu
bCase Comprehensive Cancer Center, Case Western Reserve University, Cleveland, Ohio, USA
cDepartment of Chemistry, Case Western Reserve University, Cleveland, Ohio, USA
First published on 1st September 2021
Abstract
Iron oxide nanoparticles (IONPs) have often been investigated for tumor hyperthermia. IONPs act as heating foci in the presence of an alternating magnetic field (AMF). It has been shown that hyperthermia can significantly alter the tumor immune microenvironment. Typically, mild hyperthermia invokes morphological changes within the tumor, which elicits a secretion of inflammatory cytokines and tumor neoantigens. Here, we focused on the direct effect of IONP-induced hyperthermia on the various tumor-resident immune cell subpopulations. We compared direct intratumoral injection to systemic administration of IONPs followed by application of an external AMF. We used the orthotopic 4T1 mouse model, which represents aggressive and metastatic breast cancer with a highly immunosuppressive microenvironment. A non-inflamed and ‘cold’ microenvironment inhibits peripheral effector lymphocytes from effectively trafficking into the tumor. Using intratumoral or systemic injection, IONP-induced hyperthermia achieved a significant reduction of all the immune cell subpopulations in the tumor. However, the systemic delivery approach achieved superior outcomes, resulting in substantial reductions in the populations of both innate and adaptive immune cells. Upon depletion of the existing dysfunctional tumor-resident immune cells, subsequent treatment with clinically approved immune checkpoint inhibitors encouraged the repopulation of the tumor with ‘fresh’ infiltrating innate and adaptive immune cells, resulting in a significant decrease of the tumor cell population.
1. Introduction
Hyperthermia is often used as a cancer treatment to either directly kill cancer cells or sensitize the tumor tissue to other treatments.1,2 In particular, iron oxide nanoparticles (IONPs) have been extensively investigated for tumor hyperthermia in preclinical and clinical studies.3–6 In the presence of an external alternating magnetic field (AMF), IONPs convert electromagnetic energy into thermal energy (heat), a phenomenon known as magnetic hyperthermia. On its effort to reorient its magnetic moment to the external field (Néel relaxation), IONPs need to overcome an energy barrier resulting in the production of heat due to hysteresis or relaxational losses.7 For example, in a clinical application, IONPs are injected directly into brain tumors, resulting in a high tissue concentration of iron (>30 mg mL−1 Fe) that causes hyperthermia using an external AMF.4The obvious antitumor effect of hyperthermia is typically associated to the direct killing of cancer cells. While mild increase of the intratumoral temperature (i.e., about 42–43 °C) induces apoptosis of cancer cells, temperatures above 45 °C substantially enhance tumor cytotoxicity at the increased risk of damaging surrounding healthy tissues.8,9 In addition to its direct therapeutic effect, hyperthermia also has been shown to significantly alter the immune compartment of the tumor microenvironment (TME). First, hyperthermia-induced cancer cell death releases tumor-specific antigens that can be taken up by local antigen-presenting cells (APCs), such as dendritic cells (DCs) and macrophages, leading to activation of the innate and adaptive arms of the immune system.10 Further, studies have shown that hyperthermia boosts the production of proinflammatory cytokines in the TME,11 as well as heat shock proteins (HSPs), which has been associated with activation of APCs and effector immune cells.12
Here, we focused on the direct effect of IONP-induced hyperthermia on the various immune cell subpopulations in the TME. Effective cancer immunotherapy strongly depends on overcoming the profound immunosuppression within the TME.13–16 Cancer cells are responsible for re-programming innate and adaptive immune cells into an immunosuppressive phenotype.17,18 As a result, the TME is enriched with dysfunctional and immunosuppressive APCs, myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Treg).14–16 A non-inflamed and ‘cold’ TME inhibits systemic lymphocytes from effectively trafficking into tumors, thereby shielding tumors from systemic immuno-surveillance.17,18 In this study, we sought to quantitatively assess the changes of the immune cell content in the tumor caused by hyperthermia, rather than mechanistic or functional analysis of tumor immunity. We specifically measured the hyperthermia-induced elimination of tumor-promoting immune cells. Considering that the majority of nanoparticles are often taken up by immune cells in the TME rather than by cancer cells,19–21 IONP-induced hyperthermia can decrease the levels of these inhibitory immune cells or ideally completely deplete them. We chose to test this concept using the murine 4T1 model of triple-negative breast cancer, which contains large numbers of dysfunctional immune cells, many of which possess an immunosuppressive phenotype. We compared direct intratumoral injection to systemic administration of IONPs followed by application of the external AMF. Finally, we used IONP-induced hyperthermia followed by treatment with clinically approved immune checkpoint inhibitors to initially deplete the dysfunctional tumor-resident immune cells and then encourage the infiltration of ‘fresh’ innate and adaptive immune cells into the TME.
2. Experimental section
2.1 Synthesis of iron oxide nanoparticles
Parent iron oxide nanoparticles (IONPs) were developed using a co-precipitation method with a 1 to 2 molar ratio of Fe2+ to Fe3+ ions. Briefly, FeCl2·4H2O and FeCl3·6H2O were dissolved in deoxygenated water to dissolve the iron chloride solution. The iron oxide precursor solution was further dissolved using 5 mL of a 0.4 M solution of HCl whilst stirring. To establish the co-precipitation reaction, the iron solution was added to a 50 mL solution of 0.5 M NaOH that was preheated to 80 °C in the presence of nitrogen gas. The iron oxide solution formed was further allowed to react for an additional 15 minutes. A stable ferrofluid was developed through multiple deoxygenated water washes post-iron oxide magnetic separation. To prevent aggregation and agglomeration iron oxide nanoparticles were coated in a thin layer of citric acid. Iron oxide nanoparticles were resuspended in 50 mL of deoxygenated water followed by the addition of 340 mg of anhydrous citric acid with the layering occurring upon raising the solution pH to 5.2 using ammonium hydroxide followed by heating the reaction mixture under a constant flow of inert gas at 80 °C. The citric acid coated nanoparticles were purified using Amicon® Ultra-15 centrifugal filters.Resultant iron oxide nanoparticles were further modified by adding a mPEG coating using silane–mPEG (2000 kDa). The reaction proceeded as follows; the citric acid coated iron nanoparticles were concentrated to 1 mg mL−1 in deoxygenated water and the respective pH was adjusted to 11 using ammonium hydroxide. The mPEG coating was established through the addition of an equal mass of silane–mPEG to iron oxides. The reaction solution was vigorously stirred for 24 h. To drive the reaction to completion, it was placed in an oil bath and the solution was heated to 80 °C for 2 hours to ensure the crosslinking of the silane to the iron oxide surface. The mPEG-iron oxide nanoparticles were then further purified using Amicon® Ultra-15 centrifugal filters and subsequentially stored at 4 °C. Similar methods were implemented to develop iron oxide nanoparticles with an amine functional group, using a silane–PEG–NH2 polymer in place of the silane–mPEG. To characterize the size and charge of the nanoparticles; dynamic light scattering (DLS), and zeta potential measurements were obtained, respectively. Magnetic hyperthermia measurements were obtained using a MSI Automation bench mount magnetic induction heating system. Iron oxide nanoparticles at a concentration of 24 mg Fe per mL in water were exposed to an alternating magnetic field (AMF) with an amplitude of 20 kA m−1 at a fixed frequency of 380 kHz. The temperature within the sample volume was recorded using a fiber optic temperature probe (Neoptix 1) every 5 seconds for a total time of 1 minute of radiofrequency exposure. The sample temperature profile was recorded for a short time to establish a baseline for the SAR calculation. The linear profile obtained from the 1 minute exposure was used to calculate the SAR value using the following equation:
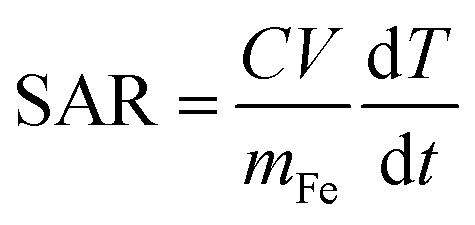
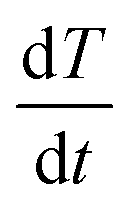 is change in temperature per unit time in the linear spectrum of the temperature profile, V is the volume of the sample and mFe is the mass in grams of the iron oxide nanoparticles.
is change in temperature per unit time in the linear spectrum of the temperature profile, V is the volume of the sample and mFe is the mass in grams of the iron oxide nanoparticles.
2.2 Functionalization of nanoparticle constructs
Nanoparticle constructs were functionalized using standard sulfo-SMCC chemistry. Nanoparticles containing amine functional groups were crosslinked by reacting sulfo-SMCC with a terminal cysteine end on the fibronectin targeting peptide CREKA. The molar ratios used in these reactions were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 amine-functional groups on the nanoparticle surface, sulfo-SMCC and CREKA peptide, respectively. Briefly, a 2 molar excess of sulfo-SMCC was added to amine functionalized nanoparticles and shaken for 30 minutes to react the N-hydroxysuccinimide (NHS ester) in sulfo-SMCC to available amines on the nanoparticles. Next, using the ratio previously described, CREKA was added to the reaction mixture to successfully crosslink the contralateral maleimide group on sulfo-SMCC to the sulfhydryl group on the CREKA peptide. The resultant CREKA functionalized nanoparticles were dialyzed against PBS using a 100 kDa MW cut-off dialysis membrane to remove excess reagents. Using previously described methodology, a Bio-Rad DC protein assay was used to deterministically identify the number of conjugated peptides. For the in vivo cell uptake studies, we used a fluorescent liposomal nanoparticle with similar size and PEG-ligand content. Briefly, the nanoparticles were developed by creating lipid films consisting of following molar ratios: 48.5% DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine, Avanti), 48.5% DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti) and 3% DSPE-PEG-NH2 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]). Lipid films were rehydrated in PBS at 60 °C for 30 minutes. Resultant liposomal nanoparticles were vortexed every 5 minutes throughout the rehydration process. Nanoparticles were sized to ∼50 nm (DLS) via ultra-sonication. Briefly, the liposomal solution was subjected to an ultra-sonication pulse sequence consisting of 10 second 20% power interval followed by 20 seconds of rest. This pulse sequence was repeated for a total 5 minutes while the solution was in an ice bath to reduce the effects of heat. The 50 nm-sized liposomal nanoparticles were dialyzed using a 100 kDa MW cut-off dialysis membrane.
:2:3 amine-functional groups on the nanoparticle surface, sulfo-SMCC and CREKA peptide, respectively. Briefly, a 2 molar excess of sulfo-SMCC was added to amine functionalized nanoparticles and shaken for 30 minutes to react the N-hydroxysuccinimide (NHS ester) in sulfo-SMCC to available amines on the nanoparticles. Next, using the ratio previously described, CREKA was added to the reaction mixture to successfully crosslink the contralateral maleimide group on sulfo-SMCC to the sulfhydryl group on the CREKA peptide. The resultant CREKA functionalized nanoparticles were dialyzed against PBS using a 100 kDa MW cut-off dialysis membrane to remove excess reagents. Using previously described methodology, a Bio-Rad DC protein assay was used to deterministically identify the number of conjugated peptides. For the in vivo cell uptake studies, we used a fluorescent liposomal nanoparticle with similar size and PEG-ligand content. Briefly, the nanoparticles were developed by creating lipid films consisting of following molar ratios: 48.5% DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine, Avanti), 48.5% DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti) and 3% DSPE-PEG-NH2 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]). Lipid films were rehydrated in PBS at 60 °C for 30 minutes. Resultant liposomal nanoparticles were vortexed every 5 minutes throughout the rehydration process. Nanoparticles were sized to ∼50 nm (DLS) via ultra-sonication. Briefly, the liposomal solution was subjected to an ultra-sonication pulse sequence consisting of 10 second 20% power interval followed by 20 seconds of rest. This pulse sequence was repeated for a total 5 minutes while the solution was in an ice bath to reduce the effects of heat. The 50 nm-sized liposomal nanoparticles were dialyzed using a 100 kDa MW cut-off dialysis membrane.
2.3 Institutional animal care and use committee statement
Animal well-being took priority over the conducted studies when it came to euthanasia or other interventions. All procedures and treatments were conducted under a protocol approved by the Institutional Animal Care and Use Committee of Case Western Reserve University (CWRU). CWRU follows the Guide for the Care and Use of Laboratory Animals, which is required by the United States Public Health Service Policy (PHS) on humane care and use of laboratory animals.2.4 Animal tumor model
4T1 tumor cells were cultured in RPMI medium (Gibco, Gaithersburg, MD) supplemented with 10% fetal bovine serum (FBS) and a 1% streptomycin/penicillin cocktail. 4T1 cancer cells were stably transfected with firefly luciferase and green fluorescent protein (GFP) for cell tracking. Briefly, 4T1 cancer cells were inoculated into the no. 9 inguinal mammary fat pad of BALB/c mice (Jackson Laboratories). Mice were subjected to 2–3% isoflurane inhalant to establish a surgical plane of anesthesia in order to surgically expose the mammary fat pad. An inoculant containing 5 × 105 4T1 cancer cells/50 μL was injected. Tumor-bearing mice were either intravenously or intratumorally injected with nanoparticle variants at a dosage of 16 mg kg−1 iron and subsequently subjected to hyperthermia treatments on day 10. For groups receiving checkpoint inhibitors, anti-PD1 and anti-CTLA-4 (BioXCell) were administered subcutaneously adjacent to the solid tumor mass at a dosage of 250 μg and 100 μg, respectively.2.5 Administration of mild hyperthermia
For hyperthermia administration studies, mice bearing orthotopic 4T1 mammary tumors were administered a dosage of 16 mg Fe per kg of iron oxide nanoparticles either i.v. or i.t. on day 10 post-inoculation. Intravenously administered IONPs were allowed to accumulate within the mammary tumor margins for 2 h prior to radiofrequency (RF) treatment. 5 minutes prior to RF treatment, mice were anesthetized with an intraperitoneal injection of a xylazine/ketamine cocktail. The mouse's core temperature was stabilized to baseline using a heating pad. Anesthetized mice receiving RF were subjected to an AMF with an amplitude of 20 kA m−1 and a frequency of 380 kHz for a total duration of 30 minutes. The mouse was placed such that the mammary tumor was located at the center of coil. Separate fiber optic temperature probes were inserted into the rectal cavity and the mammary tumor to measure whole body temperature and intratumoral temperature, respectively. The starting whole-body temperature was ∼34 °C. The schedule, dose and experimental design were identical for the studies comparing the two delivery routes of IONP (Fig. 1A) and the studies evaluating the effect of the combining checkpoint inhibitors with hyperthermia (Fig. 1B).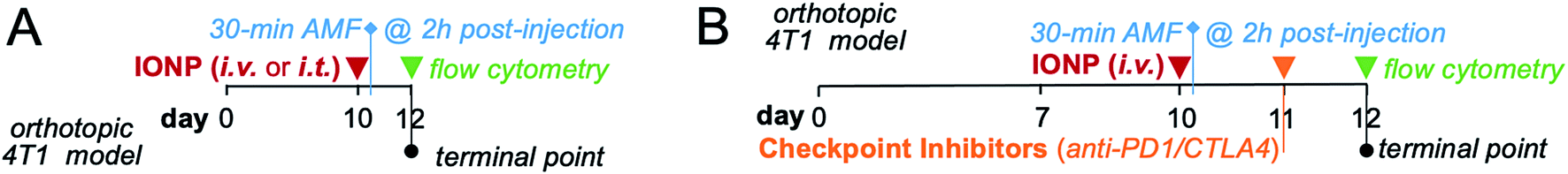 | ||
| Fig. 1 Design of in vivo studies. (A) Timeline shows administration of iron oxide nanoparticles via i.v. or i.t. injection, duration of hyperthermia treatment and flow cytometry analysis. (B) Timeline shows the schedule of administration of the combination treatment (IONP and checkpoint inhibitors) and the respective analysis. | ||
2.6 Flow cytometry analysis
For fluorescent studies, flow cytometry analysis was performed 24 h after the administration of the fibronectin targeted nanoparticle. For hyperthermia studies, flow cytometry was performed 48 h after the delivery of hyperthermia. Prior to tissue removal, blood was collected via retro-orbital bleeding. In order to conduct flow cytometry, mice were subsequently euthanized, and tumor and spleen were resected. Tumors were initially processed via digestion in a 1 mg mL−1 solution of collagenase in RPMI medium (HyClone). Collected organs, tumor and spleen, were then gently homogenized to develop a single cell suspension and then passed via a 70 μm filter. The blood was washed several times with an ACK lysis buffer (Gibco) to lyse and remove red blood cells. Single cell suspensions were placed in a solution containing an anti-mouse CD16/CD32 blocking agent (2.4G2; BD Biosciences), followed by a cell specific anti-mouse antibody stain and finally counterstained with a DAPI nuclear stain (BD Biosciences). Fluorescently tagged cells were counted using a BD LSR II flow cytometer. Analysis and cell counts were performed using FlowJo software. Anti-mouse antibodies included, CD45 (30-F11), Ly6G (1A8), Ly6C (AL-21), CD3e (145-2C11), CD8a (53–6.7), CD4 (GK1.5), F4/80 (T45-2342), CD11b (M1/70), CD11c (HL3), and CD49b (DX5). Antibodies were purchased from Biolegend and BD Biosciences with a dye-conjugated for flow cytometry.2.7 Statistical analysis
The data shown was analyzed using a one- or two-way ANOVA in conjunction with either a post hoc Tukey's or Sidak's test. The data is presented as mean ± SEM unless otherwise described. Statistical significance is observed by a P-value less than 0.05. All statistical analysis was conducted using Prism 7 (GraphPad Software).3. Results and discussion
3.1 Intratumoral administration of IONP and hyperthermia
The iron oxide nanoparticles were prepared using a previously established method.22,23 The surface of IONPs was functionalized with silane–PEG (2 kDa), resulting in about 300 PEGs per particle.22 The hydrodynamic size of IONPs was about 50 nm as measured by dynamic light scattering (Fig. 2A). Transmission electron microscopy (TEM) showed that the size of the core was about 30 nm (Fig. 2B). Zeta potential measurements indicated a nearly neutral surface charge (Fig. 2C). The heating profile of IONPs was measured in water using an external AMF at a field strength of 20 kA m−1 and a frequency of 380 kHz. The initial slope of temperature change was measured for the first 60 seconds to remain within the linear rate of temperature change (Fig. 2D). The specific absorption rate (SAR) was determined to be 123 W g−1, indicating IONPs with strong heating efficiency.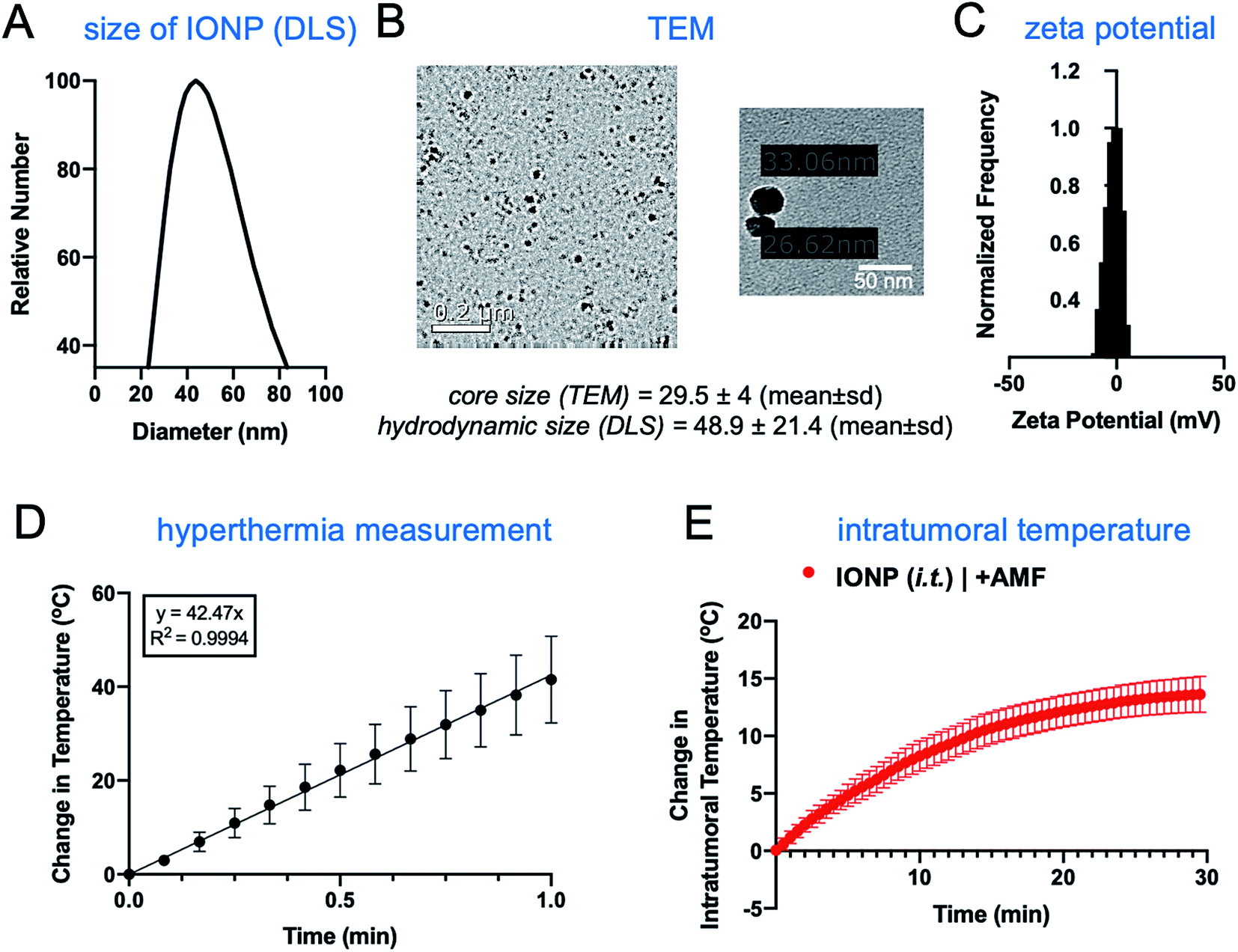 | ||
| Fig. 2 Characterization of iron oxide nanoparticles. Measurement of the nanoparticle's size using (A) DLS and (B) TEM. (C) Zeta potential of nanoparticles. (D) Linear graph illustrating the heat ramp up within the first minute of exposure to an alternating current (AC) magnetic field with an amplitude of 20 kA m−1 at a fixed frequency of 380 kHz (n = 3). Mean ± SEM. (E) Mammary tumor bearing mice were injected i.t. with a dosage of 16 mg kg−1 iron using the iron oxide nanoparticles. A fiber optic probe (Neoptix T1) was inserted into the tumor to measure bulk tumor temperature (mean ± SEM). | ||
The hyperthermic capability of IONPs was evaluated in vivo using the orthotopic mammary 4T1 mouse model, which closely resembles human aggressive triple-negative breast cancer. When the tumor size was about 500 mm3 (day 10 after tumor inoculation), IONPs were injected directly into the tumor (intratumoral or i.t.) at a dose of 16 mg kg−1 Fe. At the end of the 30 min AMF application (20 kA m−1, 380 kHz), the intratumoral temperature had increased by 13 °C (Fig. 2E), while the temperature of the rest of the body was slightly elevated (ΔT ∼ 3 °C). Two days after the hyperthermia treatment, the animals were euthanized, and flow cytometry was performed to analyze the immune cell content in the tumor and spleen, comparing mice injected with IONP with or without application of AMF. The TME of untreated mice bearing 4T1 tumors was enriched with dysfunctional immune cells. Within 10 days after 4T1 mammary tumor inoculation, CD45+ leukocytes comprised more than 50% of the cells within the TME with immune cell populations being at least 3-fold higher than cancer cells. Notably, immunosuppressive M2-like macrophages represented about 60–70% of the total macrophage population. This supports the potential impact of hyperthermia-induced depletion of these protumoral and immunosuppressive immune cells. In the case of the IONP-injected animals (no AMF), there was a mild decrease of tumor-resident dendritic cells (DCs), macrophages and natural killer (NK) cells compared to untreated mice. The application of AMF resulted in a remarkable decrease of all innate immune cell subpopulations within the tumor, including monocytes, neutrophils, DCs, and macrophages (Fig. 3A, top panel). Modest changes were observed in the spleen across the three groups (Fig. 3A, bottom panel). Changes in adaptive T cell subpopulations within the tumor followed a similar trend to those in the innate immune compartment (Fig. 3B).
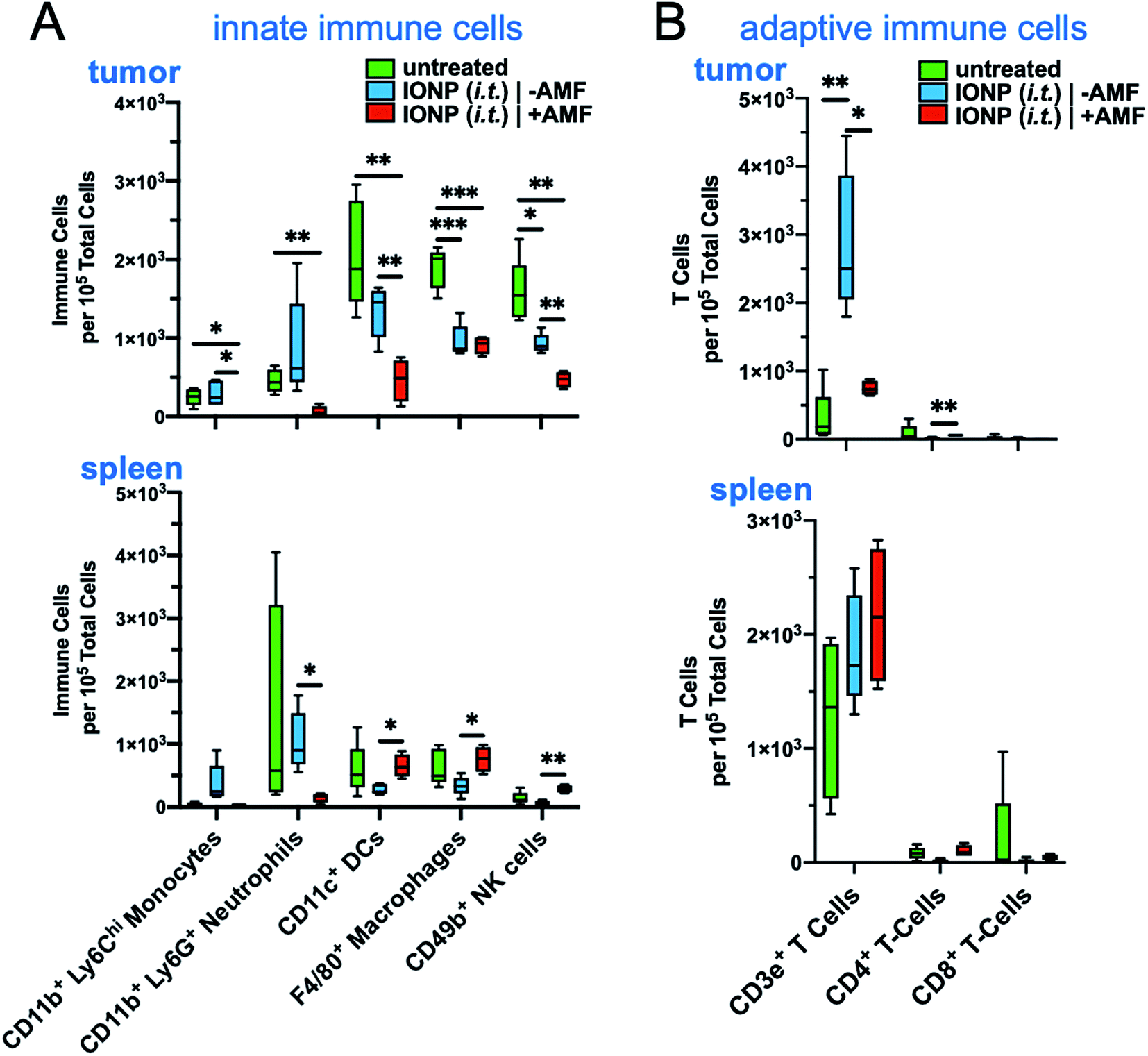 | ||
| Fig. 3 Characterization of the thermal effect of iron oxide nanoparticles on immune cell populations. Mammary tumor bearing mice were injected i.t. with a dosage of 16 mg kg−1 iron using the iron oxide nanoparticles. Flow cytometry analysis of the (A) innate and (B) adaptive immune cells was performed to identify the effects of hyperthermia on immune cell populations in both the tumor and spleen (n = 4–5 mice per group). Box and whisker plots (5–95 percentile) with statistics by one-way ANOVA with a post hoc Tukey or Sidak's test. *P < 0.05, **P < 0.01; ***P < 0.001. | ||
3.2 Systemic versus intratumoral administration of IONP
We then compared systemic to intratumoral delivery. In previous reports,24–34 we have explored various targetable receptors to direct nanoparticles to aggressive and metastatic breast cancer, resulting in high intratumoral deposition of nanoparticles. In particular, fibronectin is overexpressed in the extracellular matrix of the perivascular areas of breast cancer, which makes it a highly desirable target.26,27 To further improve the deposition of IONPs and their uptake by tumor-resident immune cells, we used fibronectin-targeting IONPs that exhibit increased deposition in the perivascular regions of tumors,26,27 where they are easily accessible by phagocytic immune cells. The schedule, doses and experimental design were identical for both the systemic and intratumoral delivery of IONPs (Fig. 1A). At the end of the 30 min AMF application, the IONP (i.v.) generated a 5 °C increase of the intratumoral temperature compared to 13 °C for the IONP (i.t.) group (Fig. 4A). Two days after the hyperthermia treatment, the animals were euthanized, and tumors and spleen were collected for flow cytometry analysis. Tumor weights in the IONP (i.t.) and IONP (i.v.) groups without the AMF application was similar to the untreated mice. However, after exposure to AMF, tumors from mice treated with either IONP (i.t.) or IONP (i.v.) exhibited a nearly 50% lower weight compared to the untreated group (Fig. 4B). Innate immune cells were significantly decreased for both IONP (i.v.) and IONP (i.t.) groups compared to untreated controls (Fig. 4C). However, the IONP (i.v.) group (+AMF) resulted in a significantly higher decrease in the tumor-resident macrophage content than the IONP (i.t.) group (+AMF). Similarly, the IONP (i.v.) group exhibited significantly lower total CD3e+ T cells, including CD4+ T cells in the tumor (Fig. 4D). What these data suggest is that the total amount of IONP delivered to the tumor site may not be as important as the degree of widespread distribution throughout the entire tumor and the uptake by the target innate immune cells. In previous studies, we have shown that systemic delivery of iron oxide nanoparticles results in significant deposition in the near-perivascular space throughout the tumor volume.33,34 This preferential deposition to the APC-rich perivascular regions of tumors led to selective and significant uptake by these phagocytic immune cells.19–21 In the case of an immunostimulatory nanoparticle, there was a significantly improved delivery and uptake by immune cells throughout a tumor in the case i.v. than i.t. administration of the same particles.21 Consistent with previous studies,19–21 we found that IONPs were primarily taken up by phagocytic immune cells (Fig. 5A). For example, nanoparticles were taken up by 35% of the macrophages in the tumor but less than 1% of the cancer cells. Similarly, IONPs were taken up by a significant portion of the phagocytic cells in blood and spleen (Fig. 5B and C).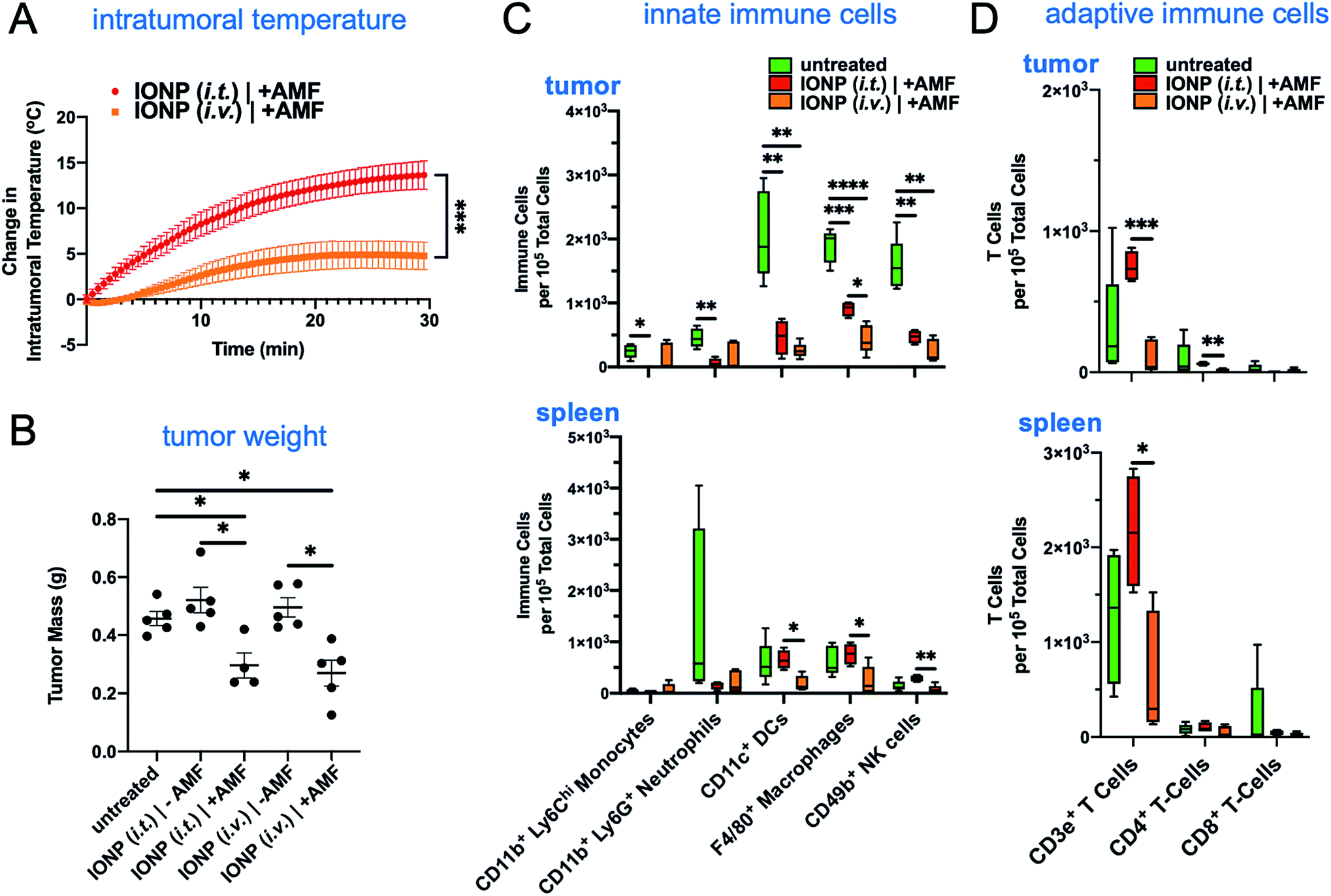 | ||
| Fig. 4 Iron oxide nanoparticles administered intravenously to target mammary tumor margins have similar effects on immune cell populations as locally administered nanoparticles. (A) Intertumoral temperature was recorded using a fiber optic temperature sensor (Neoptix T1) while administering an alternating magnetic current of 20 kA m−1 at a fixed frequency of 380 kHz. Mice were injected with a 16 mg kg−1 iron dosage. (B) Primary tumor mass obtained at the terminal point on day 12. Flow cytometry was performed on both innate (C) and adaptive (D) immune cells to comparatively observe the effects of hyperthermia from systemically and intratumorally administered iron oxide nanoparticles (n = 4–5). Box and whisker plots (5–95 percentile). Intratumoral temperature and tumor weight are plotted with mean ± SEM. Statistics are performed using a one-way ANOVA with a post hoc Tukey or Sidak's test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. | ||
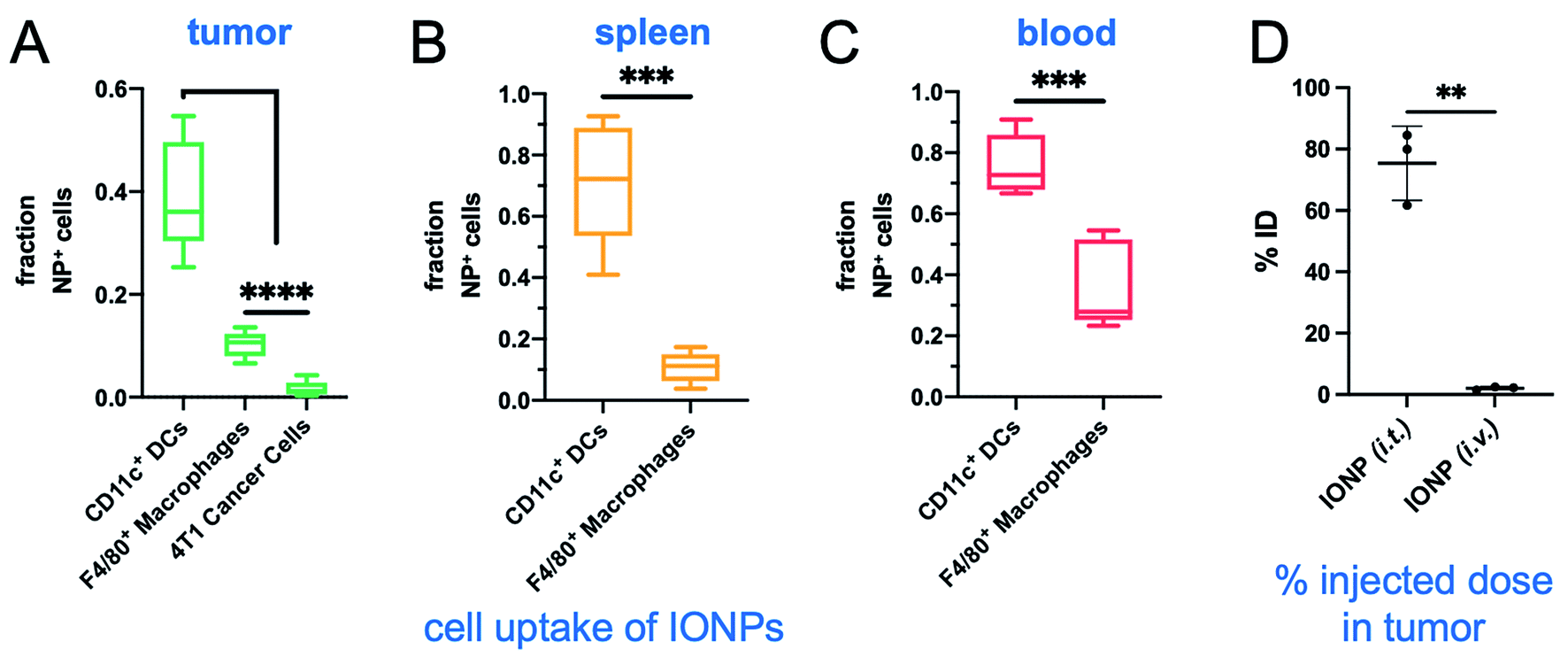 | ||
| Fig. 5 Evaluation of intratumoral deposition of IONPs after i.v. or i.t. delivery. Flow cytometry study indicating the cell uptake of IONPs in the (A) primary tumor, (B) spleen and (C) blood (n = 5 per group). (D) After i.t. or i.v. injection of IONP, tumors were perfused, excised, weighed and digested (n = 4 per group). The concentration of iron was directly measured using ICP-OES. Control mice with tumors injected with saline were used to correct for endogenous iron. Box and whisker plots (5–95 percentile). Statistics are performed using a one-way ANOVA with a post hoc Tukey or Sidak's test. ***P < 0.001; ****P < 0.0001. | ||
In a separate study, after i.t. or i.v. injection of IONP, the tumors of mice were excised and digested. The total amount of intratumoral IONPs (i.e., iron) was directly measured using inductively coupled plasma optical emission spectroscopy (ICP-OES). Control animals with 4T1 tumors were used to correct for the background iron levels in the tumor. Not surprisingly, the i.t. administration resulted in much higher intratumoral levels of IONP than the i.v. case (Fig. 5D). Overall, comparing the i.t. route of administration to i.v., the considerably higher levels of intratumoral IONP resulted in a significantly higher hyperthermic effect, which resulted in equal or lower depletion of target immune cells. This further suggests that a milder hyperthermia is preferable for the intended application. In a future study, we will use a dose for i.t. administration that achieves the same intratumoral IONP levels to i.v. administration to directly compare the effect of nanoparticle distribution in the tumor on hyperthermia.
3.3 Combination of hyperthermia with immune checkpoint inhibitors
After establishing that IONP-induced hyperthermia can achieve a significant reduction of all the immune cell subpopulations in the TME, we selected to design a therapeutic strategy to first deplete the existing dysfunctional tumor-resident immune cells and then repopulate the tumor with functional infiltrating immune cells (Fig. 1B). We selected the IONP (i.v.) option due to the overall better performance against innate and adaptive immune cells observed in the previous study. To boost the function of the infiltrating immune cells, one day after the hyperthermia treatment, the animals were intraperitoneally injected with the clinically approved immune checkpoint inhibitors anti-PD1 and anti-CTLA-4 at dose of 250 μg and 100 μg per mouse, respectively. Flow cytometry analysis 24 h after administration of the checkpoint inhibitors indicated that there were significant changes of major immune cell subsets in treated groups compared to controls in the tumor, spleen and blood (Fig. 6A–C). The rapid accumulation of innate immune cells in the tumor shows that the checkpoint inhibitors triggered a significant infiltration and expansion of monocytes, neutrophils, DCs, macrophages and NK cells (Fig. 6A). Notably, the treatment with checkpoint inhibitors alone (no IONP-medicated hyperthermia) was beneficial but was not capable of increasing the infiltration and expansion of innate immune cells into the tumor as effectively as the combination of hyperthermia and checkpoint inhibitors. Similar patterns were observed in the spleen and blood (Fig. 6B and C). Equally important was that the adaptive immune arm also exhibited significant expansion of its major cell subpopulations in the tumor, spleen and blood (Fig. 6D–F). Using tumor weight as a measure of the antitumor immune response, treatment with the checkpoint inhibitors alone resulted in tumor masses that were similar to the untreated controls (Fig. 6G). On the other hand, the combination treatment of IONP-mediated hyperthermia and checkpoint inhibitors reduced the tumor mass to half of that of the controls. The correlation between nanoparticle uptake and subsequent decrease of those cell types after AMF treatment supports our hypothesis that IONP-induced hyperthermia can facilitate immune cell depletion within tumors. When hyperthermia was combined with checkpoint inhibitors, we observed a profound influx of immune cells into the tumor coupled with a significant decrease in the number of cancer cells (∼2-fold) within 24 h compared to the group treated only with hyperthermia and the untreated group (Fig. 6H). While leukocyte content within the tumor was substantially reduced in animals treated with IONP-mediated hyperthermia (no checkpoint inhibitors), the number of tumor cells remained unaltered indicating the absence of a functional antitumor immune response. These findings suggest that a single IONP-induced hyperthermia treatment followed by standard checkpoint inhibitors was sufficient to repopulate the TME with ‘fresh’ functional immune cells resulting in a significant reduction of the cancer cell population.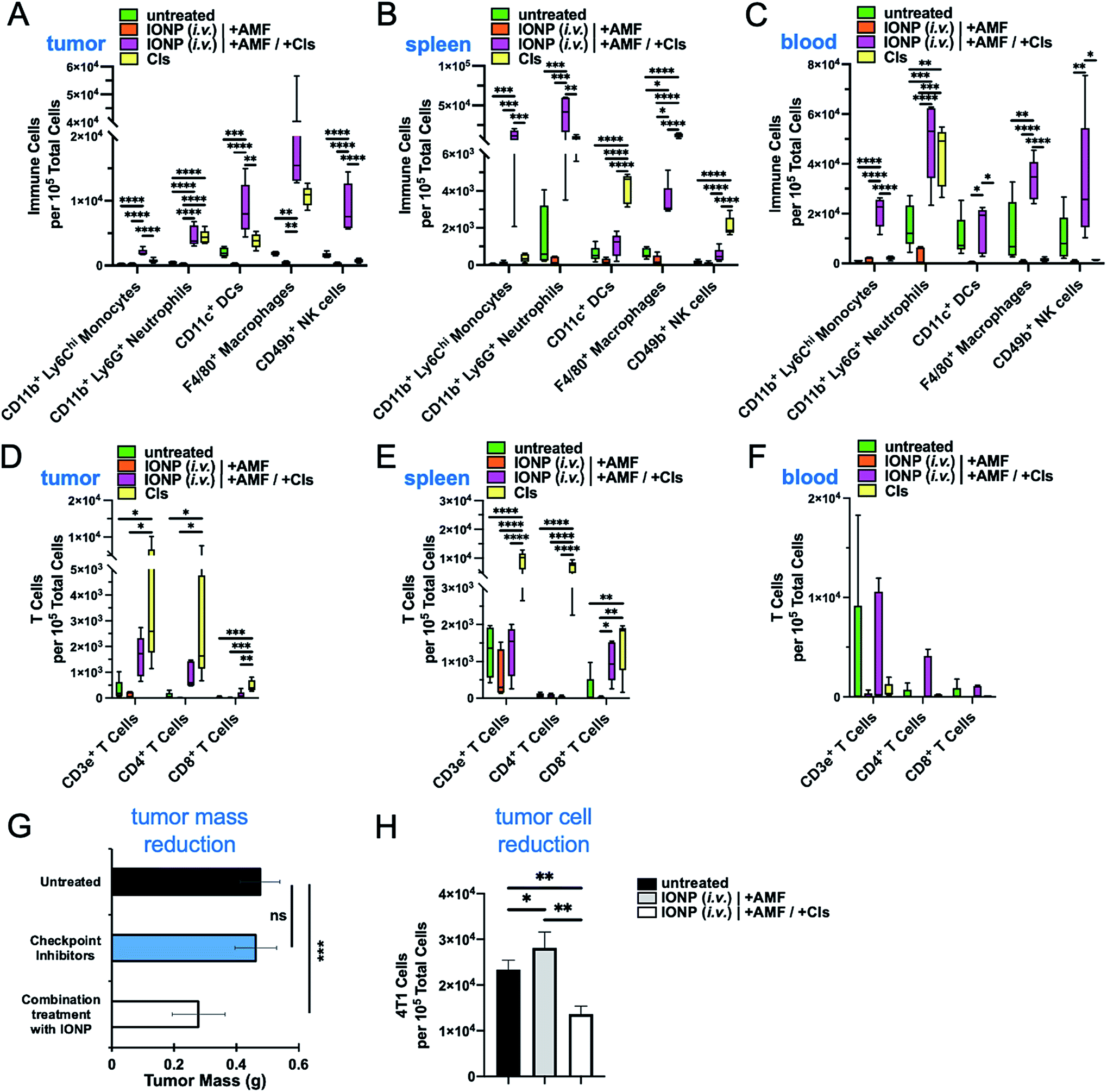 | ||
| Fig. 6 Recruitment of innate and adaptive immune cells to the tumor microenvironment post-depletion using a mild hyperthermia treatment. Innate immune cells in the (A) tumor, (B) spleen, and (C) blood were quantified using flow cytometry 48 h post-initial iron oxide hyperthermia treatment. Adaptive immune cells in the (D) tumor, (E) spleen, and (F) blood were also quantified to identify the response to mild hyperthermia followed by the administration of anti-PD1 and anti-CLTA4 checkpoint inhibitors in an effort to encourage antitumor immunity. The tumor response to the treatments was assessed in terms of (G) tumor mass and (H) content of 4T1 cancer cells post-treatment as quantified by flow cytometry. Mean ± SEM. Box and whisker plots (5–95 percentile). Statistics included a one-way ANOVA with a post hoc Tukey or Sidak's test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. | ||
4. Conclusions
In this study, our primary objective was to assess the ability of hyperthermia to alter the immune landscape in an aggressive and immunosuppressive tumor microenvironment. We show that IONP-induced hyperthermia can reduce the existing immune cell content within the TME, which then can be replaced by a fresh population of immune cells following treatment with standard immune checkpoint inhibitors. These findings present a novel route to overcoming the obstacle of immunosuppression within aggressive solid tumors. Future investigation will focus on elucidating the mechanisms of this phenomenon and explore whether depletion of immune cells is determined by the superior distribution and uptake of IONPs (i.v. vs. i.t.) or the degree of hyperthermia (mild vs. high). Special consideration also should be given to the potential role for successful hyperthermic depletion of immune cells in the context of combination therapy and development of long-term antitumor immunity.Author contributions
Guarantors of integrity of entire study: E. K., A. C. S. S, G. C., M. E. L. Study concepts/study design: E. K., A. C. S. S, G. C., M. E. L. Data acquisition or data analysis/interpretation: all authors. Manuscript drafting and editing: E. K., G. C., M. E. L., A. C. S. S. Manuscript final version approval: all authors. Experimental studies: G. C., G. L., A. R., A. C., E. A., S. W., T. J. M., M. E. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the NCI (R01CA253627, U01CA198892), the Case Comprehensive Cancer Center Support Grant (P30CA043703) and the Shiverick Family Fund, and the Alex's Lemonade Stand Foundation (to E. Karathanasis). G. Covarrubias and M. Lorkowski were supported by a fellowship from the NIH Interdisciplinary Biomedical Imaging Training Program (T32EB007509) administered by the Department of Biomedical Engineering, Case Western Reserve University.References
- L. F. Fajardo, B. Egbert, J. Marmor and G. M. Hahn, Cancer, 1980, 45, 613–623 CrossRef CAS PubMed.
- P. Wust, B. Hildebrandt, G. Sreenivasa, B. Rau, J. Gellermann, H. Riess, R. Felix and P. M. Schlag, Lancet Oncol., 2002, 3, 487–497 CrossRef CAS PubMed.
- O. Grauer, M. Jaber, K. Hess, M. Weckesser, W. Schwindt, S. Maring, J. Wolfer and W. Stummer, J. Neuro Oncol., 2019, 141, 83–94 CrossRef CAS PubMed.
- K. Maier-Hauff, F. Ulrich, D. Nestler, H. Niehoff, P. Wust, B. Thiesen, H. Orawa, V. Budach and A. Jordan, J. Neuro Oncol., 2011, 103, 317–324 CrossRef PubMed.
- K. Maier-Hauff, R. Rothe, R. Scholz, U. Gneveckow, P. Wust, B. Thiesen, A. Feussner, A. von Deimling, N. Waldoefner, R. Felix and A. Jordan, J. Neuro Oncol., 2007, 81, 53–60 CrossRef CAS PubMed.
- M. E. Lorkowski, P. U. Atukorale, K. B. Ghaghada and E. Karathanasis, Adv. Healthcare Mater., 2020, e2001044, DOI:10.1002/adhm.202001044.
- A. M. Rauwerdink and J. B. Weaver, Med. Phys., 2011, 38, 1136–1140 CrossRef PubMed.
- B. Hildebrandt, P. Wust, O. Ahlers, A. Dieing, G. Sreenivasa, T. Kerner, R. Felix and H. Riess, Crit. Rev. Oncol. Hematol., 2002, 43, 33–56 CrossRef PubMed.
- H. Dahring, J. Grandke, U. Teichgraber and I. Hilger, Mol. Imag. Biol., 2015, 17, 763–769 CrossRef CAS PubMed.
- M. H. den Brok, R. P. Sutmuller, R. van der Voort, E. J. Bennink, C. G. Figdor, T. J. Ruers and G. J. Adema, Cancer Res., 2004, 64, 4024–4029 CrossRef CAS PubMed.
- M. E. Mikucki, D. T. Fisher, A. W. Ku, M. M. Appenheimer, J. B. Muhitch and S. S. Evans, Int. J. Hyperther., 2013, 29, 464–473 CrossRef CAS PubMed.
- B. Frey, E. M. Weiss, Y. Rubner, R. Wunderlich, O. J. Ott, R. Sauer, R. Fietkau and U. S. Gaipl, Int. J. Hyperther., 2012, 28, 528–542 CrossRef CAS PubMed.
- L. Borriello, R. C. Seeger, S. Asgharzadeh and Y. A. DeClerck, Cancer Lett., 2016, 380, 304–314 CrossRef CAS PubMed.
- J. A. Joyce and D. T. Fearon, Science, 2015, 348, 74–80 CrossRef CAS PubMed.
- L. Muller, P. Aigner and D. Stoiber, Front. Immunol., 2017, 8, 304 Search PubMed.
- M. A. Swartz, S. Hirosue and J. A. Hubbell, Sci. Transl. Med., 2012, 4, 148rv149 Search PubMed.
- M. Banys, A. D. Hartkopf, N. Krawczyk, T. Kaiser, F. Meier-Stiegen, T. Fehm and H. Neubauer, Breast Cancer, 2012, 4, 183–191 CAS.
- C. M. Ghajar, Nat. Rev. Cancer, 2015, 15, 238–247 CrossRef CAS PubMed.
- P. A. Bielecki, M. E. Lorkowski, W. M. Becicka, P. U. Atukorale, T. J. Moon, Y. Zhang, M. Wiese, G. Covarrubias, S. Ravichandran and E. Karathanasis, Nanoscale Horiz., 2021, 6, 156–167 RSC.
- M. E. Lorkowski, P. U. Atukorale, P. A. Bielecki, K. H. Tong, G. Covarrubias, Y. Zhang, G. Loutrianakis, T. J. Moon, A. R. Santulli, W. M. Becicka and E. Karathanasis, J. Controlled Release, 2021, 330, 1095–1105 CrossRef CAS PubMed.
- P. U. Atukorale, S. P. Raghunathan, V. Raguveer, T. J. Moon, C. Zheng, P. A. Bielecki, M. L. Wiese, A. L. Goldberg, G. Covarrubias, C. J. Hoimes and E. Karathanasis, Cancer Res., 2019, 79, 5394–5406 CrossRef CAS PubMed.
- V. S. Perera, G. Covarrubias, M. Lorkowski, P. Atukorale, A. Rao, S. Raghunathan, R. Gopalakrishnan, B. O. Erokwu, Y. Liu, D. Dixit, S. M. Brady-Kalnay, D. Wilson, C. Flask, J. Rich, P. M. Peiris and E. Karathanasis, Nanoscale, 2017, 9, 9659–9667 RSC.
- Y. S. Kang, S. Risbud, J. F. Rabolt and P. Stroeve, Chem. Mater., 1996, 8, 2209–2211 CrossRef CAS.
- G. Covarrubias, F. He, S. Raghunathan, O. Turan, P. M. Peiris, W. P. Schiemann and E. Karathanasis, PLoS One, 2019, 14, e0220474 CrossRef CAS PubMed.
- P. U. Atukorale, S. P. Raghunathan, V. Raguveer, C. Zheng, T. J. Moon, M. L. Weiss, P. A. Bielecki, A. L. Goldberg, G. Covarrubias, C. J. Hoimes and E. Karathanasis, Cancer Res., 2019, 79, 5394–5406 CrossRef CAS PubMed.
- P. M. Peiris, F. He, G. Covarrubias, S. Raghunathan, O. Turan, M. Lorkowski, B. Gnanasambandam, C. Wu, W. P. Schiemann and E. Karathanasis, Nanoscale, 2018, 10, 6861–6871 RSC.
- G. Covarrubias, A. Cha, A. Rahmy, M. Lorkowski, V. Perera, B. O. Erokwu, C. Flask, P. M. Peiris, W. P. Schiemann and E. Karathanasis, PLoS One, 2018, 13, e0204296 CrossRef PubMed.
- P. U. Atukorale, G. Covarrubias, L. Bauer and E. Karathanasis, Adv. Drug Delivery Rev., 2017, 113, 141–156 CrossRef CAS PubMed.
- P. M. Peiris, P. Deb, E. Doolittle, G. Doron, A. Goldberg, P. Govender, S. Shah, S. Rao, S. Carbone, T. Cotey, M. Sylvestre, S. Singh, W. P. Schiemann, Z. Lee and E. Karathanasis, J. Pharm. Sci., 2015, 104, 2600–2610 CrossRef CAS PubMed.
- E. Doolittle, P. M. Peiris, G. Doron, A. Goldberg, S. Tucci, S. Rao, S. Shah, M. Sylvestre, P. Govender, O. Turan, Z. Lee, W. P. Schiemann and E. Karathanasis, ACS Nano, 2015, 9, 8012–8021 CrossRef CAS PubMed.
- P. M. Peiris, R. Toy, A. Abramowski, P. Vicente, S. Tucci, L. Bauer, A. Mayer, M. Tam, E. Doolittle, J. Pansky, E. Tran, D. Lin, W. P. Schiemann, K. B. Ghaghada, M. A. Griswold and E. Karathanasis, J. Controlled Release, 2014, 173, 51–58 CrossRef CAS PubMed.
- P. M. Peiris, M. Tam, P. Vicente, A. Abramowski, R. Toy, L. Bauer, A. Mayer, J. Pansky, E. Doolittle, S. Tucci, E. Schmidt, C. Shoup, S. Rao, K. Murray, R. Gopalakrishnan, R. A. Keri, J. P. Basilion, M. A. Griswold and E. Karathanasis, Pharm. Res., 2014, 31, 1460–1468 CrossRef CAS PubMed.
- R. Toy, E. Hayden, A. Camann, Z. Berman, P. Vicente, E. Tran, J. Meyers, J. Pansky, P. M. Peiris, H. Wu, A. Exner, D. Wilson, K. B. Ghaghada and E. Karathanasis, ACS Nano, 2013, 7, 3118–3129 CrossRef CAS PubMed.
- P. M. Peiris, R. Toy, E. Doolittle, J. Pansky, A. Abramowski, M. Tam, P. Vicente, E. Tran, E. Hayden, A. Camann, A. Mayer, B. O. Erokwu, Z. Berman, D. Wilson, H. Baskaran, C. A. Flask, R. A. Keri and E. Karathanasis, ACS Nano, 2012, 6, 8783–8795 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |