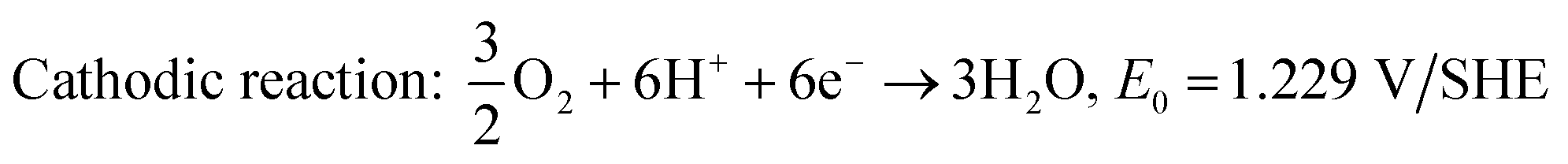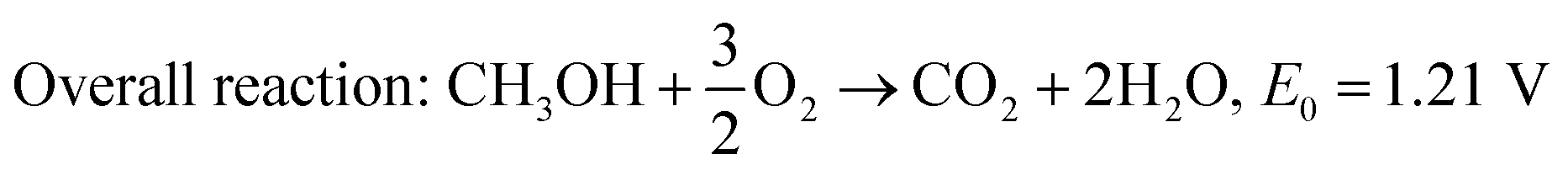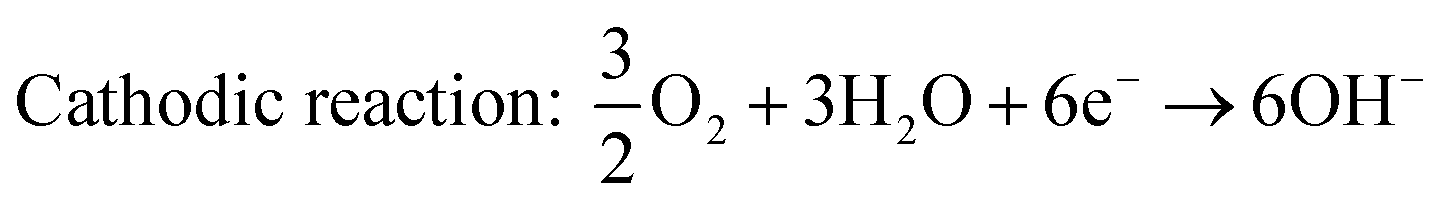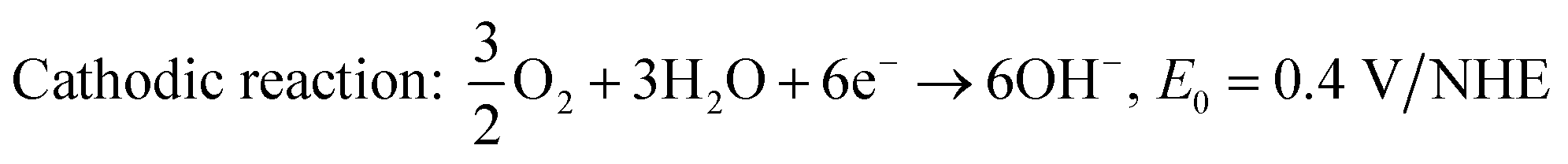DOI:
10.1039/D1NA00046B
(Review Article)
Nanoscale Adv., 2021,
3, 3681-3707
Electrochemically active site-rich nanocomposites of two-dimensional materials as anode catalysts for direct oxidation fuel cells: new age beyond graphene
Received
19th January 2021
, Accepted 24th May 2021
First published on 24th May 2021
Abstract
Direct oxidation fuel cell (DOFC) has been opted as a green alternative to fossil fuels and intermittent energy resources as it is economically viable, possesses good conversion efficiency, as well as exhibits high power density and superfast charging. The anode catalyst is a vital component of DOFC, which improves the oxidation of fuels; however, the development of an efficient anode catalyst is still a challenge. In this regard, 2D materials have attracted attention as DOFC anode catalysts due to their fascinating electrochemical properties such as excellent mechanical properties, large surface area, superior electron transfer, presence of active sites, and tunable electronic states. This timely review encapsulates in detail different types of fuel cells, their mechanisms, and contemporary challenges; focuses on the anode catalyst/support based on new generation 2D materials, namely, 2D transition metal carbide/nitride or carbonitride (MXene), graphitic carbon nitride, transition metal dichalcogenides, and transition metal oxides; as well as their properties and role in DOFC along with the mechanisms involved.
 Kashmiri Baruah | Kashmiri Baruah is currently pursuing her PhD degree under the guidance of Prof. Pritam Deb in the Department of Physics, Tezpur University (Central University). Her research area focuses on the development of 2D material-based electrocatalysts for direct oxidation fuel cells. |
 Pritam Deb | Prof. Pritam Deb is the Group Leader of Advanced Functional Materials Laboratory at the Department of Physics, Tezpur University (Central University). He obtained his M.S. degree in Physics from University of Calcutta in 1995 and accomplished his PhD in Nanoscience from Jadavpur University in 2003. He has held many visiting positions in international institutions. His current research interests include energy materials, quantum materials and applications, surfaces and interfaces, and mesoscopic physics. |
1. Introduction
The exponential increase in energy consumption in different sectors of human civilization has caused the overconsumption of fossil fuels, which is the prime reason for the rise in the CO2 level in the atmosphere. To circumvent the diminishing sources of fossil fuel and exponential rise in pollution, research is focused on finding an alternative, which is renewable and environmentally benign energy source. As the renewable energy sources solar and wind are intermittent, we need a source that is constant and accessible in any situation. Fuel cells have attracted the attention of researchers as a green alternative. They basically convert the chemical energy residing in a chemical bond to electricity. They deliver high power density and good conversion efficiency. Fuel cells are of different types depending on the electrolyte and the fuels used, which will be discussed in detail in the next section. Despite having distinct advantages, fuel cells suffer from certain drawbacks, for instance, the sluggish redox reactions on the surface of the electrodes, fuel crossover, formation of intermediates, and poisoning of the catalyst surface. Thus, in order to accelerate the redox reactions, the search for an efficient and corrosion-resistant electrocatalyst has gained momentum. In this review, we shall highlight the anode catalyst aspect and its role in electrocatalysis either actively or passively. Two-dimensional (2D) materials have recently garnered interest in electrocatalysis compared to other nanostructured materials due to their intriguing properties. Large surface area, tunable electronic states, and largely exposed active sites render them promising candidates for electrocatalysis. The large specific surface area of these materials endows them with multifarious applications in direct alcohol fuel cells, which require high surface-to-volume ratio. The outstanding electronic properties of these materials arise due to the confinement of the electrons in the 2D region. Furthermore, these materials possess excellent mechanical strength and flexibility owing to their atomic dimension and stronger in-plane covalent bonding, which make them easily fabricable into flexible thin films. The electronic structure is a big differentiator of the catalytic properties as the catalysis follows the Sabatier principle. This principle states that the adsorption energy between the reactants and the catalyst must not be too high or too low.1–3 This binding energy between the catalyst and the reactants of transition metals depends on the relative position of the d band and the Fermi level.4,5 Thus, for better catalysis, one can functionalize the surface with different entities and tune the adsorption energies by optimizing the electronic structures. Theoretical simulations such as DFT can study the correlations of the electronic structure, catalyst structure, and composition of the catalyst with their catalytic properties and help in developing highly efficient electrocatalysts.6 After the exfoliation of monolayer graphene in 2004,7 different 2D materials have gained enormous research interest in this regard. So far, at least 19 kinds of new 2D materials, including hexagonal boron nitride,8,9 transition metal dichalcogenides (TMDCs),10 phosphorene,11 graphitic carbon nitride,12 and 2D transition metal carbides/nitrides or carbonitride (MXenes),13 have been found. These 2D materials are more suitable for forming hybrid nanocomposites than their 3D counterparts and many of them exhibit good electrochemical properties. Some of their properties and application in DOFCs have been reviewed here. To increase the electrochemically active surface area, catalysts are generally supported on materials having high surface area. These supports provide a platform for the uniform distribution of active sites required for redox reactions. The supporting materials play an important role in electrocatalysis by (i) acting as an electron source with good electrical conductivity, (ii) holding the metal NPs via physical interaction, (iii) having a large surface area, (iv) providing better stability, (v) providing porosity, and (vi) having higher resistance toward corrosion under different operating conditions.14,15 The usage of a catalyst support can reduce the amount of noble metal used as the catalyst. Over the decades, a variety of carbonaceous and non-carbonaceous materials have been investigated as the catalyst support.16 Different carbonaceous supports are available depending on their structure, surface area, electrical conductivity, thermal stability, and different synthetic conditions.17 Carbonaceous supports include mesoporous carbon, graphene, carbon nanotube (CNT), carbon black, and other forms of carbon in acid and basic media. CNTs, hollow nanospheres, and carbon nanofibres (CNFs) are potential supports for anode catalysts.18–20 Despite having excellent conductivity, CNTs suffer from a poor porous structure and low surface area in comparison to Vulcan-XC.21 Multiwalled carbon nanotubes (MWCNTs) are long, twisted, and possess closed ends; similarly, CNFs possess comparatively smaller surface area, large radius, and a high aspect ratio. Thus, it becomes difficult to deposit metal catalysts with high loading (around 40% or more), leading to a reduction in the performance of MWCNTs/CNFs as the catalyst support. Hence, short and functionalized MWCNTs or CNFs can be a good catalyst support in DOFCs.22 Graphene is a better catalyst support than commercial C owing to its high electroactive behavior and durability.23 However, the corrosion of carbonaceous supports hinders their performance. To solve the problem of corrosion of carbonaceous supports, non-carbonaceous supports such as titania, tungsten oxide, indium oxide, alumina silica, ceria, zirconia, and conducting polymers have been studied.24,25 Thus, the unique structure of 2D materials make them efficient supports/substrates for nanostructured materials. Moreover, 2D materials possess controllable surface structures, which aid in tuning the particle size and distribution of the deposited catalysts. New generation 2D materials such as MXenes, graphitic carbon nitride, and TMDCs have been investigated as catalysts/catalyst supports in DOFCs.26
Apart from the nanocomposite 2D materials, there are also non-nanocomposite 2D materials that are nowadays being explored as electrocatalysts in fuel cell applications. A novel 2D–3D structure of 2D NiZn oxyphosphide nanosheets (NiZnP NSs) and 3D PdRu nanoflowers (PdRu NFs) was developed,27 which exhibited enhanced MOR and EOR activity due to the abundant active sites owing to the unique structure of the catalyst. The mass activity corresponding to MOR and EOR is 1739.5 mA mg−1 and 4719.8 mA mg−1, respectively, which are higher than that obtained from commercial Pd/C. Another group developed a 1D–2D hybrid heterostructure as the support for the Pt–Sn alloy catalyst.28 They partially exfoliated CNTS to prepare the 1D–2D structure, which provided the synergistic effect of both 1D and 2D supports, thus facilitating the complete utilization of the catalyst. A power density of 568 mW cm−2 (at 60 °C) and 34 mW cm−2 (at 80 °C) was achieved for PEMFC and DEFC, respectively. Ultrathin CoNi nanosheets (NSs) deposited over 3D rGO foam29 was employed for the oxidation of sodium borohydride in alkaline medium. CoNi-NS/rGO foam anode catalyst exhibited a higher peak power density of 91.3 mW cm−2 at 95.7 mA cm−2 and also exhibited a good fuel utilization capacity. This catalyst proved to be a potential alternative to noble metal catalysts and opens the path toward high fuel utilization metal catalysts by developing novel supports such as 3D rGO foam. Hollow nanodendrites of PdAg were developed by a unique self-template method and employed as an anode catalyst in ethylene glycol oxidation fuel cell.30 This PdAg catalyst exhibited excellent ethylene glycol oxidation in alkaline medium and a current density of about 4168.8 mA mgPd−1 was obtained, which is 3.8 times higher than that of Pd/C. The unique hollow structure facilitates charge transfer and reduces the amount of catalyst loading. Similar works include 1D PdCuTe nanowires (NWs) as ethylene glycol oxidation anode catalyst,31 Ni–Co/NF,32 CoB/Ni-foam,33 Ni@Pd,34 AuCofiber/Cu catalyst,35 and CuNiPd@Ni foam catalyst36 employed as NaBH4 oxidation anode catalyst. Despite providing excellent electrochemical activity, transition metal catalysts suffer from low fuel utilization. In this context, we cannot totally reduce the usage of noble metal catalysts. In order to lessen the loading of noble metal catalysts, more interacting support materials creating more exposed active sites are required. For instance, the 2D/2D composite of Bi2WO6/MoS2 when used as a support for Pt NPs, which exhibited 1.5 times higher MOR in the presence of visible light and 2.2 times higher than that of Pt/C.37 Co3O4 nanosheets oxidized ethanol in alkaline medium with an onset potential of 1.32 V vs. RHE and a current density of 50 mA cm−2 at 1.545 V.38 Hence, in this review, we will focus on the application of new generation 2D materials and their nanocomposites as anode catalysts/supports in DOFCs.
2. Direct oxidation fuel cells and their types
Fuel cells (FCs) have been recognized as an alternative energy conversion system, particularly for low power consumption electronic devices and domestic transportation systems.39,40 A FC converts the chemical energy stored in the chemical bond of organic and inorganic fuels into electrical energy.41,42 It consists of the following components: a primary fuel electrode (anode), an electrode of oxidant (cathode), an electrolyte in between them, and an external circuit connecting the cathode and anode. Moreover, FCs have attained tremendous research interests because of their low pollutant emission, high power density, and superfast recharging.43,44 FCs are of varying nature depending on the electrolyte used, types of ions transferring through the electrolyte, types of fuels, and the operating temperature range.45,46 The classification of fuel cells is given in Table 1. At the anode/electrolyte interface, the primary fuel gets oxidized to produce protons (H+ ions) in an acidic medium, whereas in a basic medium, OH− ions (travel toward the anode from the cathode side) react with the fuel to oxidize it and the electrons generated move toward the cathode via the external circuit through a load. The electron then reduces the oxygen present at the cathode into water. This energy generated across the load is the useful electric energy output of the fuel cell. During the whole process, the heat energy generated at the interface of the electrode/contact, electrode/electrolyte interfaces, resistance caused by the migration of protons or hydroxides through the electrolyte, and electrons migrating through the external circuit is lost into the surroundings as waste.45 The electrooxidation of these fuels on the electrode/electrolyte interface is very sluggish and the complete oxidation to CO2 is not possible, which reduces the efficiency of the process. Besides, the low stability, limited availability, high cost, and CO poisoning of the mostly-used Pt catalyst have discouraged its practical implementation.47–49 Low cost, highly stable, and CO-resistant electroactive materials are becoming the quest of the day.
Table 1 Classification of fuel cells
| Types of FCs |
Description |
|
1. Classification by electrolytes
|
| Alkaline Fuel Cell (AFC) |
Aqueous alkaline solution (generally KOH) as the electrolyte |
| Proton Exchange Membrane Fuel Cell (PEMFC)/Solid Polymer Fuel Cell (SPFC)/Solid Polymer electrolyte Fuel Cell (SPEFC)/Polymer Electrolyte Fuel Cell (PEFC)/Polymer Electrolyte Membrane Fuel Cell (PEMFC) |
Solid proton conducting membrane as the electrolyte |
| Phosphoric Acid Fuel Cell (PAFC) |
Phosphoric acid as the electrolyte |
| Molten Carbon Fuel Cell (MCFC) |
Molten carbonate salt as the electrolyte |
| Solid Oxide Fuel Cell (SOFC) |
Solid oxide ion conducting ceramic as the electrolyte |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
2. Classification by type of ions passing through the electrolyte
|
| Cation transfer fuel cell |
Positively charged ions migrate through the electrolyte. Also known as acid electrolyte fuel cell |
| H+ ion transfer PAFC |
H+ ion transfer PEMFC |
| Anion transfer fuel cell |
Negatively charged ions passing through the electrolyte. Also known as alkaline type fuel cell |
| OH− (AFC) |
CO32− (MCFC) |
O2− (SOFC) |
|
|
3. Classification by operating temperature
|
| Low temperature |
60–80 °C |
| AFC (60–80 °C) |
PEMFC (80 °C) |
| Intermediate temperature (PAFC) |
∼200 °C |
| High temperature |
High temperature range |
| MCFC (650 °C) |
SOFC (∼1000 °C) |
|
|
4. Classification by type of fuels used
|
| H2–O2 fuel cell |
H2 as the fuel and O2 as the oxidant |
| H2-air fuel cell, ammonia-air fuel cell, hydrazine-air fuel cell, hydrocarbon-air fuel cell, hydrogen–chlorine fuel cell, hydrogen–bromine fuel cell, etc. |
These fuel cells are named according to the fuel used and air as the oxidant |
| Direct alcohol fuel cell (DAFC), for instance Direct Methanol Fuel Cell (DMFC), Direct Ethanol Fuel Cell (DEFC), etc. |
Different alcohols are used as the primary fuel |
Based on the mode of fuel supply to the anode, FCs can also be classified into direct and indirect FCs. Fuels are supplied directly to the anode for electrooxidation in the direct FC, whereas in the latter, the primary fuel is first reformed into hydrogen-containing gas (named as secondary fuel) and then the secondary fuel is fed to the anode for further oxidation to produce electricity.46,50 In cation exchange FC, oxygen reduction reaction is very slow, necessitating the use of noble metal as the cathode catalyst, which accelerates the electrochemical reactions at the cathode. However, the cost factor of noble metal prevents its large-scale production, which is being addressed and needs further improvement.51,52 Water, which is produced as a byproduct at the cathode, needs to be timely removed from the fuel cell, failing which causes water flooding at the cathode and hampers the fuel cell performance as the oxygen mass transfer gets affected. On the other hand, in an alkaline or anion transfer FC, the oxygen reduction kinetics is fast and does not require noble metals unlike cation exchange FC. As hydrogen molecules are smaller than oxygen molecules with a large diffusion coefficient, the water produced at the anode does not affect the mass transfer of hydrogen considerably and is consumed at the cathode to produce hydroxide ions, leading to the reduction of cathode water flooding.46,53 Although electrolytes such as cation exchange membranes, namely, Nafion and PEM, have dominated the fuel cell market, fuel crossover, high-priced membranes and noble metals, low activity, and complicated reactions involved in the oxidation of most of the organic fuels are the factors that have arrested the further development and applications of PEMFCs. Direct oxidation alkaline fuel cells, on the other hand, have gained momentum in the present years because of their potential to outperform the shortcomings of PEMFC. The direction of hydroxide ion flow in the AFC is opposite to that in the PEMFC, reducing the fuel crossover.54 Moreover, in comparison to the aqueous electrolyte, the application of solid polymer electrolytes, such as anion exchange membranes (AEMs), helps in reducing carbonate formation from CO2. PEMFCs can very well fit in with carbonaceous fuels due to their high tolerance toward CO2, whereas AEMFCs can accommodate nitrogen-based fuels, for instance, ammonia and hydrazine, which are not suitable to be used in PEMFCs.55 Pt-based electrocatalysts suffer from poisoning by the reaction intermediates such as CO in an acidic medium. This problem of poisoning can be solved by operating the fuel cell in an alkaline electrolyte, where the kinetics get improved and Pt-free catalysts can be used.56,57 The electrooxidation of fuels such as ethanol and methanol in alkaline medium is independent of the structure, which offers scope for the usage of non-precious metals, such as Ni and Pd, and perovskite oxides in alkaline fuel cells.58–60 Thus, the usage of alkaline electrolytes in fuel cells carries ample merits, for instance, improved efficiency, better reaction environment, reduced poisoning effects, minimal sensitivity to surface structure, and a broader range of affordable non-noble metal electrode materials to choose from.61 Mixed transition metal oxides, such as Co-based oxides (MnCo2O4, CoCo2O4, or NiCo2O4), are stable in alkaline electrolytes as their surfaces remain covered by OH− ions.62,63 In alkaline medium, during alcohol oxidation, these adsorbed OH− ions cause the dehydrogenation of adsorbed alcohols and assist in the desorption of the poisoning intermediates.64,65 In this way, alcohol oxidation becomes more efficient in alkaline medium than in acidic medium. As the conductivity of the metal oxides are lesser than the Pt-based catalysts, the alcohol oxidation reaction can be compensated by making composites with other conducting 2D materials having a large surface area.
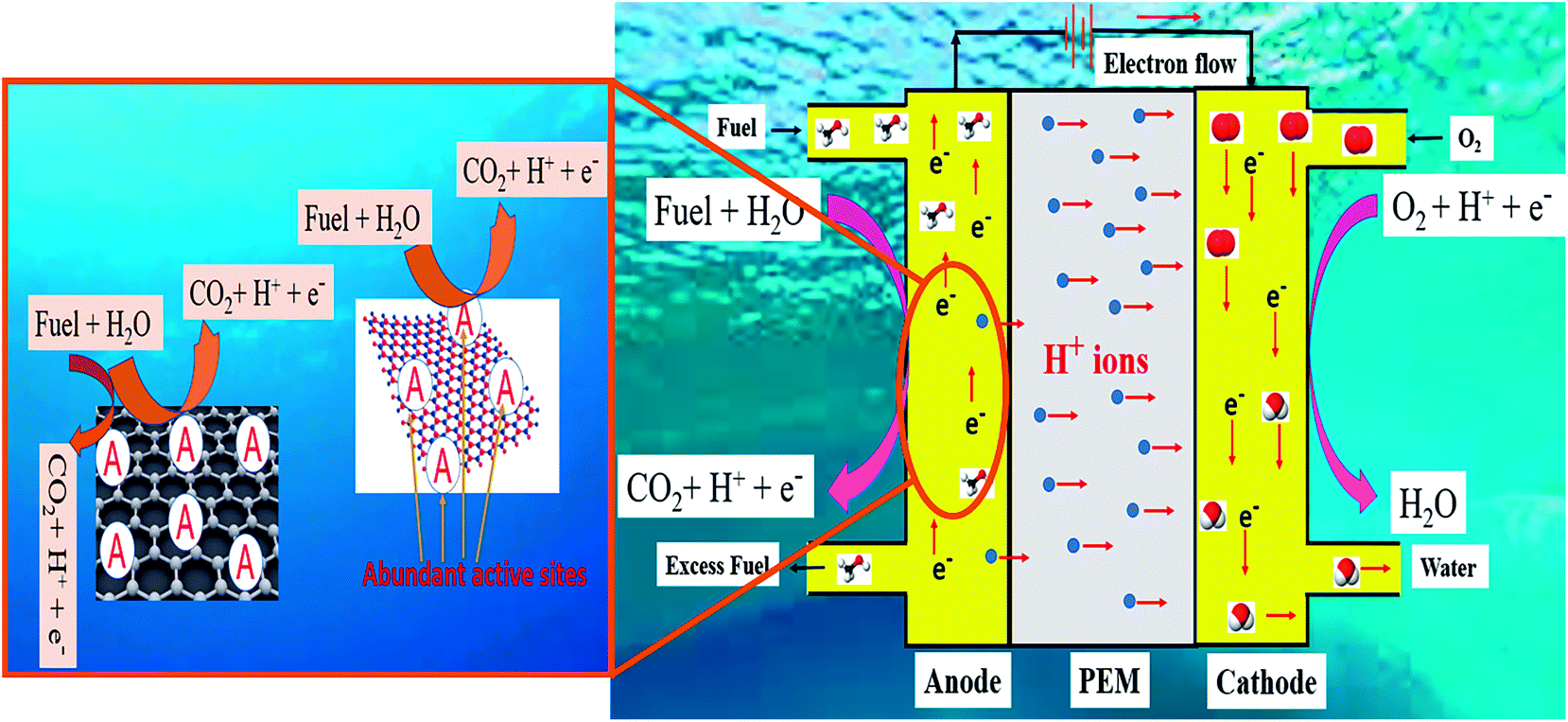
Hence, keeping the conditions alike, alkaline fuel cells provide better performance than acid/proton conducting fuel cells. The schematic of DAFC in acidic medium is shown in Fig. 1. In search of sustainable energy resources, hydrogen has proved to be a better option in reducing CO2 emissions.66,67 Hydrogen fuel cell is envisioned to meet the energy challenges as the only byproduct is water, thus making “hydrogen economy” viable. Although PEMFC has been commercialized in multifarious applications such as portable power, backup power, and domestic transport, the poisoning of platinum group elements as the catalyst and the difficulty in storage and transportation of hydrogen has reduced its feasibility.68,69 In a H2–O2 fuel cell, the electrochemical reactions are explained in eqn (1)–(3).
| | | Anodic reaction (HOR): 2H2 → 4H+ + 4e− | (1) |
| | | Cathodic reaction (ORR): O2 + 4H+ + 4e− → 2H2O | (2) |
| | | Overall reaction: 2H2 + O2 → 2H2O + W + Waste heat | (3) |
where
W represents the useful electric energy supplied by the fuel cell.
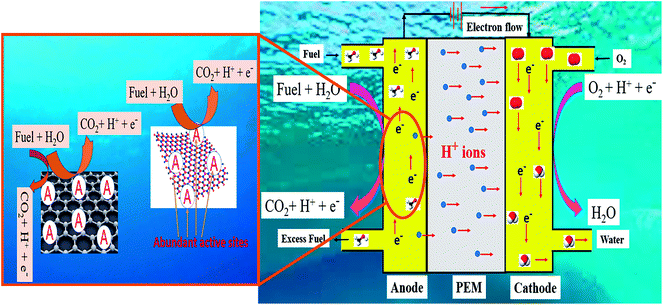 |
| | Fig. 1 Schematic of the mechanism of DAFC in acidic medium and electrochemical fuel oxidation on the 2D surface. | |
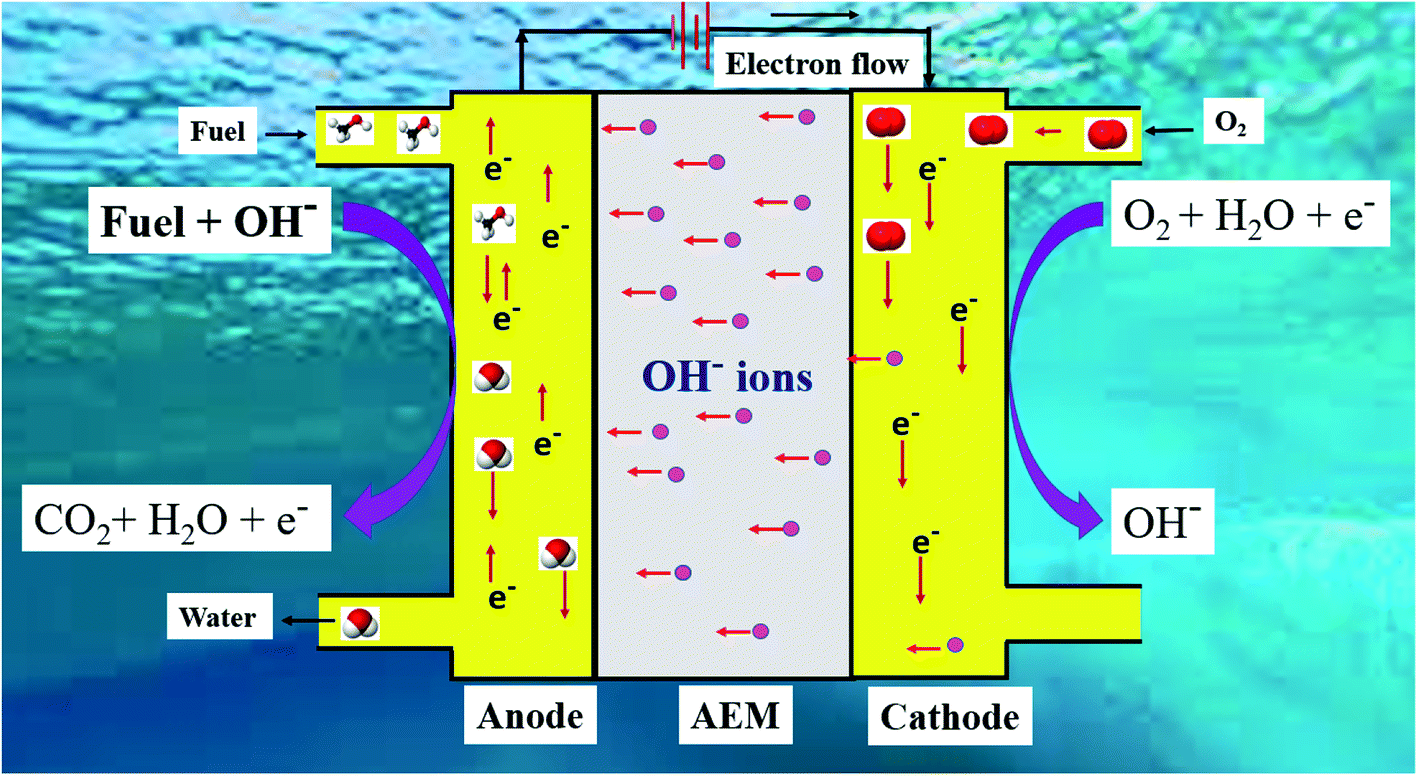
The schematic of DAFC in basic medium is shown in Fig. 2.
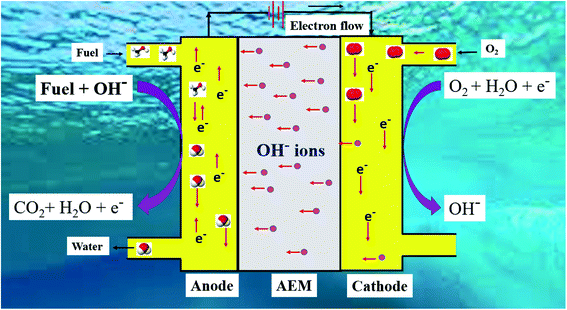 |
| | Fig. 2 Schematic of the mechanism of DAFC in basic medium. | |
Unlike hydrogen, alcohols such as methanol, ethanol, and ethylene glycol have much lower oxidation kinetics; however, they have received attention as they possess higher energy density and easier storage and transportation facility. Among direct alcohol fuel cells, DMFC is the simplest one as the complete oxidation of methanol to CO2 releases only 6 electrons,48 whereas the electrooxidation of ethanol liberates 12 electrons.70 Regardless of the sluggish kinetics, ethanol has been attractive because of its high energy density (8 kW h kg−1vs. 6.1 kW h kg−1 of methanol), less toxicity, and safety for large scale application as a renewable biofuel.71,72 The electrochemical reactions taking place in the DMFC acidic medium are shown in eqn (4)–(6), and that of the basic medium in eqn (7)–(9). These redox reactions are very sluggish and tend to produce intermediates such as carbon monoxide, formaldehyde, and formic acid,71 thereby reducing the conversion efficiency of the fuel cell. Thus, the search for an efficient, non-platinum metal-based affordable catalyst is still ongoing.
| | | Anodic reaction: CH3OH + H2O → CO2 + 6H+ + 6e−, E0 = 0.016 V/SHE | (4) |
| | 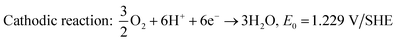 | (5) |
| | 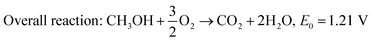 | (6) |
In basic medium, the redox reactions of DMFC are
| | | Anodic reaction: CH3OH + 6OH− → 5H2O + 6e− + CO2 | (7) |
| | 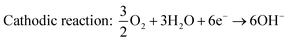 | (8) |
| | 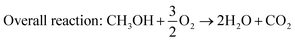 | (9) |
Urea is a non-toxic and non-flammable hydrogen-carrying compound (hydrogen content 6.71 wt%) having an energy density of 16.9 MJ L−1, which is about 10 times higher than that of hydrogen.73,74 Untreated urea-containing wastewater poses threat to the environment as it decomposes into ammonia and other nitrogen-containing pollutants. Traditional methods (such as nitrification and denitrification) used to treat this water are energy consuming.75 In this regard, direct urea fuel cell (DUFC) outperforms these methods as it simultaneously solves the problem of wastewater treatment and electricity production by oxidizing urea/human/animal urine containing water into CO2, H2O, and N2.73,76 As urea can be transported and stored with ease unlike hydrogen, treating urine-rich water will be like killing two birds with one stone. The electrochemical redox reactions involved in a urea fuel cell are shown in the following eqn (10)–(12).
| | | Anodic reaction: CO(NH2)2 + 6OH− → CO2 + 5H2O + N2 + 6e−, E0 = −0.746 V/NHE. | (10) |
| | 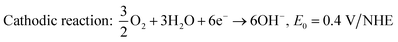 | (11) |
| | 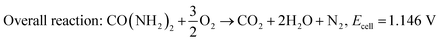 | (12) |
3. Technological aspects of graphene-based anode catalysts
After the discovery of graphene by Novoselov et al.,7 it has attracted tremendous research interest in fields where 2D materials have garnered demands. Graphene is a single layer of sp2 hybridized carbon atoms packed in a hexagonal structure with stronger in-plane σc–c bonding than the out-of-plane π bonding. This mobile network of electrons multiplies its electronic conductivity. The unique aspects of graphene include high mechanical strength of about 1 TPa Young's modulus,77 high surface area (up to 2675 m2 g−1),78 high electrical (0.5–100 S m−1) and thermal conductivity (3080–5150 W m−1 K−1), and high electron mobility at room temperature (200000 cm2 V−1 s−1),79 making it a viable candidate for electrocatalysis and other applications where good thermal and electrical conductivity is the prime requirement. Different configurations of graphene possess different properties depending on the morphology, dimensions, number of layers, and presence of defects. It shows superiority over other dimensional carbon nanostructures owing to its astonishing electrochemical properties.80 Carbon quantum dots, fullerenes, and carbon nanotubes (CNTs) are a few of the contenders of graphene; however, their scalability prevents them from outshining graphene in electrochemical applications. By rationally designing graphene, it can be used either as an electrocatalyst or a support material to enhance the catalytic process.81 Tremendous research work (both theoretical and experimental) has been devoted to graphene and/or graphene nanocomposites with noble metal nanoparticles,82 nanocrystals,83 metal oxides,84etc., as electrocatalysts/supports. As an electrocatalyst support, graphene has shown excellent durability and catalytic activity owing to its large surface area, selectivity, stability, conductivity, and tunable properties.82 Graphene-supported Pt decorated electrocatalyst performs better than the commercial catalyst Pt/C without graphene.85 Similarly, Pd-loaded graphene aerogel exhibits excellent activity compared to commercial Pd/C.86 Pt and Pd are state-of-the-art electroactive metals used in fuel oxidation; however, easy poisoning by intermediates such as CO has guided researchers to form corrosion-resistant alloys such as Pt–Co, Pt–Fe, Pt–Ni, Pd–Co, Pd–Fe, and Pd–Ni87 to improve the performance and lifetime of the catalyst. Moreover, Pt- and Pd-loaded graphene-supported electrocatalysts have also been developed for methanol oxidation fuel cells, such as PtCo/rGO, Pd/rGO, PdCo/rGO, and PtPd/rGO.88–91 Besides, replacing carbon atoms with atoms such as boron, nitrogen, sulfur, and phosphorus helps to increase the conductivity and electrochemical properties of graphene.92–95 Iqbal et al.96 reviewed the prospects and challenges of graphene-based fuel cells. Graphene's low sheet resistance, higher durability, and resistance toward corrosion make it a good candidate for bipolar plates in fuel cells. Moreover, its better stability and reduced crossover also help in attaining better polymer electrolyte membranes in PEMFCs. However, graphene-based electrocatalysts suffer from few technical drawbacks. The high degree of graphitization of carbon makes graphene chemically inert and it also lacks sufficient sites for the deposition of noble metal nanoparticles. Moreover, the catalytic mechanism of graphene-supported nanocrystals and heteroatom-doped graphene are yet not clear and need attention in order to develop highly stable and potential electrocatalysts. Usually, the large surface area of graphene tends to restack due to van der Waals forces, thus reducing the full usage of the electroactive sites. In this context, tuning the morphology such as preparing porous graphene,97 functionalizing graphene oxide with metal oxide,84 attaching it with carbon nanotubes,98 or combining it with other inorganic materials99 can be beneficial in minimizing the restacking and enhancing the electrochemical activity.
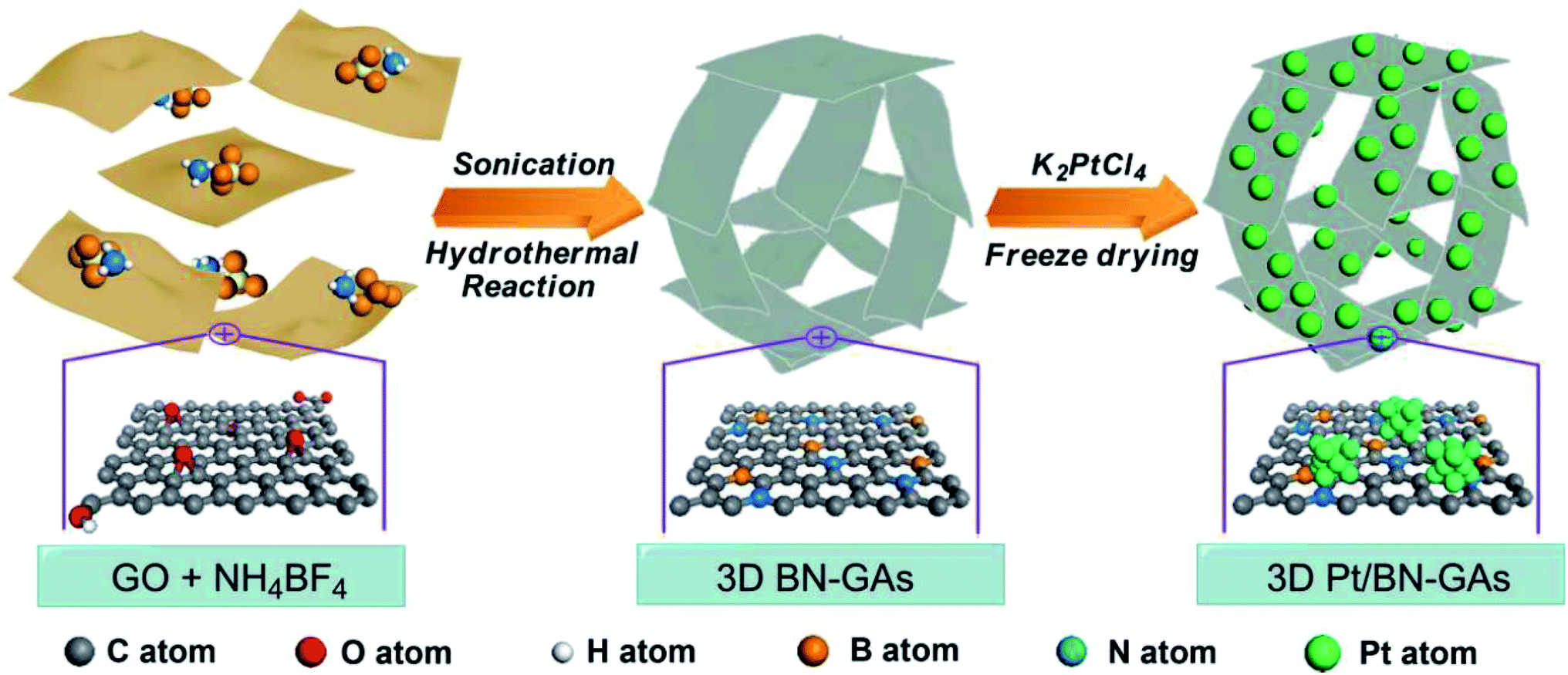
M. Li and coworkers97 constructed B- and N-doped graphene aerogel-supported Pt catalyst (Pt/BN-GA) in MOR (schematic shown in Fig. 3). This unique macro and mesoporous structure featuring large surface area with uniform boron and nitrogen distribution along with Pt nanoparticles exhibits brilliant electrical conductivity, thus making Pt/BN-GA a better electrocatalyst, surpassing traditional catalysts, namely, Pt/GA, Pt/G, and Pt/C. A carbocatalyst comprising of Pd nanoparticles deposited on carbon–rGO (Pd@C–rGO) exhibited improved oxidation of ethylene glycol.100 The smaller particle size of Pd nanoparticles and the large surface area of the rGO support contributed to the enhanced electrocatalytic activity of Pd@C–rGO. A bio-compatible electrocatalyst (Pt–Pd decorated rGO) developed from the extract of vermicast was employed for the oxidation of ethylene glycol (EG).101 A mass activity of 803.4 mA mg−1 was obtained, which is 85% retained after 500 cycles, implying improved CO tolerance. The 2D carbon support, interaction between the metal nanoparticles and the support, and the changed electronic structure of Pt after Pd encapsulation contribute to the enhanced EG oxidation, paving the path for affordable and bio-compatible electrocatalysts. The in situ carbon atom was used to increase the interaction between the MoS2/RGO support with noble metal Pd quantum dots (QDs) and this nanocatalyst was employed as a bifunctional catalyst for alcohol and water oxidation.102 It delivered higher AOR activity. The support participated in the generation of active oxygen, further oxidizing the intermediates formed during the oxidation reactions. The improved catalytic activity of the nanocatalyst is due to the smaller particle size of Pd QDs and enhanced interaction between Pd QDs and the support C@MoS2–RGO. Several reviews have been devoted to the contribution of graphene as the electrocatalyst, catalyst support,103–106 and also as the membrane in PEMFC.107,108 Hence, this timely and extensive review will focus on 2D materials beyond graphene and discuss their properties and application in DOFCs as anode catalysts or supports.
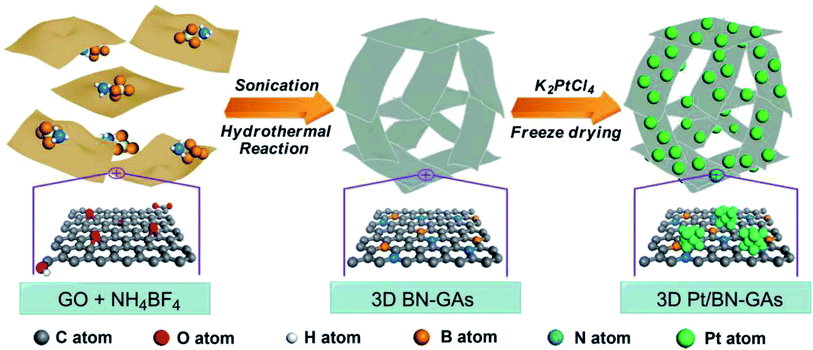 |
| | Fig. 3 Schematic of the formation of 3D Pt-doped BN-graphene aerogel. Reproduced with permission.97 Copyright 2018, ACS. | |
4. 2D materials beyond graphene
4.1 MXenes
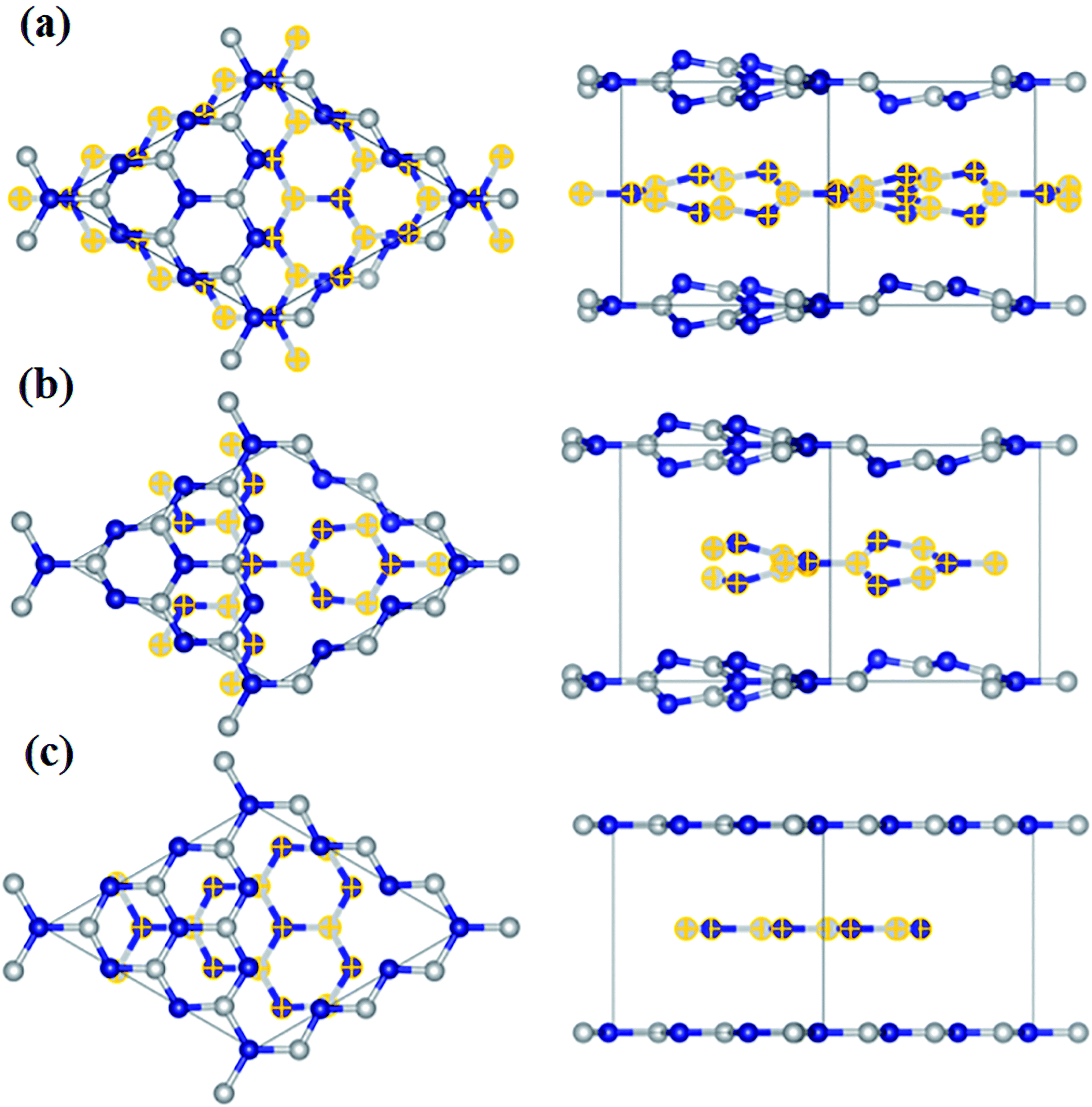
MXenes, a new member, was included in the family of 2D materials after Naguib et al.109 exfoliated MXenes by selectively etching A layer (Al) from the MAX phase (Ti3AlC2 in this case). The suffix ‘ene’ is used in analogy to graphene.110 The MAX is a transition metal carbide and nitride usually represented by Mn+1AXn, where n varies from 1 to 3, M is a transition metal, A can be any element belonging to groups 13–16 of the periodic table, and X can be either carbon, nitrogen, or their mixture.111 MAX phases have hexagonal layered structures comprising of Mn+1Xn layers held together by A layers, with X atoms occupying the octahedral sites.112 The M–X bond, being a mixture of metallic/covalent/ionic, is stronger than the M–A bond, which is generally metallic.113 Unlike the 2D inorganic analogues where the layers are held together by weak van der Waals forces, strong interlayer bonding in MAX renders it difficult to be exfoliated mechanically.114,115 However, because of the differences in the reactivity of the M–X and M–A bonds, it is possible to exfoliate it by etching the A layer from Mn+1AXn using different etchants (such as HF, LiF + HCl, NH4HF2, etc.).115,116 Despite the high yield of the etchant HF, highly concentrated HF is toxic.117 To avoid the usage of corrosive HF and promote the industrial applications of MXenes, an alternative exfoliation method that is safe and fast is urgently required. Halim et al.118 in 2014 reported ammonium bifluoride, NH4HF2, as a substitute for hazardous HF. Thereafter, Ghidiu et al.119 proposed the etching of Al from Ti3AlC2 by forming HF in situ through a reaction between HCl and LiF, in which etching and intercalation were accomplished simultaneously. During etching and interaction with the acids, the transition metals on the surface get attached with anions such as OH−, F−, and O2− present in the solution, introducing the term Tx into the formula of MXene as Mn+1XnTx, where Tx represents the surface anion terminals. These anions bestow them with a hydrophilic nature. Hence, a weaker hydrogen bond replaces the metallic bond M–A, making it easier for organic intercalants (dimethyl sulfoxide, urea, water) to intercalate between the layers such that a shearing force can delaminate the multilayers into single or few layers.115,120,121 MAX phases possess a combination of ceramic and metallic properties. They exhibit stiffness, thermal stability, brittleness, strength like ceramics, and good thermal and electronic conductivity like metals.122 Corresponding to the value of n in MAX, three lattice configurations are possible for MXene, viz., M2X, M3X2, and M4X3, as shown in Fig. 4.123 There are two ways in which more than one metal atom can be present in the M position, viz., the ordered phase and the solid solutions. In the ordered phase, single or bilayer of one metal is sandwiched between another one (e.g., Mo2TiC2 and Ti2Ta2C3), whereas in a solid solution, the metals are randomly placed in the M position (e.g., (Ti,V)2C and (Ti,Nb)4C3).124 To date, more than 70 MAX phases have been reported, out of which only 20 MXenes have been successfully exfoliated. Generally, MXenes possess a hexagonally close packed (hcp) structure. However, the crystal structure varies with the value of n in MXene as the M atoms in M2X possess an hcp structure, whereas M3X2 and M4X3 possess the fcc structure.125 MXenes possess a plethora of fascinating properties such as high electrical and metallic conductivity of about 6000–8000 S cm−1 along with hydrophilicity, large surface area, tunable structure, and good thermal conductivity.126,127
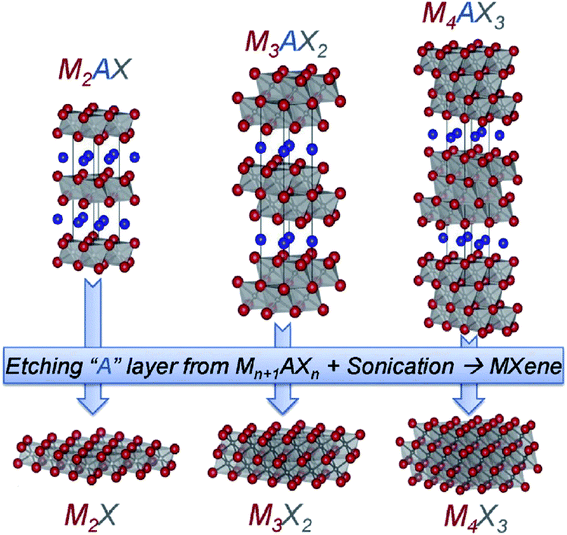 |
| | Fig. 4 Structure of different MAX phases depending upon the number of n. Reproduced with permission.123 Copyright 2014, Wiley. | |
These intriguing properties make MXene an interesting candidate for numerous applications such as electrochemical energy storage devices,128,129 electromagnetic shielding,130 sensors,131 FETs,132 catalysis,126,133 and biomedical applications.134 Recently, MXene has gained momentum in the domain of energy conversion and environmental application as a photocatalyst and an electrocatalyst.
4.2 Graphitic carbon nitride (g-C3N4)
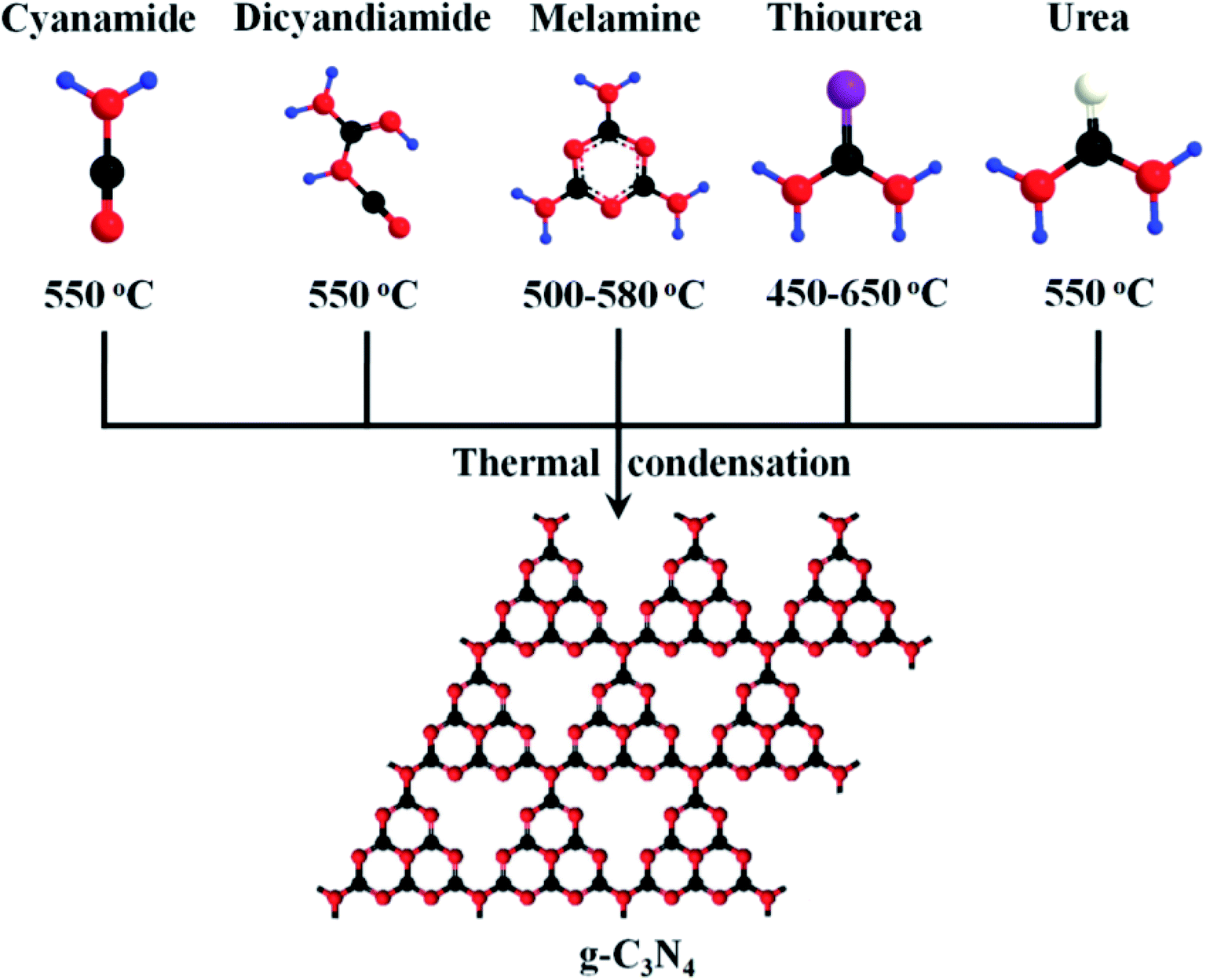
Graphitic carbon nitride (g-C3N4) is similar to graphene with the sp2 hybridized system having van der Waals interaction between the layers.135,136 It is one of the most stable allotropes of carbon nitrides, consisting of tri-s-triazines connected by tertiary amines.137 g-C3N4 is an environmentally benign material as it is composed of earth-abundant elements carbon and nitrogen, and ideally, a monolayer of g-C3N4 could possess a theoretical surface area of about 2500 m2 g−1.138 It can be synthesized by the pyrolysis or polycondensation of various nitrogen-containing precursors (explained in Fig. 5) such as cyanamide, dicyandiamide, trithiocyanuric acid, triazine, heptazine, melamine, urea, and thiourea.139–145 A different degree of condensation and synthetic processes affect the electronic structure, quality, and performance of graphitic carbon nitride.146–148 The polymeric nature of g-C3N4 renders it flexibility in surface engineering by modification at the molecular level and also provides flexibility to the structure, which makes it a good host to other inorganic nanomaterials. g-C3N4 possesses high thermal stability up to 600 °C even in air, which is mainly due to the aromatic C–N heterocycles. This thermal stability value is the highest among organic materials and is also higher than high-temperature polymers and aromatic polyamides and polyimides.149–151
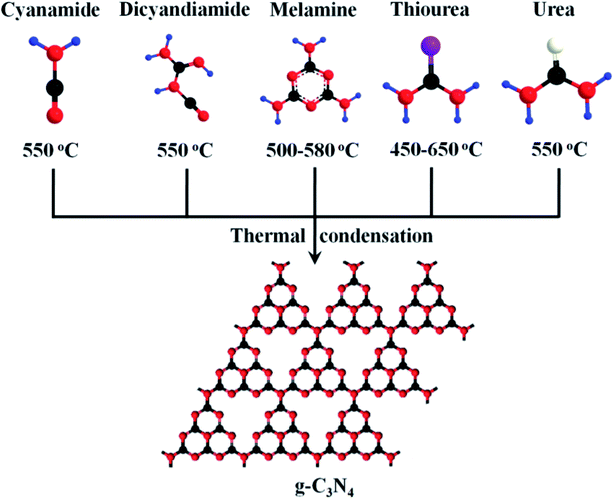 |
| | Fig. 5 Schematic diagram of the synthetic method of g-C3N4 by the condensation of different precursors-cyanamide, dicyandiamide, melamine, thiourea, and urea; here, different colors represent different atoms: black-C atom; red-N atom; blue-H atom; purple-S atom; white-O atom. Reproduced with permission.144 Copyright 2015, Wiley. | |
Besides, g-C3N4 is chemically stable in most solvents such as water, alcohols, tetrahydrofuran, N,N-dimethylformamide, toluene, glacial acetic acid, diethyl ether, and aqueous sodium hydroxide solution owing to the strong van der Waals interactions found between its layers.152,153 The electronic structure of g-C3N4 and its bandgap depend upon the degree of condensation and are tuneable by protonating it or including complex metal cations such as Fe3+ and Zn2+.154,155 Wang and co-workers156 predicted heptazine-based g-C3N4 as the most stable structure using ab initio studies. They also predicted the structure formed by thermal polycondensation and salt melt synthesis method (Fig. 6). Three structures were confirmed and their phase stability is in the order phase 1 > phase 2 > phase 3.
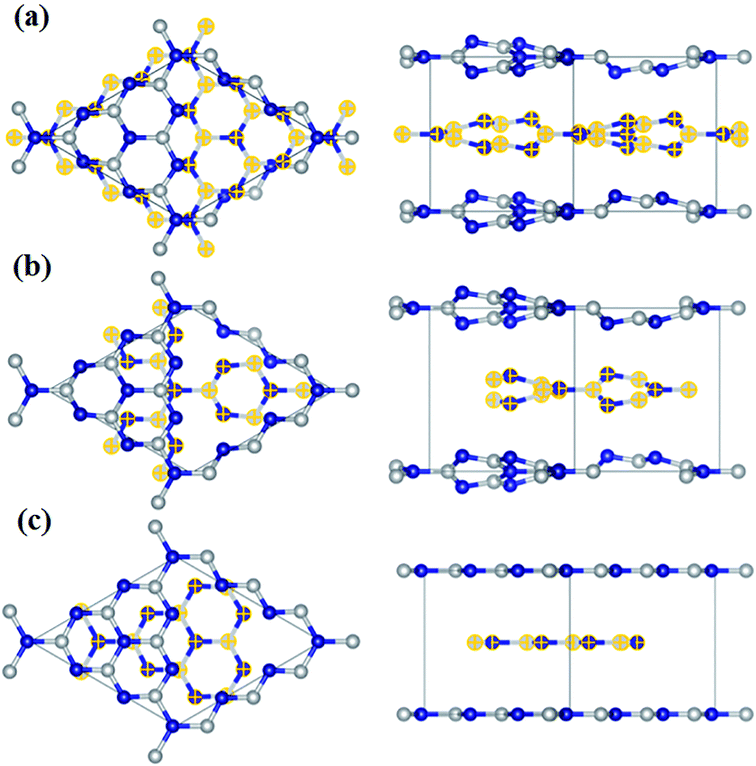 |
| | Fig. 6 Predicted structures of heptazine-based g-C3N4, (a) phase 1, (b) phase 2, (c) phase 3. Middle layer atoms are represented by yellow crosses. Reproduced with permission.156 Copyright 2017, ACS. | |
Here, phase 1 has a direct bandgap (2.87 eV), whereas phase 2 and 3 have indirect bandgaps (3.14 and 2.27 eV, respectively). g-C3N4 has grabbed attention in the fields of catalysis such as photocatalysis and electrocatalysis, viz., HER, OER, and MOR, owing to its 2D structure, low cost, facile preparation methods, and N-rich composition.157–159 However, the condensation method involved in the synthesis of g-C3N4 leads to a number of holes within the structure of g-C3N4, which further decreases its conductivity.160 This decreased conductivity, on the other hand, has an adverse effect on the electrochemical performance of pristine g-C3N4. Thus, developing composites of g-C3N4 with conducting materials can improve its performance as an electrocatalyst.161 Besides, g-C3N4 can be used as a metal-support in FCs. In order to be a good catalyst support, it should possess certain properties such as high stability, electronic conductivity, porous structure, presence of functional groups on the surface with which the catalyst particle can be attached, easy synthesis, and low cost.162 Among different catalyst supports, carbonaceous materials have been studied the most and have shown better performance. However, the most used commercial one, i.e., carbon black, suffers from corrosion and reduction in its electroactivity due to the detachment of the catalyst nanoparticles.163 Nitrogen, which is considered as an n-type dopant, can solve the corrosion-related problems of carbon-based electrocatalysts by increasing their durability.164,165 Hence, g-C3N4, which contains a high amount of N, can resolve this issue as the lone electron pairs of nitrogen act as Bronsted acid and Lewis base,162 capable of anchoring the catalyst and provide it with high resistance in acid as well as basic medium.166 Moreover, surface protonation and doping with B, S, or any metal particles can enhance the conductivity of g-C3N4 as well as its catalytic activity.161,167
4.3 Transition metal dichalcogenides (TMDC)
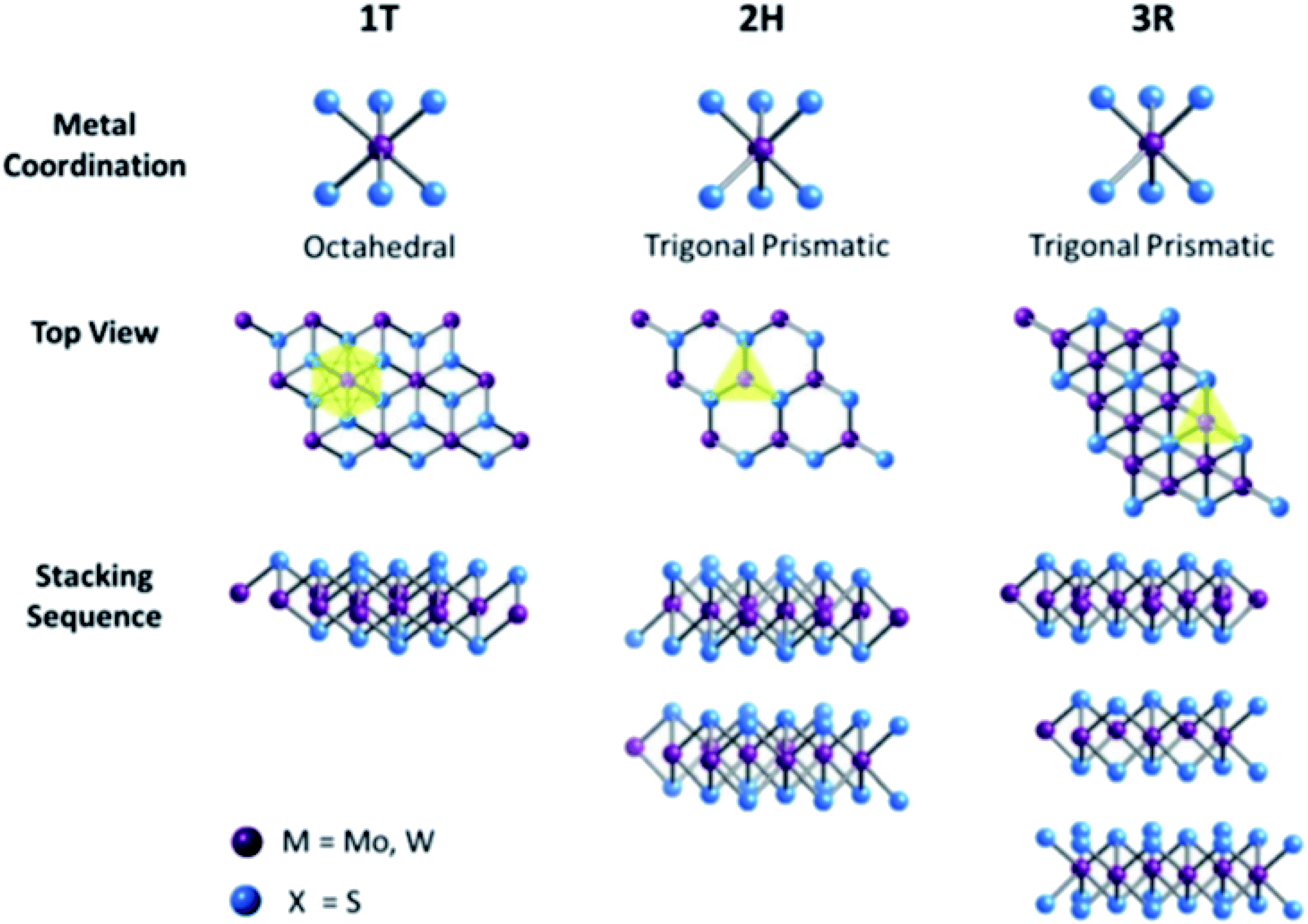
Transition metal dichalcogenides (TMDCs) are another class of 2D materials that have gained interest in many fields such as optoelectronic and electronic devices,168–170 gas sensing,171,172 energy storage,173–175 and energy conversion devices10,176,177 owing to their intriguing layer-dependent properties. TMDCs consist of transition metal (M) atoms with the formula MX2, where M can be any transition element belonging to groups IV, V, and VI, and X is a chalcogen (S, Se, Te). The transition metals are hexagonally structured and inserted between two layers of X atom. Similar to graphene, trilayers in TMDCs are held together by interlayer van der Waals interaction, whereas strong covalent bonding exists within the layers. Almost 40 different types of TMDCs have been explored depending on the combination of M and X atoms.178,179 Depending on the stacking configuration of the atoms, monolayer TMDCs can be found in two different forms, namely, trigonal prismatic phase (2H) and octahedral phase (1T). Thus, the overall crystal structure of TMDCs is hexagonal or rhombohedral with the M atoms in 2H or 1T coordination.178–181 For instance, MoS2 exists in two forms 2H and 1T, which can be changed from 2H to 1T by intercalating Li or K or by the transversal displacement of any of the sulfur planes. Depending on the polytype, the electronic properties of TMDCs vary, for instance, 2H MoS2, WS2, MoSe2, and WSe2 are semiconductors, whereas 1T MoS2 and NbSe2 are metallic.182–184 The oxidation state of the metal and chalcogen atoms in TMDC is +4 and −2, respectively. Similarly, in multi-layered TMDCs, a variety of polymorphs exist depending on the coordination phase each layer possesses. The most commonly found polymorphs are 1T, 2H, and 3R (explained in Fig. 7), where 1, 2, and 3 represent the layer number in the unit cell; T, H, and R represent the tetragonal (D3d group), hexagonal (D3h group), and rhombohedral (C53v group) symmetry of the crystal, respectively; 1T exhibits the metallic property, whereas 2H and 3R are semiconducting in nature.185
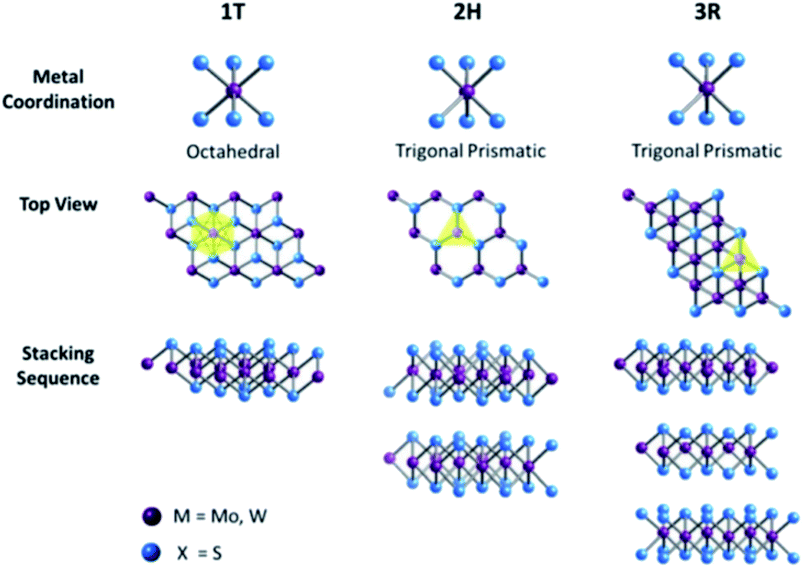 |
| | Fig. 7 Metal coordination and stacking sequence. The octahedral coordination gives tetragonal symmetry (1T), whereas distinct stacking sequences of trigonal prismatic layers yield different symmetries: 2H and 3R. Reproduced with permission under the terms of Creative Commons Attribution 3.0 Unported License (CC BY 3.0, facilitated open access).185 Copyright 2017 RSC. | |
Depending upon the filling of the non-bonding d bands of groups 4 to 10 by the M atoms of TMDC, different electronic properties arise. TMDCs exhibit metallic properties when the d bands are partially filled; on the other hand, they display a semiconducting nature when the orbitals are fully occupied.186,187 Although the impact of chalcogen atoms on the electronic properties of TMDCs is small in comparison to the transition metals, however, the atomic number of chalcogens affects the band gap in a way that the bandgap decreases when the atomic number of the chalcogens increases. The variance in the size of chalcogen and metal ions determines the length of the M–X bond, which varies in the range of 3.15–4.03 Å.187 Monolayer and few-layered TMDCs exhibit layer-dependent properties different from their bulk counterparts due to surface properties and quantum confinement. For instance, the Raman peaks of MoS2, viz., E2g1 and A1g, get blue- and red-shifted when the number of layers decreases due to partial coulombic interaction and probable stacking induced charge due to bonding between the layers.188–190 The indirect bandgap of bulk MoS2 (1.3 eV) converts to the direct bandgap (of 1.8 eV) in monolayer MoS2.191 The application of TMDCs in electrocatalysis has increased due to the edge-exposed surfaces of MoS2, WS2, WSe2, and MoSe2; hence, designing electrocatalysts with exposed edge sites with respect to the basal surfaces can enhance the electrochemical activity. Besides, electrical conductivity is another factor that determines the catalytic activity of TMDCs.192–194 Different polymorphs of TMDCs possess distinct structure and electronic properties, which can be explored to understand the effect of different polymorphs in enhancing the electrocatalytic activity of TMDCs by rationally designing the catalyst. For instance, Ambrosi et al.195 reported that 1T TMDCs are better HER electrocatalysts than their bulk counterparts semiconducting 2H because of more exposed edge sites, increased electrical conductivity, and reduced sheet size of the metallic 1T phase. Single or few-layered TMDCs can be obtained by top-down methods such as mechanical cleavage and chemical exfoliation method,196,197 and bottom-up methods such as chemical vapor deposition and direct wet chemical synthesis methods.198,199
4.4 Transition metal oxides
Among various 2D nanomaterials, transition metal oxides (TMOs) have emerged as a feasible alternative to precious noble metals as electrocatalysts in direct oxidation fuel cells200,201 due to their exceptional physical and electrochemical properties such as low cost, high stability, environment-friendliness, and tunable properties. Among the various metal oxides, NiO and spinel oxides MCo2O4 (M = Ni, Cu, Mn, Zn) have been attracting attention.202 These spinel oxides are potential electrocatalysts owing to their brilliant electrochemical activity and electrical conductivity, which originate from the synergistic effect of their individual metal oxides. In the spinel structure, oxygen possesses fcc packing and the cations occupy both octahedral and tetrahedral sites. The spinel structure can be thought of as the rocksalt structure, where the cations in the octahedral interstices move to the tetrahedral ones to make the spinel. Their thermal and chemical stability, as well as acid–base and redox properties make them suitable for catalytic applications, especially those involving oxygen species.203 Almost 30 different elements having valency in the range +1 to +6 are known to occupy the cation sites.204 Alkaline electrolytes are more suitable for TMOs as they are more stable in basic than acidic ones. Different transition metals such titanium (Ti), cobalt (Co), copper (Cu), and manganese (Mn) are included in TMOs. The presence of oxygen ions in the TMOs manifests them the ability to hold on to the charged ions present on their surfaces without any intermixing. Moreover, one can alter the stoichiometry of 2D TMOs by reducing them, which leads to non-linear and non-uniform charge distribution within the TMOs. It further screens the electric field at the nanoscale range. Thus, TMOs possess unique interfacial properties, which are beneficial for coulombic interactions.205–207 Besides, when two or more TMOs are placed aside, their electric fields interact and a potential develops at the interface due to Fermi level shifting, which affects the electronic properties of the composites. Semiconductors can be synthesized from 2D TMOs by aligning their d orbitals normal to their planes, which leads to the re-alignment of spins and orbitals.205 The cations in TMOs can possess different oxidation states and binding configurations, depending on which different electronic properties of the TMOs arise.208 TMOs are potential candidates for different applications, namely, sensors,209 energy storage,210,211 hydrogen production,212 electronics,213 and biosystems.214 There are two types of 2D TMOs: layered and non-layered. The layered TMOs are metal trioxides, e.g., MoO3, TaO3, and WO3, which possess a layered structure and can be exfoliated by top-down methods.215,216 On the other hand, non-layered TMOs are generally 3D crystals having strong interlayer chemical bonds, which cannot be exfoliated by top-down methods.217 Metal oxides based non-noble metal catalysts have been a potential candidate in PEFCs because of few characteristics: (i) possess higher corrosion resistance than carbon as the metals in the oxides possess different oxidation states and when present in a high oxidation state, they do not get oxidized easily; (ii) used as cocatalysts or supports as they share strong interactions with the metal catalyst, which prevent their agglomeration. They can also tune the electrocatalytic properties of metals; (iii) they possess several hydroxyl groups on their surfaces, which functions as the cocatalyst of noble metal particles.218,219
5. 2D materials beyond graphene: application in DOFC as the anode catalyst/support
5.1 MXenes nanocomposites
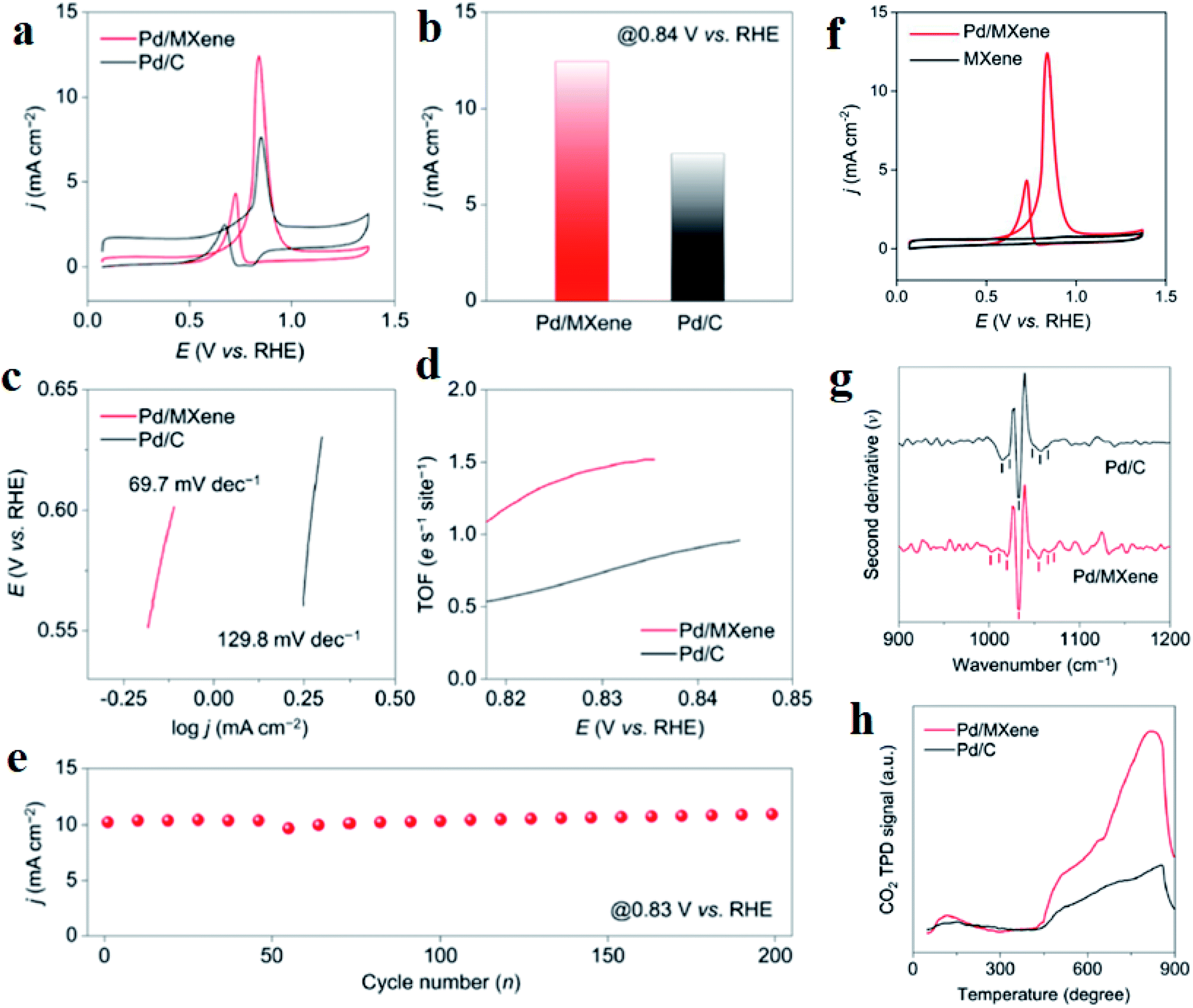
MXene has been identified as a potential support material for fuel cell electrocatalysts and other renewable energy applications. The hybridization of metal nanoparticles with MXene makes a competitive electrocatalyst for DEFC.220 Porous trimetallic nanospheres of PtRhFe loaded over ultrathin (1–2 layers, as confirmed by AFM) Ti3C2Tx nanosheets, named as PtRhFe-PNS@MXene, is an excellent MOR electrocatalyst. The optimized stoichiometry of Pt:Rh:Fe was found to be 69:8:23, as mesoporous Pt69Rh8Fe23-PNS@MXene exhibited the highest current density (3407.7 mA mg−1), which was 4.23-fold that of commercial Pt/C. Intrinsic MXene could not show much ethanol oxidation reaction (EOR) activity, which increased manifold when used as a support material for metal nanospheres Pt, Rh, and Fe. Besides, the addition of Rh and Fe also had a synergistic contribution with Pt. The difference in the Raman shift between Pt69Rh8Fe23-PNS@MXene and the pure MXene samples clearly indicated the presence of a strong interaction between the metal alloys and Ti3C2Tx. Moreover, the XPS results confirm the electronic interactions between them. The mesoporous nature of the electrode Pt69Rh8Fe23-PNS@MXene provides channels for seamless electrolyte ion flow, which enhances the EOR activity. Pt69Rh8Fe23-PNS@MXene had a faradaic efficiency of 19.9% for ethanol, much smaller than that of Pt/C TKK (49.5%), Pt69Rh8Fe23-PNS (38.6%), Pt69Rh8Fe23-PNS@C (35.9%), and Pt75Fe25-PNS@MXene (41.8%), implying better capability of splitting the carbon–carbon bond. The lowest onset potential and better cycling stability of the electrode further confirm that Pt69Rh8Fe23-PNS@MXene is the best catalyst. Moreover, due to the coexistence of Rh and MXene, the Pt69Rh8Fe23-PNS@MXene catalyst generated the highest current density, further confirming that they can promote the C1 pathway for EOR. The excellent stability and resistance to CO poisoning of the catalyst are due to the combined effect of the shifting of the Pt d-band center toward the Fermi level and the MXene surface containing hydrophilic terminations such as –O, –OH, and –F, which strongly connects with ternary alloys and decreases the bond of adsorbed CO with Pt. Thus, MXene has proved to be a great support for catalysts because of its hydrophilicity, rich functional groups attached to its surface, and chemical stability. MXene-supported Pd catalyst exhibits an MOR current density of 12.4 mA cm−2, which is higher than that of Pd/C (7.6 mA cm−2) (Fig. 8a and b).221 Although pristine Ti3C2Tx MXene does not show MOR activity (Fig. 8f), this increase in the MOR activity is due to the interaction between MXene and Pd, which is further established by the Tafel slope and turnover frequencies (TOFs) of the catalysts. The smaller Tafel slope (Fig. 8c) and higher TOF (Fig. 8d) of Pd/MXene than those of Pd/C suggest that MXene as a support increases the MOR kinetics of Pd. The durability of Pd/MXene remains intact even after 200 CV cycles (Fig. 8e). Second derivative analysis of the adsorption spectra (Fig. 8g) also adds to the point that Pd/MXene possesses more active Pd sites for methanol adsorption, originating mainly due to the surface-induced strong metal-support interaction effects. MXene as the catalyst support leads to more methanol adsorption as compared to carbon black because more Lewis basic sites are available on the surface of MXene, which is confirmed by the large and broad CO2 temperature-programmed desorption (CO2-TPD) peaks of Pd/MXene than that of Pd/C in the range of 450–900 °C (Fig. 8h).
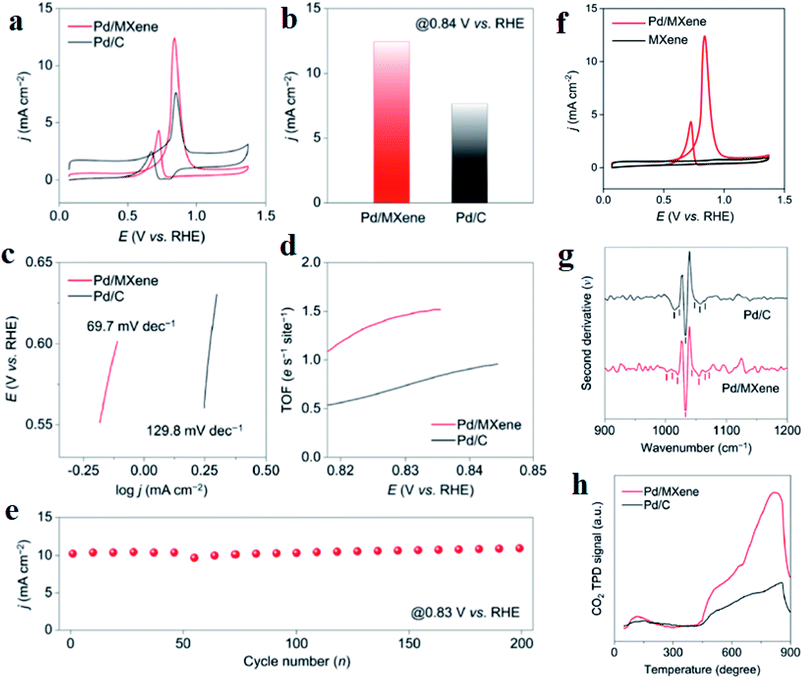 |
| | Fig. 8 (a) CVs of Pd with MXene and C as the support in 1 M methanol + 1 M potassium hydroxide, (b) current densities of Pd/C and Pd/MXene, (c) Tafel plots, (d) TOF plots, (e) current densities in the forward scan vs. the cycle number, (f) CVs of Pd/MXene and MXene in 1 M methanol + 1 M potassium hydroxide solution, (g) second derivative spectra of the catalysts after methanol adsorption, (h) CO2-TPD spectra. Reproduced with permission.221 Copyright 2020, ACS. | |
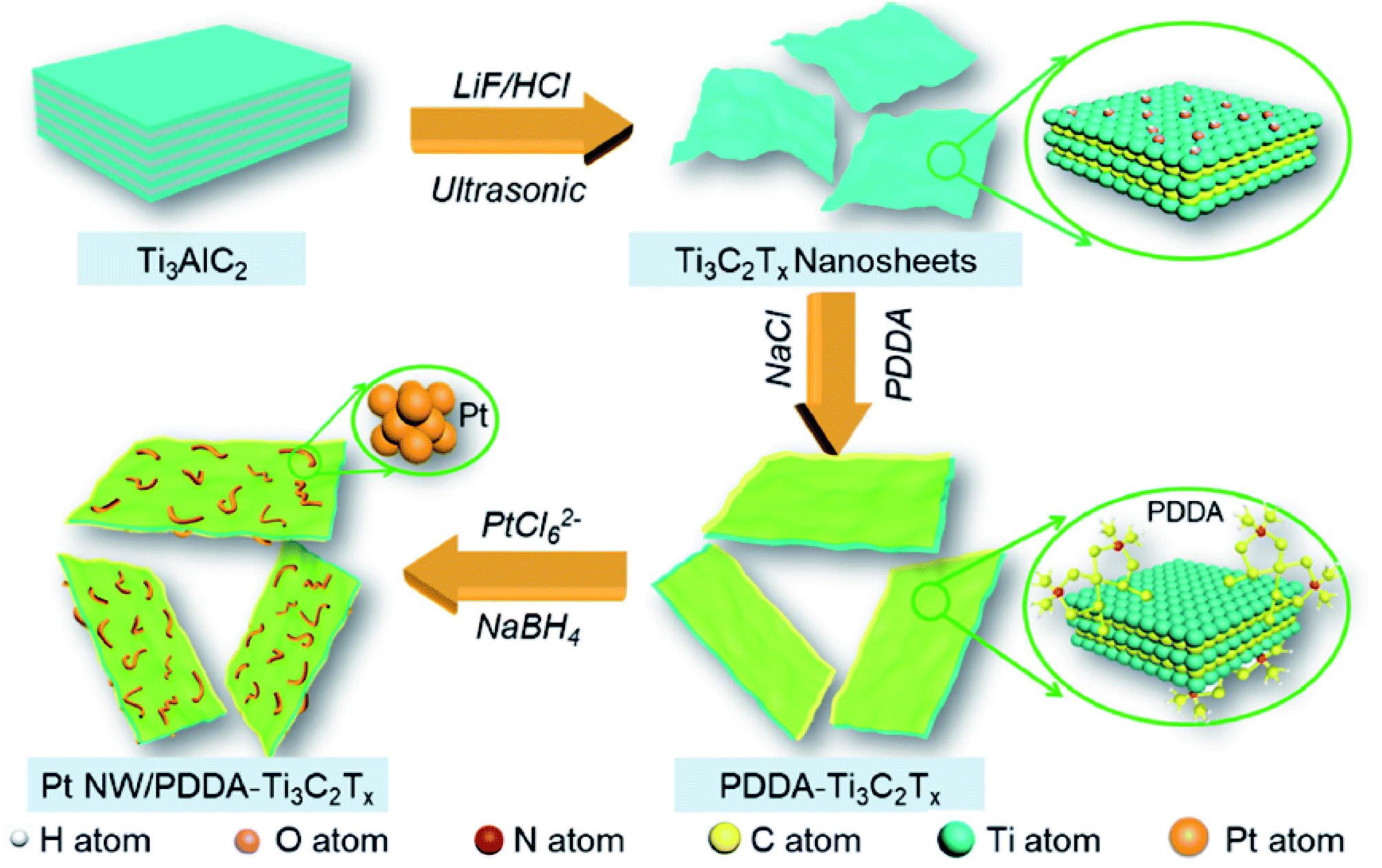
Thus, more methanol molecules get adsorbed on these basic sites, leading to higher MOR activity. DFT calculations further confirm the metal-support interactions using electron density difference map (EDDM). The EDDM results imply that electrons migrate from the top surface of the MXene to the bottom atomic layers of the Pd nanoparticles. The –OH and –F terminations present on the surface of MXene behave as electron donors. In this way, MXene activates Pd active sites and enhances the MOR activity. Besides, Pd/MXene exhibits the most favorable methanol adsorption energy of −0.58 eV, which is higher than that of pristine MXene and Pd. Thus, the introduction of the MXene support provides more scope of hydrogen bonding between the oxygen atom of methanol and the hydrogen atom of the hydroxyl group on the MXene surface, thereby improving the oxidation kinetics of the catalysts. For the first time, an MOR electrocatalyst has been developed by immobilizing Pt nanoparticles on MXene support.222 The higher anodic current density for Pt/Ti3C2 MXene (about 3 times higher than that of Pt/C) and lower onset potential proves that MXene increases the MOR kinetics. The low charge transfer resistance of Pt/Ti3C2 MXene (as obtained from the EIS spectra) also contributes to the magnified MOR activity. CO poisoning of the Pt active sites takes place on both Pt/Ti3C2 and Pt/C; however, the current density of Pt/Ti3C2 is larger than that of Pt/C even after 7200 s. Besides, 85.1% ECSA of Pt/Ti3C2 is retained after 1000 CV cycles, with only 53.2% retention of ECSA of Pt/C, demonstrating the high stability of the catalyst due to the reduced agglomeration of Pt nanoparticles on the MXene support, excellent stability of MXene under harsh environments such as strong acidic medium, and strong bonding between the MXene support and the metal nanoparticles. Despite having improved electrocatalytic activity and anti-poisoning effects, MXenes obtained using LiF/HCl etchant consist of negatively charged –O and –F terminations whose ability to trap noble metal precursors (such as PtCl42− and PdCl42) reduces because of the repulsion offered by the terminations. Decorating the basal planes of the MXenes by positive charges, one can tune the surface charge, which acts as uniform dispersion sites for the metal nanoparticles. PDDA-modified Ti3C2Tx MXene nanosheets modify the surface charge of MXene to positive, which act as growing sites for 1D worm-shaped Pt nanocrystals (named Pt NW/PDDA-Ti3C2Tx) (schematically shown in Fig. 9)223 and reduce the reaggregation of the MXene nanosheets. Yang et al.223 devoted this electrocatalyst Pt NW/PDDA-Ti3C2Tx for MOR and it exhibited superior MOR activity owing to its high specific activity, good anti-poisoning, and stability for methanol oxidation, outshining traditional Pt catalysts loaded on carbon, CNT, reduced graphene oxide (RGO), and MXene supports. It delivered a high anodic current density of 17.2 mA cm−2, outshining other electrodes such as Pt NPs-supported Ti3C2Tx, RGO, CNT, and C, which is due to the electroactive PDDA-Ti3C2 support, which increases the interfaces for triple-phase reactions, and homogeneously distributed grain boundary of the Pt nanoworms. The XPS results confirm the strong interaction between Pt and PDDA-Ti3C2Tx than that with other carbonaceous supports. Lower onset potential (0.52 V) and peak potential (0.61 V) of CO oxidation compared to other electrodes ascertain the enhanced anti-poisoning ability of Pt NW/PDDA-Ti3C2Tx (which is further ascertained by the Mulliken population surrounding the CO molecule). The reduction of CO poisoning of Pt NW/PDDA-Ti3C2Tx is also confirmed by the downshifting of the d-band center relative to the Fermi energy. The uniform distribution of PDDA chains provides conductive pathways for the easy and fast migration of charges.
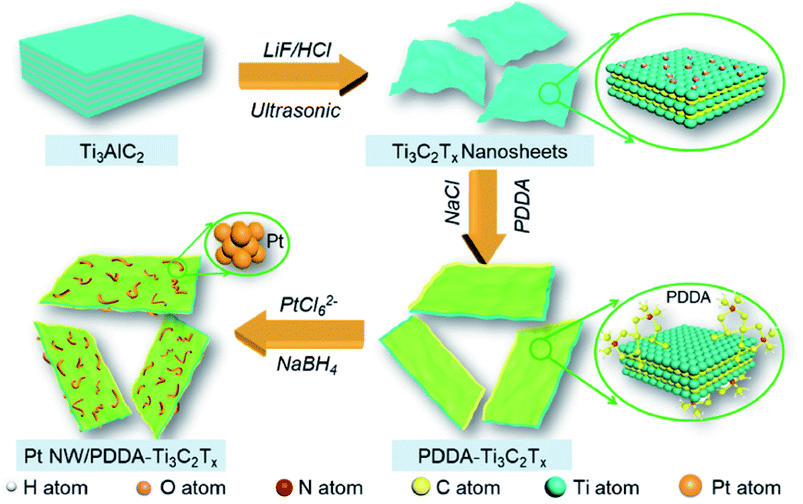 |
| | Fig. 9 Fabrication route of Pt NW/PDDA-Ti3C2Tx. Reproduced with permission.223 Copyright 2020, ACS. | |
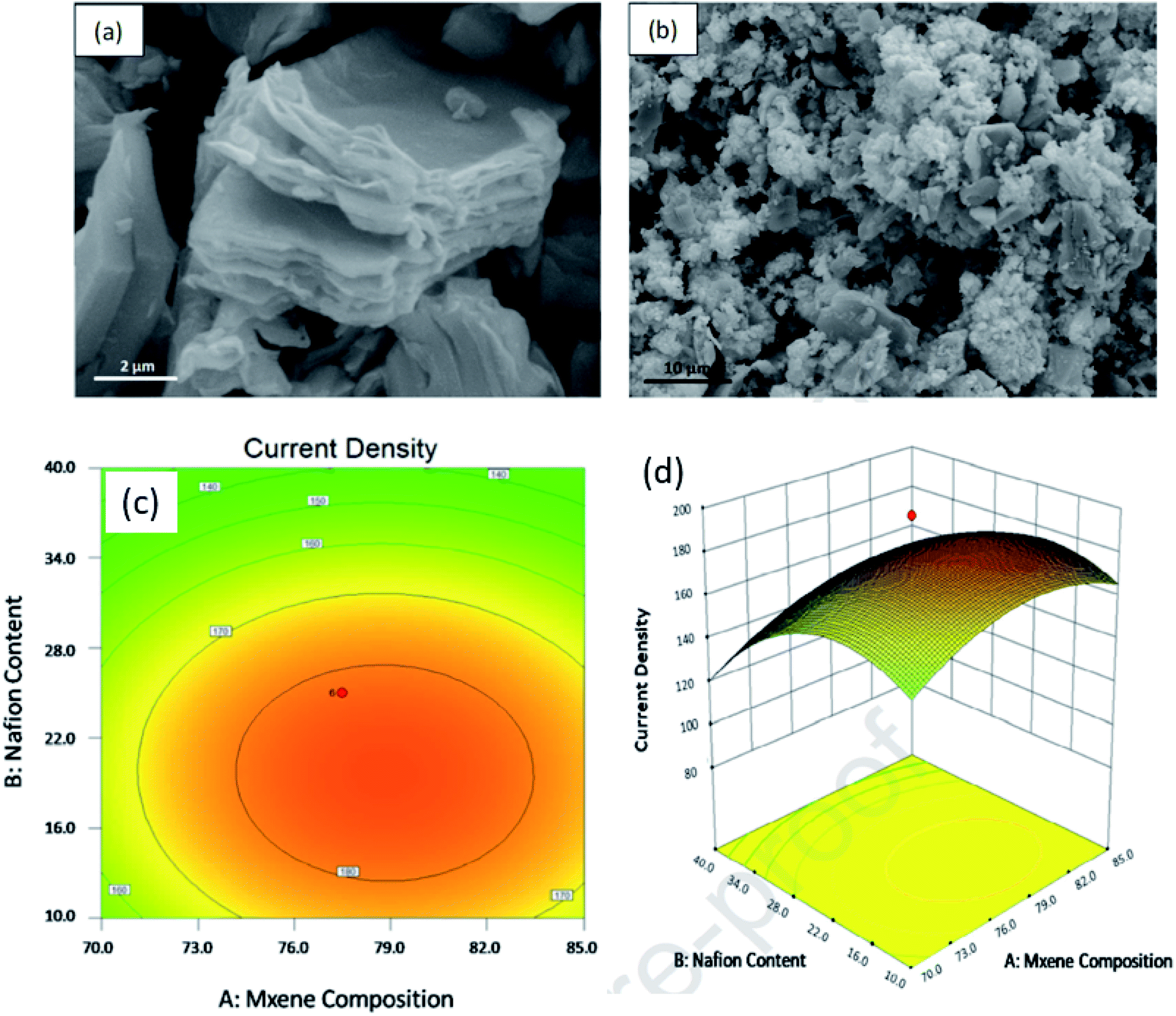
Another group224 designed for the first time PtRu-decorated 2D MXene as the DMFC catalyst. They evaluated the performance of the electrocatalyst using response surface morphology (RSM) method, considering three factors (MXene composition, amount of Nafion loaded, and methanol concentration denoted by A, B, C) and current density as the response. The strong bond between PtRu and MXene offers better MOR activity (current density about 2.34 times higher than that of commercial PtRu/C), benefitting from the large active sites present on the surface of the catalyst (morphology in Fig. 10a and b). Moreover, the unique 2D surface of MXene furnishes fast charge/ion transfer, thus increasing the conductivity. They found from a survey that the PtRu/MXene electrocatalyst offers the highest current density among other catalysts owing to the uniform distribution of bimetals on the 2D structure of MXene. The response surface analysis is presented in a graphical form as a 2D contour and a 3D surface plot (Fig. 10c and d).
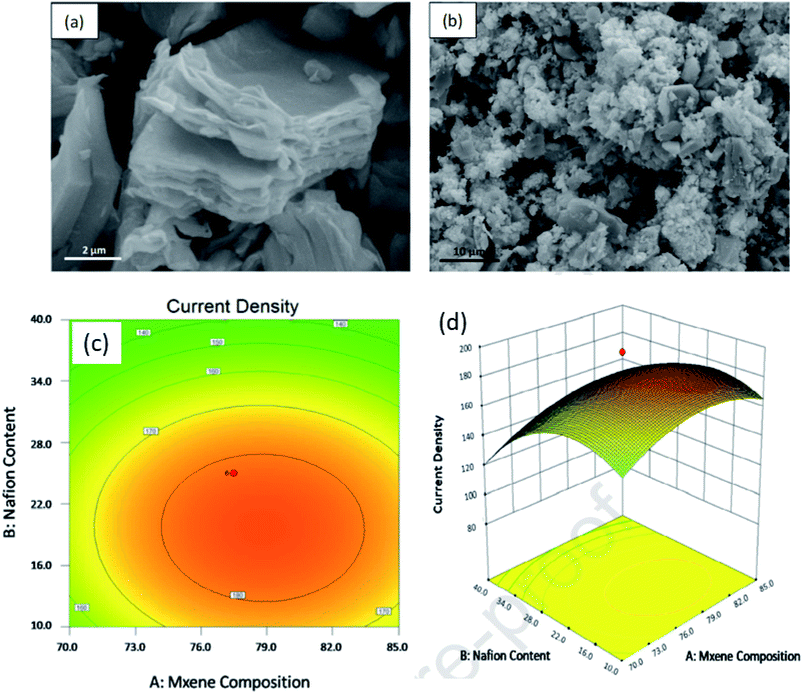 |
| | Fig. 10 SEM images of (a) MXene and (b) PtRu/MXene; response surface between the factors with response, (c) 2D contour and (d) 3D surface plot. Reproduced with permission.224 Copyright 2020, Elsevier. | |
In the 2D contour, the response, i.e., the current density, increases when the factors A and B increase; thereafter, it starts decreasing after achieving a maximum value. These values of the factors A and B corresponding to the maximum response value are obtained within the red area of the contour (Fig. 10c), known as the high response value area. The 3D surface plot (Fig. 10d) also illustrates the same optimum factor values. The validation test of the model obtained by the RSM method using the optimum factors exhibits a current density of 187.05 mA mgPtRu−1, with only 0.25% error, which clearly indicates the successful development of the RSM model. Hence, MXene is a potential material support emerging in fuel cell applications. To overcome the problem of restacking and low charge transportation of MXene, composites with conducting materials have been developed.225,226 An MOR electrocatalyst Ti3C2Tx–rGO composite support for Pd has been developed using the self-assembly of negatively charged Ti3C2Tx and positively charged CTAB-modified rGO.226 Pd/Ti3C2Tx–rGO of different stoichiometric ratios (1:1), (1:2), and (2:1) were developed and it was observed that the (1:1) ratio provided the highest ECSA value and peak current density. It thus demonstrates that an equal content of MXene and rGO provides an excess of accessible sites for Pd. Moreover, the interaction between MXene and Pd particles improves the stability. Besides, the activities (both mass and specific) of Pd/Ti3C2Tx–rGO (1:1) in alkaline medium are 446 mA mg−1 and 16.3 mA mg−1, which are 1.7 and 1.57 times higher than that of the commercial Pd/C catalyst, respectively. Moreover, the mass and specific activity of Pd/Ti3C2Tx–rGO (1:1) is 3.04 and 2.83 times higher than that of Pd/Ti3C2Tx, respectively. The increased electrical conductivity of Pd/Ti3C2Tx–rGO (1:1) (obtained from the Nyquist plot) is due to the symbiotic effect of both MXene and rGO. The current density also remains 212 mA mg−1 even after 7200 s, which is 9-fold that of the Pd/C current density. This high stability of the catalyst is due to the composite of Ti3C2Tx–rGO, which protects the defect sites from being corroded owing to the high corrosion resistance of Ti3C2Tx. This architecture inspires other MXene-based support materials for electrocatalysts in the field of FC applications. A porous 3D cross-linked nanoarchitecture Pd/MX–rGO was developed from nanosheets of Ti3C2Tx and GO, with Pd NPs deposited over them.227 A low onset potential of about 0.65 V and a peak potential of 0.71 V were obtained from formic acid oxidation using Pd/MX–rGO as the anode catalyst. It exhibited the highest current density (12 mA cm−2) among other catalysts, viz., Pd/MX (3.3 mA cm−2), Pd/rGO (6.1 mA cm−2), and Pd/C (1.8 mA cm−2). The strong interaction between Pd NPs and Ti3C2Tx weakens the CO adsorption on the Pd active sites, further increasing formic acid oxidation. Moreover, low charge transfer resistance (10.1 Ω) of the nanocomposite in comparison to other electrodes such as Pd/MX, Pd/rGO, and Pd/C is due to the 3D network, which provides multiple channels for the redox reactions to occur, which increase the triple-phase boundaries, leading to enhanced formic acid oxidation. High activity toward formic acid oxidation and better durability of the nanocomposite is obtained owing to the large surface area, 3D cross-linked porous structure, uniform dispersion of the Pd NPs, and the high conductivity of the nanocomposite. Another composite Pt–MXene–TiO2 (ref. 228) was developed and employed for MOR by a photoactive-mediated electrocatalytic method. The nanocomposite Pt–MXene–TiO2 consisted of TiO2/MXene as the photoactive component, Pt as the MOR electrocatalyst, and MXene as the catalyst that administered a photoinduced electron–hole to lift the MOR activity of Pt and activate the CH3OH molecules. A current density of 2750 mA mgPt−1 was obtained under UV light irradiation, which is 3.9-fold higher than that in dark and 4.1 times that of Pt–MXene. This high current density reduces slightly to 1269.81 mA mgPt−1 after 5000 s. This high current density is the highest among the reported photo-induced electrocatalysts. MXene here acts as the Ti source for the fabrication of MXene–TiO2. This work offers novel pathways for developing stable and efficient anode catalysts for PEMFCs by exploiting the photo energy.
5.2 g-C3N4 nanocomposites
Nanoporous g-C3N4-supported PtRu MOR catalyst was developed for the first time, which delivered 78–83% higher power density than PtRu supported on Vulcan XC-72.229 The peak current density of 60 wt% PtRu/C3N4 is more than twice that of 60 wt% PtRu/Vulcan XC-72 catalyst, commercially known as Pt–Ru/E-TEK. The superior performance of PtRu/C3N4 is accredited to the porous structure of C3N4. Well-developed pores provide a pathway for the effortless diffusion of fuels and intermediates into the catalyst surface, accelerating the reaction kinetics. Vulcan XC-72, on the other hand, consists of pores with random distribution, which do not favor fuel diffusion. Nitrogen atoms act as electron donors and increase the electrical conductivity, which further eases electron transfer and enhances the performance of the PtRu/C3N4 electrocatalyst. Moreover, the repeated triazine units of g-C3N4 undertake the coordination complex to anchor metal atoms or their alloys, increasing metal-support interaction. Despite having high electrical conductivity and high surface area, carbon black Vulcan XC-72R is not stable at high potential and acidic medium, causing corrosion, which further accelerates the lumping of Pt NPs and causes detachment from the support, ultimately hampering the ECSA and electrocatalytic activity. Besides, Pt NPs get stuck inside the pores, thus reducing the active interface of the triple-phase needed for electrochemical reactions. Carbon nanotubes, nanohorns, nanofibers, and nanocoils are different forms of graphitized carbon materials that have been explored as support materials. The oxygen functional groups present on the surface of carbon promote its corrosion, thereby reducing the durability of the catalyst. As suggested by the literature, the incorporation of nitrogen into carbon reduces the surface functional oxygen groups and leads to enhanced tolerance toward oxidation.
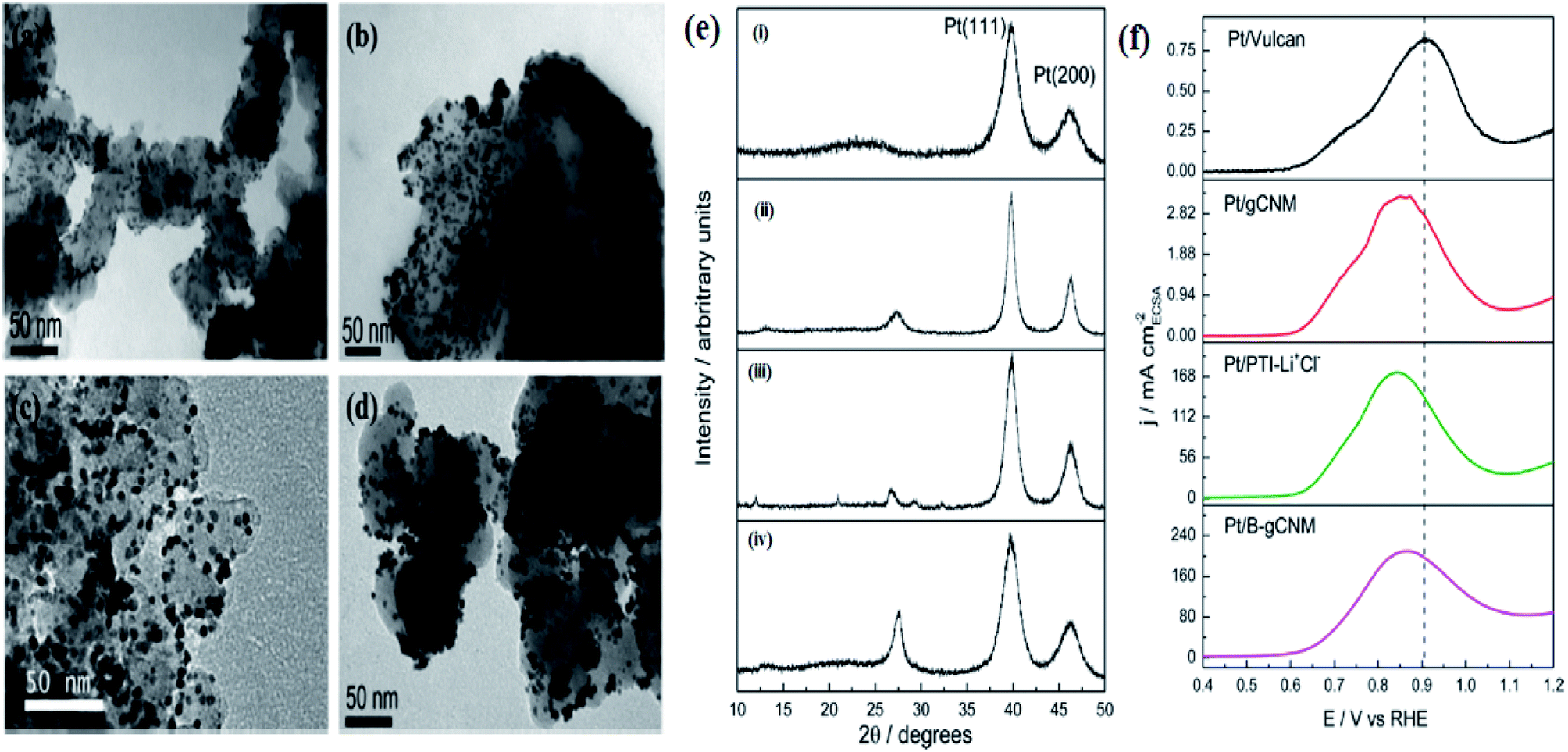
Hence, doping carbon materials with O or N has been a trending method to obtain highly durable catalyst supports. Nitrogen as the dopant alters the electronic structure of the catalyst and decreases the interaction between Pt and the poisoning intermediates (confirmed by the increasing binding energy, as supported by XPS results). Moreover, N-doped carbon possesses high specific capacitance, influencing more and more charged species to be present within the Helmholtz layer, which provides additional sites for the removal of adsorbed poisonous intermediates by the surface groups. Inspired by all these potentials of nitrogen-doped carbon materials, g-C3N4 has been opted as a potential catalyst support. Mansor et al.230 studied the effect of crystallinity, porosity, and composition on the electrocatalytic properties of the support by synthesizing three different g-C3N4 materials: polymeric carbon nitride (gCNM), poly(triazine) imide carbon nitride (PTI/Li+Cl−), and boron-doped graphitic carbon nitride (B-gCNM). gCNM was synthesized using thermolysis and condensation of dicyandiamide and melamine, B-gCNM was prepared using ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate to tune the electronic properties of gCNM by replacing carbon with boron, and PTI/Li+Cl− was prepared by the ionothermal method. PTI/Li+Cl− is the most crystalline as clear hexagonal shaped crystallites are observed, gCNM is the least crystalline, and B-gCNM lies in between them (Fig. 11e), whereas, B-gCNM possesses higher porosity (pore size of about 50–75 nm) than gCNM and PTI/Li+Cl−. Pt nanoparticles disperse in all the samples with more agglomeration in the g-C3N4 samples than in Vulcan (Fig. 11a–d), indicating that the catalyst deposition method is not optimized. B-gCNM and PTI/Li+Cl− exhibit the highest stability, proposing that the crystallinity has a deeper connection with the stability of the catalyst, also suggesting that the presence of B and Li+, Cl− as dopants may increase the stability of the catalyst. All the g-C3N4 supported Pt catalysts possess a lower initial ECSA value than that of commercial Pt catalysts because of their larger particle size and agglomeration problem. Pt–PTI/Li+Cl− exhibits the best durability with only 19% ECSA loss after 2000 cycles, which is lower than the ECSA loss of Pt/Vulcan (36%), Pt/gCNM (81%), and Pt/B-gCNM (100%). The dependence of durability of g-C3N4-supported Pt catalysts on the initial ECSA value is manifested by the observation that Pt/B-gCNM, having the lowest ECSA value, exhibited the lowest durability. On the other hand, two other g-C3N4-supported electrocatalysts having higher ECSA value exhibited better durability, proposing the relation between metal-support interaction and electrochemical durability. All g-C3N4 supported electrocatalysts exhibited superior MOR activity than Pt/Vulcan. Pt/PTI–Li+Cl− exhibited the lowest overpotential and Pt/B-gCNM the highest peak current density (Fig. 11f). Generally, a smaller particle size of the catalyst favors the MOR as it decreases the overpotential; however, in this case, despite having large particle sizes than the commercial catalyst, the g-C3N4-supported electrocatalyst exhibited the lowest overpotential, suggesting the fact that nitrogen present within or on the support material leads to enhanced MOR activity. This work proves g-C3N4 as a potential alternative catalyst support to carbon as they exhibit higher corrosion resistance and enhanced MOR activity. A novel EOR electrocatalyst g-C3N4/graphite carbon/Pd nanocomposite (Pd@g-C3N4/GC) was developed.231 The ultrathin g-C3N4 present as nano-islands on the nanosheet of GC in a discrete manner and the Pd nanoparticles are decorated over the GC, forming a three-phase boundary (confirmed by the HR-TEM images). The CV of different samples, namely, Pd/GC, Pd/AC, Pd@g-C3N4/GC, and Pd@g-C3N4/GC (with the composition of g-C3N4 varying from n = 1 to 0.0625) was done to evaluate the electrochemical performance in the absence and presence of ethanol. The CV peaks in the absence of ethanol are the characteristic peaks of hydrogen adsorption and desorption, and the development of palladium oxides and its reduction during the positive and negative scan, respectively, as well as the formation of the electrical double layer. The absence of CV peaks for Pd@g-C3N4 is due to the poor conductivity of g-C3N4 (ascertained by EIS), which leads to its poorer EOR performance. The higher ethanol oxidation peak current density of Pd@g-C3N4(0.0625)/GC (2156 A g−1 Pd), which is higher than that of the other three catalysts, and lower onset potential is due to the suitable amount of g-C3N4, which improves the catalytic performance of Pd by increasing the Pd fixation sites owing to electron-coupling interaction between g-C3N4 and GC. Moreover, the nano-islands of g-C3N4 deposited on the GC surface favors the electron/ion diffusion of the electrolyte and the pyridine-type nitrogen-rich structure of g-C3N4 induces several electrochemically active sites when interacted with palladium nanoparticles. High graphitization degree and lesser defects of GC than AC increases its electronic conductivity, which contributed to the faster charge transfer of Pd@g-C3N4(0.0625)/GC. Besides, Li et al. also established a relation between the EOR performance and the g-C3N4 content. They reported that excess of g-C3N4 content can cause blockage of the electronic transmission channel for the catalyst and excess of nitrogen may have a tendency of forming hydrogen bonding with ethanol, thereby reducing the EOR performance of Pd by hindering their contact with Pd. The addition of metal carbides and metal oxides as catalyst promoters into the noble metal catalyst increases the concentration of adsorbed OH groups on the metal surface, which favors the aldehyde to carboxylic acid transformation, thereby promoting ethanol oxidation. Compared to Pd/AC and Pd/GC, Pd@g-C3N4 (0.0625)/GC offers the highest electrochemical stability (after 200 CV cycles, the peak current density of Pd@g-C3N4 (0.0625)/GC is 1904 A g−1 Pd), which is about two times of Pd/GC (966 A g−1 Pd) and Pd/AC (954 A g−1 Pd), on account of firstly, graphite carbon exhibiting more stable crystal structure than the amorphous one and, secondly, the electron-coupling of g-C3N4 and GC promoting the loading of Pd on the support surface. Thus, the Pd@g-C3N4/GC nanocomposite has proved to be a promising EOR anode catalyst.
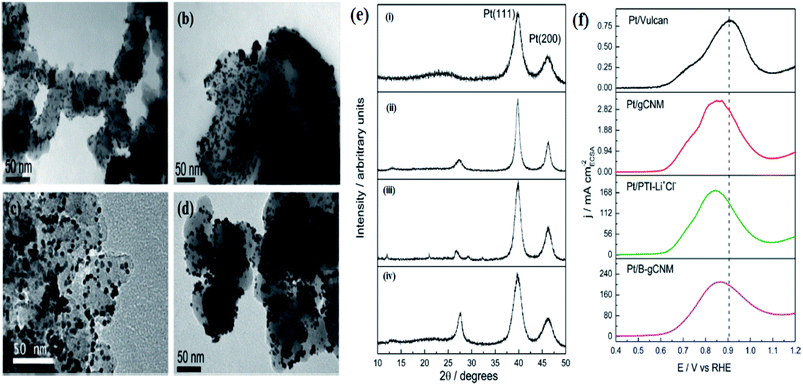 |
| | Fig. 11 TEM of Pt supported on (a) Vulcan, (b) gCNM, (c) PTILi+Cl−, (d) B-gCNM; (e) XRD pattern of the Pt catalyst supported on (i) Vulcan, (ii) gCNM, (iii) PTI/Li+Cl−, (iv) B-gCNM; (f) MOR of the Pt electrocatalysts supported all the three graphitic carbon nitrides in 1 M methanol + 0.1 M perchloric acid at a scan rate of 2 mV s−1. Reproduced with permission.230 Copyright 2014, ACS. | |
Ni-based 2D g-C3N4 (named as Ni/gCN) supported EOR catalyst232 outperforms its platinum counterpart Pt/g-C3N4 (ref. 233) and most of the reported electrocatalysts. The enhancement in the EOR activity of Ni/gCN is due to the formation of Ni(OH)2, which adsorbs water to form OH species, which further reacts with adsorbed CH3CO, leading to the formation of acetate and releasing the active sites on the catalyst surface. However, after adding MoS2 to Pt/g-C3N4, the peak current density increases manifold due to the formation of the 2D–2D heterostructure of MoS2 and g-C3N4. They varied the ethanol concentration and observed that with an increase in the alcohol concentration, the charge transfer resistance decreases; hence, the best results (peak current density ∼122.8 mA g−1 and onset potential ∼0.46 V) are obtained for 3 M ethanol. Normally, the carbonaceous intermediates get adsorbed on the catalyst surface, leading to a decrease in the current density after few seconds and it reduces by 50% in the first 1000 s, whereas a completely opposite trend is observed for Ni/gCN, where the current density first increases from 77 to 86 A g−1 and then remains constant up to 1000 s. This unique trend of Ni/gCN is attributed to the less amount of adsorption of carbonaceous species on the active sites of the catalyst and its ability to regenerate the active sites.
5.3 TMDCs nanocomposites
TMDCs have attracted attention as a potential Pt alternative owing to their natural abundance and unique catalytic ability due to the excellent stability and favorable interactions with metal catalysts.234 Among them, MoS2 possesses several fascinating properties such as easy synthesis, longer active sites, low cost, and basal planes, making it a probable candidate for developing hybrid electrocatalysts. 3D nanodendrites of Au@Pt core–shell nanostructures were decorated over MoS2 nanosheets to form Au@Pt/MoS2.235 Nanocomposites with a thick Pt shell (denoted as Au@Pt/MoS2-2) exhibited higher ECSA and electrocatalytic activity than the catalysts Pt/C, Pt/MoS2, and thin Pt shell (Au@Pt/MoS2-1). Au@Pt/MoS2-2 outperformed the other three electrocatalysts in the MOR performance with a mass activity of 6.24 A mg−1, which is about 3389, 20.3, and 4.5-fold of Pt/C, Pt/MoS2, and Au@Pt/MoS2-1, respectively. The low charge transfer resistance of the thick shell reflects the faster reaction kinetics, which is ascribed mainly to the interruption of adsorption of the intermediate on the catalyst surface. The synergistic effects of the large surface area of MoS2, the special porous structure of the Au@Pt core–shell, and the better catalytic activity of Pt nanoshells lead to the outstanding MOR performance of Au@Pt/MoS2 electrocatalyst, which is comparable to or better than several other reported performances. MoS2 nanosheets prepared by other methods such as mechanical exfoliation, chemical vapor deposition, and liquid exfoliation are normally semiconducting (2H phase) in nature, whereas chemically exfoliated MoS2 nanosheets (ce-MoS2 NSs) are generally in the metallic phase. 0D–2D functional hybrid of water-dispersible noble metal nanocrystals (NM NCs) modified ce-MoS2 was prepared using carboxymethyl cellulose as a stabilizer in an aqueous solution.236 The electron transferability of ce-MoS2 NSs increases after the incorporation of the NM NCs, which is also supported by the decrease in the charge transfer resistance. The MOR performance of Pd–MoS2 NSs was found to be better than that of Pd/C in alkaline medium (anodic peak current density is 433.5 mA mg−1, about 2.8 times that of the Pd/C catalyst). The metallic 1T phase present in ce-MoS2 is the reason behind such increased conductivity and MOR activity.
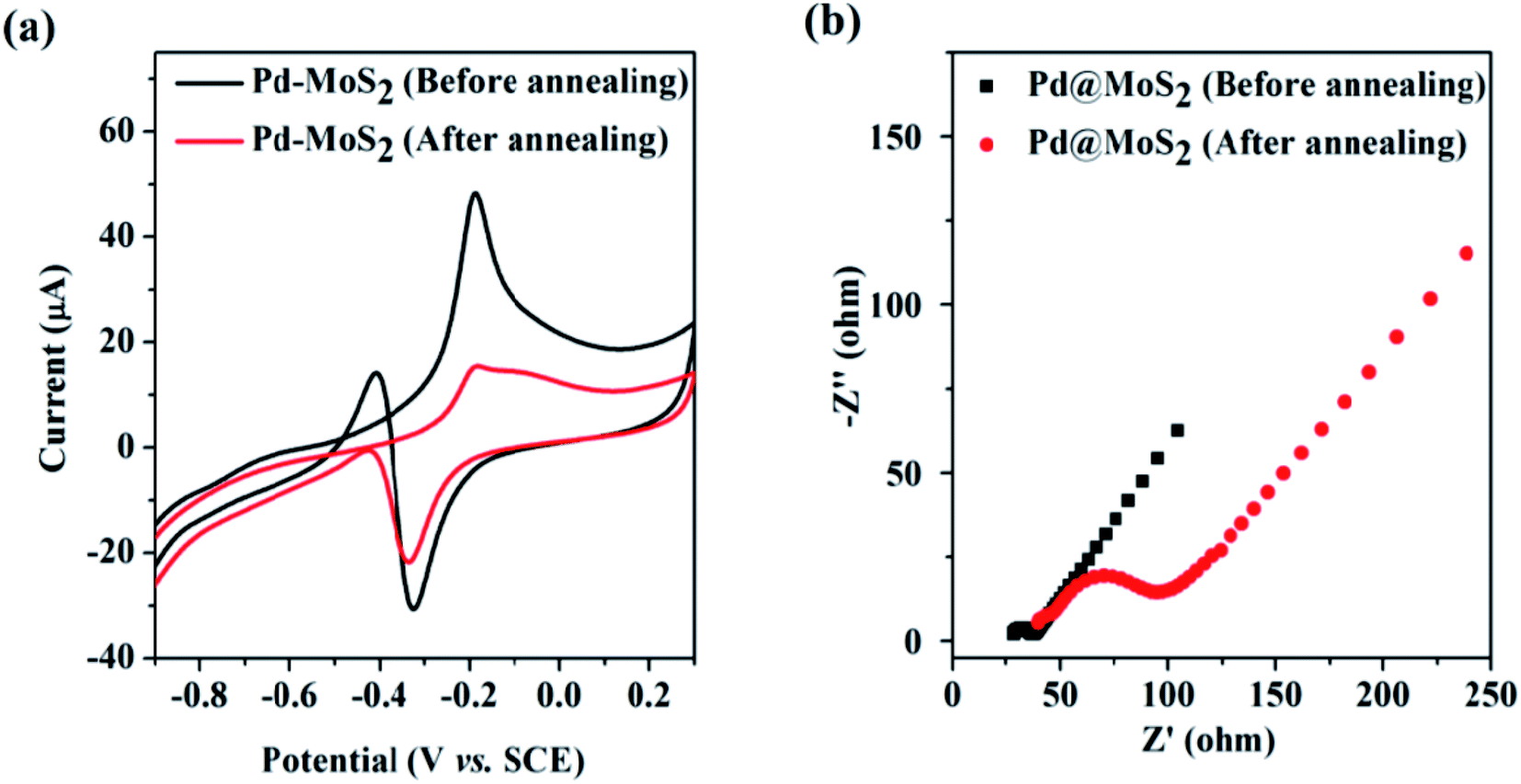
After annealing at 300 °C for 1 h under an argon environment, the 1T phase transforms to the semiconducting 2H phase, which leads to a decrease in methanol oxidation (reduces from 48 mA to 16 mA) (Fig. 12a) and increases the charge transfer resistance (Fig. 12b). Thus, a little variance in the polymorphic forms of MoS2 can predominantly affect the electrocatalytic activity. Moreover, the homogeneous distribution of Pd NCs on ce-MoS2 NSs increases their surface area, contributing to higher MOR activity. The semiconducting nature of pure MoS2 nanosheets limits their catalytic activity, which can be optimized by combining it with conducting graphene,237 reduced graphene oxide (rGO),238,239etc. Graphene-like 2D structural MoS2 nanosheets act as co-building blocks to develop 3D hybrid nanostructures, thereby lowering the charge transfer resistance of the catalyst. Ultrafine Pt nanoparticles decorated on the 3D architecture of graphene (G) and MoS2 nanosheets (Pt/G–MoS2) exhibited higher MOR and HER activity along with long-term stability and anti-toxic capacity, outshining catalysts such as Pt/G, Pt/MoS2, and commercial Pt/C with the same Pt loading.237 Such 3D porous architecture provides multiple channels for the reactants to the interior of the catalyst and improves the electron transfer between the active sites and the matrix. The hydroxyl and epoxide groups present on the GO surfaces provide active sites for Pt2+ adsorption and the 3D structure offers large, accessible pores of varying sizes for the fast transportation of Pt2+ to the nucleation sites, resulting in the uniform distribution of Pt in the hybrid material. Pure MoS2 nanosheets do not possess sufficient nucleation sites for Pt, hence leading to the formation of irregular shaped aggregates of Pt on its surface. On the other hand, comparatively low surface area and large micropores on carbon black-supported material lead to poor dispersion of Pt. Different G/MoS2 ratio 9:1, 7:3, 5:5, and 3:7 have been considered, out of which Pt/G5–(MoS2)5 provides the lowest oxidation potential, highest ECSA value, and better anti-poisoning ability. Thus, the porous 3D hybrid architecture helped in realizing a better electrocatalyst by preventing the agglomeration of MoS2 nanosheets and utilizing its benefit to the fullest. When rGO is incorporated with MoS2 and Pt nanoparticles, the peak current density of MOR (7.35 mA cm−2) and formic acid oxidation (6.8 mA cm−2) are several folds higher than that of Pt–MoS2 and Pt/C electrocatalysts,238 suggesting that MoS2/RGO is a promising Pt support in practical fuel cell application. Similarly, by inserting flower-like MoS2 between ultrafine Pt nanoparticles and nitrogen-doped rGO (Pt@MoS2/NrGO),239 a superior alcohol oxidation electrocatalyst and oxygen evolution reaction (ORR) catalyst was obtained owing to the symbiotic effect of Pt and NrGO. The Pt@MoS2/NrGO catalyst manifested superior catalytic activity, with a mass activity of 448.0 mA mgPt−1 for methanol, 158.0 mA mgPt−1 for ethylene glycol (EG), and 147.0 mA mgPt−1 for glycerol, which are greater than that of commercial Pt–C by several folds. It exhibited high stability with only 18.92, 17.98, and 14.76% activity loss rate in the case of oxidation of methanol, EG, and glycerol, respectively, which is superior to Pt@NrGO and Pt/C. The existence of hydrophilic groups on the edge of NrGO and sulphur planes in MoS2 stimulate water adsorption on the catalyst surface, which further assists the oxidation of CO to CO2, thereby reactivating the Pt surface by removing the adsorbed CO species. In this way, the synergistic effect promotes the oxidation activity of Pt@MoS2/NrGO. Bagasse is a bio-waste that creates environmental problems; thus, deriving carbon-based materials from bagasse can make the process energy-efficient and environment-friendly. MoS2-supported porous nitrogen-doped carbon (CNx) obtained from bagasse acts as a conductive support for Pt and is an excellent MOR catalyst.240 The edge sites of MoS2 that get covered due to agglomeration can be exposed by the formation of a composite with CNx, which provides binding sites for the growth of MoS2. Moreover, the combination of CNx with MoS2 makes the edge sites and defects on MoS2/CNx electrochemically active, in addition to the edge sites of unsaturated bonding atoms. The mass current density of Pt/MoS2/CNx (1030.2 mA mgPt−1) and Pt/MoS2/C (710.1 mA mgPt−1) is higher than that of Pt/MoS2 (162.6 mA mgPt−1) and the commercial catalyst Pt/C (405.4 mA mgPt−1), suggesting that the combination of MoS2 and CNx improves the conductivity and enhances the catalytic activity. Pt/MoS2/CNx possesses better durability among other catalysts, leading to the fact that N doping in the carbon skeleton changes the asymmetricity of the hexagonal carbon network and introduces defects, in addition, supplying more anchoring sites for MoS2 and Pt, leading to the improvement of electron transfer and MOR performance in acidic medium. Abundant Mo and S sites, oxophilic C–N defects, and the large porous structure assist in adsorbing more water molecules and recovering the Pt sites. Besides, the fast transport of COads on various faces of MoS2 imparts it resistance against CO poisoning. The electrochemical reactions involved in the MOR activity are explained in eqn (13)–(16).
| | | MoS2 + H2O → MoS2 − (OH)ads + H+ + e− | (13) |
| | | Pt − COads + MoS2 − (OH)ads → Pt + MoS2 + CO2 + H+ + e− | (14) |
| | | CNx + H2O → CNx − (OH)ads + H+ + e− | (15) |
| | | Pt − COads + CNx − (OH)ads → Pt + CNx + CO2 + H+ + e− | (16) |
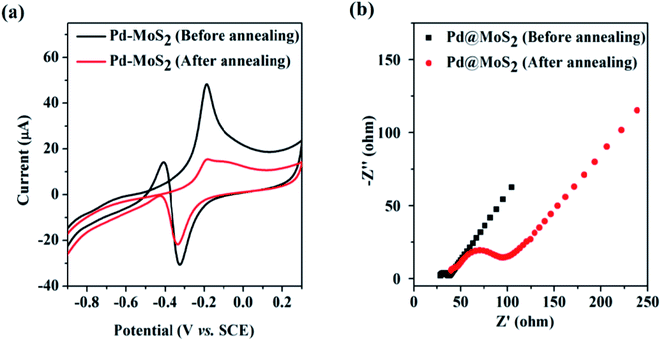 |
| | Fig. 12 (a) CV of Pd–MoS2-modified ITO electrodes in 0.5 M potassium hydroxide + 1.0 M methanol solution before and after annealing, (b) EIS measurement of Pd–MoS2-modified ITO electrodes before and after annealing. Reproduced with permission.236 Copyright 2014, RSC. | |
5.4 Transition metal oxides nanocomposites
The electrocatalytic activity of oxide materials depends on different parameters such as surface area, morphology, and porosity, which are tunable using different synthetic methods, generally by varying the surfactants in the hydrothermal method. Taking advantage of the fact that binder-free electroactive material grown on Ni foam (NF) enhances the electrochemical properties of the material, binder-free Co3O4 was grown on NF and the effect of different counter anions on the morphology was observed. Chloride ions used in the solution lead to the formation of Co3O4 microflower, sulfate ions lead to a microsphere-like structure, and acetate ions lead to a nanograss-like structure (Fig. 13).241 The difference in the morphology of Co3O4 arises due to the variation in the anionic sizes and electrostatic interactions between the precursor phases because the growth of a particular crystallographic plane is related to the surface energy, which is governed by the adsorption of species during the synthetic process.242,243 Thus, different anions prefer to adsorb on different crystallographic planes, forming different morphology of the oxide materials. The microsphere and nanograss-like Co3O4/Ni exhibit higher oxidation peak than microflower Co3O4/Ni due to their large surface area and porosity.
 |
| | Fig. 13 FESEM images of Co3O4 grown on NF using different anions, (a–c) microflower, (d–f) microsphere, and (g–i) nanograss-like Co3O4. Reproduced with permission.241 Copyright 2018, Elsevier. | |
The lower onset potential of these morphologies than the reported materials is because of (i) direct growth of Co3O4 on NF, (ii) binder-free synthesis, and (iii) high surface area and porosity, leading to the better diffusion of electrolyte ions. At 0.6 V, the anodic peak current density lies in the order: microflower (28 A g−1) < nanograss (34.9 A g−1) < microsphere (36.2 A g−1). The current decay after 1000 cycles and current retention in microsphere-Co3O4 and nanograss-Co3O4 after 1000 cycles are higher than that in the microflower morphology. Thus, by varying the morphology, we can tune the electrocatalytic properties of a catalyst and make the best use of it.
Cu-based oxide catalysts have received special attention as DMFC catalysts because of their low cost and earth abundance. In situ grown CuO nanowires (NWs) on Cu substrate exhibit current density (50 A g−1 or 100 mA cm−2) and stability (up to 1800 s) superior to other reported Cu-based electrodes.244 The electrochemical reactions taking place on the CuO NW surface are explained as follows.
| | | CuO + OH− ↔ CuOOH + e− | (17) |
| | | CuOOH + CH3OH + 1.25O2 ↔ Cu(OH)2 + CO2 + 1.5H2O | (18) |
Porous MnCo2O4 nanoflakes grown on conducting NF substrate outperform Co3O4/NF and other reported catalysts as the DMFC anode catalyst.245 MnCo2O4/NF covers larger CV area and anodic current than Co3O4/NF (in 1 M KOH + 0.5 M methanol solution). Compared to Co3O4/NF, MnCo2O4/NF displayed higher stability, holding 96 A g−1 and 91.5% retention after 2000 s. Negligible contribution from bare NF suggests NF to be an ideal substrate for the MOR electrocatalyst. Moreover, 66% current density retention is obtained after 1000 CV cycles, thus, making MnCo2O4 a promising electrocatalyst, opening the pathway for other metal oxide DMFC electrocatalysts. The mechanism of MOR of MnCo2O4 is as follows.
| | | MnCo2O4 + OH− + H2O + 3e− → MnOOH + 2CoOOH | (19) |
| | | MnOOH + CH3OH + 1.25O2 → Mn(OH)2 + CO2 + 1.5H2O | (20) |
| | | 2CoOOH + 2CH3OH + 2.5O2 → 2Co(OH)2 + 2CO2 + 3H2O | (21) |
The electrochemical oxidation activity of different 2D material-based anode catalysts of DOFC are compared in Table 2.
Table 2 Comparison of the electrochemical performance of different anode catalysts for DOFC
| Serial no. |
Material (anode catalyst) |
Fuel used |
Onset potential (V) |
Peak anode current density |
Electrolyte medium |
Ref. |
| 1 |
Pt69Rh8Fe23-PNS@MXene |
Ethanol (1 M) |
0.433 |
3407.7 mA mg−1 |
Alkaline (KOH) |
220
|
| 2 |
Pd/Ti3C2Tx |
Methanol (1 M) |
— |
12.4 mA cm−2 |
Alkaline (KOH) |
221
|
| 3 |
Pt/Ti3C2 |
Methanol (0.5 M) |
— |
1.137 mA cm−2 |
Acidic (H2SO4) |
222
|
| 4 |
Pt NW/PDDA-Ti3C2Tx |
Methanol (1 M) |
0.52 |
17.2 mA cm−2 |
Acidic (H2SO4) |
223
|
| 5 |
PtRu/MXene |
Methanol (2 M) |
— |
12.46 mA cm−2 |
Acidic (H2SO4) |
224
|
| 6 |
Pd/Ti3C2Tx–rGO |
Methanol (1 M) |
— |
Mass activity (446 mA mg−1), specific activity (16.3 mA mg−1) |
Alkaline (KOH) |
226
|
| 7 |
Pd/MX–rGO |
Formic acid (0.5 M) |
0.65 |
12 mA cm−2 |
Acidic (H2SO4) |
227
|
| 8 |
Pt–MXene–TiO2 |
Methanol (1 M) |
0.33 |
2750 mA mgPt−1 |
Acidic (H2SO4) |
228
|
| 9 |
PtRu/C3N4 |
Methanol |
— |
130 mA cm−2 (at 400 mV) |
— |
229
|
| 10 |
Pt/gCNM |
Methanol (1 M) |
— |
3.21 mA cm−2, 174 mA cm−2, 209 mA cm−2 |
Acidic (HClO4) |
230
|
| Pt/PTI–Li+Cl− |
| Pt/B-gCNM |
| 11 |
Pd@g-C3N4/GC |
Ethanol (1 M) |
0.32 |
2156 A g−1 Pd |
Alkaline (KOH) |
231
|
| 12 |
Ni/gCN |
Ethanol (3 M) |
0.46 |
122.8 mA g−1 |
Alkaline (NaOH) |
232
|
| 13 |
Pt/g-C3N4/MoS2 |
Ethanol |
— |
76.9 mA mgPt−1 |
Alkaline (KOH) |
233
|
| 14 |
Au@Pt/MoS2 |
Methanol (1 M) |
−0.24 |
6.24 A mg−1 |
Alkaline (NaOH) |
235
|
| 15 |
Pd–MoS2 NSs |
Methanol (1 M) |
— |
433.5 mA mg−1 |
Alkaline (KOH) |
236
|
| 16 |
Pt/G–MoS2 |
Methanol (2 M) |
— |
13.3 mA cm−2 |
Acidic (H2SO4) |
237
|
| 17 |
Pt–MoS2/RGO |
Methanol (1 M), formic acid (0.5 M) |
— |
7.35 mA cm−2 (methanol) |
Acidic (H2SO4) |
238
|
| 6.8 mA cm−2 (formic acid) |
| 18 |
Pt@MoS2/NrGO |
Methanol (1 M), ethylene glycol (1 M), glycerol (1 M) |
— |
448.0 mA mgPt−1 for methanol, 158.0 mA mgPt−1 for ethylene glycol (EG), and 147.0 mA mgPt−1 for glycerol |
Acidic (H2SO4) |
239
|
| 19 |
Pt/MoS2/CNx |
Methanol (0.5 M) |
— |
1030.2 mA mgPt−1 |
Acidic (H2SO4) |
240
|
| 20 |
Co3O4/Ni |
Methanol (0.5 M) |
0.32–0.34 |
Microflower (28 A g−1), nanograss (34.9 A g−1), microsphere (36.2 A g−1) |
Alkaline (KOH) |
241
|
| 21 |
CuO NWs/Cu |
Methanol (0.5 M) |
— |
50 A g−1 or 100 mA cm−2 |
Alkaline (KOH) |
244
|
| 22 |
MnCo2O4/NF |
Methanol (0.5 M) |
1.3 |
96 A g−1 |
Alkaline (KOH) |
245
|
| 23 |
Pt/BN-GA |
Methanol (1 M) |
— |
1184.5 mA mg−1 |
Acidic (H2SO4) |
97
|
| 24 |
Pd@C–rGO |
Ethylene glycol (0.5 M) |
−0.5 |
∼3 mA cm−2 |
Alkaline (KOH) |
100
|
| 25 |
Pt–Pd decorated rGO |
Ethylene glycol (0.5 M) |
0.418 |
803.4 mA mg−1 |
Acidic (H2SO4) |
101
|
| 26 |
Pd–C@MoS2/RGO |
Methanol (1 M) |
— |
11.2 mA cm−2 (methanol), 2 mA cm2 (ethylene glycol) |
Alkaline (KOH) |
102
|
6 Summary and outlook
In this review, we encapsulated a detailed description of fuel cells along with their mechanism and condensed different types of fuel cells depending on the types of electrolytes, ions, fuels, mode of fuel supply, and temperature range used in the fuel cell operation. We also outlined the problems pertaining to different types of fuel cells. Large specific surface area, tunable electronic properties, and other intriguing properties make 2D materials outshine other nanostructured materials as anode catalysts in direct alcohol fuel cell application. Considering some of the drawbacks of graphene such as chemical inertness, insufficient loading sites for noble-metal nanoparticles, and restacking problem, this review focused on 2D materials (mainly MXene, graphitic carbon nitride, transition metal dichalcogenides, and transition metal oxides) beyond graphene and summarized their properties and application in direct alcohol fuel cell either as an active material or as a support/substrate. To date, MXene or composites of MXenes with conducting materials have been used as catalyst supports for noble metal nanoparticles or alloys in direct alcohol fuel cells. MXene as a support provides hydrogen bonding between alcohol molecules and hydroxyl groups on its surface, thereby increasing the catalytic activity. In MXene, (i) surfaces possess Lewis basic sites, which help in adsorbing more and more alcohol molecules, leading to enhanced electrochemical activity, (ii) hydrophilic terminations such as OH− and F− act as electron donors and promote the interaction between the metal and the support, further increasing the oxidation reaction, (iii) by garnishing the basal planes of MXene, its surface charge can be altered, which further behave as uniform distribution sites for the metal nanoparticles. Graphitic carbon nitride is considered a promising candidate as an electrocatalyst and has been used as the catalyst support for noble metal NPs or non-noble metals due to the following reasons: (i) its porous structure accelerates the diffusion of ions into the catalyst surface, making fuel oxidation effortless, (ii) nitrogen present in its structure increases the conductivity and acts as a electron donor, further increasing the electron transfer, (iii) its repeated triazine units anchor the metal atoms/alloys, thereby promoting metal-support interaction, (iv) N-doped carbon or g-C3N4 possess high specific capacitance and help in removing the adsorbed poisonous intermediates by providing extra sites for the surface groups. Transition metal dichalcogenides exhibit polymorphism and different polymorphs lead to different electrocatalytic activity. Mostly, MoS2 has been explored as an anode catalyst support and composites with other conducting materials such as graphene and reduced graphene oxide have been explored to reduce its agglomeration, further increasing the electrocatalytic activity. The Mo and S atoms in MoS2, C–N defects, and porous nature of MoS2 help in adsorbing more H2O molecules and assist in recovering the Pt sites. Besides, the various faces of MoS2 speed up the transportation of the adsorbed CO species and improves their CO poisoning. 2D transition metal oxides are a good replacement of the noble metals due to the presence of multiple oxidation states of the transition metal. Spinel oxides are frequently studied anode catalysts. By varying the anions used in the synthetic process, one can obtain different morphology of metal oxides and can tune the electrochemical properties. Thus, we can conclude that for a better and faster electrochemical reaction and efficient electrocatalyst, one needs to keep in mind the following factors: (i) porosity of the catalyst as a porous structure enhances the electrocatalytic reactions, (ii) hydrophilicity of the material for ensuring durability and anti-poisoning ability, (iii) presence of Lewis basic sites, (iv) metal-support interaction, (v) anti-poisoning of the noble-metal and electroactive material, (vi) a good choice of support/substrate, which can reduce the agglomeration of the active material. The 2D materials are mostly used as catalyst support and have come a long way; however, scalability and the synthetic methods need to be ameliorated. More novel 2D structures and nanocomposites with other nanostructured materials can be more explored as non-noble electrocatalysts. As we have seen that unique heterostructures can provide scope for increasing the surface area, porosity, interactions between the components, and expose more electrochemically active sites, thus, one can explore a variety of heterostructures such as 1D–2D, 2D–3D, 0D–1D, 0D–2D, and 0D–3D as anode catalysts for DOFCs.
Author contributions
Kashmiri Baruah prepared the original draft of the manuscript. Pritam Deb contributed to methodology, supervision, reviewing, and editing of the manuscript.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
KB is grateful to UGC for providing the financial support (UGC NFOBC). KB sincerely thanks the late supervisor Prof. Ashok Kumar for his moral support and guidance during his lifetime. P. Deb would like to acknowledge N-BIOS award project, Department of Biotechnology, Government of India.
References
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 6321 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- A. Nilssona, L. G. M. Pettersson, B. Hammer, T. Bligaard, C. H. Christensen and J. K. Nørskov, Catal. Lett., 2005, 100, 111–114 CrossRef.
- S. K. Behera, P. Deb and A. Ghosh, Phys. Chem. Chem. Phys., 2016, 18, 23220–23230 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- H. Zhang, ACS Nano, 2015, 9, 9451–9469 CrossRef CAS PubMed.
- M. Fan, J. Wu, J. Yuan, L. Deng, N. Zhong, L. He, J. Cui, Z. Wang, S. K. Behera, C. Zhang, J. Lai, B. I. Jawdat, R. Vajtai, P. Deb, Y. Huang, J. Qian, J. Yang, J. M. Tour, J. Lou, C. W. Chu, D. Sun and P. M. Ajayan, Adv. Mater., 2019, 31, 1805778 CrossRef PubMed.
- S. K. Behera, P. Deb and A. Ghosh, ChemistrySelect, 2017, 2, 3657–3667 CrossRef CAS.
- K. Baruah, S. K. Behera and P. Deb, Bull. Am. Phys. Soc., 2017, 62, 1121 Search PubMed.
- F. Fina, S. K. Callear, G. M. Carins and J. T. S. Irvine, Chem. Mater., 2015, 27, 2612–2618 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- B. Smitha, S. Sridhar and A. A. Khan, J. Membr. Sci., 2005, 259, 10–26 CrossRef CAS.
- S. S. Ozdemir, M. G. Buonomenna and E. Drioli, Appl. Catal., A, 2006, 307, 167–183 CrossRef CAS.
- S. Sharma and B. G. Pollet, J. Power Sources, 2012, 208, 96–119 CrossRef CAS.
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- Y. Y. Xu, C. Gao, H. Kong, D. Y. Yan, Y. Z. Jin and P. C. P. Watts, Macromolecules, 2004, 37, 8846–8853 CrossRef CAS.
- X. Wang, C. Hu, Y. Xiong, H. Liu, G. Du and X. He, J. Power Sources, 2011, 196, 1904–1908 CrossRef CAS.
- J. Lobato, P. Canizares, D. Ubeda, F. J. Pinar and M. A. Rodrigo, Appl. Catal., B, 2011, 106, 174–180 CAS.
- A. Ermete, J. Power Sources, 2007, 170, 1–12 CrossRef.
- Y. Kim, S. M. Kwon, D. Y. Kim, H. S. Kim and H. J. Jin, Curr. Appl. Phys., 2009, 9, 100–103 CrossRef.
- J.-Y. Choi, D. Higgins and Z. Chen, J. Electrochem. Soc., 2012, 159, 86–89 CrossRef.
- D. J. Guo, X. P. Qju, W. T. Zhu and L. Q. Chen, Appl. Catal., B, 2009, 89, 597–601 CrossRef CAS.
- A. S. Karakoti, J. E. S. King, A. Vincent and S. Seal, Appl. Catal., A, 2010, 388, 262–271 CrossRef CAS.
- X. Jin, T.-H. Gu, K.-G. Lee, M. J. Kim, M. S. Islam and S.-J. Hwang, Coord. Chem. Rev., 2020, 415, 213280 CrossRef CAS.
- H. Xu, P. Song, Y. Zhang and Y. Du, Nanoscale, 2018, 10, 12605–12611 RSC.
- P. Chandran, D. Puthusseri and S. Ramaprabhu, Int. J. Hydrogen Energy, 2019, 44, 4951–4961 CrossRef CAS.
- B. Li, Q. Yan, C. Song, P. Yan, K. Ye, K. Cheng, K. Zhu, J. Yan, D. Cao and G. Wang, Appl. Surf. Sci., 2019, 491, 659–669 CrossRef CAS.
- F. Gao, Y. Zhang, P. Song, J. Wang, C. Wang, J. Guo and Y. Du, J. Power Sources, 2019, 418, 186–192 CrossRef CAS.
- L. Jin, H. Xu, C. Chen, T. Song, C. Wang, Y. Wang, H. Shang and Y. Du, J. Colloid Interface Sci., 2019, 540, 265–271 CrossRef CAS PubMed.
- M. Guo, Y. Cheng, Y. Yu and J. Hu, Appl. Surf. Sci., 2017, 416, 439–445 CrossRef CAS.
- S. Guo, J. Sun, Z. Zhang, A. Sheng, M. Gao, Z. Wang, B. Zhao and W. Ding, J. Mater. Chem. A, 2017, 5, 15879–15890 RSC.
- M. G. Hosseini and R. Mahmoodi, New J. Chem., 2017, 41, 13408–13417 RSC.
- A. Balinait, A. Zabielait, L. Tamaauskait-Tamainait and E. Norkus, J. Electrochem. Soc., 2018, 165, 1249–1253 CrossRef.
- C. Song, G. Wang, B. Li, C. Miao, K. Ma, K. Zhu, K. Cheng, K. Ye, J. Yan, D. Cao and J. Yin, Electrochim. Acta, 2019, 299, 395–404 CrossRef CAS.
- H. Zhang, J. He, C. Zhai and M. Zhu, Chin. Chem. Lett., 2019, 30, 2338–2342 CrossRef CAS.
- L. Dai, Q. Qin, X. Zhao, C. Xu, C. Hu, S. Mo, Y. O. Wang, S. Lin, Z. Tang and N. Zheng, ACS Cent. Sci., 2016, 2, 538–544 CrossRef CAS.
- D. J. L. Brett, A. R. Kucernak, P. Aguiar, S. C. Atkins, N. P. Brandon, R. Clague, L. F. Cohen, G. Hinds, C. Kalyvas, G. J. Offer, B. Ladewig, R. Maher, A. Marquis, P. Shearing, N. Vasileiadis and V. Vesovic, ChemPhysChem, 2010, 11, 2714–2731 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Winter and R. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- C. Rice, S. Ha, R. I. Masel and A. Wieckowski, J. Power Sources, 2003, 115, 229–235 CrossRef CAS.
- G. Che, B. B. Lakshmi, E. R. Fisher and C. R. Martin, Nature, 1998, 393, 346 CrossRef CAS.
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577 CrossRef CAS PubMed.
- K. Sundmacher, Ind. Eng. Chem. Res., 2010, 49, 10159–10182 CrossRef CAS.
-
X. Li, Principles of Fuel Cells, Taylor & Francis, 2005, p. 572 Search PubMed.
- L. Yang, J. Ge, C. Liu, G. Wang and W. Xing, Curr. Opin. Electrochem., 2017, 4, 83–88 CrossRef CAS.
- J. N. Tiwari, R. N. Tiwari, G. Singh and K. S. Kim, Nano Energy, 2013, 2, 553–578 CrossRef CAS.
- M. D. Bhatt and J. Y. Lee, Energy Fuels, 2020, 34, 6634–6695 CrossRef.
- L. V. Mattos and F. B. Noronha, J. Catal., 2005, 233, 453–463 CrossRef CAS.
- K. Jukk, N. Alexeyeva, P. Ritslaid, J. Kozlova, V. Sammelselg and K. Tammeveski, Electrocatalysis, 2013, 4, 42–48 CrossRef CAS.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- N. Ramaswamy and S. Mukerjee, Chem. Rev., 2019, 119, 11945–11979 CrossRef CAS PubMed.
- E. H. Yu and K. Scott, Electrochem. Commun., 2004, 6, 361–365 CrossRef CAS.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- Y. Zhao, L. Zhan, J. Tian, S. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967–1972 CrossRef CAS.
- J. H. Kim, H. K. Kim, K. T. Hwang and J. Y. Lee, Int. J. Hydrogen Energy, 2010, 35, 768–773 CrossRef CAS.
- R. R. Adzic, M. L. Avramovivic and A. V. Tripkovic, Electrochim. Acta, 1984, 29, 1353–1357 CrossRef CAS.
- K. Miyazaki, N. Sugimura, K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, J. Power Sources, 2008, 178, 683–686 CrossRef CAS.
- R. Kannan, A. R. Kim and D. J. Yoo, J. Appl. Electrochem., 2014, 44, 893–902 CrossRef CAS.
- W. Wang, R. Li, R. Zhang, J. Ma and B. Wang, J. Electroanal. Chem., 2015, 742, 110–121 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- M. Hamdani, R. N. Singh and P. Chartier, Int. J. Electrochem. Sci., 2010, 5, 556–577 CAS.
- R. Awasthi and R. N. Singh, Carbon, 2013, 51, 282–289 CrossRef CAS.
- Y. Wang, Z. M. Sheng, H. Yang, S. P. Jiang and C. M. Li, Int. J. Hydrogen Energy, 2010, 35, 10087–10093 CrossRef CAS.
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864–890 CrossRef CAS.
- A. Zuttel, A. Remhof, A. Borgschulte and O. Friedrichs, Philos. Trans. R. Soc., A, 2010, 368, 3329–3342 CrossRef CAS PubMed.
- Y. J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed.
- D. Banham and S. Ye, ACS Energy Lett., 2017, 2, 629–638 CrossRef CAS.
- Y. Hong, H. J. Kim, H. J. Lee, J. Kim and S.-I. Choi, Front. Chem., 2019, 7, 608 CrossRef CAS PubMed.
- A. M. I. N. Azam, S. H. Lee, M. S. Masdar, A. M. Zainoodin and S. K. Kamarudin, Int. J. Hydrogen Energy, 2019, 44, 8566–8574 CrossRef CAS.
-
T. Iwasita, Methanol and CO electrooxidation, in Handbook of Fuel Cells-Fundamentals, Technology and Applications Handbook of Fuel Cells, ed. Wea. Vielstich, 2003, p. 603 Search PubMed.
- M. Zeng, J. Wu, Z. Li, H. Wu, J. Wang, H. Wang, L. He and X. Yang, ACS Sustainable Chem. Eng., 2019, 7, 4777–4783 CrossRef CAS.
- A. N. Rollinson, J. Jones, V. Dupont and M. V. Twigg, Energy Environ. Sci., 2011, 4, 1216–1224 RSC.
- J. Wang, B. Gong, W. Huang, Y. Wang and J. Zhou, Bioresour. Technol., 2017, 228, 31–38 CrossRef CAS PubMed.
- B. K. Boggs, R. L. King and G. G. Botte, Chem. Commun., 2009, 32, 4859–4861 RSC.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
-
H. Yang, S. Kannappan, A. S. Pandian, J.-H. Jang, Y. S. Lee and W. Lu, 2013, arXiv preprint, arXiv:1311.1413.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- D. S. Hecht, L. Hu and G. Irvin, Adv. Mater., 2011, 23, 1482–1513 CrossRef CAS PubMed.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357–390 RSC.
- A. Ali and P. K. Shen, J. Mater. Chem. A, 2019, 7, 22189–22217 RSC.
- S. Cui, S. Mao, G. Lu and J. Chen, J. Phys. Chem. Lett., 2013, 4, 2441–2454 CrossRef CAS.
- M. Lei, Z. B. Wang, J. S. Li, H. L. Tang, W. J. Liu and Y. G. Wang, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- H. Huang, S. Yang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2014, 26, 5160–5165 CrossRef CAS PubMed.
- C.-H. A. Tsang, K. Hui, K. Hui and L. Ren, J. Mater. Chem. A, 2014, 2, 17986–17993 RSC.
- M. Mansor, S. N. Timmiati, K. L. Lim, W. Y. Wong, S. K. Kamarudin and N. H. Nazirah Kamarudin, Int. J. Photoenergy, 2019, 44, 14744–14769 CAS.
- R. Baronia, J. Goel, S. Tiwari, P. Singh, D. Singh, S. P. Singh and S. K. Singhal, Int. J. Hydrogen Energy, 2017, 42, 10238–10247 CrossRef CAS.
- J. C. Ng, C. Y. Tan, B. H. Ong, A. Matsuda, W. J. Basirun, W. K. Tan, R. Singh and B. K. Yap, Mater. Res. Bull., 2019, 112, 213–220 CrossRef CAS.
- R. Kiyani, S. Rowshanzamir and M. J. Parnian, Energy, 2016, 113, 1162–1173 CrossRef CAS.
- G. V. Reddy, P. Raghavendra, P. S. Chandana and L. S. Sarma, RSC Adv., 2015, 5, 100522–100530 RSC.
- Z.-S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2013, 125, 3192–3198 CrossRef.
- T. Mori, H. Tanaka, A. Dalui, N. Mitoma, K. Suzuki, M. Matsumoto, N. Aggarwal, A. Patnaik, S. Acharya, L. K. Shrestha, H. Sakamoto, K. Itami and K. Ariga, Angew. Chem., 2018, 130, 9827–9831 CrossRef.
- C. N. R. Rao, K. Gopalakrishnan and U. Maitra, ACS Appl. Mater. Interfaces, 2015, 7, 7809–7832 CrossRef CAS.
- M. Z. Iqbal, A. U. Rehman and S. Siddique, J. Energy Chem., 2019, 39, 217–234 CrossRef.
- M. Li, Q. Jiang, M. Yan, Y. Wei, J. Zong, J. Zhang, Y. Wu and H. Huang, ACS Sustainable Chem. Eng., 2018, 6, 6644–6653 CrossRef CAS.
- D. Rajesh, P. I. Neel, A. Pandurangan and C. Mahendiran, Appl. Surf. Sci., 2018, 442, 787–796 CrossRef CAS.
- X. X. Yuan, X. L. Ding, C. Y. Wang and Z. F. Ma, Energy Environ. Sci., 2013, 6, 1105–1124 RSC.
- R. Kannan, A. R. Kim, K. S. Nahm, H.-K. Leed and D. J. Yoo, Chem. Commun., 2014, 50, 14623–14626 RSC.
- T. R. Kumar, D. J. Yoo, A. R. Kim and G. G. Kumar, New J. Chem., 2018, 42, 14386–14393 RSC.
- R. Kannan, C. D. Lim, A. R. Kim, M. H. Lee and D. J. Yoo, J. Nanosci. Nanotechnol., 2019, 19, 4520–4528 CrossRef CAS PubMed.
- M. Mansor, S. N. Timmiati, K. L. Lim, W. Y. Wong, S. K. Kamarudin and N. H. N. Kamarudin, Int. J. Hydrogen Energy, 2019, 44, 14744–14769 CrossRef CAS.
- J. Liu, H. J. Choi and L. Y. Meng, J. Ind. Eng. Chem., 2018, 64, 1–15 CrossRef CAS.
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS PubMed.
- N. Seselj, C. Engelbrekt and J. Zhang, Sci. Bull., 2015, 60, 864–876 CrossRef.
- N. Shaari and S. K. Kamarudin, Renew. Sustain. Energy Rev., 2017, 69, 862–870 CrossRef CAS.
- S. Hossain, A. M. Abdalla, S. B. H. Suhaili, I. Kamal, S. P. S. Shaikh, M. K. Dawood and A. K. Azad, J. Energy Storage, 2020, 29, 101386 CrossRef.
- M. Naguib and Y. Gogotsi, Acc. Chem. Res., 2015, 48, 128–135 CrossRef CAS PubMed.
- A. Feng, Y. Yu, Y. Wang, F. Jiang, Y. Yu, L. Mi and L. Song, Mater. Des., 2017, 114, 161–166 CrossRef CAS.
- M. W. Barsoum, Prog. Solid State Chem., 2000, 28, 201–281 CrossRef CAS.
- W. Yang, K. R. Ratinac, S. P. Ringer, P. Thordarson, J. J. Gooding and F. Braet, Angew. Chem., Int. Ed., 2010, 49, 2114–2138 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- X. D. Zhang, J. G. Xu, H. Wang, J. J. Zhang, H. B. Yan, B. C. Pan, J. F. Zhou and Y. Xie, Angew. Chem. Int. Ed., 2013, 52, 4361–4365 CrossRef CAS PubMed.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29, 7633–7644 CrossRef CAS.
- N. K. Chaudhari, H. Jin, B. Kim, D. S. Baek, S. H. Joo and K. Lee, J. Mater. Chem. A, 2017, 5, 24564–24579 RSC.
- X. Zhao, M. Radovic and M. J. Green, Chem, 2020, 6, 544–546 CAS.
- J. Halim, M. R. Lukatskaya, K. M. Cook, J. Lu, C. R. Smith, L. A. Naslund, S. J. May, L. Hultman, Y. Gogotsi, P. Eklund and M. W. Barsoum, Chem. Mater., 2014, 26, 2374–2381 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall’ Agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, Nat. Commun., 2013, 4, 1716 CrossRef PubMed.
-
M. W. Barsoum, The Mn+1AXn phases and their properties, in Ceram. Sci. Technol, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, p. 299 Search PubMed.
- T. Ouisse and M. W. Barsoum, Mater. Res. Lett., 2017, 5, 365–378 CrossRef CAS.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- H. Wang, Y. Wu, X. Yuan, G. Zeng, J. Zhou, X. Wang and J. W. Chew, Adv. Mater., 2018, 30, 1704561 CrossRef.
- Q. Jiang, N. Kurra, M. Alhabeb, Y. Gogotsi and H. N. Alshareef, Adv. Energy Mater., 2018, 8, 1703043 CrossRef.
- J. Nan, X. Guo, J. Xiao, X. Li, W. Chen, W. Wu, H. Liu, Y. Wang, M. Wu and G. Wang, Small, 2019, 1902085 Search PubMed.
- Z. Hu, X. Kuai, J. Chen, P. Sun, Q. Zhang, H. H. Wu and L. Zhang, ChemSusChem, 2019, 12, 1–7 CrossRef.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, H. S. Man, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137 CrossRef CAS PubMed.
- A. Sinha, Dhanjai, H. Zhao, Y. Huang, X. Lu, J. Chen and R. Jain, Trends Anal. Chem., 2018, 105, 424–435 CrossRef CAS.
- B. Xu, M. Zhu, W. Zhang, X. Zhen, Z. Pei, Q. Xue, C. Zhi and P. Shi, Adv. Mater., 2016, 28, 3333–3339 CrossRef CAS PubMed.
- X. Q. Li, C. Y. Wang, Y. Cao and G. X. Wang, Chem.–Asian J., 2018, 13, 2742–2757 CrossRef CAS PubMed.
- M. Soleymaniha, M. A. Shahbazi, A. R. Rafieerad, A. Maleki and A. Amiri, Adv. Healthcare Mater., 2019, 8, 26 Search PubMed.
- J. J. Zhu, P. Xiao, H. L. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717 RSC.
- T. Sano, S. Tsutsui, K. Koike, T. Hirakawa, Y. Teramoto, N. Negishi and K. Takeuchi, J. Mater. Chem. A, 2013, 1, 6489 RSC.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Z. Fu and X. C. Wang, Energy Environ. Sci., 2011, 4, 675 RSC.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355 CrossRef CAS.
- Y. Ji, H. Dong, H. Lin, L. Zhang, T. Hou and Y. Li, RSC Adv., 2016, 6, 52377–52383 RSC.
- G. Algara-Siller, N. Severin, S. Y. Chong, T. Bjçrkman, R. G. Palgrave, A. Laybourn, M. Antonietti, Y. Z. Khimyak, A. V. Krasheninnikov, J. P. Rabe, U. Kaiser, A. I. Cooper, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2014, 53, 7450–7455 CrossRef CAS PubMed.
- J. Liu, T. Zhang, Z. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398–14401 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem. Int. Ed., 2010, 49, 441 CrossRef CAS PubMed.
- H. J. Yan, Y. Chen and S. M. Xu, Int. J. Hydrogen Energy, 2012, 37, 125–133 CrossRef CAS.
- F. Dong, Y. J. Sun, L. W. Wu, M. Fu and Z. B. Wu, Sci. Technol., 2012, 2, 1332–1335 CAS.
- D. Mitoraj and H. Kisch, Chem.–Eur. J., 2010, 16, 261–269 CrossRef CAS PubMed.
- T. Komatsu and T. Nakamura, J. Mater. Chem., 2001, 11, 474–478 RSC.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS.
- D. R. Miller, J. J. Wang and E. G. Gillan, J. Mater. Chem., 2002, 12, 2463–2469 RSC.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- G. P. Dong, Y. H. Zhang, Q. W. Pan and J. R. Qiu, J. Photochem. Photobiol., C, 2014, 20, 33–50 CrossRef CAS.
- X. F. Chen, J. S. Zhang, X. Z. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- X. C. Wang, X. F. Chen, A. Thomas, X. Z. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- J. Wang, D. Hao, J. Ye and N. Umezawa, Chem. Mater., 2017, 29, 2694–2707 CrossRef CAS.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- H. Huang, S. Yang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2014, 26, 5160–5165 CrossRef CAS PubMed.
- J. Tian, R. Ning, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 1011–1017 CrossRef CAS PubMed.
- J. Yuan, Q. Gao, X. Li, Y. Liu, Y. Fang, S. Yang, F. Peng and X. Zhou, RSC Adv., 2014, 4, 52332–52337 RSC.
- I. S. Pieta, A. Rathi, P. Pieta, R. Nowakowski, M. Hołdynski, M. Pisarek, A. Kaminska, M. B. Gawande and R. Zboril, Appl. Catal., B, 2019, 244, 272–283 CrossRef CAS.
- N. Mansor, T. S. Miller, I. Dedigama, A. B. Jorge, J. Jia, V. Brázdová, C. Mattevi, C. Gibbs, D. Hodgson, P. R. Shearing, C. A. Howard, F. Corà, M. Shaffer, D. J. L. Brett and P. F. McMillan, Electrochim. Acta, 2016, 222, 44–57 CrossRef CAS.
- M. Huang, G. Dong, N. Wang, J. Xu and L. Guan, Energy Environ. Sci., 2011, 4, 4513–4516 RSC.
- S. Zhang, S. Tsuzuki, K. Ueno, K. Dokko and M. Watanabe, Angew. Chem., 2015, 127, 1318–1322 CrossRef.
- S. A. Shaik, A. Goswami, R. S. Varma and M. B. Gawande, Curr. Opin. Green Sustain. Chem., 2019, 15, 67–76 CrossRef.
- Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao and P. K. Wong, Adv. Mater., 2018, 30, 1706108 CrossRef PubMed.
- M. Inagaki, T. Tsumura, T. Kinumoto and M. Toyoda, Carbon, 2019, 141, 580–607 CrossRef CAS.
- H. S. Lee, S.-W. Min, Y.-G. Chang, M. K. Park, T. Nam, H. Kim, J. H. Kim, S. Ryu and S. Im, Nano Lett., 2012, 12, 3695–3700 CrossRef CAS PubMed.
- C. Zhong, C. Duan, F. Huang, H. Wu and Y. Cao, Chem. Mater., 2011, 23, 326–340 CrossRef CAS.
- J. Pu, Y. Yomogida, K.-K. Liu, L.-J. Li, Y. Iwasa and T. Takenobu, Nano Lett., 2012, 12, 4013–4017 CrossRef CAS PubMed.
- S. Wu, Z. Zeng, Q. He, Z. Wang, S. J. Wang, Y. Du, Z. Yin, X. Sun, W. Chen and H. Zhang, Small, 2012, 8, 2264–2270 CrossRef CAS PubMed.
- Q. He, Z. Zeng, Z. Yin, H. Li, S. Wu, X. Huang and H. Zhang, Small, 2012, 8, 2994–2999 CrossRef CAS PubMed.
- G. Du, Z. Guo, S. Wang, R. Zeng, Z. Chen and H. Liu, Chem. Commun., 2010, 46, 1106–1108 RSC.
- Y. Luo, Y. Zhang, Y. Zhao, X. Fang, J. Ren, W. Weng, Y. Jiang, H. Sun, B. Wang, X. Cheng and H. Peng, J. Mater. Chem. A, 2015, 3, 17553–17557 RSC.
- M. H. Yang, J. M. Jeong, Y. S. Huh and B. J. Choi, Compos. Sci. Technol., 2015, 121, 123–128 CrossRef CAS.
- C. Zhai, M. Zhu, D. Bin, F. Ren, C. Wang, P. Yang and Y. Du, J. Power Sources, 2015, 275, 483–488 CrossRef CAS.
- Z. Gao, M. Li, J. Wang, J. Zhu, X. Zhao, H. Huang, J. Zhang, Y. Wu, Y. Fu and X. Wang, Carbon, 2018, 139, 369–377 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- R. J. Smith, P. J. King, M. Lotya, C. Wirtz, U. Khan, S. De, A. O'Neill, G. S. Duesberg, J. C. Grunlan, G. Moriarty, J. Chen, J. Wang, A. I. Minett, V. Nicolosi and J. N. Coleman, Adv. Mater., 2011, 23, 3944–3948 CrossRef CAS PubMed.
- L. F. Matthesis, Phys. Rev. B: Solid State, 1973, 8, 3719–3740 CrossRef.
- R. Lv, J. A. Robinson, R. E. Schaak, D. Sun, Y. Sun, T. E. Mallouk and M. Terrones, Acc. Chem. Res., 2015, 48, 56–64 CrossRef CAS PubMed.
- X. Li and H. Zhu, J. Materiomics, 2015, 1, 33–44 CrossRef.
- Y. Ding, Y. Wang, J. Ni, L. Shi, S. Shi and W. Tang, Physica B, 2011, 406, 2254–2260 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS.
- R. J. Toh, Z. Sofer, J. Luxa, D. Sedmidubsky and M. Pumera, Chem. Commun., 2017, 53, 3054–3057 RSC.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- S. A. Han, R. Bhatia and S.-W. Kim, Nano Convergence, 2015, 2, 17 CrossRef.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695–2700 CrossRef CAS PubMed.
- A. Kuc, N. Zibouche and T. Heine, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 245213 CrossRef.
- C. Tsai, K. Chan, F. Abild-Pedersen and J. K. Norskov, Phys. Chem. Chem. Phys., 2014, 16, 13156–13164 RSC.
- D. Y. Chung, S. K. Park, Y. H. Chung, S. H. Yu, D. H. Lim, N. Jung, H. C. Ham, H. Y. Park, Y. Piao, S. J. Yoo and Y. E. Sung, Nanoscale, 2014, 6, 2131–2136 RSC.
- D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2018, 28, 6197–6206 CrossRef PubMed.
- A. Ambrosi, Z. Sofer and M. Pumera, Chem. Commun., 2015, 51, 8450–8453 RSC.
- R. F. Frindt, J. Appl. Phys., 1966, 37, 1928–1929 CrossRef CAS.
- H. R. Gutiérrez, N. Perea-López, A. L. Elías, A. Berkdemir, B. Wang, R. Lv, F. López-Urías, V. H. Crespi, H. Terrones and M. Terrones, Nano Lett., 2013, 13, 3447–3454 CrossRef PubMed.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- A. L. Elias, N. Perea-Lopez, A. Castro-Beltran, A. Berkdemir, R. T. Lv, S. M. Feng, A. D. Long, T. Hayashi, Y. A. Kim, M. Endo, H. R. Gutierrez, N. R. Pradhan, L. Balicas, T. E. M. Houk, F. Lopez- Urias, H. Terrones and M. Terrones, ACS Nano, 2013, 7, 5235–5242 CrossRef CAS PubMed.
- Z. Yin, Y. Zheng, H. Wang, J. Li, Q. Zhu, Y. Wang, N. Ma, G. Hu, B. He, A. Knop-Gericke, R. Schlog and D. Ma, ACS Nano, 2017, 11, 12365–12377 CrossRef CAS PubMed.
- P. Manivasakan, P. Ramasamy and J. Kim, Nanoscale, 2014, 6, 9665–9672 RSC.
- H. S. Jadhav, S. M. Pawar, A. H. Jadhav, G. M. Thorat and J. G. Seo, Sci. Rep., 2016, 6, 31120 CrossRef CAS PubMed.
- F. Carraro, O. Vozniuk, L. Calvillo, L. Nodari, C. La Fontaine, F. Cavani and S. Agnoli, J. Mater. Chem. A, 2017, 5, 20808–20817 RSC.
-
R. J. Lad, Surface Structure of Crystalline Ceramics, Physical Structure, 1996, pp. 185–228, DOI:10.1016/s1573-4331(96)80010-2.
- K. Kalantar-zadeh, J. Z. Ou, T. Daeneke, A. Mitchell, T. Sasaki and M. S. Fuhrer, Appl. Mater. Today, 2016, 5, 73–89 CrossRef.
- J. Mannhart and D. Schlom, Science, 2010, 327, 1607–1611 CrossRef CAS PubMed.
- D. Chimene, D. L. Alge and A. K. Gaharwar, Adv. Mater., 2015, 27, 7261–7284 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- C. Wang, L. Yin, L. Zhang, D. Xiang and R. Gao, Sensors, 2010, 10, 2088–2106 CrossRef CAS PubMed.
- K. Bhattacharya and P. Deb, Dalton Trans., 2015, 44, 9221–9229 RSC.
- G. Rajeshkhanna, E. Umeshbabu and G. R. Rao, J. Chem. Sci., 2017, 129, 157–166 CrossRef CAS.
- R. van de Krol, Y. Liang and J. Schoonman, J. Mater. Chem., 2008, 18, 2311–2320 RSC.
- J. Meyer, S. Hamwi, M. Kröger, W. Kowalsky, T. Riedl and A. Kahn, Adv. Mater., 2012, 24, 5408–5427 CrossRef CAS.
- K. Kalantar-zadeh, J. Z. Ou, T. Daeneke, A. Mitchell, T. Sasaki and M. S. Fuhrer, Appl. Mater. Today, 2016, 5, 73–89 CrossRef.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- K. Kalantar-zadeh, J. Z. Ou, T. Daeneke, A. Mitchell, T. Sasaki and M. S. Fuhrer, Appl. Mater. Today, 2016, 5, 73–89 CrossRef.
- W. Yang, X. Zhang and Y. Xie, Nano Today, 2016, 11, 793–816 CrossRef CAS.
- K. W. Park, J. H. Choi, B. K. Kwon, S. A. Lee, Y. E. Sung, H. Y. Ha, S. A. Hong, H. Kim and A. Wieckowski, J. Phys. Chem. B, 2002, 106, 1869–1877 CrossRef CAS.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933–936 CrossRef CAS PubMed.
- P. Wang, H. Cui and C. Wang, Nano Energy, 2019, 66, 104196 CrossRef CAS.
- Z. Lang, Z. Zhuang, S. Li, L. Xia, Y. Zhao, Y. Zhao, C. Han and L. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 2400–2406 CrossRef CAS.
- Y. Wang, J. Wang, G. Han, C. Dua, Q. Deng, Y. Gao, G. Yin and Y. Song, Ceram. Int., 2018, 45, 2411–2417 CrossRef.
- C. Yang, Q. Jiang, H. Huang, H. He, L. Yang and W. Li, ACS Appl. Mater. Interfaces, 2020, 12, 23822–23830 CrossRef CAS PubMed.
- N. Abdullah, R. Saidur, A. M. Zainoodin and N. Aslfattahi, J. Clean. Prod., 2020, 277, 123395 CrossRef CAS.
- C. Yang, Q. Jiang, W. Li, H. He, L. Yang, Z. Lu and H. Huang, Chem. Mater., 2019, 31, 9277–9287 CrossRef CAS.
- P. Zhang, C. Fan, R. Wang, C. Xu, J. Cheng, L. Wang, Y. Lu and P. Luo, Nanotechnology, 2019, 31, 09LT01 CrossRef PubMed.
- C. Yang, H. He, Q. Jiang, X. Liu, S. P. Shah, H. Huang and W. Li, Int. J. Hydrogen Energy, 2021, 46, 589–598 CrossRef CAS.
- Y. Sun, Y. Zhou, Y. Liu, Q. Wu, M. Zhu, H. Huang, Y. Liu, M. Shao and Z. Kang, Nano Res., 2020, 13, 2683–2690 CrossRef CAS.
- M. Kim, S. Hwang and J.-S. Yu, J. Mater. Chem., 2007, 17, 1656–1659 RSC.
- N. Mansor, A. B. Jorge, F. Corà, C. Gibbs, R. Jervis, P. F. McMillan, X. Wang and D. J. L. Brett, J. Phys. Chem. C, 2014, 118, 6831–6838 CrossRef CAS PubMed.
- Z. Li, R. Lin, Z. Liu, D. Li, H. Wang and Q. Li, Electrochim. Acta, 2016, 191, 606–615 CrossRef CAS.
- A. Lewalska-Graczyk, P. Pieta, G. Garbarino, G. Busca, M. Holdynski, G. Kalisz, A. Sroka-Bartnicka, R. Nowakowski, M. Naushad, M. B. Gawande, R. Zbořil and I. S. Pieta, ACS Sustainable Chem. Eng., 2020, 8, 7244–7255 CrossRef CAS.
- C. Zhai, M. Sun, L. Zeng, M. Xue, J. Pan, Y. Du and M. Zhu, Appl. Catal., B, 2019, 243, 283–293 CrossRef CAS.
- H. Wang, H. Feng and J. Li, Small, 2014, 10, 2165–2181 CrossRef CAS PubMed.
- S. Su, C. Zhang, L. Yuwen, X. Liu, L. Wang, C. Fan and L. Wang, Nanoscale, 2016, 8, 602–608 RSC.
- L. Yuwen, F. Xu, B. Xue, Z. Luo, Q. Zhang, B. Bao, S. Su, L. Weng, W. Huang and L. Wang, Nanoscale, 2014, 6, 5762–5769 RSC.
- Z. Gao, M. Li, J. Wang, J. Zhu, X. Zhao, H. Huang, J. Zhang, Y. Wu, Y. Fu and X. Wang, Carbon, 2018, 139, 369–377 CrossRef CAS.
- C. Zhai, M. Zhu, D. Bin, F. Ren, C. Wang, P. Yang and Y. Du, J. Power Sources, 2015, 275, 483–488 CrossRef CAS.
- S. Ramakrishnan, M. Karuppannan, M. Vinothkannan, K. Ramachandran, O. J. Kwon and D. J. Yoo, ACS Appl. Mater. Interfaces, 2019, 11, 12504–12515 CrossRef CAS PubMed.
- B. Tang, Y. Lin, Z. Xing, Y. Duan, S. Pan, Y. Dai, J. Yu and J. Zou, Electrochim. Acta, 2017, 246, 517–527 CrossRef CAS.
- G. Rajeshkhanna and G. Ranga Rao, Int. J. Hydrogen Energy, 2018, 43, 4706–4715 CrossRef CAS.
- S. K. Meher, P. Justin and G. Ranga Rao, Nanoscale, 2011, 3, 683–692 RSC.
- G. Rajeshkhanna, E. Umeshbabu and G. Ranga Rao, J. Colloid Interface Sci., 2017, 487, 20–30 CrossRef CAS PubMed.
- S. M. Pawara, B. S. Pawar, A. I. Inamdar, J. Kim, Y. Jo, S. Cho, S. S. Mali, C. K. Hong, J. Kwak, H. Kim and H. Im, Mater. Lett., 2017, 187, 60–63 CrossRef.
- H. S. Jadhav, A. Roy, W. J. Chung and J. G. Seo, New J. Chem., 2017, 41, 15058–15063 RSC.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Pritam
Deb
and
Pritam
Deb