
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNano-manganese oxide and reduced graphene oxide-incorporated polyacrylonitrile fiber mats as an electrode material for capacitive deionization (CDI) technology†
I. W.
Siriwardane
ab,
N. P. W.
Rathuwadu
 b,
D.
Dahanayake
b,
Chanaka
Sandaruwan
b,
Rohini M.
de Silva
a and
K. M. Nalin
de Silva
*ab
b,
D.
Dahanayake
b,
Chanaka
Sandaruwan
b,
Rohini M.
de Silva
a and
K. M. Nalin
de Silva
*ab
aCentre for Advanced Materials and Devices (CAMD), Department of Chemistry, University of Colombo, Colombo 00300, Sri Lanka. E-mail: kmnd@chem.cmb.ac.lk
bSri Lanka Institute of Nanotechnology (SLINTEC), Nanotechnology and Science Park, Mahenwatte, Pitipana, Homagama, Sri Lanka
First published on 11th March 2021
Abstract
Capacitive deionization (CDI) is a trending water desalination method during which the impurity ions in water can be removed by electrosorption. In this study, nano-manganese dioxide (MnO2) and reduced graphene oxide (rGO)-doped polyacrylonitrile (PAN) composite fibers are fabricated using an electrospinning technique. The incorporation of both MnO2 and rGO nanomaterials in the synthesized fibers was confirmed by transmission electron microscopy (TEM), scanning electron microscopy (SEM), and energy dispersive X-ray spectroscopy (EDX). The electrochemical characteristics of electrode materials were examined using cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and constant current charge–discharge cycles (CCCDs). The specific capacitance of the PAN electrode increased with increasing MnO2 and rGO contents as well as when thermally treated at 280 °C. Thermally treated composite fibers with 17% (w/w) MnO2 and 1% (w/w) rGO (C-rGOMnPAN) were observed to have the best electrochemical performance, with a specific capacitance of 244 F g−1 at a 10 mV s−1 scan rate. The electrode system was used to study the removal of sodium chloride (NaCl), cadmium (Cd2+) and lead (Pb2+) ions. Results indicated that NaCl showed the highest electrosorption (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 472 C g−1) compared to two heavy metal salts (14260 C g−1 for Pb2+ and 6265 C g−1 for Cd2+), which is most likely to be due to the ease of mass transfer of lighter Na+ and Cl− ions; When compared, Pb2+ ions tend to show more electrosorption on these fibers than Cd2+ ions. Also, the C-rGOMnPAN electrode system is shown to work with 95% regeneration efficiency when 100 ppm NaCl is used as the electrolyte. Hence, it is clear that the novel binder-free, electrospun C-rGOMnPAN electrodes have the potential to be used in salt removal and also for the heavy metal removal applications of water purification.
472 C g−1) compared to two heavy metal salts (14260 C g−1 for Pb2+ and 6265 C g−1 for Cd2+), which is most likely to be due to the ease of mass transfer of lighter Na+ and Cl− ions; When compared, Pb2+ ions tend to show more electrosorption on these fibers than Cd2+ ions. Also, the C-rGOMnPAN electrode system is shown to work with 95% regeneration efficiency when 100 ppm NaCl is used as the electrolyte. Hence, it is clear that the novel binder-free, electrospun C-rGOMnPAN electrodes have the potential to be used in salt removal and also for the heavy metal removal applications of water purification.
Introduction
With the ever-increasing need for fresh water supplies worldwide, desalination of hard and sea water has become a popular option in several countries over past few decades. Capacitive deionization (CDI) technology, first introduced by Murphy et al.1 in the 1960s, is emerging as a promising water desalination method. CDI involves the application of low voltage (typically around 1.0 V) between two oppositely charged electrodes, and when the water containing a high amount of dissolved solids is pumped through high surface area electrodes, charged ions in the water stream are attracted to electrodes with opposite charges, resulting in an effluent stream with a smaller amount of dissolved salts. Usually, CDI electrodes used are made of a porous material with high surface area; this can be either in powder or monolith form. Carbon-based powder materials used for CDI electrodes include activated carbon,2–5 carbon nanotubes6–8 and graphene derivatives;7,9–12 whereas carbon monoliths used in CDI electrodes are carbon aerogels,13,14 carbon cloth15–17 and carbon fibers.18,19 However, currently, advanced carbon materials with high nanoporosities20,21 are being tested to produce CDI electrodes with the objective of achieving higher salt removal capacities.Carbon fibers are an allotrope of carbon that are continuously being improved and tested for CDI electrode applications due to their low cost,22 ease of manufacturing23 and high power density.24 Carbon fibers can be mass fabricated using various techniques, such as electrospinning and vapour growth. Electrospun carbon fiber webs have been widely explored for their salt removal capacity in CDI applications.25,26 Further, modification of these electrospun carbon fibers in order to improve their capacitive and conductive properties and achieve higher salt removal capacities has been studied by several researchers. For example, the composite formation of electrospun carbon fibers with advanced carbon materials such as graphene,27 graphene oxide (GO),9 reduced graphene oxide (rGO),28 carbon nanotubes (CNTs)29 and carbon black19 to form hybrid carbon structures has been shown to afford carbon fibers with improved CDI performance. Out of the two most conductive counterparts, rGO and graphene, rGO remains more attractive in improving CDI electrodes due to its ease of preparation compared to graphene30 as well as due to its high surface area and high conductivity.31 Importantly, rGO itself has been widely explored as an electrode material for CDI technology.31 However, its two-dimensional (2D) structure, with smaller specific capacitance, high electrical resistance between layers and high hydrophobicity, demands further modifications for its application as an electrode material for CDI.32 Consequently, the formation of metal oxide (MO) composites of rGO has been widely explored;33 titanium dioxide (TiO2),34,35 magnetite (Fe3O4),32 tin oxide (SnO2),36 MnFe2O4 (ref. 37) and CoTiO2 (ref. 38) are some materials used for this purpose. The common rationale of improving the CDI performance of rGO electrodes by MO includes the intercalation of rGO layers by the MO nanoparticles,35,38 thereby increasing the surface area34 and specific capacitance of the composite,32,36 and improving the hydrophilicity,32,34 whereas pseudocapacitance has also been identified as a possible cause.37
On the other hand, the incorporation of MO, which possesses capacitive properties,39–41 in carbon fiber-based CDI electrodes has been shown to improve the salt removal efficiency of the carbon matrix as well. For example, carbon fiber sheets dip-coated with SiO2 and Al2O3 have been shown to possess better Ca2+ removal capacities, which is attributed to their site-specific adsorption, electrical double layer formation and faradaic reactions.42 In addition, this study shows the better electrode regeneration properties and better endurance to higher voltages of MO-incorporated carbon fiber CDI electrodes.42 In a different study, activated carbon cloth modified with metal alkoxides of titanium, silicon, aluminum and zirconium showed an increase in electric field adsorption over the physical adsorption, thereby resulting in a significant enhancement of CDI performance.15 Further, a study by Myint et al. is reported to have better CDI performance when activated carbon cloth modified with ZnO nanoparticles is used as electrodes, where it is shown that the particle size and hence the surface area of the fibers can be impacted by the formation of MO-carbon fiber hybrids.17
Manganese dioxide (MnO2) is a versatile metal oxide due to its low cost, abundance and environmental friendliness.43 Whilst MnO2 has been used in modifying CDI electrodes with carbon aerogels,44 activated carbon45 and carbon particles,46 a study by Li et al.47 shows the enhanced Ni2+ and Mn2+ removal capacity of carbon fiber paper electrodes electrodeposited with α-MnO2. The improved performance of the α-MnO2/carbon fiber paper composites was attributed to their high surface area, mesoporosity, and pore-size distribution, which allow enhanced access to ions, and the pseudocapacitive property of MnO2, which contributes to a more efficient electron/charge transfer using faradaic reactions.47
Several other studies report the use of MnO2-carbon fiber-like composite structures as sorbents for water purification applications in addition to CDI technology. A study by Chao et al. reports the preparation of a hierarchical nanofiber mat of polyacrylonitrile and polypyrrole by electrospinning that is subsequently treated with KMnO4 solution to deposit MnO2 and used as adsorbent for Pb2+ removal from aqueous solution.48 Further, polydopamine-assisted MnO2-loaded PAN fibers have been shown to possess higher adsorption capacity for Pb2+ ion removal applications.49 A study by Lee et al. presents the use of hierarchically structured PAN/MnO2/graphene fibers to form heteroatom-rich carbon fibers, thereby obtaining considerably high power and energy densities for energy storage applications.50 However, a PAN fiber-based nanocomposite doped with both rGO and MnO2 nanoparticles as a potential CDI electrode material has not been reported to date.
The work reported here includes the fabrication of a PAN fiber matrix which is simultaneously doped with rGO and MnO2 nanoparticles using an electrospinning method. The electrodes developed using these nanofiber matrices were analyzed to evaluate not only their chemical and structural properties, but also their electrochemical properties, namely specific capacitance, electrochemical impedance and galvanostatic charge–discharge cycles, in order to understand their potential to be used as CDI electrode materials in future. We believe that the precursor fiber material we developed in this work will set the base for developing functionalized carbon fibers to be used in CDI applications in future.
Results and discussion
Characterization of MnO2 and rGO
Fig. S1a and b† show SEM images of the MnO2 nanoparticles and rGO, respectively. According to the image, aggregates of MnO2 nanoparticles with a nearly spherical morphology and an average size of around 90–100 nm can be seen. Generally, nanoparticle aggregation at 1–100 nm length scales occurs due to Brownian motion, resulting in ‘local aggregates’ with typical sizes of 0.1–10 μm.51 However, the TEM image obtained for MnO2 (Fig. 1a) clearly shows the continuity of interconnected particles similar to birnessite, an allotrope of MnO2 that contains infinite two-dimensional sheets of edge-shared [MnO6] octahedra.52 The intensity profile of the cross section of the zoomed area of the HRTEM (given in the inset of Fig. 1b) shows that the interatomic layer distance is 3.16 Å (Fig. 1c).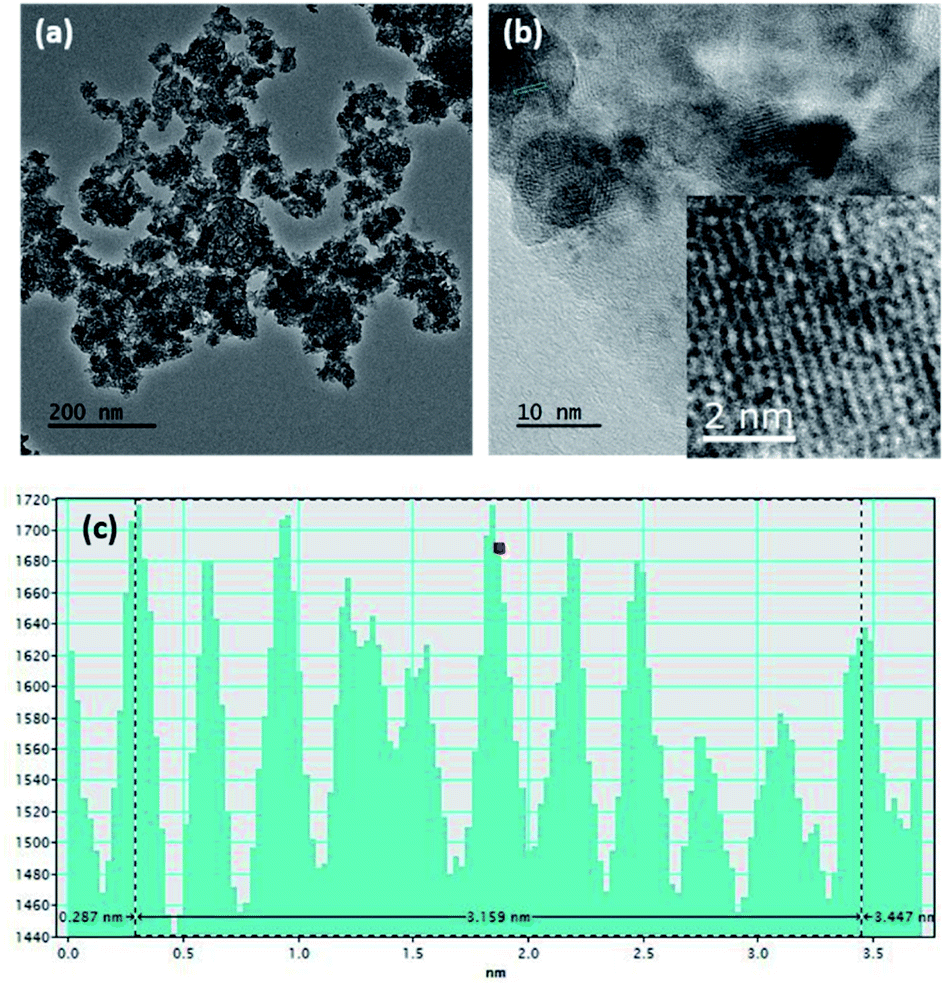 | ||
| Fig. 1 (a) TEM image, (b) HRTEM image, and (c) intensity profile along the area given in the inset of (b) of the MnO2 nanostructures. | ||
In order to confirm the oxidation state of the synthesized manganese oxide nanoparticles, an XPS study was carried out. Fig. S2† shows the XPS spectra recorded for the synthesized amorphous manganese oxide nanoparticles. The Mn (2p3/2) peak of synthetic manganese oxide consists of a narrow peak with a maximum at 642.05 eV and a distinct shoulder at 643 eV, which is characteristic of MnO2 nanoparticles, and a broad shoulder at 644.3 eV. However, there is also a long binding energy shoulder at 640.6 eV that cannot be attributed to Mn4+ species, but is rather assigned to Mn3+. The presence of such Mn3+ species is possible because the synthesis procedure involves the oxidation of Mn2+ species to form Mn4+ nanoparticles, as described in previous literature.53 Further, the Mn (2p1/2) peak is observed at 653.8 eV with a splitting of 11.75 eV with the Mn (2p3/2) peak. In the literature, a splitting energy around 11 eV is reported for Mn4+ species,54,55 and this further confirms that the synthesized nanoparticles contain MnO2 species. The O (1s) of the synthesized MnO2 nanoparticles has a maximum at 529.65 eV and two shoulders at high binding energies of 531.5 eV and 533 eV. Overall, the binding energies recorded in the XPS data matched more closely with the reported XPS data of birnessite, which is an allotrope of MnO2 with a layered structure53,55 and good supercapacitive properties.56
The sheet separation of rGO used for the fiber synthesis can be clearly observed in the SEM image (Fig. S1b†). Further analysis of the chemical structure of rGO was performed using Raman spectroscopy. Raman spectroscopy is generally used to characterize graphite-related materials such as rGO. In the Raman spectrum of a graphite-related material, two prominent peaks are observed, which are typically known as the G band and D band. The G band appears at around 1580 cm−1 and is attributed to in-plane vibrational stretching of sp2 hybridized carbon. More specifically, it is due to a doubly degenerate phonon mode with E2g symmetry at the Brillouin zone center.57 The D band appears at around 1348 cm−1 and originates due to a doubly resonant disorder-induced mode with A1g symmetry; this is indicative of the amount of defects in the material, which arises due to covalent bonds of carbon atoms with oxygen or other functional groups.58 Hence, the intensity ratio of the D and G bands (ID/IG) is used as a measure of the extent of defects present in the structure. Typically, the ID/IG ratio is around 0.13 for graphite and around 0.99 and 1.07 for graphene oxide and reduced graphene oxide, respectively.57 According to the Raman spectrum, the rGO sample used for this study shows an ID/IG ratio of 1.02 (Fig. S3†), implying the presence of a higher number of defects; in other words, the functional groups have been introduced into the structure by the thermal treatment process that occurs during the synthesis. In addition to the G and D bands, two weak peaks at ∼2718 cm−1 (2D-band) and ∼2906 cm−1 (G + D) were observed. The 2D-band is regarded as an overtone of the D-peak, whereas the G + D band is attributed to the combination of D and G peaks. Hence, the Raman spectrum confirms the chemical characteristics of the rGO used for the study and the successful reduction process used for its synthesis.
The survey scan of the XPS spectrum of rGO showed two prominent peaks for C and O at 285 and 533 eV, respectively (Fig. S4a†). The deconvolution of the C 1s spectrum of rGO (Fig. S4b†) shows characteristic peaks corresponding to sp2 hybridized C (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), sp3 hybridized C (C–C and C–H), C–O and CO at the binding energies of 285 eV, 286 eV, 288 eV and 289 eV, respectively, which agree with previously reported data.59 Further, the deconvoluted O 1s XPS spectra (Fig. S4c†) show peaks corresponding to C–O and CO functionalities at 533 eV and 531 eV, respectively. According to the XPS analysis, the total C content was 85% in rGO, whereas that of O was 15% (Table S1 in the ESI section†).
C), sp3 hybridized C (C–C and C–H), C–O and CO at the binding energies of 285 eV, 286 eV, 288 eV and 289 eV, respectively, which agree with previously reported data.59 Further, the deconvoluted O 1s XPS spectra (Fig. S4c†) show peaks corresponding to C–O and CO functionalities at 533 eV and 531 eV, respectively. According to the XPS analysis, the total C content was 85% in rGO, whereas that of O was 15% (Table S1 in the ESI section†).
The XRD pattern of pure PAN exhibits two crystalline peaks at the 2θ values of 17° and 26.5°, which correspond to the (100) and (002) planes of the hexagonal lattice of polyacrylonitrile.60 The XRD pattern of the rGOMnPAN fibers also has a very similar pattern to that of PAN, as the incorporation of amorphous MnO2 and rGO material in the fiber structure is not visible in the XRD pattern. However, upon carbonization, the peak observed for the PAN structures at 26.5° intensified, implying the increased crystallization of the PAN fibers due to the formation of cyclic structures, as shown in Fig. S5 in the ESI section.† The broad peak that developed at the 2θ range of 10°–25° provides evidence to support that complete polymerization of the nitrile groups has occurred, resulting in a primarily amorphous polyimine residue.61
The SEM image of the MnPAN fibers (Fig. 3a) shows diameters even smaller than 100 nm; however, fibers with a broad diameter range were also observed. The fibers grew broader in order to hold MnO2 nanoparticles within the fibers; this was further clarified using EDX measurements, as given in Fig. 4d. Fig. 3b–d show the morphological characteristics of the rGOMnPAN and C-rGOMnPAN fibers. As can be seen in the SEM image (3b), the fibers have an average diameter of around 200 nm. However, as is visible in the TEM image (Fig. 4), the fiber diameters are broadly distributed and contain some beaded structures. Fig. 4b and c clearly show the incorporation of MnO2 and rGO into the composite fiber structure. As can be seen in Fig. 3c and d, the fiber diameters further reduced during the thermal stabilization, thereby showing diameters less than 100 nm. This is a common feature observed for PAN-based fibers, where upon heating to 280 °C, the ring cyclization of nitrile groups occurs by evolving H2 gas (Fig. S5†). Further, the EDX spectra (Fig. 4d) show the elemental composition of fibers to contain Mn and O concentrated at the same overlapping sites, thereby confirming the immobilization of MnO2 nanoparticles in the fiber structures.
 | ||
| Fig. 2 X-ray diffraction patterns of rGO, the MnO2 nanoparticles, PAN fibers, 1% rGOMnPAN fibers and C-rGOMnPAN fibers. | ||
 | ||
| Fig. 3 SEM images of (a) 17% MnPAN fibers, (b) 1% rGO17%MnPAN (rGOMnPAN), (c) and carbonized rGOMnPAN (C-rGOMnPAN); (d) enlarged image of the selected area in (c). | ||
 | ||
| Fig. 4 TEM images of the (a) fiber morphology, (b) embedded MnO2 nanoparticles, (c) embedded rGO, (d) EDX elemental mapping data and EDX spectrum of the rGOMnPAN fibers. | ||
Structural characterization of the PAN-fiber based CDI electrode material
Fig. 2 compares the XRD patterns of the C-rGOMnPAN and rGOMnPAN fibers with those of the MnO2 nanoparticles, rGO and pure PAN fibers. As can be seen, neither the MnO2 nor the rGO nanomaterial shows distinguished peaks in the 2θ range of 5° to 45°, confirming their amorphous characteristics. In previous studies reported in the literature, the observation of no peaks in the XRD pattern of rGO was attributed to the higher degree of exfoliation of the rGO structure.62,63 Also, as-prepared hydrated MnO2 powders are generally reported to have amorphous structures in many of the cases reported in the literature,52 and this study accounts for a similar observation. The capacitive properties of MnO2 nanoparticles have been proven to be at their maximum in the amorphous state and tend to reduce with increasing carbonization temperature as the carbonization process reduces the surface area, the secondary pores and the water content in the nanomaterial structure.52 Hence, the as-synthesized amorphous MnO2 nanoparticles were immobilized in the PAN fibers by the electrospinning process, with the ultimate target of using them in capacitive desalination applications. The surface area and pore structure are also vital factors for the ion adsorption and capacitive performance of an electrode material. The surface area of C-rGOMnPAN calculated using the BET isotherm in the relative pressure (P/P0) range of 0.09–0.3 was 1108 m2 g−1. The total pore volume calculated at the P/P0 of 0.99 was 0.85 cm3 g−1. Moreover, according to the water absorption studies, the time required to absorb a definite amount of water by the C-rGOMnPAN mat is highly dependent on the thickness of the material (Table S2 in the ESI section†). However, the average time taken to absorb an amount of 50 μL of distilled water remains less than 10 seconds (Fig. S7 in the amended ESI section†). This observation confirms the hydrophilic nature of this novel CDI electrode material, ensuring its potential to be used in water purification applications.Electrochemical characterization of PAN fiber-based CDI electrodes
The electrochemical characterization of the electrospun, binder-free, PAN-based electrodes included cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and constant current charge–discharge (CCCD) cycles.Capacitance
Fig. 5a, b and c demonstrate the CV of the electrodes at a scan rate of 10 mV s−1 with variation of the MnO2 and rGO content and the effect of carbonization, respectively. Fig. 5d, e and f summarize the variations of the specific capacitance values at scan rates of 10, 50, and 200 mV s−1 for varying MnO2 and rGO contents and the effect of carbonization, respectively. The CV data showing the variations at different scan rates for a given sample are summarized in the ESI section.† Fig. S8† shows the CV curves of the binder-free, as-spun 0%-MnPAN (pure PAN), 17%-MnPAN, 33%-MnPAN and 50%-MnPAN fiber electrodes, measured at the scan rates of 10, 50 and 200 mV s−1. Similarly, Fig. S9 and S10† demonstrate the CVs at scan rates of 10, 50 and 200 mV s−1 for varying rGO contents (0.2% and 1%) and thermal conditions (room temperature and 280 °C), respectively. The CV cycles observed for the four systems with varying MnO2 content (Fig. S8†) were rectangular in shape and symmetrical with respect to the X axis, indicating the capacitive nature of the process and, hence, a reliable process within the selected operating voltages. However, at the high scan rate of 200 mV s−1, the rectangular shape becomes slightly slanted due to the ohmic resistance of the electrolyte motion affecting the double layer formation. Similar trends were observed when varying the rGO content (Fig. S9†) and thermal conditions (Fig. S10†). As shown in Fig. 5a, the incorporation of MnO2 increased the capacitance of the PAN fibers, and the effect increased with increasing MnO2 content. This observation can be attributed to the increase of the electrosorption of ions by the charged fiber structures due to the abundance of metal oxide functionalities.64 The incorporation of metal oxide nanoparticles into electrospun fibers can increase the surface area of the fibers65 as well as the available functional groups present in the fibers. These new functional groups introduced into the structure can form intermediate bonds between water molecules and the porous sorbent material, thereby allowing better contact between water and the fiber adsorbent.66Fig. 5d shows the specific capacitance values at different scan rates when varying the MnO2 content. For instance, at 10 mV, the specific capacitance values are 8.8, 31.2, 56.7 and 133.3 F g−1 and x for the 0%, 17%, 33% and 50% samples, respectively. A similar trend can be seen at other scan rates. Fig. 5b shows the CV curves of binder-free, as-spun 17%-MnPAN fiber electrodes with 0%, 0.2% and 1% doped rGO measured at 10 mV s−1. Even though the 0.2% rGO does not show a significant improvement in the capacitive performance of MnPAN electrodes, rGO as a 1% dopant tripled the capacitive performance of the electrode. This can be attributed to the increase of the conductivity and the surface area of the fiber structure by the rGO sheets, as described in previous studies.31Fig. 5e shows the specific capacitance values at different scan rates when varying the rGO content. For instance, at 10 mV, the specific capacitance values are 31.2, 27.9 and 87.3 F g−1 for the samples with 0%, 0.2%, and 1% rGO. A similar trend can be seen at other scan rates. It is evident that the effect of 1% rGO is prominent at all scan rates compared to that of 0.2% rGO. Fig. 5c shows the CV curves of the carbonized 17%-MnPAN fiber electrode (CMnPAN) and the carbonized 17%-MnPAN electrode with 1% rGO (C-rGOMnPAN), measured at 10 mV s−1. Both the CMnPAN and C-rGOMnPAN electrodes show increased capacitance values compared to their uncarbonized counterparts (17% MnPAN and rGOMnPAN); this can be attributed to the increase of the conductivity of the fiber mat due to the structural changes of the polymer that occur during the carbonization, as explained later. Fig. 5f shows the specific capacitance values at different scan rates when varying the thermal conditions. For instance, at 10 mV, the specific capacitance values are 244.4 and 43.4 F g−1 for the C-rGOMnPAN and CMnPAN samples. A similar trend can be seen at other scan rates. The effect of thermal treatment was significant at all scan rates. The capacitance value of 244 F g−1 at a scan rate of 10 mV s−1 reported for the C-rGOMnPAN electrode is higher than the value reported previously for GO-embedded carbon nanofibers,67 which was calculated to be 151.6 F g−1 using charge discharge curves obtained at 0.5 A g−1, and that for an MnO2-doped nanoporous carbon composite,68 which was 204.7 F g−1 at a scan rate of 1 mV s−1.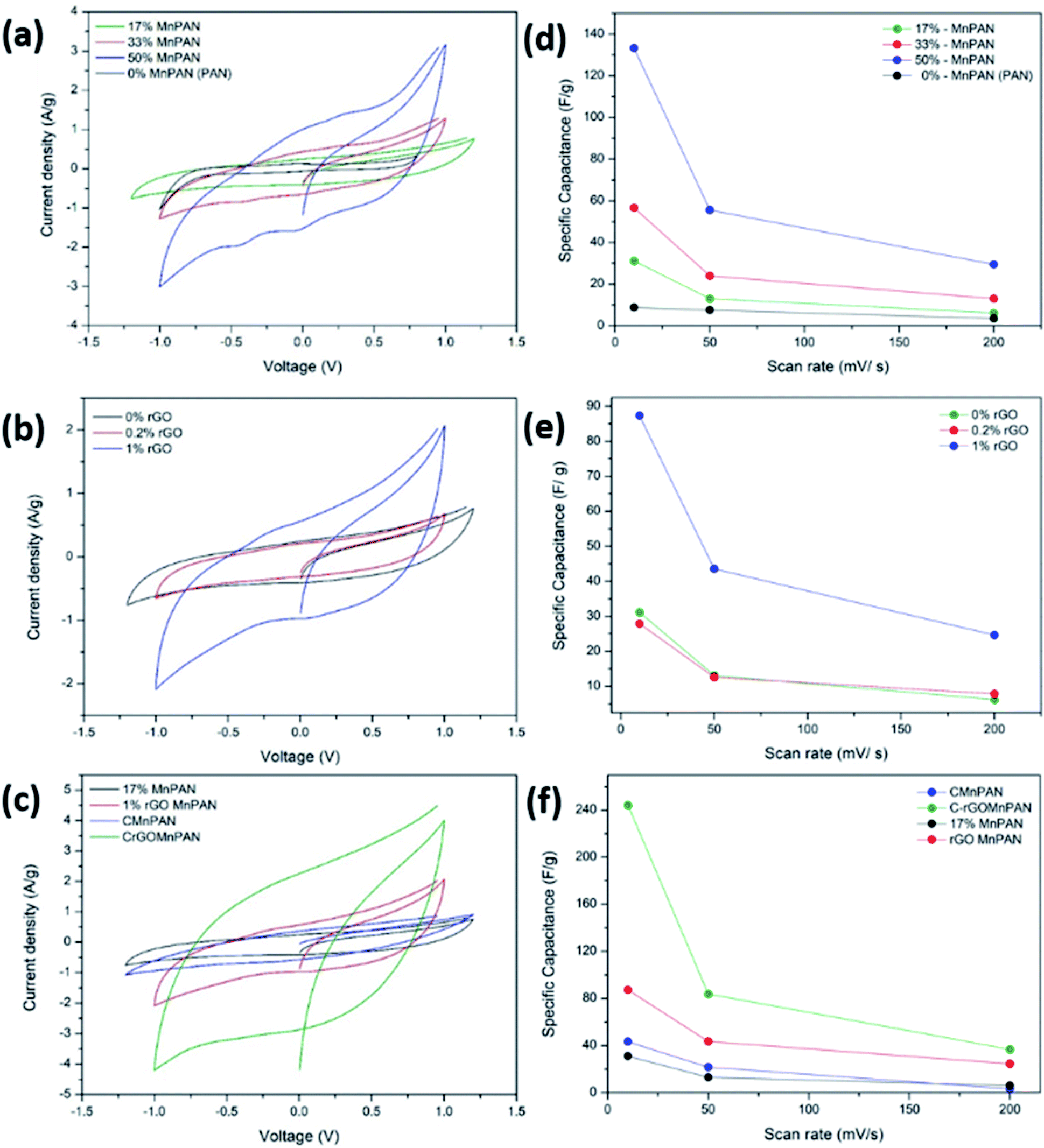 | ||
| Fig. 5 Cyclic voltammograms at 10 mV s−1 in degassed 0.1 M KNO3 using an Ag/AgCl reference electrode and Pt sheet counter electrode recorded for materials coated on graphene sheets with varying (a) MnO2 contents of 17%, 33%, 50%, and 0%, (b) rGO contents of 0.2%, 1%, and 0% and (c) thermal stabilization conditions at room temperature and 280 °C. The variation of the specific capacitance values at the scan rates of 10, 50 and 200 mV s−1, with varying (d) MnO2 contents of 17%, 33%, 50%, and 0%, (e) rGO contents of 0.2%, 1%, and 0% and (f) thermal stabilization conditions at room temperature and 280 °C. | ||
It was observed that carbonization increased the capacitance of both the CMnPAN and C-rGOMnPAN electrodes. This can be attributed to the formation of cyclic structures from the nitrile groups during the thermal stabilization of PAN fibers around 280 °C, evolving N2 gas.69 This cyclic structure with conjugated imine bonds can be considered to be more favourable in electron transportation than the pure PAN structure. The effect is significant in the C-rGOMnPAN electrode due to the presence of rGO, which has a similar structure to cyclized PAN. The general trend is that the capacitance shown by the electrodes at low scan rates remains higher than that recorded at high scan rates. This can be attributed to the fact that at low scan rates, it has sufficient time for the charging and discharging processes; therefore, the electrical double layer formation is more effective and vice versa. Additionally, the MnPAN system preserved its rectangular shape in a broader sweep potential range compared to the pure PAN fibers. The “rectangular” shape of the CV curve is indicative of the capacitive properties of the electrode under study. For ideal capacitors, the CV curve is almost rectangular in shape, and any deviation from the rectangular shape is considered to be due to charging resistance.70 The specific capacitance values were calculated using eqn (1), where the specific capacitance is C, average capacitive current is I, time spent for the scan is Δt, electrode active material mass is m and voltage window is ΔV. Table 1 summarizes the calculated specific capacitance values for each sample at a 10 mV s−1 scan rate.
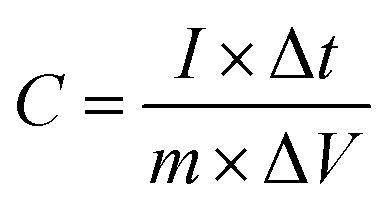 | (1) |
| Sample | Specific capacitance (F g−1) @ 10 mV s−1 |
|---|---|
| 0%-MnPAN (PAN) | 9 |
| 17%-MnPAN | 31 |
| 33%-MnPAN | 57 |
| 50%-MnPAN | 133 |
| 0.2%-rGO-17%MnPAN | 28 |
| 1%-rGO-17%MnPAN | 87 |
| CMnPAN (17% MnO2) | 43 |
| C-rGOMnPAN | 244 |
Furthermore, it is important to note the potential range in the CVs in Fig. 5a–c and S8–S10† spans up to +1.2 V to −1.2 V, ensuring that no prominent water oxidation and reduction occur in the said potential range. Therefore, it is anticipated that the electrode material can be used for electroanalytical applications in aqueous solution that require operations in a wider potential window.
Impedance
The total impedance and the phase shifts are plotted as a function of frequency in the Bode plots. Considering the Bode phase plot, if the phase change is zero, the presence of a resistive element is considered. The existence of a capacitor element is reflected by observing a −90° phase shift, and a phase change below 90° is believed to arise due to the presence of a constant phase element (CPE); this usually arises due to imperfect capacitors and inhomogeneity of the electrode material, such as surface roughness and porosity.71 The plot of the real part of the impedance against the imaginary part gives the Nyquist plot. Here, the Nyquist plots were used to derive the equivalent circuits and, thereby, the interpretations of the kinetically and diffusionally controlled processes. Fig. 6a summarizes the Bode total impedance plots of the 17% MnPAN, 1% rGOMnPAN and C-rGOMnPAN electrodes, and Fig. 6b summarizes the Bode phase plots of the same electrodes.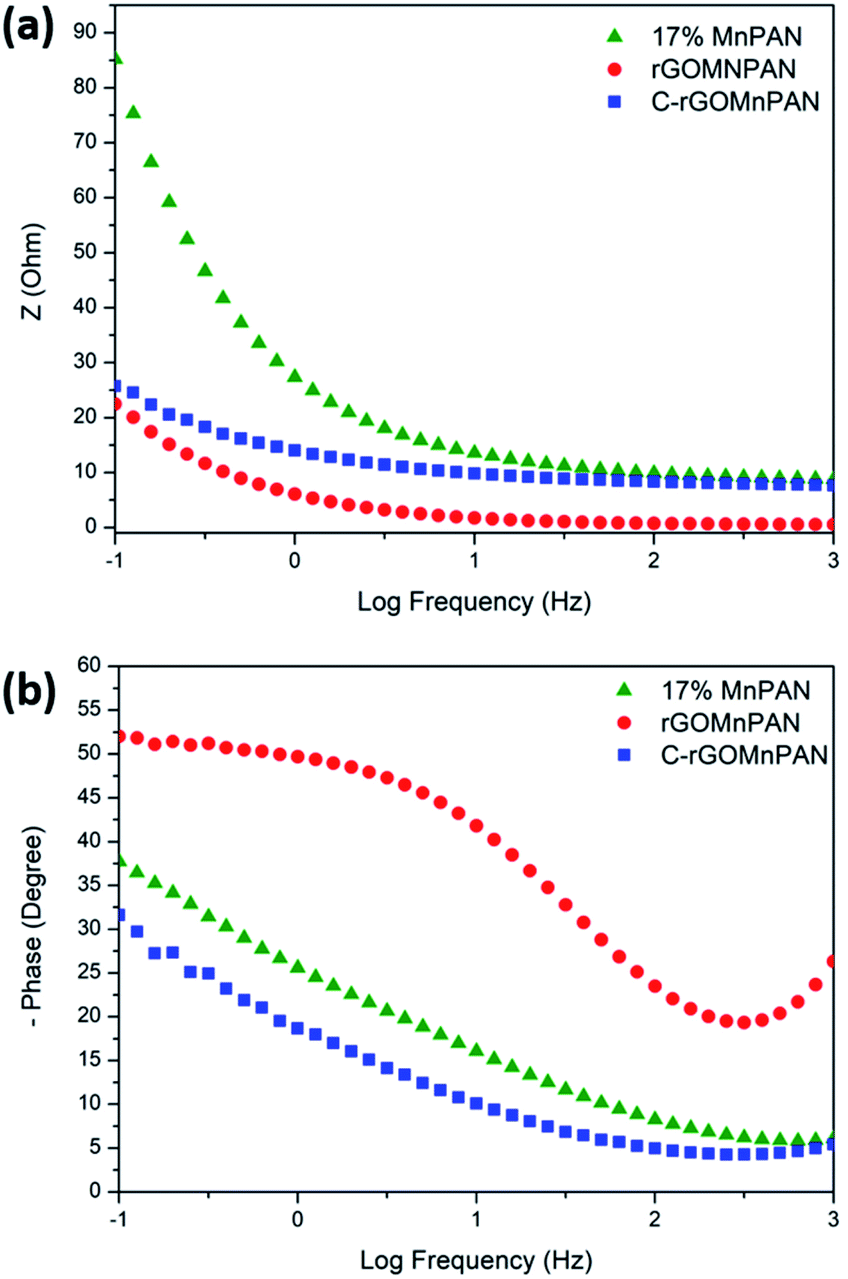 | ||
| Fig. 6 (a) Bode total impedance plots and (b) Bode phase plots of the 17% MnPAN, 1% rGOMnPAN and C-rGOMnPAN electrodes. | ||
The As shown in Fig. 6a, the total impedance at a lower frequency range, which corresponds to the capacitive behavior of the material, is significantly reduced upon addition of rGO compared to 17% MnPAN. An interesting observation is that with the introduction of rGO into the fiber structures, the impedance is reduced to a value very close to zero. This can be attributed to the inherited vacant π electron cloud of the reduced graphene structure, which affects the electrical conductance properties of rGO. Similar effects in increasing the electrical conductivity in polyvinyl alcohol72 and polyethylene73 polymer matrixes have been reported previously. Hence, the incorporation of rGO into MnPAN polymer structures is beneficial for their use in CDI-based applications. As shown in Fig. 6b, the phase change of 0° to −20° in the high frequency region gradually reaches −50 to −30 in the low frequency region. The 0° to −20° phase change at high frequency can be attributed to the resistive behavior of the material. The −50° to −30° phase change at low frequency can be attributed to the capacitive behavior of the material. According to the Bode phase plot (Fig. 6b), as none of the systems show a phase change of −90° in the low frequency region, these systems can be interpreted as imperfect capacitors with distorted capacitive behavior due to factors such as non-homogeneity or surface roughness of the fabricated electrodes. The resistive behavior of the two electrodes, 17% MnPAN and C-rGOMnPAN, are prominent at higher frequencies.
The Nyquist plot (Fig. 7a) shows the prominent capacitive behavior of the materials upon addition of MnO2 and rGO. As shown in Fig. 7a, lowered resistance with the addition of rGO (rGOMnPAN) compared to 17% MnPAN can be observed at high frequency. However, C-rGOMnPAN has increased resistance, and this could be due to poor attachment of the fiber mat on the current collector. These material exhibits diffusion and/or capacitive behavior in the low frequency range, which is advantageous in using this material as an electrode material in CDI technology. Equivalent circuit fitting was performed to understand the charging/discharging behavior of the materials. The equivalent circuits were predicted for the 17% MnPAN (Fig. 7b), rGOMnPAN (Fig. 7a), and C-rGOMnPAN electrodes (Fig. 7b), with goodness of fit (χ2) values of 0.005103, 0.01237, and 0.01132, respectively.
 | ||
| Fig. 7 (a) Nyquist plots of the 17% MnPAN, 1% rGOMnPAN and C-rGOMnPAN electrodes. (b) Equivalent circuit of the 17% MnPAN and C-rGOMnPAN electrodes. (c) Equivalent circuit of the 1% rGOMnPAN electrode. | ||
The equivalent circuit in Fig. 7b consists of a series resistor, capacitor, and constant phase element, which was the general type of circuit obtained for all the electrode materials except for the rGO-doped materials, which corresponds to the general behavior of a supercapacitor. The resistor corresponds to the equivalent series resistance of the system due to electrode material and connections. The capacitor corresponds to the capacitive nature of the material. The constant phase element (N ∼ 0.6) is attributed to the imperfect capacitive nature due to surface effects from roughness and inhomogeneity. The rGO-doped electrode material has an equivalent circuit with an R//C parallel component in series with a constant phase element (Fig. 7c). The series constant phase element (N ∼ 0.6) can be attributed to the combination of equivalent series resistance due to the polyacrylonitrile-based electrode material and connections and the imperfect capacitive behavior due to surface roughness and inhomogeneity. The capacitor element corresponds to the capacitive nature of the electrode material. The presence of R//C indicates that a prominent charge transfer process occurs upon addition of rGO. Resistance in R//C can be considered as the charge transfer resistance.
CDI performance
CDI performance was determined by CCCD cycles and CDI measurements carried out as described in Sections 2.5 and 2.6, respectively. For the CDI performance determination, the C-rGOMnPAN electrode was used, as it showed the highest capacitance value when studied by CV. Fig. 8a shows the CCCD cycles observed for the C-rGOMnPAN electrode using degassed 0.1 M KNO3 using a three-electrode system. The average equivalent series resistance (ESR) value of the C-rGOMnPAN electrode for five repetitive cycles was 21.9 Ω at 5 A g−1 current density. Fig. 8b shows the CDI performance of the C-rGOMnPAN electrode and Fig. 8c shows the total accumulated charge for each electrolyte for five repetitive cycles measured using the three electrolytes: 100 ppm NaCl, 100 ppm Cd(NO3)2 and 100 ppm Pb(NO3)2. The charge densities on the C-rGOMnPAN electrode after the third charging cycle (at 313 s) when using NaCl, Cd(NO3)2 and Pb(NO3)2 electrolytes are 21798, 14678 and 6474 C g−1, respectively. The highest electrosorption was observed for NaCl compared to the two heavy metal salts. The most likely reason for this is the ease of mass transfer of lighter Na+ and Cl− ions compared to Cd2+ and Pb2+. The electrosorption capacity of NaCl calculated based on the charge accumulation on the electrodes is 6 mg g−1, which is comparable to those of some similar systems reported previously for CDI applications.67,68,74 When comparing the two heavy metals, Pb2+ ions tend to show more electrosorption on C-rGOMnPAN fibers than Cd2+ ions. Previously, MnO2 nanoparticles and its composites have been used to absorb both Pb2+ (ref. 72) and Cd2+.75,76 When simultaneous absorption of Pb2+ and Cd2+ by MnO2 occurs, less Cd2+ adsorption occurs because Cd2+ has less physisorption ability than Pb2+, which mainly adsorbs via a physisorption mechanism.77 In addition, retarded sorption of Cd2+ has been observed in the presence of other co-ions, such as Ca2+ and Mg2+.78 The system was shown to operate with >95% regeneration efficiency with respect to accumulation throughout the five repeated cycles and release of the charge (Fig. 8b) for all three heavy metal cations. Hence, it is clear that the proposed binder-free, electrospun MnPAN electrodes have potential to be used in desalination as well as in heavy metal removal applications.
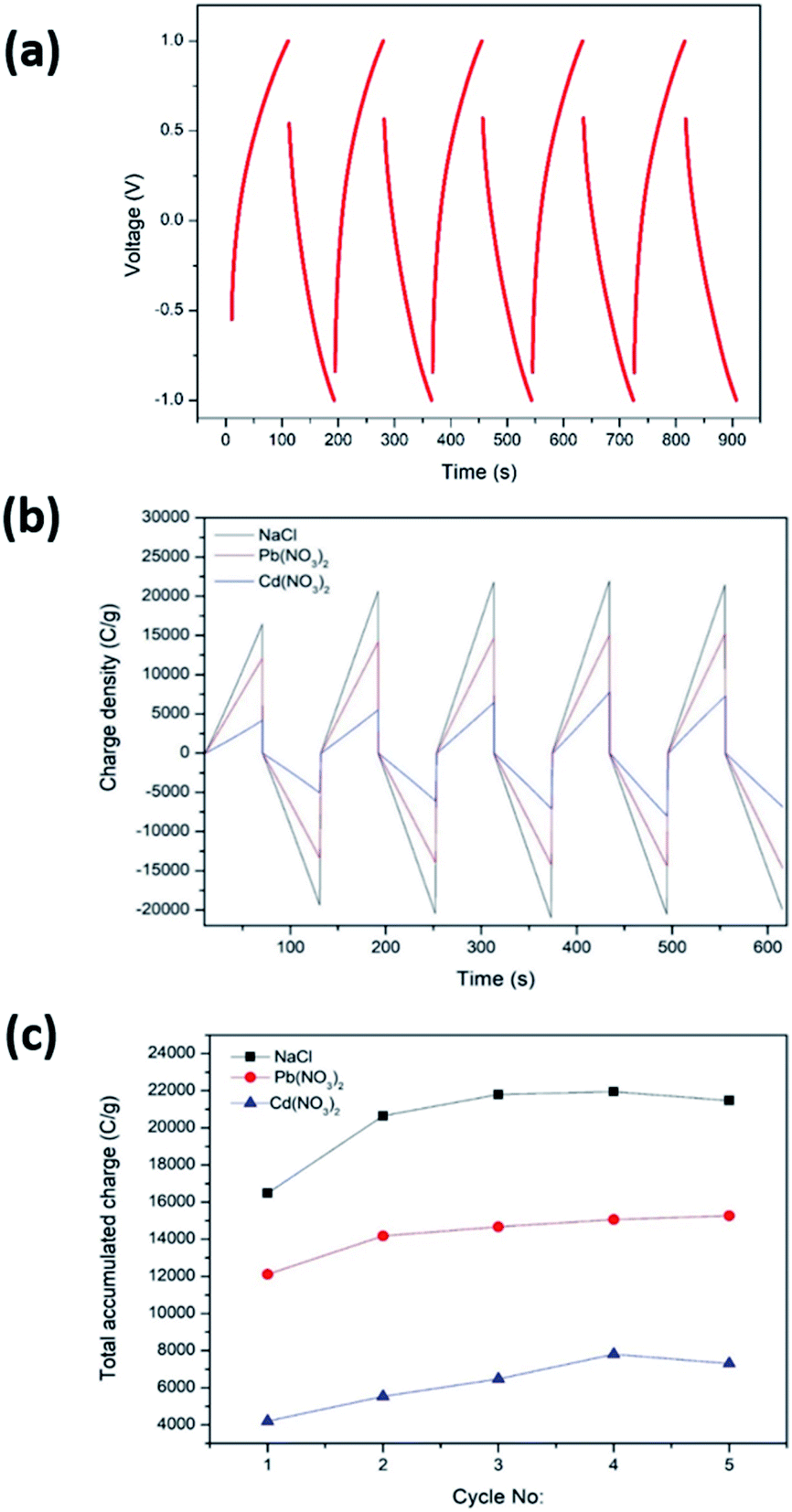 | ||
| Fig. 8 (a) CCCD cycles in degassed 0.1 M KNO3 using C-rGOMnPAN-deposited graphene sheets as the working electrode, Ag/AgCl as the reference electrode and the Pt sheet as the counter electrode and (b) CDI performance using 100 ppm NaCl, 100 ppm Pb(NO3)2 and 100 ppm Cd(NO3)2 electrolytes; (c) salt removal capacity of the C-rGOMnPAN electrode. | ||
Experimental
Materials
All chemicals used in this study were of analytical grade. For the fabrication of binder-free electrospun electrodes, polymer suspensions of polyacrylonitrile (150000 g mol−1, PAN, Sigma Aldrich) in dimethylformamide (99.5%, DMF, Sigma Aldrich) were used. The current collector used was a graphite sheet (thickness 0.25 mm, Shenzhen Union Tenda Technology Co. Ltd). Potassium permanganate (99%, 158.03 g mol−1, Central Drug House), potassium iodide (99.8%, 166.00 g mol−1, Sri Chem) and sodium dodecyl sulphate (288.37 g mol−1, Merck SDS) surfactant were used for the synthesis of manganese oxide nanoparticles. The rGO used for the study was obtained from Ceylon Graphene Technology Lanka Pvt. Ltd (CGTL). Electrolyte solutions used for electrochemical characterization and CDI measurements were potassium nitrate (≥99.5%, 101.10 g mol−1, Technopharma Chem), sodium chloride (AR 99.9%, 58.44 g mol−1, Technopharma Chem), lead nitrate (>99%, 331.21 g mol−1, Sigma Aldrich) and cadmium nitrate tetrahydrate (Cd(NO3)2·4H2O, 99%, Riedel-de-Haen). All aqueous solutions were prepared using deionized water (EVAQUA, <0.055 μS cm−1).
Synthesis of MnO2 nanoparticle
Potassium permanganate (1 g) was dissolved in 100 mL of pure water to which SDS was added, and the mixture was stirred for 30 min. To the dissolved mixture, KI (1 g) was added dropwise with stirring, and the stirring was continued for 30 more min. The black precipitate obtained was washed with pure water to remove any impurities and was dried in an oven at 105 °C.Preparation of polymer suspensions for electrospinning
The main paragraph text follows directly here. The PAN/DMF solutions with polymer loadings of 5% (w/v) and 1% (w/v) of MnO2 nanoparticles (MnPAN) were prepared. The fibers prepared by these suspensions (denoted as MnPAN) contained a theoretical maximum of 17% (w/w) of MnO2 nanoparticles in the final fiber (17% MnPAN). To prepare the polymer suspensions, first, the PAN in DMF solution was stirred at 400 rpm at 90 °C for 30 min until a transparent polymer solution was obtained. For the preparation of polymer suspensions with various concentrations of metal oxide-PAN, a known weight of nanoparticles was dispersed in a known amount of DMF by stirring for 15 min followed by ultrasonication (UP400S Hielscher™ probe tip sonicator with a diameter of 1.3 cm, operating at 430 W power output at the frequency of 24 kHz) for 15 min. Then, concentrated PAN in DMF solution (more than 5% w/v) was added to the nanoparticle dispersion until a final polymer suspension was achieved with a total solid content of 6% (w/v). The final mixture was sonicated for another 30 min. In preparing MnPAN polymer suspensions with 33% (w/w) (33% MnPAN) and 50% (w/w) (50% MnPAN) MnO2 in the final dry composite fiber, 2% (w/v) and 3% (w/v) MnO2 in 4% (w/v) and 3% (w/v) PAN were suspended in 10 mL of DMF, respectively. For the preparation of rGO-doped MnPAN fibers (denoted as rGOMnPAN), 0.01% (w/v) and 0.1% (w/v) rGO were added to polymer suspensions containing 5% (w/v) PAN and 1% (w/v) MnO2 (17% MnPAN). The two polymer suspensions prepared in this manner contained 0.2% (w/w) (0.2% rGOMnPAN) and 1% (w/w) (1% rGOMnPAN) of rGO, respectively, in the dry fiber form.Synthesis of fibers
The main paragraph text follows directly on here. The fiber synthesis followed the electrospinning of the prepared polymer suspensions. The polymer solutions prepared as described above were loaded in a 5 mL syringe equipped with a 0.3 mm (23 G) stainless steel needle. The needle was connected to a high voltage power supply (MATSUSADA Precision Inc.). The grounded counter electrode was a flat piece of aluminum foil. The solution was constantly and controllably supplied using a syringe pump (New Era Pump systems, NE 300). The electrospinning process was carried out at 25 °C and ∼55% relative humidity. The relative humidity in the chamber containing the setup was controlled by purging compressed air. The fibers of each suspension were spun at 25 kV at a polymer flow rate of 10 μL min−1 and a working distance of 20 cm. The fiber material was deposited on a graphite sheet, which was used as the current collector of the electrodes. The fibers with optimized MnO2 and rGO (17% MnO2 and 1% rGO) were thermally stabilized at 280 °C in air (denoted as C-rGOMnPAN). For the comparison of the effects of the thermal stabilization of the fibers on the capacitance properties, thermally stabilized MnPAN fibers (denoted as CMnPAN) were also prepared under the same conditions used for C-rGOMnPAN.Characterization of MnO2 nanoparticles, rGO and electrospun fibers
The morphological characteristics of rGO, synthesized MnO2 nanoparticles and composite fibers of MnO2 + rGO + PAN (rGOMnPAN) were studied using SEM (Hitachi SU 6600) and TEM (JEOL JEM 2100 HRTEM). Both MnO2 nanoparticles and rGO were further analyzed using XPS (Thermo Fischer ESCALAB Xi+ with Al-Kα radiation). Raman spectroscopy (Bruker Senterra) of rGO was also carried out using a 532 nm wavelength in order to obtain more details about the chemical structure. XRD (Bruker D8 FOCUS with Cu-Kα radiation with X-ray operating at 25 kV and 30 mA) analyses were performed for all materials in order to obtain an idea about the fiber constituents and crystallinity. EDX (Ametek-EDAX EDX detector) analyses were also carried out to ensure the incorporation of the nanomaterial within the fiber structures. The surface area and the total pore volume of the C-rGOMnPAN material were investigated by N2 adsorption at 77 K (Quantachrome Autosorb iQ Surface Area Analyzer). The sample was degassed at 120 °C for 30 min prior to analysis. To understand the hydrophilic/hydrophobic nature of the electrospun electrode material, the time to completely absorb a known amount (50 and 100 μL added using a micropipette) of distilled water by fiber mats with known thicknesses (thickness measured using a thickness gauge – SYLVAC μs229) were measured.Electrochemical characterization of the fibers
The electrodes with various amounts of MnO2 and rGO were characterized electrochemically with cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and constant current charge–discharge cycles (CCCD). Autolab PGSTAT 302N was used to obtain the measurements. A three-electrode system was used for the initial characterization, where the MnO2 and MnO2/rGO-doped PAN fiber mats deposited on graphite current collectors were used as the working electrodes, Ag/AgCl electrode was used as the reference electrode and Pt sheet electrode was used as the counter electrode. The electrolyte used for the characterization of electrodes was 0.1 M KNO3 solution prepared using deionized water, which was purged with N2 gas to remove dissolved O2 traces prior to each measurement. The potential windows used for CV measurements were between 1.2 V and −1.2 V at 10, 50 and 200 mV s−1 scan rates. The EIS was performed in the frequency range of 1000–0.1 Hz at an AC perturbation of ±10 mV amplitude for each sample. During the EIS experiments, no DC bias was applied. The CCCD was carried out at 0.5, 1, 3, and 5 A g−1 current densities for each sample within the respective potential ranges derived from the CV.CDI performance of C-rGOMnPAN electrodes
In order to assess the CDI performance of C-rGOMnPAN electrodes (with 17% (w/w) MnO2 and 1% (w/w) rGO, also thermally stabilized), the CCCD measurements were conducted in a two-electrode configuration at a constant voltage of (±)1.0 V by sandwiching two electrodes with a cellulose-based aqueous electrolyte spacer in between the two electrodes. The electrolyte solutions used were 100 ppm NaCl, 100 ppm Cd(NO3)2 and 100 ppm Pb(NO3)2. The spacer was soaked in the respective electrolyte prior each measurement.Conclusions
In this work, rGO and nano-MnO2-doped novel PAN fiber composite was synthesized for the first time. The incorporation of nano-MnO2 and rGO in the electrospun PAN fiber structure is well confirmed by the TEM and EDX analyses. The fiber composite can be applied in water desalination, and removing other prominent heavy metals via the capacitive deionization technique was explored. The capacitive properties were studied with varying MnO2 contents, rGO contents, and thermal conditions. The specific capacitance of the PAN electrode increased with increasing MnO2 and rGO contents as well as when thermally treated at 280 °C. The PAN fibers with optimized nano-MnO2 content were chosen as 17% (w/w), which were then further doped with 1% (w/w) reduced graphene oxide (rGO) to improve their electrical properties. The introduction of rGO significantly improved the capacitive properties and reduced the electrical resistance of the system. Composite fibers with 17% (w/w) MnO2 and 1% (w/w) rGO were observed to have the best electrochemical performance, with a specific capacitance of 244 F g−1 at a scan rate of 10 mV s−1. The electrode material was used to study the removal of NaCl, Cd(II) and Pb(II) ions. The results indicated that NaCl shows higher electrosorption compared to the two heavy metal salts. Of the two heavy metal ions, Pb(II) and Cd(II), Pb(II) tends to show more electrosorption on these fibers. Also, the MnPAN electrode system has been shown to work with 95% regeneration efficiency when 100 ppm NaCl is used as the electrolyte. Therefore, this study managed to deliver a novel binder-free electrode material for CDI application for water desalination.Author contributions
IWS conceptualised this work, conducted methodology and wrote the manuscript. NW conducted methodology and edited the manuscript. DD conducted the TEM analysis and CS conducted XPS analyses. WRMDS and KMNDS supervised the work and edited the manuscript.Conflicts of interest
There are no conflicts to declare.References
- G. W. Murphy and D. D. Caudle, Electrochim. Acta, 1967, 12(12), 1655–1664 CrossRef CAS.
- R. Broséus, J. Cigana, B. Barbeau, C. Daines-Martinez and H. Suty, Desalination, 2009, 249(1), 217–223 CrossRef.
- Y.-J. Kim, J. Hur, W. Bae and J.-H. Choi, Desalination, 2010, 253(1), 119–123 CrossRef CAS.
- L. Zou, G. Morris and D. Qi, Desalination, 2008, 225(1), 329–340 CrossRef CAS.
- G. Wang, B. Qian, Q. Dong, J. Yang, Z. Zhao and J. Qiu, Sep. Purif. Technol., 2013, 103, 216–221 CrossRef CAS.
- H. Li and L. Zou, Desalination, 2011, 275(1), 62–66 CrossRef CAS.
- Y. Wimalasiri and L. Zou, Carbon, 2013, 59, 464–471 CrossRef CAS.
- S. Vafakhah, G. J. Sim, M. Saeedikhani, X. Li, P. Valdivia y Alvarado and H. Y. Yang, Nanoscale Adv., 2019, 1(12), 4804–4811 RSC.
- Y. Bai, Z.-H. Huang, X.-L. Yu and F. Kang, Colloids Surf., A, 2014, 444, 153–158 CrossRef CAS.
- B. Jia and L. Zou, Carbon, 2012, 50(6), 2315–2321 CrossRef CAS.
- H. Li, L. Zou, L. Pan and Z. Sun, Sep. Purif. Technol., 2010, 75(1), 8–14 CrossRef CAS.
- H. Li, L. Pan, C. Nie, Y. Liu and Z. Sun, J. Mater. Chem., 2012, 22(31), 15556–15561 RSC.
- T. J. Welgemoed and C. F. Schutte, Desalination, 2005, 183(1), 327–340 CrossRef CAS.
- J. C. Farmer, T. D. Tran, J. H. Richardson, D. V. Fix, S. C. May and S. L. Thomson, The application of carbon aerogel electrodes to desalination {ampersand} waste treatment, Lawrence Livermore National Lab, CA, USA, 1997, p. 13, medium, ED, other: FDE: PDF, PL Search PubMed.
- M.-W. Ryoo and G. Seo, Water Res., 2003, 37(7), 1527–1534 CrossRef CAS.
- M. W. Ryoo, J. H. Kim and G. Seo, J. Colloid Interface Sci., 2003, 264(2), 414–419 CrossRef CAS PubMed.
- M. T. Z. Myint, S. H. Al-Harthi and J. Dutta, Desalination, 2014, 344, 236–242 CrossRef CAS.
- H. Li, L. Pan, Y. Zhang, L. Zou, C. Sun, Y. Zhan and Z. Sun, Chem. Phys. Lett., 2010, 485(1), 161–166 CrossRef CAS.
- G. Wang, Q. Dong, Z. Ling, C. Pan, C. Yu and J. Qiu, J. Mater. Chem., 2012, 22(41), 21819–21823 RSC.
- B. Han, G. Cheng, Y. Wang and X. Wang, Chem. Eng. J., 2019, 360, 364–384 CrossRef CAS.
- Y. Wu, G. Jiang, G. Liu, G. Lui, Z. P. Cano, Q. Li, Z. Zhang, A. Yu, Z. Zhang and Z. Chen, J. Mater. Chem. A, 2019, 7(26), 15633–15639 RSC.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5(1), 72–88 RSC.
- Y. He, W. Chen, C. Gao, J. Zhou, X. Li and E. Xie, Nanoscale, 2013, 5(19), 8799–8820 RSC.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7(5), 1597–1614 RSC.
- M. Wang, Z.-H. Huang, L. Wang, M.-X. Wang, F. Kang and H. Hou, New J. Chem., 2010, 34(9), 1843–1845 RSC.
- G. Wang, C. Pan, L. Wang, Q. Dong, C. Yu, Z. Zhao and J. Qiu, Electrochim. Acta, 2012, 69, 65–70 CrossRef CAS.
- G. Wang, Q. Dong, T. Wu, F. Zhan, M. Zhou and J. Qiu, Carbon, 2016, 103, 311–317 CrossRef CAS.
- Q. Dong, G. Wang, B. Qian, C. Hu, Y. Wang and J. Qiu, Electrochim. Acta, 2014, 137, 388–394 CrossRef CAS.
- Q. Dong, G. Wang, T. Wu, S. Peng and J. Qiu, J. Colloid Interface Sci., 2015, 446, 373–378 CrossRef PubMed.
- Z. Li, B. Song, Z. Wu, Z. Lin, Y. Yao, K.-S. Moon and C. P. Wong, Nano Energy, 2015, 11, 711–718 CrossRef CAS.
- T. N. Tuan, S. Chung, J. K. Lee and J. Lee, Curr. Appl. Phys., 2015, 15(11), 1397–1401 CrossRef.
- N. T. Trinh, S. Chung, J. K. Lee and J. Lee, J. Energy Chem., 2016, 25(3), 354–360 CrossRef.
- A. S. Yasin, A. Y. Mohamed, I. M. A. Mohamed, D.-Y. Cho, C. H. Park and C. S. Kim, Chem. Eng. J., 2019, 371, 166–181 CrossRef CAS.
- H. M. Moustafa, M. Obaid, M. M. Nassar, M. A. Abdelkareem and M. S. Mahmoud, Sep. Purif. Technol., 2020, 235, 116178 CrossRef CAS.
- A. G. El-Deen, J.-H. Choi, C. S. Kim, K. A. Khalil, A. A. Almajid and N. A. M. Barakat, Desalination, 2015, 361, 53–64 CrossRef CAS.
- S. K. Sami, J. Y. Seo, S.-E. Hyeon, M. S. A. Shershah, P.-J. Yoo and C.-H. Chung, RSC Adv., 2018, 8(8), 4182–4190 RSC.
- H. Younes, F. Ravaux, N. El Hadri and L. Zou, Electrochim. Acta, 2019, 306, 1–8 CrossRef CAS.
- H. M. Moustafa, M. M. Nassar, M. A. Abdelkareem, M. S. Mahmoud and M. Obaid, J. Environ. Chem. Eng., 2019, 7(6), 103441 CrossRef CAS.
- C. An, Y. Zhang, H. Guo and Y. Wang, Nanoscale Adv., 2019, 1(12), 4644–4658 RSC.
- M. Cui and X. Meng, Nanoscale Adv., 2020, 2(12), 5516–5528 RSC.
- A. S. Yasin, I. M. A. Mohamed, M. T. Amen, N. A. M. Barakat, C. H. Park and C. S. Kim, J. Alloys Compd., 2019, 772, 1079–1087 CrossRef CAS.
- J. J. Wouters, J. J. Lado, M. I. Tejedor-Tejedor, R. Perez-Roa and M. A. Anderson, Electrochim. Acta, 2013, 112, 763–773 CrossRef CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40(3), 1697–1721 RSC.
- S. Hand and R. D. Cusick, Environ. Sci. Technol., 2017, 51(20), 12027–12034 CrossRef CAS PubMed.
- P. J. Walker, M. S. Mauter and J. F. Whitacre, Electrochim. Acta, 2015, 182, 1008–1018 CrossRef CAS.
- G. Tan, S. Lu, N. Xu, D. Gao and X. Zhu, Environ. Sci. Technol., 2020, 54(9), 5843–5852 CrossRef CAS PubMed.
- P. Li, Y. Gui and D. J. Blackwood, ACS Appl. Mater. Interfaces, 2018, 10(23), 19615–19625 CrossRef CAS PubMed.
- C. Luo, J. Wang, P. Jia, Y. Liu, J. An, B. Cao and K. Pan, Chem. Eng. J., 2015, 262, 775–784 CrossRef CAS.
- Y. Li, R. Zhao, S. Chao, B. Sun, C. Wang and X. Li, Chem. Eng. J., 2018, 344, 277–289 CrossRef CAS.
- D. G. Lee, Y. A. Kim and B.-H. Kim, Carbon, 2016, 107, 783–791 CrossRef CAS.
- M. Carpineti, F. Ferri, M. Giglio, E. Paganini and U. Perini, Phys. Rev. A, 1990, 42(12), 7347–7354 CrossRef CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40(3), 1697–1721 RSC.
- H. W. Nesbitt and D. Banerjee, Am. Mineral., 1998, 83(3–4), 305 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257(7), 2717–2730 CrossRef CAS.
- H. Z. Chi, S. Yin, D. Cen, K. Chen, Y. Hu, H. Qin and H. Zhu, J. Alloys Compd., 2016, 678, 42–50 CrossRef CAS.
- S. Zhu, W. Huo, X. Liu and Y. Zhang, Nanoscale Adv., 2020, 2(1), 37–54 RSC.
- J. H. Warner, F. Schäffel, A. Backmatiuk and M. R. Rummeli, Graphene: Fundamentals and emergent applications, Elsevier, USA, 1st edn, 2013 Search PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143(1), 47–57 CrossRef CAS.
- K. Dave, K. H. Park and M. Dhayal, RSC Adv., 2015, 5(116), 95657–95665 RSC.
- C. Pirlot, I. Willems, A. Fonseca, J. B. Nagy and J. Delhalle, Adv. Eng. Mater., 2002, 4(3), 109–114 CrossRef CAS.
- N. Chatterjee, S. Basu, S. K. Palit and M. M. Maiti, J. Polym. Sci., Part B: Polym. Phys., 1995, 33(12), 1705–1712 CrossRef CAS.
- N. Morimoto, T. Kubo and Y. Nishina, Sci. Rep., 2016, 6, 21715 CrossRef CAS PubMed.
- K. Zhang, Y. Zhang and S. Wang, Sci. Rep., 2013, 3, 3448 CrossRef PubMed.
- M. W. Ryoo, J. H. Kim and G. Seo, J. Colloid Interface Sci., 2003, 264(2), 414–419 CrossRef CAS.
- M. T. Z. Myint, S. H. Al-Harthi and J. Dutta, Desalination, 2014, 344, 236–242 CrossRef CAS.
- L. Zou, G. Morris and D. Qi, Desalination, 2008, 225(1), 329–340 CrossRef CAS.
- Y. Bai, Z.-H. Huang, X.-L. Yu and F. Kang, Colloids Surf., A, 2014, 444, 153–158 CrossRef CAS.
- J. Yang, L. Zou, H. Song and Z. Hao, Desalination, 2011, 276(1), 199–206 CrossRef CAS.
- M. S. A. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92(8), 1421–1432 CrossRef CAS.
- B.-H. Park, Y.-J. Kim, J.-S. Park and J. Choi, J. Ind. Eng. Chem., 2011, 17(4), 717–722 CrossRef CAS.
- A. Eftekhari, J. Mater. Chem. A, 2018, 6(7), 2866–2876 RSC.
- F. Adib, A. Bagreev and T. Bandosz, Langmuir, 2000, 16(4), 1980–1986 CrossRef CAS.
- H.-x. Zhang, J.-H. Park and K.-B. Yoon, Compos. Sci. Technol., 2018, 154, 85–91 CrossRef CAS.
- G. Wang, Q. Dong, Z. Ling, C. Pan, C. Yu and J. Qiu, J. Mater. Chem., 2012, 22(41), 21819–21823 RSC.
- J. Liang, X. Li, Z. Yu, G. Zeng, Y. Luo, L. Jiang, Z. Yang, Y. Qian and H. Wu, ACS Sustainable Chem. Eng., 2017, 5(6), 5049–5058 CrossRef CAS.
- S. Mallakpour and F. Motirasoul, Ultrason. Sonochem., 2017, 37, 623–633 CrossRef CAS PubMed.
- Z. Huang, L. Lu, Z. Cai and Z. J. Ren, J. Hazard. Mater., 2016, 302, 323–331 CrossRef CAS.
- L. Dong, Z. Zhu, H. Ma, Y. Qiu and J. Zhao, J. Environ. Sci., 2010, 22(2), 225–229 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na01075h |
| This journal is © The Royal Society of Chemistry 2021 |