
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled anionic and cationic Au nanoparticles with Au nanoclusters for the exploration of different biological responsiveness in cancer therapy†
Jingjing
Yang
a,
Lu
Zhang
b,
Qian
Zhou
c,
Fan
Chen
d,
Martina
Stenzel
 d,
Fucheng
Gao
a,
Chao
Liu
e,
Huiqing
Yuan
*b,
Hui
Li
*a and
Yanyan
Jiang
*afg
d,
Fucheng
Gao
a,
Chao
Liu
e,
Huiqing
Yuan
*b,
Hui
Li
*a and
Yanyan
Jiang
*afg
aKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials, Ministry of Education, Shandong University, Jinan, Shandong, China. E-mail: lihuilmy@hotmail.com; yanyan.jiang@sdu.edu.cn
bInstitute of Medical Sciences, The Second Hospital of Shandong University, 247 Beiyuan Dajie Street, Jinan, Shandong, China. E-mail: lyuanhq@sdu.edu.cn
cDepartment of Obstetrics, Shandong Provincial Hospital Affiliated to Shandong First Medical University, 324 Jingwu Street, Jinan, Shandong, China
dCentre for Advanced Macromolecular Design (CAMD), School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia
eDepartment of Oromaxillofacial Head and Neck Oncology, Shanghai Ninth People's Hospital, College of Stomatology, Shanghai Jiao Tong University School of Medicine, Shanghai, China
fSuzhou Institute of Shandong University, Room 522, Building H of NUSP, NP.388 Ruoshui Road, SIP, Suzhou, Jiangsu, China
gShenzhen Research Institute of Shandong University, Shenzhen, Guangdong, China
First published on 8th March 2021
Abstract
Self-assembly overcomes the biodegradation resistance of some traditional inorganic drug carriers. Herein, we prepared self-assembled Au nanocluster-based nanoparticles with different sizes and charges based on solvent- and cation-induced self-assembly nanotechnology as anti-cancer drug vehicles to solve the potential metabolism problems of solid gold nanoparticles. We also systematically explored the responsiveness of cancer cells to self-assembled Au nanocluster-based nanoparticles with different sizes and surface modified properties. We discovered that self-assembled nanoparticles inherited molecular-like properties of small-size Au NCs and exhibited an aggregation-induced emission (AIE) phenomenon with intense luminescence. Self-assembled Au nanocluster-based nanoparticles (Au NPs and cAu NPs) taking advantage of their size and positive charge exhibited better cell uptake than Au NCs. Encouraged by the excellent biological compatibility and cell uptake of these nanomaterials, we prepared drug-loaded nanomaterials by diffusion absorption and hydrophobic-induced embedding. cAu NPs@DOX showed an excellent anti-cancer effect owing to efficient cell internalization; Au NPs@DOX exhibited slow release of cargo drugs which might be significant to in vivo drug delivery. This work plays a crucial role in the rational design of self-assembled multifunctional gold-based nanoparticles in the application of nanomaterial-assisted multifunctional drug delivery systems (DDSs).
1 Introduction
The precise release of cargo anti-cancer drugs at specific lesions is the main focus of nanomaterial-assisted drug delivery systems (DDSs).1 It has been demonstrated that the proliferation of tumor cells induces malformation around tissues, such as the irregular shape of blood vessels and the loose arrangement of endothelial cells, providing an opportunity to develop specific treatment options.2–4 Based on these features, various nanoparticles have been designed to improve the efficacy of anticancer drugs. For examples, hydrophilic Au nanoparticles (Au NPs) with suitable sizes can take advantage of the enhanced permeability and retention (EPR) effect to passively target tumor tissues, thus achieving site-specific drug delivery and precise drug release.5 However, Au NPs are difficult to completely metabolize and remain in metabolic organs with non-negligible burdens due to their large size.6Bovine serum albumin stabilized gold nanoclusters (BSA-Au NCs) with much smaller sizes than that of Au NPs have received increasing attention since they were first reported in 2009.7,8 The properties of size-dependent fluorescence, well-defined surface modification, large stokes shift and unique structural characteristics make Au NCs more suitable for applications as molecular fluorescence nanoprobes and drug delivery vehicles.9,10 Ultra-small Au nanoclusters have great potential to be wholly metabolized thanks to their small size (<10 nm).11,12 However, they cannot meet the size requirements of passive targeting for precise drug release. Fortunately, given the advancement of nanotechnology, nanomaterial-assisted DDSs can be designed purposefully with their unique physical and chemical properties, to integrate bio-sensing, bio-imaging and drug delivery for diagnosis, and monitoring and treatment of diseases simultaneously in biomedical fields.13–15 Solvent- and cation-induced self-assemblies as emerging nanotechnologies are of great significance to fabricate particles with a larger size to achieve passive targeting via the EPR effect, while causing fewer side-effects to normal cells or tissues.16 Nanoparticles prepared from Au NCs by self-assembly nanotechnology are believed to be promising vehicles that can better adapt to tumor microenvironments with the targeting effect.
The prerequisite for the widespread applications of nanomaterials is that they possess basic biological properties such as non-toxicity, high biosafety and excellent biocompatibility.17 Thus, the most urgent issue of nanomaterials is toxicological studies in a biological system. In addition, an in-depth understanding of the relationship between the unique properties of nanomaterials and their effects on cancer cells is of great significance for a rational design. For example, the surface charge and size of Au NPs are generally considered as essential factors in determining their biological activities.18
In this regard, the influence of the size and surface charge of Au NPs on toxicological effects is also worth investigating. Several studies have provided evidence that various Au NPs with the desired size are non-toxic when taken up by cells.19 Shukla et al.20 focused on the stress-induced immunological response of macrophage cells to Au NPs, and proved the biocompatibility of Au NPs. Yu et al.21 investigated the size-dependence cytotoxicity of water-soluble triphenylphosphine derivative stabilized Au NPs, demonstrating that 15 nm Au NPs were relatively non-toxic while 1.4 nm ones showed more sensitive cellular response with different uptake kinetics. Also, Au NPs with a size of 50 nm have shown the maximum cellular uptake without cytotoxicity.22 However, Pernodet et al.23 revealed that Au NPs showed adverse influences on cell proliferation via induction of actin stress fibers disappearing after internalized by fibroblasts. Ma et al.24 found out that Au NPs could induce size-dependent Golgi rupture and cause Golgi disordering, leading to abnormal protein processing with Golgi dysfunction. Therefore, the size effect of nanoparticles causes a paradox toxic effect and affects the cell uptake kinetics and clearance mechanism.25 Besides, the electrostatic interaction between nanomaterials and the cell membrane also affects the cellular uptake and viability. Generally, the surface of the cell membrane is slightly negatively charged, expelling anionic nanoparticles from entering the cell, leading to a lower cellular uptake of these nanoparticles.26 Cationic nanoparticles, on the other hand, are prone to binding to cell membranes to enhance cell internalization due to electrostatic interactions between them. However, cationic nanoparticles are reported to cause more damage to the plasma membrane and multiple organelles than anionic ones.27 Wang et al.28 also found that the surface charge of nanoparticles greatly influenced in vivo distribution and excretion, and cationic nanoparticles may cause a transient cytotoxic effect on metabolic systems. Hence, the investigation of the effects of the size and surface charge of Au NPs on cytotoxicity and cellular uptake behaviors towards cancer cells is helpful to rationally design nanomaterial-assisted DDSs.
Herein, we prepared biofunctional Au nanoparticles with different sizes and surface charges based on the aforementioned solvent- and cation-induced self-assembly nanotechnology. BSA-Au NCs were designed as self-assembling precursors to meet metabolic requirements. Anionic Au nanoclusters (Au NCs), self-assembled anionic Au nanoparticles (Au NPs) and cationic Au nanoparticles (cAu NPs) were evaluated through extensive characterization of physicochemical properties and consequential behaviors in biological systems. The effects of the size and surface charge of self-assembled nanomaterials on cancer cells were explored. Furthermore, the drug delivery ability and potential applications of self-assembled nanomaterial-assisted DDSs were evaluated in cancer chemotherapy. It is of great value in the future evaluation of this type of nanoparticle from performance to applications in the biomedical industry.
2 Experimental section
2.1 Materials
All glassware and stirring magnets were washed with aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 volume ratio = 3:1) and rinsed with ultrapure water thoroughly. Gold(III) chloride trihydrate (HAuCl4·3H2O, Macklin, China), bovine serum albumin (BSA, Macklin, China), sodium hydroxide (NaOH, Macklin, China), ethanol (Macklin, China), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, Macklin, China), ethylenediamine (EDA, Macklin, China), doxorubicin hydrochloride (DOX·HCl, Macklin, China), triethylamine (TEA, Macklin, China), dimethyl sulfoxide (DMSO, Macklin, China) and tetrahydrofuran (THF, Macklin, China) were used as received.
:HNO3 volume ratio = 3:1) and rinsed with ultrapure water thoroughly. Gold(III) chloride trihydrate (HAuCl4·3H2O, Macklin, China), bovine serum albumin (BSA, Macklin, China), sodium hydroxide (NaOH, Macklin, China), ethanol (Macklin, China), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, Macklin, China), ethylenediamine (EDA, Macklin, China), doxorubicin hydrochloride (DOX·HCl, Macklin, China), triethylamine (TEA, Macklin, China), dimethyl sulfoxide (DMSO, Macklin, China) and tetrahydrofuran (THF, Macklin, China) were used as received.
2.2 Preparation of BSA-stabilized Au nanoclusters
BSA-stabilized Au nanoclusters were synthesized following the mature method reported by Xie et al.8 Firstly, 10 mL of aqueous HAuCl4 (10 mM) was added to 10 mL of aqueous BSA solution (50 mg mL−1) under vigorous stirring at 37 °C. After two minutes, 1 mL of aqueous NaOH solution (1 M) was added to the mixture to adjust the pH to 12.0. Then, the temperature was raised to 70 °C and the above mixture was allowed to incubate for 60 min under vigorous stirring. The solution color changed from bright yellow to light brown and finally to deep brown, indicating the formation of Au NCs. The solution of Au NCs was entirely dialyzed against ultrapure water using dialysis tubing (MWCO = 14000 Da) for 48 h and the ultrapure water was changed every 4 h to removed excessive HAuCl4 and NaOH.
2.3 Preparation of self-assembled anionic Au nanoparticles
Self-assembled anionic Au NPs were fabricated following a previously reported method.29 Briefly, 3.5 mL of ethanol was added dropwise into 4 mL purified BSA-stabilized anionic Au NC solution via an automatic injection pump at a speed of 1 mL min−1 under stirring at 750 rpm. After 2 h, the mixture was purified by dialysis against pure water.2.4 Preparation of self-assembled cationic Au nanoparticles
BSA-stabilized Au NPs were cationized by modifying the known methods to obtain cationic Au NPs.30 4 mL of pure Au NC solution was added into ethylenediamine–HCl solution (0.5 M, 5 mL, and pH 4.75) under stirring at 750 rpm at room temperature. After being stabilized for 30 s, 10 mg of EDC powder was added into the above mixture and reacted for 20 min to stabilized the cationic Au NPs. The solution was dialyzed against ultrapure water to remove non-reacted EDA and EDC in dialysis tubing (MWCO = 14000 Da).
2.5 Preparation of doxorubicin-loading nanomaterials
In order to neutralize DOX·HCl, 50 mg of DOX·HCl was dissolved in a solution mixture of 20 mL THF and 200 μL triethylamine (TEA). The mixture was then stirred for 3 hours in the dark. The solvent was removed using a Rotavapor (RE-201D, China) to obtain base-free DOX powder. The DOX powder was dissolved in dimethyl sulfoxide (DMSO, 20 mg mL−1) for further use.31To prepare DOX-loaded Au NCs and cationic BSA-Au NPs, DOX solution (25 μL, 20 mg mL−1) was added dropwise to 4 mL dialyzed Au NC solution and cationic Au NP solution, respectively. The solutions were then stirred at 750 rpm and kept in the dark for 3 h. DOX-loaded anionic Au NPs were fabricated by DOX induced self-assembly of Au NCs. DOX solution (25 μL, 20 mg mL−1) was added dropwise into 4 mL dialyzed Au NC solution under stirring at 750 rpm and stabilized for 3 h at room temperature in the dark. Then, 3.5 mL of ethanol was added dropwise into the mixture at a speed of 1 mL min−1. After that, the mixture was stirred for 2 h to form self-assembled anionic Au NPs.
To remove free DOX completely in minimum time and avoid over-dialysis, drug-loaded nanomaterial solutions were split into 5 aliquots. Each aliquot was purified by dialysis against ultrapure water in dialysis tubing (MWCO = 14000 Da) for 6 h. Water was changed every 2 h.
2.6 Evaluation of the drug-loading ability of three types of nanomaterials
To quantify the DOX loading capacity,29 dialyzed drug-loaded samples were lyophilized (SCIENTZ, China) to obtain solid powders of DOX-loaded Au NCs, DOX-loaded anionic Au NPs and DOX-loaded cationic Au NPs. Each powder was dissolved in 2 mL of DMSO/ethanol (9:1, v/v) and sonicated (SB-5200DT, China) for 10 min respectively to extract DOX completely. After that, the suspension was centrifuged at 9000 rpm for 20 min (TG16-WS, China) to separate the supernatant and precipitate completely. The supernatant was collected for drug-loading capacity examination. The concentration of DOX was measured by using a UV-vis spectrophotometer (Specord 200 plus, Germany). The maximum absorption of DOX was examined at 480 nm and calculated reference standard curve. The E.E. and L.C. were calculated using the formulations:| Encapsulation efficiency E.E. (%, w/w) = entrapped DOX weight/total DOX weight × 100 |
| Loading capacity L.C. (%, w/w) = entrapped DOX weight/total nanoparticle weight × 100 |
2.7 TEM characterization of the three types of nanomaterials
The purified Au NC, anionic Au NP and cationic Au NP solutions were added dropwise on a copper grid three times for three replicates and dried under infrared light for 30 min. The morphology of Au NC, Au NP and cAu NP samples was observed by high-resolution transmission electron microscopy (HRTEM) (JEM-2100, Japan) and transmission electron microscopy (TEM) (HT-7700, Japan), respectively.2.8 Size and zeta potential characterization of the three types of nanomaterials
The particle size and zeta potential of three purified aqueous solutions of nanomaterials were measured by dynamic light scanning (DLS) (Nano ZS, UK).2.9 Luminescence-property characterization of the three types of nanomaterials
The purified Au NC, anionic Au NP and cationic Au NP solutions were lyophilized (SCIENTZ-10N, China). 20 mg of lyophilized powder was dissolved in 5 mL ultrapure water and sonicated for 20 min (SB-5200 DT, China) to dissolve completely. The fluorescence spectra of the three samples were recorded by using a fluorescence spectrometer (FLS1000, UK). The excitation spectra of the samples were obtained with an emission wavelength of 654 nm, and emission spectra were obtained with an excitation wavelength of 505 nm. Digital photographs of the three samples were obtained under ultraviolet light (MODEL CM-10A, USA). The measurements were performance at least three times.2.10 UV-vis spectral characterization of the three types of nanomaterials
15 mg lyophilized powder of the three samples was dissolved in 3 mL ultrapure water completed and diluted to 0.5 mg mL−1 for detection. The UV-vis spectra were obtained by using a UV-vis spectrophotometer (Specord 200 plus, Germany). The measurements were performance at least three times.2.11 FTIR spectrum of the three types of nanomaterials
Trace lyophilized powders were mixed in potassium bromide pellets to perform Fourier transform infrared spectroscopy (FTIR) (Tensor 37, Germany) in the range of 400 to 4000 cm−1. The measurements were performance at least three times.2.12 SDS-PAGE of the three types of nanomaterials
SDS-PAGE of the three types of nanomaterials was carried out according to the manufacture protocol in the presence of 12% SDS-polyacrylamide gel.2.13 Cytotoxicity assay (MTT)
Briefly, cancer cells were seeded in 96-well plates with a cell density of 5 × 103 per well in 200 μL of culture medium and incubated at 37 °C overnight to allow cells to settle. After that, the cells were treated with different drug solutions. After 48 h incubation, the culture medium was replaced by serum-free media, and then 20 μL of MTT (5 mg mL−1) was added to each well and incubated for another 4 h. Then, the solution in each well plate was discarded, and 150 μL of dimethyl sulphoxide (DMSO) was added to dissolve formazan crystals, and the absorbance of DMSO solution was measured at 570 nm with a microplate reader (Infinite M200 Pro, Switzerland).The relative cell viability (%) was calculated by using the following equation:
| Cell viability (%) = [(ODsample − ODblank)/(ODcontrol − ODblank)] × 100% |
2.14 Cellular uptake experiments
Confocal laser scanning microscopy (CLSM, ZEISS LSM800, Germany) and flow cytometry (NovoCyte 3130) were used to evaluate the cellular uptake of the nanoparticles. A549 and HCT116 cells were cultured in 6-well plates with a cover glass in each well (1 × 105 cells per well). The cells were incubated for 12 h at 37 °C and 5% CO2. After that, the cells were treated with different nanomaterials at 2 mg mL−1 for 4 h. Then, the culture medium was discarded, and the cells were washed with PBS (pH 7.4) three times to remove excess nanomaterials. After that, the cells were stained with DAPI (or Hoechst) before images were taken by CLSM.To enhance the sensitivity of the flow cytometry experiment, fluorescein FITC (5 μL, 7.0340 mg mL−1 in DMSO) was added to each equal volume of nanomaterials (12 mL, 1 mg mL−1) under stirring at 400 rpm for 2 h in an ice bath. Then, the samples were placed at 4 °C overnight. Each sample was purified by dialysis against ultrapure water in dialysis tubing (MWCO = 14000 Da) for 6 h. All the processes were carried out in the dark to avoid photobleaching of fluorescein FITC.
For the flow cytometry experiment, A549 and PC3 cells were seeded into 6-well plates at 50 × 104 cells per well and cultured for 2 days. The medium was then replaced with 3 mL fresh serum-free medium containing 1 mg mL−1 of Au NCs-FITC, Au NPs-FITC and cAu NPs-FITC. After another incubation for 4 h in the dark, the cells were washed three times with cold PBS. The cells were then collected and re-suspended in 1 mL PBS for flow cytometric analysis. A total of 200000 events were tested for each sample.
3 Results and discussion
3.1 Characteristics of the three types of nanomaterials
We initially prepared Au nanoparticles with different sizes and surface charges (Fig. 1). Briefly, ultra-small sized Au nanoclusters (Au NCs) were synthesized using bovine serum albumin (BSA) and aqueous HAuCl4 solution.8 After that, self-assembled anionic Au nanoparticles (Au NPs) and cationic Au nanoparticles (cAu NPs) were then obtained by solvent- and cation-induced self-assembly techniques. The spherical morphology of Au NCs was determined by transmission electron microscopy (TEM) as shown in Fig. 1b. The size of Au NCs was measured by dynamic light scattering (DLS) (Fig. 1e), and a diameter of ∼3 nm was obtained with a narrow polydispersity index (PDI = 0.243). Ethanol-induced self-assembly of Au NPs was performed with precursor Au NCs after the aggregation of hydrophobic domains on BSA ligands.29 The formation of self-assembled Au NPs was successfully achieved as evidenced by increased size and a change in the morphology. The results in Fig. 1c indicated that the spherical nanoparticles have an average size of ∼46 nm in the TEM image, which was in good agreement with the result from DLS, a Z-average hydrodynamic diameter of ∼52 nm (Fig. 1f). The surface of pre-synthesis Au NCs contained many reactive residues that are readily decorated by primary amino groups to convert the negatively charged surface of Au NCs into a positively charged surface. Therefore, the self-assembly of the cationic Au nanoparticles (cAu NPs) was driven by the electrostatic interaction between Au NCs and polyelectrolytes, resulting in larger nanoparticles in size and controllable positive charges.32 The size of the spherical shape of cAu NPs was around 90 nm (Fig. 1d), consistent with the DLS analysis result that the hydrodynamic diameter was around 95 nm with a relatively narrow size distribution in ultrapure water (Fig. 1f). The surface charge of nanomaterials is another essential indicator to explore the interaction between nanomaterials and cells. The zeta potential of Au NCs, self-assembled Au NPs and cAu NPs was around −22.9 mV, −23.9 mV, and 17.8 mV, respectively, as shown in Fig. 1g.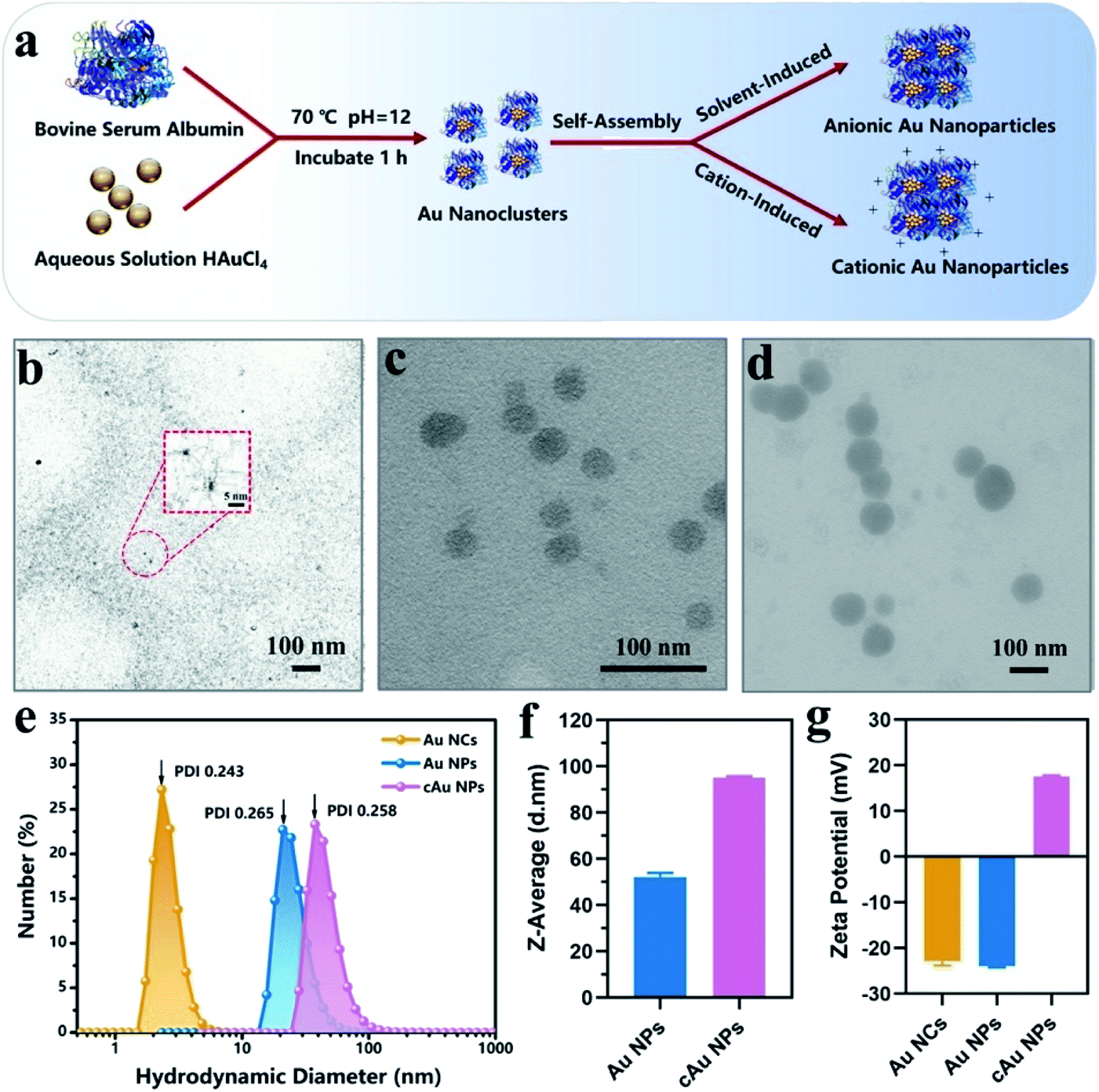 | ||
| Fig. 1 Characterization of anionic BSA-Au Nanoclusters (Au NCs), self-assembled anionic Au Nanoparticles (Au NPs) and cationic Au Nanoparticles (cAu NPs). (a) Schematic illustration of the preparation process of the three types of nanomaterials. Transmission electron microscopy (TEM) image of (b) Au NCs, (c) Au NPs and (d) cAu NPs. Scale bars, 100 nm. Inset: enlarged micrographs. Scale bar, 5 nm. (e) The size distribution of the three types of nanoparticles (n = 3). (f) Z-Average of self-assembled Au NPs and cAu NPs. The data are shown as mean ± s.d. (n = 3). (g) Zeta potential histogram of the three types of nanoparticles in water determined by dynamic light scattering (DLS). The data are shown as mean ± s.d. (n = 3). | ||
Interestingly, ultraviolet-visible (UV-vis) absorbance revealed that an absorbance peak at ∼275 nm was observed for the three types of nanomaterials, as shown in Fig. 2a. This peak generally appears in ultra-small nanoclusters relevant to their molecular-like properties, which is attributed to the spatial limitation of free electrons in nanoclusters leading to discrete electronic transitions.33 Notably, no absorbance band at ∼520 nm, a typical feature of surface plasmon resonance (SPR) for large nanoparticles,32,34 was found in UV-vis spectra for both self-assembled Au NPs and cAu NPs. It was clarified that solvent- and cation-induced self-assembled nanoparticles retained the molecular-like properties of ultra-small nanoclusters. Their simple self-assembled structure created favorable conditions for the disintegration in the cellular environment. Furthermore, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) assay was used to analyze the theoretical molecular weight of Au NCs, Au NPs and cAu NPs. As shown in Fig. 2b, although two self-assembled Au nanoparticles showed dispersed bands, an apparent central band at 66 kDa was evidenced, which was similar to the theoretical molecular weight of Au NCs,35 indicating that no significant change was observed in the molecular weight of Au NCs after self-assembly. The Fourier-transform infrared (FTIR) spectra of Au NCs exhibited a firm peak at ∼1670 cm−1 in the amide I region caused by –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O– tensile vibration, –NH bending and –CN extension at ∼1530 cm−1 to the amide II region as shown in Fig. 2c, and the absorbance pattern was similar to that of BSA. Taken together, self-assembled Au NPs and cAu NPs exhibited similar characteristics to Au NCs, implying that Au NCs were the main building units of self-assembled Au NPs and cAu NPs.
O– tensile vibration, –NH bending and –CN extension at ∼1530 cm−1 to the amide II region as shown in Fig. 2c, and the absorbance pattern was similar to that of BSA. Taken together, self-assembled Au NPs and cAu NPs exhibited similar characteristics to Au NCs, implying that Au NCs were the main building units of self-assembled Au NPs and cAu NPs.
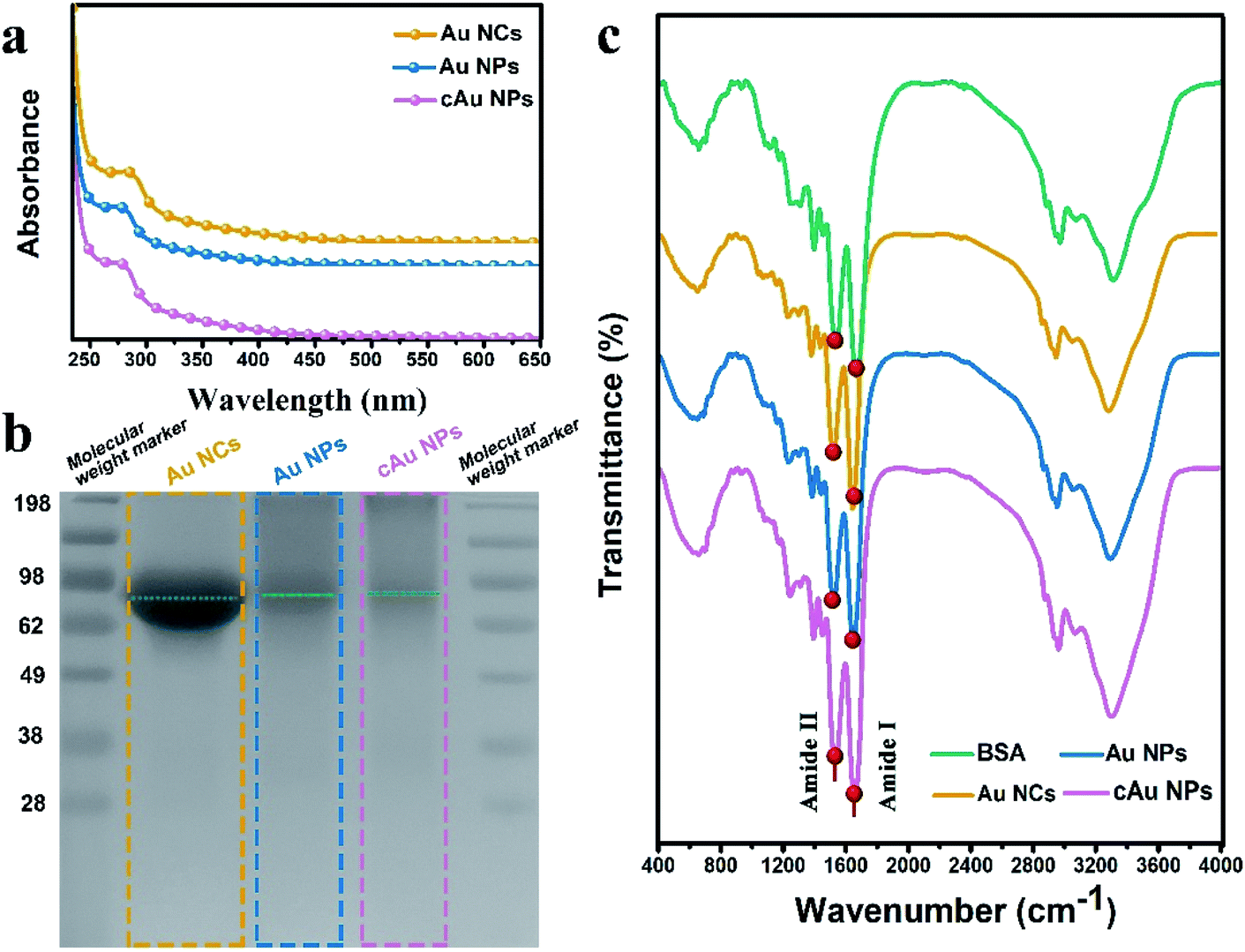 | ||
| Fig. 2 Characterization of the three types of nanomaterials. (a) Ultraviolet-visible (UV-vis) spectroscopy, (b) the sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analyses and (c) Fourier-transform infrared (FTIR) spectroscopy of Au NCs, self-assembled Au NPs and cAu NPs. | ||
3.2 Luminescence properties of the three types of nanomaterials
The fluorescence properties of the three types of nanomaterials are displayed in Fig. 3. The Au NC, self-assembled Au NP and cAu NP aqueous solutions emitted intense bright-red fluorescence under ultraviolet (UV) light of 365 nm (inset, Fig. 3). For all samples, two excitation peaks of 365 nm and 505 nm appeared simultaneously followed by emission at 654 nm, which may be attributed to multiple electronic interactions inside the metal core and ligands and between the core and ligands.32,36 The maximum emission was found at around 654 nm when excited at 505 nm and the fluorescence intensity of Au NPs and cAu NPs was higher than that of Au NCs. By virtue of the size advantages of Au NCs, the electronic transition between the highest occupied molecular orbital and lowest unoccupied molecular orbital (HOMO–LUMO) energy levels and the electronic interactions of its inside cause Au NCs emitting bright red luminescence under excitation.37 Self-assembled nanoparticles with stronger luminescence contained two types of aggregation-induced emission (AIE) phenomenon, namely solvent-induced aggregation and cationic-induced aggregation.38 The hydrophilic shell of Au NCs was destroyed by ethanol or cations. Dense and rigid self-assembled aggregates were then formed under hydrophobic or electrostatic interactions. The robust internal interaction inhibited the molecular vibration thus emitting stronger luminescence. The degree of aggregation of Au NPs and cAu NPs also caused the blue shift phenomenon. The blue shift distance was around 8 nm. The relative fluorescence intensity position was almost unchanged under 365 nm excitation (Fig. S1†). The phenomenon further verified the aggregation-induced emission was the main reason for fluorescence enhancement.32 The results indicated that the three types of nanomaterials were qualified to explore the interaction of nanoparticles with different size and charge towards cancer cells.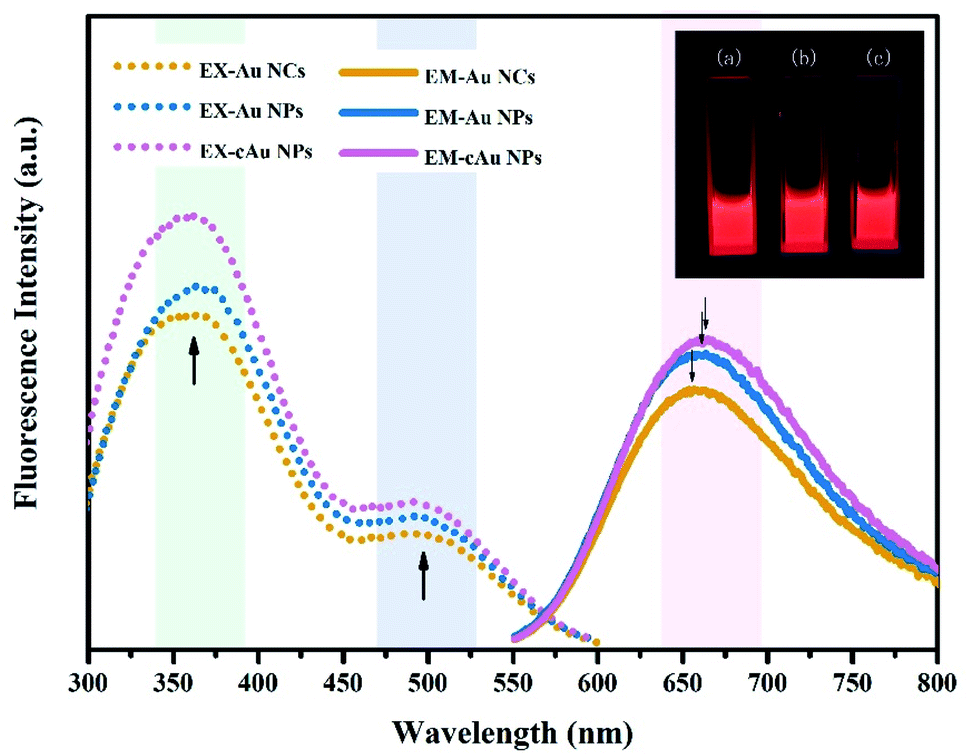 | ||
| Fig. 3 Excitation spectra (dotted line) with λem = 654 nm and emission spectra (solid line) with λex = 505 nm of Au NCs, self-assembled Au NPs and cAu NPs. The inset shows the digital photographs of (a) Au NCs, (b) Au NPs and (c) cAu NPs emitting strong red fluorescence under excitation of ultraviolet (UV) light (365 nm). | ||
3.3 In vitro biological performance of the three types of nanomaterials
In order to evaluate the biocompatibility of Au NCs, Au NPs and cAu NPs, MTT (3-(4,5-dimethyl thiazolyl-2)-2,5-diphenyltetrazolium bromide) assays were performed to examine cell viability by incubating A549 and PC3 cells with broad interval concentrations of the three types of nanomaterials (Fig. 4). After incubation with the three types of nanomaterials for 48 h, the cell viability of A549 and PC3 cell lines exhibited certain similar features that Au NC, Au NP and cAu NP nanomaterials had limited effect on cell viability in A549 and PC3 cells at the examined concentration. This indicated that the aforementioned three types of Au nanocluster-based nanomaterials, to some extent, showed biocompatibility with cancer cells. Interestingly, from the cell viability results, all three types of nanomaterials were found to be non-toxic to both A549 and PC3 cells. The high cell viabilities (>120%) are probably attributed to the BSA in the nanoparticles, which might improve the cell growth.39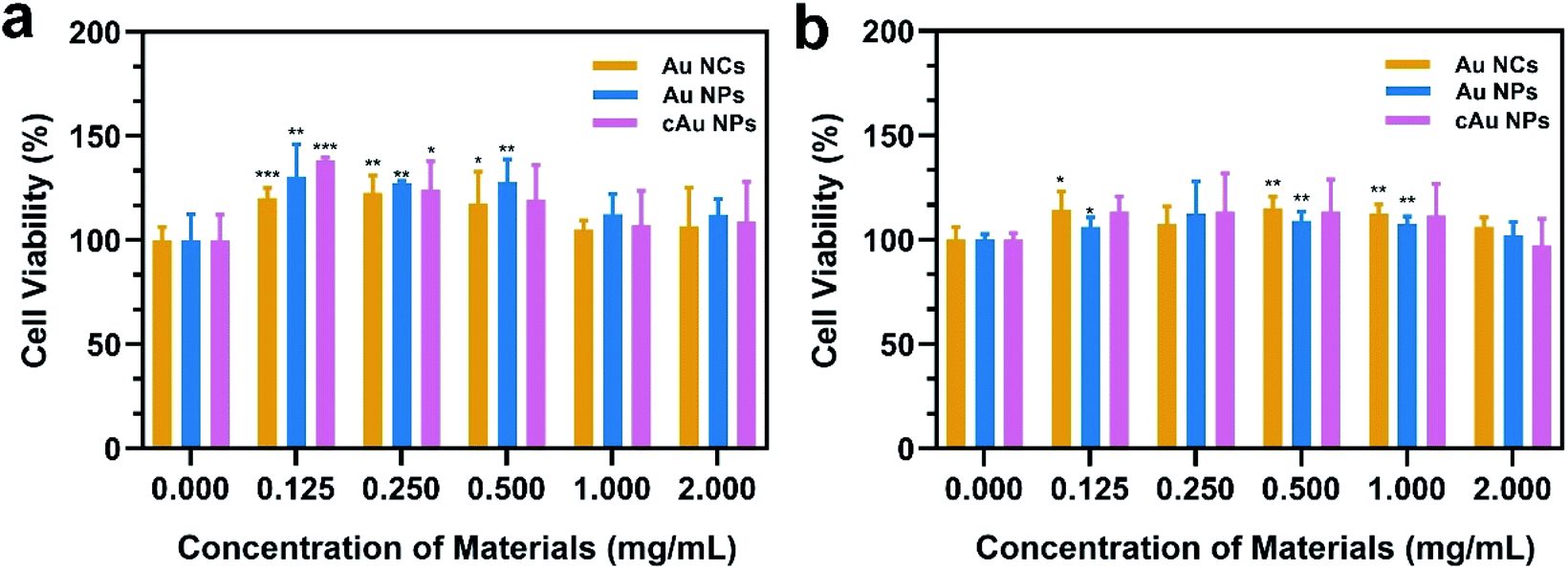 | ||
| Fig. 4 In vitro cytotoxicity of Au NCs, Au NPs and cAu NPs in (a) A549 and (b) PC3 cell lines. The data are shown as mean ± s.d. (n = 5). *p < 0.05, **p < 0.01, and ***p < 0.001 (Student's t-test); they represent statistical differences between experimental groups and the control group. | ||
Cell internalization is the premise of drug delivery candidates in DDSs. We therefore examined the three types of nanomaterials' cellular uptake behavior. After the treatment of A549 with Au NCs, self-assembled Au NPs and cAu NPs for 4 h, cell images were taken by confocal laser scanning microscopy (CLSM). It was noticeably observed that all nanoparticles were evidenced in the A549 cells (Fig. 5a). The images showed that most of the red fluorescence emitted from the three nanomaterials was distributed around the cytoplasm region with a tiny fraction in the nuclei. It appeared that the fluorescence intensity of self-assembled Au NPs and cAu NPs in the A549 cells was higher than that of Au NCs, indicating a better cellular uptake of these nanoparticles.
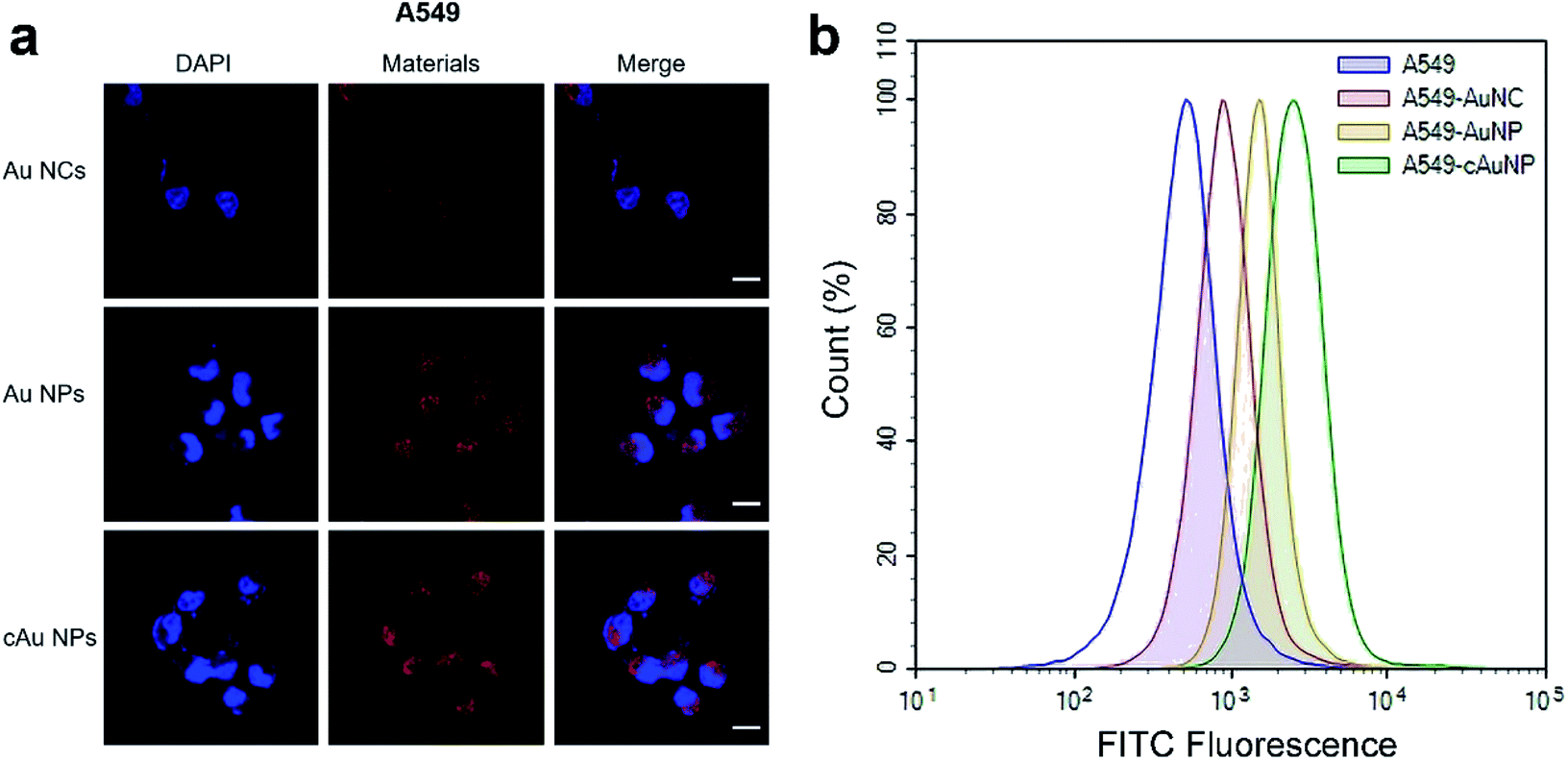 | ||
| Fig. 5 Cellular uptake behaviors of Au NCs, Au NPs and cAu NPs. (a) Confocal laser scanning microscopy (CLSM) images of A549 cells treated with the three types of nanomaterials emitting red luminescence. Red and blue fluorescence emission signals come from nanoplatforms and stained nuclei with DAPI. Scale bars, 20 μm. (b) Flow cytometric analysis of fluorescence activity in A549 cells after incubation with FITC-Au NCs, FITC-Au NPs and FITC-cAu NPs for 4 h. | ||
Furthermore, the quantitative detection of nanomaterials in A549 cells was investigated by flow cytometry (Fig. 5b). Compared to the Au NCs, the results showed that fluorescence intensity was significantly enhanced for both Au NPs and cAu NPs in A549 cells, and that of cAu NPs was a bit higher than that of the other two nanomaterials, consistent with the results obtained from CLSM. Similar results were observed in PC3 cells exposed to these three nanoparticles (Fig. S2†). As expected, self-assembled Au NPs and cAu NPs, by virtue of their size or positive charge advantages, exhibited efficient internalization by cells. Thus, in vitro cytotoxicity and cellular uptake studies indicated that although different cancer cell lines have different nanomaterial responses, self-assembled nanomaterials exhibited satisfactory cell uptake with strong luminescence and could be used to probe the cellular uptake clearly besides being promising drug delivery candidates. Besides, combined with cytotoxicity assay, it could be concluded that cation modified Au nanocluster-based nanoparticles could promote cell uptake without a strong toxic proton sponge effect via rational design.
3.4 Drug-loading ability and therapeutic effects of the three types of nanomaterials
Since the launch of Abraxane® in the United States in 2009, nanomaterial-drug delivery systems of albumin-based nanoparticles have received widespread attention.40 Doxorubicin hydrochloride, a widely used anti-tumor drug in clinical practice, inhibits the synthesis of the genetic material by embedding the DNA of cancer cells, resulting in a robust toxic effect to a variety of cancer cells.41 It was loaded into nanoparticles by diffusion absorption and hydrophobic-induced embedding with hydrophobic characters after neutralization treatment (Fig. 6). DOX was extracted from nanoparticles and the loading efficiency/encapsulation efficiency was evaluated depending on the typical absorption peak at 480 nm. The results showed that the encapsulation efficiency of Au NCs and cAu NPs was 36.61% and 14.47%, respectively, whereas the encapsulation efficiency of Au NPs was the highest among the three, with a value of 55.24% (Table 1). DOX was attached to the surface of Au NCs and cAu NPs after thoroughly mixing, driven by electrostatic interaction and hydrogen bonding. However, the electrostatic repulsion effect also led to a lower drug loading and encapsulation efficiency of DOX (pKa = 8.4) in cAu NPs. Under hydrophobic interaction, DOX is embedded into nanoparticles along with the self-assembly process to form stable drug-loaded nanoparticles with a hydrophobic drug core and hydrophilic protein shell, and the encapsulation efficiency of Au NPs was the highest one (Table 1). Therefore, all three types of nanomaterials were successfully loaded with DOX with satisfactory loading efficiency and encapsulation efficiency, based on their unique structure and properties, laying the fundamental basis for their activity in cancer cells.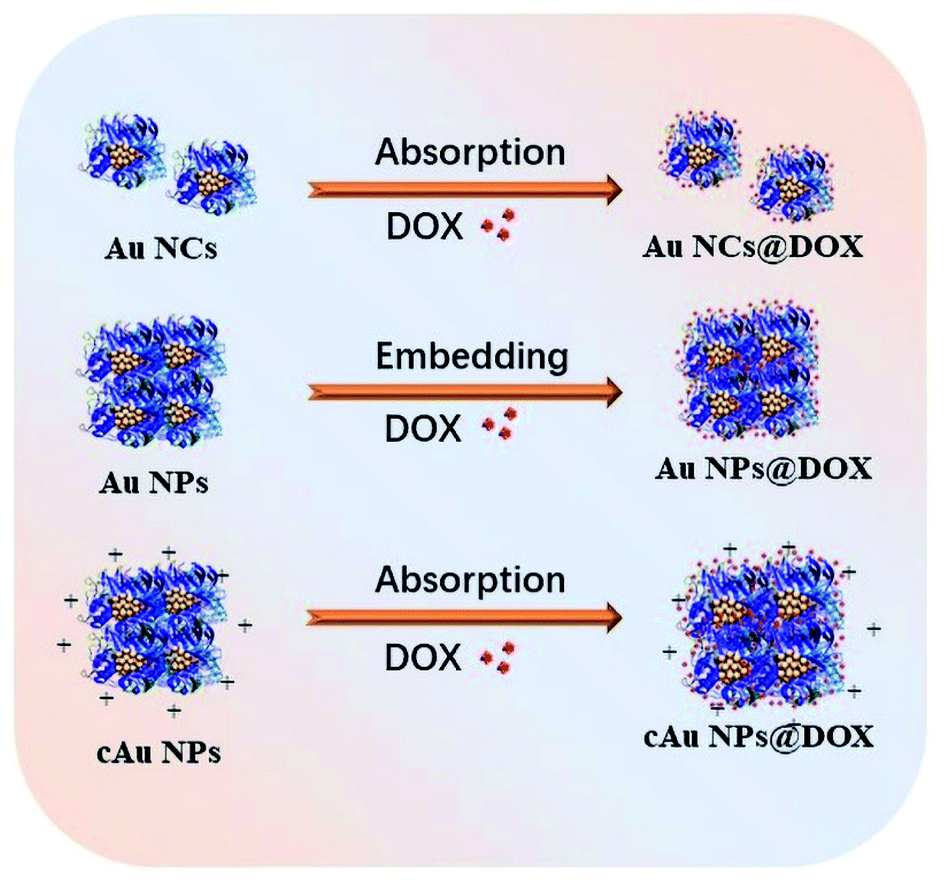 | ||
| Fig. 6 The scheme of loading of Au NCs, self-assembled Au NPs and cAu NPs with DOX to form drug delivery systems. | ||
| Encapsulation efficiency, E.E. (%, w/w) | Loading capacity, L.C. (%, w/w) | |
|---|---|---|
| Au NCs@DOX | 36.61 | 0.36 |
| Au NPs@DOX | 55.24 | 0.55 |
| cAu NPs@DOX | 14.47 | 0.14 |
The cell viability was evaluated using an MTT assay. The initial concentrations of the nanomaterials are kept constant, followed by a serial dilution. The concentration of DOX was calculated based on the loading capacity of the nanoparticle type. The relative cell viability at each concentration of DOX is displayed in Fig. 7. As shown in Table 2 and Fig. 7, cytotoxicity assessment revealed that DOX treatment exerted dose-dependent cytotoxicity on A549 cells. Drug-loaded nanomaterials present a much higher inhibitory effect than the free DOX, as indicated by the dramatically reduced IC50 values (Table 2). It was ascribed to the increased DOX in cells transported by nanomaterials. Despite the relatively low drug-loading capacity of cAu NPs, DOX-trapped cAu NPs achieved a much better inhibitory effect on cancer cells with excellent cell uptake, supporting the potential of nanoparticles in drug delivery. In addition, although self-assembled Au NPs possessed excellent drug loading efficiency, the IC50 value of DOX-loaded Au NPs was a bit higher than that of the other two nanomaterials. This may be explained by the fact that hydrophobic drugs are firmly aggregated within the core of nanomaterials, causing a slow release of cargo drugs into cells,42 which will make it more useful in clinical practice. Between the two types of cancer cells, A549 cells exhibited more sensitive than PC3 cells both in terms of free DOX and DOX-loaded nanoparticles, somehow to a large extent (Fig. 7). Taken together, cAu NPs increase DOX delivery efficiency with excellent cell uptake after positive charge surface modification, creating a better cancer treatment effect with a minimized DOX dosage. Au NPs are more suitable for long-term drug delivery as for the slow-release effect. Self-assembled Au-based nanomaterials are potential drug delivery vectors for improving the anti-cancer efficiency of DOX.
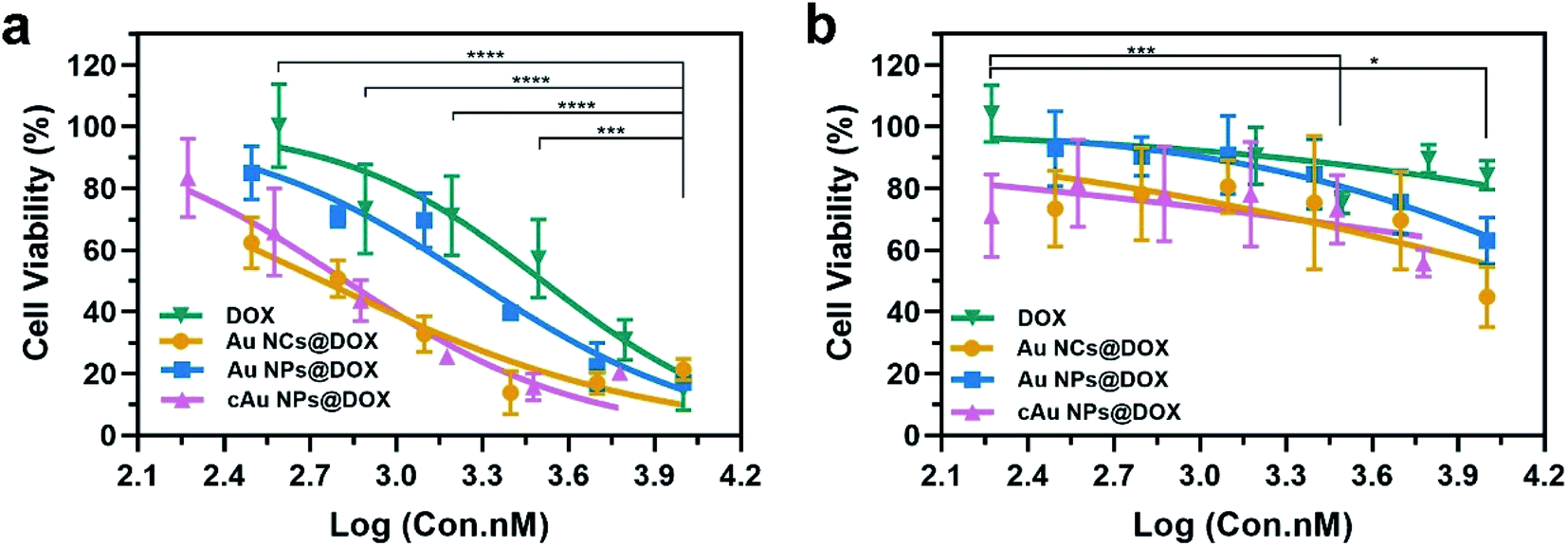 | ||
| Fig. 7 Cell viability assays of free anti-cancer drug DOX and drug-loaded nanomaterials in (a) A549 and (b) PC3 cells for 48 h. The data are shown as mean ± s.d. (n = 5). *p < 0.05, **p < 0.01,***p < 0.001, and ****p < 0.0001 (one-way ANOVA). | ||
| Materials | IC50 value (μM) in | |
|---|---|---|
| A549 cells | PC3 cells | |
| DOX | 3.23 | 84.69 |
| Au NCs@DOX | 0.55 | 7.69 |
| Au NPs@DOX | 1.87 | 16.38 |
| cAu NPs@DOX | 0.66 | 12.45 |
4 Conclusions
In this work, we used solvent- and cation-induced self-assembly nanotechnology to fabricate nanomaterials with different sizes and surface charges from the precursor Au nanoclusters to explore the relationship between the properties of Au nanomaterials and the corresponding responsiveness of cancer cells. We found that self-assembled nanomaterials inherit the molecular-like properties of ultra-small Au nanoclusters with strengthened fluorescence properties due to the aggregation-induced emission effect. Notably, all three types of nanomaterials exhibited significant biocompatibility and enhanced cellular uptake, which is of great importance to practical anti-cancer applications. Besides, it has been proven that large size and positive charge surface-modification of Au-based nanomaterials increases the cell uptake without extra cytotoxicity. Furthermore, we established three types of nanomaterial-assisted drug delivery systems as direct anti-cancer therapies. Self-assembled cAu NPs took advantage of surface-modified positive charge with better cell uptake to delivery DOX, exhibiting an excellent therapeutic effect. Besides, the unique hydrophobic cavity of Au NPs enabled high drug loading efficiency and sustained drug release. Overall, we believe that the self-assembly formulation of nanomaterials and their biological performances can provide a promising approach to the combination of bio-imaging, drug delivery and cancer therapy with high potential to clinical translations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support from the National Natural Science Foundation of China (Grant No. 51671114 and U1806219), the Natural Science Foundation of Shandong Province (ZR2019BEM024), the Shenzhen Fundamental Research Program (JCYJ20190807092803583), the Natural Science Foundation of Jiangsu Province (Grant No. BK20190205), the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2019A1515110846) and the Fundamental Research Funds of Shandong University (Grant No. 2018CJ047). This work is also supported by Special Funding from the Project of the Qilu Young Scholar Program of Shandong University.References
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H. S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS.
- D. Kalyane, N. Raval, R. Maheshwari, V. Tambe, K. Kalia and R. K. Tekade, Mater. Sci. Eng., C, 2019, 98, 1252–1276 CrossRef CAS PubMed.
- S. Thakkar, D. Sharma, K. Kalia and R. K. Tekade, Acta Biomater., 2020, 101, 43–68 CrossRef CAS PubMed.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS PubMed.
- J. Lipka, M. Semmler-Behnke, R. A. Sperling, A. Wenk, S. Takenaka, C. Schleh, T. Kissel, W. J. Parak and W. G. Kreyling, Biomater, 2010, 31, 6574–6581 CrossRef CAS PubMed.
- G. Zuber, E. Weiss and M. Chiper, Nanotechnol, 2019, 30, 352001 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS.
- J. Yang, F. Wang, H. Yuan, L. Zhang, Y. Jiang, X. Zhang, C. Liu, L. Chai, H. Li and M. Stenzel, Nanoscale, 2019, 11, 17967–17980 RSC.
- M. Matulionyte, D. Dapkute, L. Budenaite, G. Jarockyte and R. Rotomskis, Int. J. Mol. Sci., 2017, 18, 378 CrossRef PubMed.
- X. D. Zhang, D. Wu, X. Shen, P. X. Liu, F. Y. Fan and S. J. Fan, Biomater, 2012, 33, 4628–4638 CrossRef CAS PubMed.
- C. N. Loynachan, A. P. Soleimany, J. S. Dudani, Y. Lin, A. Najer, A. Bekdemir, Q. Chen, S. N. Bhatia and M. M. Stevens, Nat. Nanotechnol., 2019, 14, 883–890 CrossRef CAS PubMed.
- R. Sinha, G. J. Kim, S. Nie and D. M. Shin, Mol. Cancer Ther., 2006, 5, 1909–1917 CrossRef CAS PubMed.
- A. J. McGoron, Bioconjugate Chem., 2020, 31, 436–447 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS.
- L. Zhou, T. Qiu, F. Lv, L. Liu, J. Ying and S. Wang, Adv. Healthcare Mater., 2018, 7, e1800670 CrossRef PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- A. Albanese, P. S. Tang and W. C. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS PubMed.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS PubMed.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- N. Pernodet, X. Fang, Y. Sun, A. Bakhtina, A. Ramakrishnan, J. Sokolov, A. Ulman and M. Rafailovich, Small, 2006, 2, 766–773 CrossRef CAS PubMed.
- X. Ma, J. Sun, L. Zhong, Y. Wang, Q. Huang, X. Liu, S. Jin, J. Zhang and X.-J. Liang, Nano Lett., 2019, 19, 8476–8487 CrossRef CAS PubMed.
- B. D. Chithrani and W. C. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- K. D. Lee, S. Nir and D. Papahadjopoulos, Biochemistry, 1993, 32, 889–899 CrossRef CAS PubMed.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- J. Y. Wang, J. Chen, J. Yang, H. Wang, X. Shen, Y. M. Sun, M. Guo and X. D. Zhang, Int. J. Nanomed., 2016, 11, 3475–3485 CrossRef CAS PubMed.
- L. Liu, F. Hu, H. Wang, X. Wu, A. S. Eltahan, S. Stanford, N. Bottini, H. Xiao, M. Bottini, W. Guo and X. J. Liang, ACS Nano, 2019, 13, 5036–5048 CrossRef CAS PubMed.
- K. Eisele, R. A. Gropeanu, C. M. Zehendner, A. Rouhanipour, A. Ramanathan, G. Mihov, K. Koynov, C. R. Kuhlmann, S. G. Vasudevan, H. J. Luhmann and T. Weil, Biomater, 2010, 31, 8789–8801 CrossRef CAS PubMed.
- N. V. Cuong, J. L. Jiang, Y. L. Li, J. R. Chen, S. C. Jwo and M. F. Hsieh, Cancers, 2010, 3, 61–78 CrossRef PubMed.
- A. Yahia-Ammar, D. Sierra, F. Mérola, N. Hildebrandt and X. Le Guével, ACS Nano, 2016, 10, 2591–2599 CrossRef CAS PubMed.
- R. Han, M. Zhao, Z. Wang, H. Liu, S. Zhu, L. Huang, Y. Wang, L. Wang, Y. Hong, Y. Sha and Y. Jiang, ACS Nano, 2020, 14, 9532–9544 CrossRef CAS PubMed.
- L. Shang, N. Azadfar, F. Stockmar, W. Send, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Small, 2011, 7, 2614–2620 CrossRef CAS PubMed.
- H. Lin, L. Li, C. Lei, X. Xu, Z. Nie, M. Guo, Y. Huang and S. Yao, Biosens. Bioelectron., 2013, 41, 256–261 CrossRef CAS PubMed.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- S. Govindaraju, S. R. Ankireddy, B. Viswanath, J. Kim and K. Yun, Sci. Rep., 2017, 7, 40298 CrossRef CAS PubMed.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- L. Dong, M. Li, S. Zhang, J. Li, G. Shen, Y. Tu, J. Zhu and J. Tao, Small, 2015, 11, 2571–2581 CrossRef CAS PubMed.
- E. Miele, G. P. Spinelli, E. Miele, F. Tomao and S. Tomao, Int. J. Nanomed., 2009, 4, 99–105 CAS.
- I. Blazkova, K. Smerkova, L. Blazkova, M. Vaculovicova, M. Stiborova, T. Eckschlager, M. Beklova, V. Adam and R. Kizek, Electrophor, 2015, 36, 1282–1288 CrossRef CAS PubMed.
- J. G. Shiah, C. Konák, J. D. Spikes and J. Kopecek, Drug Delivery, 1998, 5, 119–126 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na01066a |
| This journal is © The Royal Society of Chemistry 2021 |