
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Prediction of unexpected BnPn structures: promising materials for non-linear optical devices and photocatalytic activities†
Zabiollah
Mahdavifar
 *
*
Department of Chemistry, Faculty of Science, Shahid Chamran University of Ahvaz, Ahvaz, Iran. E-mail: z_mahdavifar@scu.ac.ir; Fax: +98-611-3331042
First published on 26th March 2021
Abstract
In the present work, a modern method of crystal structure prediction, namely USPEX conjugated with density functional theory (DFT) calculations, was used to predict the new stable structures of BnPn (n = 12, 24) clusters. Since B12N12 and B24N24 fullerenes have been synthesized experimentally, it motivated us to explore the structural prediction of B12P12 and B24P24 clusters. All new structures were predicted to be energetically favorable with negative binding energy in the range from −4.7 to −4.8 eV per atom, suggesting good experimental feasibility for the synthesis of these structures. Our search for the most stable structure of BnPn clusters led us to classify the predicted structures into two completely distinct structures such as α-BnPn and β-BnPn phases. In α-BnPn, each phosphorus atom is doped into a boron atom, whereas B atoms form a Bn unit. On the other hand, each boron atom in the β-phase was bonded to a phosphorus atom to make a fullerene-like cage structure. Besides, theoretical simulations determined that α-BnPn structures, especially α-B24P24, show superior oxidation resistance and also, both α-BnPn and β-BnPn exhibit better thermal stability; the upper limit temperature that structures can tolerance is 900 K. The electronic properties of new compounds illustrate a higher degree of absorption in the UV and visible-region with the absorption coefficient larger than 105 cm−1, which suggests a wide range of opportunities for advanced optoelectronic applications. The β-BnPn phase has suitable band alignments in the visible-light excitation region, which will produce enhanced photocatalytic activities. On the other hand, α-BnPn structures with modest band gap exhibit large second hyperpolarizability, which are anticipated to have excellent potential as second-order non-linear optical (NLO) materials.
1. Introduction
In the past decade, many attempts have been made to discover non-carbon fullerene-like structures based on their unique properties.1–9 The structural and electronic properties of clusters form an intermediate state between the individual atoms and crystals. Clusters have a remarkable impression in understanding the chemical bonding and the rational design of molecules with tailored physicochemical properties. In recent years, scientists have focused on studying boron-based materials because a significant number of unfamiliar structures of boron and its compounds have been explored.10–16 The electron configuration of boron is 2s2 2p1; therefore, boron with one electron less than carbon has different characteristics compared to carbon, which can form fascinating and strong bonds with nearly all periodic table elements.16–18 On the other hand, phosphorus clusters with large pore sizes have received much attention in the past decade.18 Phosphorus is the diagonal neighbor of carbon in the periodic table; therefore, the chemical bonding and structural chemistry of phosphorus are also interesting. Elemental phosphorus exists in different forms such as white, yellow, and black phosphorus but phosphorus is never found as a free element on earth because of its high reactivity.19The group III–V compounds such as boron phosphide (BP) have been most generally investigated in their cubic crystal phase (c-BP).20 Also, BP compounds can exist in planar (two-dimensional), tubular (one-dimensional), or spherical shapes (zero-dimensional), quite the same as graphene, carbon nanotubes, and fullerenes, respectively. All structures of BP compounds are prominent as large band-gap semiconductor materials having many advanced applications including microelectronics and optoelectronics,21,22 high thermal conductivity23 hardness, chemical stability,23–25 and catalysis.20,26,27 Both boron and phosphorus elements can form 2D materials; it is interesting to determine whether the binary compound of boron and phosphorus can form stable clusters, which may display unusual structural and electronic properties better than both boron and phosphorus clusters.28 Hence, it is desirable to consider the structures of boron phosphide clusters with different sizes.
Numerous computational and experimental studies have considered the structural and electronic properties of group III–V compounds.18,19,29–32 In recent years, non-carbon (inorganic) (XY)n fullerenes of group III–V are of great scientific interest based on their promising candidates as optoelectronic devices33,34 and light-emitting diodes (LEDs).35 X12Y12, X16Y16, X24Y24, and X28Y28 clusters, where X = group III atom and Y = group V atom, are predominant inorganic (XY)n fullerenes.8 Computational investigations on (XY)n fullerenes have demonstrated that (XY)12 has the highest thermodynamic stability, which is known as a magic cluster.36–38 In this regard, Strout et al. ascertained that the X12Y12 nano-cages are the most stable structures between all fullerene-like (XY)n (X = B, Al, Ga,… and Y = N, P, As,…) structures.36 B12N12 and B24N24 fullerene-like clusters were synthesized by Oku et al.39–41 and detected using time-of-flight mass spectrometry. The B12N12 cluster is shown as the most stable structure among B12N12, B16N16, and B28N28.42 Other (XY)12 clusters such as Al12N12, Al12P12, B12N12, and B12P12 are of much interest owing to their unique chemical and physical properties. In this regard, many theoretical studies had been focused on ascertaining the relative stabilities of different sizes of these nano-cages.18,19,30–32,38,43
Boron phosphide (BP)n clusters are an attractive member of the XnYn nano-cage family with unique physical and electrochemical properties. Since B12N12 and B24N24 fullerenes have been synthesized experimentally,39–41 it has motivated researchers to investigate the structural and electronic properties of B12P12 and B24P24 fullerene-like clusters. In all the previous studies, X12Y12 and X24Y24 clusters have been considered as fullerene-like structures, which consist of four-, six-, and eight-membered rings, and have large HOMO–LUMO gaps, almost zero dipole moment, and nearly zero hyperpolarizability.31 It should be mentioned that in all the previous studies, (XY)n clusters (X = B, Al, Ga, and Y = P, As) were made directly by replacing B and N atoms of the already known structures of the (BN)n cages with other atoms, then the bonding length and angle are adjusted by further calculations. Based on this strategy, the main questions are that whether this method is correct and whether this method give the lowest energy structure. We believe that structural prediction can answer these questions. Structural prediction based on first-principles calculation has served as a very useful tool in materials research. Exploring 0, 1, and 2-dimensional (0, 1, 2-D) materials from molecular design and global search have been a hot-topic. Based on our knowledge, no study has been conducted to predict the structure of boron phosphide (BP)n clusters. Also, all previous studies on these materials have been based on the structure of boron nitride (BN)n clusters. The structure of BnPn in all previous investigations by other researchers has been reported to be cage-shaped, which is exactly similar to the (BN)n clusters, in which the P atoms are replaced by N atoms. It motivated us to use an evolutionary algorithm to predict the structures of boron phosphide clusters. For the first time, we used the evolutionary algorithm as implemented in the USPEX code to search the ground state structure of the BnPn clusters, including B12P12 and B24P24 clusters. It should be noted that the B12N12 and B24N24 fullerenes have been synthesized experimentally; hence, the B12P12 and B24P24 structures were chosen.
2. Computational details
We used the evolutionary algorithm as executed in the USPEX code to search for the potential energy surface of the B12P12 and B24P24 clusters. USPEX code by evolutionary algorithm solves the central problem of crystal structure prediction of theoretical crystal chemistry.44–47 An adequate number of initial structures, more than 1800 configurations in 50 generations by the USPEX method, was used to ensure that the global minimum is attained. The geometrical relaxation, stability assessment, and optoelectronic properties of the predicted BnPn clusters were carried out by the Vienna ab initio simulation package (VASP).48,49 The generalized gradient approximation (GGA) scheme, developed by Perdew–Burke–Ernzerhof (PBE) and projector-augmented wave (PAW)50 potentials were used. The dispersion-corrected DFT-D3 method with Beack-Jansoon damping was used in all the structure optimizations to account for the weak van der Waals interactions.51,52 The low-lying energy isomers, whose relative energies are in the range of 0–2.5 eV with respect to the most stable structure, were carefully re-optimized to distinguish the correct global minimum structure of each BnPn cluster. The screen hybrid HSE06 method53 with the mixing parameter alpha value of 0.25 was carried out to consider the electronic properties and optical absorption of the predicted clusters. An energy cutoff of 400 eV was used for the plane-wave expansion. All geometries were relaxed until the convergence criteria of energy (10−6 eV) and force on each atom (0.02 eV Å−1) were satisfied. It should be pointed out that the structures were located in the middle of a box with a vacuum spacing of ∼10 Å in all the directions to neglect the interactions between the adjacent cells. To further examine the thermal stability proposed for the BnPn clusters, ab initio molecular dynamics (AIMD) simulations using the canonical (NVT) ensemble were performed. The simulations are accomplished using a Nosé–Hoover thermostat at finite 300, 600, and 900 K temperatures for 5000 fs with a time step of 1.0 fs.3. Results and discussion
3.1. Geometry and stability
Based on the USPEX method conjugated with spin-polarized first-principles calculations, a comprehensive study to explore their ground state structures of BnPn (n = 12, 24) and low-lying isomers of these clusters was performed employing the evolutionary algorithm. The USPEX method has been successful used before to explore a wide range of materials.54–56 Among the structure search by evolutionary algorithm, more than 1800 different structures for each cluster were predicted. We mainly focused on the low-lying isomers in the energy range of 0–2.5 eV with respect to the most stable structure. We re-optimized these structures to get the precise total energy and the atomic structures. The lowest-energy structures and low-lying energy isomers of the BnPn (n = 12, 24) clusters are displayed in Fig. 1 and 2.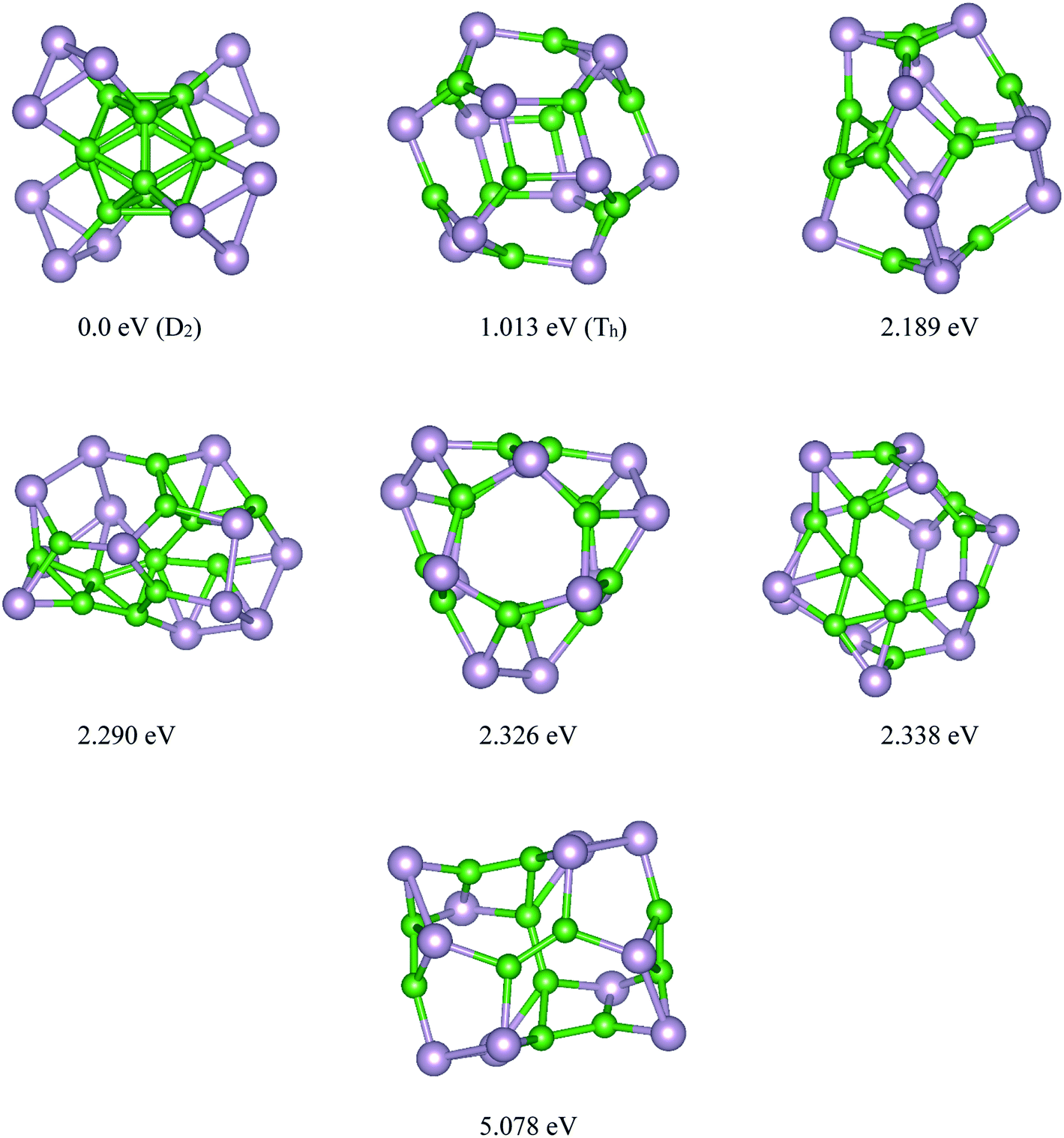 | ||
| Fig. 1 Lowest-energy structures and low-lying energy isomers of the P12B12 clusters. The green and gray balls represent the B and P atoms, respectively. | ||
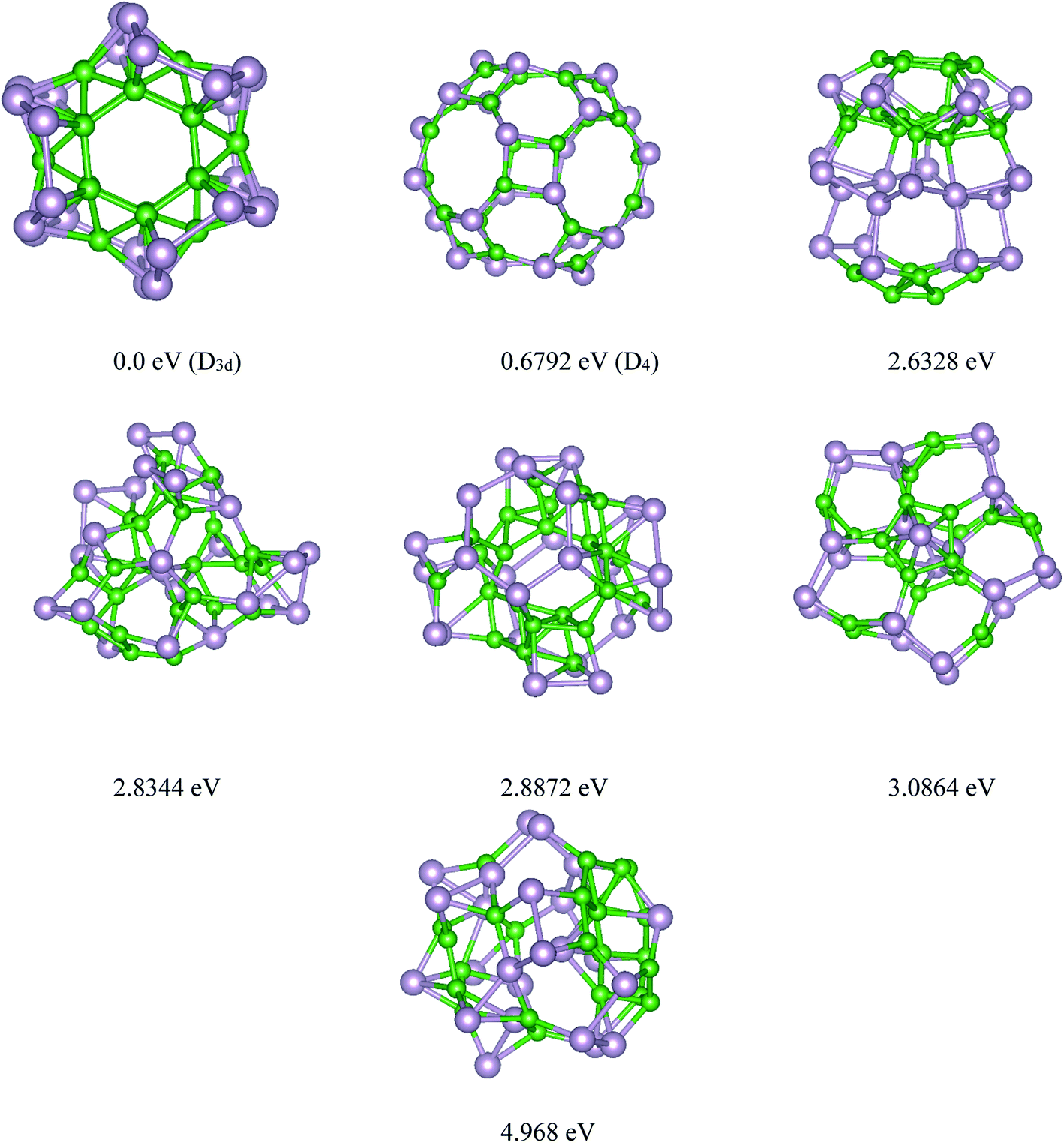 | ||
| Fig. 2 Lowest-energy structures and low-lying energy isomers of the P24B24 clusters. The green and gray balls represent the B and P atoms, respectively. | ||
Our search for the most stable structure of BnPn clusters led us to classify the predicted structures into two completely distinct structures. Interestingly, it can be classified into two α- and β-phases. In the α-phases, each phosphorus atom is doped into a boron atom while the B atoms form a Bn unit. This type of predicted structure named α-BnPn structure, is reported for the first time. On the other hand, in the β-phase, each boron atom is bonded to each phosphorus atom to make a cage cluster, which is named the β-BnPn structure. The final relaxed geometry of the α-BnPn and β-BnPn systems with their symmetries, their low-lying energy isomers, and relative stabilities are displayed in Fig. 1 and 2. To assist comprehension, two different views of the ground-state structures, as well as the first low-lying energy isomers of α-BnPn and β-BnPn are shown in Fig. S1 and S2,† respectively.
Based on our global structure search, the most stable structure of the B12P12 cluster with D2 symmetry is alpha rhombohedral-shaped, named as α-B12P12. In this structure, the arrangement of 12 boron atoms in the alpha-tetragonal boron structure made a B12 building block, while each phosphorus atom bonded to each boron atom of the alpha tetragonal B12 unit. To date, this is the first time that this type of structure has been reported for the B12P12 structure. In this beautifully predicted structure, each P atom is covalently bonded to each boron atom of the B12 unit with an average B–P bond length of ∼1.98 Å. In other words, the surface of the B12 unit is covered with phosphorus atoms. The average B–B bond lengths in the B12 unit of α-B12P12 (∼1.77 Å) is nearly the same as that of the average B–B bond in the B12 icosahedron (1.75 Å).57 As seen in Fig. 1, each phosphorus atom is covalently bonded to two neighboring phosphorus atoms to make a three-membered ring, in which the phosphorus atoms are lying in a plane. It can be imagined that the triangular rings of phosphorus have grown on the B12 unit, while each of these P atoms are bonded to each of the B atoms. In this situation, the hybridization of each P atom is sp3 The calculated average P–P bond length and the P–P–P bond angles of α-B12P12 are about 2.36 Å and 60°, respectively, which is comparable to the P–P bond length (2.23 Å) in phosphorene.58 The experimental evidence for our predicted α-B12P12 structure is the alpha tetragonal allotrope of boron determined in 1951 by Hoard et al.59 The same as α-B12P12 structure, this structure is made up of just one building block of the alpha-tetragonal B12 boron structure. Another evidence for the new predicted structure is B13P2.60 The crystal structure of B13P2 shows that this is an alpha rhombohedral boron structure with two P atoms in the unit cell. Interestingly, B13P2 was made up of just one B13 building block, in which P atoms connected each building block.60
The first low-lying energy structure of B12P12 is fullerene-like with a large cavity. This type of structure was named as the β-B12P12 structure. The structure of β-B12P12 is a polyhedral boron-phosphide structure composed of four tetragons and hexagons. As seen in Fig. 1, the stability of the low-lying energy β-B12P12 structure is less than the α-B12P12 configuration with a large amount of energy of about 1.013 eV. Based on our calculations, it has an average diameter of 0.586 nm, while the C60 fullerene has an average diameter of ∼0.7 nm.61,62 Due to the large cavity of β-B12P12, the same as that of C60 fullerene, it can be suitable for various applications such as gas storage, encapsulation of atoms, or molecules inside its cage, and endohedral metallofullerenes. As we know, the endohedral metallofullerenes have attracted much attention due to their unique potential applications such as superconductors and non-linear optical (NLO) devices. The relaxed geometry of the β-B12P12 cluster indicated that only B–P bonds with an average bond length of ∼1.911 Å were found in this nano-cage. It should be noted that two types of B–P bonds were found in this structure; the first is the B–P bond in the hexagonal ring, which is named I (B–P), and the second is the B–P bond in the tetragonal rings, which is named II (B–P). These results are collected in Table 1 and are shown in Fig. S1.†
| Type | B–P (Å) | P–P (Å) | B–B (Å) | |||
|---|---|---|---|---|---|---|
| I(B–P)h | II(B–P)T | III(B–P)O | I(B–B)h | II(B–B)Tr | ||
| α-B12P12 | 1.98 | — | — | 2.36 | 1.77 | — |
| β-B12P12 | 1.900 | 1.922 | — | — | — | — |
| α-B24P24 | 1.972 | 2.012 | — | 2.231 | 1.746 | 1.788 |
| β-B24P24 | 1.918 | 1.905 | 1.871 | — | — | — |
The final relaxed geometry of the ground-state structure and the low-lying isomers of the B24P24 cluster, together with their symmetries and relative stabilities, are shown in Fig. 2 and S2.† By looking at the predicted structures of B24P24, it was found that the ground state structure of B24P24 is α-B24P24 with a high symmetry of D3d, in which all the boron atoms are arranged as B24 unit, while the phosphorus atoms cover the B24 surface. It should be pointed out that the B24 motif in α-B24P24 is composed of two hexagons on the bottom and the top, six hexagons surrounding the waist, and 12 trigons alternatively connected to each hexagon. To date, this has been the first time that this type of structure has been reported for the P24B24 cluster. In this unique predicted structure, each P atom is bonded to one or more boron atoms of the B24 motif as the surface of the B24 unit is covered with phosphorus atoms. The average B–P bond length is obtained as ∼1.99 Å, which is comparable with the B–P bonds in phosphinoboranes and related compounds.63 In detail, two types of B–P bonds such as types I and II can be found in the α-B24P24 structure (Fig. S2†), and the corresponding values of these bond lengths are collected in Table 1. On the other hand, the calculated values of the P–P and B–B bond lengths are summarized in Table 1. The average P–P bond length is obtained as 2.231 Å while for the B–B bonds, we found two different types of B–B in τηε α-B24P24 structure. I (B–B)h represents the bonded B atoms to each other in hexagons (1.746 Å) and II (B–B)T shows the bond length between the boron atoms in trigonal geometry with a bond length of about 1.788 Å. These results are comparable with the average-B–B bond in the B12 icosahedra (1.75 Å).57 For simplicity of visualization, two different views of the B24 unit without P atoms are shown in Fig. S3.† This result is contradictory to that reported for pristine B24 clusters, where the quasi-planar and rather irregular polyhedrons are prevalent.64 It should be reminded that the ionic quasi-planar B24 structure has been experimentally synthesized.64 The evidence for our predicted α-B24P24 cluster is the encapsulation of a transition metal in the fullerene-like boron cages.65–67 Yanming Ma and coworkers65 predicted transition metal-doped B24 clusters using first-principles swarm-intelligence-based structure searches. They found that the low-lying energy structures were generally a cage-like structure. It can be concluded that doping is a significant factor in determining the geometry of boron-based materials.
Fig. 2 represents the selected low-lying energy B24P24 structures with their symmetry and energy relative to the ground-state D3d α-B24P24 structure. The first low-lying energy structure belongs to the β-B24P24 fullerene-like structure. As seen from this figure, the β-B24P24 structure with D4 symmetry is less stable than the ground-state α-B24P24 structure by a large amount of energy (∼0.68 eV). It should be reminded that the fullerene-like structure is proposed as the ground-state structure of the X24Y24 clusters. The investigated structures of X24Y24 in all the previous studies19,30–34,38,43 are based on the strategy in which B and N atoms are directly replaced with X and Y atoms in the already known structures of the (BN)n nano-cages, and then the bonding length and angle are adjusted for further calculation. In the case of BnPn, our global search indicated that the BnPn fullerene-like structure is a low-lying isomer. The β-B24P24 fullerene-like structure with a 0.859 nm cavity, which is larger than the C60 cavity (0.7 nm), includes 12 tetragonal, eight hexagonal, and six octagonal BP rings. Table 1 summarizes different boron–phosphorus (B–P) bond lengths of the most stable structures and the first low-lying energy structures of B24P24. As can be seen in Fig. 2 and Table 1, two kinds of B–P bonds are recognized for the β-B24P24 structures with B–P bonds between tetragonal/hexagonal rings, named type I (B–P), with an average bond length of about 1.918 Å, B–P bonds between tetragonal/octagonal rings, named as type II (B–P) (∼1.905 Å), and the B–P bonds between hexagonal/octagonal ring, named type III, with bond length of about 1.871 Å.
The energy difference between the most stable structure and other competing structural isomers of each compound are essential to figure out the possibility of their synthesis under different experimental conditions. The energy difference between the lowest energy structure of α-B12P12/α-B24P24 and the first low-lying energy β-B12P12/β-B24P24 structures is 0.059/0.061 eV per atom, respectively, implying that the α-BnPn structures are more stable than the β-BnPn structures (≥5.7 kJ mol−1). Based on our predictions, β-B12P12 and β-B24P24 clusters may not coexist with the α-BnPn structures at room temperature when compared with the thermal energy at 300 K (0.026 eV). Hence, it can be concluded that the BnPn fullerene-like structures can exist at higher temperatures (>300 K). In the case of other low-lying energy isomers of B12P12 and B24P24 structures, the second and third metastable structures are less stable than the global minimum α-B12P24 and α-B24P24 structures by a large amount of energy (>2.2 eV).
Now, we return to investigate the thermodynamic stability of the α-BnPn and β-BnPn clusters, which need to certify their stability and facility experimental synthesis. To estimate the thermodynamic stability of the predicted clusters, the binding energy per atom (Eb, eV per atom) is calculated. The relative binding energy (Eb) is an effective parameter to show the thermodynamic stability of the clusters and it is defined as below.
| Ebin = [E(BP)n − n/2(EB + EP)]/n | (1) |
As seen in Table 2, the calculated binding energies of the α-BnPn and β-BnPn structures are in the range from −4.723 to −4.803 eV per atom, which is much better or comparable to that of other 2D materials such as phosphorene,68 carbon phosphide,69 Al2C sheet,70 and silicene,71 having 3.48, 5.32, 3.94, and 3.55 eV per atom, respectively, indicating the very good stability of these materials. Furthermore, the binding energy of the predicted clusters was compared with that of different 2D-BP allotropes. The calculated binding energies for the α-BnPn and β-BnPn structures is comparable with the binding energy of 2D α0-BP (4.91 eV per atom), α1-BP (4.82 eV per atom), β0-BP (4.72 eV per atom), β1–BP (4.54 eV per atom), and γ-BP (4.44 eV per atom).72 Moreover, the binding energies for these new configurations are even close to the already synthesized B40,73 M@Bn,66 and B24 clusters.64 Hence, it can be concluded that the α-BnPn and β-BnPn structures have excellent stability and possible experimental synthesis. As seen in Table 2, the α-BnPn structures are more stable than the corresponding β-BnPn structure and the maximum binding energy of −4.803 eV per atom belongs to the α-B24P24 structure.
| Type | α-B12P12 | β-B12P12 | α-B24P24 | β-B24P24 |
|---|---|---|---|---|
| HOMO (eV) | −5.563 | −6.186 | −4.315 | −5.572 |
| LUMO (eV) | −4.895 | −2.977 | −3.691 | −2.592 |
| VBM (eV) | −6.120 | −6.787 | −5.313 | −6.311 |
| CBM (eV) | −5.532 | −3.579 | −4.689 | −3.331 |
| E g (eV) | 0.668 | 3.208 | 0.624 | 2.98 |
| E b (eV per atom) | −4.782 | −4.723 | −4.803 | −4.742 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Total partial charge (esu) on all B and P atoms | ||||
| B | 0.577 | 3.555 | 2.313 | 8.242 |
| P | −0.577 | −3.555 | −2.313 | −8.242 |
3.2. Dynamic and chemical stability
Next, the dynamic stability of the predicted clusters was also investigated. The vibrational frequencies at the gamma points for the most stable structures as well as the first low-lying isomers were calculated. Based on these calculations, no negative vibration frequency could be found, confirming that the most stable and the first low-lying structures are dynamically stable. Furthermore, the thermal stability of the most stable structure and the first low-lying energy isomers of the studied BnPn clusters was performed using ab initio molecular dynamic simulations (AIMD) at three different (300, 600, and 900 K) temperatures by the NVT-ensemble with the Nosé–Hoover thermostat. As represented in Fig. 3 and 4, the predicted structures equilibrated at 300 K from the AIMD simulations. Furthermore, there is no broken bond and the initial clusters are well maintained during the AIMD simulation for 5 ps with a time-step of 1 fs. As shown in these figures, the total potential energy only fluctuates around a certain constant magnitude, which indicates the most stable structures of the BnPn clusters and the first low-lying isomers are thermodynamically stable and could hold their structural integrity at room temperature. It can be seen from Fig. 3 and 4 that after heating both α-BnPn and β-BnPn structures, the basic structure of the clusters is also well maintained at 600 and 900 K. Furthermore, the thermal stability of the α-B24P24 cluster at a higher temperature such as 1200 K was also examined (Fig. S4†). Based on the AIMD simulations, when the temperature reaches as high as 900 K, the bond elongation of the 6-membered ring occurs. It can be anticipated that as the temperature increases, bond stretching is also increased. Hence, the upper limit of temperature that the structures can tolerate is 900 K and the bond stretching occurs between 900 and 1200 K.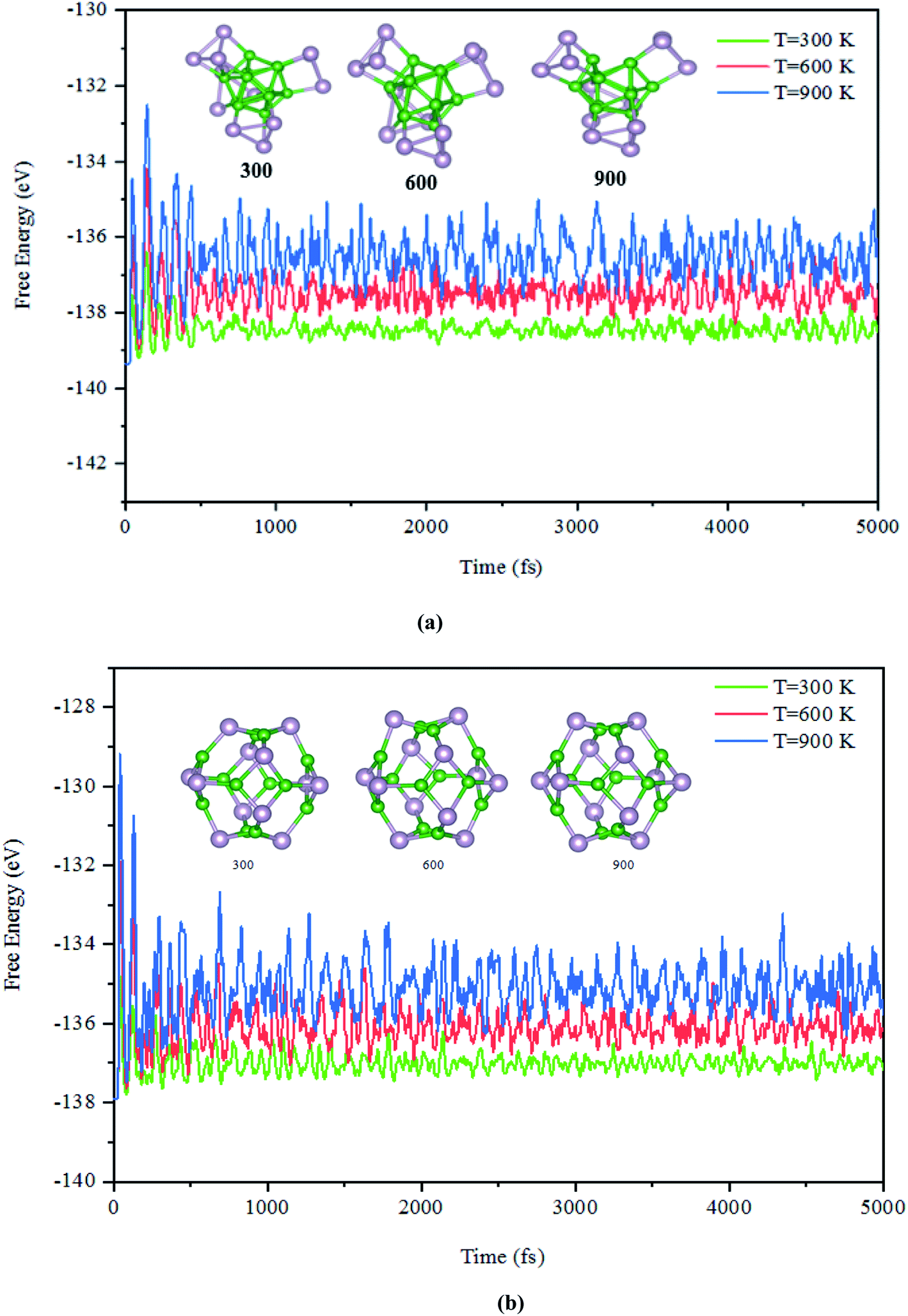 | ||
| Fig. 3 The snapshots of the most stable structures for a time of 5 ps of (a) α-B12P12 and (b) β-B12P12. The temperature of the AIMD was set to 300, 600, and 900 K. | ||
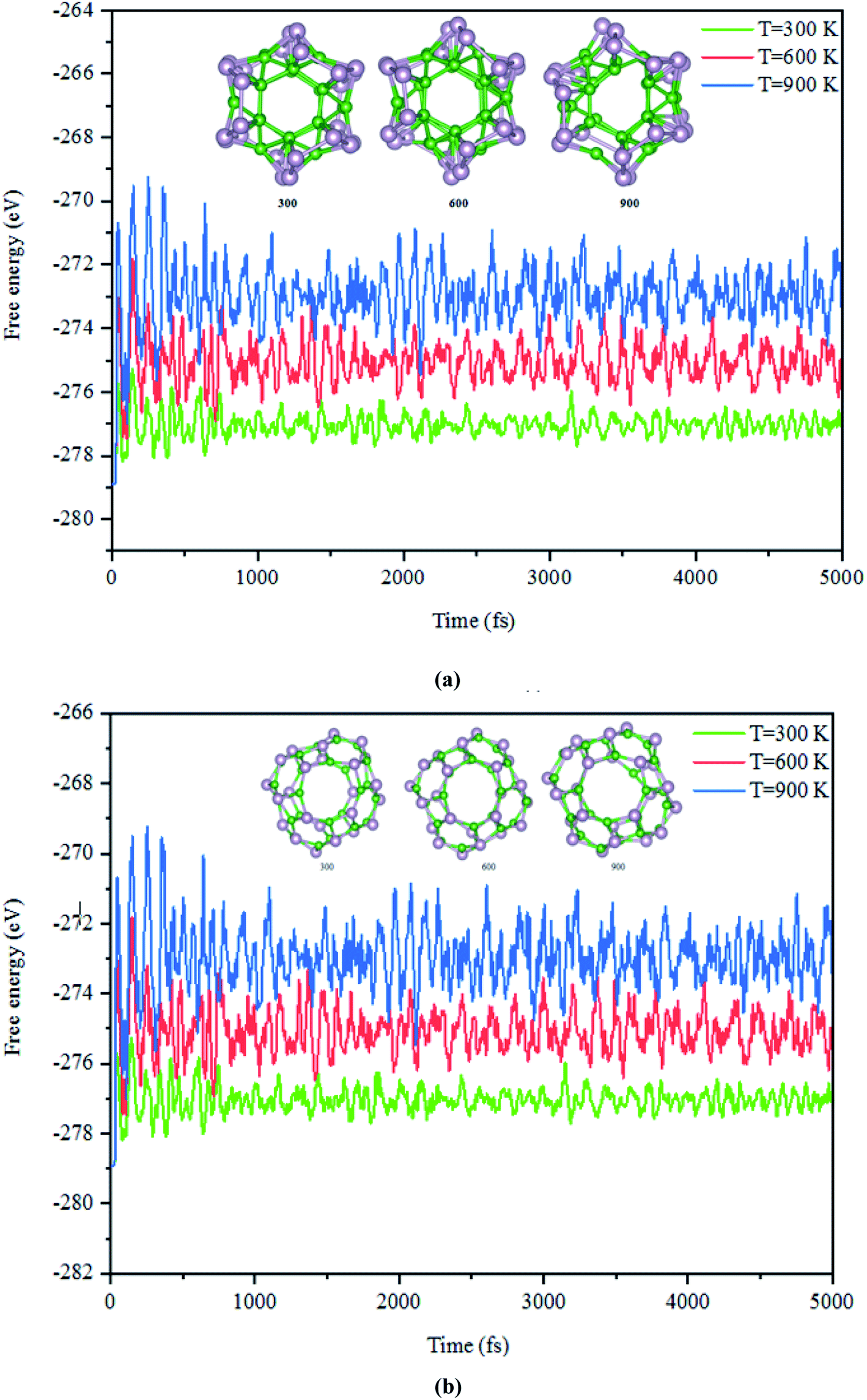 | ||
| Fig. 4 The snapshots of the most stable structures for a time of 5 ps of (a) α-B24P24 and (b) β-B24P24. The temperature of the AIMD was set to 300, 600, and 900 K. | ||
Chemical stability in air and other environments is an important issue that may limit the future applications of materials. Furthermore, a major problem with atoms located on the surface of the materials is their reaction with oxygen, nitrogen, and water in air; hence, the surface chemistry of these materials is essential in determining the intrinsic properties.74,75 Some phosphorus-based materials such as 2D-phosphorene and black phosphorus are known to oxidize and break down in ambient conditions because of the lone atomic pairs of the P atoms at the surface.76–80 Generally, P atoms favor the formation of the trigonal pyramidal sp3 bonded configuration.81 It is found that in the α-B12P12 cluster, each P atom with sp3 hybridization bonding with two P and one B neighbor atoms has one lone pair of electrons; hence, it motivates us to investigate the effects of oxygen, nitrogen, and water–gas molecules on the stability of the α-BnPn structures.
The AIMD simulation was used to investigate the interaction between α-BnPn and gaseous phase O2, N2, and H2O molecules at room temperature. As an initial model, randomly, eight O2, N2, and H2O molecules were added around the α-B12P12 andα-B24P24 clusters with 1 × 1 × 1 unit cells at an initial distance of 3.5–4 Å from the cluster surface (see Fig. 5 and 6). The lattice parameters of the optimized α-BnPn structures at the PBE + D3 level are a = 19.4 Å, b = 19.4 Å, and c = 19.4 Å. The AIMD simulations were performed for 7 ps with a time step of 1.0 fs under 300 K. Based on our AIMD simulations, it can be seen that generally the α-BnPn clusters have relatively high stability in the presence of O2, N2, and H2O molecules. Obviously, after 7 ps of contact, some O2 molecules dissociated into O atoms and chemisorbed onto the α-B12P12 surface. The P–O bonds with a bond length of about 1.524 Å are short and could be polar due to the difference in the electronegativity between the P and O atoms. Apart from the negative outcomes due to the α-B12P12 interaction with the O2 molecule, controlled passivation might lead to new stable structures for gas sensing. It should be reminded that the basic structure of the α-B12P12 cluster is also well maintained after 7 ps of contact. In contrast, the phenomena of O2 dissociation has not been found on the α-B24P24 surface in the AIMD simulation (see Fig. 6), which indicated the high oxidation resistance of the α-B24P24 structure. This result is very exciting and is different from the result we expected. Furthermore, the simulations identify that no interaction is observed between the α-B12P12/α-B24P24 structures with N2 and H2O molecules, which indicates the high stability of this type of structure. To confirm the chemical stability of the α-BnPn clusters, the adsorption of O2, N2, and H2O gas molecules onto the α-BnPn surfaces was performed. The adsorption energy (Eads; eV per gas molecule) can be calculated as follows.
| Eads = (Ecluster–gas − (Ecluster + nEgas)/n |
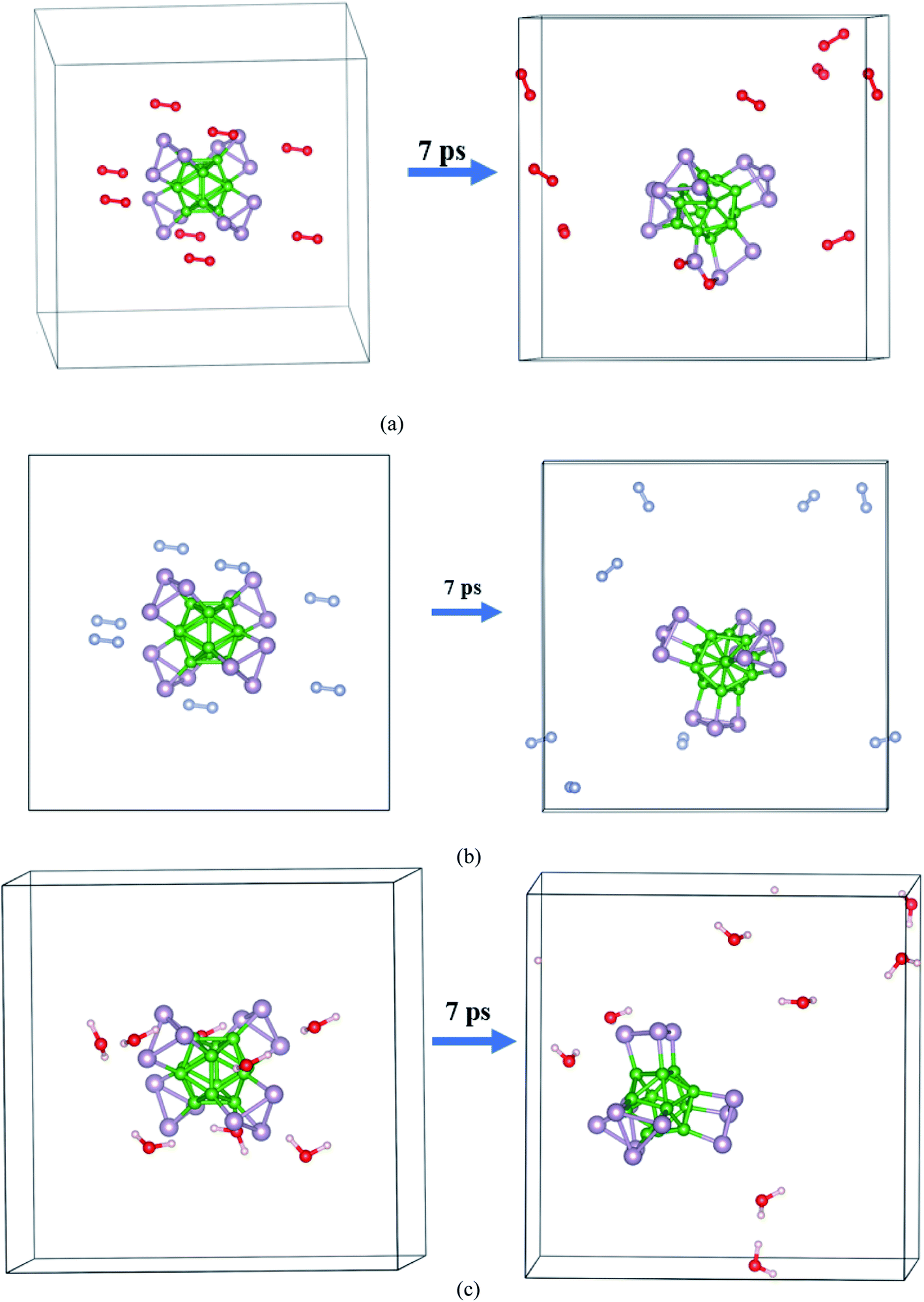 | ||
| Fig. 5 Initial and final snapshots of the α-B12P12 structure with gaseous phase (a) O2, (b) N2, and (c) H2O after 7 ps AIMD simulations at the temperature of 300 K. | ||
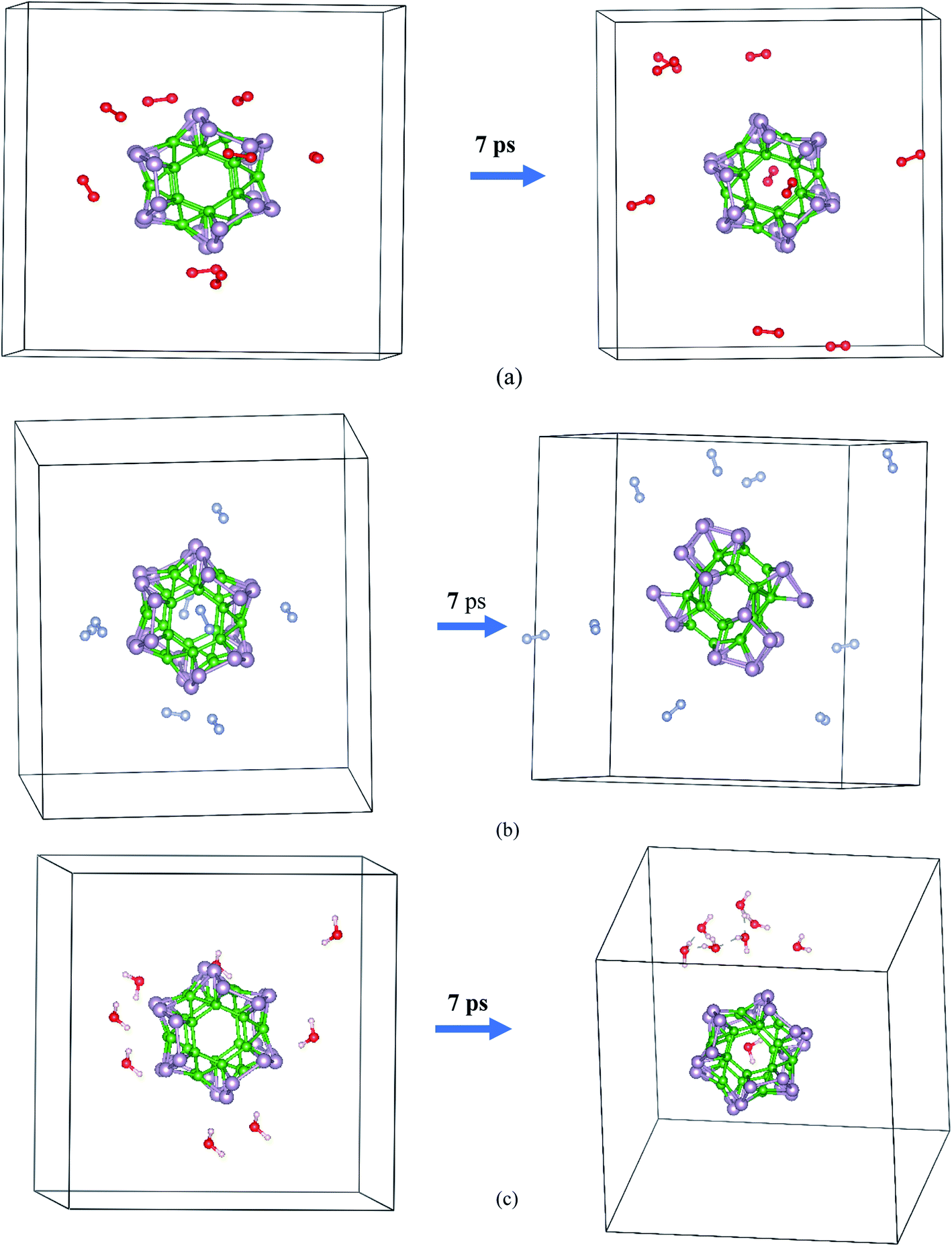 | ||
| Fig. 6 Initial and final snapshots of the α-B24P24 structure with gaseous phase (a) O2, (b) N2, and (c) H2O after 7 ps AIMD simulations at the temperature of 300 K. | ||
3.3. Electronic and optical properties
Based on the relaxed geometries, the electronic properties of the most stable structure, along with the first low-lying isomer of the BnPn structures, were also investigated. The HOMO–LUMO (H–L) energy gap (Egap) is a fruitful parameter for examining the kinetic stability in molecules. The Egap is the energy needed to excite the electron from the HOMO to the LUMO, where a large energy gap implies more kinetic stability and less reactivity. The HSE06 energy gaps of the ground-state structures and the first low-lying isomer of BnPn are summarized in Table 2. As represented in Table 2, the H–L energy gaps of the α-BnPn and β-BnPn clusters are in the range of ∼0.67–3.2 eV, while the energy gaps in the β-BnPn fullerene-like structures are higher than those of the corresponding α-BnPn structures. It is revealed that α-B12P12 and α-B24P24 are semiconductor structures with the HSE06 band gap of 0.67 and 0.62 eV respectively, which is comparable with the band gap of the 2D-BP structure.28 In contrast, β-B12P12 and β-B24P24 fullerene-like structures have large band gaps of 3.2 and 2.98 eV, respectively. The small band gap value suggests modest chemical stability while the sizeable HOMO–LUMO energy gap is representative of the sufficient chemical stability of the studied clusters. The charge density distribution of the HOMO and LUMO for both the α-BnPn and β-BnPn structures are presented in Fig. 7. As seen, the HOMO and LUMO orbitals are generally composed of p-orbitals of phosphorus and boron atoms, and they have been distributed over the boron units and B–P bonds. In case of α-BnPn, each P atom with sp3 configuration has one lone pair of electrons in the fourth orbital (bonded with three neighbors of two P and one B atoms).81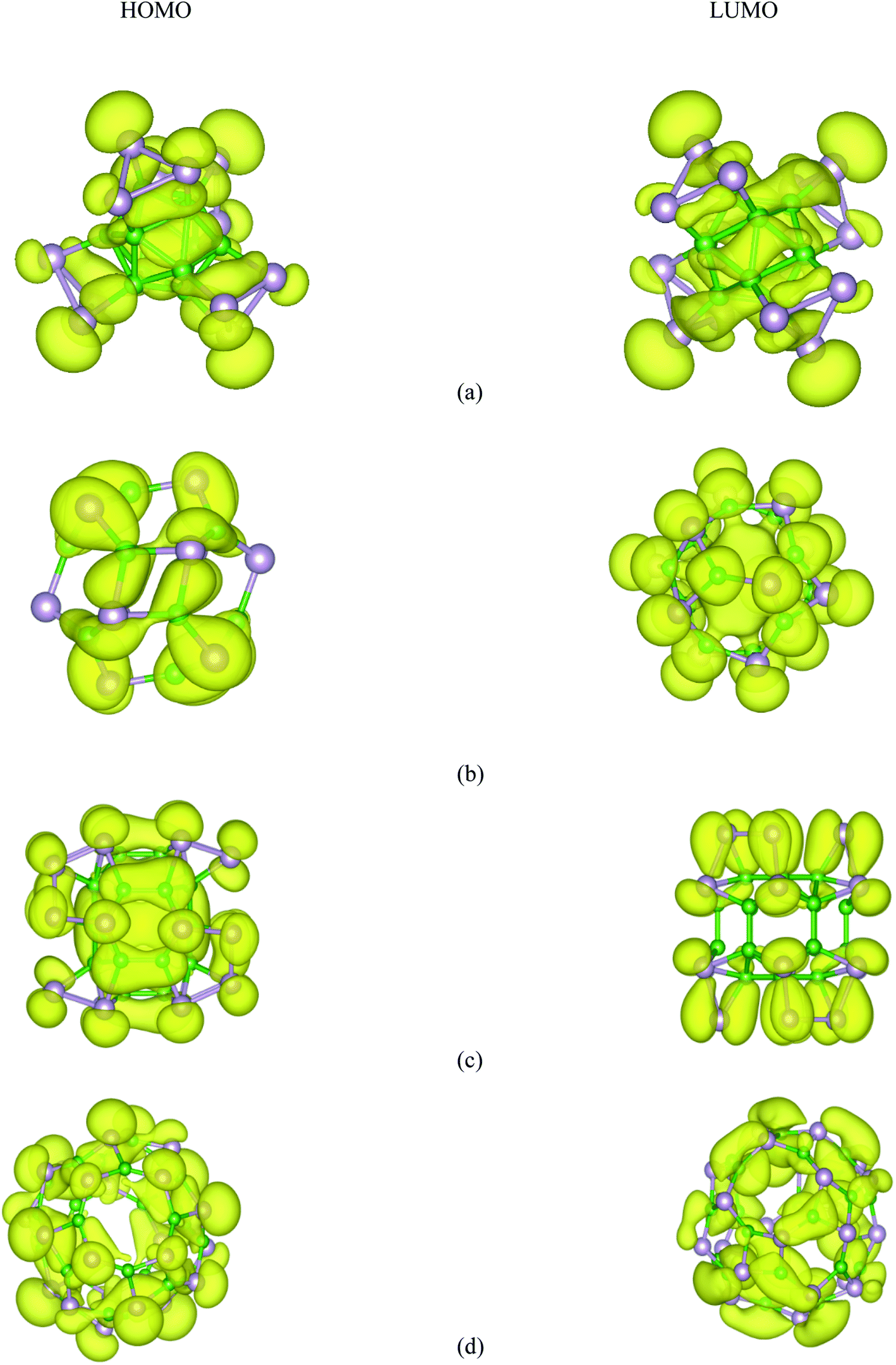 | ||
| Fig. 7 HSE06-DFT calculated contour plots of the HOMO and LUMOs orbitals of (a) α-B12P12, (b) β-B12P12, (c) α-B24P24, and (d) β-B24P24. | ||
Phosphorus has a higher electronegativity than the boron atom; hence, it is expected that the charge will be transferred from the B to the P atoms in the BP clusters. Bader charge analysis method is evaluated using the HSE06 functional82 and the corresponding values are collected in Table 2. As expected, boron atoms become slightly positively charged with the loss of electron. The amount of total charge transfer from the B atoms to the P atoms in the β-BnPn structures is more significant than the corresponding α-BnPn structures. Due to the charge transfer between B and P atoms, it is demonstrated that the BnPn clusters have polar properties, especially for the β-BnPn structures. It can be concluded that charge transfer between the B and P atoms demonstrates that electrostatic interactions have a significant effect on the stability of these types of structures.
To understand more about the nature of bonds in the BnPn structures, the electron localization function (ELF) calculated by VASP is also investigated. ELF can represent electron localization in a molecule and in the solid-state, and its values span from 0 to 1, which illustrates the degree of electron localization. Generally, the considerable value (∼1) of ELF is consistent with the strong covalent bonds or lone pair of electrons. On the other hand, a small value of ELF points out ionic or metallic bonds, and it shows the low electron density localization. Fig. 8 and S5† represent the ELF (isovalue = 0.8 au) of the BnPn structures. From these results, it can be realized that electron localization around the P atoms is slightly more than that of the B atoms, which in good agreement with Bader charge analysis. Moreover, the line plots of the ELF values for the B–P bonds indicates the covalent nature of these bonds in the BnPn structures (Fig. S6†).
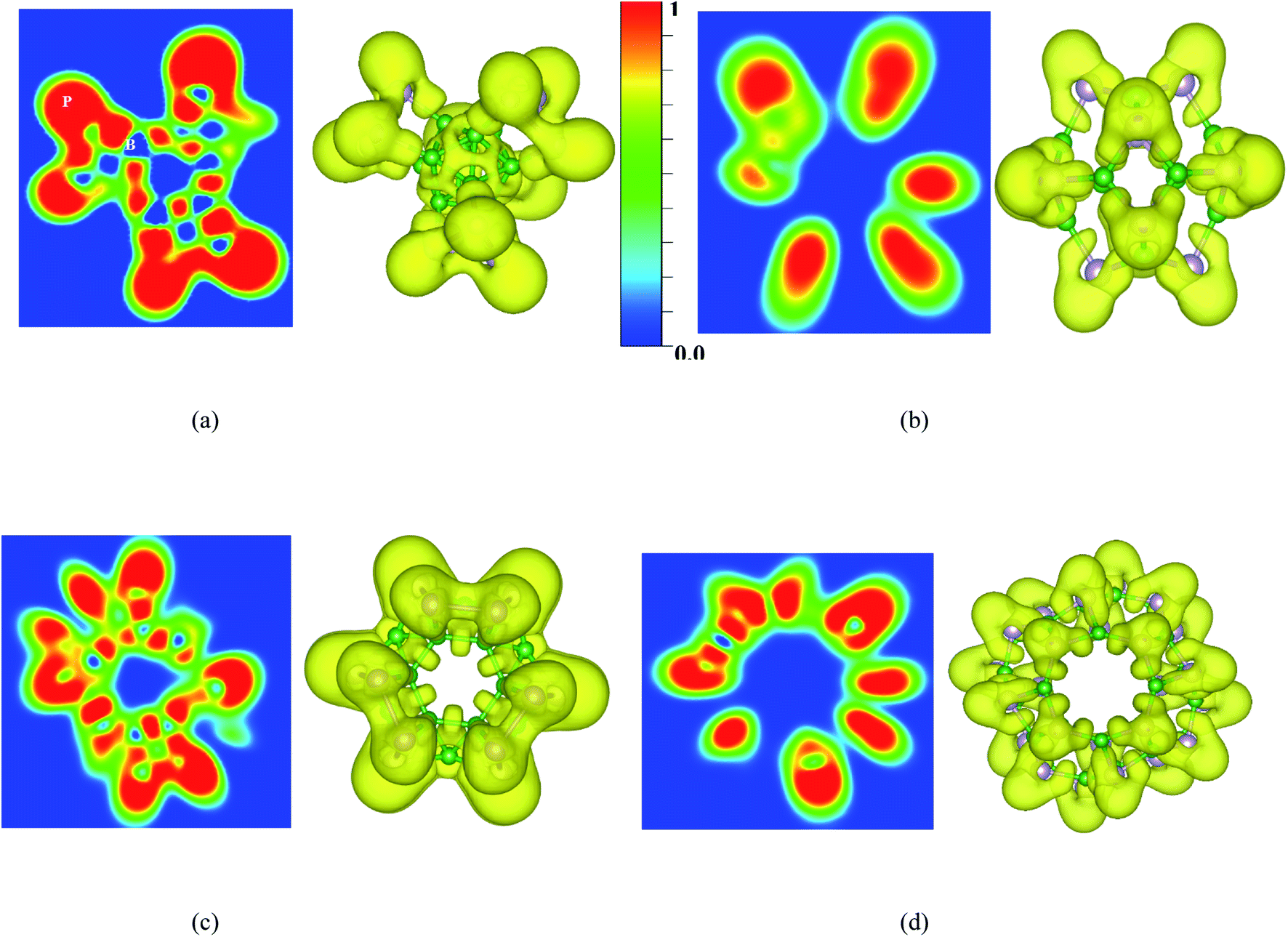 | ||
| Fig. 8 Contour and 3D plots of the electron localization function (ELF) (isovalue = 0.8 au) for (a) α-B12P12, (b) β-B12P12, (c) α-B24P24, and (d) β-B24P24 calculated by the HSE06 functional. | ||
The high stability and moderate band gap feature of the investigated BnPn clusters motivate us to explore the optical properties of the entitled structures. The optical properties of the BnPn clusters have also been studied using the HSE06 functional. To investigate the performance under light, the optical absorption coefficient α was calculated according to the equation below.75,83
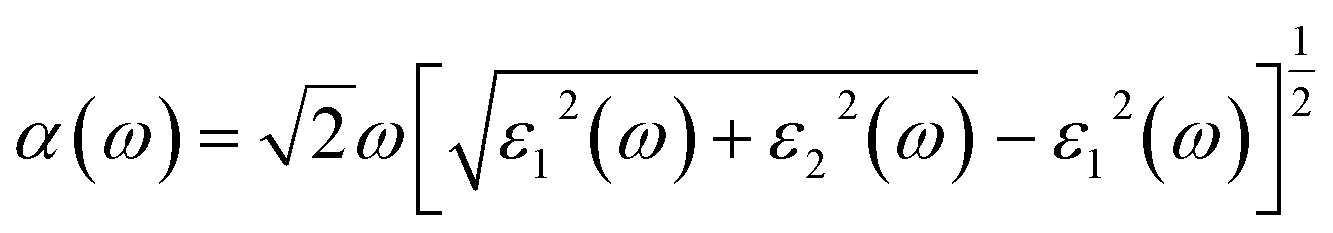 | (2) |
 | ||
| Fig. 9 Optical absorption spectrum of (BP)n with n = 12 and 24 clusters calculated by the HSE06 functional. The energy range corresponding to visible light is indicated by vertical red-dashed lines. | ||
Besides, we focused on the potential applications of these compounds for photocatalytic water-splitting. First, the HSE06 position of the valence band maximum (VBM) and the conduction band minimum (CBM) was calculated from the difference between the electrostatic potential at the vacuum region and is shown in Fig. 10. As discussed above, the β-B12P12 and β-B24P24 structures have a band gap of 3.2 and 2.98 eV, respectively, which is located in the visible-light region. The optical absorption of the β-B12P12 and β-B24P24 fullerene-like structures, as shown in Fig. 9, represents that the β-B12P12 and β-B24P24 clusters have expectantly optical absorption in the visible-light region. The optical absorption spectrum of β-B24P24 starts from ∼550 nm, belonging to 2.25 eV, where the band gap of β-B24P24 is 2.98 eV. On the other hand, the band edge alignments concerning the oxygen and hydrogen evolution potential levels should be located at proper potentials. Our calculations demonstrate that the VBMs and CBMs of the β-B12P12 and β-B24P24 clusters meet both conditions for photocatalytic water splitting. As seen from Fig. 10, their valence bands lie at more positive potentials than the water oxidation potential (O2/H2O potential) and their conduction bands (CBMs) are more negative than the hydrogen reduction potential (H+/H2 potential). These results clearly show that the β-BnPn structures have great potential for applications as a visible-light photocatalyst for water splitting. In contrast, the strong absorption coefficient of the α-B24P24 (which can reach a higher order of 5 × 105 cm−1) and α-B12P12 systems is comparable to that of the organic perovskite solar cell. These results reveal that α-B24P24 and α-B12P12 have great potential for applications as optical absorbent materials in solar cells and optoelectronic devices.
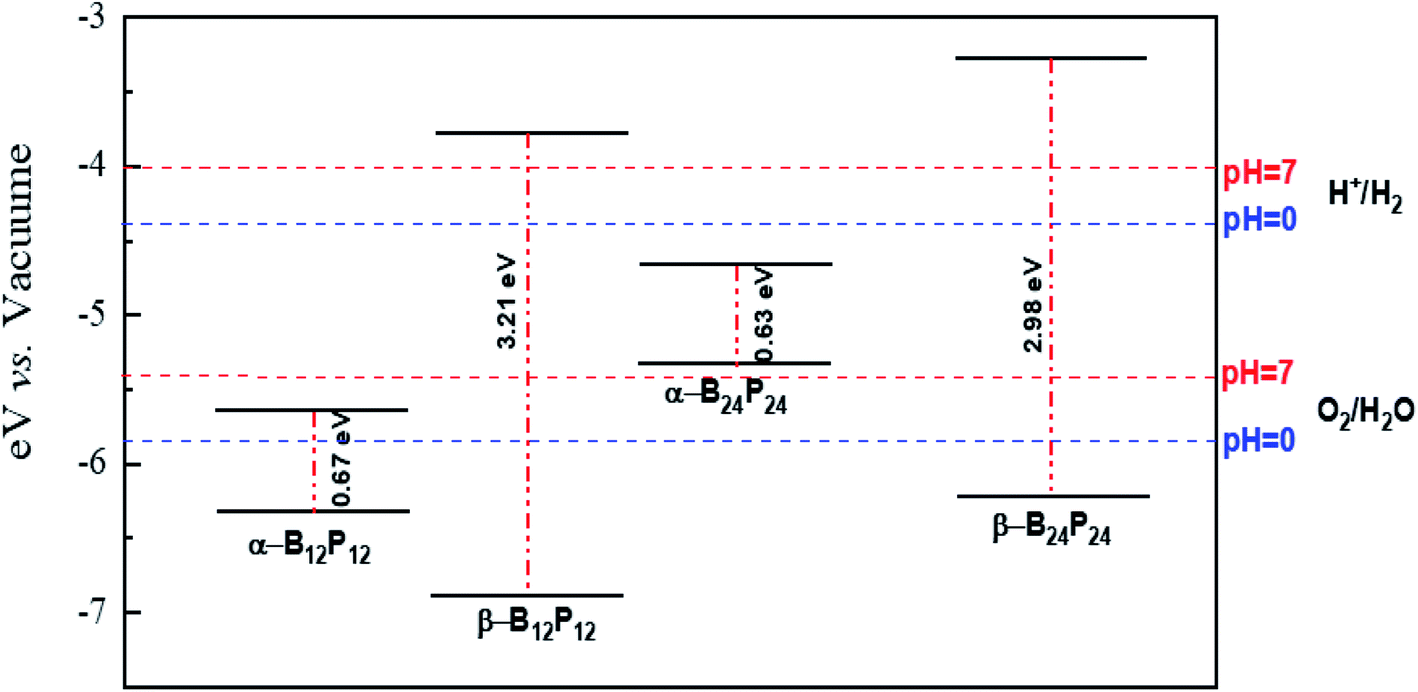 | ||
| Fig. 10 The local location of VBM and CBM with respect to the vacuum level, calculated with the HSE06 functional. The redox potentials of water splitting at pH = 0 (blue dashed line) and pH = 7 (red dashed line) are shown for comparison. | ||
3.4. NLO properties
Based on the research conducted to investigate the ability of (XY)n fullerene-like clusters as NLO materials,31,38,85,86 it has motivated us to investigate the second-order NLO properties of both α-BnPn and β-BnPn structures. Second-order NLO materials that can alter the wavelength of lasers are of considerable interest because of the potential of utilization of such materials in optical switching, optical computing, and photonic technology.31,38,85,86 Quartz crystal (α-SiO2) is a first second-order NLO material, as demonstrated by Franken et al. in 1961.87,88 In most recent studies, the ability of metal encapsulated (XY)n nano-cages (X = B, Al, and Y = N, P) as NLO materials has been considered. They found that the (XY)n nano-cages have zero dipole moment and zero hyperpolarizability. It should be noted that none of these studies have examined the second-order NLO properties. Herein, for both α-BnPn and β-BnPn structures, the average polarizability (α), first hyper-polarizabilities (β0), and second hyperpolarizability (γ) were calculated at the CAM-B3LYP/def2-tzvp level of theory using the Gaussian 09 package.89 As seen in Table 3, the mean polarizabilities (α) of α-BnPn are higher than those of the corresponding β-BnPn structures. The maximum value of α is related to the α-B24P24 structure with 965.24 a.u., which is higher than that of the many encapsulated metal–(XY)n nano-cages.31,86 Furthermore, our calculations proved that both α-BnPn and β-BnPn structures have nearly zero first hyper-polarizabilities (β0). The second hyper-polarizabilities (γ) of α-BnPn and β-BnPn are also summarized in Table 3. The second hyper-polarizabilities of all the predicted structures are higher than the experimental values of γ for C60 and C70 fullerenes,90,91 and comparable to that of the best NLO materials reported in the literature. On the other hand, α-B12P12 and α-B24P24 have larger γ values than the corresponding β-BnPn clusters. The γ value of α-B24P24 is 29 times greater than the second hyper-polarizability value of the β-B24P24 clusters. In conclusion, the strong interactions, effective intermolecular charge transfer, and cluster shape affect the magnitude of the second hyper-polarizabilities of the α-B12P12 and α-B24P24 structures. Furthermore, the α-B12P12 and α-B24P24 clusters show not only high stability but also have large second hyperpolarizability. Hence, they are anticipated to be excellent potential unprecedented candidates for second-order NLO materials.| Type | 〈α〉 | β 0 | 〈γ〉 |
|---|---|---|---|
| α-B12P12 | 476 | 10.11 | 79172.77 |
| β-B12P12 | 393.22 | 0.05 | 31780.03 |
| α-B24P24 | 965.24 | 385.97 | 2581589 |
| β-B24P24 | 867.12 | 0.78 | 89540.24 |
4. Conclusion
In this work, we explored the new stable structure of BnPn (n = 12, 24) clusters with a modern method of crystal structure prediction, USPEX conjugated with spin-polarized first-principles calculations. Our prediction demonstrates that the most stable structures of B12P12 and B24P24 are α-B12P12 and α-B24P24. Based on the relaxed geometries of the α-BnPn structures, each phosphorus atom is doped into a boron atom while the B atoms form a Bn unit. In contrast, the first low-lying β-BnPn isomers of the (BP)n clusters are shaped like fullerenes, where each boron atom is bonded to each phosphorus atom to make cage structures. The calculated binding energies for these novel structures are in the range from −4.723 to −4.803 eV per atom, which is much better or comparable to that of other 2D materials and clusters, indicating their excellent stability and possible experimental synthesis. Furthermore, α-BnPn structures are more stable than the corresponding β-BnPn structures, and the maximum binding energy of −4.803 eV per atom belongs to the α-B24P24 structure. Based on our AIMD simulations, it was found that α-BnPn structures have relatively high stability in the presence of O2, N2, and H2O molecules, in particular, α-B24P24 shows superior oxidation resistance. In the case of thermal stability, α-BnPn and β-BnPn exhibit better thermal stability, where the upper limit temperature that the structures can tolerate is 900 K.It is revealed that α-B12P12 and α-B24P24 are semiconductor structures with the HSE06 band gap of 0.67 and 0.62 eV, respectively, which is comparable with the band gap of the 2D-BP structure. In contrast, β-B12P12 and β-B24P24 fullerene-like structures have large band gaps of 3.2 and 2.98 eV, respectively. The calculated Bader charges illustrate their ionic characters with charge transfers from the B to P atoms. The electronic properties of these novel compounds illustrate a higher degree of absorption in the UV and visible-region with an absorption coefficient larger than 105 cm−1, which suggests a wide range of opportunities for advanced optoelectronic applications. The β-BnPn phase has suitable band alignments in the visible-light excitation region, which will produce enhanced photocatalytic water splitting. On the other hand, α-BnPn structures with high absorption coefficient exhibit large second hyperpolarizability, which are expected to have excellent potential as second-order NLO materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Shahid Chamran University of Ahvaz for their support of this scientific research. Also, we are grateful to the Research Council of Shahid Chamran University of Ahvaz for financial support (SCU.SC98.669).References
- L. Leszczynski and I. Yanov, J. Phys. Chem. A, 1999, 103, 396–401 CrossRef.
- L. O. Jones, M. A. Mosquera, B. Fu, G. C. Schatz, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2019, 141, 1672–1684 CrossRef CAS PubMed.
- S. Ostrowski, P. Garnuszek and J. C. Dobrowolski, Spectrochim. Acta, Part A, 2020, 231, 117791 CrossRef CAS PubMed.
- A. K. Srivastava, S. K. Pandey and N. Misra, J. Nanostruct. Chem., 2016, 6, 103–109 CrossRef CAS.
- S. Yoo, N. Shao and X. C. Zeng, J. Chem. Phys., 2008, 128, 104316 CrossRef PubMed.
- Z. Mahdavifar, M. Afshari, A. Bagheri and A. Arab, Polyhedron, 2021, 193, 114874 CrossRef CAS.
- Z. G. Fthenakis, R. W. A. Havenith, M. Menon and P. W. Fowler, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 155435 CrossRef.
- J. Beheshtian, A. A. Peyghan and Z. Bagheri, Appl. Surf. Sci., 2012, 259, 631–636 CrossRef CAS.
- Z. Mahdavifar, Z. Nomresaz and E. Shakerzadeh, Chem. Phys., 2020, 530, 110606 CrossRef CAS.
- L. C. Bo, G. S. Wei, Y. K. Xiao, L. Cheng, X. X. Xin, X. S. Hong and M. George, Inorg. Chem., 2018, 57, 343–350 CrossRef CAS PubMed.
- Z. Cui, C. Chen, Q. Wang, L. Zhao, M. Wang and Y. Ding, New J. Chem., 2020, 44, 17705–17713 RSC.
- M. S. Messina, J. C. Axtell, Y. Q. Wang, P. Chong, A. I. Wixtrom, K. O. Kirlikovali, B. M. Upton, B. M. Hunter, O. S. Shafaat, S. I. Khan, J. R. Winkler, H. B. Gray, A. N. Alexandrova, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 6952–6955 CrossRef CAS PubMed.
- Y. Wang, M. M. Guo, G. L. Wang, C. Q. Miao, N. Zhang and T. D. Xue, Phys. Chem. Chem. Phys., 2020, 22, 20362–20367 RSC.
- C. Wu, H. Wang, J. J. Zhang, G. Y. Gou, B. Pan and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 2526–2532 CrossRef CAS PubMed.
- P. Li, X. Du, J. J. Wang, C. Lu and H. Chen, J. Phys. Chem. C, 2018, 122, 20000–20005 CrossRef CAS.
- Z. Mahdavifar and F. Shojaei, Phys. Chem. Chem. Phys., 2019, 21, 22618–22628 RSC.
- W. Huang, A. P. Sergeeva, H. J. Zhai, B. B. Averkiev, L. S. Wang and A. I. Boldyrev, Nat. Chem., 2010, 2, 202–206 CrossRef CAS PubMed.
- S. Munsif and K. Ayub, Chem. Phys. Lett., 2018, 698, 51–59 CrossRef CAS.
- Z. Liu, R. Huang and L. Zheng, Z. Phys. D: At., Mol. Clusters, 1996, 38, 171–177 CrossRef CAS.
- S. P. Huber, V. V. Medvedev, E. Gullikson, B. Padavala, J. H. Edgar, R. W. E. Kruijs, F. Bijkerk and D. Prendergasta, Phys. Chem. Chem. Phys., 2017, 19, 8174–8187 RSC.
- H. L. Alves and K. Kunc, J. Phys.: Condens. Matter, 1992, 4, 6603 CrossRef CAS.
- V. Ferreira and H. L. Alves, J. Cryst. Growth, 2008, 310, 3973–3978 CrossRef CAS.
- B. Padavala, C. Frye, Z. Ding, R. Chen, M. Dudley, B. Raghothamachar, N. Khan and J. H. Edgar, Solid State Sci., 2015, 47, 55–60 CrossRef CAS.
- B. Padavala, C. D. Frye, X. Wang, Z. Ding, R. Chen, M. Dudley, B. Raghothamachar, P. Lu, B. N. Flanders and J. H. Edgar, Cryst. Growth Des., 2016, 16, 981–987 CrossRef CAS.
- G. Li, J. K. Abbott, J. D. Brasfield, P. Liu, A. Dale, G. Duscher, P. D. Rack and C. S. Feigerle, Appl. Surf. Sci., 2015, 327, 7–12 CrossRef CAS.
- L. Shi, P. Li, W. Zhou, T. Wang, K. Chang, H. Zhang, T. Kako, G. Liu and J. Ye, Nano Energy, 2016, 28, 158–163 CrossRef CAS.
- A. N. Caruso, J. Phys.: Condens. Matter, 2010, 22, 443201 CrossRef CAS PubMed.
- Z. Zhu, X. Cai, C. Niu, C. Wang and Y. Jia, Appl. Phys. Lett., 2016, 109, 153107 CrossRef.
- A. Shokuhi Rad and K. Ayub, J. Mol. Liq., 2018, 255, 168–175 CrossRef.
- A. A. Salari, Inorg. Chim. Acta, 2017, 456, 18–23 CrossRef CAS.
- K. Ayub, J. Mater. Chem. C, 2016, 4, 10919–10934 RSC.
- Y. Qu, W. Ma, X. Bian, H. Tang and W. Tian, Int. J. Quantum Chem., 2006, 106, 960–967 CrossRef CAS.
- A. K. Kandalam, M. A. Blanco and R. Pandey, J. Phys. Chem. B, 2002, 106, 1945–1953 CrossRef CAS.
- J. Wu, J.-H. Li and Y.-X. Yu, Phys. Chem. Chem. Phys., 2020, 22, 7633–7642 RSC.
- A. Costales, A. K. Kandalam, R. Franco and R. Pandey, J. Phys. Chem. B, 2002, 106, 1940–1944 CrossRef CAS.
- D. L. Strout, J. Phys. Chem. A, 2000, 104, 3364–3366 CrossRef CAS.
- R. Wang, D. Zhang and C. Liu, Chem. Phys. Lett., 2005, 411, 333–338 CrossRef CAS.
- Maria, J. Iqbal, R. Ludwig and K. Ayub, Mater. Res. Bull., 2017, 92, 113–122 CrossRef CAS.
- T. Oku, M. Kuno, H. Kitahara and I. Narita, Int. J. Inorg. Mater., 2001, 3, 597–612 CrossRef CAS.
- T. Oku, A. Nishiwaki and I. Narita, Sci. Technol. Adv. Mater., 2004, 5, 635–638 CrossRef CAS.
- T. Oku, A. Nishiwaki, I. Narita and M. Gonda, Chem. Phys. Lett., 2003, 380, 620–623 CrossRef CAS.
- G. Seifert, P. W. Fowler, D. Mitchell, D. Porezag and T. Frauenheim, Chem. Phys. Lett., 1997, 268, 352–358 CrossRef CAS.
- A. Shokuhi Rad, S. M. Aghaei, V. Poralijan, M. Peyravi and M. Mirzaei, Comput. Theor. Chem., 2017, 1109, 1–9 CrossRef.
- A. R. Oganov and S. Ono, Nature, 2004, 430, 445–448 CrossRef CAS PubMed.
- A. O. Lyakhov and A. R. Oganov, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 092103 CrossRef.
- S. V. Lepeshkin, V. S. Baturin, Y. A. Uspenskii and A. R. Oganov, J. Phys. Chem. Lett., 2019, 10, 102–106 CrossRef CAS PubMed.
- A. J. Mannix, X. F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154122 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106–224115 CrossRef PubMed.
- K. A. Tikhomirova, C. Tantardini, E. V. Sukhanova, Z. I. Popov, S. A. Evlashin, M. A. Tarkhov, V. L. Zhdanov, A. A. Dudin, A. R. Oganov, D. G. Kvashnin and A. G. Kvashnin, J. Phys. Chem. Lett., 2020, 11(10), 3821–3827 CrossRef CAS PubMed.
- I. A. Kruglov, A. Yanilkin, A. R. Oganov and P. Korotaev, Phys. Rev. B: Condens. Matter Mater. Phys., 2019, 100, 174104 CrossRef CAS.
- A. G. Kvashnin, C. Tantardini, H. A. Zakaryan, Y. A. Kvashnina and A. R. Oganov, Chem. Mater., 2020, 32(16), 7028–7035 CrossRef CAS.
- J. He, E. Wu, H. Wang, R. Liu and Y. Tian, Phys. Rev. Lett., 2005, 94, 015504 CrossRef PubMed.
- Z. Lu, Y. Pang, S. Li, Y. Wang, Z. Yang, D. Ma and R. Wu, Appl. Surf. Sci., 2019, 479, 590–594 CrossRef CAS.
- J. L. Hoard, S. Geller and R. E. Hughes, J. Am. Chem. Soc., 1951, 73(4), 1892–1893 CrossRef CAS.
- E. P. Domashevskaya, N. E. Solovjev, V. A. Terekhov and Y. A. Ugai, J. Less-Common Met., 1976, 47, 189–193 CrossRef CAS.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- C. H. L. Quay, J. Cumings, S. J. Gamble, A. Yazdani, H. Kataura and D. Goldhaber-Gordon, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 073404 CrossRef.
- D. C. Pestana and P. P. Power, J. Am. Chem. Soc., 1991, 113, 8426–8437 CrossRef CAS.
- I. A. Popov, Z. A. Piazza, W.-L. Li, L. S. Wang and A. I. Boldyrev, J. Chem. Phys., 2013, 139, 144307 CrossRef PubMed.
- J. Lv, Y. Wang, L. Zhang, H. Lin, J. Zhao and Y. Ma, Nanoscale, 2015, 7, 10482–10489 RSC.
- A. B. Rahane, P. Saha, N. Sukumar and V. Kumar, arXiv:1907.12611, 2019..
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Chem. Rev., 2020, 120, 9021–9163 CrossRef CAS PubMed.
- Q. Wei and X. Peng, Appl. Phys. Lett., 2014, 104, 251915 CrossRef.
- D. Singh, S. Kansara, S. K. Gupta and Y. Sonvane, J. Mater. Sci., 2018, 53, 8314–8327 CrossRef CAS.
- Y. Li, Y. Liao, P. v. R. Schleyer and Z. Chen, Nanoscale, 2014, 6, 10784–10791 RSC.
- A. Fleurence, R. Friedlein, T. Ozaki, H. Kawai, Y. Wang and Y. Yamada-Takamura, Phys. Rev. Lett., 2012, 108, 245501 CrossRef PubMed.
- Z. Zhu, X. Cai, C. Niu, C. Wang and Y. Jia, Appl. Phys. Lett., 2016, 109, 153107 CrossRef.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727–731 CrossRef CAS PubMed.
- N. Lu, Z. Zhuo, Y. Wang, H. Guo, W. Fa, X. Wu and X. C. Zeng, J. Phys. Chem. Lett., 2018, 9, 6568–6575 CrossRef CAS PubMed.
- S. Jiang, J. Li, W. Chen, H. Yin, G. P. Zheng and Y. Wang, Nanoscale, 2020, 12, 5888–5897 RSC.
- K. L. Kuntz, R. A. Wells, J. Hu, T. Yang, B. Dong, H. Guo, A. H. Woomer, D. L. Druffel, A. Alabanza, D. Tománek and S. C. Warren, ACS Appl. Mater. Interfaces, 2017, 9, 9126–9135 CrossRef CAS PubMed.
- J. O. Island, G. A. Steele, H. S. J. van der Zant and A. Castellanos-Gomez, 2D Mater., 2015, 2, 11002 CrossRef.
- Y. Huang, J. Qiao, K. He, S. Bliznakov, E. Sutter, X. Chen, D. Luo, F. Meng, D. Su, J. Decker, W. Ji, R. S. Ruoff and P. Sutter, Chem. Mater., 2016, 28, 8330–8339 CrossRef CAS.
- J. Pei, X. Gai, J. Yang, X. Wang, Z. Yu, D.-Y. Choi, B. Luther-Davies and Y. Lu, Nat. Commun., 2016, 7, 10450 CrossRef CAS PubMed.
- W. Luo, D. Y. Zemlyanov, C. A. Milligan, Y. Du, L. Yang, Y. Wu and P. D. Ye, Nanotechnology, 2016, 27, 434002 CrossRef PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, CA, 2006 Search PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef.
- S. Saha, T. P. Sinha and A. Mookerjee, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 8828–8834 CrossRef CAS.
- Z. Mahdavifar and F. Shojaei, J. Alloys Compd., 2021, 854, 157220 CrossRef CAS.
- F. Ullah, N. Kosar, K. Ayub, M. A. Gilani and T. Mahmood, New J. Chem., 2019, 43, 5727–5736 RSC.
- M. Niu, G. Yu, G. Yang, W. Chen, X. Zhao and X. Huang, Inorg. Chem., 2014, 53, 349–358 CrossRef CAS PubMed.
- H. Liu, B. Zhang and Y. Wang, Chem. Commun., 2020, 56, 13689–13701 RSC.
- P. A. Franken, A. E. Hill, C. W. Peters and G. Weinreich, Phys. Rev. Lett., 1961, 7, 118–119 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts,B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov,J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini,F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson,D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega,G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota,R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi,J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09 Revision D.01, GaussianInc., Wallingford CT, 2009 Search PubMed.
- D. S. Sabirov, RSC Adv., 2014, 4, 44996–45028 RSC.
- A. A. Quong and M. R. Pederson, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 12906–12909 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na01040e |
| This journal is © The Royal Society of Chemistry 2021 |