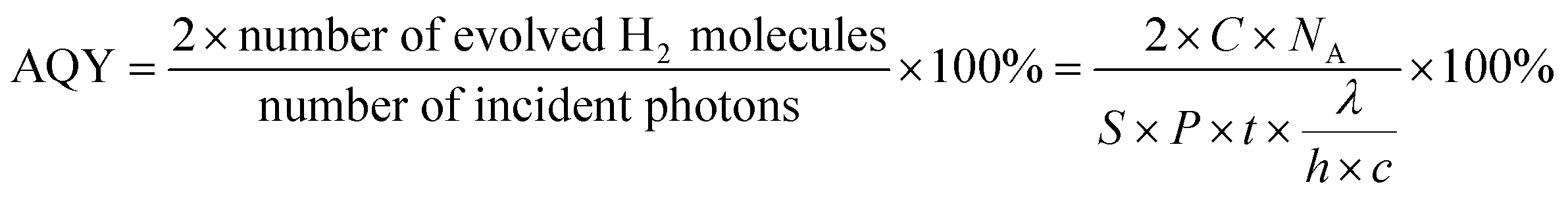DOI:
10.1039/D0NA01011A
(Paper)
Nanoscale Adv., 2021,
3, 1699-1707
Construction of polymeric carbon nitride and dibenzothiophene dioxide-based intramolecular donor–acceptor conjugated copolymers for photocatalytic H2 evolution†
Received
2nd December 2020
, Accepted 28th January 2021
First published on 28th January 2021
Abstract
Polymeric carbon nitride (g-C3N4) has succeeded as a striking visible-light photocatalyst for solar-to-hydrogen energy conversion, owing to its economical attribute and high stability. However, due to the lack of sufficient solar-light absorption and rapid photo-generated carrier recombination, the photocatalytic activity of raw g-C3N4 is still unsatisfactory. Herein, new intramolecular g-C3N4-based donor–acceptor (D–A) conjugated copolymers have been readily synthesized by a nucleophilic substitution/condensation reaction between urea and 3,7-dihydroxydibenzo[b,d]thiophene 5,5-dioxide (SO), which is strategically used to improve the photocatalytic hydrogen evolution performance. The experimental results demonstrate that CNSO-X not only improves light utilization, but also accelerates the spatial separation efficiency of the photogenerated electron–hole pairs and increases the wettability with the introduction of SO. In addition, the adsorption energy barrier of CNSO-X to H* has a significant reduction via theoretical calculation. As expected, the CNSO-20 realizes the best photocatalytic H2 evolution activity of 251 μmol h−1 (50 mg photocatalyst, almost 8.5 times higher than that of pure CN) with an apparent quantum yield of 10.16% at 420 nm, which surpasses most strategies for the organic molecular copolymerization of carbon nitride. Therefore, this strategy opens up a novel avenue to develop highly efficient g-C3N4 based photocatalysts for hydrogen production.
1. Introduction
Hydrogen energy is a clean secondary energy source with high energy density, high heating value, zero pollution, and zero carbon emissions. It is conducive to resolving the energy crisis and environmental pollution, and widely known as the “ultimate energy” in the 21st century.1–4 Using semiconductor-based photocatalytic technology for water-splitting to produce hydrogen can convert low-density and clean solar energy into high-density chemical energy, which has far-reaching application prospects in solving the problem of energy shortage.5–7 However, solar hydrogen energy conversion is limited by a variety of dynamic and thermodynamic factors. At present, the highest solar energy conversion hydrogen energy efficiency achieved by semiconductor materials is still far from the requirements of industrialization. To solve the critical problem of the application of solar water splitting hydrogen production technology, the key lies in developing efficient and visible-light responsive photocatalysts.8–10
In recent years, from the perspective of cost reduction, the research and development of non-metal photocatalysts has attracted widespread attention. As the most stable species structure of carbon nitride compounds, g-C3N4 has been mirrored to possess visible-light catalytic activity.11–15 The g-C3N4 photocatalyst has an energy gap of 2.7 eV, and can capture light below 460 nm. Its conduction band (CB) and valence band (VB) are located at −1.10 V and +1.60 V, respectively, which meet the thermodynamic requirements of hydrogen produced by water splitting.16–20 Unfortunately, the photocatalytic performance of pure g-C3N4 for H2 production from water is quite unsatisfactory.21–25 This is because the photo-generated electrons and holes are easily and quickly recombined in g-C3N4, which is released as heat or light.26–30 In addition, the insufficient ability to capture visible light is a significant reason for its low photocatalytic activity.31–33 Until now, many strategies to improve the photocatalytic hydrogen evolution activity of g-C3N4 have been reported, including doping modification technology,34–36 semiconductor composite modification technology,37,38 and specific surface area modification technology.39 In particular, the copolymerization strategy of organic molecules and CN to construct intramolecular donor–acceptor (D–A) conjugated copolymers not only ameliorates the light capture capacity, but also improves the carrier separation efficiency, thereby enhancing the photocatalytic activity.40,41 According to previous reports,42 nitrogen atoms containing lone pair electrons in g-C3N4 are considered to be good donor groups. Therefore, it is necessary to find an optimal acceptor group and introduce it into the g-C3N4 skeleton to construct the intra-D–A structure.
Dibenzothiophene dioxide, which has a rigid structure and excellent electron deficiency, has been widely selected as the candidate for the electron acceptor to construct D–A type conjugated polymers with high photocatalytic activity. For instance, the Cooper group designed and synthesized a series of linear polymer photocatalysts containing dibenzothiophene dioxide acceptors, achieving a H2 evolution rate of up to 3.68 mmol h−1 g−1. Such high activity is even comparable with many inorganic semiconductor photocatalysts.43 In addition, due to the good hydrophilic nature of the dibenzothiophene dioxide group, the introduction can improve the dispersion of the polymer in the aqueous solution, thereby improving the photocatalytic hydrogen evolution activity.44 Nevertheless, there are no reports on the use of dibenzothiophene dioxide and CN copolymers for photocatalytic hydrogen production.
Herein, polymeric carbon nitride-based intramolecular donor–acceptor conjugated copolymers (record as CNSO-X) have been readily obtained through the nucleophilic substitution reaction of urea and 3,7-dihydroxydibenzo[b,d]thiophene 5,5-dioxide (abbreviated as SO) at high temperature, followed by condensation polymerization. As expected, compared with pristine CN, the CNSO-X copolymer photocatalysts display significant redshifted absorption edges, more efficient electron–hole separation, better hydrophilicity and a smaller adsorption H* energy barrier. Furthermore, the energy gap of CNSO-X depends on the SO content, which can be facilely adjusted by varying the addition amount of SO. In particular, the CNSO-20 reveals the supreme H2 evolution activity (251 μmol h−1, 50 mg photocatalyst), which is about 8.5 times greater than that of pure CN. The corresponding apparent quantum yield (AQY) is 10.2% at 420 nm, and the value is superior to most previously reported SO-based D–A conjugated polymers.45–50 To the best of our knowledge, this is the first report on the application of SO and CN-based copolymers for efficient photocatalytic H2 evolution. This work provides a new perspective for the design and development of highly efficient D–A conjugated polymer photocatalysts.
2. Experimental
2.1 Instrumentation and chemicals
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were received in deuterated acetone on a Bruker AM-400 MHz with tetramethyl-silane as an internal standard. High-resolution mass spectral (HRMS) characterization was obtained from the Waters LCT Premier XE spectrometer. The solid-state 13C cross polarization magic angle spinning (13C-CP/MAS) NMR spectra were measured on the Bruker Advance 600 MHz spectrometer. The Fourier transform infrared (FTIR) spectra were determined on a NICOLET 380 spectrometer in the wavenumber range of 4000–400 cm−1. DRS UV-Vis characterizations were obtained on a Varian Cary 500 spectrophotometer. Photoluminescence (PL) spectra were tested on a Hitachi F-4500 fluorescence spectrophotometer. Powder X-ray diffraction (XRD) patterns were recorded on a RigakuD/MAX 2550 diffractometer (Cu K radiation, λ = 1.5406 Å), which operated at 40 kV and 100 mA. Field-emission scanning electron microscope (FE-SEM) images were recorded on the Nova Nanosem 450. Energy-dispersive X-ray spectra (EDS) were conducted employing a SEM equipped with an EDS detector. X-ray Photoelectron Spectroscopy (XPS) spectrograms were recorded on a Perkin-Elmer PHI 5000C ESCA system with Al Kα radiation operating at 250 W. All binding energies were referenced to the C 1s peak at 284.6 eV. Specific surface areas and pore size distributions were systematically analysed via the N2 adsorption–desorption curve at 77.3 K with an ASAP 2420-4 (Micromeritics) volumetric adsorption analyser. Powder samples were degassed offline at 120 °C for 12 h under a dynamic vacuum (10−5 bar) before analysis.
Unless otherwise noted, the materials obtained from commercial suppliers were used without further purification. Glassware were dried in the oven prior to use. All chemical reactions were carried out under an inert Ar atmosphere. The 3,7-dihydroxydibenzo[b,d]thiophene 5,5-dioxide was prepared according to the published literature.51
2.2 Synthesis of 3,7-dihydroxydibenzo[b,d]thiophene 5,5-dioxide
3,7-Dibromodibenzo[b,d]thiophene 5,5-dioxide (500 mg, 1.34 mmol), KOH (329 mg, 5.88 mmol), Pd2(dba)3 (49 mg, 0.053 mmol), t-Bu XPhos (91 mg, 0.214 mmol) were charged in a dried Schlenk flask under N2 atmosphere. Dioxane (1.5 mL) and H2O (1 mL) were sequentially added to this mixture. The resulting solution was continuously stirred at 100 °C for 16 h. The reaction mixture was cooled to room temperature, and acidified with dilute aqueous HCl. The resulting solution was extracted multiple times with ethyl acetate (EA). The EA phase was separated in a separatory funnel, then dried over anhydrous Na2SO4, and the filtrate was filtered and then distilled under reduced pressure. The obtained crude product was further purified with column chromatography on silica gel (PE/EA = 4/1, v/v) to provide 3,7-dihydroxydibenzo[b,d]thiophene 5,5-dioxide (253 mg, yield 76%) as a pale yellow solid. 1H NMR (400 MHz, acetone-d6) δ (TMS, ppm): 9.52 (s, 2H), 7.79 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 7.15–7.12 (m, 2H). 13C NMR (acetone-d6, 100 MHz), δ (TMS, ppm): 159.47, 140.23, 124.04, 123.40, 121.82, 109.08. HRMS (ESI-MS) m/z calcd for C12H8O4S: 247.0065. [M + H+]. Found: 247.0062.
2.3 Preparation of CN and CNSO-X
The CNSO-X photocatalysts were prepared by the copolymerization strategy. Typically, 20 g of urea and X mg of SO (X = 10, 20, 30, 40 and 50) were fully mixed, and then put the mixture into a crucible with a cover by thermally decomposing at 550 °C for 4 h in static air; the heating rate was 2.3 °C min−1. The resulting powder was then transferred to a rectangular crucible without a lid, and subjected to a thermal etching treatment at 500 °C for 2 h at a heating rate of 10 °C min−1 in a muffle furnace. After natural cooling to RT, the obtained photocatalysts were grated and collected for further use. Comparatively, raw g-C3N4 was obtained via the same process without adding SO. Possible thermal polymerization processes during the synthesis of CNSO-X are shown in Scheme S1.† Notably, in order to further improve the photocatalytic activity, 3.0 wt% of Pt was deposited on CNSO-X by photochemical reduction strategy before the photocatalytic reaction.52
2.4 Photoelectrochemical tests
Transient photocurrent analysis (I–t) and electrochemical impedance spectra (EIS) measurements were carried out using a CHI650E electrochemical workstation with a three-electrode system: prepared membrane electrode, Pt plate, and Ag/AgCl as the working electrode, counter electrode, and reference electrode, respectively. A solution of 0.5 M Na2SO4 was used as the supporting electrolyte, and a 300 W Xe-lamp (CEL-HXF 300) with a cut-off filter (420 nm ≤ λ ≤ 780 nm) served as the light source. EIS was determined in the frequency range of 102 to 106 Hz, and the AC amplitude under the open circuit voltage at room temperature was 10 mV.
2.5 Photocatalytic H2 evolution and AQY measurement
The photocatalytic experiments were conducted in a glass gas-closed-circulation system (CEL-SPH2N) under irradiation. Briefly, 50 mg sample was ultrasonically dispersed in a mixed solution containing 60 mL water and 10 mL triethanolamine (TEOA) as the hole quencher. Before light irradiation, the system was always kept in a vacuum. Then, a 300 W xenon lamp connected with a cut-off filter (420 nm ≤ λ ≤ 780 nm) was used to illuminate the reactor from the top. The amount of evolved H2 was detected by an online gas chromatograph (GC 2060, TCD detector and Ar carrier). In addition, the apparent quantum yield (AQY) for H2 production under monochromatic light irradiation (λ = 420, 450, 500, 550, 600, 630 nm) was estimated as the following equation.| |  | (1) |
where C is the H2 production amount (μmol) per hour; NA is the Avogadro constant (6.02 × 1023 mol−1); S is the irradiation area (12.56 cm2); P is the monochromatic light intensity (W cm−2) (P is detected by optical power meter); t is the light exposure time (1 h); λ is the monochromatic light wavelength (nm); h is the Planck constant (6.626 × 10−34 J s); c is the speed of light (3 × 108 m s−1). The light intensity of λ = 420, 450, 500, 550, 600, 630 nm are 1.96, 2.03, 2.46, 2.41, 2.19 and 2.12 mW cm−2, respectively.
2.6 Calculation methods
Density functional theory (DFT) calculations were performed with the Vienna Ab initio Simulation Package (VASP).53 In our calculations, the spin-polarized projector augmented wave (PAW) methods54 were applied together with the Perdew–Burke–Ernzerhof (PBE)55 electron exchange correlation functional of the generalized gradient approximation (GGA).56 The energy cut-off of the wave function in the plane-wave basis was set to 400 eV, and the calculations were implemented until the maximum force upon each relaxed atom was less than 0.05 eV Å−1. To optimize the different molecules, the unit cell was set to 20 Å × 20 Å × 15 Å to avoid the interactions between the neighboring molecules, and a Monkhorst–Pack grid of (2 × 2 × 1) k points was used. In order to study the adsorption of hydrogen and water molecules on the g-C3N4 surfaces, we used the following models: a 3 × 1 unit of monolayer g-C3N4, and all atoms were allowed to relax. The Brillouin-zone integration was performed along with a 2 × 2 × 1 Monkhorst–Pack grid for the different surface slabs, and the vacuum height was set to 15 Å to eliminate the interaction between neighboring slabs. The adsorption energy (Eads) was computed as below:| |  | (2) |
where Etotal is the calculated total energy of the adsorption system, Esubstrate is the calculated energy of the clean substrate and Egas-phase adsorbate is the calculated energy of the gas-phase molecule.
3. Results and discussion
3.1 Morphology and structure of the obtained samples
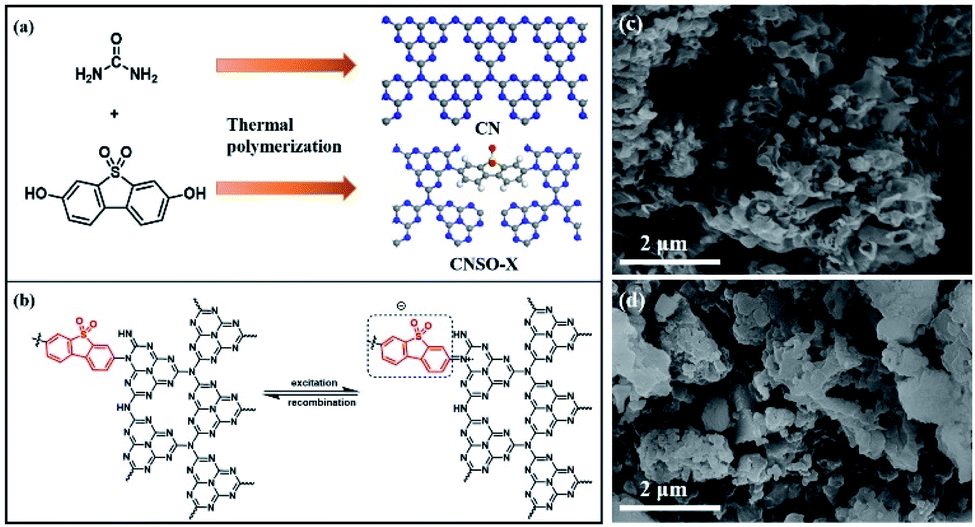
For the SO introduced copolymers, they were marked as CNSO-X, where X (mg) represents the amount of SO added. The CNSO-X were synthesized by the classical nucleophilic substitution organic reaction of urea and 3,7-dihydroxydibenzo[b,d]thiophene 5,5-dioxide at high temperature, followed by condensation polymerization (Fig. 1a). The possible thermal polymerization processes during the synthesis of CNSO-X are shown in Scheme S1.† As indicated in Fig. 1b, the N atoms that have lone pair electrons act as the electron donor (D), and dibenzothiophene-S,S-dioxide acts as the acceptor (A). In the CNSO-X skeletons, the formation of the D–A structure can promote the transfer of the photogenerated carriers, which will be beneficial to the photocatalytic process. As depicted in Fig. 1c and d, the morphologies of the CN-based polymer were investigated by SEM analyses. A similar layered structure of CN and CNSO-20 was found, which indicates that the introduction of SO in the CN framework has little effect on its surface morphology.
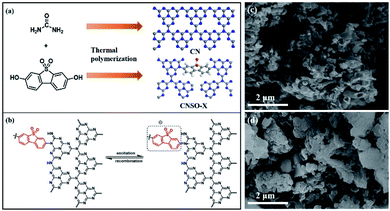 |
| | Fig. 1 (a) The schematic view of the construction; (b) diagram of the two electron transition modes in CNSO-X, and (c and d) SEM images of CN and CNSO-20. | |
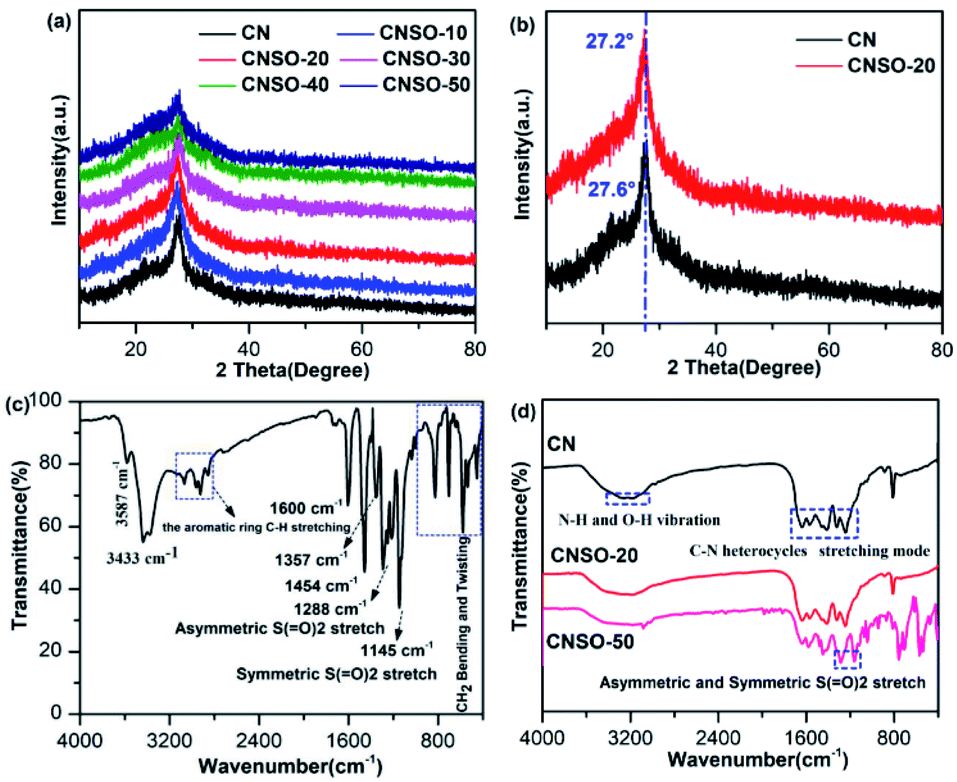
Known from the literature,57 the crystal structure of the CN photocatalyst has a great influence on its photocatalytic H2 evolution activity. Therefore, PXRD was used to characterize the crystal structure of CNSO-X. As shown in Fig. 2a, when a small amount (0–20 mg) of SO is introduced into the CN framework, it hardly affects the crystal form of CN. However, as the amount of SO increases (30–50 mg), the crystal structure of CN is distorted, which is manifested as a wide diffraction peak deformation and gradually weakens in strength. In CNSO-20, the representative peak of the periodic stacking of layers at 27.6° of CN shifted to 27.2° (Fig. 2b). This result reveals that SO may be copolymerized with urea, resulting in a highly ordered CN-based network structure. Experimental results show that there is an optimal value for the copolymerizing amount of SO, which needs to be further optimized.
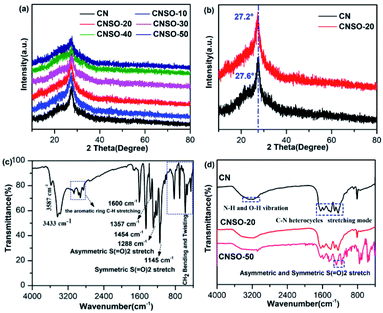 |
| | Fig. 2 (a) PXRD patterns of CNSO-X (X = 0, 10, 20, 30, 40, 50); (b) XRD patterns of CNSO-20 and CN; (c) FTIR spectra of the as-prepared 3,7-dihydroxydibenzo[b,d]thiophene 5,5-dioxide; (d) FTIR spectra of CN, CNSO-20 and CNSO-50. | |
The FTIR spectra of SO, CN and CNSO-X were further investigated to characterize their chemical structures. As shown in the FTIR of SO in Fig. 2c, the peaks around 1288 and 1145 cm−1 denote the characteristics asymmetric stretching vibration and symmetric stretching vibration peak of the sulfone group (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO), respectively.47–49 The peak at 3587 cm−1 is the stretching vibration of the free hydroxyl group, and 3433 cm−1 represents the hydroxyl peak associated with hydrogen bonding. The characteristic peaks of the aromatic ring are located at 3000 cm−1, 1600 cm−1, 1357 cm−1, 1454 cm−1 and 800 cm−1. As shown in Fig. 2d, the chemical structure of CNSO-20 is almost identical to that of pure CN because the amount of doping is relatively too small. A series of peaks in the range of 1200–1700 cm−1 were presented, which are attributed to the typical tensile vibration mode of the C–N heterocyclic ring. The sharp peak at 813 cm−1 is attributed to the out-of-plane bending vibration of the heptazine ring units. The broad peak of 3000–3500 cm−1 belongs to the uncondensed N–H vibration peak in CN, and the surface adsorbs the O–H vibration peak in water.32–36 Notably, for CNSO-50, the SO copolymerizing content is greater, and the FTIR is mostly consistent with CN. The only difference is that the characteristic peak (1281 and 1152 cm−1) of the sulfone group in SO appears. This indicates that SO is successfully incorporated into the CN framework, and does not interfere with its chemical framework. This can be further proved by the solid-state 13C NMR spectra. As shown in Fig. 3a, two obvious resonance peaks can be differentiated in each spectrum of CN and CNSO-20. The characteristic peak at a chemical shift of 168 ppm is attributed to the sp2 carbon atoms attached to the terminal –NH2, while the other peak at 160 ppm is marked as the sp2 carbon atoms in heptazine units connected to the bridging N atoms.32 In particular, compared with CN, CNSO-20 shows two new peaks at 128 ppm and 119 ppm, which belong to the characteristic carbons bonded to the sulphone group. The 13C NMR result further proved that SO was successfully introduced into the CN framework.
SO), respectively.47–49 The peak at 3587 cm−1 is the stretching vibration of the free hydroxyl group, and 3433 cm−1 represents the hydroxyl peak associated with hydrogen bonding. The characteristic peaks of the aromatic ring are located at 3000 cm−1, 1600 cm−1, 1357 cm−1, 1454 cm−1 and 800 cm−1. As shown in Fig. 2d, the chemical structure of CNSO-20 is almost identical to that of pure CN because the amount of doping is relatively too small. A series of peaks in the range of 1200–1700 cm−1 were presented, which are attributed to the typical tensile vibration mode of the C–N heterocyclic ring. The sharp peak at 813 cm−1 is attributed to the out-of-plane bending vibration of the heptazine ring units. The broad peak of 3000–3500 cm−1 belongs to the uncondensed N–H vibration peak in CN, and the surface adsorbs the O–H vibration peak in water.32–36 Notably, for CNSO-50, the SO copolymerizing content is greater, and the FTIR is mostly consistent with CN. The only difference is that the characteristic peak (1281 and 1152 cm−1) of the sulfone group in SO appears. This indicates that SO is successfully incorporated into the CN framework, and does not interfere with its chemical framework. This can be further proved by the solid-state 13C NMR spectra. As shown in Fig. 3a, two obvious resonance peaks can be differentiated in each spectrum of CN and CNSO-20. The characteristic peak at a chemical shift of 168 ppm is attributed to the sp2 carbon atoms attached to the terminal –NH2, while the other peak at 160 ppm is marked as the sp2 carbon atoms in heptazine units connected to the bridging N atoms.32 In particular, compared with CN, CNSO-20 shows two new peaks at 128 ppm and 119 ppm, which belong to the characteristic carbons bonded to the sulphone group. The 13C NMR result further proved that SO was successfully introduced into the CN framework.
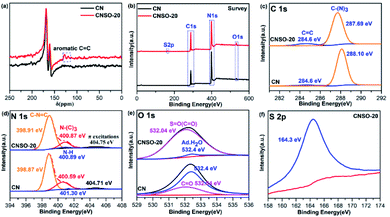 |
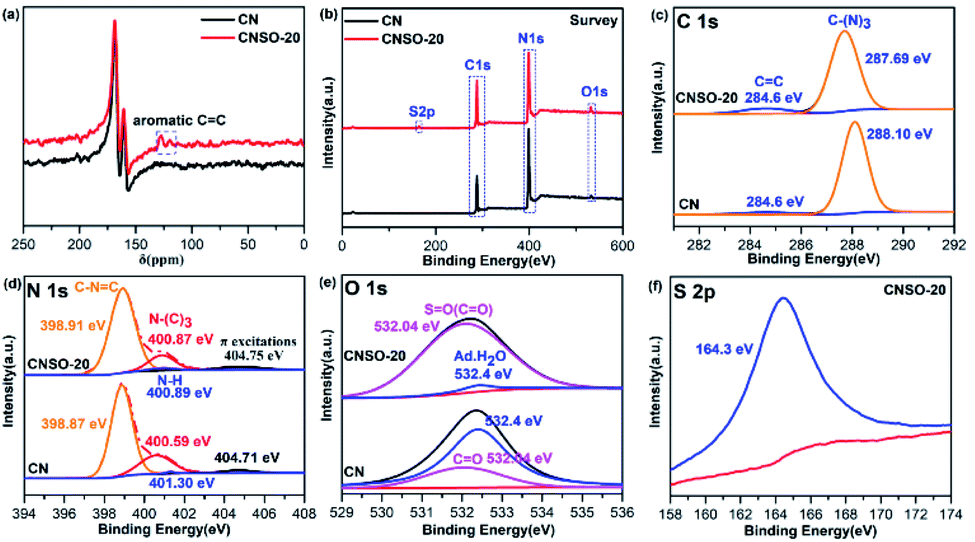
| | Fig. 3 (a) The solid-state 13C NMR of CN and CNSO-20; high-resolution XPS spectra of CN and CNSO-20: (b) survey; (c) C 1s; (d) N 1s; (e) O 1s; (f) S 2p. | |
Subsequently, the surface chemical state and element structure of CN and CNSO-20 were profoundly analysed by XPS. As displayed in the survey spectra (Fig. 3b), CN is predominantly composed of C and N elements and some O, which comes from water molecules adsorbed on the surface. In contrast, the CNSO-20 consisted of four elements: C, N, O, and S. Part of O and all of S come from the dibenzothiophene-S,S-dioxide unit. As can be seen from Table S1,† compared with CN (content ratio: C/N is 0.76), the C/N value in CNSO-20 gradually increased (0.84), and the content of S and O elements also increased. This is consistent with the incorporation of the SO unit, which contains aromatic carbon and sulfone groups. In addition, for the deconvoluted C 1s spectra of CNSO-20 (Fig. 3c), the peaks at 284.60 eV and 287.69 eV are assigned to graphite sp2 carbon (CC) and sp2-hybridized carbon (N–CN). Obviously, CNSO-20 shows a higher CC peak area ratio (Table S2†), which proves the successful introduction of the SO unit. Fig. 3d displays the deconvoluted N 1s spectra of CNSO-20 with peaks located at 398.91, 400.87, 400.89 and 404.75 eV, corresponding to the sp2-hybridized N (CN–C), the tertiary N (–N–(C)3), terminal amino groups (C–NH) and π excitation peak, respectively. The respective peak areas are summarized in Table S3.† Interestingly, the N 1s peak of CNSO-20 shifts to a higher binding energy compared with CN. This shift should be attributed to the decrease of the electron cloud density around N atoms in CN. Because of the introduction of a stronger electron-drawing sulfone group, which forms a local D–A structure inside the photocatalyst, this results in an enhanced intramolecular charge transfer and electron transfer from CN to the SO unit (Fig. 1b). The O 1s XPS spectrum (Fig. 3e) of CNSO-20 can be deconvoluted into two peaks at 532.04 and 532.4 eV, which belong to SO and H–O–H, respectively. The SO bond comes from the doped dibenzothiophene-S,S-dioxide unit, while the H–O–H originated from the surface adsorbed water. What is more, the S 2p XPS spectrum (Fig. 3f) also confirms the sulfone group in CNSO-20 with the distinct peak at 164.3 eV.
The above comprehensive analysis results prove that the SO unit was successfully introduced into the CN framework through the copolymerization strategy. The formed D–A structure in CNSO-X will benefit to enhance the photocatalytic performance.
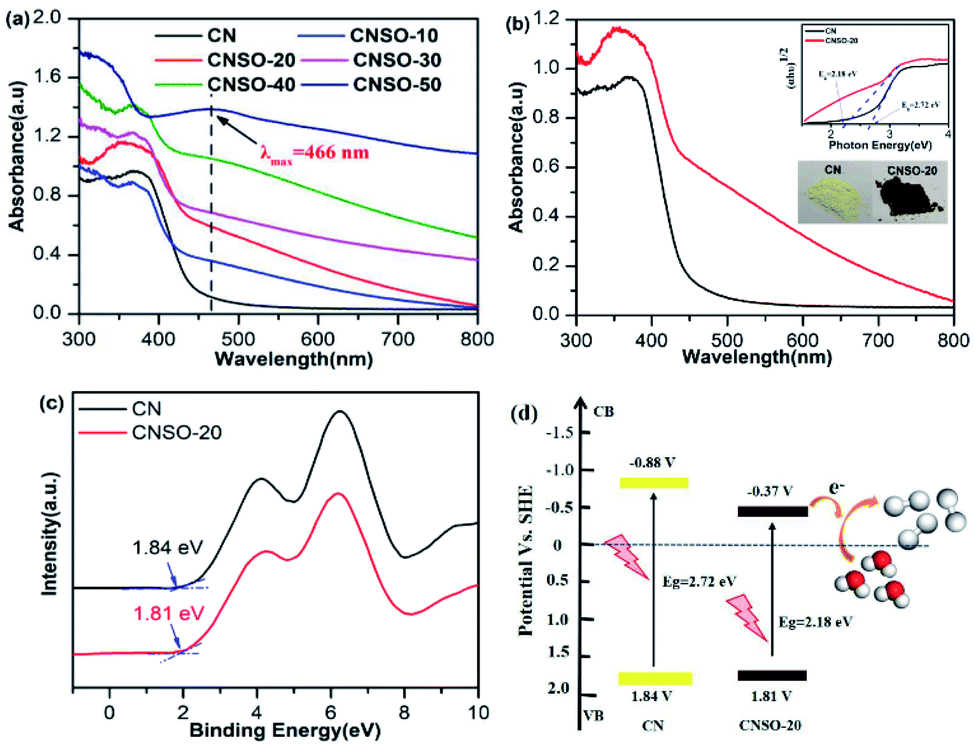
The effect of copolymerizing SO on the CN electronic band structure was further investigated by UV-Vis diffuse reflectance spectra (DRS). As shown in Fig. 4a, with the increasing amount of SO copolymerization, the entire wavelength range absorption becomes stronger, which indicates that the SO incorporation can greatly expand the range of electron delocalization in the CN networks. According to their UV-Vis absorption spectra with a more and more obvious “shoulder” and the obvious absorption peak at 466 nm in the visible light region, we can see that an additional electron-transition mode gradually appears, except for the electron transition from the intrinsic HOMO to LUMO. Considering the structure of these copolymers, the intramolecular charge transfer transition from the N atoms (donor) to the dibenzothiophene-S,S-dioxide (acceptor) unit will most probably occur in it. Comparatively, CNSO-20 displays a significantly increased visible light absorption up to near 800 nm with a brown color in appearance. The corresponding band gap (Eg) of CNSO-20 is estimated by the fitting of the Tauc plots to be 2.18 eV, which is 0.54 eV narrower than that of CN (2.72 eV) (Fig. 4b). The corresponding valence bands (VB) of CN and CNSO-20 were obtained through testing the XPS valence band spectrum (Fig. 4c). The VB values of CN and CNSO-20 are close at 1.84 V and 1.81 V (vs. NHE), respectively. The conduction band (CB) values were calculated by the difference between the respective Eg and VB, which are −0.88 V and −0.37 V (vs. NHE), respectively. The schematics of the relative positions of HOMO and LUMO for CN and CNSO-20 are shown in Fig. 4d. According to the calculation results, the Eg reduction after the incorporation of SO into the CN network is mainly due to the CB move down.
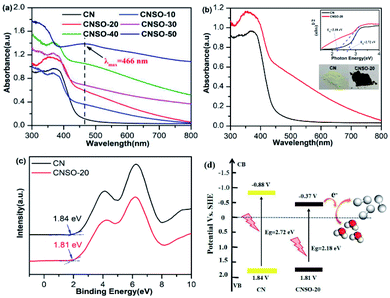 |
| | Fig. 4 (a) UV-Vis absorption spectra of different amounts of dibenzothiophene-S,S-dioxide doped CN (CNSO-X (X = 0, 10, 20, 30, 40, 50)); (b) UV-Vis absorption spectra of CN and CNSO-20 nanosheets (inset shows the Tauc plots of CN and CNSO-20); (c) valence band XPS spectra of CN and CNSO-20; (d) schematics of the relative positions of HOMO and LUMO for CN and CNSO-20. | |
3.2 Photocatalytic hydrogen evolution performance
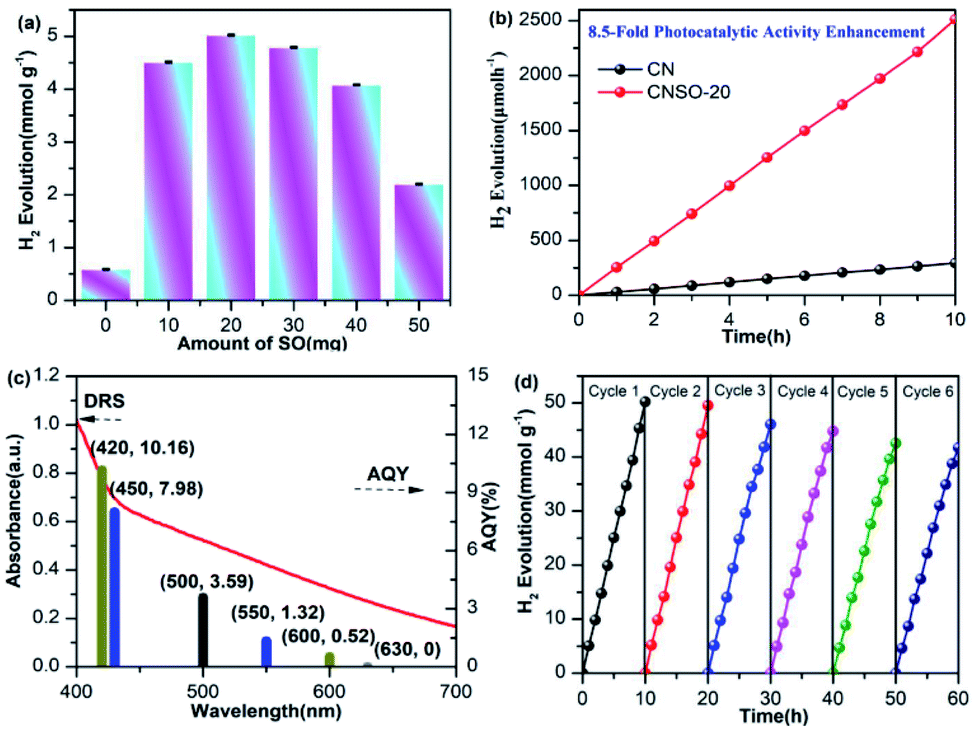
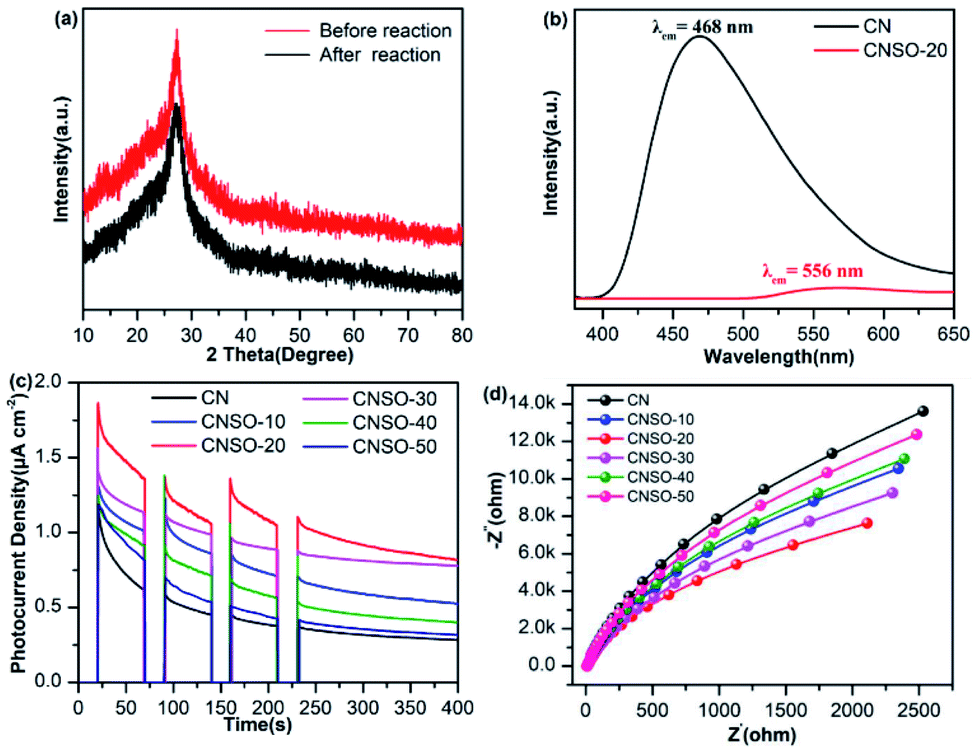
In order to evaluate the photocatalytic activity of the as-obtained samples, the visible-light driven H2 generation was systematically investigated in the presence of 3.0 wt% Pt cocatalyst and triethanolamine hole quencher. As given in Fig. 5a, the H2 evolution activity of CNSO-X has been greatly improved by doping a moderate amount of SO into the CN framework. Strikingly, as shown in Fig. 5b, CNSO-20 displays the highest H2 evolution activity of 251 μmol h−1, which is almost 8.5 times higher than that of CN (29.5 μmol h−1). Compared with the recent reports, CNSO-20 is one of the most outstanding g-C3N4-based photocatalysts for H2 evolution.31–42 The enhanced photocatalytic hydrogen evolution activity may be attributed to the following two aspects: on the one hand, the introduction of the SO unit broadens the π-conjugation degree of the CN skeleton, and improves the photo-response ability. On the other hand, SO as a strong electron-withdrawing group and N containing a lone pair of electrons form a D–A configuration to significantly induce the separation of the e−/h+ pairs. However, the amount of SO is increased from 20 mg to 50 mg, and the H2 evolution activity of the photocatalyst decreases instead. This is because the addition of excessive SO will damage the crystallinity to a certain extent, which is not conducive to the separation of photo-generated carriers (Fig. 6). Fig. 5c compares the DRS and wavelength-dependent AQY value for CNSO-20 under various monochromatic light irradiations (420, 450, 500, 550, 600 and 630 nm). The AQY values of CNS0-20 at different monochromatic wavelengths are similar to the changing tendency of its DRS curves. For CNSO-20, the AQY values of 10.16, 7.98, 3.59, 1.32, 0.52 and 0% were obtained under monochromatic illumination of λ = 420, 450, 500, 550, 600 nm and 630 nm, respectively. Besides the high hydrogen evolution activity and AQY, the resulting CNSO-20 also reveals satisfactory photocatalytic stability even after six cycles in 60 h (Fig. 5d), which is very important in practical applications. The XRD of the CNSO-20 sample before and after photocatalytic reaction has no obvious characteristic peak changes, which further proves the moderate stability of the prepared photocatalyst (Fig. 6a).
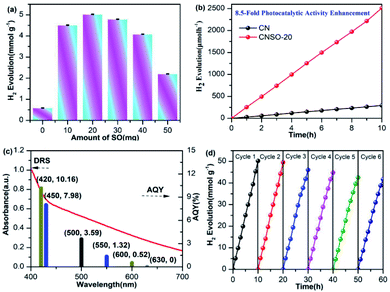 |
| | Fig. 5 (a) Photocatalytic H2 evolution rates of CNSO-X; (b) time-dependent photocatalytic hydrogen evolution over CN and CNSO-20; (c) wavelength-dependent AQY and DRS spectrum of CNSO-20 (λ = 420, 450, 500, 550, 600 and 630 nm); (d) photostability for H2 production of the CNSO-20. Reaction conditions: 50 mL water, 10 mL TEOA, 50 mg photocatalyst under visible-light. | |
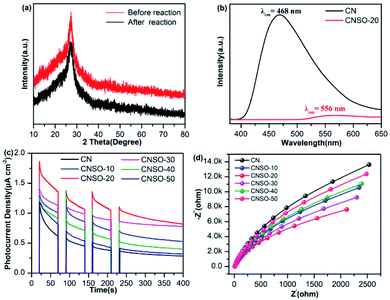 |
| | Fig. 6 (a) The XRD spectra of CNSO-20 before and after irradiation under visible-light for 60 h in a triethanolamine/water mixture; (b) static fluorescence emission spectra of CN and CNSO-20; (c) transient current responses to on–off cycles and full-time photocurrent of illumination on the CN and CNSO-X membrane electrodes, respectively (0 bias); (d) EIS Nyquist plots of CN and CNSO-X at the open circuit voltage. | |
To better understand the influence of introducing SO into the CN framework to form a D–A conjugated polymer on the interface electron transfer, the photoluminescence (PL) spectra, transient photocurrent curve (I–t) and electrochemical impedance curve (EIS) were conducted. As revealed in Fig. 6b, pure CN presents a broad and strong emission peak at 468 nm, which is attributed to the rapid recombination of electron–hole pairs at the interface, resulting in poor photocatalytic hydrogen evolution activity. On the contrary, CNSO-20 displays a much weaker emission peak and a significant red shift (556 nm) emerged, indicating that the electron–hole pair separation has been significantly improved through the incorporation of SO, leading to better photocatalytic hydrogen evolution activity. I–t curve measurements are shown in Fig. 6c. The photocurrent density increases sequentially with the addition of SO (0–20 mg). However, when the amount of SO further increases (20–50 mg), the photocurrent density shows a downward trend. Importantly, the transient photocurrent of CNSO-20 is 0.82 μA cm−2, which is 2.93 times higher than that of pure CN (0.28 μA cm−2). The results also show that CNSO-20 possessed higher carrier transfer efficiency. The EIS tests (Fig. 6d) were further carried out to study the charge transfer resistance of pure CN and CNSO-20 in the low-frequency region. It was observed that the radius of the Nyquist curve of CNSO-20 was significantly reduced, indicating that SO copolymerization improves the electron transfer kinetics during the photocatalytic hydrogen production process. Synthetically, all of the above results confirm that CNSO-20 exhibited excellent photocatalytic hydrogen evolution activity due to the extended light absorption, effective charge separation and rapid charge transfer.
3.3 Theoretical calculations
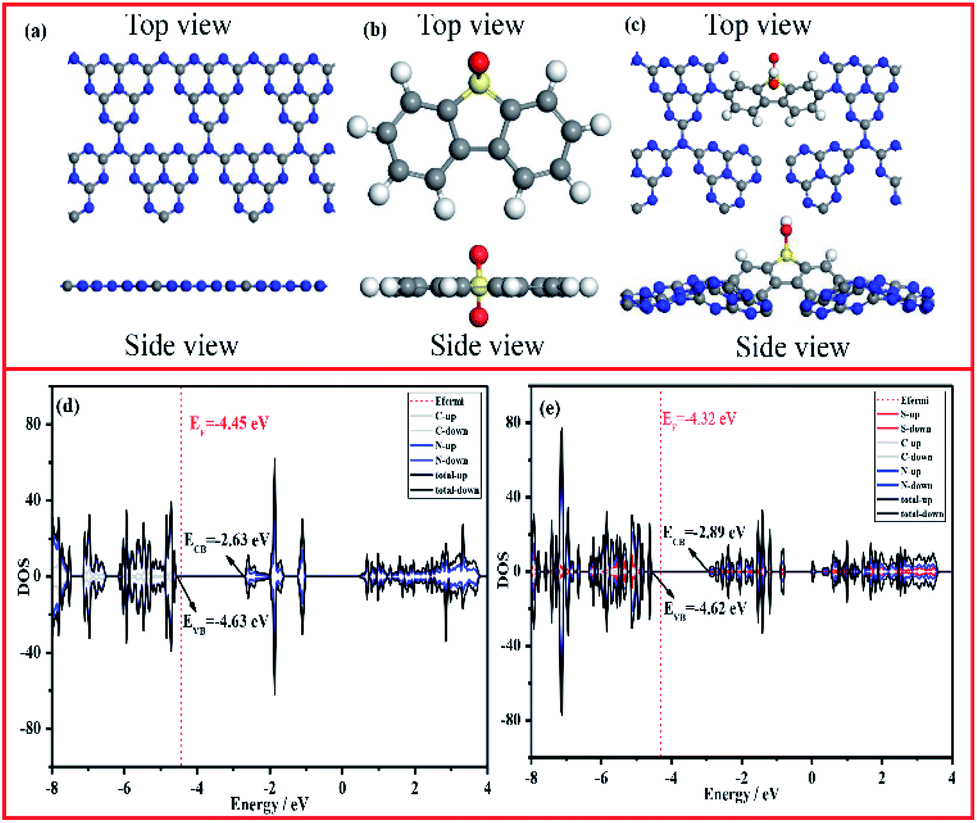
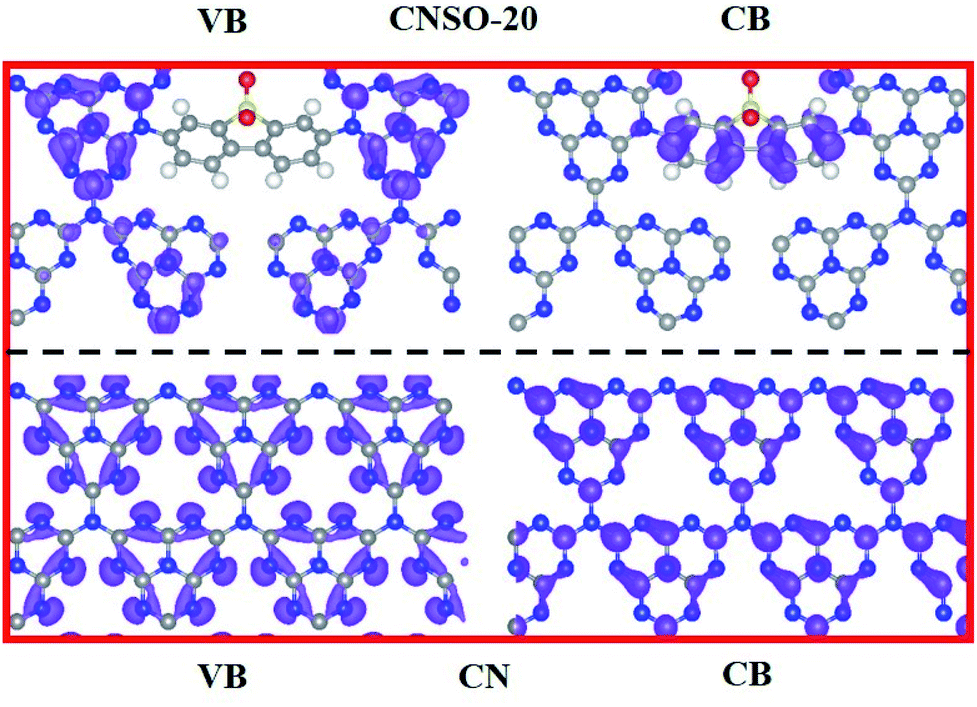
To further disclose the inner relationship between the doping SO and the superior photocatalytic performance, the energy and electronic structure were theoretically calculated using the Vienna ab initio simulation package (VASP).54Fig. 7a–c illustrate the side and top views of the optimized models, from which the total electronic density of states (DOS) of CN and CNSO-20 were further optimized. It can be seen from Fig. 7d and e that the main difference between their electronic structures is that the CB moves down, while the VB is basically unchanged for CNSO-20. It then shows that the copolymerization of SO in the CN framework leads to the decrease of Eg and increase of the visible light response range. Then, the molecular orbitals of CN and CNSO-20 were also analysed. As shown in Fig. 8a, for CN, the charge distribution was mainly on the nitrogen atom in the ground state. After being excited, the charge was distributed on the carbon and nitrogen atoms without obvious charge separation, indicating an easy recombination process. However, for CNSO, the charge in the VB is still mainly concentrated on the nitrogen atom, while the charge in the CB is largely concentrated on the SO aromatic ring, indicating that the copolymerized SO can induce a significant spatial separation of charges. This means that SO generally exhibits the acceptor characteristics, and the tertiary nitrogen atoms gives the donor characteristics, which is similar to the previously reported D–A polymer photocatalyst.45–50 The obvious spatial separation of charges in the CNSO photocatalyst is beneficial to the photocatalytic hydrogen evolution process, which is consistent with the research results of charge transfer kinetics.
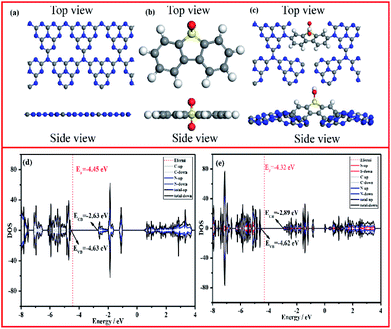 |
| | Fig. 7 The top and side views of the constructed cluster model of (a) CN, (b) 3,7-dihydroxydibenzo[b,d]thiophene-5,5-dioxide, (c) CNSO (gray: C atoms, blue: N atoms, yellow: S atoms, red: O atoms), and the calculated density of states (DOSs) of (d) CN and (e) CNSO. | |
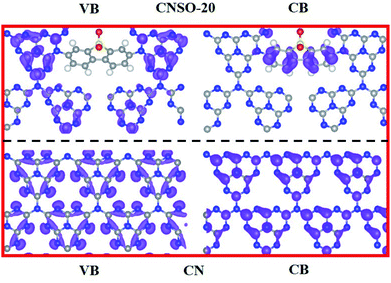 |
| | Fig. 8 The calculated distributions of VB (left) and CB (right) (gray: C atoms, blue: N atoms, yellow: S atoms, red: O atoms) of CN (below) and CNSO-20 (above). | |
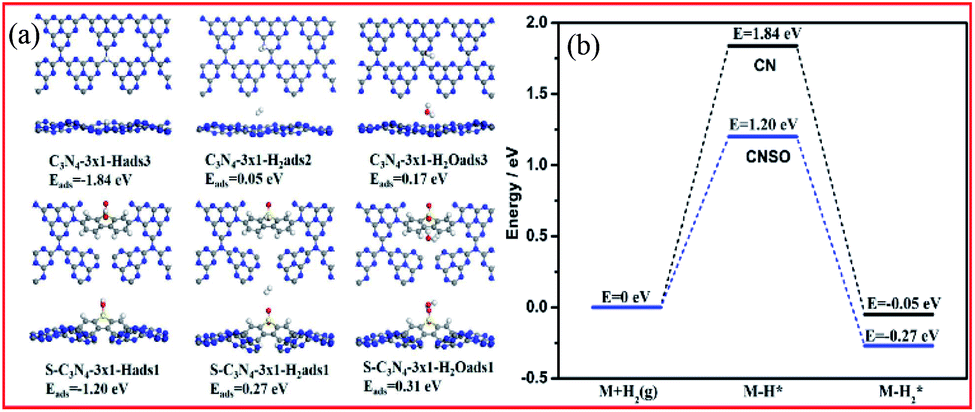
In the process of photocatalytic water hydrogen evolution, the adsorption of the hydrogen atoms and water on the photocatalyst has a non-negligible effect on the H2 evolution kinetics. Therefore, the difference in the adsorption energies of the participating species at CN and CNSO was further measured. As shown in Fig. 9a and b and Table 1, compared with the adsorption of water at CN (Eads = 0.17 eV), that at CNSO is clearly much stronger (Eads = 0.31 eV). In addition, compared with that at CN, the calculated barrier for the adsorption process of H at CNSO drastically increases by 0.64 eV. The reason may be that SO contains a sulfone group, which is a hydrophilic polar group, and can help the capture of water and its H atoms, thereby promoting the photocatalytic H2 evolution activity of CN.
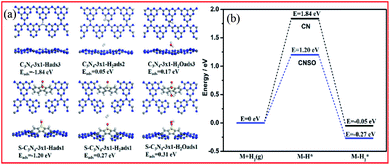 |
| | Fig. 9 (a) Calculated adsorption structures and the corresponding adsorption energies. (b) Calculated energy profiles for the adsorption of H2 at CN and CNSO. | |
Table 1 Calculated adsorption energies (in eV) of various species at CN and CNSO
| Adsorbed species |
H2O* |
H* |
H2* |
| CN |
0.17 |
−1.84 |
0.05 |
| CNSO |
0.31 |
−1.20 |
0.27 |
3.4 Possible H2 evolution mechanism
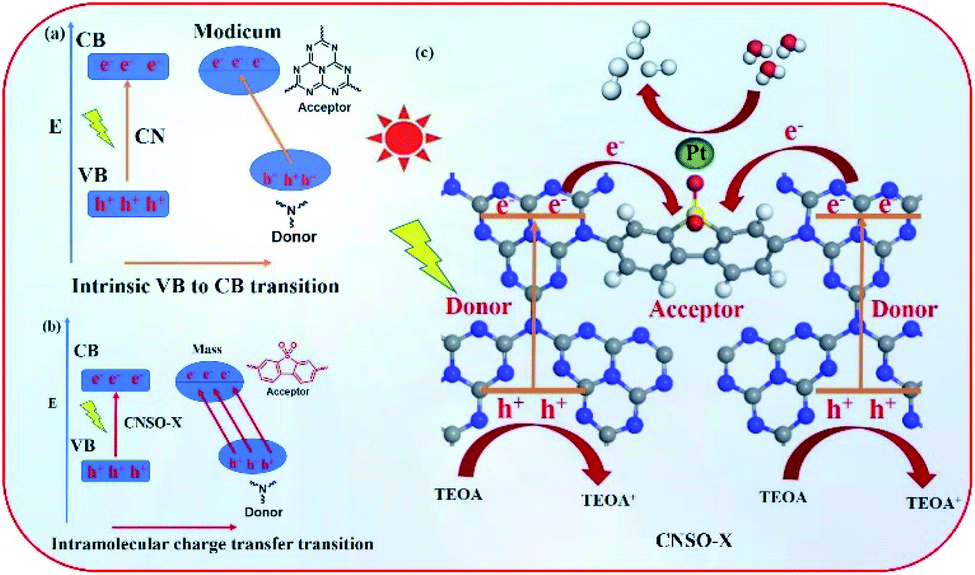
Based on the above experimental results, a possible mechanism for the significantly improved photocatalytic activity and electron transfer processes of CNSO-X are proposed. As shown in Fig. 10a, the bulk g-C3N4 can generate photogenerated holes and electrons under visible-light irradiation, where the holes and electrons are located on N and C in the heptaazine ring, respectively. Unfortunately, they are easy to recombine during the photocatalytic reaction process, resulting in its very low photocatalytic hydrogen evolution performance. In contrast, for CNSO-20, in the case of intramolecular charge transfer transitions, holes and electrons are located at the two separate tertiary nitrogen (donor) and SO (acceptor) groups, respectively (Fig. 10b). It not only broadens the visible-light response range, but also promotes the electron–hole separation. Moreover, in order to further clearly and quantitatively describe the charge transfer and separation process of CNSO-X, the Bader charge was calculated and the result is displayed in Fig. S1.† The results reveal that there is 0.964e being transferred from CN to the SO at CNSO-X. In addition, a possible photocatalytic hydrogen evolution mechanism of CNSO-X is shown in Fig. 10c. After absorbing visible light, the generated electron–holes are effectively separated, and the electrons immediately migrate to the Pt nanoparticles on the surface for photocatalytic hydrogen evolution. The holes left in the VB react with TEOA through an oxidation reaction.
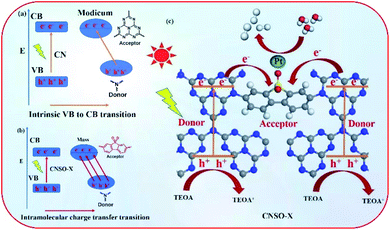 |
| | Fig. 10 (a and b) Diagram of the two electron transition modes in CN and CNSO-X; (c) the proposed mechanism for H2 evolution with the intramolecular CNSO-X D–A conjugated copolymers. | |
4. Conclusions
In conclusion, a facile strategy has been successfully developed to construct 3,7-dihydroxydibenzo-[b,d]-thiophene-5,5-dioxide doped g-C3N4 nanosheets for photocatalytic H2 evolution under visible light. Experimental and theoretical calculations comprehensively confirm that the SO copolymerized g-C3N4 displayed a significant enhancement of the photocatalytic hydrogen evolution activity because of the formation of the D–A structure, which broadens the visible light response range, promotes the electron–hole separation, increases the wettability and lowers the adsorption H* energy barrier. Notably, this is the first report on the use of dibenzothiophene dioxide and CN copolymers for photocatalytic hydrogen production. As a result, the AQY values of CNSO-20 is 10.16% under λ = 420 nm monochromatic illumination, which is a very competitive efficiency. This work provides a new perspective for the design and development of highly efficient D–A conjugated polymer photocatalysts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
For financial support of this research, we thank the National Natural Science Foundation of China (21971064, 21825301 and 21772040), Shanghai Science and Technology Committee (17520750100), Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03), the Fundamental Research Funds for the Central Universities (222201717003 and 50321101918001) and the Programme of Introducing Talents of Discipline to Universities (B16017). The authors thank the Research Center of Analysis and Test of East China University of Science and Technology for the help with the characterization.
Notes and references
- G. Liao, Y. Gong, L. Zhang, H. Gao, G. J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- G. G. Hu, H. B. Liu, Y. Liu, J. F. Chen, Y. L. Li and L. M. Dai, Nano Energy, 2019, 63, 103874–103881 CrossRef.
-
(a) N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS;
(b) F. T. Yu, F. R. Song, R. Z. Wang and M. Xu, Polym. Chem., 2021 10.1039/D0PY01656J.
- J. Xu, S. M. Bi, W. Q. Tang, Q. Kang, D. F. Niu, S. Hu, S. Z. Hu, S. L. Zhao, L. M. Wang, Z. Xin and X. S. Zhang, J. Mater. Chem. A, 2019, 7, 18100–18108 RSC.
- Z. Z. Wu, B. Z. Fang, A. Bonakdarpour, A. Sun, D. P. Wilkinson and D. Z. Wang, Appl. Catal., B, 2012, 125, 59–66 CrossRef CAS.
- F. T. Yu, Z. Q. Zhu, S. P. Wang, Y. K. Peng, Z. Z. Xu, Y. Tao, J. B. Xiong, Q. W. Fan and F. Luo, Chem. Eng. J., 2020 DOI:10.1016/j.cej.2020.127558.
- J. Xu, W. Q. Tang, F. T. Yu, S. L. Zhao, D. F. Niu, X. S. Zhang, Z. Xin and R. J. Chen, J. Mater. Chem. A, 2020, 8, 19001–19010 RSC.
- F. T. Yu, Z. Q. Wang, S. C. Zhang, H. N. Ye, K. Y. Kong, X. Q. Gong, J. L. Hua and H. Tian, Adv. Funct. Mater., 2018, 28, 1804512–1804524 CrossRef.
- H. N. Ye, Z. Q. Wang, F. T. Yu, S. C. Zhang, K. Y. Kong, X. Q. Gong, J. L. Hua and H. Tian, Appl. Catal., B, 2020, 267, 118577–118588 CrossRef CAS.
- F. T. Yu, Z. Q. Wang, S. C. Zhang, H. N. Ye, K. Y. Kong, X. Q. Gong, J. L. Hua and H. Tian, Appl. Catal., B, 2018, 237, 32–42 CrossRef CAS.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- Y. Zheng, L. H. Lin, B. Wang and X. C. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS.
- F. T. Yu, Z. W. Yu, Z. Z. Xu, J. B. Xiong, Q. W. Fan, X. F. Feng, Y. Tao, J. L. Hua and F. Luo, Mol. Syst. Des. Eng., 2020, 5, 882–889 RSC.
- X. B. Li, J. Xiong, X. M. Gao, J. T. Huang, Z. J. Feng, Z. Chen and Y. F. Zhu, J. Alloys Compd., 2019, 802, 196–209 CrossRef CAS.
- X. B. Li, J. Xiong, Y. Xu, Z. J. Feng and J. T. Huang, Chin. J. Catal., 2019, 40, 424–433 CrossRef CAS.
- W. Ren, J. J. Cheng, H. H. Qu, C. J. Huang, M. Anpo and X. C. Wang, J. Catal., 2020, 389, 636–646 CrossRef CAS.
- J. J. Huang, Y. Lu, H. Zhang, L. S. Guan, Z. G. Mou, J. H. Sun, S. P. Sun, J. H. He and W. W. Lei, Chem. Eng. J., 2021, 405, 126685–126693 CrossRef CAS.
- J. Xu, F. T. Yu, J. L. Hua, W. Q. Tang, C. Yang, S. Z. Hu, S. L. Zhao, X. S. Zhang and Z. Xin, Chem. Eng. J., 2020, 392, 123694–123704 CrossRef CAS.
- W. J. Wang, C. Y. Zhou, Y. Yang, G. M. Zeng, C. Zhang, Y. Zhou, J. N. Yang, D. L. Huang, H. Wang, W. P. Xiong, X. P. Li, Y. K. Fu, Z. W. Wang, Q. Y. He, M. Y. Jia and H. Z. Luo, Chem. Eng. J., 2021, 404, 126540–126582 CrossRef CAS.
- Y. X. Li, S. X. Ouyang, H. Xu, W. S. Hou, M. Zhao, H. Y. Chen and J. H. Ye, Adv. Funct. Mater., 2019, 29, 1901024–1901031 CrossRef.
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS.
- X. C. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- Z. W. Zhao, Y. J. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- G. G. Zhang, J. S. Zhang, M. W. Zhang and X. C. Wang, J. Mater. Chem., 2012, 22, 8083–8091 RSC.
- X. Q. Fan, L. X. Zhang, R. L. Cheng, M. Wang, M. L. Li, Y. J. Zhou and J. L. Shi, ACS Catal., 2015, 5, 5008–5015 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2011, 2, 1826–1830 Search PubMed.
- A. Du, S. Sanvito, Z. Li, D. Wang, Y. Jiao, T. Liao, Q. Sun, Y. Hau Ng, Z. Zhu, R. Amal and S. C. Smith, J. Am. Chem. Soc., 2012, 1344, 393–4397 Search PubMed.
- G. G. Zhang, Z. A. Lan, L. H. Lin, S. Lin and X. C. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS.
- S. Bellamkonda, R. Shanmugam and R. R. Gangavarapu, J. Mater. Chem. A, 2019, 7, 3757–3771 RSC.
- C. H. Han, L. L. Du, M. Konarova, D. C. Qi, D. L. Phillips and J. S. Xu, ACS Catal., 2020, 10, 9227–9235 CrossRef CAS.
- Q. Han, B. Wang, J. Gao, Z. Cheng, Y. Zhao, Z. Zhang and L. Qu, ACS Nano, 2016, 10, 2745–2751 CrossRef CAS.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS.
- J. Ran, T. Y. Ma, G. Gao, X.-W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708–3717 RSC.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS.
- Y. Wang, W. Yang, X. Chen, J. Wang and Y. Zhu, Appl. Catal., B, 2018, 220, 337–347 CrossRef CAS.
- Y. F. Li, R. X. Jin, Y. Xing, J. Q. Li, S. Y. Song, X. C. Liu, M. Li and R. C. Jin, Adv. Energy Mater., 2016, 6, 1601273–1601277 CrossRef.
- J. Liu, Y. Yu, R. L. Qia, C. Y. Cao, X. Y. Liu, Y. J. Zheng and W. G. Song, Appl. Catal., B, 2019, 224, 459–464 Search PubMed.
- J. S. Zhang, M. W. Zhang, S. Lin, X. Z. Fu and X. C. Wang, J. Catal., 2014, 310, 24–30 CrossRef CAS.
- J. J. Tian, L. X. Zhang, X. Q. Fan, Y. J. Zhou, M. Wang, R. L. Cheng, M. L. Li, X. T. Kan, X. X. Jin, Z. H. Liu, Y. F. Gao and J. L. Shi, J. Mater. Chem. A, 2016, 4, 13814–13821 RSC.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Y. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968–4978 CrossRef.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, I. McCulloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- Y. Bai, D. J. Wood, L. Wilbraham, C. M. Aitchison, M. A. Zwijinenburg, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 8700–8705 RSC.
- R. S. Sprick, O. J. Cheetham, Y. Bai, J. A. Fernandes, M. Barnes, J. W. Bradley and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7125–7129 RSC.
- Z. J. Wang, X. Y. Yang, T. J. Yang, Y. B. Zhao, F. Wang, Y. Chen, J. H. Zeng, C. Yan, F. Huang and J. X. Jiang, ACS Catal., 2018, 8, 8590–8596 CrossRef CAS.
- Y. B. Zhao, W. Y. Ma, Y. F. Xu, C. Zhang, Q. Wang, T. J. Yang, X. M. Gao, F. Wang, C. Yan and J. X. Jiang, Macromolecules, 2018, 51, 9502–9508 CrossRef CAS.
- C. H. Dai, S. D. Xu, W. Liu, X. Z. Gong, M. Panahandeh-Fard, Z. T. Liu, D. Q. Zhang, C. Xue, K. P. Loh and B. Liu, Small, 2018, 14, 1801839–1801844 CrossRef.
- X. M. Gao, C. Shu, C. Zhang, W. Y. Ma, S. B. Ren, F. Wang, Y. Chen, J. H. Zeng and J. X. Jiang, J. Mater. Chem. A, 2020, 8, 2404–2411 RSC.
- M. Bhanuchandra, K. Murakami, D. Y. Vasu, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 10234–10238 CrossRef CAS.
- X. H. Zhang, T. Y. Peng, L. J. Yu, R. J. Li, Q. Q. Li and Z. Li, ACS Catal., 2015, 5, 504–510 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- M. P. Teter, M. C. Payne and D. C. Allan, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 12255–12263 CrossRef.
- W. Ren, J. J. Cheng, H. H. Ou, C. J. Huang, M. M. Titirici and X. C. Wang, ChemSusChem, 2019, 12, 3257–3262 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experiments and additional figures. See DOI: 10.1039/d0na01011a |
| ‡ Authors are equal to contribution. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Haonan
Ye
a,
Haoran
Ding
a,
Xueqing
Gong
*a,
Haonan
Ye
a,
Haoran
Ding
a,
Xueqing
Gong