
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the coalescence degree in the growth of Pt–Pd nanoalloys
Diana
Nelli
a,
Manuella
Cerbelaud
 b,
Riccardo
Ferrando
*c and
Chloé
Minnai
*d
b,
Riccardo
Ferrando
*c and
Chloé
Minnai
*d
aDipartimento di Fisica dell'Università di Genova, Via Dodecaneso 33, Genova 16146, Italy
bUniversité de Limoges, CNRS, IRCER, UMR 7315, F-87000 Limoges, France
cDipartimento di Fisica dell'Università di Genova and CNR-IMEM, Via Dodecaneso 33, Genova 16146, Italy. E-mail: ferrando@fisica.unige.it
dNanoparticles by Design Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, Kunigami-gun, Okinawa, Japan 904-0495. E-mail: chloe.minnai@oist.jp
First published on 9th December 2020
Abstract
Coalescence is a phenomenon in which two or more nanoparticles merge to form a single larger aggregate. By means of gas-phase magnetron-sputtering aggregation experiments on Pt–Pd nanoalloys, it is shown that the degree of coalescence can be tuned from a growth regime in which coalescence is negligible to a regime where the growth outcome is dominated by coalescence events. This transition is achieved by varying both the length of the aggregation zone and the pressure difference between the aggregation and the deposition chamber. In the coalescence-dominated regime, a wide variety of coalescing aggregates is produced and analyzed by TEM. The experimental results are interpreted with the aid of molecular-dynamics simulations. This allows to distinguish four different steps through which coalescence proceeds towards equilibrium. These steps, occurring on a hierarchy of well-separated time scales, consist in: (i) alignment of atomic columns; (ii) alignment of close-packed atomic planes; (iii) equilibration of shape; (iv) equilibration of chemical ordering.
1 Introduction
Binary metallic clusters and nanoparticles (also known as nanoalloys)1–5 are receiving increasing attention in recent years due to their versatility in applications which range from catalysis, to optics and plasmonics, to data storage and biomedicine. In view of building functional devices, nanoparticles are often assembled on substrates to constitute cluster assembled structures which can reflect at the macroscopic scale, the peculiar properties of their nanocomponents. This requires a fine control on the properties of individual nanoparticles as well as of the size distribution in the overall nanoparticle population.A very important phenomenon which may occur during the formation process of nanoparticles is coalescence. In coalescence, two or more nanoparticles collide and begin to merge to form a single, larger aggregate. This phenomenon is driven by the minimisation of surface area and the resultant decrease in surface energy that occurs when two smaller particles form a single aggregate, whose surface to volume ratio will be finally smaller than that of the initial colliding units.6 Coalescence may be useful as a tool for producing specifically tailored cluster structures.7 For example, coalescence of clusters of different elements may be used to produce nanoalloys with core–shell8 or Janus structures.9,10 Sintering can also improve the electrical conductivity11 and mechanical stability12 of nanoparticles. Moreover, it has been shown that it is also beneficial for the catalytic activity and the hydrogen adsorption capacity of Pd nanoparticles. Both are indeed enhanced by surface anomalies, such as steps and high index facets which emerge when nanoparticles coalesce forming necks, which are believed to be their most chemically active sites.6,13,14
Different subsequent stages can be distinguished in the processes involving the encounter of two nanoparticles. Following the terminology in ref. 15, collision generally refers to the initial translational motion of the nanoparticles eventually leading to their contact. Aggregation describes the formation of weak bonding such as that due to van der Waals forces, or simple physical entanglement between the nanoparticles. Sintering or coalescence indicates hard aggregation of clusters with the formation of chemical bonds between them, while fusion refers to the full coalescence in which a single nanoparticle, with a rounded shape close to that of the equilibrium structure, is finally formed. Due to kinetic trapping, two nanoparticles may stick to an intermediate stage without being able to complete the process leading to equilibration.16
To optimize the functionalities of a distribution of nanoparticles, it is also necessary to control their degree of coalescence.15 For instance, whereas full fusion of small clusters can degrade the performance of a catalyst, partial coalescence can also be beneficial,17 providing additional active sites for catalysis via necking.18 To this end, it is necessary to have a deep understanding theoretical and experimental of the coalescence phenomena at the nanoscopic scale.
In this paper we study the gas-phase growth of Pt–Pd nanoparticles. Both platinum and palladium are widely used in the chemical industry, for automobile exhaust purification and fuel cells,19–21 for hydrogen oxidation,22 oxygen reduction,23,24 and several other electrocatalytic reactions.25–28 Pt–Pd nanoparticles are widely studied because of their excellent catalytic activity, which has been demonstrated as being superior to that of the corresponding elemental Pt or Pd nanoparticles in several cases.23–26,29 We recently showed that PtPd nanoparticles with tunable core–shell structure (i.e. (PtPd)@Pd and (PtPd)@Pt nanoparticles) can be produced by gas-phase aggregation, by controlling the composition of the atomic vapor.30
The aim of this paper is to investigate the coalescence of Pt–Pd both experimentally and by computer simulations. We show that the percentage of coalescing aggregates in the experimentally grown samples can be controlled by tuning the dwell time i.e. the time that cluster spend growing before landing on the substrate. We show that there are experimental conditions in which a significant part of the growing nanoparticles presents some degree of coalescence. This allows to produce several coalescing aggregates which are representative of the different stages of the process, showing also that coalescence can be used to form peculiar structures, such as for example (PtPd)@Pd nanoparticles with asymmetrically placed off-center cores. The coalescence process is then analyzed by computer simulations, which allow to single out the key steps through which the coalescence process takes place and to understand the relevant atomic-level mechanisms.
2 Methods and model
Pt–Pd nanoparticles are synthesized by inert gas-aggregation sputtering deposition (Nanogen-Trio source, Mantis Deposition Ltd, UK). This technique is based on the condensation of an atomic vapor produced by DC simultaneous magnetron-sputtering of targets.As schematically shown in Fig. 1, and described in detail in ref. 31 and 32, the Nanogen-Trio source employs three separate circular 1′′ targets mounted on the same linear translator within one region of inert gas aggregation, called aggregation zone. The sputtering current applied to the different targets can be adjusted separately, thereby providing control over the mole fraction of metals incorporated into the nanoparticles. Here, two Pt and Pd highly pure (99.99%) single metal targets are co-sputtered in an Ar flux of 60 sccm (standard cubic centimeters per minute) which originates in a Nanogen pressure of 2–10−1 mbar. The applied powers are set to PPt = 8 W and PPd = 24 W. This creates a Pd-rich atomic vapor, whose composition is estimated by dividing the power applied to the target by the cohesion energy of the specific element (Ec,Pd = 3.89 eV per atom and Ec,Pt = 5.84 eV per atom), which gives Pd0.8Pt0.2. The composition of individual nanoparticles is also checked by performing quantitative analysis of the EDX maps. As deeply described in ref. 30, this composition of the atomic vapor produces a distribution of Pd-rich nanoparticles with (PtPd)@Pd core–shell chemical ordering. The actual composition of individual nanoparticles is rather variable, but still in the Pd-rich range, and, on average, well corresponds to the estimated composition of the vapor. The length of the aggregation zone can be mechanically varied in the range 35–125 mm by linear translation of the magnetron gun and multitarget holder. The deposition chamber has a backing pressure of PDC ∼ 10−8 mbar, which increases to ∼10−4 mbar in standard operating conditions. This value can be further increased to ∼10−1 mbar by flowing a controlled amount of Ar at the aperture of the aggregation zone, as indicated in Fig. 1. When the pressure in deposition chamber in raised by flowing argon at the exit orifice, we do not observe a change in the absolute value of the pressure measured inside the aggregation zone. Specifically, this value fluctuates during the experiments between 2.0 × 10−1 and 2.2 × 10−1 mbar. The difference of pressure causes the nanoparticle extraction and acceleration from the aggregation zone to the deposition chamber where they soft land onto substrates held at ground potential.
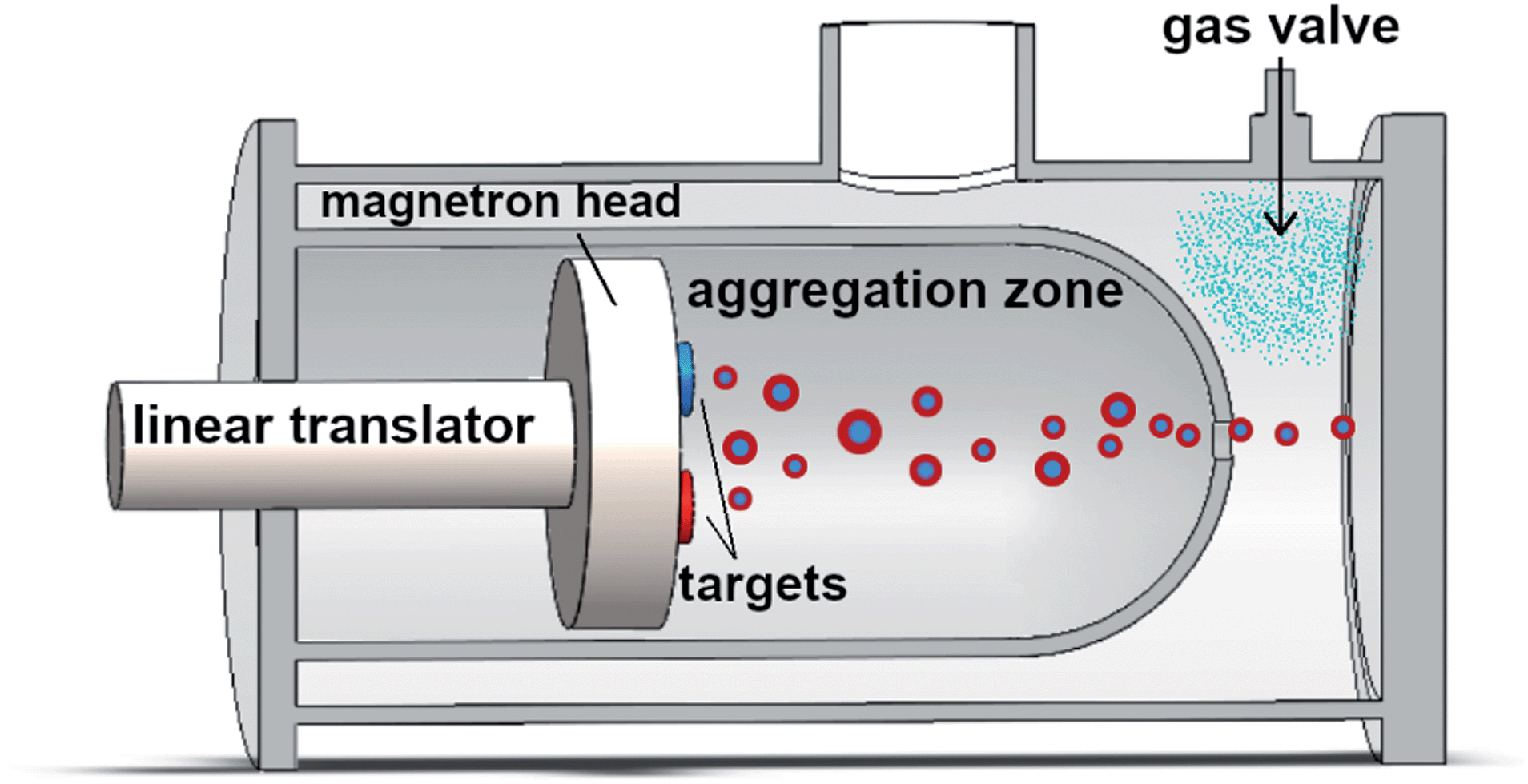 | ||
| Fig. 1 Schematic representation of the Nanogen-Trio nanoparticle source used in this study. The linear translator can mechanically vary the length of the aggregation zone in a range of 90 mm. The gas valve allows to introduce a controlled flow of Ar at the exit of aggregation zone (not in scale). | ||
To unravel the features of the Pt–Pd distribution the nanoparticles are deposited directly on amorphous carbon coated TEM grids to yield random arrays of binary nanoparticles. Each deposition lasts 60 seconds to avoid coalescence phenomena due to overlapping of the nanoparticles on the grid. The average composition, size and morphology of the nanoparticles are examined using an aberration-corrected scanning transmission electron microscopy (Cs-corrected-STEM), JEOL JEM-ARM 200F (200 kV). The nanoparticles are characterized using high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM).
The Molecular-Dynamics (MD) simulations are performed by our own codes. The simulations for smaller sizes (below 1000) atoms are made by the code version running on CPUs, whereas for larger sizes we use the GPU version, which allows to simulate the coalescence of aggregates of a few thousand atoms on the time scale of several μs. The time step for the solution of the equations of motions is set to 5 fs. Simulations are performed at constant temperature, controlled by an Andersen thermostat, by using the same protocol of ref. 16: the nanoparticles are initially placed at a distance of 0.5 nm with zero relative velocity and random orientation with respect to each other, and then they are left to the subsequent evolution.
The interactions between atoms are modelled by the Gupta potential.33 The form and parameters of the potential can be found given in ref. 30, where this model was shown to reproduce the correct chemical ordering and cluster shape in growth experiments of PtPd nanoparticles.
In the following we report the results of coalescence simulations at constant temperature. In the experiments, temperature is not constant during the growth process, since growth starts in a hot environment and continues in the inert gas atmosphere which cools down the growing nanoparticles. The actual temperature at which growth is taking place depends indeed on the efficiency of the inert gas in subtracting the condensation heat of the metal atoms, and it is difficult to determine. For this reason, we consider a range of temperatures from 550 to 900 K, and check whether the evolution is qualitatively of the same type at these different temperatures. Moreover, since our simulation time scale is limited to ∼10 μs, we choose to simulate at rather high temperatures, so that evolution is sufficiently fast to be observed. This is the usual choice in coalescence simulations by MD (see ref. 16 and references therein). We remark that here we present the longest coalescence simulations ever made by MD.
3 Experimental results
Coalescence is a complex phenomenon, which occurs through different stages. For this reason we first provide an overview of the typologies of coalesced nanoparticles in order to single out these different stages. The origin of these different typologies will be then discussed with the aid of MD simulations in Section 4.After having described the typical coalesced structures, we analyze the properties of the overall nanoparticle distributions produced in the aggregation experiments, discussing the experimental parameters which allow to control the resulting percentages of nanoparticles belonging to different stages of coalescence. This demonstrates that the degree of coalescence can be tuned in two ways, i.e. by changing the length of the aggregation zone and by changing the pressure difference between the aggregation chamber and the deposition chamber.
3.1 Structure of coalesced (PtPd)@Pd nanoparticles
Let us consider first a low magnification image of a distribution of (PtPd)@Pd nanoparticles, see Fig. 2, where we can see several nanoparticles grown in standard sputtering conditions i.e. with LAZ = 125 mm and PDC = 6 × 10−4 mbar. We can observe that 21/34 nanoparticles are well isolated monomers, which do not have any physical entanglement with other nanoparticles. In addition, two pairs of collided nanoparticles are highlighted in yellow. Their mutual distance is smaller than their diameter. Three examples of aggregated nanoparticles are highlighted in red. These structures consist of pairs of nanoparticles softly bonded to each other, presenting misoriented faces. In the bottom right case, one of the two particles has a diameter significantly larger than the other. An early stage of coalescence is observable between the three nanoparticles in the blue rectangle. Here, comparing with the aggregated structures, we can see the monomers have rotated to create a more compact structure. We note that under these sputtering conditions we generally do not observe fused nanoparticles. After undergoing fusion, faceted nanoparticles become ovoidal or quasi spherical, with a size obviously larger than their components. | ||
| Fig. 2 Cs-corrected STEM-HAADF images of (Pt–Pd)@Pd nanoparticles grown in a Pd-rich atmosphere (Pd0.8Pt0.2) at a PDC = 6 × 10−4 mbar and LAZ = 125 mm. (a) Low-magnification image showing several nanoparticles. (b) Sequences of three similar nanoparticle assemblies (dimers and trimers) which underwent different stages of coalescence in-flight. From left to right: collision, aggregation and coalescence. | ||
In the following we focus on coalescing nanoparticles, trying to single out the different steps in the process towards fusion.
In Fig. 2b, representative images of Pt–Pd dimers and trimers are presented to summarize the different stages that we observe in the experiments. From left to right: collision, aggregation and coalescence. We note that, going from left to right, the images do not correspond to the same nanoparticle assemblies at different time stages, but they represent three similar nanoparticle assemblies, which happened to have undergone different stages of coalescence in-flight. These images highlight that, at more advanced stages, the monomers first tend to orient themselves parallel to each other and subsequently tend to form more compact structures as the ones shown in the third column.
A more precise characterization of the structures can be achieved by looking at the orientation of lattice planes, which can be better analyzed at higher magnification. An example is shown in Fig. 3 where we present three stages of coalescence occurring between three pairs of (PtPd)@Pd nanoparticles. In the first row we present STEM-HAADF images of the nanoparticles. Diffraction patterns acquired in the sintering neck and in the core zone of one of the coalescing units are shown in the bottom row. The areas from which the patterns are calculated are marked with green and red squares for the neck and the core, respectively.
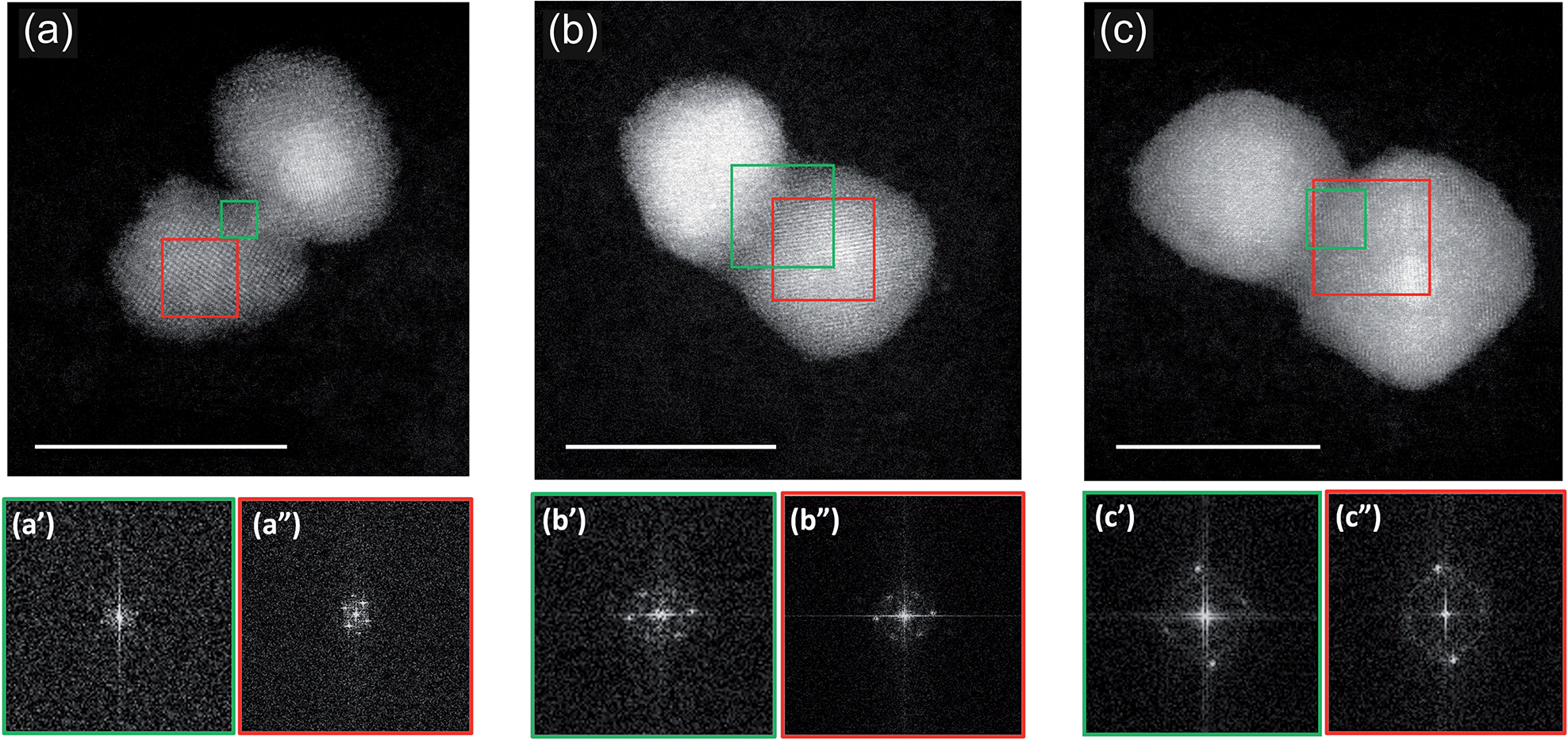 | ||
| Fig. 3 Cs-corrected STEM-HAADF images and diffracting patterns of coalesced (Pt–Pd)@Pd nanoparticles. The scattered intensity scale is associated with the atomic number Z of the elements. This makes easy to distinguish the heaviest Pt (brighter) from Pd (darker). The three nanoparticle assemblies illustrates coalescence occurred in flight at different stages: from (a) an early one happened between a (PtPd)@Pd nanoparticle and a nanoparticle constituted mainly of Pd to (b and c) more and more mature stages of coalescence occurred between two (PtPd)@Pd nanoparticle assemblies. In the bottom row, diffraction patterns acquired in the sintering neck (a′, b′ and c′) and in the core region of one of the units (a′′, b′′ and c′′) are shown. The areas from which the patterns are calculated are marked with a green and red square for neck and core, respectively. Scale bar 10 nm. | ||
The initial stage of coalescence is shown in Fig. 3a, where we can observe a thin sintering neck of about 1 nm formed between these two units, which clearly still keep their individuality. From a crystallographic point of view, we observed that both units exhibit crystallinity, while the sintering neck is mainly amorphous (see the diffraction patterns reported in the bottom row). Fig. 3b presents a subsequent stage of coalescence: the Pd neck is reduced in favour to a more compact structure. The original nanoparticles still remain misoriented with respect to each other. The original units, and the sintering neck present now a crystalline structure (see FFT data in Fig. 3b′ and 3b′′).
A further stage is shown in Fig. 3c. There, the structure is a little more compact, but, what is more important, the two original nanoparticles are parallel to each other. This latter structure is generally associated to a more mature stage of sintering as the original nanoparticles had time to reduce the interparticle neck and orient their facets to reduce their energy. As in the previous assembly, the sintering neck presents a very well defined crystalline structure, indicating that more advanced stage of coalescence occurred within this assembly.
Fig. 4 reports images from further coalescence stages. In this case, we consider the coalescence between core–shell (PtPd)@Pd nanoparticles and nearly pure Pd nanoparticles. These different aggregates are grown with LAZ = 40 mm and PDC = 1 × 10−1 mbar and can be considered as representative of the likely evolution of the nanoparticles shown in the previous Fig. 3. From (a to d), we see that the Pd nanoparticle progressively reduces the sintering neck, and wets the shell of the (PtPd)@Pd nanoparticle, thus making that shell thicker on one side of the aggregate. In the last stage, shown in Fig. 4d, a Pt-rich core is asymmetrically placed off-center within a Pd-rich shell. This feature indicates the particle is the result of full coalescence (i.e. of fusion), in which the nanoparticle shape becomes close to spherical, as expected at equilibrium. However, chemical ordering is not yet equilibrated, since at equilibrium the Pd atoms would be symmetrically distributed within the core.30,34 This indicates that chemical ordering equilibrates on a longer time scale than nanoparticle shape. This point will be discussed in Section 4 with the aid of simulation results.
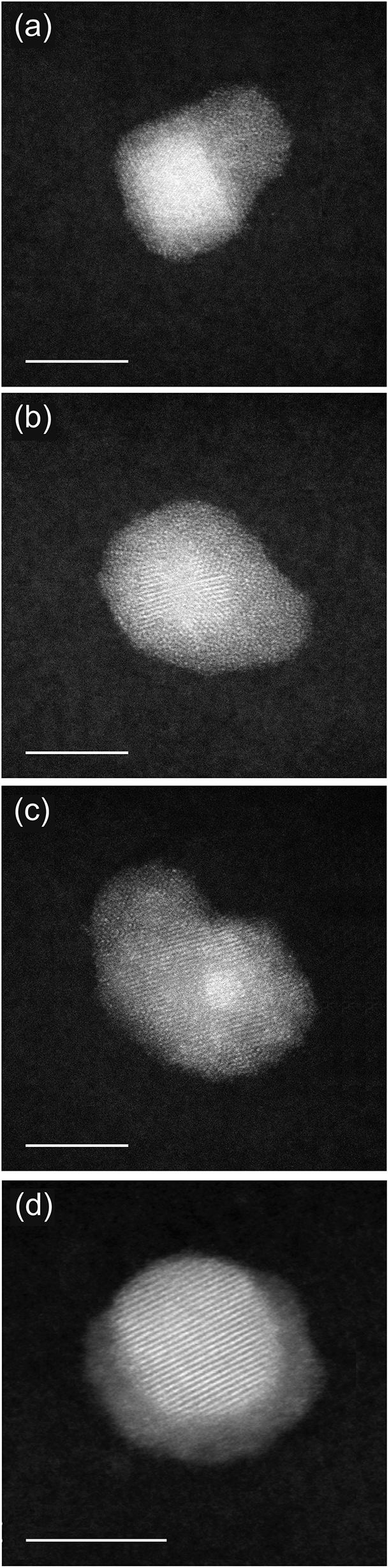 | ||
| Fig. 4 (a)–(d) Sequence of HAADF-STEM images showing the evolution in the coalescence between (PtPd)@Pd nanoparticles and almost pure Pd ones. These nanoparticles are deposited with a (PDC = 1 × 10−1 mbar). Scale bar 5 nm. | ||
In summary, the images presented so far show that the production of Pt–Pd nanoparticles from the gas phase leads to the in-flight formation of coalescing aggregates. These aggregates show different features according to their degree of advancement in the sintering process.
3.2 Tuning the coalescence of a distribution of Pt–Pd nanoparticles
In this section, we show that adjusting the residence time in the aggregation zone (dwell time), it is possible to control the degree of coalescence of the nanoparticles. In particular we show that increasing the dwell time the distribution shifts from being constituted mainly by isolated nanoparticles to aggregate and coalesced structures. The dwell time can be controlled by adjusting both the length of the aggregation zone (LAZ) and the pressure difference ΔP between the source and the deposition chamber (PDC).L AZ is well known in literature to strongly affect the size of mono and multi-metallic systems.35,36 On the contrary, the effect of ΔP is poorly characterized. Intuitively, an increase in this parameter allows a faster motion of the nanoparticles towards the deposition chamber, hence decreasing their dwell time.31 Here we study the combined effect of these two parameters on the fraction of coalescing nanoparticles observed in the final distribution. Three values of LAZ are tested: 45, 70 and 125 mm; for each of them, we consider three different PDC (6 × 10−4, 1 × 10−2 and 1 × 10−1 mbar). The distributions of nanoparticles are then observed by HRTEM/STEM and classified according to their structures. Table 1 summarises the results of this analysis. In Table 1, the growth conditions are reported in the first two columns while the following columns report the percentages of monomers (identified as isolated nanoparticles with compact shape), of collided particles (i.e. particles that are very close to each other but do not merge, as in the first column of Fig. 2b) and of coalescing particles.
| L AZ (mm) | P DC (mbar) | % monomers | % collided | % coalescing |
|---|---|---|---|---|
| 45 | 6 × 10−4 | 82 | 10 | 8 |
| 70 | 77 | 7 | 16 | |
| 125 | 74 | 4 | 20 | |
| 45 | 1 × 10−2 | 89 | 1 | 10 |
| 70 | 70 | 1 | 29 | |
| 125 | 38 | 1 | 61 | |
| 45 | 1 × 10−1 | 76 | 0 | 24 |
| 70 | 77 | 0 | 23 | |
| 125 | 69 | 0 | 31 |
Considering the distributions grown with PDC = 6 × 10−4 mbar we note that an increase in LAZ causes a slight decrease in the percentage of monomers from 82% (@LAZ = 45 mm) to 77% (@LAZ = 70 mm) and 74% (@LAZ = 125 mm). Among the sintered nanoparticles, the increase of LAZ causes a quite drastic decrease in the percentage of collided in favour of coalescing structures. At this PDC the structures constituted by three or more monomers represent are almost absent at LAZ = 45 mm while, they are a small but non negligible proportion (∼1 out of 10 coalescing nanoparticles) at LAZ = 125 mm.
At a PDC of 1 × 10−2 mbar, we observe that an increase in LAZ causes a decrease in the percentage of monomers which represent now the 89%, 70% and 38% of the distribution grown with LAZ = 45, 70, 125 mm respectively. While the first two values are comparable with those obtained at lower PDC, a notable increase is observed at LAZ = 125 mm. Concurrently, we observe an increasing of the degree of coalescence among the deposited dimers and trimers, with collided nanoparticles almost disappearing. The proportion of coalescing structures comprising structures with three or more units also increases, becoming about 1/4 of the total coalescing nanoparticles in the cases LAZ = 70 and 125 mm. Interestingly, none of these multi-particle aggregates is found at LAZ = 45 mm, as noted also at lower PDC.
Increasing PDC to 1 × 10−1 mbar the percentage of monomers deposited with LAZ = 40 and 70 mm further decreases but by a small amount compared to the distributions obtained at lower pressures. On the other hand at LAZ = 125 mm the monomers recorded constitute the 69% of the deposition. This might lead to think about an inversion of the trend observed at lower pressures for the same LAZ, according to which the increase in PDC reinforces the coalescence process. However, by observing at higher magnification, we note that the monomers grown in these conditions present an important difference compared to those deposited at lower pressures. First, at this high pressure we observe several (PtPd)@Pd core–shell structures with off-center cores, as the one shown in Fig. 4d. Second, the average monomer size is 10.3 ± 0.6 nm, which is almost twice as much as it was for the distribution grown at a PDC of 6 × 10−4 mbar (5.2 ± 0.5 nm). Both these elements indicate that the monomers observed at LAZ = 125 mm and PDC to 1 × 10−1 mbar are indeed the result of completed fusion processes which finally lead to compact nanoparticle shapes.
Regarding the non-monomer nanoparticles, collided structures are not found at PDC = 1 × 10−1 mbar. Coalescing nanoparticles consist either in large core–shells with smaller Pd nanoparticles attached, or in two or more large core–shells in an advanced stage of coalescence: the interfacing faces are parallel to each other, the sintering necks not visible anymore, as shown in Fig. 3b.
In summary these results show that both LAZ and ΔP cause drastic changes in the distribution of Pt–Pd nanoparticles and allow to tune the degree of coalescence of the whole distribution from collided to aggregated to more and more compact structures. In particular, an increase in LAZ causes a reduction in the percentage of monomers with consequent increase in the sintered structures, an increase of the degree of coalescence of these sintered structures and the formation of trimers at LAZ larger that 45 mm. On the other hand, an increase of PDC causes the formation of increasingly compact structures which typically consist in large off-centered monomeric core–shells, core–shells with well oriented faces, and complex multimeric structures. This confirms that the reduction of ΔP slows down the motion of the nanoparticles from the source to the deposition chamber, hence increasing the time they spend growing in the aggregation zone.
4 Simulation results
4.1 Shape evolution during coalescence
In order to analyze the evolution of the coalescence process, we first describe the results of a representative simulation in which the total size of the aggregate is of about 3000 atoms (linear sizes of 5–6 nm during evolution) (see Fig. 5). The simulation extends to a time scale which is much longer than that of previous molecular-dynamics simulations in the size range of a few thousand atoms,6,16,37–39 and allows to qualitatively distinguish different steps during the coalescence stage. Simulations of this type are quite cumbersome from a computational point of view so that we were able to run only a few of them. Therefore, in order to accumulate a more complete statistics allowing quantitatve estimates, we performed coalescence simulations for smaller sizes, of about 800 atoms in total (linear sizes of 3–4 nm during the coalescence process).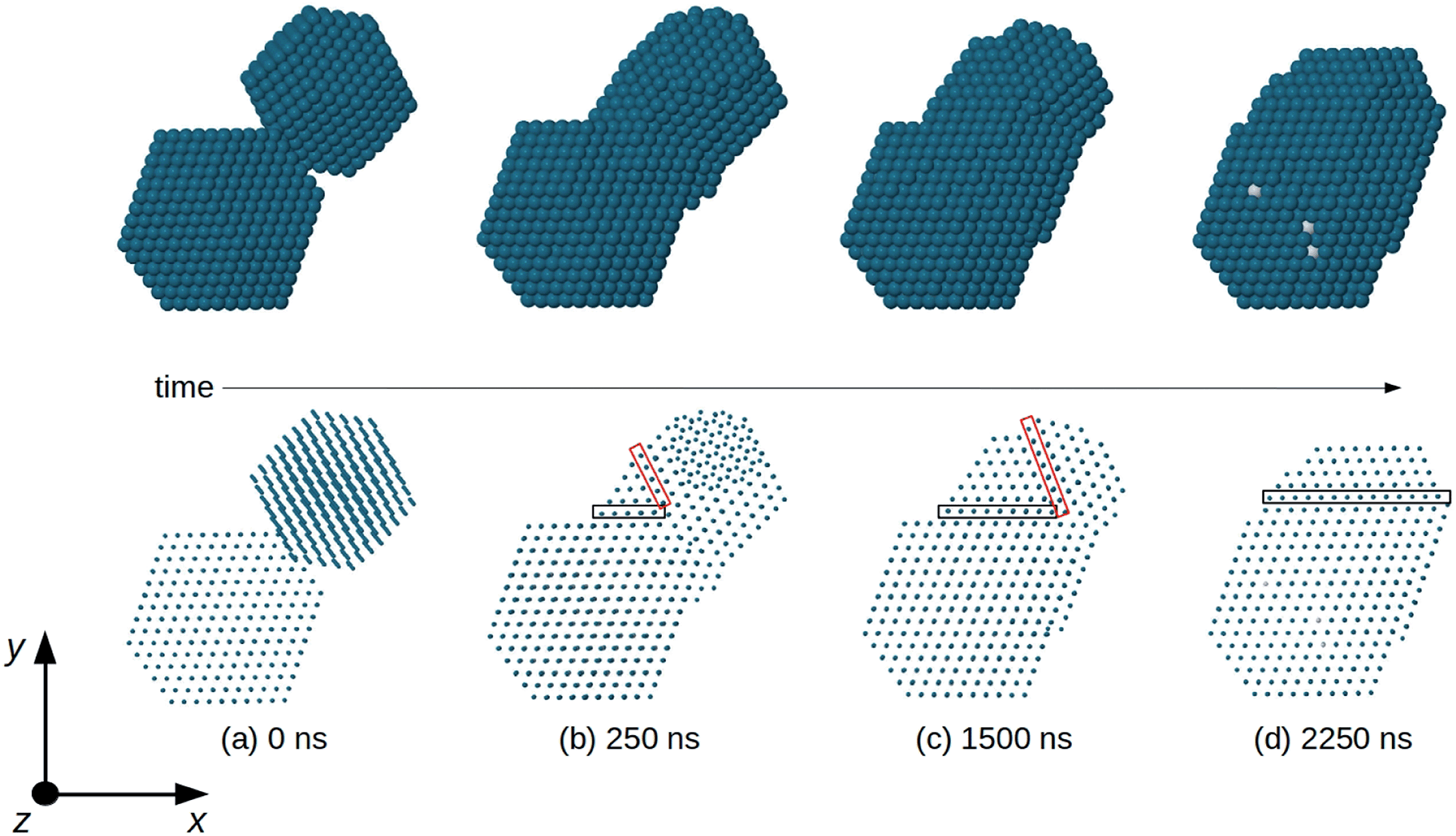 | ||
| Fig. 5 Snapshots from a coalescence simulation of two truncated octahedra at T = 750 K. The truncated octahedra have compositions Pd976 and Pt645Pd1280. Here and in the following figures Pd and Pt atoms are colored in blue and white, respectively. In the top row, we show the nanoparticle surface, which is almost completely made of Pd atoms. In the bottom row, the same images are shown by representing the atoms by small spheres, so that the atomic columns can be seen. (a) Initial configuration: the nanoparticles are randomly oriented, so that atomic columns of Pd976 are not parallel to those of Pt645Pd1280. (b) Very quickly the two nanoparticles rearrange their orientations so that all atomic columns become parallel to the z direction. This happens well before the time (250 ns) of this snapshot. Two non-parallel twin planes (enclosed by the black and red rectangles) separate different fcc fragments of the aggregate. The close-packed planes to the right of the red rectangle do not present the same orientation as the planes of the remaining part of the aggregate. (c) The twin plane enclosed by the red rectangle shifts to the right, so that the fragment with misoriented planes becomes smaller and smaller and finally (d) disappear. In (d) all close-packed planes are parallel (to the x–z plane), but the twin plane enclosed by the black rectangle is still there. In (d), top row, some isolated white spots show very the few Pt atoms appearing at the nanoparticle surface. | ||
In Fig. 5 we report snapshots from a coalescence of two fcc truncated octahedral nanoparticles of larger size at T = 750 K. The first is pure Pd976 and the second is Pt645Pd1280 (with (PtPd)@Pd chemical ordering30). The initial configuration is of 0.5 nm distance between the nanoparticles, random orientations and zero relative velocity, as in Fig. 5a. The first rearrangement step in the coalescence process leads to the alignment of atomic columns. An atomic column is a straight row of atoms connected by nearest-neighbour bonds. This first step is quite fast, since the atomic columns align along a common direction (z-direction in Fig. 5), on a time scale of a few ns, much shorter than the time at which the snapshot of Fig. 5b is taken. Even though all columns are aligned along z, in Fig. 5b and c the close-packed atomic planes that are perpendicular to x–y do not form a sequence of parallel planes extending to the whole aggregate. In fact, in Fig. 5b and c three zones, separated by two non-parallel twin planes, can be singled out. There is a first larger zone on the bottom left part of the coalescing aggregate, which derives from the original Pt645Pd1280 unit. In that zone, all close-packed planes are parallel to the x–z plane. The second zone is smaller and it is between the two twin planes (which are enclosed by the black and red rectangles in the figure). In this second zone, all close-packed planes are again parallel to the x–z plane. In the third zone, which derives from Pd976, the close packed planes are tilted with respect to those of the other zones, so that they are not parallel to the x–z plane. As the simulation proceeds, the twin plane in the red rectangle progressively shifts to the right, so that the third zone becomes smaller and smaller, and finally disappears (Fig. 5d). This process ends with an elongated aggregate with all close-packed planes parallel to x–z. It takes place on a time scale of a few 103 ns, being therefore much slower than the alignment of atomic columns, but faster than the scale of shape rounding. This hierarchy of time scales is experimentally confirmed by the frequent observation of nanoparticles in which the close-packed planes are very well aligned, but the overall shape is quite elongated, as in the TEM image shown in Fig. 6.
 | ||
| Fig. 6 HRTEM-STEM image of a coalescing nanoparticle of elongated shape in which atomic close-packed planes are very well aligned, forming a single sequence of planes spanning the whole nanoparticle. | ||
We note also that nanoparticles with multiple-twin structures such as decahedra and icosahedra are energetically unfavorable for Pt and Pd already for very small sizes.40 These multiple-twin structures present several non-parallel twin planes. Their elimination in our coalescing aggregates is therefore likely to be driven by energy optimization.
The sequence shown in Fig. 5 suggests that the evolution of the shape of the coalesced aggregate takes place through a three-steps process: (i) alignment of the atomic columns of the two subunits (Fig. 5b); (ii) alignment of close-packed planes (Fig. 5d); (iii) structural rearrangement in a compact geometric shape (not shown in the figure). To validate this scheme we perform a further set of simulations, in which a pure Pd201 and a Pt100Pd305 nanoparticle collide. Even in this case, both nanoparticles have truncated octahedral shape and the bigger one exhibits a core–shell (PtPd)@Pd chemical ordering. The smaller size of the colliding units allows us to perform several simulations and thus to study the coalescence stages with a more complete statistics. The simulations are performed at 500, 600, 650 and 700 K. For each temperature we run ten short simulations of at least 10 ns and five long simulation of 10 μs.
For each simulation, we identify the times at which the three different evolution steps are completed. The alignment time of atomic columns and of closed-packed planes are the times in which configurations such as Fig. 5b and d are achieved for the first time, respectively. To identify the characteristic time of the third step, i.e. the achievement of a compact shape, we consider the evolution of the asphericity of the aggregate. It is a geometric parameter defined from the eigenvalues of the gyration tensor, as b = λ12 − 1/2(λ22 + λ32), where λ1 is the largest of the three eigenvalues. The asphericity is always non-negative and zero only when the eigenvalues are all equal, i.e. when the aggregate is spherically symmetric. The use of this parameter is particularly appropriate in this case, since the equilibrium structure of the coalesced nanoparticle is the quasi-spherical truncated octahedral shape.
In Fig. 7a we show the behaviour of the asphericity of the coalescing aggregates for all temperatures, whereas in Fig. 7b–d we show some snapshots from an evolution sequence obtained at 650 K. These structures exhibit aligned close-packed planes and are therefore representative of the final stage of the shape evolution process. The corresponding asphericity values are reported below each structure. The figure shows that the asphericity decreases during the simulations, as the aggregate goes from the initially elongated shape towards a more compact one. We point out that the difference in the initial values of the curves in Fig. 7a are simply due to statistical fluctuations in the very first steps of the simulations.
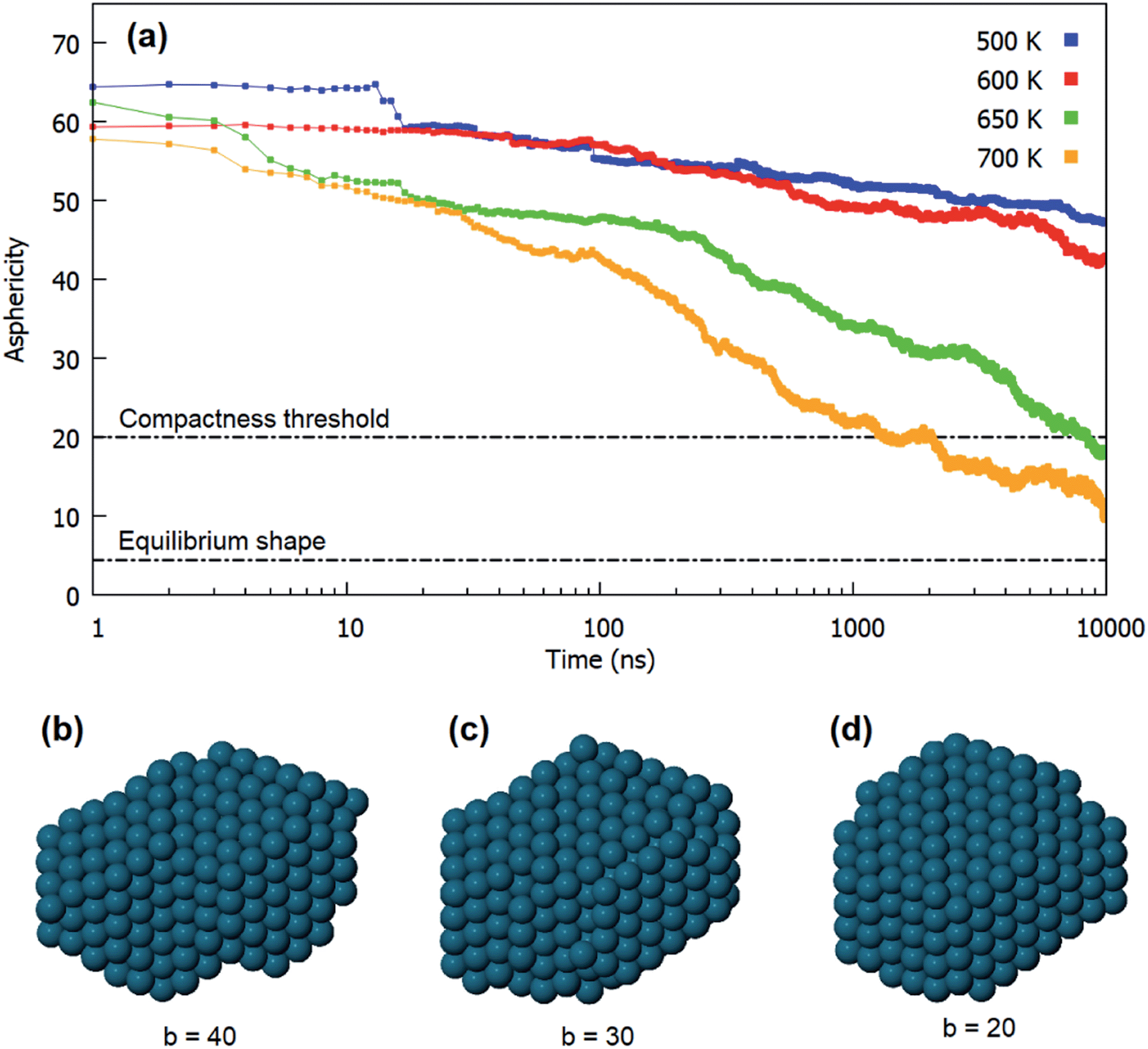 | ||
| Fig. 7 Shape evolution in the coalescence of Pd201 and Pt100Pd305. (a) Evolution of the asphericity of the aggregate at different temperatures. For each temperature we show the average over all the performed simulations. (b–d) Structures obtained during a simulation at 650 K, corresponding to decreasing values of the asphericity. Structure in (d) corresponds to the chosen compactness threshold of b = 20. | ||
Here we assume that the third step of the evolution is reasonably completed when the asphericity has reached the value b = 20, i.e. when structures such as the one in Fig. 7d are achieved. This structure is still rather different from the equilibrium truncated octahedral shape, for which we have calculated b = 4.44. However, it exhibits a fairly good degree of compactness, especially in comparison to the previous stages of the evolution (see Fig. 7b and d). We notice that the chosen threshold is reached only at the highest temperatures (650 and 700 K) in the time scale of our simulations.
The results are summarized in Fig. 8, in which the characteristic times of the three different steps of the evolution are shown as a function of the simulation temperature. For each temperature we show the average values over all the performed simulations. The graph shows that the three different steps previously identified are well-separated in time at all temperatures.
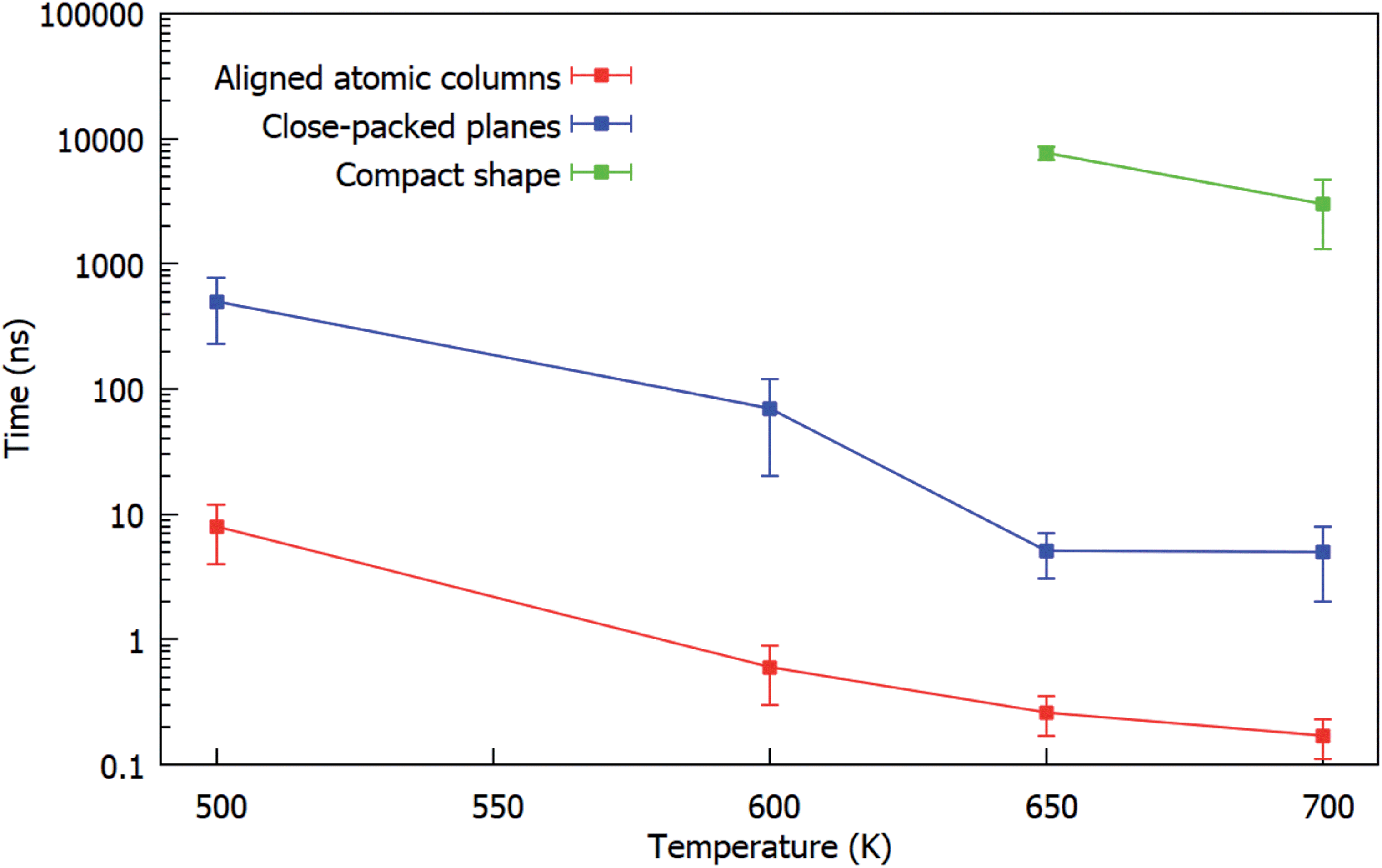 | ||
| Fig. 8 Characteristic times of the three steps (alignment of columns, alignment of close-packed planes, rounding of the shape) of the evolution during the coalescence of fcc truncated octahedral Pd201 with Pt100Pd305. For each temperature we show the average over all the performed simulations. The graph shows that the three steps are well-separated in time at all temperatures. | ||
4.2 Formation of nanoalloys with off-center cores
Pd-rich nanoalloys with Pd shell and PtPd cores were commonly formed in our experiments of ref. 30. In most cases, the cores were placed in the center of the nanoparticles in a rather symmetric way, as the result of a kinetically trapped growth mode in which coalescence is not playing an important role.30In the present experiments we have shown evidence for the formation of nanoalloys in which the core is placed asymmetrically in an off-center position, as shown in Fig. 4. Here we show, by means of MD simulations, that these asymmetric structures can be the result of a coalescence process between a PtPd nanoalloy and an almost pure Pd nanoparticle of smaller size.
In Fig. 9 we show representative snapshots of the coalescence process between a truncated octahedral nanoalloy of composition Pt645Pd1280 and a truncated octahedral pure Pd976 nanoparticle. The Pt645Pd1280 nanoparticle has a Pd surface layer and a randomly intermixed inner part. The colliding units are initially placed at a distance of 0.5 nm with a randomly selected orientation and zero relative velocity. In the two simulations of Fig. 9, the system is evolved at a constant temperature of 900 and 800 K for 1.5 and 3 μs, respectively.
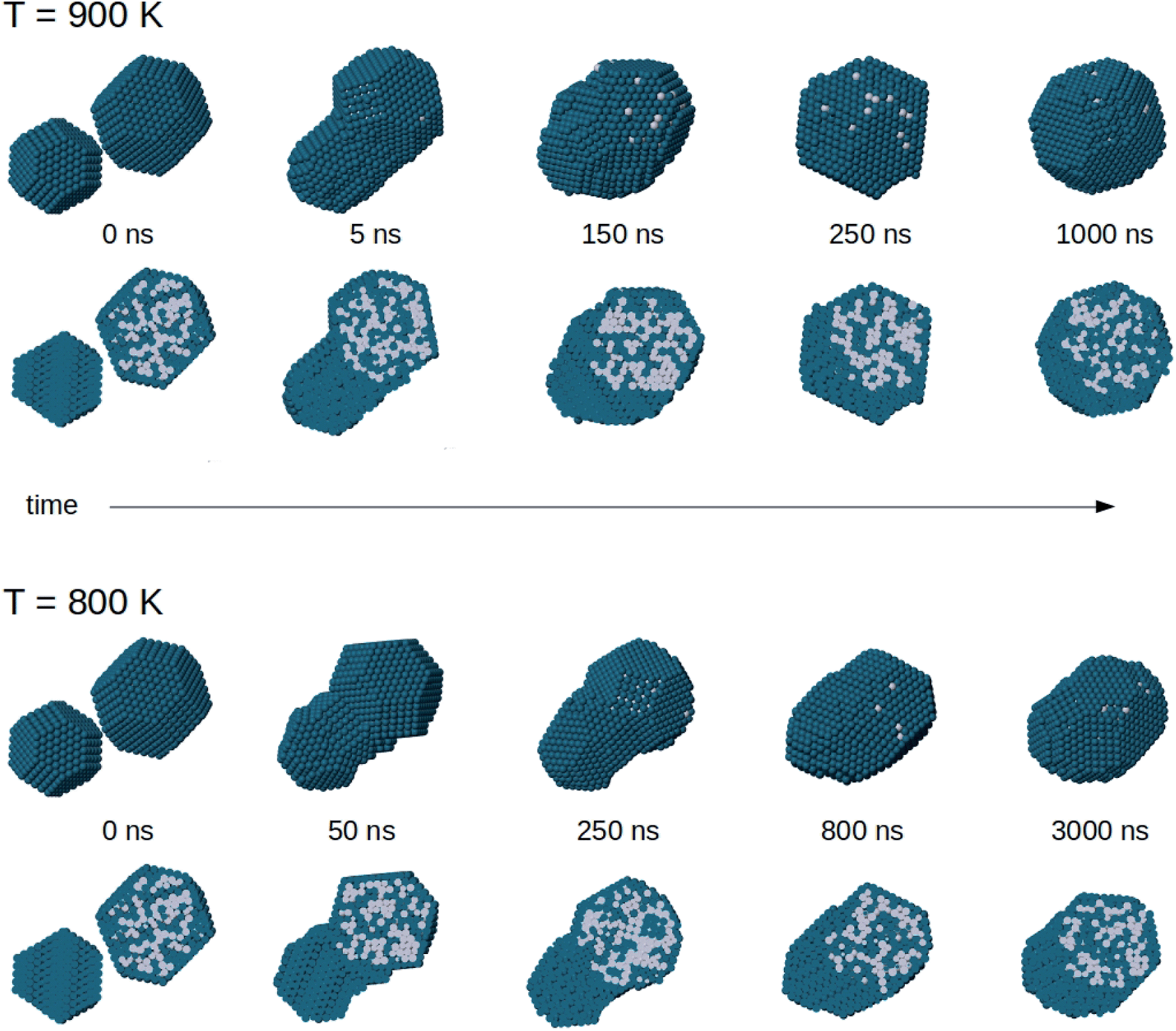 | ||
| Fig. 9 Snapshots from coalescence simulations at T = 900 and 800 K. The initial configuration is the same in both cases. For each structure, two views are shown: in the top row we present the cluster surfaces, in the bottom row we present cross sections to show chemical ordering inside the nanoparticles. | ||
The sequence at 900 K shows a complete evolution of the nanoparticle shape towards a truncated octahedral structure. The nanoparticle becomes more and more compact with increasing time, and reaches a truly compact shape already on a time scale of ∼250 ns. However, equilibrium is not completely reached. Chemical ordering is not the optimal one, because the Pd-rich core still remains in off-center position. The equilibration of chemical ordering is therefore quite slow even at this temperature, so that coalescence can lead to structures which closely resemble the experimental image of Fig. 4d. In the sequence at 800 K, shape equilibration is not yet completed after 3000 ns. The nanoparticle structure is still somewhat ellipsoidal at the end of the simulation. The Pt core is clearly off-center also in this case. We note also that the intermediate images of both simulation sequences qualitatively resemble different coalescence stages that are experimentally represented by the images of Fig. 4a–c.
The same behaviour is observed in the coalescence of smaller nanoparticles. In all simulations of the coalescence of smaller nanoparticles, the final structure after 10 μs exhibits core–shell chemical ordering with off-centered Pt–Pd mixed core. To quantify this effect, we calculate the distance between the geometric center of the whole aggregate and the geometric center of the distribution of Pt atoms, which roughly corresponds to the center of the Pt–Pd mixed core. We also monitor the mean distance of Pt atoms from Pt geometric center, as a measure of the core compactness. We analyse the simulations at 650 and 700 K, i.e. the ones in which the coalesced aggregate has achieved a fairly compact shape (see Section 4.1). After 10 μs, the distance between the centers of the core and of the whole nanoparticle is 5.81 Å and 5.11 Å on average at 650 and 700 K, respectively. It is a significant off-center displacement compared to the nanoparticle radius, which is in the range of 15–16 Å for these nanoparticles. Besides, the mean distance of Pt atoms from Pt geometric centre does not change during the evolution at 650 K and increases only slightly at 700 K (plus 1.9% by the end of the simulation, on average), meaning that the Pt–Pd mixed core keeps the same size as at the beginning of the coalescence process, without significant diffusion of its atoms within the coalescing aggregate.
In summary, these simulations show that equilibrium shape is attained well before equilibrium chemical ordering. This is due to the fact that shape can attain equilibrium by atomic-level processes which involve the displacement of surface and subsurface atoms, while chemical ordering needs also the occurrence of diffusion events in the inner part of the aggregate. The latter are much slower than the former, as already shown in simulations of the equilibration phase-separating systems.8
5 Conclusions
The experimental results obtained in this work show that the degree of coalescence can be controlled in gas-phase nanoparticle synthesis. We were able to single out different and well controlled growth conditions in which the role of coalescence can be tuned from negligible to dominant. This was achieved both by varying the length of the aggregation zone in the source, and the pressure difference between aggregation and deposition chambers. Both variations influence the dwell time. Long aggregation zones and small pressure differences increase the dwell time which in turn increases the probability of encounters between growing particles and therefore of coalescence.By growing the nanoparticles in conditions in which coalescence is dominant, a considerable proportion of coalescing nanoparticles was produced, containing representative examples of the different steps in the coalescence process. These steps were further analyzed with the aid of molecular-dynamics simulations, which were employed to gain understanding of atomic-level processes.
The combined analysis of experiments and simulations showed that the coalescence process in PtPd nanoparticles occurs through four steps, with a clear hierarchy of well-separated time scales. From the shortest to the longest: (i) alignment of close-packed atomic columns along a common direction; (ii) alignment of close-packed atomic planes into a sequence of parallel planes which spans the nanoparticle; (iii) equilibration of the nanoparticle shape which becomes more and more compact; (iv) equilibration of chemical ordering. Examples of nanoparticles at step (ii) are given in Fig. 6 (experiment) and Fig. 5 (simulation), whereas nanoparticles at step (iii) may present the off-center PtPd cores as in Fig. 4d (experiment) and Fig. 9 (simulation).
In conclusion, our results demonstrate that coalescence in gas-phase growth can be used as an effective tool to produce specific nanoparticle shapes and chemical orderings, paving the way to the controlled production of new types of structures.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research performed at OIST was supported by funding from the Okinawa Institute of Science and Technology Graduate University. DN and RF acknowledge support from the project PRIN2017 UTFROM of the Italian Ministry of University and Research. The authors thank the OIST Imaging Section for providing access to TEM and Dr Toshio Sasaki for support, and Ella Joyce Minnai for technical drawing. The authors acknowledge networking support from the International Research Network Nanoalloys of CNRS.Notes and references
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS.
- Nanoalloys from Fundamentals to Emergent Applications, ed. F. Calvo, Elsevier, 2013 Search PubMed.
- R. Ferrando, Structure and Properties of Nanoalloys, Elsevier, 2016, Frontiers of Nanoscience, vol. 10, pp. 1–337 Search PubMed.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS.
- P. Grammatikopoulos, C. Cassidy, V. Singh and M. Sowwan, Sci. Rep., 2014, 4, 5779 CrossRef CAS.
- P. Grammatikopoulos, J. Kioseoglou, A. Galea, J. Vernieres, M. Benelmekki, R. E. Diaz and M. Sowwan, Nanoscale, 2016, 8, 9780–9790 RSC.
- D. Nelli and R. Ferrando, Nanoscale, 2019, 11, 13040–13050 RSC.
- M. M. Mariscal, S. A. Dassie and E. P. M. Leiva, J. Chem. Phys., 2005, 123, 184505 CrossRef.
- S. A. Paz, E. P. M. Leiva, J. Jellinek and M. M. Mariscal, J. Chem. Phys., 2012, 134, 094701 CrossRef.
- N. Riefler and L. Mädler, J. Nanopart. Res., 2010, 12, 853–863 CrossRef CAS.
- A. Tricoli, M. Graf, F. Mayer, S. Kuühne, A. Hierlemann and S. E. Pratsinis, Adv. Mater., 2008, 20, 3005–3010 CrossRef CAS.
- J. Han, W. Hu and H. Deng, Surf. Interface Anal., 2009, 41, 590–594 CrossRef CAS.
- A. Mohanty, N. Garg and R. Jin, Angew. Chem., 2010, 122, 5082–5086 CrossRef.
- P. Grammatikopoulos, M. Sowwan and J. Kioseoglou, Adv. Theory Simul., 2019, 2, 1900013 CrossRef.
- D. Nelli, G. Rossi, Z. Wang, R. E. Palmer and R. Ferrando, Nanoscale, 2020, 12, 7688–7699 RSC.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer and P. C. Redfern, et al. , Science, 2010, 328, 224–228 CrossRef CAS.
- M. L. Eggersdorfer, D. Kadau, H. J. Herrmann and S. E. Pratsinis, J. Aerosol Sci., 2012, 46, 7–19 CrossRef CAS.
- R. Huang, Y.-H. Wen, Z.-Z. Zhu and S.-G. Sun, J. Phys. Chem. C, 2012, 116, 8664–8671 CrossRef CAS.
- N. V. Long, T. D. Hien, T. Asaka, M. Ohtaki and M. Nogami, Int. J. Hydrogen Energy, 2011, 36, 8478–8491 CrossRef.
- M. Mougenot, A. Caillard, P. Brault, S. Baranton and C. Coutanceau, Int. J. Hydrogen Energy, 2011, 36, 8429–8434 CrossRef CAS.
- S. J. Yoo, H. Y. Park, T. Y. Jeon, I. S. Park, Y. H. Cho and Y. E. Sung, Angew. Chem., Int. Ed., 2008, 47, 9307–9310 CrossRef CAS.
- Z. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542–7543 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS.
- H. Zhang, M. Jin and Y. Xia, Chem. Soc. Rev., 2012, 41, 8035–8049 RSC.
- H. Lee, S. E. Habas, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2008, 130, 5406–5407 CrossRef CAS.
- D.-B. Huang, Q. Yuan, H.-H. Wang and Z.-Y. Zhou, Chem. Commun., 2014, 50, 13551–13554 RSC.
- L. Wang, Y. Nemoto and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 9674–9677 CrossRef CAS.
- D. Nelli, A. Krishnadas, R. Ferrando and C. Minnai, J. Phys. Chem. C, 2020, 124, 14338–14349 CrossRef CAS.
- G. E. Johnson, R. Colby and J. Laskin, Nanoscale, 2015, 7, 3491–3503 RSC.
- A. Mayoral, S. Mejia-Rosales, M. M. Mariscal, E. Perez-Tijerina and M. Jose-Yacaman, Nanoscale, 2010, 2, 2647–2651 RSC.
- R. P. Gupta, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 6265 CrossRef CAS.
- A. D. Clercq, S. Giorgio and C. Mottet, J. Phys.: Condens. Matter, 2016, 28, 064006 CrossRef.
- S. Pratontep, S. J. Carroll, C. Xirouchaki, M. Streun and R. E. Palmer, Rev. Sci. Instrum., 2005, 76, 045103 CrossRef.
- Y. Huttel, L. Martinez, A. Mayoral and I. Fernández, MRS Commun., 2018, 8, 947–954 CrossRef CAS.
- S. Arcidiacono, N. Bieri, D. Poulikakos and C. Grigoropoulos, Int. J. Multiphase Flow, 2004, 30, 979–994 CrossRef CAS.
- K. Yun, J. Lee and H.-S. Nam, Electron. Mater. Lett., 2019, 15, 133–139 CrossRef CAS.
- P. Lu, M. Chandross, T. J. Boyle, B. G. Clark and P. Vianco, Modell. Simul. Mater. Sci. Eng., 2014, 22, 075012 CrossRef.
- F. Baletto, R. Ferrando, A. Fortunelli, F. Montalenti and C. Mottet, J. Chem. Phys., 2002, 116, 3856 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |