
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidics for the rapid and controlled preparation of organic nanotubes of bent-core based dendrimers†
Martín
Castillo-Vallés
 a,
Pilar
Romero
a,
Víctor
Sebastián
*bc and
M. Blanca
Ros
*a
a,
Pilar
Romero
a,
Víctor
Sebastián
*bc and
M. Blanca
Ros
*a
aDepartment of Organic Chemistry, Instituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza, 50009, Spain. E-mail: bros@unizar.es
bDepartment of Chemical Engineering and Environmental Technology, Instituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza, 50009, Spain. E-mail: victorse@unizar.es
cNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine, CIBER-BBN, 28029-Madrid, Spain
First published on 6th January 2021
Abstract
Recently, bent-core molecules have emerged as excellent building blocks for the obtaining of nanostructures in solvents. Herein, we report the use of a coaxial microfluidic system as a promising tool to control the self-assembly of non-conventional bent-core amphiphiles. Moreover, a TEM study to comprehend the hierarchical self-assembly process in solution was carried out. The proposed tool provides both a cost-effective platform to save hard-to-synthesise reagents and a rapid method to screen a plethora of different parameters, i.e., THF/water ratio, residence time, concentration of the amphiphile, temperature and pH. The experiments allowed to test for the first time the suitability of microfluidics for the self-assembly of bent-core molecules, as well as the study of a range of conditions to control the assembly of different nanostructures in a rapid and controlled manner. Additionally, organic nanostructures were combined with gold nanoparticles to prepare nanocomposites with enhanced properties. Both organic and hybrid nanostructures were also obtained in the solid state. These results may inspire scientists working on supramolecular chemistry and bent-core molecules expanding the scope of microfluidic systems for the self-assembly of other low-molecular-weight compounds.
Introduction
Supramolecular chemistry, also known as the chemistry beyond the molecule, is a rich and mature research field that is still under intense investigation.1 The non-covalent interactions (van der Waals forces, π–π stacking, hydrogen bonding, etc.) established within molecules can be employed as the driving force for the bottom-up construction of nanostructures.Among the large variety of molecular designs that are capable of self-assembling at the nanoscale, bent-core molecules warrant special attention as they are responsible for opening the door to the newest family of liquid crystalline phases in the bulk with very appealing properties, i.e., polar organisations, the induction of supramolecular chirality from achiral molecules, non-linear optical response, and ferroelectric/antiferroelectric or piezoelectricity behaviour.2 Attractively, these mesogens have recently proven to be versatile supramolecular build-blocks capable of organising not only in the bulk but also in solution if their chemical structure allows interaction with selected solvents.3 Thus, in our previous research, we demonstrated that the introduction of hydrophobic promesogenic bent-core moieties into a hydrophilic poly(propylene) imine (PPI) dendrimer provided molecules with an amphiphilic character that allowed them to self-assemble in THF/water mixtures. These materials gave rise to a wide range of nanostructures (rods, non-twisted or twisted fibres, helical ribbons and nanotubes) in which compact packing that characterises bent-core mesophases was also present.4,5 Interestingly, these results offer innovative possibilities for bent-core based associations.
Many supramolecular assemblies have been explored as functional materials in different applications.6 Both the size and shape of nanostructures greatly influence the properties of the materials and dictates their potential applications.,7–9 and consequently great effort has been devoted by the scientific community to develop and control a wide range of nanostructures with different chemical natures and with various morphologies (micelles, vesicles, nanoparticles or nanotubes, amongst others). Despite the fact that most of these nanostructures can fulfil nanotechnological requirements, nanotubular morphologies are of special interest in many applications owing to their unidimensional and hollow structure.10 These characteristics make them excellent candidates as photoconductive materials or molecular wires as well as for the storage and liberation of small molecules such as hydrogen or bioactive compounds. For this reason, precise control of the fabrication process of the assemblies is a key point. Moreover, the development of an optimized processing method for the materials in order to obtain large amounts of high-quality nanostructures is a prerequisite in the search for real applications.
In this target, microfluidics has been successfully implemented in the last few years to overcome these two issues, that is, control over the morphology and the scale-up of production.11–13 Many of the advantages of these systems, when compared to conventional batch syntheses, can be exploited in the self-assembly of nanostructures.14–17 For example, the confined space within the microchannel allows fine control over mass transfer as mixing is only mediated by diffusion. Moreover, numerous parameters can be easily modified in order to define standard protocols17–19 and to achieve kinetically favoured assemblies that differ from thermodynamic nanostructures obtained in flask-mixing procedures.20,21 Finally, the process can be automated and this removes the lack of reproducibility in batch syntheses and allows the preparation of the materials in a continuous flow.22–24 This possibility can increase the productivity and allows the fabrication of nanomaterials to be scaled-up, which is a major issue in nanoscience. Nevertheless, from a basic research point of view, microfluidics, unlike the time-consuming co-solvent method, allows the rapid screening of a large number of experimental conditions. Additionally, the small volume of the microreactor means that only small amounts of reactants are needed. This is especially important when using compounds that are difficult to synthesise.
Although microfluidics has been widely used to obtain molecular assemblies such as polymerosomes14 and liposomes17 for drug delivery systems, it is noticeable that other types of organic soft materials formed by low-molecular-weight compounds have been much less studied to date.16,21,25–28 There is still a vast and scarcely explored field of research concerning the continuous and scalable production of nanomaterials built from small organic molecules, such as bent-core molecules.
Herein we report a pioneering approach to the formation of a range of nanostructures (twisted fibres, helical ribbons, nanotubes and large non-twisted fibres) from a bent-core dendrimer by using a coaxial microfluidic reactor as a fast, continuous and highly reproducible way to prepare nanostructures from non-conventional small organic molecules.
The scope of this manuscript focuses on the demonstration of the suitability of a microfluidic device for the self-assembly of bent core molecules. For the analysis of a range of processing conditions, the ionic bent-core dendrimer PPI1-B1-4-8 previously reported by us (Scheme 1) was employed. To this aim, we evaluated the influence of several preparation parameters, i.e., residence time, THF/water rate, concentration of organic molecules, temperature and pH. Moreover, we also deposited AuNPs onto the surface of assemblies to obtain hybrid nanomaterials. Finally, in order to obtain nanotubes and fibres in a solid state, the materials were also deposited onto different surfaces.
 | ||
| Scheme 1 Chemical structure of the ionic bent-core dendrimer PPI1-B1-4-8. | ||
Results and discussion
Processing conditions
The co-solvent method is one of the most common techniques to prepare molecular aggregates. This methodology, a conventional batch synthesis, involves the dropwise addition of water (poor solvent) to an organic solution that contains an amphiphile,29 an approach that has proven to be an easy procedure for the fabrication of nanostructures from bent-core based dendrimers in previous studies.4,5 However, the co-solvent method is time-consuming and concentration gradients are formed as the water content is higher in the locations where the drop comes into contact with the organic phase. In contrast, microfluidic devices allow fluid flows to be handled at the microscale very precisely and this leads to highly reproducible syntheses and mixing conditions. Moreover, the ability of microfluidic systems to manage and process reduced volumes (at the nanolitre scale) diminishes the amount of reagents required when compared to batch-to-batch synthesis. This, together with the short self-assembly times in the microreactor allows the rapid screening of a large number of experimental conditions.With the aim of exploiting the benefits of this technology in the study of the self-assembly of amphiphilic bent-core based dendrimers, we employed a capillary-based flow-focusing device with coaxially aligned microcapillaries composed of a 150 μm-diameter inner capillary that contains the organic phase (a 0.5 wt% solution of the dendrimer in THF) and a 500 μm-diameter outer capillary supplied with the aqueous phase (poor solvent). The microfluidic system volume was 150 μL. The inner capillary was coaxially localized under an optical microscope to obtain an axisymmetric flow-focusing device in which the organic phase is surrounded symmetrically by the aqueous sheath phase. The fast and efficient mixing of the two streams causes aggregation of the bent-core compound as it is exposed to the poor solvent, and the resulting nanostructures were collected in a vial (see ESI† for additional experimental details and pictures of the setup).
Since the experimental conditions may strongly affect the self-assembly process, in the first stage we studied by TEM the influence of several parameters on the morphology of nanostructures obtained in the microreactor. First of all, we carried out experiments at different THF/water ratios in order to check whether the self-assembly mechanism was equivalent to that observed in the co-solvent method or not. Subsequently, we took advantage of the ease of modifying the conditions of the microreactor to explore the effect of several new experimental conditions in the self-assembly process, i.e., flow rates, bent-core amphiphile concentration, temperature and pH. The results are summarised in Table 1.
| Parameter | Water ratio | Residence time | Amphiphile concentration | Temperature | pH |
|---|---|---|---|---|---|
| Effect | Increasing the water proportion in the reactor stimulates the evolution from fibres to nanotubes | No relevant influence above 9 s of residence time | Lower concentrations retard the formation of nanotubes | Heating the reactor favours the formation of nanotubes | Acid pH makes the amphiphile self-assemble into new structures whereas basic pH hinders the self-assembly |
 | ||
| Fig. 1 (a) TEM images of the nanostructures of PPI1-B1-4-8 fabricated in the coaxial microreactor at different THF/water ratios at a total flow rate of 500 μL min−1. (b) Enlarged images of twisted fibres, helical ribbons and nanotubes. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μL min−1) resulted in twisted fibres, helical ribbons and nanotubes depending only on the THF/water ratio. However, TEM images of samples prepared at the shortest residence time of 0.9 s (10000 μL min−1) revealed the formation of amorphous agglomerates in addition to the well-defined nanostructures described previously (see ESI†). The sudden mixing obtained at the fastest flow rate (10000 μL min−1) caused all of the bent-core molecules to be exposed to the aqueous phase in a short period of time. This can create a large number of nucleation points that cannot grow further and this may be the reason for the observation of ill-defined aggregates.
000 μL min−1) resulted in twisted fibres, helical ribbons and nanotubes depending only on the THF/water ratio. However, TEM images of samples prepared at the shortest residence time of 0.9 s (10000 μL min−1) revealed the formation of amorphous agglomerates in addition to the well-defined nanostructures described previously (see ESI†). The sudden mixing obtained at the fastest flow rate (10000 μL min−1) caused all of the bent-core molecules to be exposed to the aqueous phase in a short period of time. This can create a large number of nucleation points that cannot grow further and this may be the reason for the observation of ill-defined aggregates.
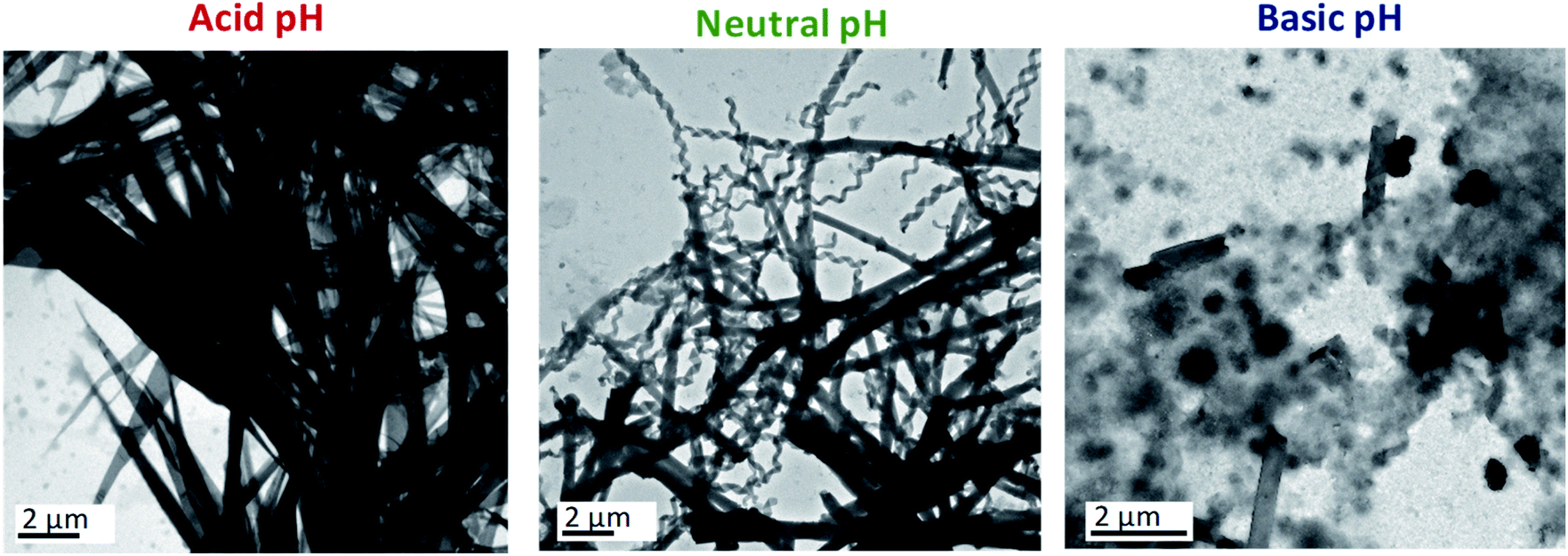 | ||
| Fig. 2 TEM images of the samples of PPI1-B1-4-8 prepared at THF/water ratios of 1/1 and at a total flow rate of 500 μL min−1 at variable pH. Acid conditions: diluted HCl aqueous solutions ([HCl] = 2 × 10−3, pH ≈ 4.5); basic conditions: diluted NaOH aqueous solutions ([NaOH] = 2 × 10−3, pH ≈ 8.5). | ||
Basic aqueous phases completely disfavoured the self-assembly of ionic dendrimers. At pH ≈ 8.5, aggregates of indeterminate morphology were observed along with a small number of twisted fibres. In contrast to the aggregates formed at neutral and acidic pH, nanotubular structures were not formed at ratios of 1/0.70 or 1/0.85. When the amount of water was increased to 1/1 a few nanotubes were finally formed, although the amorphous precipitates still constituted the majority of the aggregates (Fig. 2, see also ESI†). These results confirmed the pH-sensitive nature of the self-assemblies.
In light of the results outlined above, NMR experiments (1H-NMR, 1H–1H COSY and 1H–13C HSQC) at variable pH were carried out in an effort to understand better the chemical modifications at the molecular level that caused this behaviour (ESI† includes the NMR spectra, experimental details and further explanations). The NMR experiments in the acidic medium revealed the protonation of the inner nitrogen atoms of the PPI dendrimer. Similar behaviour on protonation of PPI dendrimers in the presence of excess acid was reported in the liquid crystalline state.31 This change accentuates the chemical difference between the hydrophilic and hydrophobic blocks, which is the driving force for the self-assembly of the ionic dendrimer, and this may explain the formation of a new type of nanostructure when acidic aqueous phases are employed.
On the other hand, the NMR experiments performed in a basic medium revealed the rupture of the ionic bond between the PPI moiety and the bent-core carboxylate. As a consequence, the organisation of the bent-core structures is affected, since previous work by us proved that the presence of the PPI dendrimer is essential for the self-assembly,32 and amorphous aggregates are observed in TEM images.
In all of the studies detailed above, the experiments were repeated and a high level of reproducibility was obtained when different assemblies collected from different synthesis runs were compared (see ESI† for some selected examples). As a result, it can be concluded that the proposed microfluidic platform provides the required reproducibility for the preparation procedure both within and between runs. Moreover, calculations have been made in order to compare the productivity of the batch-to-batch synthesis previously reported vs. the microfluidic device. Considering that a standard co-solvent protocol takes 1 hour approximately and employs 1.6 mL of a 0.5 wt% THF solution and 1.6 mL of water, the estimated productivity equals 7.2 mg h−1. On the other hand, taking into account the most similar microfluidics conditions, i.e. ratio 1/1 and 0.5 wt%, and an intermedium flow rate, i.e. 500 μL min−1, the estimated productivity rises up to 66.75 mg h−1, almost 10 times higher than that calculated for the batch-to-batch synthesis. These values highlight the improvement in terms of productivity accomplished by microfluidic devices, along with the time- and solvent-savings of this method.
Preparation of hybrid materials
1D nanomaterials have been employed as organic scaffolds for the growth of inorganic nanoparticles33,34 to obtain nanocomposites with enhanced mechanical properties35 and for applications in catalysis,36 photonics,37 and other fields. Additionally, very recently helical liquid crystal phases have also been employed as a template for the growth of AuNPs.38 Among others, gold nanoparticles stand out owing to their remarkable physical and biomedical properties. Taking into account that dendritic structures, especially those based on PAMAM and PPI dendrimers, have been widely reported to interact with gold nanoparticles, we decided to prepare Au-decorated nanostructures as a proof-of-concept of the possibilities of these materials. With this objective in mind, different approaches were tested. Firstly, we addressed the most straightforward protocol, i.e., the introduction of citrate-capped AuNPs in the aqueous phase. These nanoparticles had a mean particle size of 12 ± 3 nm and were prepared by a modified Turkevich method.39 TEM images of the samples collected at the outlet of the reactor showed that the nanoparticles had poor affinity for the nanostructures. Very few AuNPs were attached to the surface of the twisted fibres and the nanotubes. Instead, they accumulated predominantly in zones where the ionic dendrimers had not been able to self-assemble properly and this gave rise to amorphous aggregates (Fig. 3a). | ||
| Fig. 3 TEM images of self-assembled nanostructures of PPI1-B1-4-8 covered with gold nanoparticles. Conditions: (a) samples prepared with presynthesised citrate-capped gold nanoparticles as the aqueous phase at a total flow rate of 500 μL min−1 and THF/water ratio of 1/1. (b) Samples prepared by reduction of AuCl3. The gold precursor solution was employed as the aqueous phase in the microreactor. The samples were prepared at a total flow rate of 500 μL min−1 and THF/water ratio of 1/1. The samples were subsequently reduced with CO. (c–g) Samples prepared by reduction of AuCl3. The organic nanostructures were prepared in a first step in the microreactor. The gold precursor was subsequently added and reduced with CO. Nanostructures were prepared in the microreactor at a total flow rate of 500 μL min−1 and THF/water ratios from 1/0.70 to 1/1. | ||
The samples were prepared at a total flow of 500 μL min−1 and THF/water ratio of 1/1. The samples were subsequently reduced with CO. Fig. 3c–g show samples prepared by reduction of AuCl3. The organic nanostructures were prepared in a first step in the microreactor. The gold precursor was subsequently added and reduced with CO. The nanostructures were prepared in the microreactor at a total flow of 500 μL min−1 and THF/water ratios from 1/0.70 to 1/1.
In view of the ineffectiveness of this protocol, we attempted to grow AuNPs directly on the surface of the nanoassembly. With this purpose, a 0.5 mg mL−1 solution of AuCl3 (gold precursor) was employed as the aqueous phase in the microfluidic system. The gold atoms were expected to interact with the nitrogen atoms of the poly(propylene imine) dendrimer.40 Once the AuIII-doped self-assemblies were prepared in the reactor, the suspension was subjected to a treatment with carbon monoxide41 under mild conditions (30 °C) in order to reduce the AuIII to Au0 and to obtain AuNPs without thermally affecting the assemblies (see ESI† for more details). The reduction of gold atoms after the treatment could be confirmed with the naked eye as the solution changed from the yellowish colour of the precursor to a purple suspension attributed to the formation of the nanoparticles and their surface plasmon resonance. TEM images confirmed the correct synthesis of spherical AuNPs after treatment with the reducing gas. The population of the nanoparticles was quite polydisperse, with sizes ranging from 4 to 15 nm. A few bigger nanoparticles of 30 nm in diameter were also formed. In this case, the nanoparticles adhered to a large extent to the self-assemblies, unlike the situation when the pre-synthesised nanoparticles were employed. Moreover, AuNPs were not observed independently from the organic nanostructures (Fig. 3b). This highlights the strong affinity between gold and the organic nanostructures. However, the introduction of gold ions to the reactor seemed to perturb nucleation and the growth of the assemblies. As a consequence, non-twisted fibres with diverse widths were mostly observed in the samples, whereas helical ribbons or nanotubes were not obtained.
To overcome this issue, we proceeded with the third protocol. In the first step organic nanostructures were prepared in the microfluidic system as stated previously. A solution of the gold precursor was subsequently added to the suspension of organic nanostructures (0.2 mL of AuCl3 solution per mL of nanostructure suspension) and the mixture was incubated for 45 minutes to ensure sufficient contact time to promote the diffusion of Au species to the surface of the assemblies. The sample was then treated with CO to reduce the gold in order to synthesise nanoparticles. This methodology finally allowed the formation of hybrid nanomaterials composed of organic twisted fibres, helical ribbons and nanotubes together with inorganic spherical AuNPs (Fig. 3c–g).
The nanoparticles were adhered to the surface of the self-assemblies and free nanoparticles were not observed in the medium. In this case, the synthesised AuNPs had smaller sizes and ranged from 2 to 8 nm in diameter. Under these conditions, THF/water ratios from 1/0.70 to 1/1 were tested and all of the morphologies involved in the self-assembly process (twisted fibres, helical ribbons and nanotubes) were formed and covered by the AuNPs. These experiments confirmed the ability of these organic nanostructures to act as scaffolds for the growth of homogenous inorganic nanoparticles. This finding opens the door to future applications.
Deposition onto surfaces
The self-assembly of mesogenic molecules in solvents instead of in the bulk is an interesting approach that can provide molecular organisations similar to those of the liquid crystalline state.3,42 However, for some applications it is necessary to obtain nanostructures in the solid state.43–45 Therefore, we combined the continuous fabrication of assemblies in solution through microfluidic devices with the deposition of nanostructures on different substrates to obtain liquid crystal-like organisations in the bulk. Firstly, deposition onto a glass plate was evaluated. The outlet stream of the microreactor was directly dropped onto the glass and the sample was dried before SEM investigation. Images of the surface confirmed the formation of twisted fibres with morphologies identical to those observed in TEM images of the nanostructures suspended in water (see ESI†).A continuous separation system was envisaged to enable the efficient separation of nanostructures from the assembly medium. This resulted in a multistage microfluidic system involving (1) assembly and (2) separation. This procedure enabled the deposition of nanostructures onto a PTFE filter (0.22 μm pore size). For this purpose, the outlet stream of the microreactor was connected to a syringe filter and the nanostructure suspension was passed through (see ESI†). At this point, it should be remarked that the components considered in this multi-stage microfluidic system are low-cost, modular and can be easily assembled into the flow-focusing device by inexperienced users or those without access to microfabrication facilities. SEM images revealed that the assemblies were successfully retained in the filter to obtain large surface areas covered with the material in the solid state (Fig. 4 and ESI†). At lower proportions of water, the surface was covered by a dense network of twisted fibres (Fig. 4a). On the other hand, helical ribbons and nanotubes were observed when the amount of water was increased (Fig. 4b and c).
 | ||
| Fig. 4 SEM images of nanostructures of PPI1-B1-4-8 fabricated in the coaxial microreactor and deposited onto Teflon® filters. Samples prepared at THF/water ratios of (a) 1/1.25 (twisted fibres) (b and c) 1/1.5 (helical ribbons and nanotubes). | ||
This set up served as a proof-of-concept to assess the possibility of obtaining nanostructures in the solid state and in a continuous manner. It was also confirmed that more complex self-assemblies, i.e., nanotubes, can be formed during the residence time of the bent-core based dendrimer in the microreactor.
Finally, in a similar way to the experiments discussed above, the self-assemblies retained in the filters were also treated with the gold precursor and reduced under a CO atmosphere to obtain Au-decorated nanostructures. 1 mL of a 0.5 mg mL−1 solution of AuCl3 was passed through the filter that contained the deposited nanostructures and the filter was flushed with air to remove the excess wet solution and incubated for 30 minutes. Subsequently, the filter was treated with the reducing gas to synthesise AuNPs. The formation of Au0 nanoparticles could again be observed with the naked eye as the filter was covered by a purple layer. Moreover, SEM images confirmed that this protocol allowed the AuNPs to be grown on the twisted fibres and nanotubes (Fig. 5a). The SEM analysis by backscattered electrons revealed a homogeneous deposition of the nanoparticles over the entire surface covered by the self-assemblies (Fig. 5b). Moreover, EDS experiments confirmed the gold nature of the nanoparticles deposited onto the twisted fibres and tubes (see ESI†).
 | ||
| Fig. 5 SEM images of Au-decorated organic nanotubes of PPI1-B1-4-8 deposited onto Teflon® filters. Samples prepared at a THF/water ratio of 1/1.5 and the same area was observed with (a) an in-lens detector (morphology) and (b) energy backscattered electrons (composition, white dots correspond to Au nanoparticles). | ||
Conclusions
With the aim of extending and exploring the supramolecular potential of bent-core based molecules, the utility of microfluidic systems for the preparation of nanostructures of low-molecular-weight organic molecules has been demonstrated. To the best of our knowledge, this paper constitutes the first example of the use of a microreactor to study a hierarchical self-assembly process from non-conventional bent-core based amphiphiles. The results obtained may serve as an inspiration for a large number of scientists working in the rich field of supramolecular chemistry and can expand the scope of microfluidic technology.Owing to the characteristic low consumption of reactants and rapid preparation of samples in microfluidic devices, many parameters were evaluated in order to study their influence on the self-assembly of bent-core based amphiphiles, i.e., THF/water ratio, residence time, concentration of the amphiphile, temperature and pH. These experiments are of interest at the fundamental level and they allowed us to understand better the self-assembly process and to establish optimum preparation conditions. Changes in parameters such as THF/water ratio or pH allowed us to obtain twisted fibres, helical ribbons, nanotubes and large non-twisted fibres.
Moreover, the combination of organic 1D nanostructures with AuNPs was explored. A novel procedure that consisted of the incubation of nanostructures with a gold precursor and subsequent reduction with carbon monoxide resulted in the formation of the desired hybrid materials. These nanocomposites open the door to combine inorganic materials with bent-core supramolecular materials.
Finally, both organic and hybrid nanostructures were obtained in the solid state by simply connecting a separation module (syringe filter) to the outlet stream of the microreactor. The resulting multi-stage microfluidic system operated under reproducible conditions that enabled the quality of the resulting assemblies to be preserved. The resulting nanostructures produced by the multi-stage microfluidic system retained the morphology previously observed in solvent dispersions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciate financial support from the Spanish Government (MICINN-FEDER project MAT2015-66208-C3-1-P, PGC2018-093761-B-C31, PGC2018-097583-B-I00 and RTI2018-099019-A-I00) and the Government of Aragon (GA) (project E47_20R and T57_20R). We are also grateful to the BES-2016-078753 MINECO-FEDER (M. C.-V.) fellowship program for support. We acknowledge the nuclear magnetic resonance, mass spectrometry, and thermal analysis services of the INMA (Uni. Zaragoza-CSIC), the LMA (Uni. Zaragoza) for TEM images, the ICTS NANBIOSIS and Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza for SEM images.Notes and references
- A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó’Máille and T. Gunnlaugsson, Chem, 2017, 3, 764–811 CAS.
- J. Etxebarria and M. B. Ros, J. Mater. Chem., 2008, 18, 2919–2926 RSC.
- M. Castillo-Vallés, A. Martínez-Bueno, R. Giménez, T. Sierra and M. B. Ros, J. Mater. Chem. C, 2019, 7, 14454–14470 RSC.
- M. Cano, A. Sanchez-Ferrer, J. L. Serrano, N. Gimeno and M. B. Ros, Angew. Chem., Int. Ed., 2014, 53, 13449–13453 CrossRef CAS.
- M. Castillo-Vallés, M. Cano, A. Bermejo-Sanz, N. Gimeno and M. B. Ros, J. Mater. Chem. C, 2020, 8, 1998–2007 RSC.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- S. Wang, W. Lu, O. Tovmachenko, U. S. Rai, H. Yu and P. C. Ray, Chem. Phys. Lett., 2008, 463, 145–149 CrossRef CAS.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS.
- P. C. Ray, Chem. Rev., 2010, 110, 5332–5365 CrossRef CAS.
- N. Kameta, H. Minamikawa and M. Masuda, Soft Matter, 2011, 7, 4539–4561 RSC.
- P. M. Valencia, O. C. Farokhzad, R. Karnik and R. Langer, Nat. Nanotechnol., 2012, 7, 623–629 CrossRef CAS.
- T. W. Herling, A. Levin, K. L. Saar, C. M. Dobson and T. P. J. Knowles, Lab Chip, 2018, 18, 999–1016 RSC.
- L. Wang and J. Wang, Nanoscale, 2019, 11, 16708–16722 RSC.
- J. Il Park, A. Saffari, S. Kumar, A. Günther and E. Kumacheva, Annu. Rev. Mater. Res., 2010, 40, 415–443 CrossRef.
- Y. Dou, B. Wang, M. Jin, Y. Yu, G. Zhou and L. Shui, J. Micromech. Microeng., 2017, 27, 113002 CrossRef.
- S. Sevim, A. Sorrenti, C. Franco, S. Furukawa, S. Pané, A. J. Demello and J. Puigmartí-Luis, Chem. Soc. Rev., 2018, 47, 3788–3803 RSC.
- D. Carugo, E. Bottaro, J. Owen, E. Stride and C. Nastruzzi, Sci. Rep., 2016, 6, 1–15 CrossRef.
- V. Sebastian, C. D. Smith and K. F. Jensen, Nanoscale, 2016, 8, 7534–7543 RSC.
- G. Lou, G. Anderluzzi, S. Woods, C. W. Roberts and Y. Perrie, Eur. J. Pharm. Biopharm., 2019, 143, 51–60 CrossRef CAS.
- T. O. Mason, T. C. T. Michaels, A. Levin, E. Gazit, C. M. Dobson, A. K. Buell and T. P. J. Knowles, J. Am. Chem. Soc., 2016, 138, 9589–9596 CrossRef CAS.
- A. Sorrenti, R. Rodriguez-Trujillo, D. B. Amabilino and J. Puigmartí-Luis, J. Am. Chem. Soc., 2016, 138, 6920–6923 CrossRef CAS.
- V. Sebastian, S. A. Khan and A. A. Kulkarni, J. Flow Chem., 2017, 7, 96–105 CrossRef CAS.
- S. Garcia-Salinas, E. Himawan, G. Mendoza, M. Arruebo and V. Sebastian, ACS Appl. Mater. Interfaces, 2018, 10, 19197–19207 CrossRef CAS.
- U. Laura, M. Arruebo and V. Sebastian, Dalton Trans., 2018, 47, 1693–1702 RSC.
- M. Numata and T. Kozawa, Chem.–Eur. J., 2014, 20, 6234–6240 CrossRef CAS.
- M. Numata, A. Sato and R. Nogami, Chem. Lett., 2015, 44, 995–997 CrossRef CAS.
- D. Hermida-Merino, M. Trebbin, S. Foerster, F. Rodriguez-Llansola and G. Portale, Macromol. Symp., 2015, 358, 59–66 CrossRef CAS.
- M. Numata, Y. Nishino, Y. Sanada and K. Sakurai, Chem. Lett., 2015, 44, 861–863 CrossRef CAS.
- W. S. Saad and R. K. Prud'Homme, Nano Today, 2016, 11, 212–227 CrossRef CAS.
- S. Hernández-Ainsa, J. Barberá, M. Marcos and J. L. Serrano, Soft Matter, 2011, 7, 2560–2568 RSC.
- R. Martín-Rapún, M. Marcos, A. Omenat, J. Barberá, P. Romero and J. L. Serrano, J. Am. Chem. Soc., 2005, 127, 7397–7403 CrossRef.
- J. Vergara, N. Gimeno, M. Cano, J. Barberá, P. Romero, J. L. Serrano and M. B. Ros, Chem. Mater., 2011, 23, 4931–4940 CrossRef CAS.
- A. K. Awasthi, S. D. Bhagat, R. Ramakrishnan and A. Srivastava, Chem.–Eur. J., 2019, 12905–12910 CrossRef CAS.
- P. Choudhury, S. Dinda and P. K. Das, Soft Matter, 2019, 16, 27–53 RSC.
- Y. Cao, S. Bolisetty, G. Wolfisberg, J. Adamcik and R. Mezzenga, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4012–4017 CrossRef CAS.
- S. Wu, Y. Li, S. Xie, C. Ma, J. Lim, J. Zhao, D. S. Kim, M. Yang, D. K. Yoon and M. Lee, et al. , Angew. Chem., Int. Ed., 2017, 56, 11511–11514 CrossRef CAS.
- M. Golla, S. K. Albert, S. Atchimnaidu, D. Perumal, N. Krishnan and R. Varghese, Angew. Chem., Int. Ed., 2019, 58, 3865–3869 CrossRef CAS.
- M. Baginski, M. Tupikowska, G. González-Rubio, M. Wójcik and W. Lewandowski, Adv. Mater., 2020, 32, 1904581 CrossRef CAS.
- E. Luque-Michel, A. Larrea, C. Lahuerta, V. Sebastian, E. Imbuluzqueta, M. Arruebo, M. J. Blanco-Prieto and J. Santamaría, Nanoscale, 2016, 8, 6495–6506 RSC.
- K. Torigoe, A. Suzuki and K. Esumi, J. Colloid Interface Sci., 2001, 241, 346–356 CrossRef CAS.
- M. Sancho-Albero, B. Rubio-Ruiz, A. M. Pérez-López, V. Sebastián, P. Martín-Duque, M. Arruebo, J. Santamaría and A. Unciti-Broceta, Nat. Catal., 2019, 2, 864–872 CrossRef CAS.
- A. Zep, M. Salamonczyk, N. Vaupotič, D. Pociecha and E. Gorecka, Chem. Commun., 2013, 49, 3119–3121 RSC.
- E. Gomar-Nadal, J. Puigmartí-Luis and D. B. Amabilino, Chem. Soc. Rev., 2008, 37, 490–504 RSC.
- T. Wolf, A. Niazov-Elkan, X. Sui, H. Weissman, I. Bronshtein, M. Raphael, H. D. Wagner and B. Rybtchinski, J. Am. Chem. Soc., 2018, 140, 4761–4764 CrossRef CAS.
- V. Novotná, V. Hamplová, L. Lejček, D. Pociecha, M. Cigl, L. Fekete, M. Glogarová, L. Bednárová, P. W. Majewski and E. Gorecka, Nanoscale Adv., 2019, 1, 2835–2839 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed data on every protocol employed in this work, additional TEM and SEM images of samples prepared under different conditions, NMR spectra at variable pH and EDS experiments. See DOI: 10.1039/d0na00744g |
| This journal is © The Royal Society of Chemistry 2021 |