
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced synthesis method of graphene oxide†
Zineb
Benzait
 *a,
Pengwan
Chen
b and
Levent
Trabzon
acd
*a,
Pengwan
Chen
b and
Levent
Trabzon
acd
aNanosicence and Nanoengineering Department, Istanbul Technical University, Maslak, Istanbul 34469, Turkey. E-mail: zineb@itu.edu.tr
bSchool of Mechatronic Engineering, Beijing Institute of Technology, Beijing 100081, China
cDepartment of Mechanical Engineering, Istanbul Technical University, Istanbul 34437, Turkey
dNanotechnology Research and Application Center – ITUnano, Istanbul Technical University, Istanbul 34437, Turkey
First published on 9th November 2020
Abstract
The synthesis of high quality graphene oxide (GO) in large quantities is a matter of great importance for both research institutes and industries. In the present study, we report an improvement in the so-called “improved method” reported by Tour et al., which had already improved the very famous “Hummers method” to a certain extent. Through an important pre-treatment step, GO with larger sheets, better structural integrity, and a higher yield of monolayers was obtained. Furthermore, both the oxidation time and temperature were reduced without reducing the degree of high oxidation. Even though a low temperature is known to be a prerequisite for obtaining less defective GO in its reduced form (rGO), we found through this research that the pre-treatment step minimizes the negative effect of the moderate temperature (35 °C) needed to enhance the reaction rate, without altering the basal graphitic plane, which was also preserved at a low temperature (<10 °C). Both the mechanical and electrical properties confirm the enhancement of the GO quality obtained through improving the improved method, and make the rGO films produced attractive for practical applications.
Introduction
No scientist can dispute the importance of graphene and its derivatives, and the potential of its current and future use in different applications. Among these derivatives, graphene oxide (GO) has been widely investigated either alone for constructing diverse devices,1,2 or as an additive for enhancing the performance of materials,3,4 or even as a precursor for synthesizing graphene through either physical or chemical reduction.5–7 However, to synthesize GO, several methods have been proposed since 1855; the most famous one is Hummers' method8 which uses potassium permanganate (KMnO4) and sodium nitrate (NaNO3). In 2010, Tour et al.9,10 reported an improvement on Hummers' method through excluding NaNO3 to prevent the generation of toxic gases, using ice instead of liquid water to prevent the high temperature rise, thus promoting better and easier control of the process, and through increasing the yield and degree of oxidation, together with promoting the retention of carbon rings in the basal plane by introducing phosphoric acid (H3PO4) to the reaction media. The Tour group's article on “Improved Synthesis of Graphene Oxide”9 has received significant interest from the research community, which proves the importance and the ease of use of their method; and since then only three alternative methods have been reported, to the best of our knowledge.11–13Among these methods, Peng et al.11 reported a fast, scalable and green method to produce GO within 1 h using potassium ferrate (K2FeO4) as a strong oxidant instead of KMnO4, however, the reliability of this method is doubted as almost no researchers, as far as we know, have used it, and Sofer et al.14 have questioned whether it is a myth or reality. Through investigation and their critical replication, they claimed that K2FeO4 is unsuitable for graphite oxidation because of the high instability of ferrate(VI) in an acidic environment. Another novel method attracted our attention owing to the ease of washing the GO obtained using it, through a so-called “monolithic crystalline swelling” strategy reported by Lu et al.12 However, after trying the same method with the same reactant quantities and reaction conditions, we found that the product obtained was not homogeneous and contains a large number of black particles (unreacted/not completely exfoliated graphite), as shown in Fig. S1.† After facing these problems, we contacted the first author of the debatable article who claimed that they solved this problem by extending the reaction time to 24 h, and by keeping the graphite flakes (which tend to float owing to gas generation) under the liquid surface by pushing them down every 10 min in the first hour and every 20 min over the next 3 h. Although these details make this method unattractive and non-practical, we tried to repeat it, in the same manner, several times. Unfortunately, this method was not suitable because the one-step process, together with the non-agitation approach, provide an inhomogeneous reaction medium, and thus an inhomogeneous product.
Herein, we report the use of the well-known improved method to synthesize GO, but with some parameter modifications and with an essential pre-treatment step in order to further improve it. This pre-treatment step allows the chemical expansion of graphite before its oxidation using piranha solution in an easy way. Actually, several works have reported that the use of expanded graphite (EG), prepared using different methods, as a starting material can help intercalate the graphitic layers, increasing the accessible specific surface area (SSA), and mitigating the harsh oxidation which propagates from the edges to the center of the sheets.15,16 Thermally and microwave expanded graphite were produced by first preparing a graphite intercalation compound (GIC) and then rapidly heating it to high temperatures (≥900 °C) or exposing it to a high irradiation energy, which led to gas-release and expansion of the layers.17–20 These methods have many drawbacks, such as involving many steps (natural graphite intercalation, washing, drying, expansion), and require energy and equipment, and can produce EG with a low SSA. On the other hand, chemically expanded graphite can be prepared through gentle, gas-releasing oxidation–reduction21 or catalytic22 reactions between a GIC and its surrounding solution. Both chemicals used (CrO3 and FeCl3) are toxic and are environmental pollutants. Furthermore, the multi-steps required, especially the washing needed, make these methods tedious.
In this work, EG was prepared in an environmentally friendly one-step convenient method that does not involve heating or the use of any sophisticated devices. This method requires only simple stirring and standing of the graphite in cooled piranha solution without washing and drying. The subsequent oxidation reaction at a lower temperature (35 °C instead of 50 °C) and the shorter time (6 h instead of 12 h) resulted in GO with a higher degree of oxidation, larger sheets, and fewer defects. Using the industrially suitable doctor-blade technique and hydroiodic acid (HI) reduction, the reduced graphene oxide (rGO) film obtained through this enhanced method achieved a tensile strength of 190 MPa, a toughness of 5.7 MJ m−3, and a conductivity of 470 S cm−1. The pre-treatment of graphite with piranha solution minimized the negative effects of the moderate temperature (35 °C) used during oxidation, as it did not strongly affect the GO structural integrity, and it gave good results close to those of graphene oxide synthesized at a low temperature (<10 °C).
Experimental
The same raw material as that used in the Tour group's work (natural graphite flakes (Sigma-Aldrich, cat #332461, +100 mesh)) was used in this work. Potassium permanganate (KMnO4) with a purity higher than 99% was obtained from NeoFroxx (LC-7081). Sulfuric acid (H2SO4) 95–97%, phosphoric acid (H3PO4) 85%, and hydrogen peroxide (H2O2) 35% were obtained from Merck. HI, 57 wt% stabilized aqueous solution was obtained for analysis from Acros (19837).Control sample synthesis
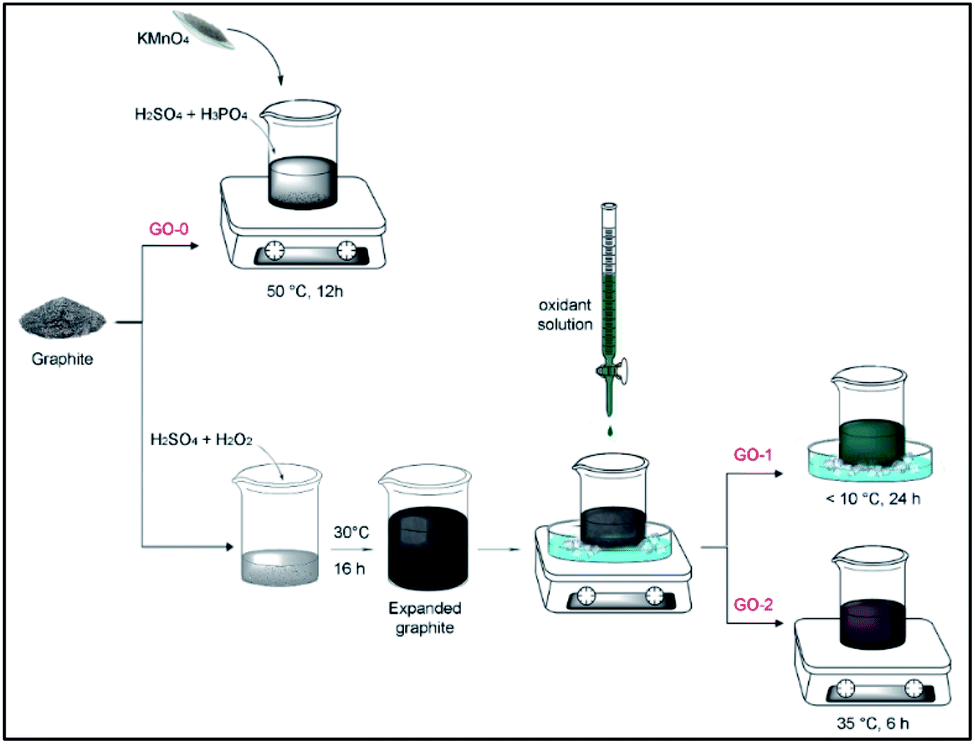
As a reference sample, graphene oxide GO-0 was obtained through direct oxidation of graphite as described by Tour et al.9 Briefly, 1 g of graphite, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acidic solution containing 120 ml of H2SO4 and 13.4 ml of H3PO4 were stirred at room temperature. 6 g of the oxidant (KMnO4) was added slowly to avoid it becoming explosive.10 The reaction mixture was then heated and kept under magnetic stirring (300 rpm) at an oxidation temperature (T) equal to 50 °C, throughout an oxidation time of 12 h (Fig. 1).
:1 acidic solution containing 120 ml of H2SO4 and 13.4 ml of H3PO4 were stirred at room temperature. 6 g of the oxidant (KMnO4) was added slowly to avoid it becoming explosive.10 The reaction mixture was then heated and kept under magnetic stirring (300 rpm) at an oxidation temperature (T) equal to 50 °C, throughout an oxidation time of 12 h (Fig. 1).
 | ||
| Fig. 1 Schematic illustration of the synthesis of GO. | ||
Expanded graphite preparation
Fresh piranha solution (100 ml, 9:1 of H2SO4/H2O2) was prepared and cooled using an ice-water bath. 1 g of graphite was added and stirred for 10 min at 300 rpm. The magnetic rod was then removed and the mixture was left overnight at room temperature at approximately 25–30 °C (Fig. S2†).
Enhanced GO synthesis
The enhanced GO samples were prepared using the same recipe as the improved method (Tour's method) but using fresh EG as a starting material: a 9:1 mixture of concentrated H2SO4/H3PO4 (120:13.4 ml) was cooled using an ice-water bath, 6 g of KMnO4 was added and stirred at 500 rpm for 30 min. This cold oxidant solution was slowly added to EG over approximately 15 min while mixing it mechanically and keeping the bath temperature lower than 10 °C.
GO-1 was obtained by keeping this mixture at a low temperature (<10 °C) over 24 h as the low temperature induces a low reaction rate. On the other hand, GO-2 was synthesized using the same method as GO-1, but at a temperature of less than 10 °C for only 2 h in order to allow the oxidant to gently penetrate between the graphitic layers, then by increasing the temperature to 35 °C to enhance the oxidation rate and degree of oxidation, the total oxidation time (t) for GO-2 was fixed at 6 h (Fig. S3a†).
Post-processing
After completion, each reaction was stopped by adding approximately 400 ml ice cubes to promote a better temperature control (Fig. S3b†), and approximately 3 ml H2O2 (Fig. S3c†) in order to decompose the insoluble manganese salts.23 A few unreacted black particles were found floating above the acidic solution and were thus removed using a plastic pipette. The GO was separated from the acidic solution, then washed with HCl, ethanol and deionized (DI) water by spontaneous sedimentation (Fig. S4a†) which was found to be faster in the case of GO-1 and GO-2, thus facilitating the purification step. Later, GO dispersions were subjected to 3–4 cycles of high-speed centrifugation until the pH reached approximately 6. After each centrifugation and re-dispersion cycle, the GO swelled further. Finally, each GO aqueous solution was further exfoliated by magnetic stirring at 300 rpm for 5 min (Fig. S4b and Video S1†) and bath-sonication at approximately 74 W L−1 for 10 min. Three cycles of low-speed centrifugation at 2000 rpm for 30 min were then performed in order to remove the unexfoliated sheets (Fig. S4c†).Chemical and morphological characterization
To determine the concentration of the GO dispersions, 15 ml of the diluted solution (5 ml of the stock solution) was filtered using a vacuum-assisted filtration (VAF) set with approximately 0.22 μm PTFE hydrophilic membranes (Haining Yibo, China). The GO films were peeled off easily after wetting the membrane on the opposite film side using ethanol. The obtained GO films were stored in a desiccator for 1 day before being weighed.50 μg ml−1 of each GO solution was prepared using serial dilutions to perform UV/visible characterization using a Scinco Neosys 2000 spectrophotometer. Similarly, approximately 2 μg ml−1 solutions were prepared and drop cast onto freshly cleaved mica surfaces for atomic force microscopy (AFM) analysis using a Bruker Multimode 8.
Approximately 10 μL of 5 μg ml−1 and 2 μg ml−1 GO solutions were drop cast onto 300 nm-thick-SiO2/Si substrates and holey carbon-coated Cu 300 mesh grids for scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analysis using a TESCAN VEGA 3 and JEOL JEM-2100F respectively. The substrates and grids were first made hydrophilic using oxygen plasma to facilitate the spreading of each solution drop. To facilitate the visualization of sheets under an optical microscope (OM) instead of SEM, the GO sheets prepared on wafers were reduced using hydroiodic/trifluoro-acetic acid at 80 °C for 10 min and then washed and dried at room temperature.
The GO films were directly characterized using a Shimadzu IRTracer-100 Fourier transform infrared (FTIR) spectrometer, a Bruker D8 advance X-ray diffractometer (XRD), a NETZSCH TG 209 F1 thermogravimetric analyser (TGA), a PHI QUANTERA-II SXM X-ray photoelectron spectroscope (XPS), and a JEOL JNM-ECZ600R 13C-solid state nuclear magnetic resonance (NMR) spectrometer. Raman analysis was performed using a RENISHAW inVia spectrometer with a 532 nm excitation laser, 1% laser power (0.6 mW), 40 s acquisition time, and ×50 objective lens. The Raman curves given are the average of analyzing each film at three different points.
Samples tests
For the rheological test, the GO dispersions were concentrated via 10000 rpm centrifugation to obtain viscous solutions of approximately 1.45 wt% and then submitted to a shear rate from 0.01 to 10 s−1 under 25 °C using Thermo Haake ReoStess 300 with a 1 mm plate–plate gap.
GO-2 viscous solution was further heat-treated at 70 °C for 36 h, cast onto a glass substrate using a film applicator by the so-called doctor-blade technique, dried overnight at room temperature, peeled-off the substrate and stored in a desiccator. This film was designated GO-2′, and together with the films obtained using VAF, was reduced using 57% HI acid over 3 h (Fig. S5†) followed by washing and drying steps in order to obtain the free-standing rGO films. rGO-1, 2 and 2′ have a metallic gray color, whereas rGO-0 is darker.
For the mechanical tests, each rGO film was cut into 3 × 32 mm strips attached to paper frames with a 10 mm square window at the center (Fig. S6a†). These frames facilitate the attachment of the strips to the grips without damage, then subsequent cutting of the legs of the frames release the samples for the tensile tests (Fig. S6b†) conducted using a Zwick/Roell Z0.5 universal testing machine with 5 bar pneumatic grips and a strain rate of 0.3 mm min−1. The width of each tested strip was measured using a Mitutoyo Absolute micrometer with a resolution of 0.01 mm, and the thickness was measured using a Mitutoyo 293-100-10 micrometer with a resolution of 0.1 μm. Noting that the average mechanical properties for each film and their standard deviations were obtained from four sample strips.
The electrical conductivity values of the rGO films were determined using the relationship C (S m−1) = 1/(R × w), in which R is the average square resistance (unit: ohms per sq) of each sample measured using a M-3 handheld four-point probe tester at room temperature, and w is the average thickness.
Results and discussion
Graphite characterization
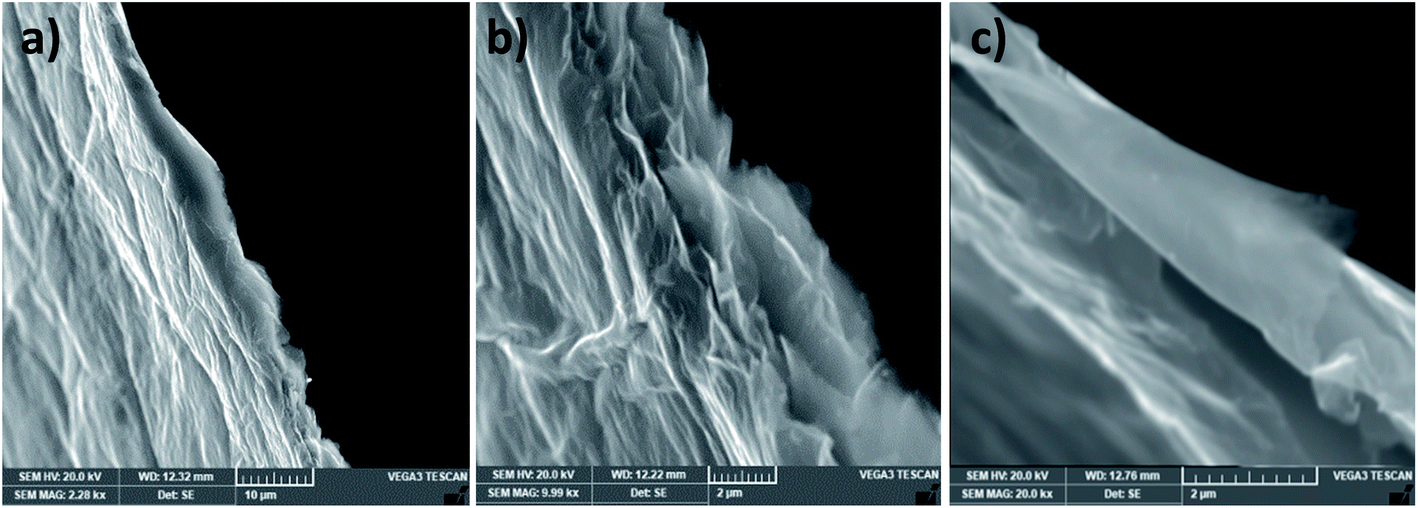
The pristine graphite (Fig. 2) has a flake-like morphology and the lateral sizes of the flakes vary from 150 to 700 μm. The enlarged SEM image of a vertical flake shows that its thickness is around 20 μm. This compacted structure will be expanded by the piranha solution as follows.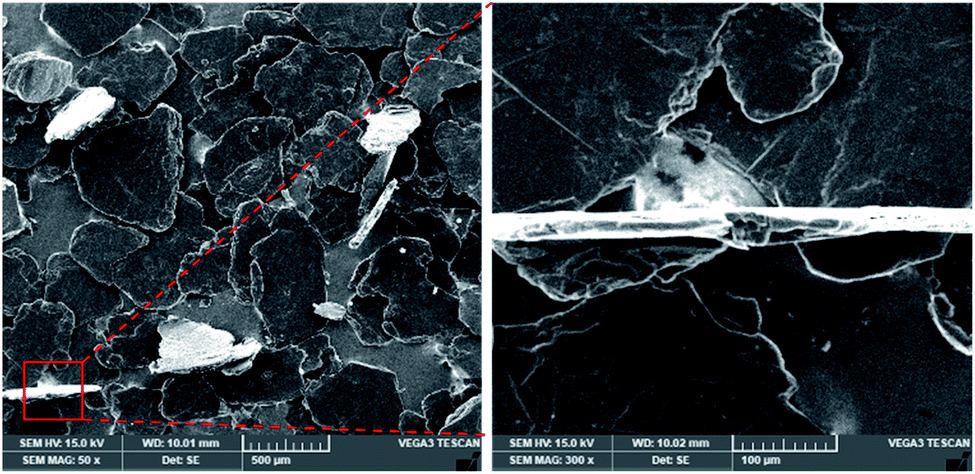 | ||
| Fig. 2 SEM image of graphite. | ||
EG characterization
The piranha-treated graphite has been expanded and its apparent volume has increased by a factor of four (Fig. S2†) owing to the intercalated H2SO4 and molecular oxygen produced from H2O2 decomposition. The entire reaction can be expressed by:20| H2O2 → H2O + O |
| 24nC + mH2SO4 + ½O → C24n+(H2SO4)−(m − 1)H2SO4 + ½H2O |
Noting that this reaction is environmentally friendly and no toxic gases are released. The graphitic layers become swollen giving an accordion-like structure, as shown in Fig. 3a and b. This worm-like morphology obtained through an easy chemical expansion provides a large accessible surface for the oxidant solution to interact with, thus promoting further homogeneous oxidation with a higher reaction rate and exfoliation yield.21
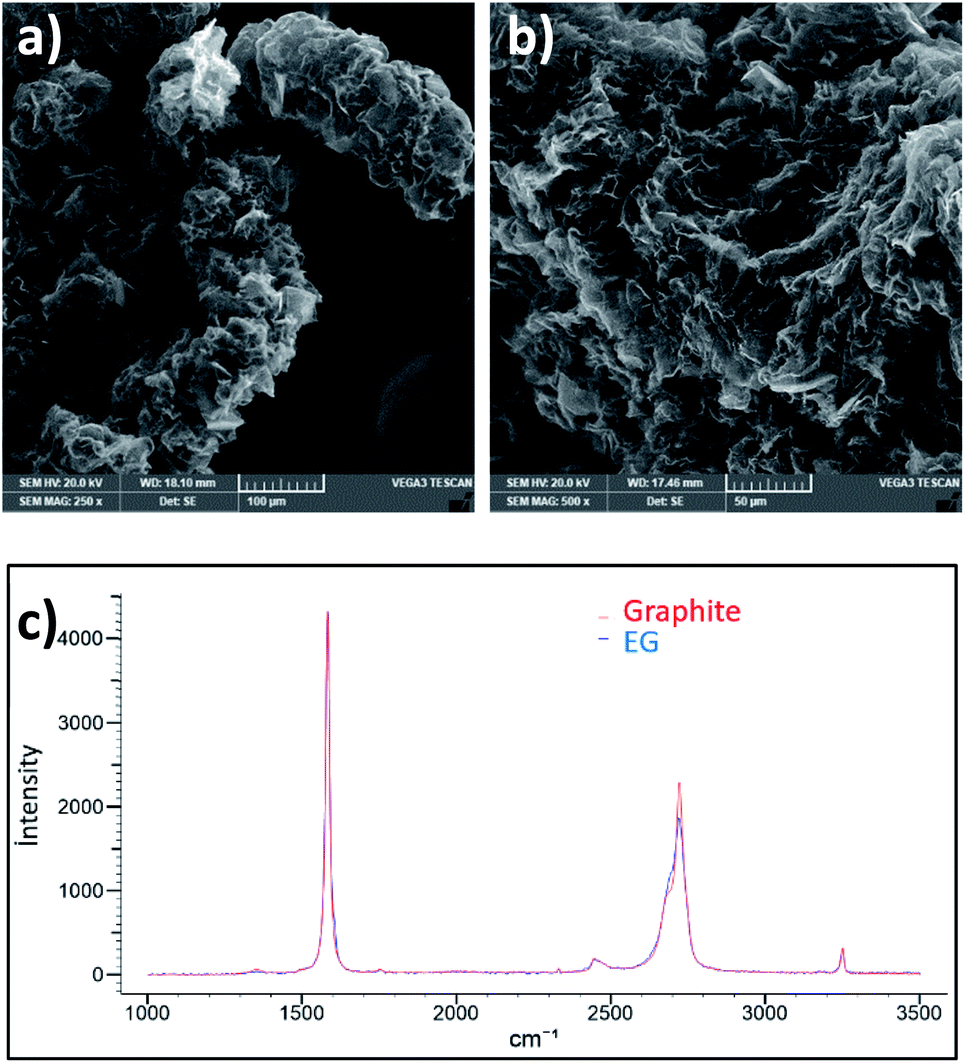 | ||
| Fig. 3 (a) and (b) SEM image of EG, and (c) Raman spectra of graphite and EG. | ||
Moreover, this expansion did not create defects as the ID Raman peak has not increased (Fig. 3c).
Chemical and morphological characterization
UV-visible spectra were recorded for an equal concentration (50 μg ml−1) of GO dispersions. All samples have a peak at 229 nm and a shoulder at 300 nm attributed to the π–π* and n–π* transitions (Fig. 4a), suggesting that the different graphene oxide materials synthesized have grossly similar structures, as indicated by the FTIR, Raman, AFM and TGA analysis (Fig. S7 and S8†). However, the extinction coefficient (i.e. UV light absorbed at λmax/concentration) of GO-1 and 2 is higher than GO-0, which reveals that they have a greater number of retained aromatic rings.9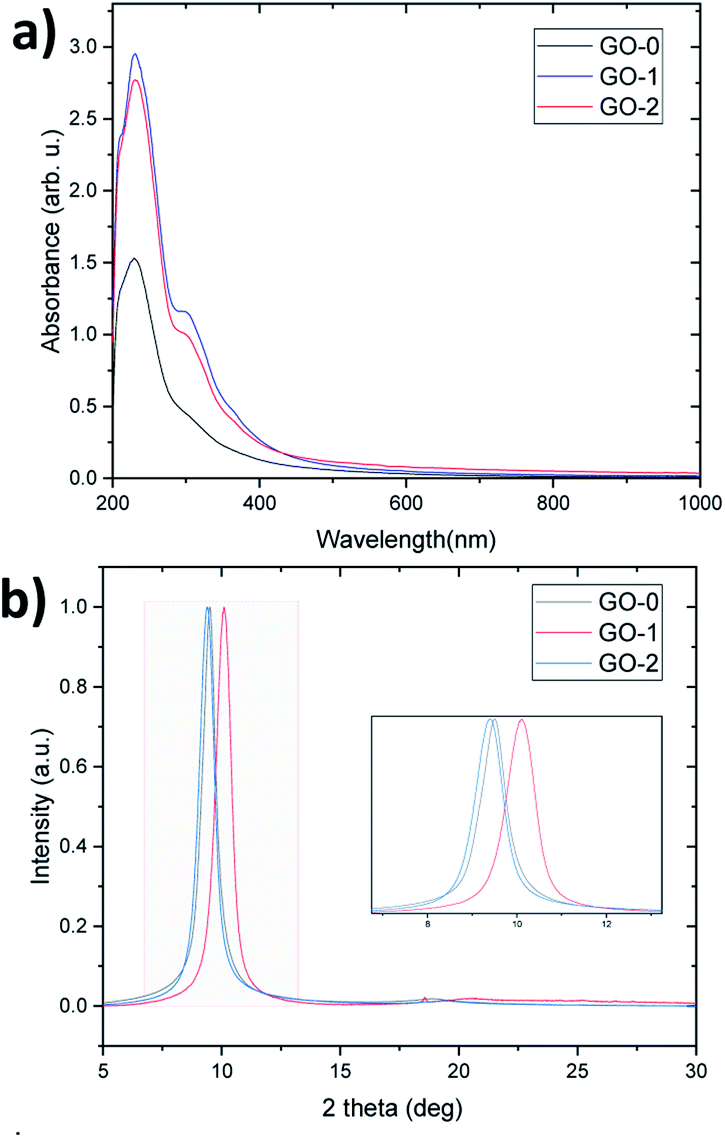 | ||
| Fig. 4 (a) UV/vis spectra and (b) XRD pattern of GO-0, 1, and 2. | ||
FTIR spectroscopy is a very useful tool that provides important information about the nature and abundance of the functional groups introduced by oxidation. KnowItAll software has been used to analyze the peaks of the spectra. As illustrated in Fig. S7a,† all the GO samples exhibit mainly O–H stretching vibrations in the range of 3000 to 3700 cm−1 which arise from either the hydroxyl groups in GO or from adsorbed water molecules, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of the carbonyl and carboxyl groups at 1730 cm−1, the CC vibration at 1620 cm−1 from the sp2 bonds of the aromatic rings of GO, as well as from the possible scissoring mode of the water present in the samples, and finally the C–O vibration at 1223 and 1050 cm−1 which corresponds to the hydroxyl and epoxy groups.
O vibration of the carbonyl and carboxyl groups at 1730 cm−1, the CC vibration at 1620 cm−1 from the sp2 bonds of the aromatic rings of GO, as well as from the possible scissoring mode of the water present in the samples, and finally the C–O vibration at 1223 and 1050 cm−1 which corresponds to the hydroxyl and epoxy groups.
Fig. 4b reveals the normalized XRD patterns of the GO films. As the XRD interlayer spacing is proportional to the degree of oxidation,9 GO-2 has a slightly higher degree than GO-0, followed by GO-1 (d-spacing equals 9.39, 9.50 and 10.11 Å respectively). Despite allowing it to react for a whole day, GO-1 still has a low oxidation level owing to a low reaction rate at low temperature. This low temperature was suggested by several researchers to protect the graphitic basal plane,16,23,24 however, it has the drawback of leading to a very slow and less-economical process because it requires continuous cooling. On the other hand, GO-2 has a similar degree of oxidation to GO-0, but thanks to the graphite pretreatment, the temperature can be reduced to 35 °C instead of 50 °C and the oxidation time can be reduced by half, which improves the process efficiency in terms of the cost and production rate.
The NMR and XPS analysis confirm the same oxidation order, that is, the percentage of graphitic carbon and oxidized carbon were determined by calculating the NMR peak areas of carbon sp2 (CC, ∼130 ppm), and of epoxy/hydroxyls (C–O, ∼60 and 70 ppm) as shown in Fig. 5a.
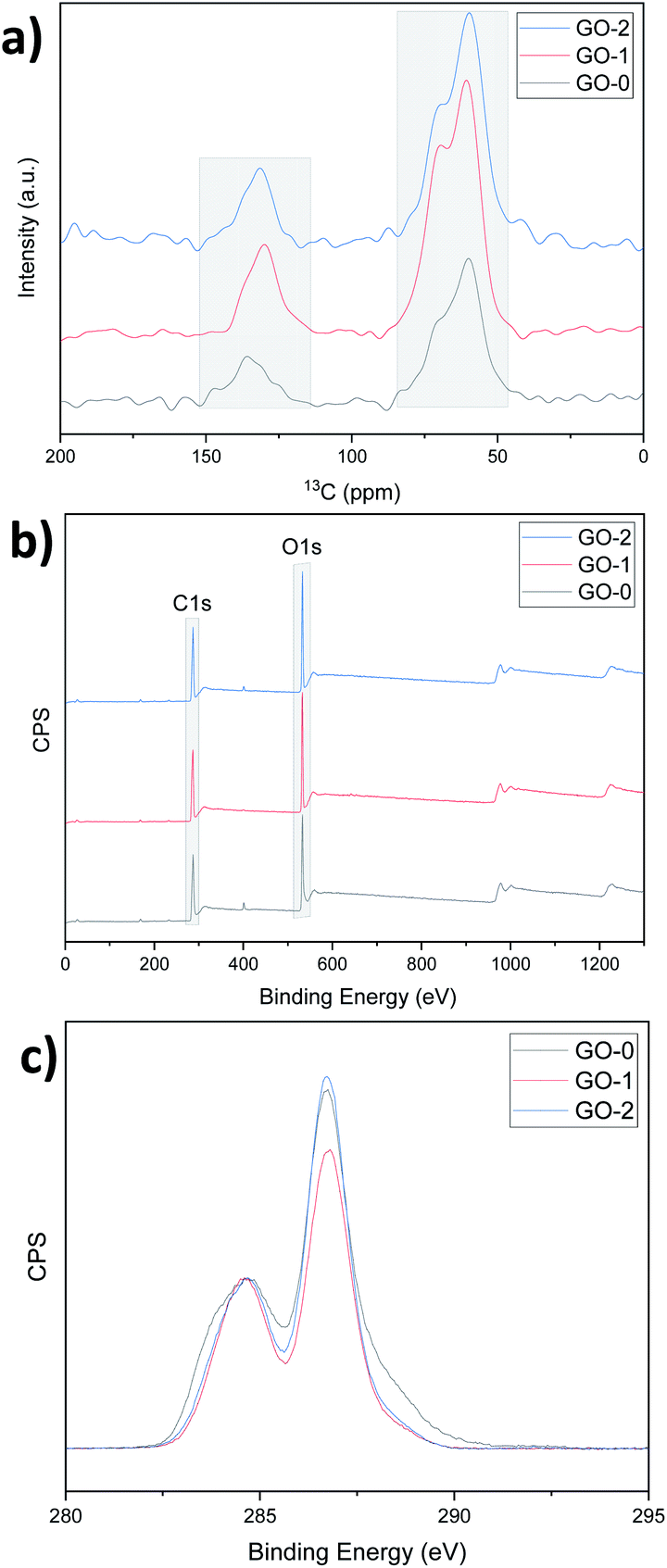 | ||
| Fig. 5 (a) NMR analysis, (b) XPS survey and (c) XPS C 1s spectra of GO-0, 1, and 2. | ||
GO-0 and GO-2 have high oxidized carbon contents (81% and 83% respectively). Similarly, C/O calculated using the XPS survey curves gave values of 2.39, 2.41 and 2.35 for GO-0, 1 and 2 respectively (Fig. 5b). Moreover, after normalizing the C 1s spectra with respect to the CC peak at approximately 285 eV, the oxidized carbon peak at approximately 287 eV is sharper for GO-1 and 2, indicating that for similar levels of overall oxidation, GO-2 has a more regular structure than GO-0 (ref. 9 and 25) owing to the graphite pretreatment. The deconvolution of the C 1s spectra peaks was performed using CasaXPS software and the results are displayed in Fig. S9 and Table S1.† This deconvolution suggests that GO-1 and -2 have more hydroxyl (C–OH) and epoxy (C–O–C) groups compared to GO-0, which has more carbonyl (CO) and carboxyl (O–CO) groups (22.35% for GO-0 and only 12.37% and 11.21% for GO-1 and 2). These CO related groups are known to reduce the mechanical properties of the resulting GO, whereas the C–O related groups can be removed upon reduction, leading to a better recovery of the graphitic structure, that is, reduction of the number of defects in the final rGO films.26,27
This important aspect was further confirmed by Raman analysis which revealed a higher ID/IG-apparent intensity ratio, that is, a higher LD (distance between two defects, stage II23) and a sharper 2D peak characteristic for the sp2 hybridized carbon bonds in graphene28 in both rGO-1 and 2 (Fig. 6a). The deconvolution of the D and Gapparent peaks, as shown in Fig. 6b–d and Table S2,† reveals larger areas under the defects peaks such as D*, D**,29,30 and D′31 in rGO-0, which again demonstrates its high defectiveness compared to rGO1 and 2. Calculating the ratios of the areas of these peaks to that of the G peak gives values of 3, 2.1 and 2.4 for rGO-0, 1 and 2 respectively, which is in accordance with order of the UV results, that is rGO-0 > rGO-2 > rGO-1 in terms of defects. Furthermore, the large D′,31 D and Gapparent intensity values32 of rGO-0 (Fig. S10†) confirm its low C/O ratio, that is, the existence of residual oxide groups, which approves the persistence of a larger defective region in the resulting rGO-0 film. All these results demonstrate that the pre-treatment of graphite with piranha solution is a very important step in obtaining a less-defective rGO thanks to the easy penetration of the oxidant solution between the expanded graphitic layers, and thus the avoidance of a harsh reaction.
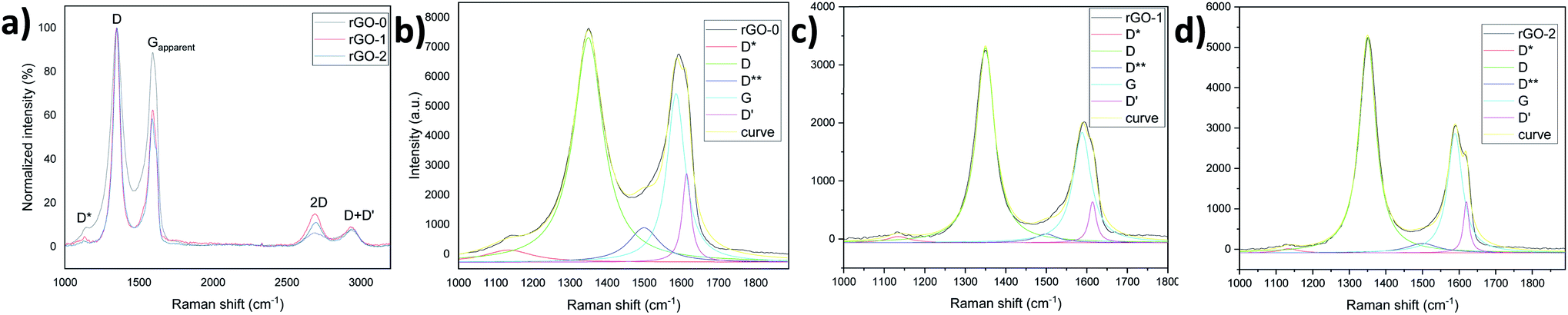 | ||
| Fig. 6 (a) Raman spectra of the rGO films and (b)–(d) their main peaks deconvolution. | ||
This avoidance of a harsh oxidation reaction has helped to avoid breakage of the GO sheets, thus preserving the large sheet size as confirmed by SEM and OM, as well as by the time required to filtrate the GO solutions (15, 49 and 33 h for GO-0, 1 and 2 respectively). Actually, global SEM images (Fig. S11†) show no obvious difference between the samples, however, ImageJ software was used to measure the size of the sheets and the statistical analysis performed and illustrated in Fig. S11† reveals that the majority of the GO-0 sheets are less than 30 μm, whereas the GO-1 and GO-2 sheets are mainly in the range of 10–50 μm and their mean size is approximately 36 μm. Interestingly, some ultra-large GO sheets (>100 μm) were visualized using OM in both GO-1 and -2, whereas the largest sheet found in GO-0 was approximately 74 μm (Fig. 7).
 | ||
| Fig. 7 (a) The largest sheet in GO-0 and (b)–(e) examples of the ultra-large sheets found in GO-1 and 2. | ||
To determine the yield of the monolayer GO in each sample, low-speed centrifugation (2000 rpm, 30 min) was used. As can be seen in Fig. S4c,† multilayered GO was precipitated in the bottom of the tubes. The monolayer yield was estimated by dividing the volume of the supernatant by the total volume. GO-2, prepared by exfoliating graphite then oxidizing it at 35 °C, has the highest yield of about 98%, whereas GO-1 prepared by exfoliating graphite, but oxidizing it at less than 10 °C, has a yield of approximately 87%. The control sample GO-0 has the lowest yield (estimated to be ∼80%), although it was prepared at a high temperature and has a high degree of oxidation. These results manifest once more the benefit of exfoliating graphite prior to its oxidation.
With regards to the TEM characterization, the GO-0 observed sheet was unfortunately a multilayer, whereas GO-1 and GO-2 were respectively bilayer and monolayer (Fig. 8). GO-0 has the most diffuse diffraction pattern, indicating a more amorphous structure, whereas GO-1 has the sharpest diffraction pattern indicating a more regular carbon framework.10
 | ||
| Fig. 8 TEM images of GO-0, 1 and 2 and their diffraction patterns. | ||
Samples tests
The rheological test results displayed in Fig. 9a reveal the non-Newtonian shear-thinning behavior of the GO dispersions, that is the viscosity decreases when the shear rate increases owing to the better alignment in the direction of the shear stress.33 At similar concentrations, GO-1 and GO-2 exhibited high zero-shear viscosity equal to 445 and 577 Pa s at a shear rate of 0.01 s−1, while it was found to be only 146 Pa s for GO-0. These results indicate that the graphite pretreatment has helped us to synthesize a better GO that is able to form at a high concentration of approximately 1.45 wt% in a dynamic 3D network, suggesting its ability to be 3D printed, and behaving like a nematic liquid crystal phase owing to the strong interactions between adjacent large sheets. In fact, GO-1 and GO-2 dispersions have shown liquid crystal behavior even at a low concentration, as can be seen in Fig. S4b and Video S1.† | ||
| Fig. 9 (a) Rheology data for GO-0, 1 and 2, and tensile curves of rGO prepared using VAF of (b) rGO-2 and (c) rGO-2′. | ||
Not only were the rheological properties enhanced, but also the mechanical and electrical properties of the graphene oxide prepared by improving the improved method. For example, the resistivity of rGO-1 and -2 prepared using VAF were found to be almost five times lower than that of rGO-0. rGO-2 has an electric conductivity of approximately 330 S cm−1, a tensile strength of 97 ± 24 MPa, a Young's Modulus of approximately 2.2 GPa, and a toughness of 1.1 ± 0.6 MJ m−3. rGO-1 has slightly higher values except for the elastic modulus (∼345 S cm−1, 103 ± 35 MPa, ∼1.3 GPa and 1.4 ± 0.9 MJ m−3 respectively). Whereas the control sample rGO-0, produced using the improved method, exhibited a tensile strength of only 28 ± 7 MPa, and a toughness of 0.3 ± 0.1 MJ m−3 (Fig. 9b).
By comparing rGO-2′ with rGO-2, that is the film obtained by the doctor-blade technique with that obtained using VAF, we found that the mechanical properties were doubled (Fig. 9c) owing to the alignment of the sheets during the hydrogel casting.33 The strength and toughness reached 190 ± 28 MPa and 5.7 ± 1.1 MJ m−3 respectively. The electrical conductivity was also enhanced, reaching approximately 470 S cm−1. These values are determined based on measuring the thickness of the film with a high-resolution micrometer (i.e. 5.2 ± 0.7 μm). However, if they are determined, as in some previously published articles, based on the thickness measured through the SEM fracture cross-section (Fig. 10), that is. 3.6 ± 0.8 μm, the strength, toughness, and conductivity equal approximately 275 MPa, 8 MJ m−3 and 680 S cm−1 respectively. It should be noted that the high flexibility and large failure strain (∼7%) of this film are due to the toughening mechanism observed in Fig. 10c, which leads to rupturing of the sheets and pulling-out of the layers under tensile force.
 | ||
| Fig. 10 (a)–(c) SEM images of the rGO-2′cross-sections with different magnifications. | ||
Conclusions
The synthesis of graphene oxide using the famous “improved method” has been further improved by adding a pre-treatment step which provides expanded graphite using piranha solution. Thanks to this simple, one-step and environmentally friendly expansion, the oxidation temperature can be reduced to 35 °C and the oxidation time can be reduced by half. XRD, XPS, and NMR analysis have shown that GO obtained through our method called the “enhanced method” has a high degree of oxidation, while UV, XPS, and Raman analysis have shown that it retains more aromatic rings. Moreover, the morphological study demonstrates the production of monolayer GO large sheets by mitigating the cutting effect resulting from the diffusion-controlled oxidation reaction. The rheological, mechanical and electrical properties of the resulting rGO films confirm the improvement of the improved Tour's method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Istanbul Technical University – Scientific Research Projects Unit under the BAP project number 40587.References
- S. Porro and I. Roppolo, in Graphene Oxide, John Wiley & Sons, Ltd, 2016, pp. 231–256 Search PubMed.
- C. M. Hayner, in Graphene Oxide, John Wiley & Sons, Ltd, 2016, pp. 257–295 Search PubMed.
- M. M. Gudarzi, S. H. Aboutalebi and F. Sharif, in Graphene Oxide, John Wiley & Sons, Ltd, 2016, pp. 314–363 Search PubMed.
- I. V. Pavlidis, in Graphene Oxide, John Wiley & Sons, Ltd, 2016, pp. 382–409 Search PubMed.
- S. Eigler, Chem.–Eur. J., 2016, 22, 7012–7027 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- K. K. H. De Silva, H.-H. Huang, R. K. Joshi and M. Yoshimura, Carbon, 2017, 119, 190–199 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. S. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2018, 12, 2078 CrossRef CAS.
- L. Peng, Z. Xu, Z. Liu, Y. Wei, H. Sun, Z. Li, X. Zhao and C. Gao, Nat. Commun., 2015, 6, 5716 CrossRef CAS.
- J. Zhang, Q. Liu, Y. Ruan, S. Lin, K. Wang and H. Lu, Chem. Mater., 2018, 30, 1888–1897 CrossRef CAS.
- S. Pei, Q. Wei, K. Huang, H.-M. Cheng and W. Ren, Nat. Commun., 2018, 9, 145 CrossRef.
- Z. Sofer, J. Luxa, O. Jankovský, D. Sedmidubský, T. Bystroň and M. Pumera, Angew. Chem., Int. Ed., 2016, 55, 11965–11969 CrossRef CAS.
- A. M. Dimiev and J. M. Tour, ACS Nano, 2014, 8, 3060–3068 CrossRef CAS.
- L. Dong, J. Yang, M. Chhowalla and K. P. Loh, Chem. Soc. Rev., 2017, 46, 7306–7316 RSC.
- Z. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS.
- S. H. Aboutalebi, M. M. Gudarzi, Q. B. Zheng and J.-K. Kim, Adv. Funct. Mater., 2011, 21, 2978–2988 CrossRef CAS.
- J. Rouhollah, S. H. Aboutalebi, D. Esrafilzadeh, R. L. Shepherd, J. Chen, A. -Y. Sima, K. Konstantin, I. Minett Andrew, M. Razal Joselito and G. Wallace Gordon, Adv. Funct. Mater., 2013, 23, 5345–5354 CrossRef.
- F. Kang, Y. Leng and T.-Y. Zhang, J. Phys. Chem. Solids, 1996, 57, 889–892 CrossRef CAS.
- L. Dong, Z. Chen, S. Lin, K. Wang, C. Ma and H. Lu, Chem. Mater., 2017, 29, 564–572 CrossRef CAS.
- X. Geng, Y. Guo, D. Li, W. Li, C. Zhu, X. Wei, M. Chen, S. Gao, S. Qiu, Y. Gong, L. Wu, M. Long, M. Sun, G. Pan and L. Liu, Sci. Rep., 2013, 3, 1134 CrossRef.
- S. Eigler, M. Enzelberger-Heim, S. Grimm, P. Hofmann, W. Kroener, A. Geworski, C. Dotzer, M. Röckert, J. Xiao, C. Papp, O. Lytken, H.-P. Steinrück, P. Müller and A. Hirsch, Adv. Mater., 2013, 25, 3583–3587 CrossRef CAS.
- H. Chen, M. Wu and C. Li, Carbon, 2018, 139, 216–225 CrossRef CAS.
- Y. Aoi, K. Ono and E. Kamijo, J. Appl. Phys., 1999, 86, 2318–2322 CrossRef CAS.
- Z. Miao, X. Li, X. Zhang, M. Zhou, J. Ning, L. Miao, X. Qiu, M. Jin and L. Zhi, Adv. Mater. Interfaces, 2016, 3, 1500842 CrossRef.
- H. Chen, W. Du, J. Liu, L. Qu and C. Li, Chem. Sci., 2019, 10, 1244–1253 RSC.
- J.-B. Wu, M.-L. Lin, X. Cong, H.-N. Liu and P.-H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- A. Kaniyoor and S. Ramaprabhu, AIP Adv., 2012, 2, 032183 CrossRef.
- D. López-Díaz, M. López Holgado, J. L. García-Fierro and M. M. Velázquez, J. Phys. Chem. C, 2017, 121, 20489–20497 CrossRef.
- A. A. K. King, B. R. Davies, N. Noorbehesht, P. Newman, T. L. Church, A. T. Harris, J. M. Razal and A. I. Minett, Sci. Rep., 2016, 6 DOI:10.1038/srep19491.
- P. Vecera, S. Eigler, M. Koleśnik-Gray, V. Krstić, A. Vierck, J. Maultzsch, R. A. Schäfer, F. Hauke and A. Hirsch, Sci. Rep., 2017, 7, 45165 CrossRef CAS.
- A. Akbari, P. Sheath, S. T. Martin, D. B. Shinde, M. Shaibani, P. C. Banerjee, R. Tkacz, D. Bhattacharyya and M. Majumder, Nat. Commun., 2016, 7, 10891 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00706d |
| This journal is © The Royal Society of Chemistry 2021 |