DOI:
10.1039/D0NA00647E
(Minireview)
Nanoscale Adv., 2021,
3, 24-40
Photoresponsive porous materials
Received
5th August 2020
, Accepted 11th November 2020
First published on 11th November 2020
Abstract
Molecular machines, switches, and motors enable control over nanoscale molecular motion with unprecedented precision in artificial systems. Integration of these compounds into robust material scaffolds, in particular nanostructured solids, is a fabrication strategy for smart materials with unique properties that can be controlled with external stimuli. Here, we describe a subclass of these structures, namely light-responsive porous materials metal–organic frameworks (MOFs), covalent–organic frameworks (COFs), and porous aromatic frameworks (PAFs) appended with molecular photoswitches. In this review, we provide an overview of a broad range of light-responsive porous materials focusing on potential applications.
 Wojciech Danowski | Wojciech Danowski studied chemistry at Warsaw University of Technology, where he received his MSc in 2014 working on the synthesis and functionalization of ZnO quantum dots. In December 2014 he joined the group of B. L. Feringa as a PhD student focusing on the development of light-responsive porous materials and functional interfaces. After earning his PhD in 2019, he is currently a postdoctoral researcher in the group of Prof Feringa focusing on molecular electronics and responsive-interfaces. |
 Thomas van Leeuwen | Thomas van Leeuwen studied chemistry at the University of Groningen, where he received his MSc in 2012. After an internship in the group of Prof T. Bach at the Technical University of Munich, he started his PhD under the supervision of Prof B. L. Feringa working on the synthesis and applications of molecular motors. After the completion of this PhD in 2017, he joined the group of Prof P. Melchiorre as a postdoctoral researcher working on photochemical transformations. Currently, he works in industry as a research scientist. |
 Wesley R. Browne | Wesley R. Browne completed his PhD thesis on the photochemistry and photophysics of Ru(II) polypyridyl complexes at Dublin City University (2002), in the group of Prof J. G. Vos. Following postdoctoral positions at Queen's University Belfast and the University of Groningen, in 2008, he joined the faculty at the University of Groningen as assistant professor and is currently full professor and Chair of Molecular Inorganic Chemistry at the Stratingh Institute for Chemistry. His research interests are in the application of spectroscopy, and especially Raman spectroscopy and electrochemistry to (bio)inorganic catalysis and molecular based materials. |
 Ben L. Feringa | Ben L. Feringa obtained his PhD degree in 1978 at the University of Groningen in the Netherlands under the guidance of Prof. Hans Wynberg. After working as a research scientist at Shell he was appointed full professor at the University of Groningen in 1988 and as distinguished Jacobus H. van't Hoff Professor of Molecular Sciences in 2004. He is an elected foreign honorary member of the American Academy of Arts and Sciences and member of the Royal Netherlands Academy of Sciences. His research interests include stereochemistry, organic synthesis, asymmetric catalysis, molecular switches and motors, photopharmacology, self assembly and nanosystems. |
Introduction
Nature has evolved a collection of complex molecular machinery that drives essentially all aspects of dynamic functions at the cellular level from protein synthesis to cellular locomotion.1,2 These molecules, typically proteins or multi-protein complexes, are capable of performing complex, structural motion in response to external chemical stimuli. Ribosomes3 and chaperonins4 operate in the cytoplasm but it is only when immobilized that synchronization of molecular machines can drive nanoscale motion and overcome the thermal noise to perform tasks at larger length scales.2,5 Most biological molecular machines are immobilized and synchronized temporarily to allow for amplification of their motion along multiple length scales. Motor proteins like dyneins and kinesins,6 transport cargo along microtubules towards and from the nucleus, while flagella motors are anchored in membranes and rotate in unison to achieve directional motion of bacteria7 and mechanical force in skeletal muscles is generated by myosin through cooperative action with actin filaments.8
These amazing systems have inspired synthetic chemists to create artificial molecular machines that can control nanoscale structural motion with a precision on a par with their biological counterparts.9–11 When in solution, these compounds can carry out microscopic mechanical tasks such as to control the stereochemical outcome of a catalytic reactions12 or mechanical twisting of guest molecules,13 however, they can reach their full potential at greater length scales only when able to operate in unison.10 This point is illustrated by the pioneering examples of artificial molecular machines or switches organized in mesoscopic arrays capable of delivering work and performing tasks at the micro and macroscopic level thanks to the cooperative effects.5,14,15 Monolayers of responsive molecules can bend a microscopic cantilever16 or move droplets of organic liquids across a surface,17,18 polymerized liquid crystalline films containing various photoresponsive azobenzenes change shape,19 contract20 or move21,22 when exposed to light, while molecular motors can rotate microscopic glass rods deposited on a liquid crystalline film,23,24 actuate muscle-like self-assembled gel fibers25 or mechanically contract organogels.26,27
Beyond these soft-matter based assemblies, hard-matter assemblies offer advantageous mechanical properties and robustness. Therefore, integration of artificial molecular machines or switches within hard-matter based materials provides the combination of the rigidity of solids with the flexibility of the switchable molecules to generate new classes of responsive solids, with unique properties that can be tuned dynamically by the collective action of the molecules precisely organized and positioned in three-dimensions in the material.28 Despite the astonishing progress in the development of soft responsive materials based on artificial molecular machines and switches, incorporation of these molecules in solid scaffolds without impairing their function is a major challenge. In the pioneering work by the groups of Stoddart, Heath, Flood and others a significant decrease in the rate of pirouetting and shuttling motion of bistable rotaxanes and catenanes embedded in self-assembled monolayers (SAMs) and polymer matrices was observed.29 Similarly, Feringa and co-workers observed a large decrease in the rate of the thermal helix inversion and efficiency of photoswitching for molecular motors densely packed in SAMs on gold and quartz substrates.30,31 This observed decrease in the rate of molecular motion is rationalised by crowding, i.e. tight packing of the molecules in SAMs or the polymer matrix, which limits the free volume crucial for unobstructed motion.32 In contrast, in the typical solid structures, for example crystals, the free volume available for molecular motion is even lower than in densely packed SAMs, which in combination with the rigidity of the solid lattice blocks the larger-amplitude molecular motion. Therefore, perhaps unsurprisingly, initial attempts to incorporate molecular machines in hard-matter based materials resulted in a loss of function.33,34 The few notable exceptions that could operate in the solid state include dithienylethene derivatives, which undergo minor geometrical changes upon isomerization and therefore show efficient photocyclization even in densely packed molecular crystals,35 some azobenzene derivatives functionalized with bulky or polar substituents36,37 and anthracene derivatives.38
Pioneering studies by the groups of Garcia-Garibay and Michl on molecular rotors, demonstrated that these moieties can undergo fast rotations, reaching up to gigahertz frequencies for C3 symmetrical rotors (bicyclo[2.2.2]octane derivatives), when embedded in the molecular crystals that were engineered to possess sufficient free volume for the rotational motion.39–41 Hence, incorporation of artificial molecular machines and switches in porous solids such as metal–organic (MOFs), covalent–organic (COFs), and porous aromatic (PAFs) frameworks offer an opportunity to overcome constraints imposed on molecular motion by confinement in the solid environment and concomitantly to organize them in three-dimensions.28,42,43 The inherent porosity of these structures can potentially provide sufficient free volume for unobstructed motion of incorporated molecular machines.10,28 Recent reports by the groups of Garcia-Garibay, Yaghi, Sozzani, Comotti and others, on molecular rotors incorporated in MOFs and PAFs established their rapid, nearly barrierless rotary motion in the activated frameworks, thus indicating that the microporous environment in these materials can be considered equivalent to a low density liquid or high-density gas rather than a typical solid.44–48 Indeed, this remarkable feature of MOFs enabled fabrication of frameworks capable of supporting large-amplitude conformational dynamics of the incorporated machines, in particular of pirouetting or shuttling motion of crown ether encircling [2]rotaxane struts49–53 or of the unidirectional rotary motion of overcrowded-alkene based molecular motor.54,55 In addition to their porosity, MOFs and other porous structures offer high structural diversity and design flexibility making them a suitable platform to exploit the cooperative nanoscale motion of synthetic molecular machines for fabrication of interactive and responsive materials.10,15,56
In this review only a subclass of the functional solid materials, namely photoresponsive porous materials will be discussed. First, we an overview of photoresponsive molecules will be given, followed by a discussion of applications of these structures. Finally, some intrinsic limitations, key challenges and future prospects of these functional materials will be discussed. Thus far these materials have received limited attention mainly owning to their often challenging synthesis and interpretation of characterisation data. Nevertheless, we firmly believe that this review will provide a fundamental overview of these materials to the broader community, shed light on the major challenges and opportunities associated with these materials and will help to stimulate further research in this area.
Molecular photoswitches
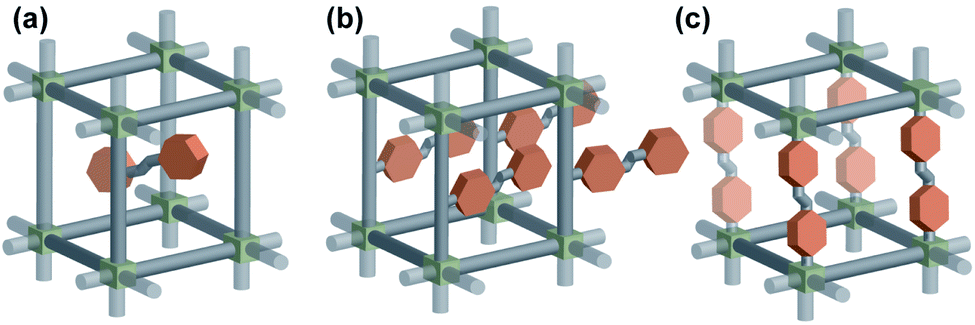
The most straightforward approach to impart a photoresponsive function to a solid material is to incorporate the photoresponsive molecules in the rigid material scaffold. It can be achieved either by incorporation of photoswitches as a guest within pores (Fig. 1a) of the material or integration of the functional molecules in the material's scaffold (Fig. 1b and c). While the first approach is a reliable strategy and was widely used to fabricate photo-responsive functional porous materials, the latter strategy is more challenging but potentially leads to more robust and stable materials and therefore this review will be focused solely on the materials featuring photoswitches installed in the scaffold. The reader is referred also to other recent reviews on responsive porous materials57–62 and molecular machines embedded in MOFs.63
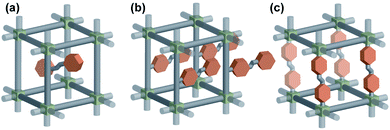 |
| | Fig. 1 Illustration of three distinct modes of incorporation of a photoswitch in a solid material scaffold (a) as a guest in the pores, (b) pendant to the linker, (c) in the backbone of the linker. | |
Azobenzenes
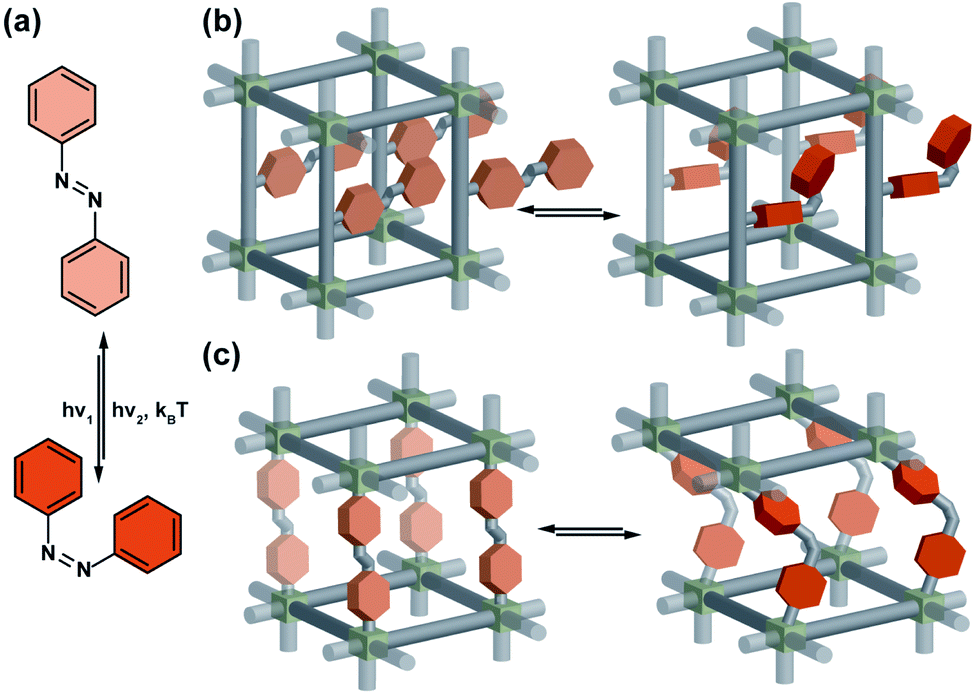
Azobenzenes are by far the most commonly studied and extensively used photoswitches in material science, most probably due to their relatively simple synthesis, photostability, and reliability. Upon irradiation, the planar E-isomer undergoes isomerization to the non-planar, more bulky Z-isomer. The reverse Z → E isomerization is accomplished typically either thermally, or photochemically by irradiation at longer wavelengths (Fig. 2a). In general, azobenzenes show high quantum yields for both Z → E and E → Z photoisomerizations, and high photostationary state ratios. In addition, nearly all the photophysical and photochemical properties of azobenzenes, in particular quantum yield, thermal stability of Z-isomer, photostationary state ratios, excitation wavelengths, can be tuned easily by introducing appropriate substituents at the azobenzene core and the underlying structure–property relationships are well studied and documented in the literature.64 Azobenzenes can be integrated in the solid material scaffold either as a pendant or in a backbone of the linker. In the former case, size aperture or polarity of the pores can be modulated by isomerization of the pendant azobenzene (Fig. 2b). The latter mode of incorporation in principle can allow for fundamental light-induced changes in the material's structure, however typically it prohibits azobenzene isomerization and non-responsive frameworks are obtained (Fig. 2b).60 Nevertheless, there is some precedent in azobenzenes and acylhydrazones incorporated in the backbone of the 2D COFs indicated that such a mode of incorporation is feasible without impairment of the light-induced isomerization of the photoswitches.65,66
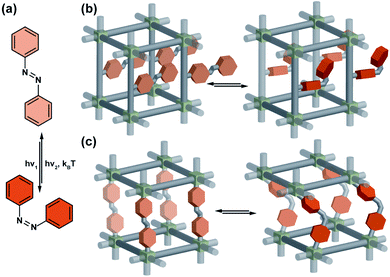 |
| | Fig. 2 (a) Light and heat induced structural changes in an archetypical azobenzene photoswitch. (b) Schematic representation of the structural changes induced by azobenzene incorporated in a porous material as pendants (c) and backbone of the linker. | |
Spiropyrans
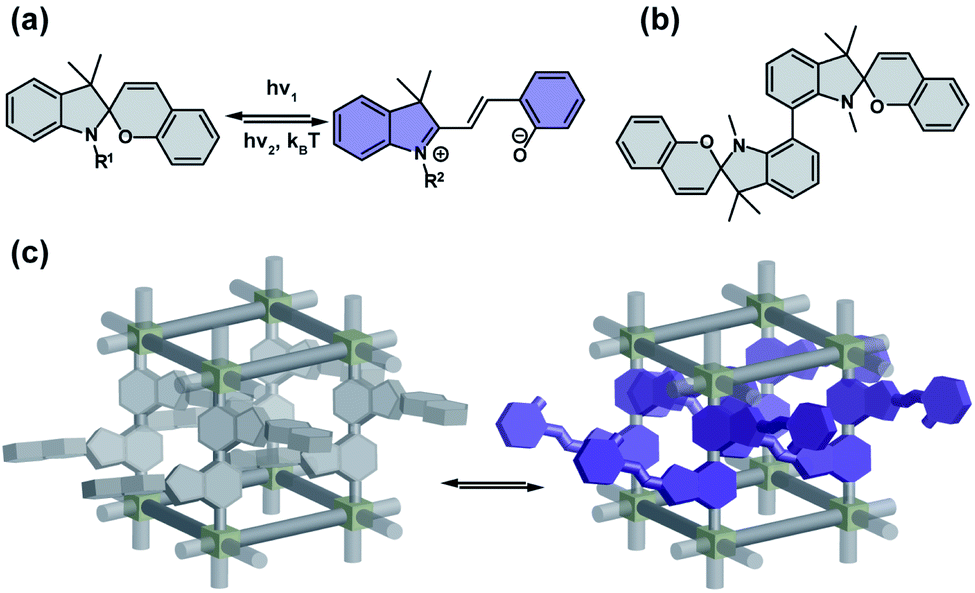
Colourless spiropyrans undergo UV light induced isomerization to the zwitterionic, coloured merocyanine form (Fig. 3a).67,68 The first step in the photochemical isomerization is cleavage of the Cspiro–O bond in the singlet excited state followed by rotation on π–π* and finally relaxation to the ground state furnishing either cis- or trans-oid merocyanine isomers, with the latter form the more thermodynamically stable.69 Substitution of spiropyrans with nitro groups at the 6- or 8-position of the pyran ring, results in an increase in quantum yield of intersystem crossing to a triplet state that significantly lowers their photochemical stability by facilitating bimolecular degradation and decomposition with oxygen.67,70 The spiropyran form can be regenerated by irradiation at longer wavelengths or by heating and the transoid merocyanine form can be stabilized by protonation. Apart of photochromism, these compounds are often thermo-, acido-, solvato-, and mechano-chromic. Spiropyrans properly integrated as pendants or guests in porous solid materials can retain this remarkable multi-stimuli responsivity (Fig. 3b and c).71
 |
| | Fig. 3 (a) Light and heat induced structural changes in archetypical spiropyran photoswitch. (b) Example of dimeric spiropyran that can be incorporated in the framework. (c) Schematic representation of the structural changes induced by spiropyran depicted on panel (b) incorporated in a porous material as pendants. | |
Dithienylethenes
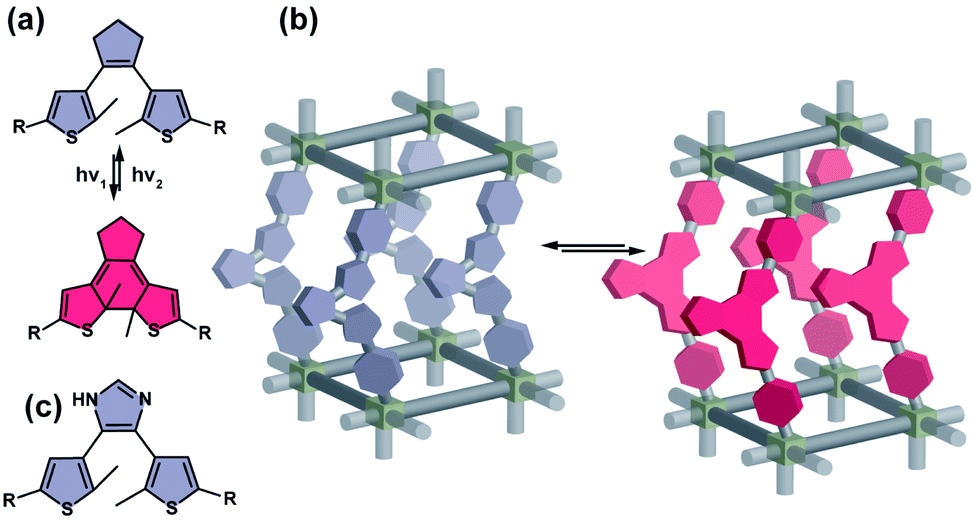
Dithienylethenes (DTEs) comprise a broad class of mostly P-type (photochemically reversible) photoswitches. Exposure of the ring-opened isomer (colourless) to UV light triggers a photochemical 6π electrocyclization and formation of the coloured, ring-closed isomer, while the reverse (cycloreversion) isomerization can be induced with visible light (Fig. 4a). Properly designed derivatives show half-lifes at room temperature of the ring-closed isomer reaching 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years72 and a remarkably high photostability even over 10000 isomerization cycles.73 Notably, while ring-closing isomerization of DTEs is temperature independent, the ring-opening photoisomerization is typically blocked at lower temperatures (77 K) due to an energy barrier along the trajectory towards the conical intersection on the excited state potential energy surface.74–76 As mentioned above, reversible photochromism of dithienylethenes is well documented in molecular crystals, however it requires close spatial proximity between reactive carbons (<4 Å) and an antiparallel conformation of the thiophene rings in the crystal structure.35 While ring-closure of dithienylethenes leads to only minor changes in shape or dipole moment of the molecule, it increases rigidity yielding a fully conjugated structure. Due to the small geometrical differences between ring-closed and opened isomers, DTEs typically readily undergo isomerization when incorporated in MOFs as backbone (Fig. 4b) or pendant (for example through imidazole moiety in ZIF structures, see (Fig. 4c)) of the linker.77–79
000 years72 and a remarkably high photostability even over 10000 isomerization cycles.73 Notably, while ring-closing isomerization of DTEs is temperature independent, the ring-opening photoisomerization is typically blocked at lower temperatures (77 K) due to an energy barrier along the trajectory towards the conical intersection on the excited state potential energy surface.74–76 As mentioned above, reversible photochromism of dithienylethenes is well documented in molecular crystals, however it requires close spatial proximity between reactive carbons (<4 Å) and an antiparallel conformation of the thiophene rings in the crystal structure.35 While ring-closure of dithienylethenes leads to only minor changes in shape or dipole moment of the molecule, it increases rigidity yielding a fully conjugated structure. Due to the small geometrical differences between ring-closed and opened isomers, DTEs typically readily undergo isomerization when incorporated in MOFs as backbone (Fig. 4b) or pendant (for example through imidazole moiety in ZIF structures, see (Fig. 4c)) of the linker.77–79
 |
| | Fig. 4 (a) Light and heat induced structural changes in archetypical dithienylethene photoswitch. (b) Schematic representation of the structural changes induced by dithienylethene incorporated in a porous material in a backbone of the linker. (c) Representation of a structure of a dithienylethene photoswitch that can be incorporated in a material scaffold as pendants. | |
Anthracenes
Anthracenes undergo a formal [4 + 4] photocycloaddition dimerization upon exposure to UV light, while the monomerization of the photodimer can be achieved thermally (Fig. 5). This bimolecular reaction is known to proceed smoothly in molecular crystals and thin films provided proper stacking and intermolecular distance between reactive carbons (<4.5 Å) is present.38
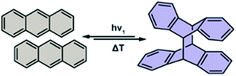 |
| | Fig. 5 Schematic depiction of anthracene photoswitching. | |
Stilbenes and molecular motors
Stilbenes undergo either of two competing reactions upon exposure to light. The first pathway involves photochemical E/Z isomerization of the olefinic bond to yield the Z-isomer, and may be followed by a conrotatory 6π electrocyclization and finally (in the presence of oxidants) the resulting trans-dihydrophenanthrene can undergo irreversible oxidation to the corresponding phenanthrene (Fig. 6a, top row). The second reaction pathway involves bimolecular [2 + 2] photocycloaddition of the excited and ground state stilbenes leading to a mixture of the stereoisomers of substituted cyclobutane (Fig. 6a, bottom row).80 In general, both photochemical reactions are competing, however confinement (increasing effective molarity) in polymers81 or macrocyclic cavitands82 may promote the cycloaddition pathway. Furthermore, when incorporated in COFs or MOFs in the linker backbones, stilbenes, owning to the spatial proximity of olefinic bonds, tend to undergo photocycloaddition, with high selectivity dictated by the molecular arrangement in the network,83–85 which is often accompanied by contraction of the unit cell.86–88 Structurally related so-called stiff-stilbenes, in which the molecular architecture features two five- or six-membered rings attached to the double bond favours the efficient E/Z isomerization pathway (Fig. 6b).89–91 Overcrowded-alkene based molecular motors constitute a distinct class of molecular machines derived from stilbene photoswitches (Fig. 6c).92–96 These unique compounds convert light and heat into a repetitive unidirectional stereochemically-controlled rotatory motion, i.e. rotary motors, or can operate as multistate switches. The substitution patterns around the olefinic bond in these compounds precludes the competitive photodegradation pathways characteristic for stilbene derivatives and controls the rate of the rotary motion, thus providing a possibility to fine-tune the rate of rotation by synthetic modification.97
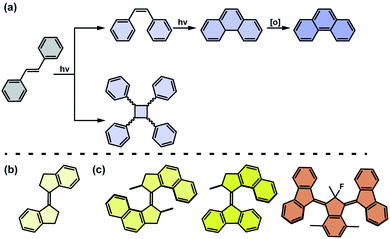 |
| | Fig. 6 (a) Photochemical reaction pathways of an archetypical stilbene photoswitch. (b) Structure of a stiff-stilbene. (c) Structures of archetypical overcrowded-alkene based light-driven unidirectional rotary molecular motors (first, second and third generation of motors are shown from left to right). | |
Applications of photoresponsive solid materials
Gas uptake, storage, separation and release
The key feature of interest in MOFs and COFs is their high porosity and surface area, which has led to their widespread use in gas storage and separation. The challenge in this field is the capture and release of CO2 produced during hydrocarbon combustion. The technologies used currently require high temperatures to release the captured CO2 and regenerate the adsorbent, which makes a major contribution (as much as 40% of costs) to the energy consumption of the processes.98,99 Therefore, the development of light-responsive porous adsorbents that can be operated in situ by remote switching between high and low CO2 adsorption capacity with light would significantly reduce the costs associated with processing of combustion generated CO2. For this reason, substantial effort has been directed to the field of light-responsive porous materials to modulate their gas uptake capacity.
Azobenzenes.
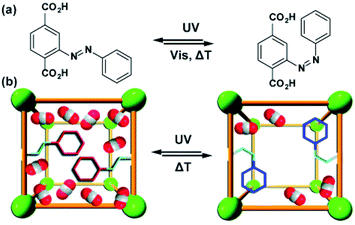
Initial attempts to incorporate azobenzene photoswitches in MOFs as a backbone of the linker were largely unsuccessful due to impartment of their photochromism the rigidity of the surrounding framework.33,34 In contrast, incorporation of azobenzenes as pendant to the linkers typically preserved their photoresponsive behaviour in MOFs of various structures and topologies.100–103 However, even this mode of incorporation does not ensure the photoresponsivity of the incorporated azobenzenes is retained, as the steric interactions the pendant azobenzenes encountered in small pores or with neighbouring linkers may still hinder the isomerization. Bléger, Castellanos and co-workers, demonstrated that with an ortho-fluoro azobenzene as pendant linker in MIL-53(AL) drastically reduced the photoisomerization efficiency of the azobenzene unit while in the more open UiO-66(Zr) scaffold unhindered isomerization was observed.104 Further studies showed that the azobenzene functionality can be post-synthetically introduced in MIL-101(Cr)–NH2 MOF, via azo coupling or amide and urea formation, thereby demonstrating that this functionality can be conveniently and easily incorporated in the material scaffold.
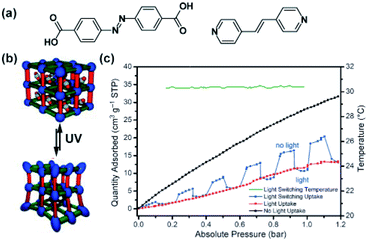
Zhou and co-workers introduced the first example of photomodulation of CO2 uptake in a MOF bearing azobenzene pendants.105 In the solvothermal synthesis from N,N-diethylformamide they obtained the stimuli-responsive material isoreticular to MOF-5 (Fig. 7b), while synthesis in DMF gave the two-fold interpenetrated framework with a considerably lower gas uptake capacity and hindered azobenzene switching behaviour. Irradiation at 365 nm to induce E → Z isomerization of pendant azobenzene resulted in a large drop in the CO2 uptake capacity of the framework, which reverted almost quantitatively upon thermally induced Z → E isomerization (Fig. 7a and b). Based on the analysis of structural data and pore size distribution of pristine and irradiated materials these changes in gas uptake were attributed to the isomerization of azobenzene pendants, for which the non-planar Z-isomer was found to shield the main CO2 adsorption sites—metal oxygen bonds, and partially block smaller pores, thereby leading to lower CO2 uptake (Fig. 7b).
 |
| | Fig. 7 (a) Structure and switching of the incorporated azobenzene in the isoreticular MOF-5 scaffold. (b) Schematic depiction of the light and heat induced structural changes in the pore structure and gas adsorption of azobenzene functionalized isoreticular MOF-5 structural reprinted with permission from ref. 105 Copyright 2013 American Chemical Society. | |
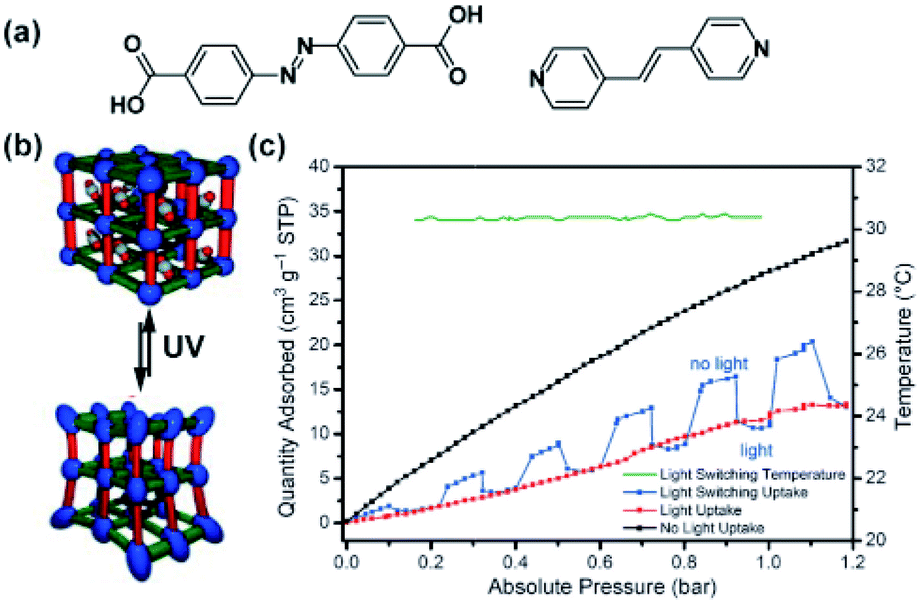
Lyndon, Hill and co-workers achieved photomodulation in CO2 uptake in a triply-interpenetrated, pillared Zn-paddlewheel framework bearing stilbene and azobenzene functionalities (Fig. 8a) incorporated in the backbone of the framework (Fig. 8a and b).106 Although, evidence for isomerization was not obtained upon exposure of the material to light, a large drop, of up to 64%, in CO2 uptake by the framework was observed upon in situ broadband illumination (Fig. 8c, black and red lines, respectively). Since the framework did not undergo structural transformation when irradiated, the observed differences in gas uptake were proposed to stem from light-induced periodic bending motion of the photoactive framework at a local scale. Nevertheless, the response of the framework was found to be almost instantaneous and the amount of the adsorbed CO2 could be modulated dynamically (Fig. 8c, blue line).
 |
| | Fig. 8 (a) Structure of the stilbene bispyridyl pillar and azobenzene dicarboxylic acid used as linkers. (b) Depiction of the active framework. (c) Changes in the CO2 adsorption isotherm of the material (black line, pristine), during in situ irradiation (red line) and upon a modulated exposure to light (blue line), temperature of the sample (green line). Reprinted with permission from ref. 106. Copyright 2013 Wiley-VHC. | |
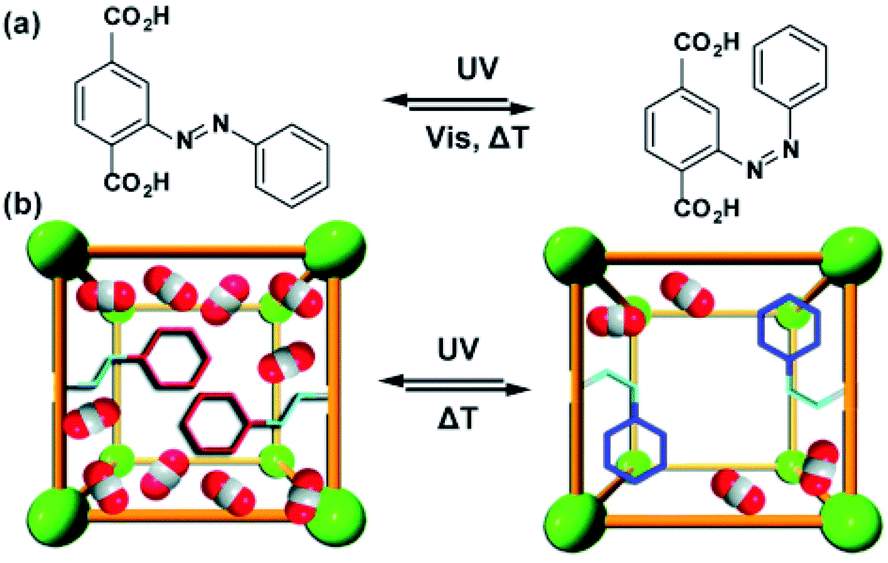
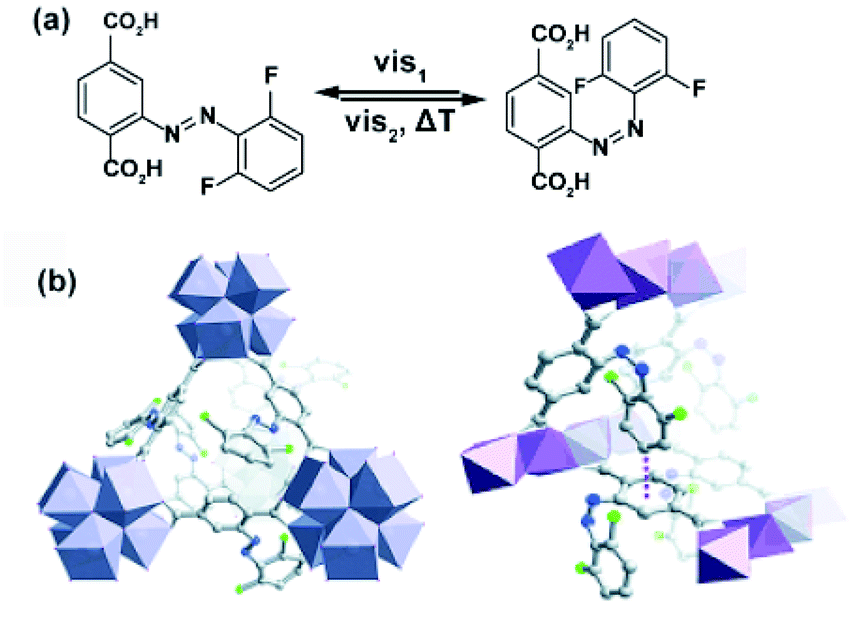
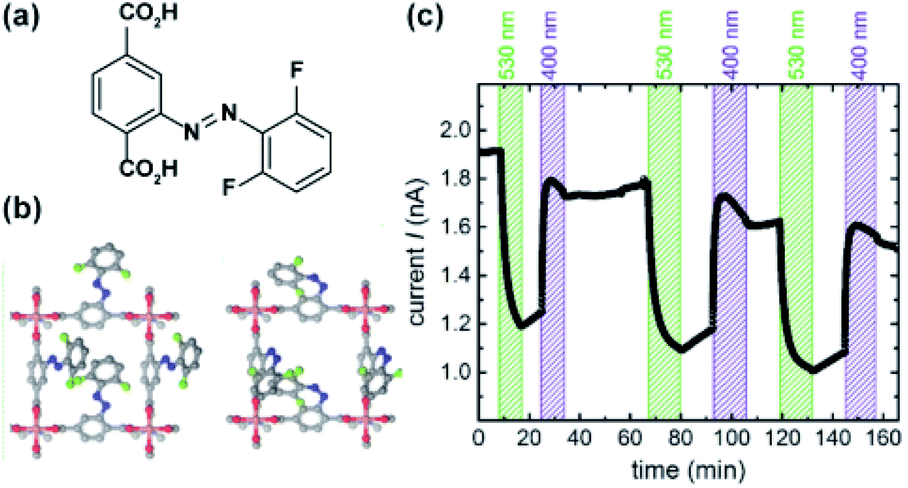
Similar effects of in situ light irradiation on CO2 adsorption in azobenzene-containing MOFs were observed by Bléger, Castellanos and co-workers.107 Two MOF scaffolds MIL-53(AL) and UiO-66(Zr) were constructed using visible-light (>500 nm) responsive ortho-fluoro azobenzene linkers (Fig. 9a). Interestingly, the azobenzene linkers incorporated in MIL-53(Al) (Fig. 9b) showed limited isomerization ascribed to steric congestion in the MIL-53 scaffold. In contrast, the azobenzene pendants UiO-66(Zr) framework (Fig. 9b) could be readily isomerized with visible light owing to reduced steric hindrance in the UiO-66 scaffold. Even though UiO-66(Zr) was proven to provide sufficient free volume to promote solid-state isomerization of the pendants, no changes in the CO2 uptake were observed upon MOF irradiation. Conversely, the non-responsive MIL-53(Al)-based MOF showed a 10% decrease in the CO2 uptake during in situ irradiation and no change in uptake upon ex situ irradiation. Since the isomerization of the azobenzene pendants was hindered by the MIL-53(Al) scaffold, the observed differences in gas absorption capacity in response to green light, were attributed to local heating of the framework induced by vibrational relaxation of the excited azobenzenes. Additional control experiments demonstrated that a 10% decrease in CO2 uptake corresponds to 10 °C increase in the overall temperature of the sample. Similar effects, albeit more pronounced (>40%) were found for non-fluorinated azobenzene-decorated UiO-66(Zr)108 and DMOFs109 upon in situ exposure to UV-light and in mixed composite materials with polymers as secondary adsorbents.110
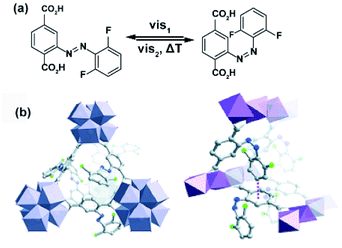 |
| | Fig. 9 (a) Structure and switching of the incorporated visible-light responsive ortho-fluoro azobenzene linker. Models of the solid-state structure of the isoreticular (b) UiO-66(Zr) (left panel) and MIL-53(Al) (right panel). Reprinted with permission from ref. 107. Copyright 2016 Wiley-VHC. | |
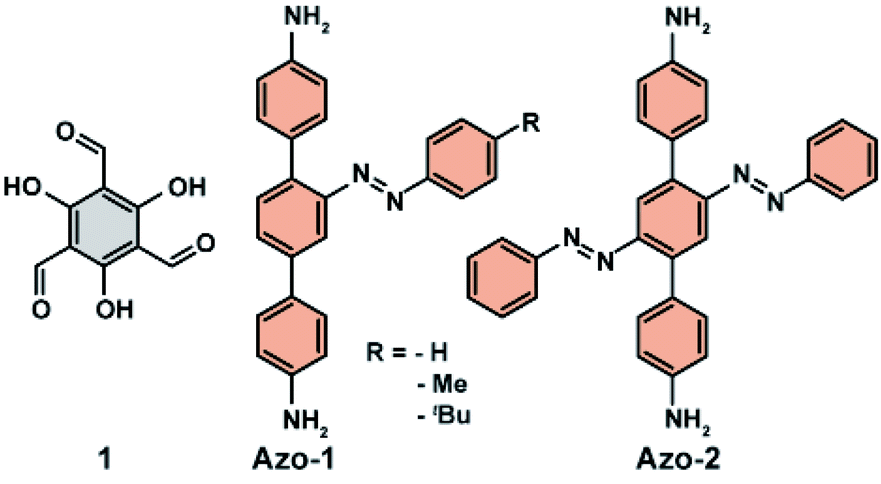
The opposite effect following azobenzene E → Z isomerization on the uptake of CO2 in a porous organic polymer decorated with pendant azobenzenes was reported by Zhang and co-workers.111 A series of functional materials was fabricated by condensation of triformylphloroglucinol (1, Fig. 10) and diamine-functionalized azobenzene (Azo-1, Fig. 10) or bis-azobenzene functionalities (Azo-2, Fig. 10). Ex situ irradiation with UV light of these materials resulted in a substantial increase in CO2 uptake and only minor change in surface area and pore size distribution. The highest difference, amounting to almost 30% of adsorption capacity between pristine and UV-treated materials, was observed for the least sterically hindered pendants. The observed changes in the CO2 adsorption were rationalized by the changes in the pores surface polarity induced by azobenzene isomerization. The considerably larger dipole moment (∼3 D) of Z-azobenzene than the E-azobenzene,64 should promote dipole–quadrupole interaction between CO2 and pendants manifested in the observed higher uptake of CO2. A similar influence of the azobenzene isomerization on polarity of the channels was observed by Aida et al. in isoreticular UiO-68(Zr) MOF bearing azobenzene pendants.112
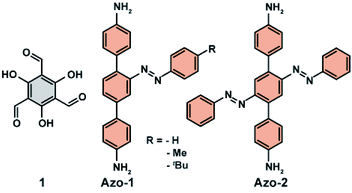 |
| | Fig. 10 Structures of triformylphloroglucinol (1) and diamine-functionalized azobenzenes (Azo-1, Azo-2) used to construct porous switchable organic polymer. | |
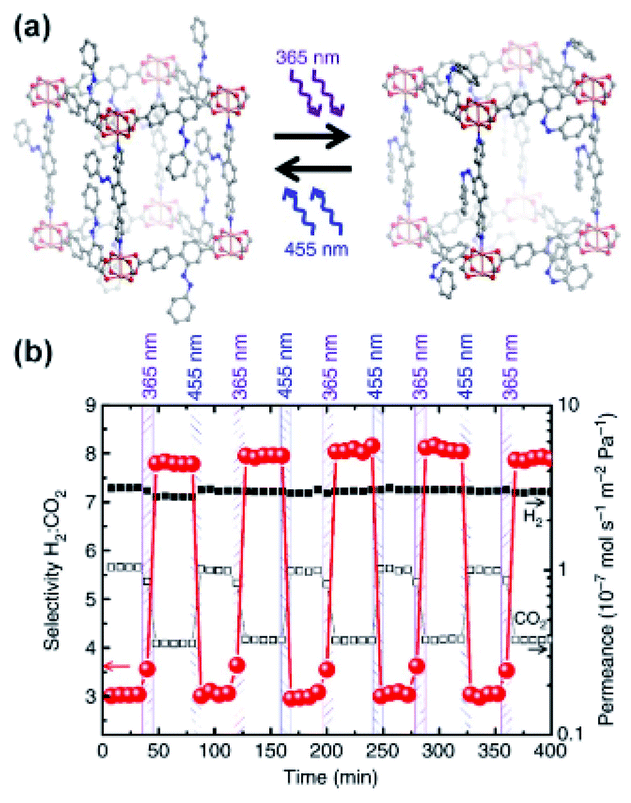
Taking advantage of the strong dipole–quadrupole interactions of CO2 with Z-azobenzenes, Heinke, Wang, Wöll and co-workers fabricated a responsive membrane for tuneable gas separation. The membrane was based on surface-mounted (SURMOF) azobenzene-appended pillared Cu(II)-paddlewheel MOF supported on mesoporous α-Al2O3 (Fig. 11a).126 Owing to the attractive interactions of CO2 quadrupole with Z-azobenzene dipole moments, the CO2 permeability through the membrane could be precisely tuned by adjusting the E/Z ratio of azobenzene linkers with UV and visible light. Conversely, N2 and H2 showed only minimal changes in the permeability through the membrane upon isomerization of the azobenzene pendants owning to their negligible interactions with MOF constituents. This discrepancy in the gases permeability behaviour allowed for reversible and remote control over the composition of the permeate flux with light adjustable separation factors. Starting from 1:1 mixture of gasses feed separation ratios ranging from 3.0 (pristine material) to 8.0 (irradiated state material) for CO2:H2 and 5.5 (pristine) to 8.5 (UV treated) for CO2:N2 gas mixtures were determined (Fig. 11b). Similar selectivity changes in a separation of H2/C2H4 and H2/C2H6 mixtures were found for analogous membrane with appended visible light responsive ortho-fluoro azobenzenes, while an effect of switching on selective permeability was not observed for CO2:H2 mixtures.127
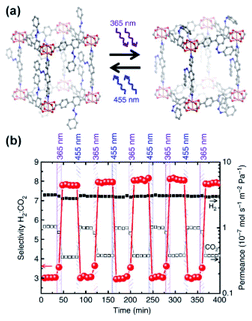 |
| | Fig. 11 (a) Representation of light induced structural changes in the SURMOF functionalized with azobenzene pendants. (b) Light-induced changes in selectivity of H2/CO2 separation on α-Al2O3-supported SURMOF membrane (red circles) and H2 (black squares) and CO2 (white squares). Adapted by permission from Springer Nature from ref. 126, Copyright 2014. | |
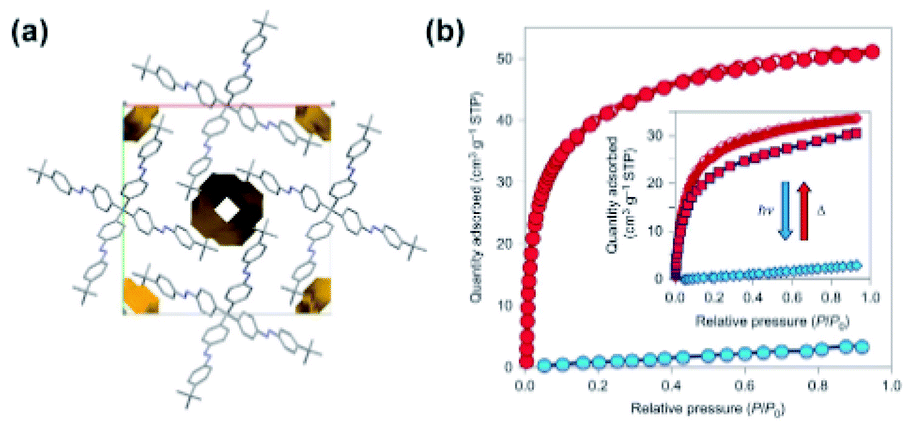
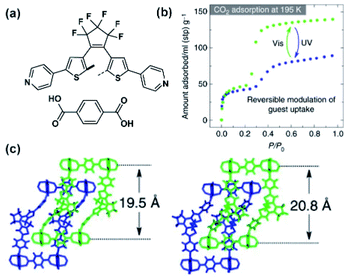
Photomodulation of gas uptake capacity in molecular crystals remains largely unexplored, mainly due to challenges faced in the design of these materials.113,114 One exception is a material based on azobenzene moieties mounted on a tetraphenyl methane core that forms a rigid, star-shaped unit (Fig. 12a). This unit was purposely designed to pack inefficiently in the solid state and form permanently extrinsically porous molecular crystals (Fig. 12a).115 Crystals formed by the all-E isomer showed a type-I Langmuir adsorption isotherm and selective uptake of CO2 over N2. Irradiation of these crystals with UV-light induced the transformation to amorphous melt phase consisting of a mixture of E/Z-azobenze diastereoisomers. This amorphous azobenzene phase showed almost negligible CO2 uptake, therefore in comparison to crystalline all-E isomer the drop in the adsorption capacity amounting up to almost 50 cm3 (STP) of CO2 at 195 K (Fig. 12b). Importantly, the crystallinity and porosity of the material recovered upon Z → E isomerization of azobenzene moieties induced by visible light and thermal treatment (Fig. 12b, inset).
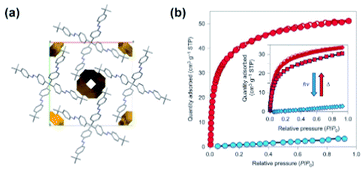 |
| | Fig. 12 (a) Packing of the tetrameric azobenzenes in the solid state along with representation of the empty channels. (b) CO2 adsorption isotherms at 195 K of pristine material (red isotherm) and after exposure to UV light (blue isotherm). Reprinted by permission from Springer Nature from ref. 115, Copyright 2015. | |
Dithienylethenes.
Dithienylethene (DTE) derivatives present only modest steric requirements for photoisomerization in molecular crystals.35 Therefore, provided that all other requirements for DTE isomerization in solids can be fulfilled, incorporation of the DTE photoswitches in the linker backbone should not impede its photocyclization. In addition, the light-induced ring closure of the DTE can potentially trigger cooperative structural transformation on a global scale leading to large changes in the framework properties.35,116,117 Pu, Guo and co-workers synthesized a five-fold interpenetrated MOF with biphenyldicarboxylate and DTE bispyridyl linkers (Fig. 13a and b).118 Despite of the large degree of the interpenetration, the resulting material had ca. 37% void volume in the unit cell, and readily responded to light with almost immediate colouration and decolouration of the crystals upon alternating irradiation with UV and visible light. In addition, the material showed a 4-fold increase in CO2 uptake capacity upon ex situ irradiation with UV light, rationalised by the favourable quadrupole–quadrupole interactions between CO2 and ring-closed isomer of DTE. In a dynamic gas adsorption experiments, an instantaneous release of 76% of adsorbed CO2 at P/P0 = 1 was observed upon exposure of the UV-pretreated (with DTE linkers cyclized) MOF to visible light. Further studies on this framework, showed almost two times more favourable adsorption of C2H2 over C2H4 at low pressure and temperature (100 kPa, 195 K) for pristine material and negligible differences in the uptake for UV treated material, which could potentially be exploited for construction of smart, responsive membranes.119
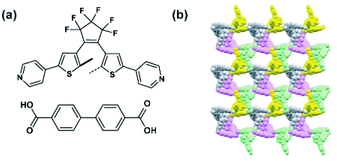 |
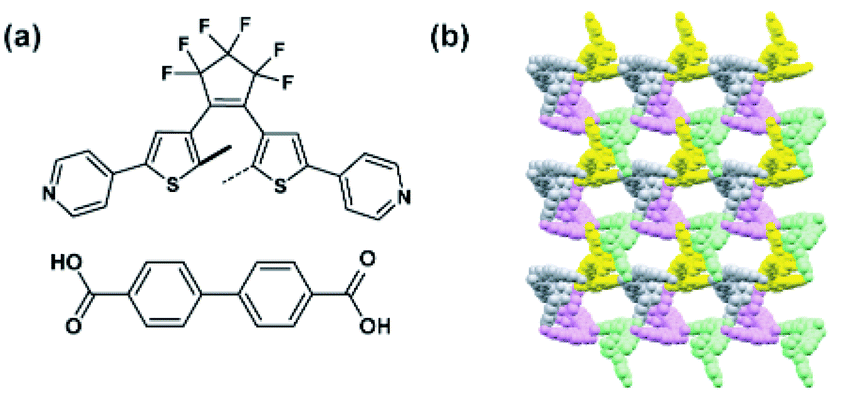
| | Fig. 13 (a) Structure of the linkers used to construct five-fold interpenetrated pillared MOF. (b) Packing of the five independent networks in the crystal structure, viewed along the b direction. Interpenetrating networks were indicated by various colours. | |
Independently, Barbour and co-workers investigated the same DTE based MOF (Fig. 14b), and reported that the photocyclization reaction of DTE linkers is limited only to the surface and does not affect the bulk of the material. No changes in CO2 uptake were observed for the material presumed to be due to incomplete linker transformation.120 This limited light-induced isomerization was hypothesized to originate from rigidity of the original network, and to bypass these constraints another two-fold interpenetrated MOF bearing a more flexible dicarboxylate linker (Fig. 14a) was prepared. Irradiation of the crystal of the flexible MOF at 365 nm led to a bulk photocyclization of DTE linkers, confirmed unequivocally by single-crystal X-ray diffraction (SC X-ray) data (Fig. 14b). In parallel, the isomerization of DTE was accompanied by a large structural deformation of the framework with a remarkable decrease of solvent accessible volume from 262 Å3 (for ring-opened DTE MOF) to 20 Å3 (for ring closed DTE MOF), thus, in essence, switching between a porous and a non-porous material (Fig. 14b).
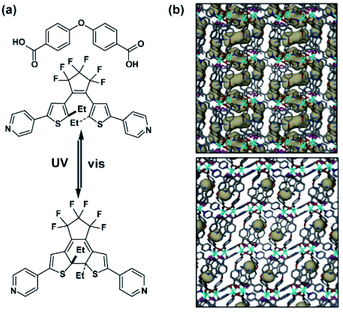 |
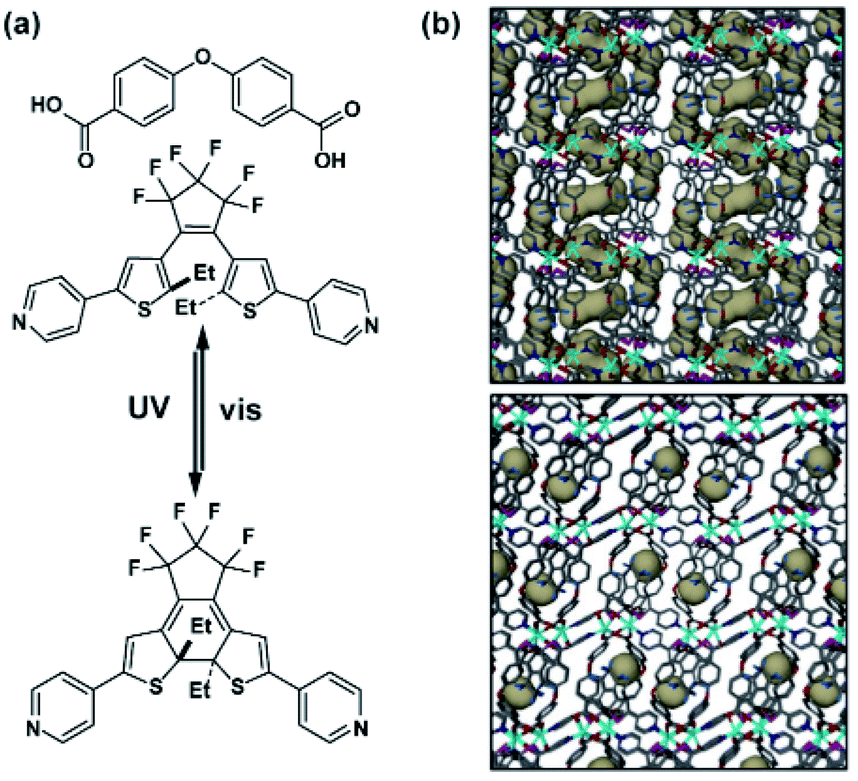
| | Fig. 14 (a) Structure of the linkers used to construct framework and switching of the incorporated DTE pillar. (b) Representation of the porosity (grey areas) of the pristine material (top panel) and UV-treated material (bottom panel). Adapted with permission from ref. 120. Copyright 2017, Royal Society of Chemistry. | |
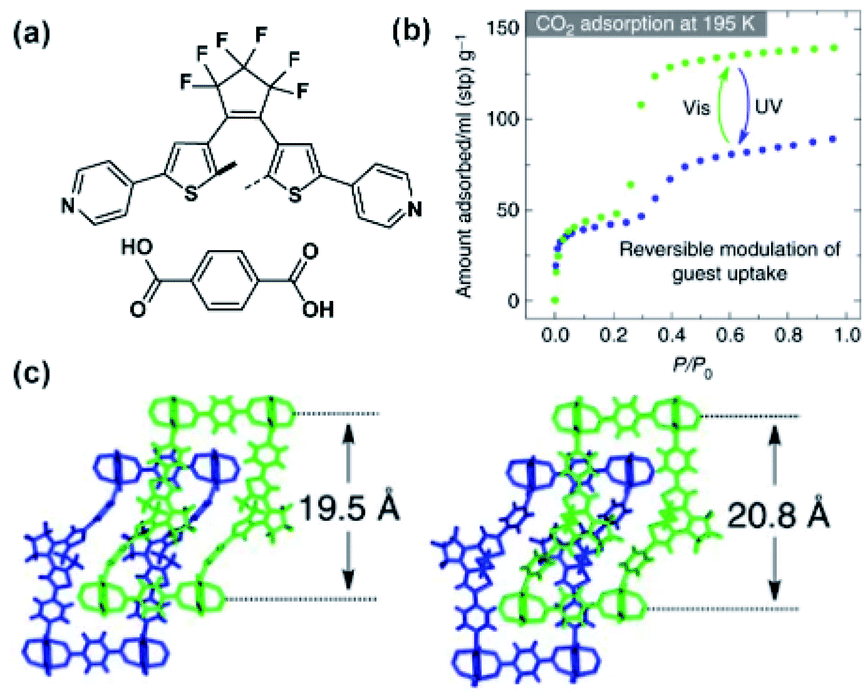
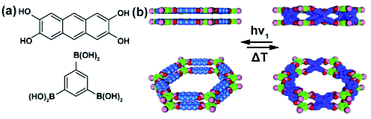
A similar example of bulk photoisomerization in a flexible two-fold interpenetrated framework was reported by Kitagawa and co-workers (Fig. 15a and b).121 The reversible photoswitching of the DTE pillars was confirmed with analysis of the SC X-ray data and 1H NMR spectra of digested crystals (Fig. 15c). Both irradiated and non-irradiated MOFs showed a type IV CO2 sorption isotherm, indicating a flexible structure with a gate-opening sorption mechanism. In the low-pressure regime, both pristine and UV-treated MOFs exhibited similar CO2 uptake. Conversely, after the inflection point the irradiated material showed a significant drop in the CO2 (from 140 ml to 89 ml STP at P/P0 = 0.95), which corresponded to the changes of the void volume in the structure.121
 |
| | Fig. 15 (a) Structure of the linkers used to construct two-fold interpenetrated framework. (b) CO2 adsorption isotherms of pristine (green points) and irradiated (blue points) materials. (c) Representation of the SC X-ray structure of pristine (left) material and UV-treated (right) materials viewed along b direction. Adapted by permission from Springer Nature from ref. 121, Copyright 2017. | |
Anthracene.
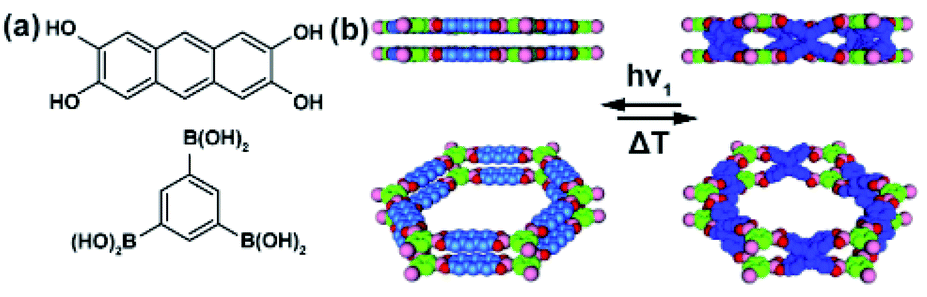
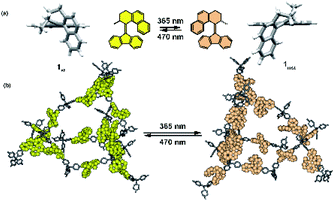
Jiang and co-workers demonstrated a photomodulation of a COF surface area taking advantage of the reversible [4 + 4] photocycloaddition of anthracene moieties in the solid COF.122 Modelling and PXRD data revealed the eclipsed stacking of the adjacent two-dimensional (Fig. 16a and b) layers, which positions neighbouring anthracene moieties in close spatial proximity crucial for the efficiency of the photocycloaddition in the solid state (Fig. 16b). Irradiation of the COF thin films at 360 nm led to a conversion of neighbouring stacked anthracenes to the corresponding photo-dimer with a 47% yield (Fig. 16b), and the reverse reaction could be achieved in almost quantitative yield upon heating at 100 °C. These light- and heat-induced structural transformations were accompanied by a change in the surface area of the material (from 1864 m2 g−1 to 1456 m2 g−1, BET values) and a decrease in pore capacity from (1.24 cm3 g−1 to 1.08 cm3 g−1) while heating of the irradiated COF reverted these changes (surface area 1684 m2 g−1, BET value). More recently, similar changes in the gas uptake were observed upon reversible [2 + 2] photocycloaddition in poly(aryl vinylene) 2D COFs.123,124
 |
| | Fig. 16 (a) Structure of the component units of anthracene-based two-dimensional COF connected by catechol boronate linkages. (b) Top and side view of structural model of pristine material (left panel) and UV-irradiated material after dimerization of anthracene building units (right panel). Reprinted with permission from ref. 122. Copyright 2015 Wiley-VHC. | |
Overcrowded alkenes
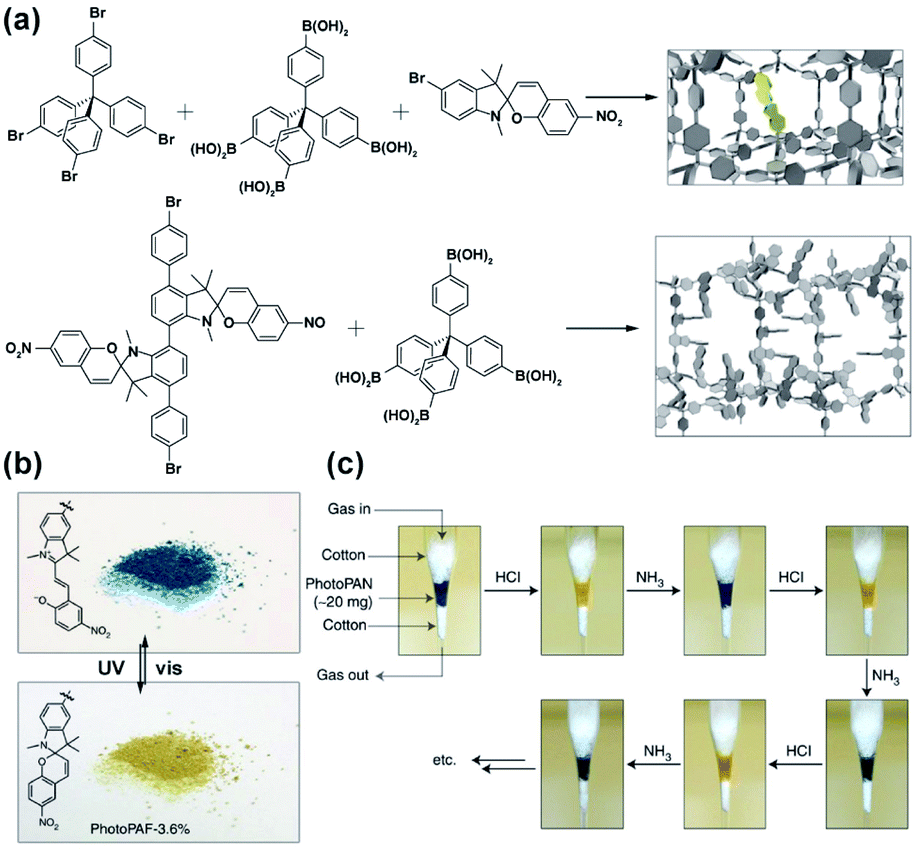
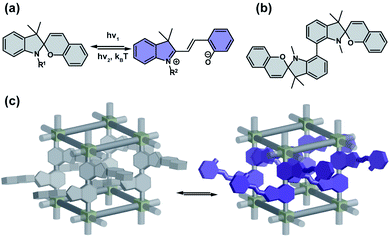
Recently, we have demonstrated photomodulation of the gas adsorption capacity in porous aromatic framework (PAF) enabled by the bulk isomerization of a incorporated bistable overcrowded alkene photoswitch.125 The responsive materials were prepared using a Yamamoto cross-coupling between tetrabromo-tetraphenylmethane and an overcrowded alkene, which enabled fabrication of the highly-porous materials with predetermined content of the light-sensitive component. Diffuse-reflectance, Raman and solid-state NMR spectroscopies confirmed that the photoisomerization of the switch was quantitative, and reversed upon irradiation with light of longer wavelength or heating. The isomerization of the alkene was accompanied by a change in N2 and CO2 uptake amounting to a drop by 20% of the initial capacity at a relative pressure (P/P0) of 0.6 bar for the materials with higher content of the photoswitch (Fig. 17).
 |
| | Fig. 17 (a) Representation of the light-induced isomerization of the bistable overcrowded alkene switch. (b) Schematic representation of the photoswitching in the solid material. Adapted by permission from Springer Nature from ref. 125, Copyright 2020. | |
Guest uptake, release and cargo delivery systems
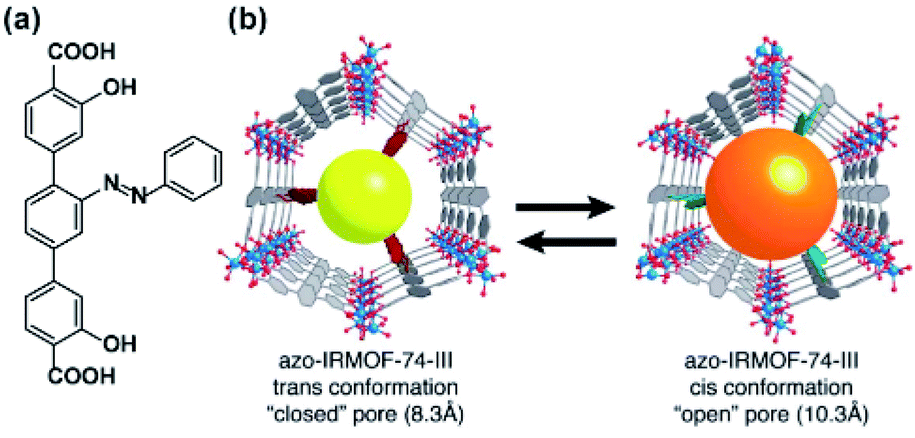
The geometrical change associated with E ↔ Z isomerization of azobenzene (Fig. 18a) can be used to induce a release of cargo trapped inside of microporous material. The first practical realization of this idea was achieved in isoreticular MOF-74, having a one-dimensional hexagonal microchannels functionalized with evenly separated azobenzene pendants pointing towards the centre of the pore.128 As a consequence of this alignment, a substantial part of the aperture could be controlled by the isomerization of the appended azobenzenes from 8.3 Å for E-azobenzene to 10.3 Å for Z-azobenzene (Fig. 18b). Consequently, this framework showed slow release of entrapped probe molecules (propidium iodide) upon irradiation at 408 nm (wavelength close to isosbestic point 402 nm) and a much lower rate of the cargo release was observed upon discontinuing the 408 nm irradiation. The acceleration of the dye expulsion from the pores of the material was attributed to the rapid E ↔ Z interconversion and associated with the wagging motion of the azobenzene pendants triggered upon irradiation at wavelengths near the isosbestic point. A similar principle of UV-light induced cargo-release from azobenzene-appended 1D channels was recently demonstrated in 2D-COF cargo delivery platform.129
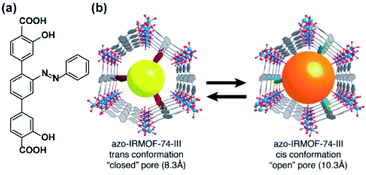 |
| | Fig. 18 (a) Structural the azobenzene linker. (b) Schematic representation of light-induced changes in pore aperture of MOF-74 upon photochemical isomerization of the appended azobenzenes. Adapted with permission from ref. 128. Copyright 2013, Royal Society of Chemistry. | |
Heinke, Wöll and co-workers developed a responsive surface coating capable of storage and on-demand release of cargo based on azobenzene functionalized SURMOF.130 The two-component system was constructed on the surface by liquid-phase epitaxy with first non-responsive layers serving as a storage tank and terminating layers, functionalized with photoresponsive azobenzenes serving as an addressable valve (Fig. 19a). The resulting surface-mounted architecture, showed a four times slower uptake of butanediol than the SURMOF without azobenzene functionalities. Moreover, in the E-state of the azobenzene pendants the uptake of the guest molecules was 15 times higher than in the Z-state (Fig. 19c). Therefore, the SURMOF could be used as a remotely controlled supramolecular container in which cargo could be loaded in E-state of active layer, contained by E → Z isomerization of the azobenzenes and then stored for a prolonged period of time to finally be released on-demand upon visible-light induced Z → E isomerization of the switches (Fig. 19d). Further work on similar azobenzene-based surface mounted MOFs established the influence of steric hindrance and lateral separation of azobenzenes pendants on the efficiency of photoswitching131 and rate of thermal Z → E isomerization132 as well as indicated that polar interactions are dominant over steric effects in the uptake and release of butanediol.133,134
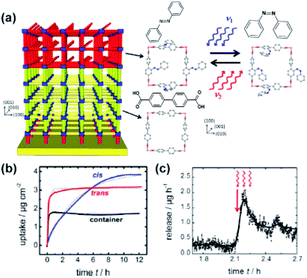 |
| | Fig. 19 (a) Structure of the layered SURMOF consisting of non-responsive, storage compartment (yellow part) and light-responsive azobenzene appended layer (red part) with structure of the SURMOF layers viewed along c direction. (b) Comparison of the butanediol uptake by the two-layered SURMOF with Z-azobenzene (blue line), E-azobenzene (red line) pendants and lone passive layer (black line) determined by quartz microbalance. (c) Butanediol release experiment monitored by quartz microbalance. Red arrow indicates start of the irradiation at 560 nm to induce Z → E isomerization of the azobenzene pendants. Reprinted with permission from ref. 130. Copyright 2017 American Chemical Society. | |
Based on a similar principle, e.g., a passive compartment for storage and active shell acting as a gate, a novel cargo delivery platform was developed by Hecht and co-workers.135 The responsive MOF was based on UiO-68 network and was fabricated using solvent-assisted linker exchange with appropriate azobenzene-derived linker, which rendered a core–shell structure with an azobenzene rich shell of uniform thickness. This approach bypassed the problems associated with light penetration depth, and as a result practically the same photostationary states isomers ratios could be achieved for the core–shell crystal as for free azobenzene in solution. Owing to the large steric bulk of the designed azobenzene linker, the Z-azobenzene rich shell could serve as a barrier for the diffusion of 1-pyrenecarboxylic acid, while for the E-azobenzene rich shell the diffusion was much faster for both uptake and release experiments. A similar approach was demonstrated by Meng, Gui and co-workers, who explored azobenzene β-cyclodextrin host–guest complexes to trap and release cargo in azobenzene functionalized UiO-68(Zr) MOF.136 Furthermore, analogues azobenzene-appended UiO-type framework was used for modulation of magnetic properties of encapsulated metallofullerene.137
Separation of chemicals
Azobenzenes.
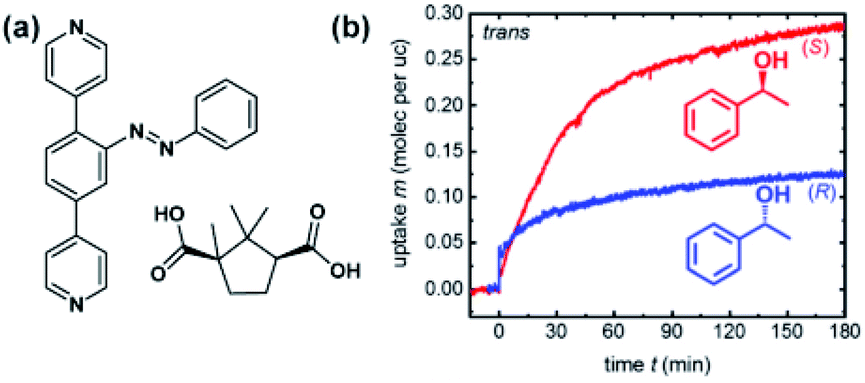
Recently, Heinke and co-workers demonstrated photocontrol over enantioselective uptake of 1-phenylethanol in azobenzene appended SURMOF.138 The pillared Cu(II)-paddlewheel MOF was based on two linkers—light-switchable azobenzene bispyridyl derivative and D-camphoric acid, giving rise to homochiral responsive framework (Fig. 20a). It was found that SURMOF has the adsorption capacity of (S)-phenyl ethanol ca. three times higher than that of (R)-phenyl ethanol. However, upon UV irradiation and E → Z isomerization of azobenzenes pillars, this difference is diminished as the polar interactions of phenyl ethanol with Z-azobenzene prevails over weaker enantiospecific interactions with the chiral camphoric acid linker (Fig. 20b). Hence, the composition of the adsorbate in the MOF could be switched from enantio-enriched (ca. ee = 50%) to racemic upon light irradiation, thus opening opportunities for light-switchable separation of enantiomers in permeable membranes.
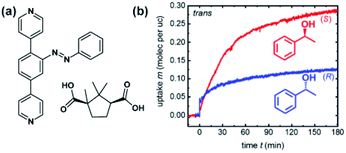 |
| | Fig. 20 (a) Structure of the linkers used to construct homochiral SURMOF. (b) Uptake of (R)- and (S)-1-phenylethanol by the Z-azobenzene MOF thin films. Adapted with permission from ref. 138. Copyright 2019, Royal Society of Chemistry. | |
Dithienylethenes.
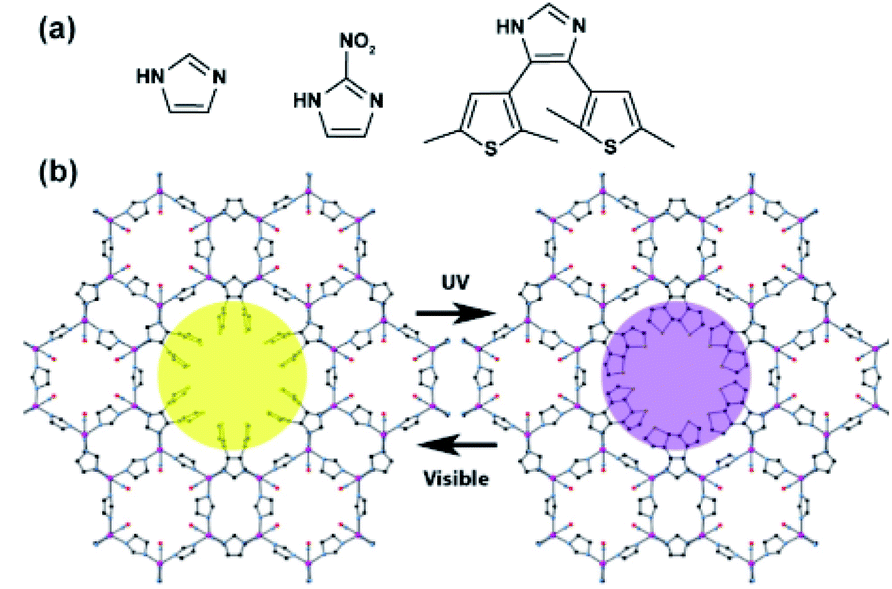
Katz and co-workers took advantage of the changes in electronic properties of DTE photoswitches and applied it to a separation of chemicals in ZIF-70 MOF bearing imidazole linkers.139 The DTE photoswitch was introduced in the pores of the ZIF-70 framework by means of solvent assisted linker exchange between ZIF-70 and mixture of 2-nitroimidazole and the DTE reaching a final composition of 0.27:1:0.7 of DTE:imidazole:2-nitroimidazole (Fig. 21a). In the obtained framework, the DTEs pendants were pointing towards the interior of the large pore (Fig. 21b), and could be readily cyclized and opened upon alternating irradiations at the appropriate wavelengths. Finally, the UV-treated MOF, containing the ring-closed DTEs linkers, showed much stronger interactions with various aromatic compounds (benzene, naphthalene, pyrene) in comparison to the pristine material, which makes it potentially useful in light-controlled adsorption of aromatic hydrocarbons from diluted solutions.
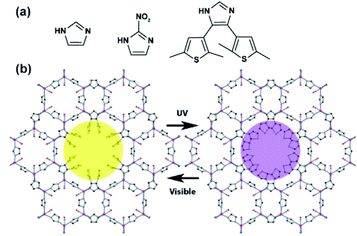 |
| | Fig. 21 (a) Structure of the imidazole derived linkers used in the synthesis of DTE functionalized ZIF-70 framework. (b) Model of DTE decorated ZIF-70 framework with ring-opened (left panel) and ring-closed (right-panel) DTE pendants. Adapted with permission from ref. 139. Copyright 2017 American Chemical Society. | |
Catalysis
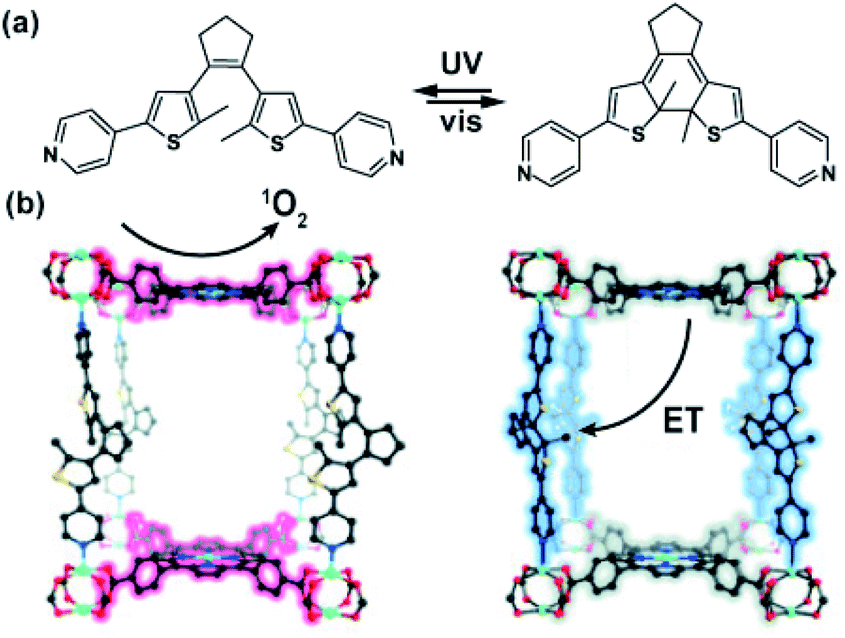
Recent studies on porphyrin MOFs as light-harvesting platforms showed fast and long-distance exciton migration between organized porphyrin moieties upon light excitation.140,141 Furthermore, studies of Shustova on porphyrin Zn-paddlewheel framework pillared with photochromic dithienylethene showed the efficient energy transfer between porphyrin linkers and ring-closed DTE pillars manifested as quenching of the framework fluorescence (Fig. 22a).142 Based on these studies, the same porphyrinic framework bearing DTE pillars was used in light-controlled singlet oxygen sensitization (Fig. 22b).143 Irradiation of the framework bearing the ring-opened DTE pillars at the Soret band of the zinc-porphyrin (405 nm) in the presence of oxygen led to the formation of 1O2 by means of the energy transfer between the porphyrin framework and triplet oxygen. Conversely, irradiation at the same wavelength of the MOF bearing photocyclized DTE led to the triplet–triplet energy transfer between the porphyrin linker and the DTE pillar as a result formation of singlet oxygen was not observed. This property was exploited in the switchable photo-oxidation of 1,5-dihydroxynaphthalene with the framework as heterogeneous catalyst. Furthermore, the same light-gated energy transfer between DTE and porphyrin was used in a multivariate UiO-66 nanoparticles, bearing both porphyrin and DTE linkers in light-controlled photodynamic therapy.144
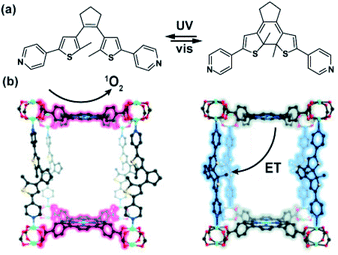 |
| | Fig. 22 (a) Structure of the DTE pillars and tetrakis(carboxyphenyl) porphyrin linker. (b) Model of the 3D structure of Zn-paddlewheel porphyrin DTE-pillared MOF. (Note that the porphyrin linker coordinates Zn cation during the solvothermal synthesis). | |
Electronic and proton conductivity
Azobenzenes.
Heinke and co-workers demonstrated photo-modulation of proton conductivity in pillared Cu(II)-paddlewheel SURMOFs bearing ortho-fluoro-azobenzene functionalized linkers (Fig. 23a).145 The light-responsive SURMOFs were infused with either butanediol or 1,2,3-triazole and showed reversible switching of proton conductivity upon isomerization of appended azobenzenes with light (Fig. 23b and c). SURMOFs with either of the guest molecules showed lower proton conductivity for Z-azobenzene pillars and higher proton conductivity for E-isomer of the incorporated azobenzene. Quantum mechanical calculations supported with infrared spectroscopic data indicated that for both guest molecules the hydrogen bonding with Z-isomer of azobenzene are stronger than with E-isomer. Thus leading to a decrease in the mobility of guest molecules and the proton conductivity.
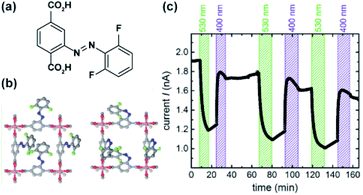 |
| | Fig. 23 (a) Structure of ortho-fluoro azobenzene linkers and (b) switchable SURMOF viewed in c direction. (c) Changes in the proton current conduction of butanediol in SURMOF at 1 V and 1 Hz upon alternating irradiation at 530 and 400 nm. Adapted with permission from ref. 145. Copyright 2018 Wiley-VHC. | |
Spiropyrans and dithienylethenes.
Shustova and co-workers investigated the impact of light-induced changes in the electronic structure of DTE and spiropyran photoswitches integrated in MOF scaffolds on electronic conductivity of bulk materials.146 For both photoswitches significant changes in the materials conductivity, amounting up to 3-fold difference between irradiated and pristine materials were observed upon exposure to UV-light (Fig. 24a and b). A combination of spectroscopic techniques and theoretical calculations allowed for correlation of these changes with the molecular state of the incorporated photoswitches. In the case of spiropyran linkers, increase in the material conductivity upon UV-irradiation was expected to originate from a decrease in the spatial separation between the adjacent linkers upon isomerization to elongated merocyanine form. In contrast, for DTE-appended MOFs the increased conductivity attributed to the decrease in the bandgap of the material. Further studies by Heinke and co-workers showed similar impact of the photoswitching on electronic147 and proton148 conductivity in spiropyran appended SURMOFs. A similar photomodulation of electronic conductivity was achieved in a hexagonal 2D-COF featuring DTE photoswitches incorporated in the backbone of the framework.149
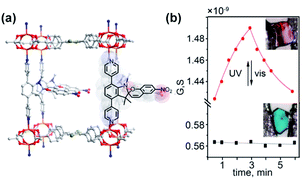 |
| | Fig. 24 (a) Single crystal X-ray structure with simulated spiropyran pillars showing isomerization to merocyanine form. (b) Light-induced changes in the conductance of the single crystal of the MOF bearing spiropyrans pillars (red line) and control non-responsive MOF (black line). Adapted with permission from ref. 146. Copyright 2019 American Chemical Society. | |
Sensors
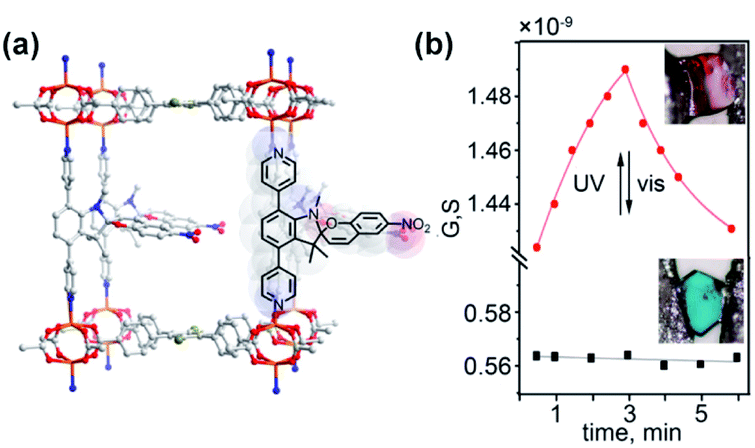
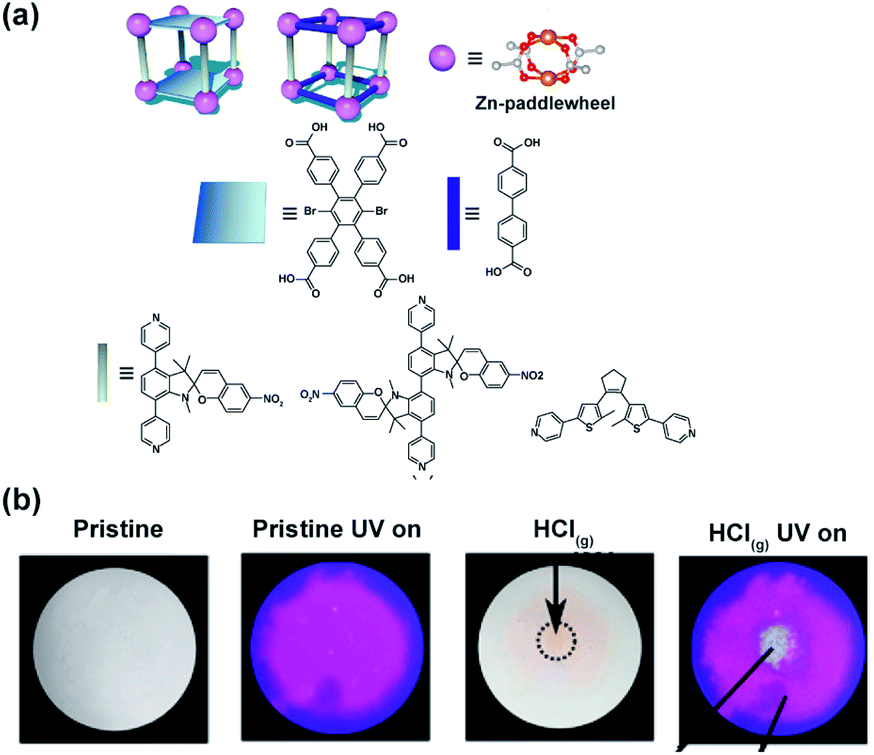
The influence of flexibility and sterics of the framework on the kinetics of photochemical cycloreversion reactions was assessed by Shustova and co-workers.150 Various spiropyrans and dithienylethene photoswitches were synthesized and incorporated in the MOFs with varying flexibility and pore aperture as pendants or backbone of the linkers (Fig. 25a). The rates of the photochemical cycloreversion reactions for these photoswitches were studied and compared in molecular solids, solutions and as linkers in MOFs which allowed to establish the link between the rate of isomerization and flexibility, sterics and structure of the surrounding environment. Notably, the spiropyran derived pillars, showed an incomplete cycloreversion in solids but nearly the same rate of the light-induced isomerization as in solution when embedded in the MOFs. On the other hand, bispyridyl DTEs derivative showed the highest rate of ring-opening isomerization in the molecular solid, intermediate rates in solution and lower rate when incorporated in rigid pillared MOF. In contrast, when incorporated in the more flexible MOF, the DTE bispyridyl pillars displayed a comparable rate of photochemical reaction to that observed in solution. Interestingly, for the dicarboxylic acid DTE derivative the same rate constants were observed for all investigated phases. These differences in rates of isomerization of the photoswitches embedded in distinct environments were further used as a marker for changes in material integrity. The intact spiropyran-appended MOF showed purple emission (λmax = 680 nm, λext = 350 nm) along with fast and reversible photoisomerization of spiropyrans pendants. However, localized exposure to HCl vapor lead to the local degradation of the framework and leaching of the spiropyrans pillars, which in turn induced irreversible changes in the framework emission and colour thus allowing for visual detection of the damaged region (Fig. 25b).
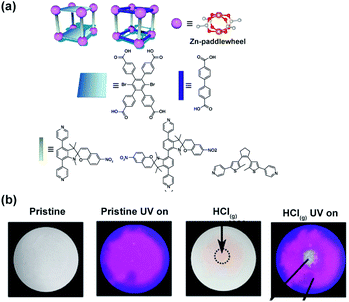 |
| | Fig. 25 (a) Structure of the linkers, nodes and schematic depiction of the frameworks structure used in the study. (b) Optical micrographs of the MOF bearing spiropyran pillars pristine material (left panel) and material subjected to HCl vapour at the point indicated by the black arrow (right panel). Adapted with permission from ref. 150. Copyright 2018 American Chemical Society. | |
Klajn and co-workers utilized the versatility of spiropyran switches for the construction of multi-purpose porous aromatic frameworks.71 Materials were based on pore-forming tetrahedral tetraphenylsilane and monotopic or ditopic spiropyrans, which were connected via Suzuki cross-coupling to form amorphous, three-dimensional porous polymers (Fig. 26a). Incorporation of the monotopic spiropyran derivative in the aromatic framework, preserved its characteristic photochromism and led to an exceptional fatigue resistance for over 100 isomerization cycles (without the presence of oxygen), which was attributed to the isolation of the switchable pendants thus excluding the bimolecular photodegradation pathway (Fig. 26b).70 In contrast, an aromatic network constructed with a ditopic spiropyran derivative showed reversible light induced spiropyran isomerization only when soaked in solvents, while pendants in the evacuated material were permanently frozen in merocyanine form. Finally, the multi-stimuli responsive properties of the porous material harbouring ditopic spiropyrans were further used for the detection of pH changes as the framework readily responded with colour change to the protonation and subsequent deprotonation of pendants (Fig. 26c) as well as switchable capture and release of Cu(II) cations from acetonitrile.
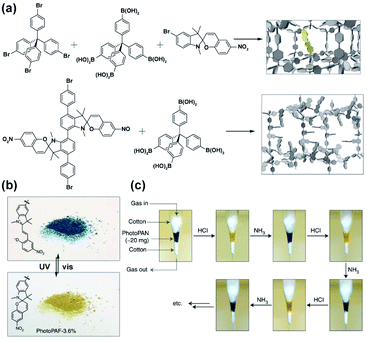 |
| | Fig. 26 (a) Structure of building blocks and monotopic and ditopic spiropyran pendants used to synthesize responsive porous organic polymers. (b) Light-induced changes in colour of the porous material bearing monotopic spiropyran photoswitch. (c) Changes in the colour of the material bearing ditopic spiropyran upon alternating protonation/deprotonation cycles of merocyanine pendants. Adapted by permission from Springer Nature from ref. 71, Copyright 2014. | |
Macroscopic actuators
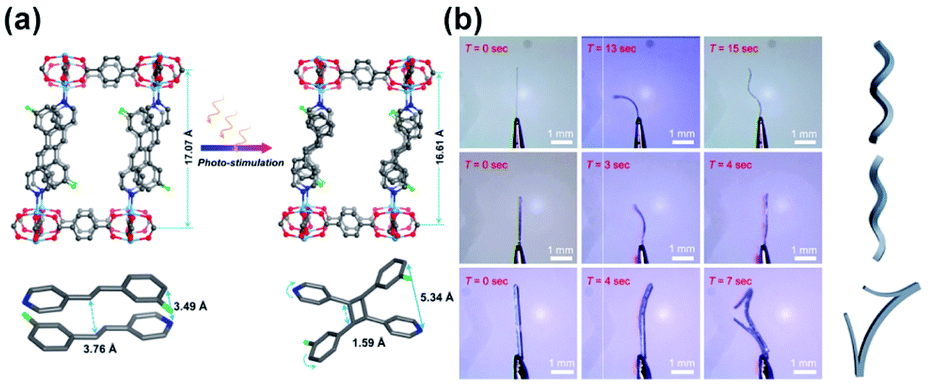
Typically, crystals of photoresponsive MOFs do not show macroscopic deformation upon isomerization of switchable linkers, mainly due to the flexibility intentionally engineered in the crystal to alleviate a mechanical stress associated with the structural changes of the responsive-linker. One exception features a four-fold interpenetrated Zn-paddlewheel framework with pyridyl-functionalized stilbene attached to the metal cluster.86 Irradiation of such crystals at 365 nm led to [2 + 2] photocycloaddition of the neighbouring stilbene moieties accompanied by the contraction of the elementary cell volume by 6.1% and shear distortion of the two-dimensional Zn-paddlewheel layers (Fig. 27a). As a consequence of this distortion, upon 365 nm light irradiation the thin crystal of the MOF bent towards the light source, with a relatively high rate of 80 s−1 and subsequently twisted in right-handed helix regardless of the direction of the irradiation (Fig. 27b, top and middle panels). Conversely, thicker crystals bent away from the light source, and upon prolonged irradiation the built-up strain was released in abrupt break-up of the crystal (Fig. 27b, bottom panel). To improve the mechanical properties of such actuators, MOF crystals were combined with a poly-vinyl alcohol membrane, which could also be actuated with light, albeit with a lower rate of ca. 10 s−1.
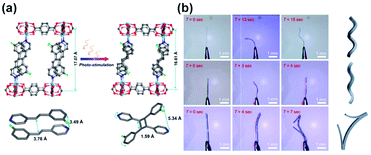 |
| | Fig. 27 (a) UV-light induced changes in the SC X-ray structure of the photoresponsive MOF bearing stilbene-pyridyl pendants (top panel) along with stacking of the pendants and structure of the stilbene dimer (bottom panel). (b) Optical micrographs of the photochemical crystals deformation, twisting and break-up of the crystals with different sizes: thin crystals (top and middle panels), thick crystal (bottom panel). Samples were irradiated from the left side from the perspective of the micrographs. Adapted with permission from ref. 86. Copyright 2019 Wiley-VHC. | |
Conclusions
The pioneering studies on porous solids, highlighted in this review, established the opportunities arising from incorporation of light-responsive switches in these scaffolds as a viable method to harness their collective motion towards responsive, dynamic materials. Starting from non-responsive solids, where light-induced structural changes of these molecules were hampered by the rigidity of the solid environment, systematic progress resulted in development of fundamental design principles allowing for integrating the responsive molecules with the solid support without impairment of their dynamic function and reforming the materials robustness. It was illustrated by the development of various, proof of concept materials with tuneable properties, including gas adsorption capacity, electronic structure or pore aperture. However, despite significant progress in this field it is still in its infancy, and there are still a number of key challenges that have to be addressed in the future to support the development of these materials and exploit their full potential.
Mode of linker incorporation
The photoswitchable units can be incorporated in the material scaffold as either pendants or backbone of the linker. It seems, that the first method of incorporation is more general and reliable to ensure preservation of the light-responsive function, however, in the latter case more pronounced changes in the material structure can be expected. The few examples presented in the recent literature of fully operational MOFs and COFs bearing photoswitches incorporated in the linker backbone, provide insight into the requirements that have to be fulfilled in order to facilitate photoswitching. It seems that in this scenario, the most important prerequisite of the switching in the solid state is the flexibility of the framework. Therefore, research in this direction should focus on incorporation of these molecules in inherently flexible MOFs or frameworks capable of promoting sheer displacement distortion.
Bulk photoisomerization
In general, a characteristic feature of most molecular photoswitches is their high molar attenuation coefficient > 104 M−1 cm−1. Therefore, owing to the relatively high density and light scattering on the periodic structure, light penetration depth in these types of solid materials is low and consequently the effect is limited to the surface of the material. Although few examples of bulk photochemical transformations are present in the literature,122,123,127 the phenomenon is not general and limited to systems displaying a large spectral separation between distinct forms. This problem can be addressed with several strategies: (i) a first possibility to circumvent these problems is the design of the surface-mounted metal organic frameworks (SURMOFs)151 which can be grown epitaxial, in layer-by-layer fashion, which allows to control the thin film thickness and minimize the absorption pathway (ii) another option is to employ core–shell135 structures, where a small crystal of non-responsive MOF (core) is covered by a thin shell of photoswitchable MOF in such a way that the molecular design of the hybrid material is fine-tuned to display the desired effect; (iii) applying a multivariate MOFs strategy,152,153 in other words dilution of the active linker to the point that the concentration of the photoswitch will be low but sufficient to induce large changes in properties of the material upon switching; (iv) or design of multifunctional materials featuring distinct incorporated photoswitchable linkers that can be addressed with orthogonal wavelengths; (v) use of the molecular photoswitches with very large spectral separation between two distinct forms e.g. DASA154 switches.
Local heating
Local heating associated with vibrational cooling of the excited state of a given photoswitch is inevitable in any system with low thermal conductivity, in particular solid state materials. Even where there is no obvious electronic absorption, vibrational absorption (overtones) in the visible region and Raman effect can result in rapid heating with NIR light. Therefore, control experiments always need to be performed in order to elucidate the origin of the light-induced changes in the properties of the photoactive solid materials. On the other hand, local heating effects can be advantageous, for example by exciting selectively certain phonon modes of the crystalline material and thus leading to unique changes in the materials properties.155
Despite the significant challenges in this field, a careful and systematic approach to the design and characterization of light-responsive porous materials will stimulate further developments of these fascinating materials. The ground-breaking studies that have been conducted thus far were focused on understanding of the fundamental aspects of the stimuli-responsive behaviour in the confined environment and its implications on material properties. In the future, development of the frameworks featuring two or more orthogonally addressable photoswitches will lead to the development of the materials in which multiple functionalities of the materials can be controlled independently and on-demand. Furthermore, the close spatial proximity between components of the frameworks may facilitate the transmission, coupling and synchronization of the light-induced motion between incorporated photoswitches and other functionalities and thereby amplify their motion and create new properties and functions beyond these achievable by isolated molecules. On the other hand, development of the strategies that would allow addressing individual photoswitches in the material would open new opportunities for the fabrication of high-density data storage devices. It is evident that the photoresponsive porous materials discussed here offer ample space, solid ground and bright prospects for future discoveries.156
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported financially by the Netherlands Organization for Scientific Research (NWO-CW), the European Research Council (ERC, advanced grant no. 694345 to B.L.F.), the Ministry of Education, Culture and Science (Gravitation Program no. 024.001.035).
Notes and references
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377–1400 CrossRef CAS.
-
M. Schliwa, Molecular Motors, Wiley-VCH, Weinheim, 2003 Search PubMed.
- G. Yusupova, L. Jenner, B. Rees, D. Moras and M. Yusupov, Nature, 2006, 444, 391–394 CrossRef CAS.
- W. A. Fenton and A. L. Horwich, Q. Rev. Biophys., 2003, 36, 229–256 CrossRef CAS.
- R. D. Astumian, Biophys. J., 2010, 98, 2401–2409 CrossRef CAS.
- R. D. Vale, Cell, 2003, 112, 467–480 CrossRef CAS.
- M. J. Pallen and N. J. Matzke, Nat. Rev. Microbiol., 2006, 4, 784–790 CrossRef CAS.
- V. Ropars, Z. Yang, T. Isabet, F. Blanc, K. Zhou, T. Lin, X. Liu, P. Hissier, F. Samazan, B. Amigues, E. D. Yang, H. Park, O. Pylypenko, M. Cecchini, C. V. Sindelar, H. L. Sweeney and A. Houdusse, Nat. Commun., 2016, 7, 12456 CrossRef CAS.
-
V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines: A Journey into the Nanoworld, Wiley-VCH, Weinheim, 2003 Search PubMed.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS.
- J. Wang and B. L. Feringa, Science, 2011, 331, 1429–1432 CrossRef CAS.
- T. Muraoka, K. Kinbara and T. Aida, Nature, 2006, 440, 512–515 CrossRef CAS.
- R. D. Astumian, Proc. Natl. Acad. Sci. U. S. A., 2006, 46, 10771–10776 Search PubMed.
- R. D. Astumian, Chem. Sci., 2017, 8, 840–845 RSC.
- T. J. Huang, B. Brough, C.-M. Ho, Y. Liu, A. H. Flood, P. A. Bonvallet, H.-R. Tseng, J. F. Stoddart, M. Baller and S. Magonov, Appl. Phys. Lett., 2004, 85, 5391–5393 CrossRef CAS.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef.
- K. Ichimura, Science, 2000, 288, 1624–1626 CrossRef CAS.
- S. Iamsaard, S. J. Aßhoff, B. Matt, T. Kudernac, J. J. L. M. Cornelissen, S. P. Fletcher and N. Katsonis, Nat. Chem., 2014, 6, 229–235 CrossRef CAS.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS.
- C. L. Van Oosten, C. W. M. Bastiaansen and D. J. Broer, Nat. Mater., 2009, 8, 677–682 CrossRef CAS.
- A. H. Gelebart, D. Jan Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Nature, 2017, 546, 632–636 CrossRef CAS.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef CAS.
- T. Orlova, F. Lancia, C. Loussert, S. Iamsaard, N. Katsonis and E. Brasselet, Nat. Nanotechnol., 2018, 13, 304–308 CrossRef CAS.
- J. Chen, F. K. C. Leung, M. C. A. Stuart, T. Kajitani, T. Fukushima, E. van der Giessen and B. L. Feringa, Nat. Chem., 2018, 10, 132–138 CrossRef CAS.
- Q. Li, G. Fuks, E. Moulin, M. Maaloum, M. Rawiso, I. Kulic, J. T. Foy and N. Giuseppone, Nat. Nanotechnol., 2015, 10, 161–165 CrossRef CAS.
- J. T. Foy, Q. Li, A. Goujon, J. R. Colard-Itté, G. Fuks, E. Moulin, O. Schiffmann, D. Dattler, D. P. Funeriu and N. Giuseppone, Nat. Nanotechnol., 2017, 12, 540–545 CrossRef CAS.
- H. Deng, M. A. Olson, J. F. Stoddart and O. M. Yaghi, Nat. Chem., 2010, 2, 439–443 CrossRef CAS.
- A. H. Flood, A. J. Peters, S. A. Vignon, D. W. Steuerman, H.-R. Tseng, S. Kang, J. R. Heath and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 6558–6564 CrossRef CAS.
- G. London, K. Y. Chen, G. T. Carroll and B. L. Feringa, Chem.–Eur. J., 2013, 19, 10690–10697 CrossRef CAS.
- G. T. Carroll, G. London, T. F. Landaluce, P. Rudolf and B. L. Feringa, ACS Nano, 2011, 5, 622–630 CrossRef CAS.
- K. Y. Chen, O. Ivashenko, G. T. Carroll, J. Robertus, J. C. M. Kistemaker, G. London, W. R. Browne, P. Rudolf and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 3219–3224 CrossRef CAS.
- Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS.
- T. M. Reineke, M. Eddaoudi, D. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 4843–4844 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS.
- H. Koshima, N. Ojima and H. Uchimoto, J. Am. Chem. Soc., 2009, 131, 6890–6891 CrossRef CAS.
- H. Koshima and N. Ojima, Dyes Pigm., 2012, 92, 798–801 CrossRef CAS.
- M. O'Donnell, Nature, 1968, 218, 460–461 CrossRef.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281–1376 CrossRef CAS.
- S. D. Karlen, H. Reyes, R. E. Taylor, S. I. Khan, M. F. Hawthorne and M. A. Garcia-Garibay, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14973–14977 CrossRef CAS.
- B. Rodríguez-Molina, S. Pérez-Estrada and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2013, 135, 10388–10395 CrossRef.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef.
- C. S. Vogelsberg, F. J. Uribe-Romo, A. S. Lipton, S. Yang, K. N. Houk, S. Brown and M. A. Garcia-Garibay, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13613–13618 CrossRef CAS.
- A. Comotti, S. Bracco, T. Ben, S. Qiu and P. Sozzani, Angew. Chem., Int. Ed., 2014, 53, 1043–1047 CrossRef CAS.
- S. Bracco, F. Castiglioni, A. Comotti, S. Galli, M. Negroni, A. Maspero and P. Sozzani, Chem.–Eur. J., 2017, 23, 11210–11215 CrossRef CAS.
- S. L. Gould, D. Tranchemontagne, O. M. Yaghi and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2008, 130, 3246–3247 CrossRef CAS.
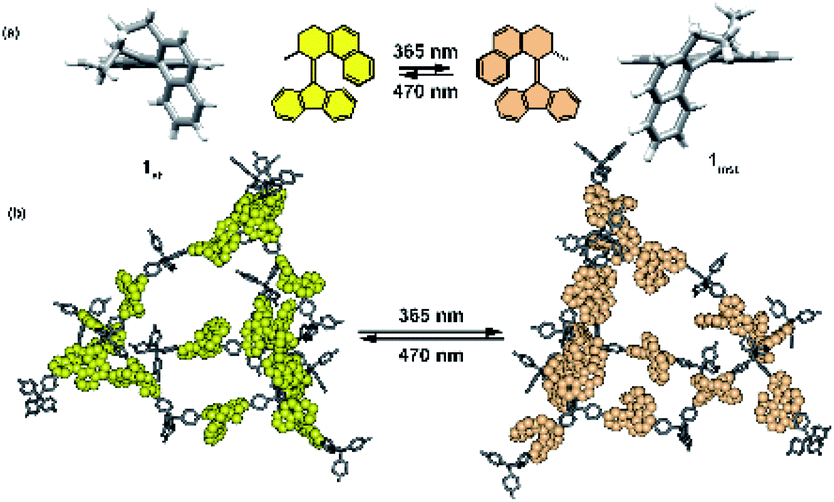
- J. Perego, S. Bracco, M. Negroni, C. X. Bezuidenhout, G. Prando, P. Carretta, A. Comotti and P. Sozzani, Nat. Chem., 2020, 12, 845–851 CrossRef CAS.
- K. Zhu, V. N. Vukotic, C. A. Okeefe, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2014, 136, 7403–7409 CrossRef CAS.
- K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514–519 CrossRef CAS.
- V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS.
- V. N. Vukotic, C. A. O. Keefe, K. Zhu, K. J. Harris, C. To, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2015, 137, 9643–9651 CrossRef CAS.
- P. Martinez-Bulit, C. A. O'Keefe, K. Zhu, R. W. Schurko and S. J. Loeb, Cryst. Growth Des., 2019, 19, 5679–5685 CrossRef CAS.
- W. Danowski, T. van Leeuwen, S. Abdolahzadeh, D. Roke, W. R. Browne, S. J. Wezenberg and B. L. Feringa, Nat. Nanotechnol., 2019, 14, 488–494 CrossRef CAS.
- W. Danowski, F. Castiglioni, A. S. Sardjan, S. Krause, L. Pfeifer, D. Roke, A. Comotti, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2020, 142, 9048–9056 CrossRef CAS.
- T. van Leeuwen, A. S. Lubbe, P. Štacko, S. J. Wezenberg and B. L. Feringa, Nat. Rev. Chem., 2017, 1, 96 CrossRef CAS.
- F. X. Coudert, Chem. Mater., 2015, 27, 1905–1916 CrossRef CAS.
- S. Castellanos, F. Kapteijn and J. Gascon, CrystEngComm, 2016, 18, 4006–4012 RSC.
- B. Gui, Y. Meng, Y. Xie, K. Du, A. C.-H. Sue and C. Wang, Macromol. Rapid Commun., 2018, 39, 1700388 CrossRef.
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2019, 32, 1905227 CrossRef.
- A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Chem. Rev., 2020, 120, 8790–8813 CrossRef CAS.
- F. Bigdeli, C. T. Lollar, A. Morsali and H. Zhou, Angew. Chem., Int. Ed., 2020, 59, 4652–4669 CrossRef CAS.
- P. Martinez-Bulit, A. J. Stirk and S. J. Loeb, Trends Chem., 2019, 1, 588–600 CrossRef CAS.
-
B. L. Feringa and W. R. Browne, Molecular Switches, Wiley-VCH, Weinheim, 2011 Search PubMed.
- J. Zhang, L. Wang, N. Li, J. Liu, W. Zhang, Z. Zhang, N. Zhou and X. Zhu, CrystEngComm, 2014, 16, 6547–6551 RSC.
- X. Guo, T. Mao, Z. Wang, P. Cheng, Y. Chen, S. Ma and Z. Zhang, ACS Cent. Sci., 2020, 6, 787–794 CrossRef CAS.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- A. K. Chibisov and H. Görner, J. Phys. Chem. A, 1997, 101, 4305–4312 CrossRef CAS.
- M. Sakuragi, K. Aoki, T. Tamaki and K. Ichimura, Bull. Chem. Soc. Jpn., 1990, 63, 74–79 CrossRef CAS.
- P. K. Kundu, G. L. Olsen, V. Kiss and R. Klajn, Nat. Commun., 2014, 5, 3588 CrossRef.
- S. Takami, S. Kobatake, T. Kawai and M. Irie, Chem. Lett., 2003, 32, 892–893 CrossRef CAS.
- M. Hanazawa, R. Sumiya, Y. Horikawa and M. Irie, J. Chem. Soc., Chem. Commun., 1992, 206–207 RSC.
- A. Yukako, M. Akinori, K. Takao, G. Alexander, G. Dominique, Y. Satoshi, I. Masahiro and N. Shinichiro, J. Am. Chem. Soc., 2004, 126, 12112–12120 CrossRef.
- T. Kudernac, T. Kobayashi, A. Uyama, K. Uchida, S. Nakamura and B. L. Feringa, J. Phys. Chem. A, 2013, 117, 8222–8229 CrossRef CAS.
- D. Dulić, T. Kudernac, A. Pužys, B. L. Feringa and B. J. van Wees, Adv. Mater., 2007, 19, 2898–2902 CrossRef.
- I. M. Walton, J. M. Cox, J. A. Coppin, C. M. Linderman, D. G. Patel and J. B. Benedict, Chem. Commun., 2013, 49, 8012–8014 RSC.
- I. M. Walton, J. M. Cox, C. A. Benson, D. G. Patel, Y. S. Chen and J. B. Benedict, New J. Chem., 2016, 40, 101–106 RSC.
- I. M. Walton, J. M. Cox, T. B. Mitchell, N. P. Bizier and J. B. Benedict, CrystEngComm, 2016, 18, 7972–7977 RSC.
- A. Galvan-Gonzalez, K. D. Belfield, G. I. Stegeman, M. Canva, S. R. Marder, K. Staub, G. Levina and R. J. Twieg, J. Appl. Phys., 2003, 94, 756–763 CrossRef CAS.
- M. Schraub, H. Gray and N. Hampp, Macromolecules, 2011, 44, 8755–8762 CrossRef CAS.
- M. Pattabiraman, J. Sivaguru and V. Ramamurthy, Isr. J. Chem., 2018, 58, 264–275 CrossRef CAS.
- I. E. Claassens, V. I. Nikolayenko, D. A. Haynes and L. J. Barbour, Angew. Chem., Int. Ed., 2018, 130, 15789–15792 CrossRef.
- I. E. Claassens, L. J. Barbour and D. A. Haynes, J. Am. Chem. Soc., 2019, 141, 11425–11429 CrossRef CAS.
- I. Park, E. Lee, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2019, 58, 14860–14864 CrossRef CAS.
- Y.-X. Shi, W.-H. Zhang, B. F. Abrahams, P. Braunstein and J.-P. Lang, Angew. Chem., Int. Ed., 2019, 58, 9453–9458 CrossRef CAS.
- Y. Shi, H. Chen, W. Zhang, G. S. Day, J. Lang and H. Zhou, Chem.–Eur. J., 2019, 25, 8543–8549 CrossRef CAS.
- T. Jadhav, Y. Fang, C.-H. Liu, A. Dadvand, E. Hamzehpoor, W. Patterson, A. Jonderian, R. Stein and D. F. Perepichka, J. Am. Chem. Soc., 2020, 142, 8862–8870 CrossRef CAS.
- B. Feringa and H. Wynberg, J. Am. Chem. Soc., 1977, 99, 602–603 CrossRef CAS.
- M. Quick, F. Berndt, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, C. Knie, R. Mahrwald, D. Lenoir, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 1389–1402 CrossRef CAS.
- S. J. Wezenberg and B. L. Feringa, Org. Lett., 2017, 19, 324–327 CrossRef CAS.
- N. Koumura, R. W. J. Zijistra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS.
- N. Koumura, E. M. Geertsema, M. B. van Gelder, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 5037–5051 CrossRef CAS.
- J. C. M. Kistemaker, P. Štacko, J. Visser and B. L. Feringa, Nat. Chem., 2015, 7, 890–896 CrossRef CAS.
- Q. Z. Yang, Z. Huang, T. J. Kucharski, D. Khvostichenko, J. Chen and R. Boulatov, Nat. Nanotechnol., 2009, 4, 302–306 CrossRef CAS.
- D. Villarón and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef.
- J. Bauer, L. Hou, J. C. M. Kistemaker and B. L. Feringa, J. Org. Chem., 2014, 79, 4446–4455 CrossRef CAS.
- A. S. Bhown and B. C. Freeman, Environ. Sci. Technol., 2011, 45, 8624–8632 CrossRef CAS.
- P. H. M. Feron, Int. J. Greenhouse Gas Control, 2010, 4, 152–160 CrossRef CAS.
- D. Jiang, L. L. Keenan, A. D. Burrows and K. J. Edler, Chem. Commun., 2012, 48, 12053–12055 RSC.
- A. Modrow, D. Zargarani, R. Herges and N. Stock, Dalton Trans., 2012, 41, 8690–8696 RSC.
- A. Modrow, D. Zargarani, R. Herges and N. Stock, Dalton Trans., 2011, 40, 4217–4222 RSC.
- S. Bernt, M. Feyand, A. Modrow, J. Wack, J. Senker and N. Stock, Eur. J. Inorg. Chem., 2011, 5378–5383 CrossRef CAS.
- S. Castellanos, A. Goulet-Hanssens, F. Zhao, A. Dikhtiarenko, A. Pustovarenko, S. Hecht, J. Gascon, F. Kapteijn and D. Bléger, Chem.–Eur. J., 2016, 22, 746–752 CrossRef CAS.
- J. Park, D. Yuan, K. T. Pham, J. R. Li, A. Yakovenko and H. C. Zhou, J. Am. Chem. Soc., 2012, 134, 99–102 CrossRef CAS.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695–3698 CrossRef CAS.
- S. Castellanos, A. Goulet-Hanssens, F. Zhao, A. Dikhtiarenko, A. Pustovarenko, S. Hecht, J. Gascon, F. Kapteijn and D. Bléger, Chem.–Eur. J., 2016, 22, 746–752 CrossRef CAS.
- N. Prasetya, B. C. Donose and B. P. Ladewig, J. Mater. Chem. A, 2018, 6, 16390–16402 RSC.
- N. Prasetya and B. P. Ladewig, ACS Appl. Mater. Interfaces, 2018, 10, 34291–34301 CrossRef CAS.
- Y. Jiang, P. Tan, S.-C. Qi, X.-Q. Liu, J.-H. Yan, F. Fan and L.-B. Sun, Angew. Chem., Int. Ed., 2019, 58, 6600–6604 CrossRef CAS.
- Y. Zhu and W. Zhang, Chem. Sci., 2014, 5, 4957–4961 RSC.
- H. Huang, H. Sato and T. Aida, J. Am. Chem. Soc., 2017, 139, 8784–8787 CrossRef CAS.
- L. J. Barbour, Chem. Commun., 2006, 1163–1168 RSC.
- J. R. Holst, A. Trewin and A. I. Cooper, Nat. Chem., 2010, 2, 915–920 CrossRef CAS.
- M. Baroncini, S. d'Agostino, G. Bergamini, P. Ceroni, A. Comotti, P. Sozzani, I. Bassanetti, F. Grepioni, T. M. Hernandez, S. Silvi, M. Venturi and A. Credi, Nat. Chem., 2015, 7, 634–640 CrossRef CAS.
- K. Uchida, M. Walko, J. J. D. de Jong, S. Sukata, S. Kobatake, A. Meetsma, J. van Esch and B. L. Feringa, Org. Biomol. Chem., 2006, 4, 1002 RSC.
- K. Uchida, S. Sukata, Y. Matsuzawa, M. Akazawa, J. J. D. de Jong, N. Katsonis, Y. Kojima, S. Nakamura, J. Areephong, A. Meetsma and B. L. Feringa, Chem. Commun., 2008, 326–328 RSC.
- F. Luo, C. Bin Fan, M. B. Luo, X. L. Wu, Y. Zhu, S. Z. Pu, W. Y. Xu and G. C. Guo, Angew. Chem., Int. Ed., 2014, 53, 9298–9301 CrossRef CAS.
- C. Bin Fan, L. Le Gong, L. Huang, F. Luo, R. Krishna, X. F. Yi, A. M. Zheng, L. Zhang, S. Z. Pu, X. F. Feng, M. B. Luo and G. C. Guo, Angew. Chem., Int. Ed., 2017, 56, 7900–7906 CrossRef.
- V. I. Nikolayenko, S. A. Herbert and L. J. Barbour, Chem. Commun., 2017, 53, 11142–11145 RSC.
- Y. Zheng, H. Sato, P. Wu, H. J. Jeon, R. Matsuda and S. Kitagawa, Nat. Commun., 2017, 8, 100 CrossRef.
- N. Huang, X. Ding, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 8704–8707 CrossRef CAS.
- A. Acharjya, P. Pachfule, J. Roeser, F. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS.
- T. Jadhav, Y. Fang, C.-H. Liu, A. Dadvand, E. Hamzehpoor, W. Patterson, A. Jonderian, R. S. Stein and D. F. Perepichka, J. Am. Chem. Soc., 2020, 142, 8862–8870 CrossRef CAS.
- F. Castiglioni, W. Danowski, J. Perego, F. K. C. Leung, P. Sozzani, S. Bracco, S. J. Wezenberg, A. Comotti and B. L. Feringa, Nat. Chem., 2020, 12, 595–602 CrossRef CAS.
- Z. Wang, A. Knebel, S. Grosjean, D. Wagner, S. Bräse, C. Wöll, J. Caro and L. Heinke, Nat. Commun., 2016, 7, 13872 CrossRef CAS.
- K. Müller, A. Knebel, F. Zhao, D. Bléger, J. Caro and L. Heinke, Chem.–Eur. J., 2017, 23, 5434–5438 CrossRef.
- J. W. Brown, B. L. Henderson, M. D. Kiesz, A. C. Whalley, W. Morris, S. Grunder, H. Deng, H. Furukawa, J. I. Zink, J. F. Stoddart and O. M. Yaghi, Chem. Sci., 2013, 4, 2858–2864 RSC.
- G. Das, T. Prakasam, M. A. Addicoat, S. K. Sharma, F. Ravaux, R. Mathew, M. Baias, R. Jagannathan, M. A. Olson and A. Trabolsi, J. Am. Chem. Soc., 2019, 141, 19078–19087 CrossRef CAS.
- L. Heinke, M. Cakici, M. Dommaschk, S. Grosjean, R. Herges, S. Bräse and C. Wöll, ACS Nano, 2014, 8, 1463–1467 CrossRef CAS.
- Z. Wang, L. Heinke, J. Jelic, M. Cakici, M. Dommaschk, R. J. Maurer, H. Oberhofer, S. Grosjean, R. Herges, S. Bräse, K. Reuter and C. Wöll, Phys. Chem. Chem. Phys., 2015, 17, 14582–14587 RSC.
- X. Yu, Z. Wang, M. Buchholz, N. Füllgrabe, S. Grosjean, F. Bebensee, S. Bräse, C. Wöll and L. Heinke, Phys. Chem. Chem. Phys., 2015, 17, 22721–22725 RSC.
- Z. Wang, S. Grosjean, S. Bräse and L. Heinke, ChemPhysChem, 2015, 16, 3779–3783 CrossRef CAS.
- Z. Wang, K. Müller, M. Valášek, S. Grosjean, S. Bräse, C. Wöll, M. Mayor and L. Heinke, J. Phys. Chem. C, 2018, 122, 19044–19050 CrossRef CAS.
- D. Mutruc, A. Goulet-Hanssens, S. Fairman, S. Wahl, A. Zimathies, C. Knie and S. Hecht, Angew. Chem., Int. Ed., 2019, 58, 12862–12867 CrossRef CAS.
- X. Meng, B. Gui, D. Yuan, M. Zeller and C. Wang, Sci. Adv., 2016, 2, e1600480 CrossRef.
- H. Meng, C. Zhao, M. Nie, C. Wang and T. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 32607–32612 CrossRef CAS.
- A. B. Kanj, J. Bürck, S. Grosjean, S. Bräse and L. Heinke, Chem. Commun., 2019, 55, 8776–8779 RSC.
- B. J. Furlong and M. J. Katz, J. Am. Chem. Soc., 2017, 139, 13280–13283 CrossRef CAS.
- H.-J. Son, S. Jin, S. Patwardhan, S. J. Wezenberg, N. C. Jeong, M. So, C. E. Wilmer, A. A. Sarjeant, G. C. Schatz, R. Q. Snurr, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 862–869 CrossRef CAS.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS.
- D. E. Williams, J. A. Rietman, J. M. Maier, R. Tan, A. B. Greytak, M. D. Smith, J. A. Krause and N. B. Shustova, J. Am. Chem. Soc., 2014, 136, 11886–11889 CrossRef CAS.
- J. Park, D. Feng, S. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 430–435 CrossRef CAS.
- J. Park, Q. Jiang, D. Feng and H.-C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7188–7193 CrossRef CAS.
- K. Müller, J. Helfferich, F. Zhao, R. Verma, A. B. Kanj, V. Meded, D. Bléger, W. Wenzel and L. Heinke, Adv. Mater., 2018, 30, 1706551 CrossRef.
- E. A. Dolgopolova, V. A. Galitskiy, C. R. Martin, H. N. Gregory, B. J. Yarbrough, A. M. Rice, A. A. Berseneva, O. A. Ejegbavwo, K. S. Stephenson, P. Kittikhunnatham, S. G. Karakalos, M. D. Smith, A. B. Greytak, S. Garashchuk and N. B. Shustova, J. Am. Chem. Soc., 2019, 141, 5350–5358 CrossRef CAS.
- S. Garg, H. Schwartz, M. Kozlowska, A. B. Kanj, K. Müller, W. Wenzel, U. Ruschewitz and L. Heinke, Angew. Chem., Int. Ed., 2019, 58, 1193–1197 CrossRef CAS.
- A. B. Kanj, A. Chandresh, A. Gerwien, S. Grosjean, S. Bräse, Y. Wang, H. Dube and L. Heinke, Chem. Sci., 2020, 11, 1404–1410 RSC.
- F. Yu, W. Liu, B. Li, D. Tian, J. Zuo and Q. Zhang, Angew. Chem., Int. Ed., 2019, 58, 16101–16104 CrossRef CAS.
- D. I. Kolokolov, A. G. Stepanov, V. Guillerm, C. Serre, B. Frick, H. Jobic, D. E. Williams, C. R. Martin, E. A. Dolgopolova, A. Swifton, D. C. Godfrey, O. A. Ejegbavwo, P. J. Pellechia, M. D. Smith and N. B. Shustova, J. Am. Chem. Soc., 2018, 140, 7611–7622 CrossRef.
- L. Heinke and C. Wöll, Adv. Mater., 2019, 31, 1806324 CrossRef.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882–885 CrossRef CAS.
- M. M. Lerch, W. Szymański and B. L. Feringa, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC.
- H. Babaei and C. E. Wilmer, Phys. Rev. Lett., 2016, 116, 25902 CrossRef.
- During the submission process few interesting reports were published online: J. Liu, S. Wang, T. Huang, P. Manchanda, E. Abou-Hamad and S. P. Nunes, Sci. Adv., 2020, 6, eabb3188 CrossRef CAS; H. Sato, T. Matsui, Z. Chen, J. Pirillo, Y. Hijikata and T. Aida, J. Am. Chem. Soc., 2020, 142, 14069–14073 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Ben L.
Feringa
*b and
Ben L.
Feringa