
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn experimental design to extract more information from MS-based histone studies†
Laura
De Clerck
,
Sander
Willems
 ,
Simon
Daled
,
Bart
Van Puyvelde
,
Sigrid
Verhelst
,
Laura
Corveleyn
,
Dieter
Deforce
and
Maarten
Dhaenens
*
,
Simon
Daled
,
Bart
Van Puyvelde
,
Sigrid
Verhelst
,
Laura
Corveleyn
,
Dieter
Deforce
and
Maarten
Dhaenens
*
Laboratory of Pharmaceutical Biotechnology, Ghent University, Ottergemsesteenweg 460, 9000 Ghent, Belgium. E-mail: maarten.dhaenens@ugent.be
First published on 13th September 2021
Abstract
Histone-based chromatin organization paved the way for eukaryotic genome complexity. Because of their key role in information management, the histone posttranslational modifications (hPTM), which mediate their function, have evolved into an alphabet that has more letters than there are amino acids, together making up the “histone code”. The resulting combinatorial complexity is manifold higher than what is usually encountered in proteomics. Consequently, a considerably bigger part of the acquired MSMS spectra remains unannotated to date. Adapted search parameters can dig deeper into the dark histone ion space, but the lack of false discovery rate (FDR) control and the high level of ambiguity when searching combinatorial PTMs makes it very hard to assess whether the newly assigned ions are informative. Therefore, we propose an easily adoptable time-lapse enzymatic deacetylation (HDAC1) of a commercial histone extract as a quantify-first strategy that allows isolating ion populations of interest, when studying e.g. acetylation on histones, that currently remain in the dark. By adapting search parameters to study potential issues in sample preparation, data acquisition and data analysis, we stepwise managed to double the portion of annotated precursors of interest from 10.5% to 21.6%. This strategy is intended to make up for the lack of validated FDR control and has led to several adaptations of our current workflow that will reduce the portion of the dark histone ion space in the future. Finally, this strategy can be applied with any enzyme targeting a modification of interest.
Introduction
Histones are the Archaeal contribution to the Eukaryote lineage.1 In fact, histone-based chromatin organization has led to eukaryotic genome complexity; their epigenetic control mechanisms allow the differentiation of controlled gene-expression patterns in various cell types and thus the very existence of multicellular organisms. Consequently, histones are among the most complexly modified molecules in the biotic world, which makes these key regulators hard to analyze.Because of the essential biological function of histones, their primary structure remained unaltered across the eukaryotic lineage, while the histone post-translational modifications (hPTM) mediating their function have evolved into an alphabet that has more letters than there are amino acids. More specifically, there are chemical reactions involving the energy-rich donors like acyl-CoA (acylations), ATP (phosphorylation), and S-adenosylmethionine (methylation) that support the notion of an ancient mechanism to directly sense the energetic state of the eukaryotic cell by translating metabolic information into gene regulation via histones.2 Still, as the number of metabolites that can modify histones keeps on growing, with e.g. lactylation recently,3 an even more complex language is surfacing. For example, it was recently discovered that histones can also sense cellular metal content4 or become e.g. dopaminylated in the brain.5 And while studying so many hPTMs is challenging in its own right, trying to understand their interplay makes the study of the histone code a daunting task.6,7
Mass spectrometry (MS) is currently the most promising technique to read out the proverbial grammar that emerges from the histone code. There are three classes of MS-based approaches to study proteins: bottom-up,8–11 middle-down12–15 and top-down.15–19 Accurate identification and quantification of hPTMs can be done by means of all three approaches, of which the performance has been thoroughly validated using internal standards or by comparing the outcome between different strategies.20,21 However, benchmarking strategies usually assess the identifications, while MS-data contains much more information. In essence, MS produces a data matrix of ion coordinates like (precursor) m/z, intensity, retention time and possibly drift time. Therefore, an unprocessed data file can be considered as a digital transcript of the sample that requires extensive processing and data interpretation to recreate a picture of the underlying biology. Although annotation rates in proteomics keep on rising,22–25 the study of the histone code requires entirely different analytical strategies. That is because it targets an additional layer of complexity, i.e. a dynamic alphabet that sits on top of the amino acid sequence. Thus, it is not surprising that a considerable part of the digital transcript, potentially harboring a wealth of insights, remains unannotated to date.
Here, we propose a quantify-first strategy based on induced hPTM changes to isolate ion populations of interest that currently remain in the dark and to verify the incorrectness of newly found annotations. This approach is akin to the Navarro benchmark in conventional proteomics, wherein two different hybrid proteome sample are used to plot fold changes of precursors as a validation that (novel) annotation algorithms annotate peptides of the correct organism.26 However, no hybrid histone mixtures can be made that reflects necessary biological complexity. This is due to the evolutionary conservation of histone proteins and the difference between the study of hPTM changes and conventional proteomics. Therefore, this strategy is intended to make up for the lack of validated false discovery rate (FDR) control and the abundant ambiguity that afflicts histone annotation and can be applied with any enzyme targeting a hPTM of interest.27,28 To this end, we analyzed an easily adoptable time-lapse enzymatic deacetylation (HDAC1) of a commercial histone extract which retains the full combinatorial complexity of a biological sample while introducing predictable changes that involve true enzyme dynamics. Thereby, we probe the depth of the dark histone ion space to drive optimizations for our own bottom-up workflow and potentially catch a glimpse of undiscovered hPTMs.
Experimental
Histone deacetylase assay
Deacetylation was done by adding 10% human HDAC-1 enzyme (Sigma, SRP0100) to 15 μg bovine histone extracts (Sigma, 10223565001), in a log-scale time-lapse series (0 min–8 h) at 37 °C. Three negative controls were included in the experiment, two of which only had buffer added for 0 min and 8 h (HDAC-) and the third had inhibited enzyme added at time point 0 min (Fig. 1). For all samples, a minimum of three replicates were generated. The reactions were conducted in an HDAC assay buffer containing 10 mM Tris, pH 8, 150 mM NaCl, and 1 mM MgCl2 according to the time line depicted in Fig. 1. Enzyme activity was inhibited by adding 0.4 N HCl (pH 1).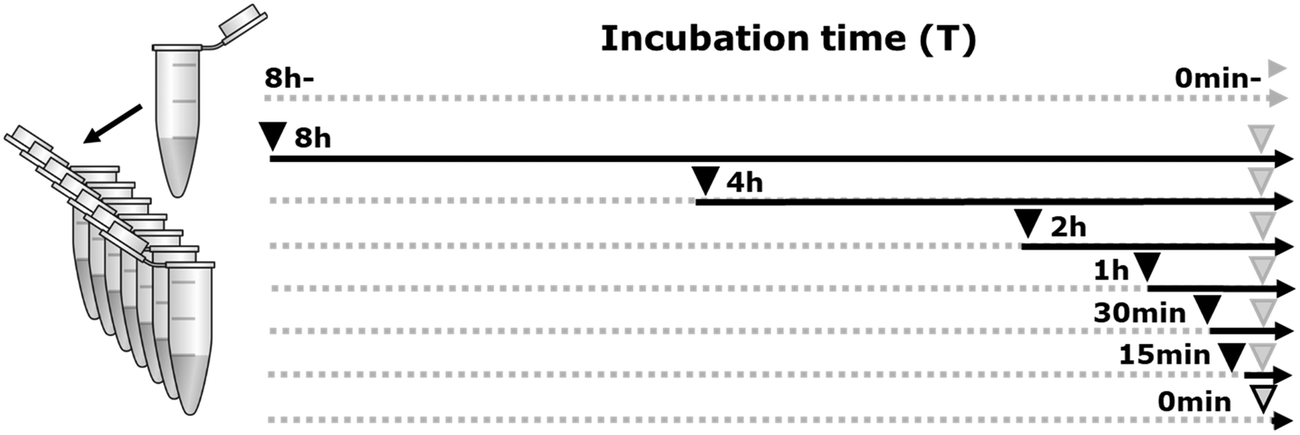 | ||
| Fig. 1 Enzymatic time-lapse changes of hPTMs as an experimental strategy to probe the depth of the dark histone ion space in untargeted MS strategies. Experimental design. Equimolar fractions of histone extracts were made from a single vial (left). All samples were incubated in the assay buffer (dotted grey line) at 37 °C. At several time points between T0 (0 min) and Tmax (8 h), HDAC1 enzyme was added (black arrowheads), except for two negative controls (−), where only buffer was added for 0 min and 8 h (top rows). Note that in this way, the time stipulation mirrors the time at which enzyme was actually added. At the final time point the enzymatic activity was stopped (grey arrowhead). As a third negative control, inhibited enzyme was added for time point 0 min (bottom row). The experimental design was performed in triplicate to enable statistical analysis before the isolation of potentially interesting ion populations. All further sample preparation steps (e.g. derivatization by propionylation) as well as the final MS sample list were randomized to minimize batch effects. | ||
Histone propionylation and digestion prior to mass spectrometry analysis
Chemical derivatization was optimized and performed as previously described.29,30 All histone samples were dissolved in 20 μL 1 M triethylammonium bicarbonate (TEAB) buffer, pH 8.5. Next, 20 μL of propionylation reagent (propionic anhydride: 2-propanol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 (v/v)) was added, for an incubation of 30 min at RT. This was followed by adding 20 μL milliQ water (Merck Millipore) for 30 min at 37 °C. Histones were then digested overnight at 37 °C using trypsin (at an enzyme/protein ratio of 1:20 (m/m)) in 500 mM TEAB, supplemented with CaCl2 and ACN to a final concentration of 1.0 mM and 5% respectively. Subsequently, the derivatization reaction was carried out again to cap peptide N-termini. Aspecific overpropionylation at serine (S), threonine (T) and tyrosine (Y) was reversed by adding hydroxylamine.
:80 (v/v)) was added, for an incubation of 30 min at RT. This was followed by adding 20 μL milliQ water (Merck Millipore) for 30 min at 37 °C. Histones were then digested overnight at 37 °C using trypsin (at an enzyme/protein ratio of 1:20 (m/m)) in 500 mM TEAB, supplemented with CaCl2 and ACN to a final concentration of 1.0 mM and 5% respectively. Subsequently, the derivatization reaction was carried out again to cap peptide N-termini. Aspecific overpropionylation at serine (S), threonine (T) and tyrosine (Y) was reversed by adding hydroxylamine.
LC-MS acquisition
Samples were resuspended in 0.1% FA to a final concentration of 500 ng/4 μl, i.e. one injection. Equal fractions of all samples were pooled to generate quality control (QC) samples, which were analyzed at fixed intervals in between the other randomized samples. All samples were spiked with digested beta-galactosidase (50 fmol on column; Sciex) and MPDS standard (50 fmol on column, Waters) to monitor chromatographic quality and variation between runs.Liquid chromatography (LC) was performed using a nanoACQUITY UPLC system (Waters). First, samples were delivered to a trap column (180 μm × 20 mm nanoACQUITY UPLC 2G-V/MTrap 5 μm Symmetry C18, Waters) at a flow rate of 8 μL min−1 for 2 min in 99.5% buffer A. Subsequently, peptides were transferred to an analytical column (100 μm × 100 mm nanoACQUITY UPLC 1.7 μm Peptide BEH, Waters) and separated at a flow rate of 300 nL min−1 using a gradient of 60 min going from 1% to 40% buffer B (0.1% formic acid in acetonitrile). MS data acquisition parameters were set according to Helm et al., with minor adaptations.31 A Q-TOF SYNAPT G2-Si instrument (Waters) was operated in positive mode for High Definition-DDA, using a nano-ESI source, acquiring full scan MS and MS/MS spectra (m/z 50–5000) in sensitivity mode. Survey MS scans were acquired using a fixed scan time of 200 ms. Tandem mass spectra of up to eight precursors with charge state 2+ or higher were generated using CID in the trapping region with intensity threshold set at 2000 cps, using a collision energy ramp from 6/9 V (low mass, start/end) up to 147/183 V (high mass, start/end). MS/MS scan time was set to 100 ms with an accumulated ion count “TIC stop parameter” of 200000 cps allowing a maximum accumulation time of 200 ms. Dynamic exclusion of fragmented precursor ions was set to 30 s. Ion mobility wave velocity was ramped from 2500 to 400 m s−1 in MSMS to enable wideband enhancement in order to obtain a near 100% duty cycle on singly charged fragment ions (no multiply charged ions are recorded in MSMS). LockSpray of Glufibrinopeptide-B (m/z 785.8426) was acquired at a scan frequency of 30 s.
Data analysis
To perform untargeted screening of all relevant hPTMs raw data from all LC-MS runs was imported and aligned in Progenesis QIP 4.2 (nonlinear dynamics, waters) for feature detection (Fig. S1, ESI†). Ion filtering was performed for the +1 ions. In addition, ions derived from polyethylene glycol (PEG), contamination coming from the enzyme (Fig. S1A, ESI†), were removed from the analysis. For identification, MS/MS spectra were ranked according to their distance to the elution apex of the extracted-ion chromatogram (XIC). The 10 MSMS spectra closest to the elution apex (i.e. 94655 out of a total of 347868 spectra) were retained per feature, the remaining MSMS spectra were considered redundant.27 A single *.mgf file containing all selected MSMS spectra was exported for searching using Mascot (Matrix Science). The Three different searches were performed, of which enzyme specificity and modifications are given in Table 1. Note that a fixed modification in Mascot is only applied when there is no variable modification detected on the amino acid. For all three searches, mass error tolerances for the precursor ions and its fragment ions were set at 10 ppm and 50 ppm respectively and a bovine histone database (downloaded from Uniprot on 12/09/19 comprising 76 proteins in total) was supplemented with contaminants from the cRAP database (http://www.thegpm.org/crap/) and internal standards.| Enzyme | Mascot searches | |||||
|---|---|---|---|---|---|---|
| Standard | Formyl/under-propionylation | Semi-ArgC | ||||
| ArgC | ArgC | Semi-ArgC | ||||
| Fixed modifications | Propionyl | K | — | — | ||
| Propionyl | N-Term | |||||
| Variable modififcations | Acetyl | K | Acetyl | K | Acetyl | K |
| Butyryl | K | Butyryl | K | Butyryl | K | |
| Dimethyl | K | Dimethyl | K | Dimethyl | K | |
| Trimethyl | K | Trimethyl | K | Trimethyl | K | |
| Methyl | R | Methyl | R | Methyl | R | |
| Crotonyl | K | Formyl | K | Formyl | K | |
| Lactylation | K | Formyl | STY | Formyl | STY | |
| Succinyl | K | Propionyl | K | Propionyl | K | |
| Deamidated | R | Propionyl | N-Term | Propionyl | N-Term | |
Of the resulting peptide-to-spectrum matches (PSMs), the highest scoring one was used to annotate the precursor ion in Progenesis QI 4.2 (Fig. S1B, ESI†). After all the identified PTMs were checked according to UniProt, all LC-MS runs were normalized against histone peptides in order to consolidate the constant protein abundance within the data and minimize the impact of the HDAC enzyme addition (Fig. S1C, ESI†). Feature detection was manually verified for acetylated histone features to resolve isobaric near-coelution. Next, all multiply charged ions (21293) were exported as *.csv and imported into Qlucore omics 3.4. Ions were filtered at a maximum projection score of 0.37.32 Briefly, it measures the informativeness of a low-dimensional representation obtained by PCA, and allows explicit comparison of representations corresponding to different variable subsets. Ions were filtered by multi group comparison q-value below 0.01.
For the analysis of the +1 ions, all the above steps were redone in a separate Progenesis project without filtering out the +1 ions.
Results and discussion
For histone samples, an overwhelming number of combinatorial PTM searches can be done in order to annotate new ions.27 However, because no FDR control strategy exists for PTM searches and because a considerable portion of the annotations are in fact undecisive, i.e. ambiguous, another layer of information is required to verify which annotations are informative. An in vitro time-lapse enzymatic treatment of histones extracted from a biological source mimics the complexity of a biological experiment both in terms of hPTM prevalence and quantitative changes. Therefore, as an adequate number of replicates was measured (minimally n = 3), this design captures biological-like variation, yet in a predictable way. By using different search parameters, novel annotations appear that can either pinpoint weaknesses in the workflow or surface novel hPTMs. In addition, the experimental design allows to isolate incorrect annotations. Therefore, this design is particularly suited to probe the depth of the dark histone ion space, while assessing result quality and allowing further optimization of the workflow (Fig. 1).Previously, we analyzed the samples on a TT5600 mass spectrometer (Sciex) in data-independent acquisition (DIA) mode. More specifically, we used this enzyme-based strategy to validate the accuracy of annotation of our histone SWATH workflow (hSWATH; sequential window acquisition of all theoretical fragment ion spectra).33 Therefore, we exclusively focused on annotated precursor ions and found that all acetylated histone peptidoforms clustered together in a hierarchical cluster of declining intensities. While this is not a true benchmark at single peptidoform resolution, it does show the overall performance of the workflow, much in a sense like fold changes in the Navarro benchmark are used to assess the overall performance of different search algorithms.26 However, more importantly, it surfaced an artefact with the calculation of relative abundance of hPTMs. Briefly, because the unmodified peptidoform (H4 (4–17)) had been lost through filtering, two acetylated residues on that peptide stretch appeared to increase following HDAC treatment, a direct effect of the interdependencies in relative abundance calculation because HDAC enzymes cannot increase acetylations. In turn, this finding inspired us to the reversed reasoning, i.e. all ions that hierarchically cluster together in a declining population should contain the ac(et)ylated histone peptidoforms and the opposing population of ions should contain the deac(et)ylated counterparts. Identification of unexpected peptidoforms in these populations mark potential pitfall in the workflow, be it during sample preparation, data acquisition or data analysis. Likewise, unannotated ions in these ion populations show which fraction of the histone ion space is still in the dark, maybe even because of unexpected or unknown biological hPTMs.
Therefore, we ran the same samples in data-dependent acquisition mode (DDA) on a SynaptG2-Si (Waters), our more commonly used acquisition strategy. Raw data from all these LC-MS runs were imported in Progenesis QIP 4.2 (Nonlinear Dynamics, Waters) and aligned at the MS1 level for feature detection. Here, a feature is defined as an ion with a restricted retention time window and at least one isotopic peak to determine the charge. Next, ion filtering was performed for the +1 ions. Before the actual analysis, the experimental design surfaced a prominent population of ions which was only present in the samples where enzyme was added. These ions turned out to be polyethylene glycol (PEG) contamination introduced together with the enzyme and were therefore removed from the analysis (Fig. S1A, ESI†). For identification, the 10 MSMS spectra that were triggered closest to the apex of each remaining precursor ion across all runs in the experiment were exported and searched in Mascot 2.7. Of the resulting peptide-to-spectrum matches (PSMs), the highest scoring one was used to annotate the precursor ion in Progenesis QI 4.2 (Fig. S1B, ESI†). Normalization was done against all annotated histone peptidoforms (Fig. S1C, ESI†), to consolidate the constant histone protein abundance within the data and to minimize the impact of the addition of the HDAC enzyme, which in turn assures that each deacetylation event is reflected by an increase in unmodified peptidoform abundance.
After peak detection in Progenesis QIP 4.2 and peptide annotation using Mascot, more than 99% of the total ion population stays in the dark (Fig. 2). To find out which of these ions are potential histone peptides, all multiply charged ions (21293) were imported in Qlucore Omics Explorer 3.6 for statistical analysis and generation of the heatmap. Fig. 3A displays some patterns differential ions can show in a heatmap. 1140 (5.4%) ions were statistically differential (q < 0.01) and were hierarchically clustered in the heat map given in Fig. 3B. From these significantly changing ions, a targeted precursor ion population was isolated that consists of two subclusters; ions that disappear only in the samples incubated with active enzyme, i.e. 108 precursor ions assumed to be acetylated (Fig. 3D) and (ii) their counterparts that rise in response to the active enzyme treatment, i.e. 63 precursor ions assumed to be their unmodified counterparts (Fig. 3C). Note that each peptide sequence potentially has several acetylated peptidoforms that will converge into one unmodified form after enzymatic treatment.
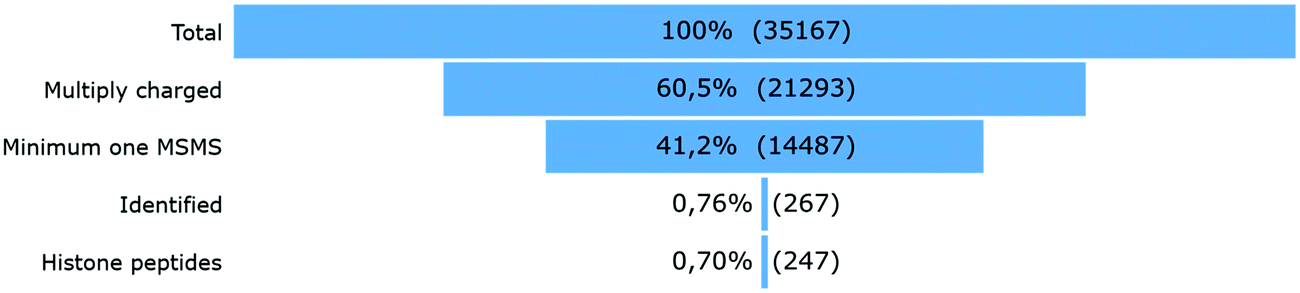 | ||
| Fig. 2 Ion population. 35167 ions were detected (PEG ions not included), 13874 (39.5%) of which were singly charged and therefore not targeted by the instrument operated in DDA mode. Thus, 21293 different multiply charged ions were detected, 14487 of which had at least one MSMS. Out of these annotatable ions, only 267 were identified with the standard search (Table 1), whereof 247 were histone peptides. | ||
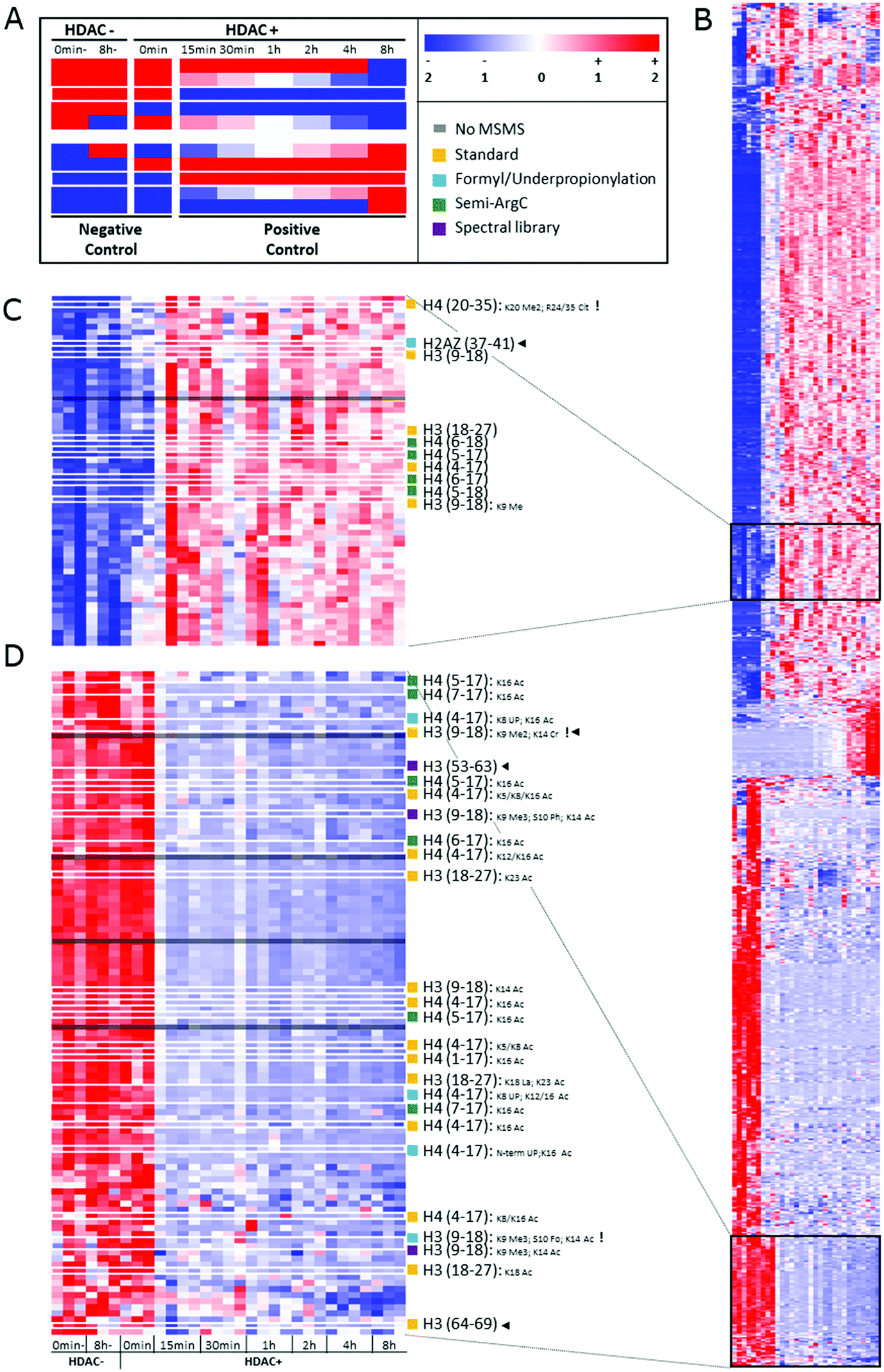 | ||
| Fig. 3 Heatmap depicting the changes in ion abundances that are found after time-lapse HDAC1 treatment of commercial histones. (A) Diagrammatic representation of some predictable ion intensity patterns. This experimental design induces changes in a closed system, i.e. for each decreasing peptide ion, its (un)modified counterpart increases and vice versa. Note that the design allows distinguishing true enzymatic effects and in vitro changes induced by buffer effects or through time (negative controls). White borders indicate the two ion populations represented in (C and D). Sample information: 0 min- and 8 h-: two negative controls incubated in enzyme buffer for 0 minutes or 8 hours, but lacking HDAC1 and the inhibiting acid; 0 min: pre-inhibited HDAC1 was added; 15 min to 8 h: incubation times of enzyme and buffer, ending with acid inhibition (0.4 N HCl) as depicted in the experimental design (Fig. 1). A minimum of three replicates of each treatment are shown. Color codes: red: high relative intensity, blue: low relative intensity, white: average intensity throughout the experiment. (B) Hierarchically clustered heatmap of all 1140 multiply charged precursor ions that have significantly differential abundance in the data according to a maximum projection score of 0.37 and a multi group comparison q value below 0.01. A minimum of triplicate measurements per time point are presented. (C and D) Highlighted ion populations of potential interest with all annotations depicted on the right. UP: underpropionylation. Black arrow heads highlight the unexpected identifications. Exclamation marks highlight the identifications containing modifications that are not listed by UniProt. | ||
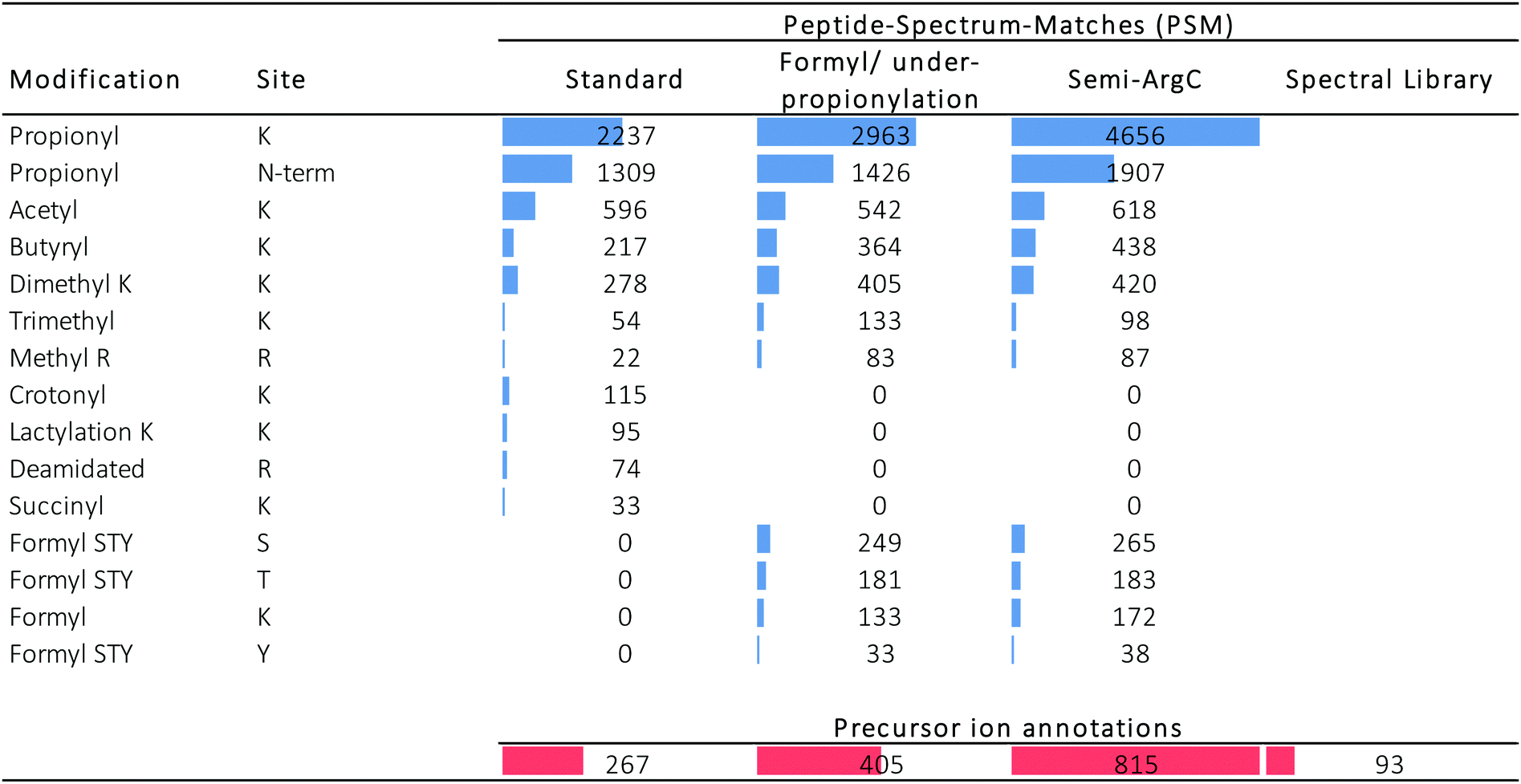
Three different searches were done in Mascot (Table 1): (i) a standard search, (ii) a search to estimate the performance of the propionylation protocol, and (iii) a search to assess the acquisition performance. In addition, we also validated the use of a completely different search strategy, i.e. spectral library matching. Table 2 distinguishes between the PSMs in the different searches (blue) and the amount of precursor ions that were annotated by these PSMs (red). Fig. S2 (ESI†) displays the coverage plots of the hPTMs on H3.1 and H4 detected by the three Mascot searches, color coded according to the number of spectra covering this residue, as presented recently elsewhere.34
|
In the standard search, 1309 PSMs were found with an N-terminal fixed propionylation, comprising a total of 596 PSMs with acetylation. From the targeted precursor ion population (171 precursor ions), 18 precursor ions got annotated in this standard search (Fig. 3), two of which were not part of the expected population: H3(64–69) and H3(9–18) K9Me2K14Cr, which do not carry an acetylation, yet are part of a declining population that is otherwise populated by acetylated peptidoforms. Indeed, manual inspection showed that the H3(64–69) feature was wrongly created by Progenesis QIP and that H3(9–18) K9Me2K14Cr was incorrectly annotated by Mascot (Fig. S3, ESI†). In addition, it was also discovered that the rising population contains one annotation (H4 (20–35) K20 Me2; R24/35 Cit) whose PTMs are not reported in UniProt, while the PSM seems credible.
In addition, unconsidered modifications can also be hidden in the data. One such example that was very recently discovered is that other acylations, like lactylation, could also be removed by HDAC enzymes.35 Therefore, lactylation was included in the standard search (Table 2). Indeed, we were able to identify one lactylation (H3 (18–24) Lac18-Ac24) in the target population of declining ions (Fig. 3C), albeit with a neighboring acetylation that could be the true modification causing the decline. Still, it surfaces yet another source of dark ions in the histone ion space, probably one of the most interesting to pursue in the future, e.g. by applying this design on large collections of histone writers and erasers.
Next, we set out to search specifically for pitfalls that cause wrong identification or loss of annotation at every stage of the workflow, i.e. sample preparation, data acquisition and data analysis. First, the search parameters were adapted to search for chemical noise derived from derivatization using propionic anhydride.29,30,34,36–38 Therefore, the data was re-searched by setting propionylation at N-terminus and at K as variable modifications (to assess underpropionylation) and by including variable formylation at STYK to detect (spontaneously) induced formylation (sample preparation search; Table 1).34,39 596 spectra were annotated with a formylation, giving rise to one additional annotation of the declining ion population because they also carried an acetylation (Fig. 3C). The respective formylation on serine is also not supported by UniProt, confirming its chemical-induced origin. In addition, three H4 (4–17) peptides were annotated with an underpropionylation (indicated with UP in Fig. 3C and D). HLKSR from H2AZ was identified in the correct, upper population, yet has none of the modifications that are part of this search. It can be seen from Fig. S4 (ESI†) that this peptide is in fact annotated in a chimeric spectrum and that because of its size, had not passed the scoring threshold in the previous search. Thus, avoiding formylation by omitting the propionylation34 or using a different acid for reversing overpropionylation would minimize the loss of signal by chemical noise. Additionally, by lack of a decoy strategy, probabilistic scoring is still the preferred workflow in histone analysis,27,28 yet herein the choice of the search parameters in itself changes the scoring threshold for reporting true hits. Finally, chimericy is and will always be an important issue in histone analysis.27
Next, we assessed the occurrence of non-specific cleavage sites by searching Semi-ArgC specificity (propionylation still set to variable, Table 1). This surfaces peptides that arise by (i) tryptic aspecificity, (ii) underpropionylated peptides that now get cleaved at lysine residues, (iii) intrinsically unstable or preprocessed peptides in the sample (not caused by trypsin) and (iv) in-source decay fragments. Indeed, 4446 PSMs in this search annotated 815 precursor ions (Table 2). All 10 semi-specific precursor ions annotated in the target precursor ion population, 4 unacetylated and 6 acetylated (Fig. 3C), were classified as expected. Surprisingly, they all derived from the H4 N-tail. Indeed, in total 27 precursor ions had been annotated as semi-ArgC H4 N-tail peptides (Table S1, ESI†), of which 13 had a propionylation at the N-terminus and no aspecific cleavage at the C-terminus, suggesting they arose before the second round of propionylation. The 11 precursors without N-terminal propionylation seem to imply considerable in-source decay, as they arose after the second round of propionylation. Three ions with no aspecific cleavage at the N-terminus do not have a C-terminal R and could have been created both by enzymatic effects or by in-source decay. Together, this implies that searching with semi-specificity yields considerable higher number of annotations, yet that some kind of post-processing step should be implemented to distinguish the different causes of the non-specifically cleaved peptidoforms. The latter is especially important when e.g. histone clipping is being studied.40,41
By performing a completely different search strategy, i.e. spectral library matching, 3 additional precursor ions from the declining target population were identified (Fig. 3C). These peptidoforms were annotated in Progenesis QIP with a spectral library from another project measured earlier on mouse stem cells.42 Indeed, the conservation of the histone backbone sequence allows using previously acquired spectra of a different organism as a library to increase annotation (e.g. Fig. S5A, ESI†). However, the unmodified H3 (53–63) with a declining intensity shows that the lack of a scoring threshold in this decoy-free library matching greatly advises against the use of this identification strategy for histones currently (Fig. S5B, ESI†).
A significant amount of relevant histone signal is in fact singly charged, as our mobile phase contains DMSO, which improves ionization, but also causes charge state reduction.43 Because +1 ions are not measured by DDA,34 they were excluded from the Progenesis QIP 4.2 analysis in the first place. The analysis was therefore redone without filtering the 13874 singly charged ions, of which 2062 turned out to have an ANOVA q-value < 0.01 and 322 showed an expression profile like the deacetylated ion population from Fig. 3C. Because Progenesis QIP can transfer annotations to other charge states by charge state deconvolution, we could isolate several annotated +1 ions from this ion population. One example that was annotated in this way is H3(19–27) K24Ac, of which 50% of the total signal is singly charged (Fig. S6, ESI†). Together, this confirms that a large part of the peptides that reacted to the enzyme treatment are singly charged. These ions might in fact be of interest to incorporate when DMSO is used, both because they might be the predominant signal for smaller peptides and because they can be used for more accurate quantification when there is an interference at the doubly charged precursor.
Finally, it is important to note that nearly 30% of the multiply charged ions escaped fragmentation on our Synapt G2-Si instrument and can therefore never be annotated. The ions without an MSMS spectrum of the targeted population are indicated with grey boxes in Fig. 3C and D. Using instruments that can sample the precursor ion space to greater depth, by adjusting the LC gradient, by reducing 1+ ion formation using alternative enzyme approaches,34 or by including them during acquisition could mediate both issues. An alternative solution is to apply DIA, where precursors ions are continuously fragmented in an unbiased fashion, as suggested earlier by us and others with the hSWATH acquisition.33,44
In summary, each adaptation in the data analysis surfaced novel and often totally unrelated phenomena that either introduced false annotations or partially shed light on the dark histonome (Fig. 4). Similar to the experimental design proposed by Navarro et al.,26 quantification in our experimental design allows isolating incorrect annotations. Using our conventional workflow with our standard search, only 10.5% of the ion population that changes in response to the treatment could be annotated. During our efforts to increase this fraction, we were cautioned that occasional misalignment of PSM to precursor can occur and that histone searches have a larger FDR than conventional proteomics approaches. Therefore, manual data curation during histone analysis is mandatory. This is additionally true because of the high degree of chimericy in the spectra. This could be alleviated in time by using novel data formats like ion-networks.45 Additionally, we still seem to suffer from formylation and some underpropionylation during sample preparation, which recently led us to propose a propionylation-free protocol.34 Still, propionylation did allow us to more clearly distinguish in-source decay from enzyme specificity. Alternatively, another derivatization strategy, like trimethylacetic anhydride could reduce the chemical noise.46 Additionally, like any other current search strategy, spectral library matching too is restricted by the lack of a decoy strategy for histones. Therefore, we are looking into novel (decoy-free) approaches as well. Finally, both singly and multiply charged precursor ions that are not targeted for MSMS during DDA are lost, but can be very informative. Therefore, apart from omitting DMSO from our LC buffer system, we are looking into acquiring singly charged precursors during DDA and into optimizing DIA acquisition strategies. Altogether, the different adaptations in the workflow doubled the annotated portion of the targeted ion population from 10.5% to 21.6% (Fig. 4). Most importantly however, this leaves another 78% that still resides in the dark. There are exciting times ahead for histone analysis.
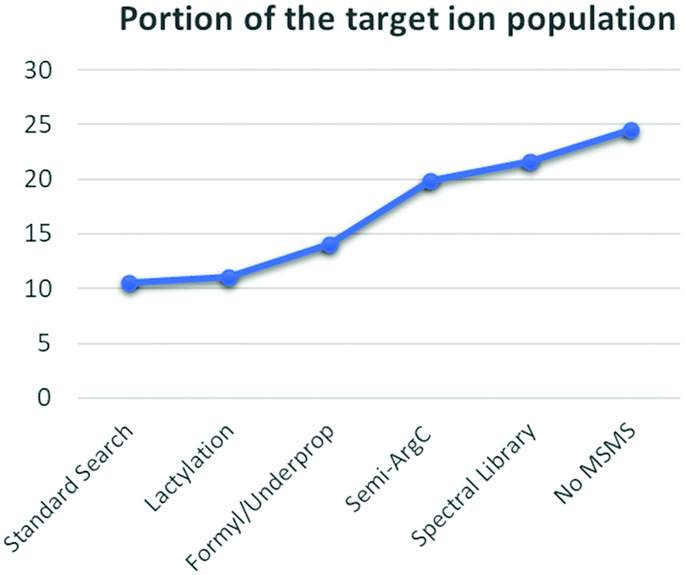 | ||
| Fig. 4 Summary of the annotated dark histone ionome. MSMS spectra of the multiply charged precursor ions were searched in Mascot using three different search parameter sets. The standard search resulted in the annotation of only 10.5% of the targeted ion population, without taking into account the one annotation with a lactylation which accounts for an extra 0.5%. The second search, targeting formylated and underpropionylated peptidoforms, provided an additional 2.9%. The third search, with Semi-ArgC specificity, increased the annotated portion from 14.0% to 19.9%. The MSMS spectra were also matched against a spectral library in Progenesis QIP 3.0, accounting for an extra 1.8%. Finally, 2.9% of the ions in the target population do not have a MSMS spectrum that can be used for annotation. | ||
Conclusion
We show that many of the ions that are formed during a DDA workflow on histone extracts are not being annotated. It remains unclear how many of these derive from as of yet undiscovered peptidoforms with biological importance and how many are impairing this annotation or interfering with the quantification because they are in fact in vitro side effects. However, the use of a controlled in vitro time-lapse removal of PTMs of commercial histones makes it possible to statistically isolate ions of interest and to investigate why most ions remain elusive. In fact, extending the experimental design to epigenetic writers and erasers of unknown function could equally surface their substrate repertoire. This subset then points the way to adjustments that can be made to the workflow in order to either increase their annotation or minimize their formation, respectively. We anticipate that novel acquisition strategies will increase our understanding of the histone code most of all.Data availability
Data are available via ProteomeXchange with identifier PXD009910.Author contributions
Laura De Clerck and Maarten Dhaenens conceived the experimental design. Laura De Clerck performed sample preparation and MS analysis. Laura De Clerck and Maarten Dhaenens performed the data analysis with support of Sander Willems, Bart Van Puyvelde, Sigrid Verhelst and Laura Corveleyn. Simon Daled created the coverage plots. Laura De Clerck and Maarten Dhaenens wrote the manuscript with input and critical feedback from all authors. Maarten Dhaenens supervised and Dieter Deforce co-supervised the experiment.Abbreviations
| DDA | Data-dependent acquisition mode |
| DIA | Data-independent acquisition |
| FDR | False discovery rate |
| HDAC1 | Histone deacetylase 1 |
| hPTM | Histone posttranslational modifications |
| hSWATH | Gistone sequential window acquisition of all theoretical fragment ion spectra |
| MS | Mass spectrometry |
| PEG | Polyethylene glycol |
| PSM | Peptide-to-spectrum matches |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank the Flanders Agency Entrepreneurship and Innovation (VLAIO) and the Research Foundation Flanders (FWO) for funding this research. This research was mainly funded by a PhD grant from the Flanders Agency Entrepreneurship and Innovation (VLAIO) awarded to LDC (SB-141209). Partial funding was received through a grant from FWO (G013916N), FWO mandates 12E9716N, 11B4518N and 3S031319 awarded to MD, BVP, and SV respectively and BOF Mandate BOF20DOC220 awarded to LC.References
- C. F. Brunk and W. F. Martin, Archaeal Histone Contributions to the Origin of Eukaryotes., Trends Microbiol., 2019, 27, 703–714 CrossRef CAS PubMed.
- J. Fan, K. A. Krautkramer, J. L. Feldman and J. M. Denu, Metabolic regulation of histone post-translational modifications., ACS Chem. Biol., 2015, 18, 90–101 Search PubMed.
- D. Zhang, et al., Metabolic regulation of gene expression by histone lactylation., Nature, 2019, 574, 575–580 CrossRef CAS PubMed.
- N. Attar, et al., The histone H3-H4 tetramer is a copper reductase enzyme., Science, 2020, 369(6499), 59–64 CrossRef CAS PubMed.
- A. E. Lepack, et al., Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking., Science, 2020, 368, 197–201 CrossRef CAS PubMed.
- R. Noberini and T. Bonaldi, A Super-SILAC Strategy for the Accurate and Multiplexed Profiling of Histone Posttranslational Modifications., Methods Enzym., 2017, 586, 311–332 CrossRef CAS PubMed.
- C. Alabert, et al., Domain Model Explains Propagation Dynamics and Stability of Histone H3K27 and H3K36 Methylation Landscapes., Cell Rep., 2020, 30, 1223–1234 CrossRef CAS.
- A. Villar-Garea and A. Imhof, The analysis of histone modifications, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764(12), 1932–1939 CrossRef CAS PubMed.
- M. C. Völker-Albert, A. Schmidt, I. Forne and A. Imhof, Analysis of Histone Modifications by Mass Spectrometry., Curr. Protoc. Protein Sci., 2018, 92, e54 Search PubMed.
- F. H. Andrews, B. D. Strahl and T. G. Kutateladze, Insights into newly discovered marks and readers of epigenetic information., Nat. Chem. Biol., 2016, 12, 662–668 CrossRef CAS PubMed.
- H. Huang, S. Lin, B. A. Garcia and Y. Zhao, Quantitative proteomic analysis of histone modifications., Chem. Rev., 2015, 115, 2376–2418 CrossRef CAS.
- B. A. Garcia, C. E. Thomas, N. L. Kelleher and C. A. Mizzen, Tissue-specific expression and post-translational modification of histone H3 variants., J. Proteome Res., 2008, 7, 4225–4236 CrossRef CAS PubMed.
- M. D. Plazas-Mayorca, et al., Quantitative proteomics reveals direct and indirect alterations in the histone code following methyltransferase knockdown., Mol. BioSyst., 2010, 6, 1719–1729 RSC.
- P. V. Shliaha, et al., Middle-Down Proteomic Analyses with Ion Mobility Separations of Endogenous Isomeric Proteoforms., Anal. Chem., 2020, 92, 2364–2368 CrossRef CAS PubMed.
- R. C. Molden and B. A. Garcia, Middle-Down and Top-Down Mass Spectrometric Analysis of Co-occurring Histone Modifications, Curr. Protoc. Protein Sci., 2014, 77, 23.7.1–23.7.28 Search PubMed.
- B. A. Garcia, What does the future hold for Top Down mass spectrometry?, J. Am. Soc. Mass Spectrom., 2010, 21, 193–202 CrossRef CAS PubMed.
- M. V. Holt, T. Wang and N. L. Young, High-Throughput Quantitative Top-Down Proteomics: Histone H4, J. Am. Soc. Mass Spectrom., 2019, 30, 2548–2560 CrossRef CAS PubMed.
- A. Khan, C. K. Eikani, H. Khan, A. T. Iavarone and J. J. Pesavento, Characterization of Chlamydomonas reinhardtii Core Histones by Top-Down Mass Spectrometry Reveals Unique Algae-Specific Variants and Post-Translational Modifications., J. Proteome Res., 2018, 17, 23–32 CrossRef CAS PubMed.
- X. Dang, et al., The first pilot project of the consortium for top-down proteomics: a status report., Proteomics, 2014, 14, 1130–1140 CrossRef CAS PubMed.
- S. Lin, et al., Stable-isotope-labeled Histone Peptide Library for Histone Post-translational Modification and Variant Quantification by Mass Spectrometry., Mol. Cell. Proteomics, 2014, 13, 2450–2466 CrossRef CAS PubMed.
- S. Sidoli, S. Lin, K. R. Karch and B. A. Garcia, Bottom-up and middle-down proteomics have comparable accuracies in defining histone PTM relative abundance and stoichiometry., Anal. Chem., 2015, 87, 3129–3133 CrossRef CAS PubMed.
- A. T. Kong, F. V. Leprevost, D. M. Avtonomov, D. Mellacheruvu and A. I. Nesvizhskii, MSFragger: ultrafast and comprehensive peptide identification in shotgun proteomics., Nat. Methods, 2017, 14, 513–520 CrossRef CAS PubMed.
- B. Van Puyvelde, et al., Removing the Hidden Data Dependency of DIA with Predicted Spectral Libraries., Proteomics, 2020, 20, e1900306 CrossRef PubMed.
- B. C. Searle, et al., Generating high quality libraries for DIA MS with empirically corrected peptide predictions., Nat. Commun., 2020, 11, 1548 CrossRef CAS PubMed.
- V. Demichev, C. B. Messner, S. I. Vernardis, K. S. Lilley and M. Ralser, DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput., Nat. Methods, 2020, 17, 41–44 CrossRef CAS PubMed.
- P. Navarro, et al., A multicenter study benchmarks software tools for label-free proteome quantification., Nat. Biotechnol., 2016, 34, 1130–1136 CrossRef CAS PubMed.
- S. Willems, et al., Flagging False Positives Following Untargeted LC-MS Characterization of Histone Post-Translational Modification Combinations., J. Proteome Res., 2017, 16, 655–664 CrossRef CAS PubMed.
- Z. F. Yuan, S. Lin, R. C. Molden and B. A. Garcia, Evaluation of proteomic search engines for the analysis of histone modifications., J. Proteome Res., 2014, 13, 4470–4478 CrossRef CAS PubMed.
- P. Meert, E. Govaert, E. Scheerlinck, M. Dhaenens and D. Deforce, Pitfalls in histone propionylation during bottom-up mass spectrometry analysis., Proteomics, 2015, 15, 2966–2971 CrossRef CAS PubMed.
- P. Meert, et al., Tackling aspecific side reactions during histone propionylation: The promise of reversing overpropionylation., Proteomics, 2016, 16, 1970–1974 CrossRef CAS PubMed.
- D. Helm, et al., Ion mobility tandem mass spectrometry enhances performance of bottom-up proteomics., Mol. Cell. Proteomics, 2014, 13, 3709–3715 CrossRef CAS PubMed.
- M. Fontes and C. Soneson, The projection score - an evaluation criterion for variable subset selection in PCA visualization., BMC Bioinf., 2011, 12 DOI:10.1186/1471-2105-12-307.
- L. De Clerck, et al., hSWATH: Unlocking SWATH's Full Potential for an Untargeted Histone Perspective, J. Proteome Res., 2019, 18, 3840–3849 CrossRef CAS PubMed.
- S. Daled, et al., Histone Sample Preparation for Bottom-Up Mass Spectrometry: A Roadmap to Informed Decisions, Proteomes, 2021, 9 DOI:10.3390/proteomes9020017.
- C. Moreno-Yruela, et al., Class I Histone Deacetylases (HDAC1-3) are Histone Lysine Delactylases, bioRxiv, 2021 DOI:10.1101/2021.03.24.436780.
- S. Sidoli and B. A. Garcia, Properly reading the histone code by MS-based proteomics., Proteomics, 2015, 15, 2901–2902 CrossRef CAS PubMed.
- S. Sidoli, et al., Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis., Proteomics, 2015, 15, 1459–1469 CrossRef CAS PubMed.
- M. Soldi, A. Cuomo and T. Bonaldi, Quantitative assessment of chemical artefacts produced by propionylation of histones prior to mass spectrometry analysis., Proteomics, 2016, 16, 1952–1954 CrossRef CAS PubMed.
- J. Lenčo, M. A. Khalikova and F. Švec, Dissolving Peptides in 0.1% Formic Acid Brings Risk of Artificial Formylation., J. Proteome Res., 2020, 19, 993–999 CrossRef.
- M. Dhaenens, P. Glibert, P. Meert, L. Vossaert and D. Deforce, Histone proteolysis: a proposal for categorization into ‘clipping’ and ‘degradation’, BioEssays, 2015, 37, 70–79 CrossRef CAS PubMed.
- H. Santos-Rosa, et al., Histone H3 tail clipping regulates gene expression., Nat. Struct. Mol. Biol., 2009, 16, 17–22 CrossRef CAS PubMed.
- G. van Mierlo, et al., Integrative Proteomic Profiling Reveals PRC2-Dependent Epigenetic Crosstalk Maintains Ground-State Pluripotency., Cell Stem Cell, 2019, 24, 123–137 CrossRef CAS PubMed.
- H. Hahne, et al., DMSO enhances electrospray response, boosting sensitivity of proteomic experiments., Nat. Methods, 2013, 10, 989–991 CrossRef CAS PubMed.
- S. Sidoli, et al., SWATH Analysis for Characterization and Quantification of Histone Post-translational Modifications., Mol. Cell. Proteomics, 2015, 14, 2420–2428 CrossRef CAS PubMed.
- S. Willems, et al., Ion-networks: a sparse data format capturing full data integrity of data independent acquisition mass spectrometry, bioRxiv, 2019 DOI:10.1101/726273.
- H. Kuchaříková, P. Dobrovolná, G. Lochmanová and Z. Zdráhal, Trimethylacetic anhydride-based derivatization facilitates quantification of histone marks at the MS1 level, Mol. Cell. Proteomics, 2021, 100114 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental file 1 contains all supplementary figures and table. See DOI: 10.1039/d1mo00201e |
| This journal is © The Royal Society of Chemistry 2021 |