
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The state of the art in plant lipidomics
Cheka
Kehelpannala
 *a,
Thusitha
Rupasinghe
b,
Thomas
Hennessy
c,
David
Bradley
c,
Berit
Ebert
a and
Ute
Roessner
a
*a,
Thusitha
Rupasinghe
b,
Thomas
Hennessy
c,
David
Bradley
c,
Berit
Ebert
a and
Ute
Roessner
a
aSchool of BioSciences, The University of Melbourne, Melbourne, VIC 3010, Australia. E-mail: clkehelpannala@gmail.com; Tel: +61 451821235
bSciex, 2 Gilda Ct, Mulgrave, VIC 3170, Australia
cAgilent Technologies Australia Pty Ltd, 679 Springvale Road, Mulgrave, VIC 3170, Australia
First published on 26th October 2021
Abstract
Lipids are a group of compounds with diverse structures that perform several important functions in plants. To unravel and better understand their in vivo functions, plant biologists have been using various lipidomic technologies including liquid-chromatography (LC)–mass spectrometry (MS). However, there are still significant challenges in LC-MS based plant lipidomics, which need to be addressed. In this review, we provide an overview of the key developments in LC-MS based lipidomic approaches to detect and identify plant lipids with emphasis on areas that can be further improved. Given that the cellular lipidome is estimated to contain hundreds of thousands of lipids,1,2 many of the lipid structures remain to be discovered. Furthermore, the plant lipidome is considered to be significantly more complex compared to that of mammals. Recent technical developments in mass spectrometry have made the detection of novel lipids possible; hence, approaches that can be used for plant lipid discovery are also discussed.
Introduction
Lipids are important components of plants; they are part of cell structures,3 act as energy reserves,4 participate in cell signaling,5 mitigate stress tolerance6 and play a role in both symbiotic and pathogenic interactions.7 The entire lipid profile of an organism, a tissue or a cell, is known as the “lipidome”,8 and the extensive study of lipid molecules including identification, quantification and elucidation of their role in biological systems is called “lipidomics”.8,9 Lipids are highly diverse both in structure and in composition.8,10 Structural diversity arises from the differences in the chemical structures of lipids10 while the ratio of different lipids in cells, tissues, organelles, membrane leaflets, membrane subdomains or within an organism is known as compositional diversity.10For many years, scientists have been studying lipids and their functions in biological systems. It has been shown that even small changes in the structure and composition of lipids can drastically affect vital biological functions.10 For example, changes in membrane lipids directly affect the function of membrane proteins and the physical properties of the cell membrane including fluidity and permeability.11 Therefore, to correctly interpret the in vivo functions of lipids they must be studied as part of an integrated system with metabolites and enzymes.12 It is also important to determine not only the type of lipid present but also its time-dependent local concentration.12
One of the major challenges in uncovering the function of lipids at the cellular level is the existence of thousands of structurally diverse lipids, some of which may be present at trace level concentrations, thus limiting their detection and identification.12 Consequently, a robust analysis workflow is necessary from sampling to extraction and subsequently to detection and identification of lipids. In this review, we discuss the key developments in liquid chromatography–mass spectrometry-based approaches for the extraction, detection, identification, quantification and discovery of plant lipids (Fig. 1). We conducted a comprehensive and in-depth survey of over 400 journal articles published on lipidomics from 1957 to 2021 to identify studies that have played pivotal roles in advancing plant lipidomics. Our survey indicated that many of the techniques in plant lipid analysis were adapted from lipidomic approaches developed for mammalian lipid analysis, which we have discussed in this review. We have also discussed the recently developed LC-MS based lipidomic approaches to analyse lipids in animal tissues, which can be adapted for plant lipid analysis in the future. The primary goal of this review is to provide an overview of the evolution of plant lipidomics, its current state and potential future developments with special focus on LC-MS based workflows.
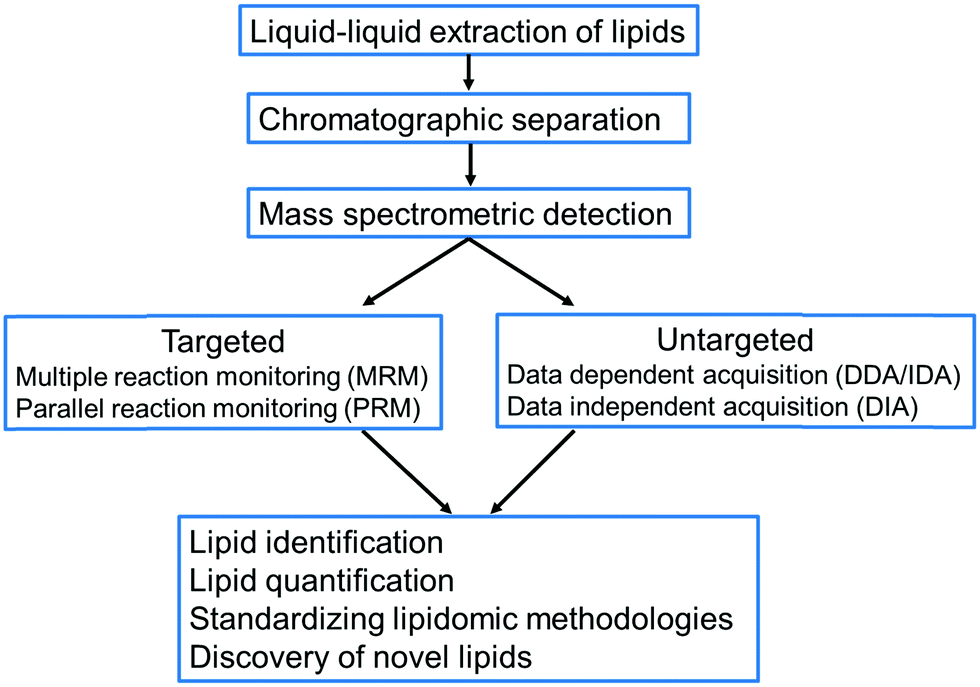 | ||
| Fig. 1 The workflow applied in liquid chromatography–mass spectrometry-based lipidomics. | ||
Plant lipids
Lipids are classified into different groups by the LIPID MAPS (LIPID Metabolites And Pathways Strategy; https://www.lipidmaps.org/) classification system based on the distinct hydrophilic and hydrophobic elements that comprise the lipid.13 The eight main groups include fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, polyketides, sterol lipids and prenol lipids, which can be distinguished by their chemically functional backbone structures (Table 1 and Fig. 2).13 The major plant lipids and their functions are summarized in Table 1.| Lipid class | Core structure | Functions in plants | Ref. |
|---|---|---|---|
| Fatty acyls | Repeating series of methylene groups | Main building blocks of complex lipids.13 | Fahy et al., 2005 |
| Function as signal transduction mediators.14 | Lim et al., 2017 | ||
| Glycerolipids | Glycerol backbone | Major constituents of the thylakoid membranes essential for photosynthesis.15–17 | Dormann and Benning, 2002 |
| Important molecules in cell communication, signal transduction and pathogen responses.18 | Holzl and Dormann, 2007 | ||
| Major storage reserves in plants.19 | Hölzl and Dörmann, 2019 | ||
| Cavaco et al., 2021 | |||
| Yang and Benning, 2018 | |||
| Glycerophospholipids | Phosphate group esterified to a glycerol backbone | Major constituents of cell membranes.20 | Reszczyńska and Hanaka, 2020 |
| Act as signaling molecules.21 | Welti et al., 2007 | ||
| Involved in plant defense mechanisms against pathogens.18 | Cavaco et al., 2021 | ||
| Sphingolipids | Sphingoid long-chain base backbone | Participate in cellular signaling,22 growth and stress responses.23,24 | Ali et al., 2018 |
| Involved in cellular trafficking25 and defense responses against microbial pathogens7 | Hou et al., 2016 | ||
| Mamode Cassim et al., 2021 | |||
| Sterol lipids | Fused four ring structure | Structural components of cell membranes.20 | Reszczyńska and Hanaka, 2020 |
| Regulate acyl chain organization whereby the domain structures of cell membranes are reinforced.26,27 | Schaller, 2004 | ||
| Participate in plant stress responses.28 | Dufourc, 2008 | ||
| Involved in resistance mechanisms against pathogens.7 | Rogowska and Szakiel, 2020 | ||
| Precursors of steroid hormones in plants.28 | Siebers et al., 2016 | ||
| Rogowska and Szakiel, 2020 | |||
| Prenol lipids | C5 unit | Participate in protective mechanisms supporting the adaptation of plants to environmental stresses.29,30 | Baczewska et al., 2014 |
| Bajda et al., 2009 | |||
| Saccharolipids | Fatty acids linked directly to a sugar backbone | Involved in plant defense responses against insects and fungal pathogen attacks.31 | Luu et al., 2017 |
| Polyketides | Polyketide backbone | Responsible for the antibacterial, antifungal, antiviral and antiparasitic properties of plants.32 | Han et al., 2018 |
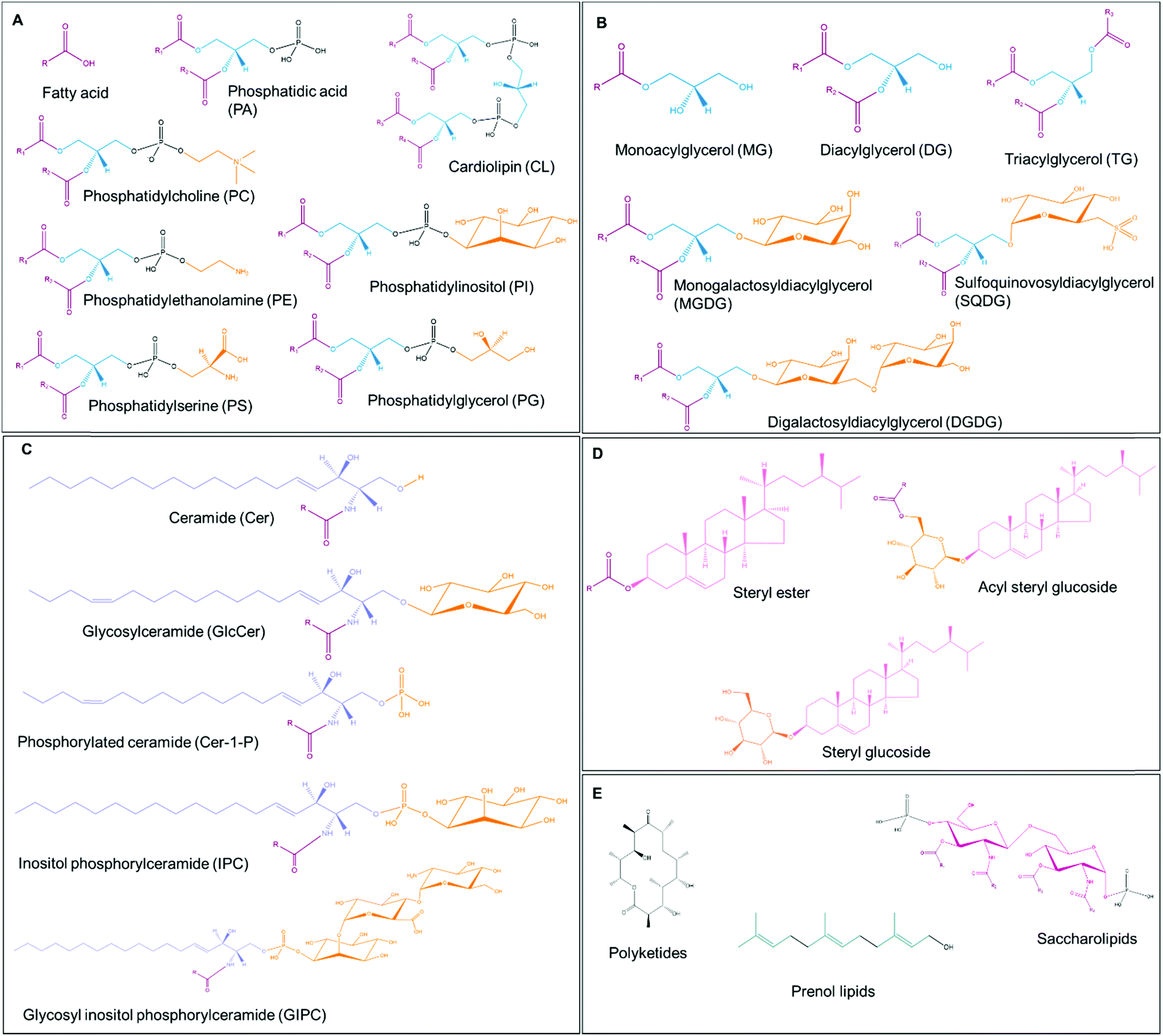 | ||
| Fig. 2 Representative lipid structures. (A) Fatty acid and glycerophospholipids. The fatty acyl chains are shown in brown, glycerol backbone is indicated in blue, phosphate groups are in black and the head group is in orange. (B) Glycerolipids. The glycerol backbone is indicated in blue, fatty acyl chains are in brown and the hexose group is in orange. (C) Sphingolipids. The long-chain base is denoted in purple, fatty acyl chains are in brown and the head group is in orange. (D) Sterol lipids. The sterol group is indicated in pink, fatty acyl chains are in brown and the sugar moiety is in orange. (E) Polyketides, saccharolipids and prenol lipids. The polyketide backbone is shown in black and the five-carbon groups in prenol lipids are indicated in green. The sugar backbone in saccharolipids is shown in magenta, the phosphate group in black and the fatty acyl chains in brown. R indicates a repeating series of methylene groups. Lipid structures are based on the LIPID MAPS structure database and were generated using ChemDraw 20.0 software. | ||
Sample preparation
Given the highly diverse nature of lipid structures (Fig. 2), a comprehensive analysis of the lipidome requires firstly an efficient sample preparation process, then an effective lipid separation strategy followed by an optimal mass spectrometric detection method.33The first step for lipid analysis is the successful extraction of lipids from the tissue of interest, for which a rapid, simple and efficient extraction method is desirable.34 The most widely used sample preparation method in lipidomics is liquid–liquid extraction.35 Most of the liquid–liquid plant lipid extraction methods are derived from extraction protocols developed for animal tissues, such as the Folch36 and the Bligh and Dyer method.37 In addition to these two, several other protocols using diverse mixtures of organic solvents, including hexane,38 butanol39 and petroleum ether,40 and different treatments such as cold41 and heat42 have been developed, compared and evaluated for plant lipid extraction. Studies comparing these methods concluded that the Folch or Bligh and Dyer methods and those derived from them using chloroform, methanol and water as the extraction solvents are the most efficient methods for plant lipid extraction,34,42–45 with the Folch method36 referred to as the “gold standard”.46 Originally, modifications to the Folch method36 led to the development of the Bligh and Dyer method.37 Later, significant improvements were made to the Bligh and Dyer protocol37 when it was adopted for plant lipid extractions, leading to several new protocols. For example, in the study by de la Roche et al.42 wheat, Triticum aestivum L. cv, seeds were boiled with isopropanol to inactivate the lipolytic enzymes and extract neutral lipids more efficiently before applying the Bligh and Dyer method.42 Ryu and Wang47 simplified the protocol published by de la Roche et al.42 and introduced 0.01% butylated hydroxytoluene (BHT) to the extraction solvent to reduce lipid oxidation.47 The Ryu and Wang47 method was adapted and modified with additional steps by Welti et al.48 and since then this method has been extensively used in plant lipidomics. Since the method by Ryu and Wang47 was relatively labor-intensive, a shorter high-throughput single-step extraction method with a 24 h extraction period was developed by Vu et al.49 Additional modifications to this single-step extraction method of Vu et al.49 resulted in an extraction protocol that uses 30 parts chloroform, 25 parts isopropanol, 41.5 parts methanol, and 3.5 parts water (v/v/v/v) with 0.01% BHT.34 This protocol developed by Shiva et al.34 has been shown to be highly efficient in extracting lipids from Arabidopsis thaliana and Sorghum bicolor leaf tissues.34 To inactivate the lipolytic enzymes, fresh leaves were harvested directly into isopropanol pre-heated to 75 °C.42,50 However, this single step protocol34 does not include steps for tissue homogenization. Tissue homogenization, however, is crucial to obtain a representative sample as it allows the extraction solvent to access lipids even in rigid plant tissues, increases the surface area in contact with the extraction solvent51 and solubilizes the lipids. While an extraction method should contain minimal sample preparation to achieve high-quality results, homogenization and metabolic quenching are vital steps.52 Thus, the efficiency of a lipid extraction method can be further improved through the addition of a mechanical grinding step.53 A recent comparison of four popular plant lipid extraction methods undertaken by us included the single step extraction method published by Shiva et al.,34 the multi-step extraction method of Welti et al.,48 the biphasic extraction method using methyl tert-butyl ether (MTBE)46 derived from the Folch method36 and a rapid method employing an extraction temperature of 4 °C.41 This study revealed that overall, the single-step extraction method34 is the most efficient for extracting most of the major lipid classes from Arabidopsis tissue. However, due to the diverse relative hydrophobicity of lipid molecules, the development of a single protocol efficient in extracting all lipid species in a plant sample is impossible.45 For example, several studies have shown that chloroform/methanol mixtures are not suitable for the extraction of plant sphingolipids such as glycosylinositol phosphorylceramides (GIPCs).34,54,55 Due to the highly amphiphilic nature of sphingolipids,55 the standard lipid extraction methods are inefficient in solubilizing them.54 It is therefore recommended to use a mixture of isopropanol, hexane and water for the extraction of plant sphingolipids55,56 as described by Markham et al.54
Even though chloroform and methanol are considered ideal solvents for lipid extraction, they can have detrimental effects on human health due to their lipo-solubility.45 Given the proven carcinogenicity of chloroform, attempts have been made to replace it with methyl-tert-butyl ether (MTBE), which extracts lipids into the organic layer while the polar compounds remain in the aqueous layer.46 However, this biphasic extraction method is labor-intensive, which limits its applicability in large-scale lipidomic studies. Furthermore, significant technical errors may be introduced during the separation of the organic layer from the aqueous layer, thus ultimately affecting the extraction reproducibility.53 Substituting the chloroform used in the single-step extraction protocol of Shiva et al.34 with MTBE and comparing the extraction efficiencies are worth testing to further improve the existing extraction protocols.
Recent studies have focused on developing lipid extraction methods using environmentally friendly “green solvents”, which can be organic, ionic liquids or supercritical fluids, such as carbon dioxide under supercritical conditions.45 Supercritical fluids are substances that are not in a distinct gas or liquid phase but can be gradually compressed from low to high density.57 By adjusting the density, its properties can be manipulated for the required process.57 Organic green solvents are low in toxicity, readily biodegradable, easily recyclable and have high boiling points and low miscibility, while ionic liquids are non-aqueous organic salts, which are liquid at room temperature.45,58 The use of green solvents is widely explored for lipid extraction in biomass processing for biofuel production where large amounts of chemicals are routinely used.59 Thus, there could be the potential to replace chloroform with one or some of these green solvents to efficiently extract plant lipids. For example, a biphasic system of cyclopentyl methyl ether (CPME):methanol:water was as efficient as the Bligh and Dyer method in extracting triacylglycerols from wet biomass of the oleaginous yeast Lipomyces starkeyi,60 thus the study concluded that CPME can be used as an alternative to chloroform.60 However, a comparative study applying the green solvents 2-methyl tetrahydrofuran (2-MeTHF) and cyclopentyl methyl ether (CPME) revealed that the Bligh and Dyer method was more effective in extracting total lipids from the microalgae Chlorella pyrenoidosa for biodiesel production.61 Although the potential of green solvents such as isoamyl acetate,62 terpenes, ionic liquids,58 2-methyltetrahydrofuran,63 supercritical and subcritical fluids64,65 for lipid extraction from yeast and microalgae has been evaluated, their applicability for plant lipid extraction is yet to be demonstrated. Future work will require the comparison of the traditional plant lipid extraction methods with the green solvent-based extraction methods to evaluate the applicability of green solvents in routine plant lipidomic analysis.
Even though current plant lipidomic analyses rely on manual extraction of lipids, automated workflows for lipid analysis will be developed in the future. Automation for sample preparation would be beneficial since the quality, reproducibility and efficiency would be enhanced when compared with manual execution.
Liquid chromatography–mass spectrometry (LC-MS) in lipidomics
Electrospray ionization (ESI) mass spectrometers are the predominant instruments used for lipid analysis.66 Lipid samples can be analyzed either by directly injecting the extracts into the mass spectrometer or by first separating them through chromatographic methods before injection into the mass spectrometer.1 In 1994, Han and Gross67 first proposed lipidomic analysis using ESI mass spectrometry, when they analyzed human erythrocyte plasma membrane phospholipids using a triple quadrupole with syringe pump sample injection. This method was further extended to characterize and quantify membrane lipids from cells or subcellular structures by Brügger et al. 1997,68 using a nano-ESI source. This is now widely known as “shotgun lipidomics” and has seen continuous improvement over the past 25 years. The strategy used by Brügger et al. 199768 for the high-throughput profiling of complex lipids from unfractionated extracts of animal cell membranes was later adapted by Welti et al.48 to profile membrane lipids in Arabidopsis tissues.48,69 Because of the relative simplicity, high sample throughput and cost-effectiveness of the shot-gun lipidomic methods34 developed by Welti and colleagues over the past 20 years,48,49,69–71 they are still widely used in plant lipidomic analysis.72,73 However, the application of direct infusion methods is not without drawbacks. They show higher ion suppression effects compared to methods with chromatographic separation, consequently hindering the analysis of low-abundant lipid species.1 The selectivity of the direct infusion approach is limited by the existence of isobaric precursor and fragment ions.74 The predominantly targeted analysis involving these methods makes the detection of unknowns impossible.74 Due to these factors, the shot-gun approach was rapidly followed by LC-MS for plant lipidomics.75The LC-MS based methods were highly sensitive with reduced ion suppression and matrix effects,76 able to detect novel lipids,77 able to yield reliable identities of lipids even at low levels and able to separate isomers and isobaric compounds.78 LC-MS is now considered a comprehensive and powerful technique for the analysis of complex lipid classes such as glycerolipids, glycerophospholipids, and glycolipids and is successfully used in plant lipid research.79 To maximize the performance of the LC-MS systems, both the LC and MS methods must be meticulously optimized.33 An ideal LC method should possess high peak capacity, good chromatographic separation and resolution, reproducibility, selectivity and sensitivity in addition to being compatible with the data acquisition cycles in mass spectrometers.33 In general, lipidomic methods take long chromatographic analysis times to separate the large number of lipids, which are present over a wide concentration range in biological samples, and to separate isomeric and isobaric compounds.80 Quicker chromatographic methods can be used; however, several limitations need to be addressed such as analyte overlap, isotopic interferences and ion suppression effects.80 To mitigate these effects, ion mobility can be introduced into the analytical workflow. For example, a separation method employing an elution time of 3.7 min per sample has resulted in a reduced number of features compared to longer chromatographic methods.80 The introduction of ion mobility to the analytical workflow, increased the peak capacity and helped to resolve co-eluting species, thereby increasing the number of detected features.80
The LC technologies used in modern lipidomics include reversed-phase chromatography (RPLC), normal phase chromatography (NPLC), hydrophilic interaction liquid chromatography (HILIC) and supercritical fluid chromatography (SFC).76 RPLC is one of the most often used techniques utilizing a non-polar stationary phase and a polar mobile phase where lipid molecules are separated based on their hydrophobicity, length of fatty acyl chain, and number and position of double bonds.76 In general, the retention time of lipids increases with increasing carbon number and decreases with increasing number of double bonds. For example, PC 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 elutes before PC 36:6 while PC 34:5 elutes after PC 34:6 (Fig. 3). Non-polar lipids, such as TGs are longer retained in the column while polar lipids such as lysophospholipids elute faster.
:6 elutes before PC 36:6 while PC 34:5 elutes after PC 34:6 (Fig. 3). Non-polar lipids, such as TGs are longer retained in the column while polar lipids such as lysophospholipids elute faster.
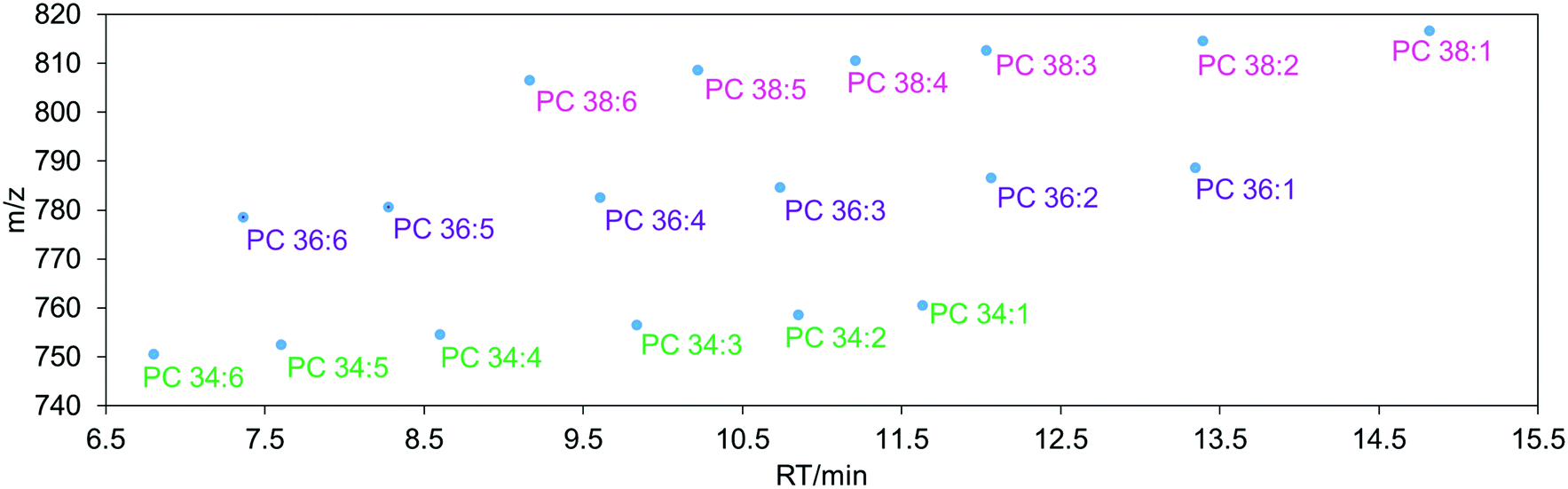 | ||
| Fig. 3 The elution order of phosphatidylcholine (PC) 34, 36 and 38 carbon lipid species in reverse-phase liquid chromatography (RPLC). | ||
The stationary phase of RPLC consists of silica microparticles containing hydrophobic functional groups such as C30, C18, C8, C4 hydrocarbon chains or ligands, which may be cyano, phenyl or amino derivatives with C18 being the most widely used.76 The polar mobile phase generally contains water and a water-miscible organic solvent such as acetonitrile, methanol and isopropanol.76 The development of ultrahigh performance liquid chromatography (UHPLC) columns and core–shell materials for columns significantly improved traditional HPLC techniques providing higher resolution and requiring less time.81
NPLC separates lipid classes based on polarity and consists of a polar stationary phase and a non-polar mobile phase such as hexane or heptane with a miscible organic solvent like chloroform, isopropanol or ethylacetate.76 NPLC can only separate non-polar lipids like DGs and TGs while phospholipids cannot be separated using this method.76 In addition, NPLC methods are lengthy and the highly non-polar organic solvent systems used in NPLC are incompatible with ESI-MS.82 HILIC is a more efficient type of NPLC technique that uses less apolar solvent systems compatible with ESI-MS and separates lipid classes based on their head group polarity.82 The polar stationary phase consists of silica or functionalized silica while the mobile phase consists of water and water-miscible organic solvents.76 A comprehensive description of the theories behind different chromatographic approaches has been recently published by Lange et al. 2019.76 HILIC and RPLC are currently the two most widely applied chromatographic separation techniques in LC-MS based lipidomics including plant lipidomics.78,83 A direct comparison of HILIC-MS and RPLC-MS based lipidomic workflows concluded that both can be equally used to accurately quantify LPCs, LPEs, PCs, PEs and sphingomyelins in human blood plasma;83 however, no such comparison has been reported for lipids from plant tissues. An evaluation of HILIC-MS and RPLC-MS based workflows for plant lipid analysis will be of great benefit for future studies on plant lipids.
In addition to traditional one dimensional LC methods, recently two dimensional LC (2DLC) methods have also been investigated for high-throughput lipidomic analysis of various samples including human plasma84 and plant tissues.85 In LC, only one liquid phase separation system is applied to the analytes in a sample while 2DLC techniques utilize two orthogonal liquid phase separation systems to separate sample components.86 A study comparing several LC and 2DLC methods coupled with high-resolution time-of-flight mass spectrometry (ToF-MS) revealed that the C18xHILIC approach was the most effective in comprehensive lipid profiling of extracts of zebrafish embryos.87 However, the data generated by 2DLC coupled with mass spectrometry are highly complex and make an untargeted analysis challenging,85 consequently, advanced chemometric data analysis has been proposed for such studies.85 For example, a 2DLC setup combining RPLC and HILIC coupled to a triple quadrupole (QQQ) mass spectrometer with chemometric data analysis was able to evaluate the effects of arsenic exposure on the rice lipidome, resolving a larger number of lipids compared to the LC system.85 However, the identification of lipids was difficult due to poor detection sensitivity, likely caused by the dilution due to two successive chromatographic separations or because it was impossible to optimize the collision energy.85 While the application of 2DLC systems in routine lipidomic analysis may be challenging,3 where simple, rapid, user-friendly and proven lipidomic data acquisition strategies are required, future developments particularly in data processing workflows may improve its applicability. It is worthwhile to also investigate if the 2DLC approach can be used to discover novel lipids.
The separation of lipids by LC is followed by transfer to a mass spectrometer. Mass spectrometers have been used in the analysis of lipids since the 1980s88 and over the past decades have seen substantial improvements in the instrumentation and data acquisition methodologies. Mass spectrometric analysis of complex biological samples can be performed using untargeted or targeted approaches.89 Untargeted methods aim to comprehensively profile all detectable compounds in a sample while targeted methods focus on analyzing a pre-defined set of metabolites.89 In general, targeted methods are more sensitive and more quantitative, however, only a limited number of compounds can be analyzed.89 On the other hand, untargeted methods can provide an unbiased analysis of the changes in a system and allow the discovery of novel lipids.90 While targeted methods require prior knowledge of sample matrix and LC-MS properties of the analytes, untargeted methods do not rely on prior information about the sample matrices and are ideal for discovery-focused lipidomic approaches.83 However, due to the complexity of the data retrieved using untargeted methods and the unspecific nature of the analysis, some analytes may go undetected. Furthermore, targeted MS conditions can specifically be tuned to detect individual compounds, for example through the selection of optimized collision energies, while this option is rather limited for the discovery approach.
The commonly used mass spectrometers in lipidomics are triple quadrupoles (QQQ), quadrupole ion trap (QIT), triple quadrupole-linear ion trap (QTRAP), orbitraps and quadrupole time-of-flight (Q-ToF) instruments.35,91 Instruments such as Q-ToF and Orbitrap provide high mass accuracy at MS and MS/MS levels and offer both accurate identification and quantification of lipids, although at a lower sensitivity.89,90 In contrast, QQQ mass spectrometers show low mass accuracy but offer high sensitivity when operated in the multiple reaction monitoring (MRM) mode.90,91 This makes them the preferred instruments for quantitative targeted studies including the analysis of low-abundant lipid species.90,91 However, parallel reaction monitoring also known as MRM high-resolution (MRMHR) performed on a high-resolution instrument such as Q-ToF is a more comprehensive, targeted quantitative strategy where all detectable product ions of a pre-selected precursor ion are scanned in the high-resolution mode.92 In parallel reaction monitoring a scheduled algorithm is used, in which a targeted/predefined set of precursor ions are isolated in the quadrupole followed by fragmentation and subsequent detection of all the product ions.92 This yields more accurate m/z and narrower peak widths, and simultaneously detects up to 100 precursor ions per duty cycle; the intensities of multiple fragments can be summed for better sensitivity.92 Untargeted LC-MS based lipidomic analyses were found to be independent of the type of high-resolution mass spectrometer used and led to nearly identical results when lipidomic data acquisitions across nine different platforms (one single ToF, one Q/orbital ion trap, and seven Q-ToF instruments) were compared.93 This suggests that the up- and down-regulation patterns of lipids in biological samples can be studied using any type of mass spectrometer.35
MS-based lipidomic analyses utilizing high-resolution mass spectrometers can apply data-dependent acquisition (DDA) or data-independent acquisition (DIA) strategies.80 With DDA methods, full scan spectra are recorded at the MS level and MS/MS spectra are recorded by automatically fragmenting the precursor ions with an intensity above a certain threshold or as specified by a user-defined precursor list,35 a given mass range or by mass defect filters.94 Since the precursor ions for fragmentation are selected based on the MS survey scan, the DDA approach is also known as information-dependent acquisition (IDA),94 data directed acquisition or data-directed analysis.95 Generally, the most abundant 10–20 precursor ions are selected for fragmentation using the DDA approach,94 which is also known as the top-N data-dependent MS/MS approach96 with N referring to the number of the most abundant precursor ions used for fragmentation. Therefore, MS/MS spectra of low-intensity ions will be excluded35 and the reproducibility of the data is low.94 To increase the coverage of low abundant lipids using the DDA approach, an iterative exclusion is proposed where precursors previously selected for fragmentation are excluded in subsequent injections.97,98 The application of the iterative exclusion to DDA workflows has shown to increase lipid identification by over 50% in some cases.98
By contrast, the DIA methods collect spectral data of all fragments without the selection of specific precursor ions, yielding highly complicated spectra.94 DIA methods include collision energy switching MS of everything (MSE) also known as MSAll or all ion fragmentation (AIF)35,99 and sequential window acquisition of all theoretical fragment ion spectra (SWATH).100 In the MSAll approach, the data-dependent acquisition is performed on all product ions regardless of the precursor ion. As all precursor ions are sent to the collision cell for fragmentation, MSAll relies on good chromatographic separation techniques, such as ultrahigh-performance liquid chromatography (UHPLC) to obtain quality MS/MS spectra.95
To reduce the complexity of the spectra generated using the MSAll approach, SWATH was introduced by Gillet et al.100 for proteomic experiments and was quickly adapted for metabolomic and lipidomic studies.94 However, SWATH still provided complex spectral data and special software tools were necessary to deconvolute the data.35 This issue was solved when an open-source data processing software called mass spectrometry-data independent analysis (MS-DIAL) software101 was introduced to deconvolute the data accumulated by SWATH, which incorporated the LipidBlast102 library to identify lipids.101 At present, the SWATH acquisition method is immensely popular for untargeted lipidomic analyses103 and has been used to analyze lipids in human platelets,94 mouse plasma,104 and human plasma.105 More recently it was adapted for plant lipidomics to analyse lipids from diverse Arabidopsis tissues.106
SWATH is considered to be advantageous over the DDA methods as it offers full coverage of all MS/MS fragments, yields more lipid identifications and shows high sensitivity, which is comparable to MRM reactions on a QQQ.99 Furthermore, the SWATH approach extracts chromatograms not only at the MS level but also at the MS/MS level whereby reliable quantification can be achieved at either the ToF-MS or SWATH-MS/MS levels using precursor or product ions, respectively.104 A comparison of the IDA, SWATH and MSAll approaches revealed that the IDA methods yield qualitatively better MS/MS spectra but a lower number of lipid annotations, while SWATH outperformed the MSAll approach with better quality MS/MS spectra and identical MS/MS acquisition hit rates.95
Here, it should be noted that the method of choice for lipidomics depends on the purpose of the study95 and available resources (Table 2). The IDA approaches such as SWATH and MSAll are ideal for untargeted lipidomic approaches.99,106,107 However, while SWATH can be used for quantitative analyses,99 lipid identification and quantification may be far more simple with targeted MRM methods where quantification of a targeted set of lipids is the primary focus.
| Plant tissue | Homogenization method | Lipid extraction method | LC conditions | MS conditions | Lipid identification | Internal standards | Lipids identified | Ref. |
|---|---|---|---|---|---|---|---|---|
| RT: retention time, THF: tetrahydrofuran, a.a: acetic acid, a.f.: ammonium formate, f.a.: formic acid, NH4Ac: ammonium acetate IT-TOF: Ion trap-time-of-flight mass spectrometer, QToF: quadrupole time-of-flight mass spectrometer, QQQ: triple quadrupole mass spectrometer, SPRM: scheduled parallel reaction monitoring, MRM: multiple reaction monitoring, DDA: data dependent acquisition, SWATH: sequential window acquisition of all theoretical fragment ion spectra, GIPC: glycosyl inositol phosphoryl ceramide, CHCl3: chloroform, MeOH: methanol, BuOH: butanol ACN: acetonitrile, IPP: isopropanol, MTBE: methyl-tert-butyl-ether. | ||||||||
| Arabidopsis thaliana rosettes, leaves, flowers, flower stalks, siliques, roots, and seeds | None | Extracted with CHCl3:H2Ofollowed by a five-fold extraction with a mixture of CHCl3:MeOH (2:1). The solvent was evaporated under nitrogen and resuspended in CHCl3 |
N/A | QQQ in positive and negative modes | MRM transitions | di14:0-PC, di24:1-PC, 13:0-LPC, 19:0-LPC, di14:0-PE, di24:1-PE, 14:0-LPE, 18:0-LPE, di14:0-PG, di24:1-PG, 14:0-LPG, 18:0-LPG, di14:0-PA, di20:0 (phytanoyl)-PA, di14:0-PS, di20:0 (phytanoyl)-PS, 16:0–18:0-PI, di18:0-PI, 16:0–18:0-MGDG, di18:0-MGDG, 16:0–18:0 DGDG and di18:0-DGDG | Phospholipids, polar glycerolipids | Welti et al., 2002, Devaiah et al., 2006150,151 |
| Arabidopsis thaliana leaves | The glass plunger of the DUALL tissue grinder, attached to a Ryobi D45CK power drill | Extracted with IPP:hexane:H2O (55:20:25). The dried extract was de-esterified using methylamine, dried under nitrogen and dissolved in THF:MeOH:H2O (2:1:2) containing 0.1% f.a. |
SUPELCOSIL ABZpPlus column (150 mm × 3 mm × 5 μm) A: THF:MeOH:5 mM a.f. (3:2:5) + 0.1% f.a. B: THF:MeOH:5 mM a.f. (7:2:1) + 0.1% f.a. |
QTRAP using MRM mode in positive and negative modes | RT of known standards and by comparison with other peaks | GM1, 12-GlcCer, C12-Cer, sphingosine (C17 base), and sphingosine1-phosphate (C17 base) | Sphingolipids | Markham and Jaworski, 200755 |
| Arabidopsis thaliana leaves | Retsch mill | Extracted with MeOH:MTBE (1:3) followed by H2O:MeOH (3:1). Upper organic phase was removed, dried in a speed-vac and resuspended in a buffer |
C8 RP column (100 mm × 2.1 mm × 1.7 μm) A: 1% 1 M NH4Ac +0.1% a.a. B: ACN:IPP (7:3) + 1% 1 M NH4Ac +0.1% a.a. |
Orbitrap with full scan and all-ion fragmentation in positive and negative modes | Matching the m/z values with previously reported data | PE 34:0 (17:0, 17:0) and PC 34:0 (17:0, 17:0) |
Phospholipids, oxidized lipids, glycerolipids, sphingolipids (less comprehensive compared to other lipid classes) | Hummel et al., 2011152 |
| Arabidopsis thaliana rosettes | Powdered using a cryogenic grinding robot | Extracted with a mixture of CHCl3:MeOH:H2O (1:2.5:1), dried down in a speed vac and resuspended in IPP:hexane:H2O (55:20:25) |
C8 RP column (150 mm × 2.1 mm × 1.8 μm) A: 1% 1 M NH4Ac +0.1% a.a. in H2O B: MeOH:IPP (5:2) + 1% 1 M NH4Ac, 0.1% a.a. |
SYNAPTTM using MSE in negative ion mode | Targeted search based on m/z values, expected RT and manual inspection of MS/MS data | Not given | Phospholipids, galactolipids | Burgos et al., 201141 |
| Arabidopsis thaliana leaves and roots. leaves of rice (Oryza sativa), | Mixer mill | Bligh and Dyer method with slight modifications. Extracted with CHCl3:MeOH:H2O and the lower layer evaporated by centrifugal concentration and reconstituted in CHCl3 |
HILIC silica column (100 mm ×2.1 mm × 3 μm) A: MeOH:H2O (95:5) + 0.2% a.f. B: ACN:MeOH:H2O (95:2:3) + 0.2% a.f. |
IT-TOF operated at scan mode in negative ion mode | m/z values and RT of authentic compounds | 10:0/10:0 PC |
Novel plant lipid, glucuronosyldiacylglycerol, phospholipids, polar glycerolipids, sterol lipids, sphingolipids | Okazaki et al., 2013a, Okazaki et al., 2013b148,153 |
| Arabidopsis thaliana leaves | Mixer mill cooled with liquid nitrogen | Extracted with a hot solvent mixture containing 60% IPP | ACQUITY UPLC HSS T3 column (100 mm × 1 mm × 1.8 μm) A: MeOH:20 mM NH4Ac (3:7) + 0.1% a.a. B: THF:MeOH:20 mM NH4Ac (6:3:1) + 0.1% a.a. |
QTRAP using MRM in positive and negative modes | MRM transitions | Not given | Phospholipids, sphingolipids, sterol lipids, glycerolipids | Tarazona et al., 201575 |
| Arabidopsis thaliana leaves | None | Extracted with CHCl3:MeOH:300 mM NH4Ac in H2O (30:41.5:3.5) |
N/A | QQQ in positive and negative modes | MRM transitions | A mixture of 22 internal standards | Phospholipids, glycerolipids, acylated galactolipids, sterol lipids | Shiva et al., 202072 |
| Arabidopsis thaliana leaves and roots. Mature leaves and new shoots of Camellia sinensis (tea) plants. | Lyophilized samples were homogenized in a Retschmill | Modified Matyash et al., 2008 method. Extracted with MeOH:MTBE:H2O (1:3:1) followed by MeOH:water (1:3). Upper phase was dried down in a speed vac and resuspended in ACN:IPP (7:3) |
C8 column (100 mm × 2.1 mm × 1.7 μm) A: H2O + 1% 1 M NH4Ac + 0.1% a.a. B: ACN + IPP (7:3)+ 1% 1 M NH4Ac + 0.1% a.a. |
Q Exactive using full scan and AIF in positive and negative modes | In-house library | None. Content of each lipid species is presented as mean intensity | Phospholipids, galactolipids and sphingolipids | Giavalisco et al., 2011, Liu et al., 2017154,155 |
| Barley (Hordeum vulgare L.) roots | Ground to a fine powder using a mortar and pestle in liquid nitrogen | Sphingolipid extraction in plant tissues as described by Markham et al., 2006. Extracted with IPP:hexane:H2O (60:26:14), evaporated under liquid nitrogen and resuspended in IPP:MeOH:H2O (4:4:1) |
Poroshell EC-C18 column (100 mm × 2.1 mm × 2.7 μm) A: MeOH:20 mM NH4Ac (3:7) B: IPP:MeOH:20 mM NH4Ac (6:3:1) |
QToF using SPRM in positive and negative modes. Oxidized lipids were analyzed based on precursor–production transitions. | Precursor-product ion transitions | PE(12:0/12:0) and Cer(d18:1/12:0) |
Oxidized lipids, phospholipids, polar and neutral glycerolipids, sterol lipids, sphingolipids | Yu et al., 2018, Yu et al., 202092,156 |
| Arabidopsis seeds | Not given | Extracted with IPP:hexane:H2O (55:20:25) according to the protocol by Markham et al., 2006 followed by alkaline hydrolysis, dried down and reconstituted in BuOH:MeOH (1:1) |
Ascentis Express RP Amide column (50 mm × 2.1 mm × 2.7 μm) H2O:MeOH:THF (50:20:30 to 5:20:75) |
QQQ using MRM transitions | MRM transitions | 16:0-d31 SM |
Ceramides, hydroxyceramides, glucosylceramides and GIPCs | Ebert et al., 201856 |
| Rice (Aerial parts and roots) | Ground to a fine powder using a mortar and pestle in liquid nitrogen and lyophilized for 24 h to dryness | Modified Matyash et al. 2008 method extraction with pre-cooled MeOH:MTBE (1:3) followed by MeOH:water (1:3), upper-phase dried down and resuspended in MeOH:water (1:0.85) |
Column 1: RP ZORBAX Eclipse XDB-C18 column (150 mm × 2.1 m × 5 μm) A: ACN:IPP (1:2) + 0.1% f.a. B: H2O + 0.1%f.a. Column 2: KINETEX HILIC column (30 mm × 3 mm × 2.6 μm) A: 5 mM NH4Ac B: 5 mM NH4Ac |
QQQ in full scan mode followed by MRM transitions in negative and positives modes. | Matching the experimental MS/MS spectra with the reference spectra available in Metlin | 17:1 LPC, 17:0 LPC, 1,3-17:0 D5 DG, 17:0 CE, 1,2,3-17:0 TG, 16:0 D31-18:1 PC, 16:0 D31-18:1 PS, N-dodecanoylsphingosine, N-dodecanoylglucosyl-sphingosine and N-dodecanoylsphingosylphosphorylcholine. |
Phospholipids, glycerolipids | Navarro-Reig et al., 201885 |
| Halophytes: Salicornia ramosissima (fresh branch tips) and Halimione portulacoides (leaves) | Ground to a powder | Bligh and Dyer protocol with modifications. Extracted with CHCl3:MeOH (1:2) followed by the addition of water. The lower phase was dried under nitrogen and resuspended in mobile phase B. |
Ascentis Si column (150 mm × 1 mm, 3 μm) A: ACN:MeOH:H2O (50:25:25) + 1 mM NH4Ac B:ACN:MeOH (60:40) + 1 mM NH4Ac |
Q-Exactive with full scan MS and DDA in positive and negative modes | MS/MS spectral data | None | Phospholipids, glycolipids and glycosphingolipids | Maciel et al., 2018157 |
| Wheat leaves | Cryo-homogenization (Cryomill) | Folch method with modifications. Extracted with MeOH, CHCl3 vacuum dried and resuspended in BuOH:MeOH (1:1) |
Ascentis Express RP-Amide column (50 mm × 2.1 mm, 2.7 μm) A: 10 mM a.f. in H2O:MeOH:THF (50:20:30) B: 10 mM a.f. in H2O:MeOH:THF (5:20: 75) |
QQQ in positive and negative modes | MRM transitions | LPC(17:0), PC(34:1), PE(34:0), PG(34:1), PI(36:2), PS(34:0). |
Phospholipids | Cheong et al., 2019, Cheong et al., 2020158,159 |
| Vascular plants (kale leaves and corn roots) | Cryo-homogenization (Cryomill) | Bligh and Dyer method. Samples were incubated in hot IPP prior to extraction. Extracted with MeOH and CHCl3 followed by the addition of H2O. The bottom organic layer was dried under a nitrogen stream and reconstituted in CHCl3:MeOH (1:1) |
HILIC column (Luna 100 mm× 2 mm × 3 μm) to screen for any novel lipid classes A: ACN:H2O (97:3) + 10 mM NH4Ac B: 10 mM NH4Ac in pure H2O C30-RPLC (150 mm × 2 × 2.6 μm) to confirm the HILIC results A: ACN:H2O (60:40) +10 mM a.f. + 0.1% f.a. B: IPP:ACN:H2O (90:10:1) + 10 mM a.f. + 0.1% f.a. |
Q-extractive orbitrap using a top-20 DDA in positive and negative modes | MS/MS fragmentation patterns | SQDG 16:0/16:0, MGDG 16:3(7Z,10Z,1 3Z)/18:3(9Z,12Z,15Z), PA 18:1(9Z)/18:1(9Z), PG 18:0/20:4(5Z,8Z,11Z,14Z), PC 18:0/20:4(5Z,8Z,11Z,14Z), PE 18:0/20:4(5Z,8Z,11Z,14Z), diether PC O-18:0/O-18:0, plasmalogen PC P-18:0/20:4(5Z,8Z,11Z,14Z), plasmalogen PE P-18:0/20:4(5Z,8Z,11Z,14Z), MMPE 16:0/16:0 and DMPE 16:0/16:0, LPA 20:4(5Z,8Z,11Z,14Z), LPC 18:1(9Z), LPE 18:0, plasmalogen LPE P-18:0; PI 18:0/20:4(5Z,8Z,11Z,14Z); DLCL 18:2(9Z,12Z)/18:2(9Z,12Z), and SM d18:1/18:0. |
Phospholipids, polar and neutral glycerolipids, phytosphingolipids, glycolipids. Acylphosphatidylglycerols, a modified class of acylphosphatidylglycerols and their isomeric forms in corn roots. Regioisomers of lysophospholipids in kale leaves | Pham et al., 201996 |
| Root, stem, leaf, and petiole tissues of Rapeseed | Intact plant tissues were used without homogenization | Plant tissue harvested directly into hot IPP and extracted with CHCl3:H2O as described by Welti et al., 2002 |
Acquity UPLCTM BEH C18 column (100 mm × 2.1 mm × 2 μm) A: H2O:MeOH:ACN:300 mM NH4Ac (20:20:20:1) B: IPP:MeOH:300 mM NH4Ac (180:20:3) |
TripleTOF using DDA and MS/MSALL scan in positive and negative modes | RT, accurate m/z, and fragmentation ion patterns | PC-12:0/12:0, PA-14:0/14:0, PE-12:0/12:0, PG-20:0/20:0, PI-16:0/18:0, PS-20:0/20:0, LPC-19:0, LPG-18:0, LPE-18:0, MGDG18:0/18:0, DGDG-18:0/18:0. TAG-17:0/17:0/17:0 DAG-17:0/17:0-d5 |
Phospholipids, glycerolipids | Lu et al., 2019120 |
| Salad (Lactuca sativa var. capitata nidus tenerimma), deep frozen spinach (Spinacia oleracea), raspberries (Rubus idaeus) and strawberries (Fragaria) | Salad was manually cut into small pieces and homogenized using an ultra-turax. Raspberries and strawberries were homogenized using a hand blender | Extracted with IPP, n-hexane and H2O according to the protocol by Markham et al., 2006, followed by alkaline hydrolysis, dried down and reconstituted in IPP:H2O (65:35) |
C18 Acquity UHPLC HSS T3 column (150 mm × 2.1 mm × 1.8 μm) A: ACN:H2O (3:2) + 0.1% f.a. + 10 mM a.f. B: IPP:ACN (9:1) + 0.1% f.a. + 10 mM a.f. |
Q-Exactive using top-10 DDA in positive and negative modes | Lipid Data Analyzer and manual inspection of data. | C16 Lactosyl(β) Ceramide (d18:1/16:0) |
GIPC | Panzenboeck et al., 2020160 |
| Green alga, Arabidopsis tissues, maize, tomato and grey poplar and common beech trees | Bead beater | Extracted with pre-cooled MeOH:MTBE (1:3) solution followed by MeOH:water (1:3) extraction. The upper-phase was dried down and resuspended in ACN:IPP (70:30) |
C8 column (100 mm × 2.1 mm × 1.7 μm) A: H2O:IPP:ACN (446 g:130 g:300 g)+ 10 ml 1 M NH4Ac+ ml a.a. B: IPP:ACN (236 g :546 g) + 10 ml 1 M NH4Ac + 1 ml a.a. |
Q-Exactive with full scan mode in positive and negative modes | Matching the exact m/z and RT to a list of potential compounds | PC 34:0 |
Phospholipids, glycerolipids | Lapidot-Cohen et al., 2020161 |
| Banana fruit peel | Crushing | Extracted with MeOH followed by MTBE and H2O. The upper phase was dried with nitrogen stream and resuspended in IPP | C18 column (100 mm × 2.1 mm × 1.7 μm) A: ACN:H2O (6:4) + 10 mM a.f. B: ACN:IPP (1:9) + 10 mM a.f. |
Q-Exactive using full scan MS in positive and negative modes | LipidSearch | None. Content of each lipid species is presented as mean intensity | Plycerophospholipids, saccharolipids, and glycerolipids | Liu et al., 2020162 |
| Arabidopsis thaliana leaves, rosettes, internodes, roots, seedlings, seeds and siliques | Cryo-homogenization (Cryomill) | Extracted with CHCl3: MeOH: 300 mM NH4Ac in H2O (30:41.5:3.5), dried down in a speed vac and resuspended in BuOH:MeOH (1:1) |
InfinityLab Poroshell C18 column (100 mm × 2.1 mm × 2.7 μm) A: ACN:H2O (6:4) + 10 mM a.f. B: IPP:ACN (9:1) + 10 mM a.f. |
QToF using SWATH in positive mode | MS-DIAL, m/z values, RT pattern and manual inspection of the MS/MS spectra | None | Phospholipids, glycerolipids, sphingolipids | Kehelpannala et al., 2020, Kehelpannala et al., 2021, Schillaci et al., 202153,106,163 |
Lipid identification
High-throughput untargeted data acquisition strategies on high-resolution mass spectrometers generate a large amount of spectral data,108 which makes the accurate annotation of the mass spectrometric features challenging. Since the launch of the LipidBlast library in 2013,102 over 25 commercial and open-source software tools have been developed to process data and identify lipids.108 However, there are currently no lipid libraries developed specifically for the identification of plant lipids. A customized library for plant lipid identification will be of great value for plant lipid researchers.Most of the developed software tools use in silico lipid libraries, which can be rapidly generated by computing all possible combinations of fatty acids, head groups, linkages and backbones with fragments predicted using a simple set of rules.108 Commercial software tools include for example SimLipid (PREMIER Biosoft), Lipid SearchTM (Thermo Fisher Scientific),109 Lipid Annotator software97 (Agilent Technologies), and LipidyzerTM platform110 (Sciex) while freely available open-access tools include lipid data analyzer 2 (LDA2),111 LipidBlast,102 Competitive Fragmentation Modeling-ID (CFM-ID),112 LIQUID,113 MS-DIAL,101 Greazy,114 lipidr,115 LipiDex,116 LipidMS117 and LipidMatch Flow.118 From these tools, MS-DIAL is the most commonly used open-source software, which is compatible with many LC-MS workflows.108 The acquisition modes, data processing pipelines and features of the above-mentioned LC-MS based lipidomic software tools have recently been reviewed and compared by Züllig and Köfeler, 202035 and Züllig et al., 2020.119 Despite all these software solutions, unambiguous structure identification is still a challenging task, mostly due to the over annotation of lipid identities that cause false-positive results and demand manual correction of the lipid annotations.35,120 This problem may be circumvented by developing a lipid library where the lipid identities have been manually curated and validated at least for a model organism such as Arabidopsis. Recently, we have compiled a manually curated database containing over 800 lipids in Arabidopsis tissues with information on the precursor m/z values, adducts, product ions, characteristic fragmentation patterns and retention times.53 The information therein can be used to confirm lipid identities and develop targeted MS methods for lipid analysis.
Lipid quantification
Qualitative analyses focus on the profiling and comparison of lipids in biological samples while quantitative analyses aim to provide the concentrations of specific lipids in a biological matrix using meticulously developed quantification methods and lipid standards.80 Qualitative comparisons between biological samples use a limited number of internal standards or no standards at all; instead, the peak intensities or area of detected lipids are compared.2 This approach is generally known as lipid profiling and can provide comprehensive comparisons between biological samples when combined with substantive statistical analyses.2,121–126For the absolute quantification of lipids, ideally, one stable isotope-labelled internal standard per compound should be used to account for the ion suppression effects that can arise from matrix effects and changes in mobile phase composition.35 Absolute quantification will immediately indicate whether a lipid is a major or a minor species and allows the direct comparison of lipidomic results across different laboratories, which increases the confidence in lipidomic data.127 However, using a lipid standard for each lipid species is almost impossible, particularly in large-scale lipidomic studies where several thousand lipids are present in the biological sample of interest.83 Therefore, instead of one lipid standard for each lipid species, the “minimum standards” established by the Lipidomics Standards Initiative128 require that at least one internal standard per class should be used for lipid quantification. However, it is important to note that even then only a relative quantification can be achieved where the increasing and decreasing patterns of lipids between two samples are compared.129 While the use of a single internal standard per lipid class may account for errors in extraction efficiency and systematic errors, concentrations of individual species cannot be established.129 This is because the instrument's response for different lipid species even within the same lipid class can differ significantly depending on various factors.129,130 For example, the instrument's response for phospholipids is dependent on acyl chain length, acyl chain unsaturation, the structure of the polar head group, total lipid concentration, solvent composition, and instrument settings.130 Consequently, multiple internal standards per lipid class distributed across the entire retention time range and acyl chain composition would be required for an accurate quantitative analysis where the concentrations of individual lipids need to be recorded.35,129 Hence, the choice of quantification strategy depends on the scope of the study, and it is necessary to carefully consider all the different strategies before undertaking a lipidomic analysis. The principles of quantification, selection of internal standards for different analytical platforms and the factors that affect the accurate quantification of lipids are extensively reviewed by Wang et al.2
Standardizing lipidomic methodologies
The lipidomics standards initiative (https://lipidomics-standards-initiative.org/) was established in 2018 to provide guidelines and standards for lipidomics.128 The first draft of guidelines is now provided, which covers the entire lipidomic workflow, including pre-analytics, lipid extraction, data acquisition, lipid identification, quantification, method validation, quality control and data reporting.128 If the guidelines introduced by the Lipidomics Standards Initiative128 are followed by the lipid research community, this could enable the cross-comparison of lipidomic data across many different laboratories. For example, we recently developed a comprehensive lipid map of Arabidopsis across development; this includes a catalogue of the major lipids observed in different Arabidopsis tissues and provides the initial framework for a plant lipid database.106 This Arabidopsis lipid database has great potential to be expanded, for example, by including profiles of sphingolipids, sterol lipids, oxylipins and prenol lipids detected in diverse Arabidopsis tissues such as leaves, stems, flowers and roots, during development. Many laboratories around the world conduct studies comparing the lipid profiles of Arabidopsis wild-type and mutants under standard and various stress conditions. If the data generated from all these studies can be collated to build a common platform similar to the Arabidopsis lipid map, this would enable an effective sharing and comparison of data and results. Following the minimum standards set by the Lipidomics Standards Initiative128 would be a first step towards achieving such a resource that assists the lipid research community.Furthermore, to enable the efficient cross-comparison of lipidomic data, it is advisable to follow the quality control (QC) guidelines set by the lipidomics standards initiative (https://lipidomics-standards-initiative.org/). QC in lipidomic analyses is an important aspect that must be used at all times to ensure data quality. QC samples are used to stabilize the analytical instrument, monitor the instrument performance131 and account for within and between batch variations in mass spectrometric responses.132 The occurrence of systemic variations of instrument's response within and between batches, for example, due to variations in mobile phase composition, system calibration, differences in MS detection sensitivity and resolution, is often encountered in untargeted LC-MS metabolomics.132 The qualitative and quantitative tools that can be used to account for batch variations have previously been evaluated and discussed by Sanchez-Illana et al., 2018132 while various QC strategies in mass spectrometry-based lipidomics have been reviewed by Xie et al., 2017.131
Identifying lipid functions
Plant scientists have studied lipids for over 50 years and their initial focus was mainly directed at understanding their biosynthesis.133 The development of Arabidopsis thaliana as a genetic plant model, and the sequencing of its genome,134 enabled the exploration of many lipid biosynthetic pathways including the synthesis of membrane glycerolipids, sphingolipids, triacylglycerols, cutins, waxes and suberins.133,135 With the advent of innovative mass spectrometric technologies in the 1990s,71 plant biologists started applying mass spectrometry to study lipid metabolic pathways involved in plant development and stress responses,71,136 thereby identifying lipids that act as substrates and products of enzymes137 and detecting lipid-metabolizing enzymes.21 To gain a clear understanding of the in vivo functions of lipids in plants, often several different approaches are required including genetic, genomic19 and biochemical studies. These studies can provide novel insights into the roles of lipids in plants and the information may help in designing engineering strategies in biotechnology.19 For example, TGs have been long known to be the major energy reserves in seeds;19,138 however, newer studies have shown that they are also involved in cell division and expansion,139 stomatal opening,140 membrane lipid remodelling,19 organ formation,19 stress response141 and pollination.142 This suggests that strategies aiming to increase the amount of TGs in plant tissues for biodiesel and industrial chemical production must be designed carefully,19 as an overaccumulation of TGs could prove detrimental to the growth of such engineered plants.19It has been estimated that the cellular lipidome comprises hundreds of thousands of diverse lipids1,143 and the plant cellular sphingolipidome alone may comprise at least 500 and perhaps thousands of different species of sphingolipids.144 Theoretically, possible lipid structures exceed 180000 without even considering all the position isomers, backbone substitutions and stereochemistry. Indeed several million potential lipids could exist when all structural differences are taken into account.98 This suggests that much of the plant lipidome is yet to be fully described and that there are likely many lipid species to be discovered. However, new species are most often present in low levels in complex biological matrices compared to other more common lipids,107 which is one of the main reasons why they are yet to be discovered. Due to their low abundance, the precursor ions will not be selected for fragmentation by data-dependent acquisition methods in the LC-MS/MS approaches,107 which makes it difficult to identify them. In shotgun lipidomics, the MS/MS spectra of novel species are difficult to interpret due to the presence of product ions of co-fragmented lipids and chemical noise.107 Furthermore, identifying the structures of all unknown lipids in a complex biological sample is challenging since thousands of unidentified mass spectrometric features are detected,92 some of which may be artifacts generated from in-source fragmentation145 while some peaks could also be the result of solvent masses.146 Multiple adduct formation by the same lipid species and isotopic peaks may further complicate the identification of novel lipids.146 Together these factors hinder the systematic discovery of novel lipids.
A strategy that can be used to detect novel lipids is to look for new peaks in the mass spectra of the treated samples, which are absent in control samples. Since plant cells often produce lipids in response to environmental stresses and during the developmental process,147 there is potential to discover new lipid species through a comparison of lipid profiles from treated and control plant tissues. Given that the respective biosynthetic genes are identified first, the functions of newly discovered lipids can be studied in the corresponding mutants. For example, a new class of lipids, glucuronosyldiacylglycerols, was discovered in phosphorus-starved Arabidopsis plants, and further studies showed that they are essential for protection against phosphorus depletion.148 The authors observed a new peak in the mass spectra of lipid extracts from the leaves of phosphorus-starved wild-type plants but not in the extracts of the plants grown with sufficient phosphorus.148 Further investigations into the new peak by tandem mass spectrometry revealed the building blocks to be a glucuronosylglycerol and two fatty acids, which suggested that the structure is a glucuronosylated diacylglycerol.148 The novel compound was purified from Arabidopsis leaf material and its structure was established by nuclear magnetic resonance spectroscopy (NMR).148 Subsequent studies revealed that the glucuronosyldiacylglycerols mitigate phosphorus depletion stress as sqd2 mutant plants, which were unable to accumulate SQDGs and glucuronosyldiacylglycerol, showed severe growth defects under phosphorus limiting conditions.148 In another study, mechanical disruption of leaf tissue caused by wounding, pathogen attack and cold stress stimulated the production of an extensive variety of oxidized and acylated galactolipids in Arabidopsis.149 The enzyme responsible for the acylation of the galactolipid head group was purified from oat leaves and subsequently identified using tandem MS. This led to the identification of the Arabidopsis ortholog, which was subsequently confirmed through lipid analysis of loss-of-function mutants.149 These examples highlight that by monitoring the lipid changes of plants subjected to stress conditions and by comparing the lipid profiles of loss-of-function mutants and wild-type plants, novel lipids essential for the survival of plants under challenging environmental conditions can be detected and their specific functions can be uncovered.
Conclusions
LC-MS based technology is now considered as a powerful tool to explore lipid biosynthetic pathways, identify lipid-metabolizing enzymes, and uncover lipid functions.Plant lipid extraction methods, which were initially adapted from animal lipid extraction methods, have also vastly improved and modified to better suit the requirements for extracting lipids from plant tissue.
Shotgun lipidomics and LC-MS based lipidomics are the two lipidomic approaches widely used to effectively analyse plant lipids. For LC-MS based plant lipidomic workflows, there is a shift towards using high-resolution mass spectrometers such as QToF and orbitrap coupled with RPLC and HILIC. Most high-resolution mass spectrometers now offer high sensitivity with higher resolution that can be used for both targeted and discovery lipidomic approaches. Nevertheless, the unambiguous identification of lipids is still extremely challenging and requires the manual curation of data sets, and yet, no lipid library specifically built for plant lipid identification is available.
The development of high-resolution mass spectrometers and comprehensive data acquisition methods enable the detection of not only known lipid species but also a plethora of unidentified features. It is currently impossible to identify all unknown mass spectrometric features that can be detected and many of them may be due to artefacts. However, recent studies have shown that by considering lipids as part of an integrated system with enzymes and metabolites and in the context of plant function, new lipids essential for the survival of plants can be identified and studied.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
C. K. is funded through a Research Training Program Scholarship provided by the Australian Commonwealth Government and the University of Melbourne. BE is supported by an Australian Research Council Future Fellowship [FT160100276] and the University of Melbourne Botany Foundation.References
- R. Harkewicz and E. A. Dennis, Annu. Rev. Biochem., 2011, 80, 301–325 CrossRef CAS PubMed.
- M. Wang, C. Wang and X. Han, Mass Spectrom. Rev., 2017, 36, 693–714 CrossRef CAS PubMed.
- F. Furt, F. Simon-Plas and S. Mongrand, in The Plant Plasma Membrane, ed. A. S. Murphy, B. Schulz and W. Peer, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011 DOI:10.1007/978-3-642-13431-9_1, pp. 3–30.
- A. Cagliari, R. Margis, F. dos Santos Maraschin, A. C. Turchetto-Zolet, G. Loss and M. Margis-Pinheiro, Int. J. Plant Biology, 2011, 2, e10 CrossRef.
- W. van Leeuwen, L. Ökrész, L. Bögre and T. Munnik, Trends Plant Sci., 2004, 9, 378–384 CrossRef CAS PubMed.
- Y. Okazaki and K. Saito, Plant J., 2014, 79, 584–596 CrossRef CAS PubMed.
- M. Siebers, M. Brands, V. Wewer, Y. Duan, G. Hölzl and P. Dörmann, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2016, 1861, 1379–1395 CrossRef CAS PubMed.
- E. Fahy, D. Cotter, M. Sud and S. Subramaniam, Biochim. Biophys. Acta, 2011, 1811, 637–647 CrossRef CAS PubMed.
- S. J. Blanksby and T. W. Mitchell, Annu Rev Anal Chem, 2010, 3, 433–465 CrossRef CAS PubMed.
- T. Harayama and H. Riezman, Nat. Rev. Mol. Cell Biol., 2018, 19, 281–296 CrossRef CAS PubMed.
- Q. Guo, L. Liu and B. J. Barkla, Int. J. Mol. Sci., 2019, 20, 4264 CrossRef CAS PubMed.
- G. van Meer, EMBO J., 2005, 24, 3159–3165 CrossRef CAS PubMed.
- E. Fahy, S. Subramaniam, H. A. Brown, C. K. Glass, A. H. Merrill, Jr., R. C. Murphy, C. R. Raetz, D. W. Russell, Y. Seyama, W. Shaw, T. Shimizu, F. Spener, G. van Meer, M. S. VanNieuwenhze, S. H. White, J. L. Witztum and E. A. Dennis, J. Lipid Res., 2005, 46, 839–861 CrossRef CAS PubMed.
- G. H. Lim, R. Singhal, A. Kachroo and P. Kachroo, Annu. Rev. Phytopathol., 2017, 55, 505–536 CrossRef CAS PubMed.
- P. Dormann and C. Benning, Trends Plant Sci., 2002, 7, 112–118 CrossRef CAS PubMed.
- G. Holzl and P. Dormann, Prog. Lipid Res., 2007, 46, 225–243 CrossRef PubMed.
- G. Hölzl and P. Dörmann, Annu. Rev. Plant Biol., 2019, 70, 51–81 CrossRef PubMed.
- A. R. Cavaco, A. R. Matos and A. Figueiredo, Cell. Mol. Life Sci., 2021, 78, 4399–4415 CrossRef CAS PubMed.
- Y. Yang and C. Benning, Curr. Opin. Biotechnol., 2018, 49, 191–198 CrossRef CAS PubMed.
- E. Reszczyńska and A. Hanaka, Cell Biochem. Biophys., 2020, 78, 401–414 CrossRef PubMed.
- R. Welti, J. Shah, W. Li, M. Li, J. Chen, J. J. Burke, M. L. Fauconnier, K. Chapman, M. L. Chye and X. Wang, Front. Biosci., 2007, 12, 2494–2506 CrossRef CAS PubMed.
- U. Ali, H. Li, X. Wang and L. Guo, Mol. Plant, 2018, 11, 1328–1343 CrossRef CAS PubMed.
- A. Mamode Cassim, Y. Navon, Y. Gao, M. Decossas, L. Fouillen, A. Grélard, M. Nagano, O. Lambert, D. Bahammou, P. Van Delft, L. Maneta-Peyret, F. Simon-Plas, L. Heux, B. Jean, G. Fragneto, J. C. Mortimer, M. Deleu, L. Lins and S. Mongrand, J. Biol. Chem., 2021, 100602, DOI:10.1016/j.jbc.2021.100602.
- Q. Hou, G. Ufer and D. Bartels, Plant, Cell Environ., 2016, 39, 1029–1048 CrossRef CAS PubMed.
- P. Kachroo and A. Kachroo, Mol. Plant, 2020, 13, 351–353 CrossRef CAS PubMed.
- H. Schaller, Plant Physiol. Biochem., 2004, 42, 465–476 CrossRef CAS PubMed.
- E. J. Dufourc, Plant Signaling Behav., 2008, 3, 133–134 CrossRef PubMed.
- A. Rogowska and A. Szakiel, Phytochem. Rev., 2020, 19, 1525–1538 CrossRef CAS.
- A. H. Baczewska, W. Dmuchowski, A. Jozwiak, D. Gozdowski, P. Bragoszewska, P. Dabrowski and E. Swiezewska, Dendrobiology, 2014, 72, 177–186 CrossRef.
- A. Bajda, D. Konopka-Postupolska, M. Krzymowska, J. Hennig, K. Skorupinska-Tudek, L. Surmacz, J. Wójcik, Z. Matysiak, T. Chojnacki, E. Skorzynska-Polit, M. Drazkiewicz, P. Patrzylas, M. Tomaszewska, M. Kania, M. Swist, W. Danikiewicz, W. Piotrowska and E. Swiezewska, Physiol. Plant., 2009, 135, 351–364 CrossRef CAS PubMed.
- V. T. Luu, A. Weinhold, C. Ullah, S. Dressel, M. Schoettner, K. Gase, E. Gaquerel, S. Xu and I. T. Baldwin, Plant Physiol., 2017, 174, 370–386 CrossRef CAS PubMed.
- J. W. Han, G. J. Choi and B. S. Kim, World J. Microbiol. Biotechnol., 2018, 34, 163 CrossRef PubMed.
- A. Criscuolo, M. Zeller, K. Cook, G. Angelidou and M. Fedorova, Chem. Phys. Lipids, 2019, 221, 120–127 CrossRef CAS PubMed.
- S. Shiva, R. Enninful, M. R. Roth, P. Tamura, K. Jagadish and R. Welti, Plant Methods, 2018, 14, 14 CrossRef PubMed.
- T. Züllig and H. C. Köfeler, Mass Spectrom. Rev., 2020, n/a.
- J. Folch, M. Lees and G. H. Sloane Stanley, J. Biol. Chem., 1957, 226, 497–509 CrossRef CAS.
- E. G. Bligh and W. J. Dyer, Can. J. Biochem. Physiol., 1959, 37, 911–917 CrossRef CAS PubMed.
- D. Shearer and R. Carson, J. Assoc. Off. Agric. Chem., 1958, 41, 414–416 CrossRef CAS.
- T. A. Macmurray and W. R. Morrison, J. Sci. Food Agric., 1970, 21, 520–528 CrossRef CAS.
- C. Brown, E. Weber and C. Wilson, Crop Science, 1970, 10, 488–491 CrossRef CAS.
- A. Burgos, J. Szymanski, B. Seiwert, T. Degenkolbe, M. A. Hannah, P. Giavalisco and L. Willmitzer, Plant J., 2011, 66, 656–668 CrossRef CAS PubMed.
- I. A. de la Roche and C. J. Andrews, Plant Physiol., 1973, 51, 468–473 CrossRef CAS PubMed.
- M. J. Fishwick and A. J. Wright, Phytochemistry, 1977, 16, 1507–1510 CrossRef CAS.
- M. Axelsson and F. Gentili, PLoS One, 2014, 9, e89643 CrossRef PubMed.
- S. S. de Jesus and R. M. Filho, Renewable Sustainable Energy Rev., 2020, 133, 110289 CrossRef CAS.
- V. Matyash, G. Liebisch, T. V. Kurzchalia, A. Shevchenko and D. Schwudke, J. Lipid Res., 2008, 49, 1137–1146 CrossRef CAS PubMed.
- S. B. Ryu and X. Wang, Biochim. Biophys. Acta, 1998, 1393, 193–202 CrossRef CAS.
- C. M. Buseman, P. Tamura, A. A. Sparks, E. J. Baughman, S. Maatta, J. Zhao, M. R. Roth, S. W. Esch, J. Shah, T. D. Williams and R. Welti, Plant Physiol., 2006, 142, 28–39 CrossRef CAS PubMed.
- H. S. Vu, S. Shiva, M. R. Roth, P. Tamura, L. Zheng, M. Li, S. Sarowar, S. Honey, D. McEllhiney, P. Hinkes, L. Seib, T. D. Williams, G. Gadbury, X. Wang, J. Shah and R. Welti, Plant J., 2014, 80, 728–743 CrossRef CAS PubMed.
- M. Kates, in Advances in Lipid Research, ed. R. Paoletti and D. Kritchevsky, Elsevier, 1970, vol. 8, pp. 225–265 Search PubMed.
- A. Krastanov, Biotechnol. Biotechnol. Equip., 2010, 24, 1537–1543 CrossRef CAS.
- P. Yin and G. Xu, J. Chromatogr. A, 2014, 1374, 1–13 CrossRef CAS PubMed.
- C. Kehelpannala, T. W. T. Rupasinghe, T. Hennessy, D. Bradley, B. Ebert and U. Roessner, Plant Methods, 2020, 16, 155 CrossRef CAS PubMed.
- J. E. Markham, J. Li, E. B. Cahoon and J. G. Jaworski, J. Biol. Chem., 2006, 281, 22684–22694 CrossRef CAS PubMed.
- J. E. Markham and J. G. Jaworski, Rapid Commun Mass Spectrom, 2007, 21, 1304–1314 CrossRef CAS PubMed.
- B. Ebert, C. Rautengarten, H. E. McFarlane, T. Rupasinghe, W. Zeng, K. Ford, H. V. Scheller, A. Bacic, U. Roessner, S. Persson and J. L. Heazlewood, Nat Plants, 2018, 4, 792–801 CrossRef CAS PubMed.
- E. Kiran, P. G. Debenedetti and C. J. Peters, Supercritical fluids: fundamentals and applications, Springer Science & Business Media, 2012 Search PubMed.
- S. P. J. Kumar, S. R. Prasad, R. Banerjee, D. K. Agarwal, K. S. Kulkarni and K. V. Ramesh, Chem. Cent. J., 2017, 11, 9 CrossRef CAS PubMed.
- S. P. Jeevan Kumar, G. Vijay Kumar, A. Dash, P. Scholz and R. Banerjee, Algal Res., 2017, 21, 138–147 CrossRef.
- K. V. Probst, M. D. Wales, M. E. Rezac and P. V. Vadlani, Biotechnol. Prog., 2017, 33, 1096–1103 CrossRef CAS PubMed.
- S. S. de Jesus, G. F. Ferreira, L. S. Moreira, M. R. Wolf Maciel and R. Maciel Filho, Renewable Energy, 2019, 143, 130–141 CrossRef CAS.
- N. Imatoukene, M. Koubaa, E. Perdrix, M. Benali and E. Vorobiev, Process Biochem., 2020, 90, 139–147 CrossRef CAS.
- S. S. de Jesus, G. F. Ferreira, L. V. Fregolente and R. Maciel Filho, Algal Res., 2018, 35, 292–300 CrossRef.
- M. Cvjetko Bubalo, S. Vidović, I. Radojčić Redovniković and S. Jokić, Food and Bioprod. Process., 2018, 109, 52–73 CrossRef CAS.
- S. Obeid, N. Beaufils, S. Camy, H. Takache, A. Ismail and P.-Y. Pontalier, Algal Res., 2018, 34, 49–56 CrossRef.
- J. A. Bowden, C. Z. Ulmer, C. M. Jones, J. P. Koelmel and R. A. Yost, Metabolomics, 2018, 14, 53 CrossRef PubMed.
- X. Han and R. W. Gross, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10635–10639 CrossRef CAS PubMed.
- B. Brügger, G. Erben, R. Sandhoff, F. T. Wieland and W. D. Lehmann, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2339–2344 CrossRef PubMed.
- R. Welti, X. Wang and T. D. Williams, Anal. Biochem., 2003, 314, 149–152 CrossRef CAS PubMed.
- S. Narayanan, P. J. Tamura, M. R. Roth, P. V. V. Prasad and R. Welti, Plant, Cell Environ., 2016, 39, 787–803 CrossRef CAS PubMed.
- R. Welti and X. Wang, Curr. Opin. Plant Biol., 2004, 7, 337–344 CrossRef CAS PubMed.
- S. Shiva, T. Samarakoon, K. A. Lowe, C. Roach, H. S. Vu, M. Colter, H. Porras, C. Hwang, M. R. Roth, P. Tamura, M. Li, K. Schrick, J. Shah, X. Wang, H. Wang and R. Welti, Plants, 2020, 9, 845 CrossRef CAS PubMed.
- Z. S. Zoong Lwe, R. Welti, D. Anco, S. Naveed, S. Rustgi and S. Narayanan, Sci. Rep., 2020, 10, 22163 CrossRef CAS PubMed.
- H. C. Köfeler, A. Fauland, G. N. Rechberger and M. Trötzmüller, Metabolites, 2012, 2, 19–38 CrossRef PubMed.
- P. Tarazona, K. Feussner and I. Feussner, Plant J., 2015, 84, 621–633 CrossRef CAS PubMed.
- M. Lange, Z. Ni, A. Criscuolo and M. Fedorova, Chromatographia, 2019, 82, 77–100 CrossRef CAS.
- H. Nygren, T. Seppänen-Laakso, S. Castillo, T. Hyötyläinen and M. Orešič, in Metabolic Profiling: Methods and Protocols, ed. T. O. Metz, Humana Press, Totowa, NJ, 2011 DOI:10.1007/978-1-61737-985-7_15, pp. 247–257.
- T. Cajka and O. Fiehn, TrAC, Trends Anal. Chem., 2014, 61, 192–206 CrossRef CAS PubMed.
- T. W. T. Rupasinghe and U. Roessner, in Plant Metabolomics: Methods and Protocols, ed. C. António, Springer, New York, New York, NY, 2018, DOI:10.1007/978-1-4939-7819-9_9, pp. 125–135.
- A. M. King, R. D. Trengove, L. G. Mullin, P. D. Rainville, G. Isaac, R. S. Plumb, L. A. Gethings and I. D. Wilson, J. Chromatogr. A, 2020, 1611, 460597 CrossRef CAS PubMed.
- M. Li, L. Yang, Y. Bai and H. Liu, Anal. Chem., 2014, 86, 161–175 CrossRef CAS PubMed.
- G. Isaac, N. Munjoma, L. A. Gethings and R. S. Plumb, 2018.
- M. Lange and M. Fedorova, Anal. Bioanal. Chem., 2020, 412, 3573–3584 CrossRef CAS PubMed.
- M. Holčapek, M. Ovčačíková, M. Lísa, E. Cífková and T. Hájek, Anal. Bioanal. Chem., 2015, 407, 5033–5043 CrossRef PubMed.
- M. Navarro-Reig, J. Jaumot and R. Tauler, J. Chromatogr. A, 2018, 1568, 80–90 CrossRef CAS PubMed.
- M. de la Guardia and S. Armenta, in Comprehensive Analytical Chemistry, ed. M. D. L. Guardia and S. Armenta, Elsevier, 2011, vol. 57, pp. 121–156 Search PubMed.
- M. Xu, J. Legradi and P. Leonards, Anal. Bioanal. Chem., 2020, 412, 4313–4325 CrossRef CAS PubMed.
- N. J. Jensen and M. L. Gross, Mass Spectrom. Rev., 1987, 6, 497–536 CrossRef CAS.
- A. V. Melnik, R. R. da Silva, E. R. Hyde, A. A. Aksenov, F. Vargas, A. Bouslimani, I. Protsyuk, A. K. Jarmusch, A. Tripathi, T. Alexandrov, R. Knight and P. C. Dorrestein, Anal. Chem., 2017, 89, 7549–7559 CrossRef CAS PubMed.
- L. Gao, A. Cazenave-Gassiot, B. Burla, M. R. Wenk and F. Torta, Metabolomics, 2020, 16, 53 CrossRef CAS PubMed.
- T. F. Jorge, A. T. Mata and C. António, Philos. Trans. R. Soc., A, 2016, 374, 20150370 CrossRef PubMed.
- D. Yu, T. W. T. Rupasinghe, B. A. Boughton, S. H. A. Natera, C. B. Hill, P. Tarazona, I. Feussner and U. Roessner, Anal. Chim. Acta, 2018, 1026, 87–100 CrossRef CAS PubMed.
- T. Cajka, J. T. Smilowitz and O. Fiehn, Anal. Chem., 2017, 89, 12360–12368 CrossRef CAS PubMed.
- J. Schlotterbeck, M. Chatterjee, M. Gawaz and M. Lämmerhofer, Anal. Chim. Acta, 2019, 1046, 1–15 CrossRef CAS PubMed.
- X. Zhu, Y. Chen and R. Subramanian, Anal. Chem., 2014, 86, 1202–1209 CrossRef CAS PubMed.
- T. H. Pham, M. Zaeem, T. A. Fillier, M. Nadeem, N. P. Vidal, C. Manful, S. Cheema, M. Cheema and R. H. Thomas, Sci. Rep., 2019, 9, 5048 CrossRef PubMed.
- J. Koelmel, M. Sartain, J. Salcedo, A. Murali, X. Li and S. Stow, Agilent Technologies Application Note, 2019, 5994-0775EN Search PubMed.
- J. P. Koelmel, N. M. Kroeger, E. L. Gill, C. Z. Ulmer, J. A. Bowden, R. E. Patterson, R. A. Yost and T. J. Garrett, J. Am. Soc. Mass Spectrom., 2017, 28, 908–917 CrossRef CAS PubMed.
- M. Raetz, R. Bonner and G. Hopfgartner, Metabolomics, 2020, 16, 71 CrossRef CAS PubMed.
- L. C. Gillet, P. Navarro, S. Tate, H. Röst, N. Selevsek, L. Reiter, R. Bonner and R. Aebersold, Mol. Cell. Proteomics, 2012, 11, O111.016717 CrossRef PubMed.
- H. Tsugawa, T. Cajka, T. Kind, Y. Ma, B. Higgins, K. Ikeda, M. Kanazawa, J. VanderGheynst, O. Fiehn and M. Arita, Nat. Methods, 2015, 12, 523–526 CrossRef CAS PubMed.
- T. Kind, K. H. Liu, D. Y. Lee, B. DeFelice, J. K. Meissen and O. Fiehn, Nat. Methods, 2013, 10, 755–758 CrossRef CAS PubMed.
- T. Xu, C. Hu, Q. Xuan and G. Xu, Anal. Chim. Acta, 2020, 1137, 156–169 CrossRef CAS PubMed.
- B. Drotleff, J. Illison, J. Schlotterbeck, R. Lukowski and M. Lämmerhofer, Anal. Chim. Acta, 2019, 1086, 90–102 CrossRef CAS PubMed.
- B. Drotleff, M. Hallschmid and M. Lämmerhofer, Anal. Chim. Acta, 2018, 1022, 70–80 CrossRef CAS PubMed.
- C. Kehelpannala, T. Rupasinghe, A. Pasha, E. Esteban, T. Hennessy, D. Bradley, B. Ebert, N. J. Provart and U. Roessner, Plant J., 2021, 107, 287–302 CrossRef CAS PubMed.
- M. Bilgin, P. Born, F. Fezza, M. Heimes, N. Mastrangelo, N. Wagner, C. Schultz, M. Maccarrone, S. Eaton, A. Nadler, M. Wilm and A. Shevchenko, Sci. Rep., 2016, 6, 27920 CrossRef CAS PubMed.
- J. P. Koelmel, X. Li, S. M. Stow, M. J. Sartain, A. Murali, R. Kemperman, H. Tsugawa, M. Takahashi, V. Vasiliou, J. A. Bowden, R. A. Yost, T. J. Garrett and N. Kitagawa, Metabolites, 2020, 10, 101 CrossRef CAS PubMed.
- R. Taguchi and M. Ishikawa, J. Chromatogr. A, 2010, 1217, 4229–4239 CrossRef CAS PubMed.
- B. K. Ubhi, A. Conner, E. Duchoslav, A. Evans, R. Robinson, P. R. Baker and S. Watkins, SCIEX Technical Application Note, 2015 Search PubMed.
- J. Hartler, A. Triebl, M. Trötzmüller, A. Ziegl, G. Rechberger, F. Spener, H. Köfeler and G. Thallinger, 2015.
- Y. Djoumbou-Feunang, A. Pon, N. Karu, J. Zheng, C. Li, D. Arndt, M. Gautam, F. Allen and D. S. Wishart, Metabolites, 2019, 9, 72 CrossRef CAS PubMed.
- J. E. Kyle, K. L. Crowell, C. P. Casey, G. M. Fujimoto, S. Kim, S. E. Dautel, R. D. Smith, S. H. Payne and T. O. Metz, Bioinformatics, 2017, 33, 1744–1746 CrossRef CAS PubMed.
- M. A. Kochen, M. C. Chambers, J. D. Holman, A. I. Nesvizhskii, S. T. Weintraub, J. T. Belisle, M. N. Islam, J. Griss and D. L. Tabb, Anal. Chem., 2016, 88, 5733–5741 CrossRef CAS PubMed.
- A. Mohamed, J. Molendijk and M. M. Hill, J. Proteome Res., 2020, 19, 2890–2897 CrossRef CAS PubMed.
- P. D. Hutchins, J. D. Russell and J. J. Coon, Cell Systems, 2018, 6(621-625), e625 Search PubMed.
- M. I. Alcoriza-Balaguer, J. C. García-Cañaveras, A. López, I. Conde, O. Juan, J. Carretero and A. Lahoz, Anal. Chem., 2019, 91, 836–845 CrossRef CAS PubMed.
- J. P. Koelmel, N. M. Kroeger, C. Z. Ulmer, J. A. Bowden, R. E. Patterson, J. A. Cochran, C. W. W. Beecher, T. J. Garrett and R. A. Yost, BMC Bioinf., 2017, 18, 331 CrossRef PubMed.
- T. Zullig, M. Trotzmuller and H. C. Kofeler, Anal. Bioanal. Chem., 2020, 412, 2191–2209 CrossRef PubMed.
- S. Lu, H. Liu, C. Jin, Q. Li and L. Guo, Plant Direct, 2019, 3, e00183 CrossRef PubMed.
- J. Liu, Q. Li, J. Chen and Y. Jiang, Foods, 2020, 9, 894 CrossRef CAS PubMed.
- A. Millner, D. Y. Lizardo and G. E. Atilla-Gokcumen, Proteomics, 2020, 20, 2000013 CrossRef CAS PubMed.
- X. Zhang, W. Liu, J. Zan, C. Wu and W. Tan, Sci. Rep., 2020, 10, 14509 CrossRef CAS PubMed.
- A. Acera, X. Pereiro, B. Abad-Garcia, Y. Rueda, N. Ruzafa, C. Santiago, I. Barbolla, J. A. Duran, B. Ochoa and E. Vecino, Mol. Vis., 2019, 25, 934–948 CAS.
- S. B. Breitkopf, S. J. H. Ricoult, M. Yuan, Y. Xu, D. A. Peake, B. D. Manning and J. M. Asara, Metabolomics, 2017, 13, 30 CrossRef PubMed.
- A. Gil, W. Zhang, J. C. Wolters, H. Permentier, T. Boer, P. Horvatovich, M. R. Heiner-Fokkema, D.-J. Reijngoud and R. Bischoff, Anal. Bioanal. Chem., 2018, 410, 5859–5870 CrossRef CAS PubMed.
- G. Liebisch, K. Ekroos, M. Hermansson and C. S. Ejsing, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1862, 2017, 747–751 Search PubMed.
- G. Liebisch, R. Ahrends, M. Arita, M. Arita, J. A. Bowden, C. S. Ejsing, W. J. Griffiths, M. Holčapek, H. Köfeler, T. W. Mitchell, M. R. Wenk, K. Ekroos and C. Lipidomics Standards Initiative, Nature Metabolism, 2019, 1, 745–747 CrossRef PubMed.
- S. K. K. Mackenzie Pearson, P. Norris and C. Hunter, SCIEX, 2018 Search PubMed.
- M. Koivusalo, P. Haimi, L. Heikinheimo, R. Kostiainen and P. Somerharju, J. Lipid Res., 2001, 42, 663–672 CrossRef.
- Y. Xie, H. Ma, F. Wei, X. Lyu, Z. Wu, B. Wu, S. Xu, X. Dong, H. Chen and F. Huang, Proc. - Soil Crop Sci. Soc. Fla., 2017, 2, 217–224 Search PubMed.
- Á. Sánchez-Illana, J. D. Piñeiro-Ramos, J. D. Sanjuan-Herráez, M. Vento, G. Quintás and J. Kuligowski, Anal. Chim. Acta, 2018, 1019, 38–48 CrossRef PubMed.
- H. Tenenboim, A. Burgos, L. Willmitzer and Y. Brotman, Biochimie, 2016, 130, 91–96 CrossRef CAS PubMed.
- I. The Arabidopsis Genome, Nature, 2000, 408, 796–815 CrossRef PubMed.
- Y. Li-Beisson, B. Shorrosh, F. Beisson, M. X. Andersson, V. Arondel, P. D. Bates, S. Baud, D. Bird, A. Debono, T. P. Durrett, R. B. Franke, I. A. Graham, K. Katayama, A. A. Kelly, T. Larson, J. E. Markham, M. Miquel, I. Molina, I. Nishida, O. Rowland, L. Samuels, K. M. Schmid, H. Wada, R. Welti, C. Xu, R. Zallot and J. Ohlrogge, Arabidopsis Book, 2013, 11, e0161 CrossRef PubMed.
- X. Wang, Plant Physiol., 2005, 139, 566–573 CrossRef CAS PubMed.
- X. Wang, W. Li, M. Li and R. Welti, Physiol. Plant., 2006, 126, 90–96 CrossRef CAS.
- J. Bewley and M. Black, 1986.
- K. Takahashi, R. Morimoto, H. Tabeta, M. Asaoka, M. Ishida, M. Maeshima, H. Tsukaya and A. Ferjani, Plant Cell Physiol., 2017, 58, 668–678 CrossRef CAS PubMed.
- D. H. McLachlan, J. Lan, C.-M. Geilfus, A. N. Dodd, T. Larson, A. Baker, H. Hõrak, H. Kollist, Z. He, I. Graham, M. V. Mickelbart and A. M. Hetherington, Curr. Biol., 2016, 26, 707–712 CrossRef CAS PubMed.
- S. D. Singer, J. Zou and R. J. Weselake, Plant Sci., 2016, 243, 1–9 CrossRef CAS PubMed.
- M. Wolters-Arts, W. M. Lush and C. Mariani, Nature, 1998, 392, 818–821 CrossRef CAS PubMed.
- M. Wang, C. Wang, R. H. Han and X. Han, Prog. Lipid Res., 2016, 61, 83–108 CrossRef CAS PubMed.
- M. O. Pata, Y. A. Hannun and C. K. Y. Ng, New Phytologist, 2010, 185, 611–630 CrossRef CAS PubMed.
- C. Hu, W. Luo, J. Xu and X. Han, Mass Spectrom. Rev., 2020 DOI:10.1002/mas.21659.
- R. M. Gathungu, P. Larrea, M. J. Sniatynski, V. R. Marur, J. A. Bowden, J. P. Koelmel, P. Starke-Reed, V. S. Hubbard and B. S. Kristal, Anal. Chem., 2018, 90, 13523–13532 CrossRef CAS PubMed.
- E. Ruelland and O. Valentova, Front. Plant Sci., 2016, 7, 324 Search PubMed.
- Y. Okazaki, Y. Kamide, M. Y. Hirai and K. Saito, Metabolomics, 2013, 9, 121–131 CrossRef CAS PubMed.
- A. K. Nilsson, O. N. Johansson, P. Fahlberg, M. Kommuri, M. Töpel, L. J. Bodin, P. Sikora, M. Modarres, S. Ekengren, C. T. Nguyen, E. E. Farmer, O. Olsson, M. Ellerström and M. X. Andersson, Plant J., 2015, 84, 1152–1166 CrossRef CAS PubMed.
- R. Welti, W. Li, M. Li, Y. Sang, H. Biesiada, H. E. Zhou, C. B. Rajashekar, T. D. Williams and X. Wang, J. Biol. Chem., 2002, 277, 31994–32002 CrossRef CAS PubMed.
- S. P. Devaiah, M. R. Roth, E. Baughman, M. Li, P. Tamura, R. Jeannotte, R. Welti and X. Wang, Phytochemistry, 2006, 67, 1907–1924 CrossRef CAS PubMed.
- J. Hummel, S. Segu, Y. Li, S. Irgang, J. Jueppner and P. Giavalisco, Front. Plant Sci., 2011, 2, 54 CAS.
- Y. Okazaki, H. Otsuki, T. Narisawa, M. Kobayashi, S. Sawai, Y. Kamide, M. Kusano, T. Aoki, M. Y. Hirai and K. Saito, Nat. Commun., 2013, 4, 1510 CrossRef PubMed.
- M.-Y. Liu, A. Burgos, L. Ma, Q. Zhang, D. Tang and J. Ruan, BMC Plant Biol., 2017, 17, 165 CrossRef PubMed.
- P. Giavalisco, Y. Li, A. Matthes, A. Eckhardt, H.-M. Hubberten, H. Hesse, S. Segu, J. Hummel, K. Köhl and L. Willmitzer, Plant J., 2011, 68, 364–376 CrossRef CAS PubMed.
- D. Yu, B. A. Boughton, C. B. Hill, I. Feussner, U. Roessner and T. W. T. Rupasinghe, Front. Plant Sci., 2020, 11, 1 CrossRef PubMed.
- E. Maciel, A. Lillebø, P. Domingues, E. da Costa, R. Calado and M. R. M. Domingues, Phytochemistry, 2018, 153, 94–101 CrossRef CAS PubMed.
- B. E. Cheong, W. W. H. Ho, B. Biddulph, X. Wallace, T. Rathjen, T. W. T. Rupasinghe, U. Roessner and R. Dolferus, Metabolomics, 2019, 15, 144 CrossRef PubMed.
- B. E. Cheong, O. Onyemaobi, W. Wing Ho Ho, T. B. Biddulph, T. W. T. Rupasinghe, U. Roessner and R. Dolferus, Cells, 2020, 9, 1309 CrossRef CAS PubMed.
- L. Panzenboeck, N. Troppmair, S. Schlachter, G. Koellensperger, J. Hartler and E. Rampler, Metabolites, 2020, 10, 375 CrossRef CAS PubMed.
- T. Lapidot-Cohen, L. Rosental and Y. Brotman, Curr Protoc Plant Biol, 2020, 5, e20109 CAS.
- J. Liu, Q. Li, J. Chen and Y. Jiang, Foods, 2020, 9, 894 CrossRef CAS PubMed.
- M. Schillaci, C. Kehelpannala, F. Martinez-Seidel, P. M. C. Smith, B. Arsova, M. Watt and U. Roessner, Metabolites, 2021, 11, 358 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |