
Ruthenium(II)–arene complexes as anti-metastatic agents, and related techniques
Chanchal
Sonkar
 a,
Sayantan
Sarkar
b and
Suman
Mukhopadhyay
*ab
a,
Sayantan
Sarkar
b and
Suman
Mukhopadhyay
*ab
aDepartment of Biosciences and Biomedical Engineering, School of Engineering, Indian Institute of Technology Indore, Khandwa Road, Simrol, Indore 453552, MP, India. E-mail: suman@iiti.ac.in
bDepartment of Chemistry, School of Basic Sciences, Indian Institute of Technology Indore, Khandwa Road, Simrol, Indore 453552, MP, India
First published on 15th September 2021
Abstract
With the discovery of cisplatin, a vast area of applications of metallodrugs in cancer treatment was opened but due to the side effects caused by the cisplatin complexes, researchers began to look for alternatives with similar anticancer properties but fewer side effects. Ruthenium was found to be a promising candidate, considering its significant anticancer properties and low side effects. Several ruthenium complexes, viz. NAMI-A, KP1019, KP1339, and TLD1433, have entered clinical trials. Some other arene ruthenium complexes such as RM175 and RAPTA-C have also entered clinical trials but very few of them have shown anti-metastatic properties. Herein, we provide information and probable mechanistic pathways for ruthenium(II)–arene complexes that have been studied, so far, for their anti-metastatic activities. Also, we discuss the techniques and their significance for determining the anti-metastatic effects of the complexes.
 Chanchal Sonkar | Chanchal Sonkar was born in Madhya Pradesh, India. She received her Bachelor of Science degree from Devi Ahilya Vishwavidyalaya, Indore, Madhya Pradesh, and Master of Science degree from Jawaharlal Nehru University, New Delhi, India. She joined the Department of Biosciences and Biomedical Engineering, IIT Indore, as a Ph.D. student in December 2016. Currently, she is working on the Anticancer Activities of Ruthenium Complexes and their Mechanistic Pathway. |
 Sayantan Sarkar | Sayantan Sarkar was born in West Bengal, India. He received his Bachelor of Science degree from Calcutta University, Kolkata, West Bengal, and Master of Science degree from Vidhyasagar University, West Bengal, India. He joined the Department of Chemistry, IIT Indore, as a Ph.D. student in July 2019. Currently, he is working on the Design, Synthesis, and Applications of Macromolecular Assemblies. |
 Suman Mukhopadhyay | Prof. Suman Mukhopadhyay was born in Kolkata. He received his Bachelor of Science and Master of Science degrees from the University of Kalyani, West Bengal, India. He carried out his doctoral research at the Indian Association for the Cultivation of Science, Kolkata, and completed it in 2004. He subsequently went on to the National University of Singapore as a post-doctoral Fellow, and in 2006 he moved to Lisbon (Portugal) as an FCT post-doctoral fellow at Instituto Superior Técnico. In 2009 he went on to EPFL in Lausanne (Switzerland) as a recipient of the Marie-Curie International Incoming Fellowship. In 2010 he joined the Indian Institute of Technology Indore, India as an assistant professor, and currently, he is a full professor at the same institute. His research interests include organometallic compounds in therapeutics and their mechanisms, bioinspired catalysis, and metallogels and their applications. |
Introduction
Cancer is the second leading cause of death, after cardiovascular disease in developing countries, mainly because of unhealthy lifestyles and the disadvantages of environmental impacts on the lives of the people. According to WHO, the death toll, which was around 14 million per year up until 2012, is expected to increase to 26 million within two decades.1,2 Thus, there is an urgent need to produce novel anticancer drugs that can only be produced through active knowledge in radiomics, transcriptomics, metabolomics, proteomics, and a thorough understanding of cancer biology.3 Currently, various methods are available to treat cancer, such as chemotherapy, radiotherapy, immunotherapy, and surgery. Chemotherapy is an approach where therapeutic molecules are utilized to obstruct the rapidly proliferating cells. Many transition metals have been used as diagnostic and therapeutic agents and have significantly impacted anticancer drug development.4 However, it was not until the discovery of cisplatin in 1965 by Rosenberg, which proved to be a milestone in the use of metallodrugs in cancer therapy due to its versatile anticancer properties, that metal complexes have been taken seriously in cancer chemotherapy.5 Cisplatin, its congeners, and other platinum complexes showed efficacy in treating various cancers including neck, ovarian, head, cervical, lymphoma, and bladder cancer. However, despite its efficacy, it showed several side effects such as nephrotoxicity, neurotoxicity, nausea, hair loss, vomiting, bone marrow suppression, and drug resistance.6–9 Thus, the researchers started looking for an alternative that had fewer side effects than cisplatin, and ruthenium (Ru) complexes appeared to be the promising candidates for next-generation metallodrugs used for cancer treatment.10–12 These ruthenium complexes have several advantages over platinum analogues as follows. (a) Ruthenium has hexacoordinated octahedral geometry and different oxidation states (mainly RuII, RuIII and RuIV), which allow the rearrangement of the ligand in different geometries.13,14 (b) Low ligand exchange kinetics allows the ruthenium complexes to remain attached to some species in the cell during the entire life cycle. (c) Ruthenium mimics iron, which facilitates its binding to the transferrin receptors; these receptors are considered to be overly expressed in the cancer cells, which aids the selectivity of the ruthenium complexes for entry into the cancer cells.15–17 Along with these properties, ruthenium and other metal complexes, especially iridium and rhodium, can also act as a fluorescent probes for biomolecules, biosensors, molecular probes and nanosensors for the determination of biomarkers in the living cells or organisms.18–21 These ruthenium and iridium complexes can also be effectively used to target cancer cells as compared to normal cells and have remarkably garnered attention in the field of bioimaging.18,22 The wide application of these complexes may be due to their unique properties in the optical field such as large stroke shifts, thermal-, chemical- and photo-stability, and significant absorption and emission spectra in the visible light range.23–25 It is also believed that Ru(III) species act as a prodrug and they are converted into Ru(II) species due to the hypoxic environment within the cancer cells. To date, four ruthenium complexes have entered clinical trials, viz. NAMI-A, KP1019, KP1339, and TLD1433.26–28 NAMI-A entered phase II trials but due to limited efficacy, it could not proceed further for clinical development. Another ruthenium complex, KP1019, entered phase I trials but its further development was halted due to its low solubility; however, the sodium salt of KP1019, KP1339, is still under clinical trials. Also, another ruthenium complex, TLD1433, has entered phase 2a trials for prospective non-muscle invasive bladder treatment by a photodynamic therapy approach.12,26,27,29 Other organoruthenium Ru(II) complexes have also shown potential anticancer activities.30 Very recently, some ruthenium complexes such RM175, RDC11 (redox organoruthenium complex), and RAPTA-C have entered phase I clinical trials.31 Also, some other clinically approved Ru(II) arene complexes, such as AH63 and AH54, are utilized in the radiosensitization of Caco cells (human colorectal cells), and RAPTA-C in A2780 (human ovarian carcinoma cells).32 These Ru(II)–arene scaffolds have been found to be very useful templates for the development of anticancer agents due to their, stability, solubility, lipophilicity, and accessibility through the synthetic routes.33 These Ru(II)–arene complexes have octahedral geometry with exclusively low d6 spin value, where the arene ligand engages three coordination sites, and other ligands may bind with the three remaining coordination sites, giving a three-legged piano stool structure. The mononuclear ruthenium complexes have shown therapeutic and anticancer properties in both in vitro and in vivo studies.34 This encouraged the researchers to directly work on the Ru(II)-based complexes, of which the Ru(II)–arene scaffolds have captured much attention.35–37Primary tumours can be treated through surgical treatment and adjuvant therapy, however, the treatment of metastatic disease is difficult because of the resistance of therapeutic agents towards the dissemination of the tumour cells. This explains why mortality rates greater than 90% are caused by metastasis as compared to primary tumours.38 Thus, the only way to treat cancer is to intercept the metastatic process or even reverse it. Metastasis may occur due to the somatic evolution of genetically diversified cancer cell populations under specific pressure, which changes the behavior of the cells. The peripheral view of the metastatic cascade involves angiogenesis, the loss of cellular adhesion, invasion, intravasation into the circulatory or lymphatic system, dodging the cellular defence mechanism, extravasation and colonization into distant organs.39 The process of metastasis is driven by several intricate pathways in the interactions of cancerous cells with neoplastic stromal cells, which play a vital role. Thus, malignant tumour cells must first breach the basement membrane, which leads to the modulation of signal transduction pathways. The signal transduction pathways further regulate integrin-mediated cell adhesion, which also alters the cellular polarity, proliferation, invasion and survival. Generally, the focus of researchers is on anti-proliferative and tumour-shrinkage drugs, which undermines the effect of cancer invasion, thus causing a lack of anti-metastatic drugs.40 However, NAMI-A is a ruthenium complex that is known to act as an anti-metastatic agent. It was found to have remarkable anti-metastatic activity against lung metastasis in solid murine metastasizing tumours.41 On in vitro drug screening, it was found that NAMI-A showed no cytotoxicity activities against a 60 cell line NCI panel.42 This non-cytotoxicity of two Ru(III) complexes, NAMI-A and KP1019, may be because of the essential requirement of activation by reduction mechanism, which operates in vivo.43,44 However it has shown anti-metastatic activity by blocking the major steps of angiogenesis (EC proliferation and migration induced by VEGF),45 and the inhibition of MMP-2 and MMP-9 matrix metalloproteinases.46,47 It has also been found that reduced cellular motility, increased cell adhesion, decreased cell penetration ability and transient blocking of cell progression at the G2/M phase may also contribute to the anti-metastatic activity of the complex.48–50 However it is worth noting that not many studies have been conducted so far in the area of Ru(II)–arene complexes acting as an anti-metastatic agents. As such it would be interesting to get an idea about the progress of these complexes to design improved molecules in anti-metastatic field.
In this review, we have highlighted the Ru(II)–arene complexes that have shown anti-metastatic properties and their mechanistic pathways (Fig. 1). We have also briefly discussed the techniques used for determining the anti-metastatic properties and their significance for accessing the various steps of metastasis, which itself is a very complex process to unravel.
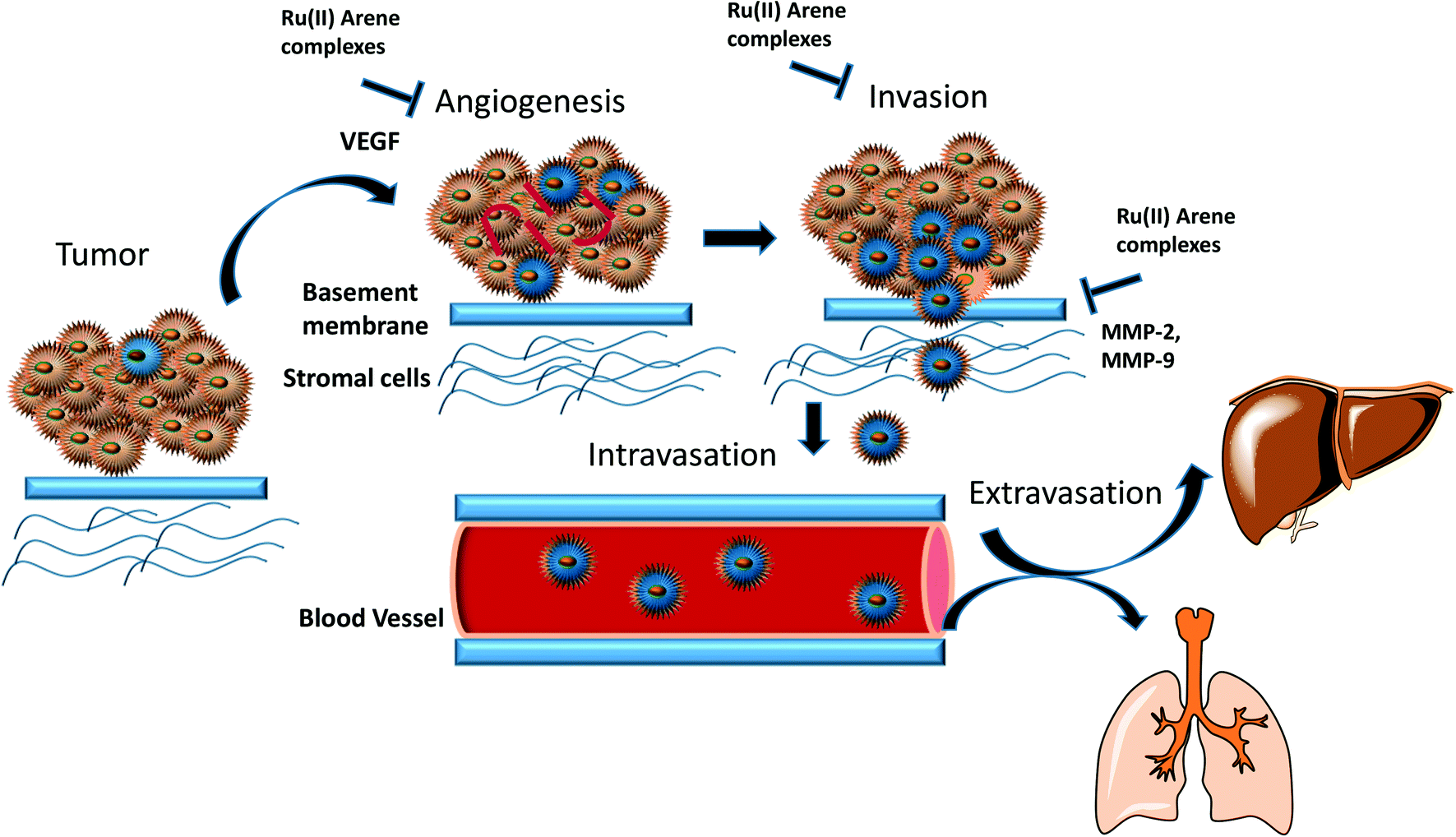 | ||
| Fig. 1 Schematic representation of the overall anti-metastatic activities of Ru(II) complexes. | ||
Complexes under clinical trials
Ru(II)–Arene complexes can show both hydrophobic and hydrophilic properties, which are expected to demonstrate additive as well as synergistic effects. The Ru(II)–arene unit, along with various diverse ligands, provides diverse structural possibilities and various modes of interactions with the biomolecules within the cells, which contribute to their good potential for anticancer drug development and therapeutics.14RAPTA complexes
RAPTA (Ru arene-PTA) complexes are susceptible to hydrolysis and bind to DNA as a crucial target.51 It was found that CBA mice having Mca mammary carcinoma lost lung metastasis numbers and weight throughout the treatment with RAPTA-C (1), RAPTA-T (4), and RAPTA-B (12).52–54 However, these complexes showed less anti-metastatic activity in comparison with NAMI-A. The RAPTA complexes (Fig. 2) were found to be effective inhibitors of thioredoxin reductase (TrxR) and cathepsin B (Cat-B).55 Among all the RAPTA complexes, only RAPTA-C was found to inhibit TrxR, and the rest inhibited Cat-B. It was noticed that one chloride derivative of the RAPTA complexes was hydrolyzed, followed by the entrance of the metal center into the active site of the cysteine residue. This metallodrug enzyme complex was further stabilized by the nitrogen of PTA and chloride of the RAPTA derivative with the remaining amino acids of Cat-B.55 It is also worth noting the selective nature of RAPTA-B, RAPTA-T, and RAPTA-C complexes, as these complexes were cytotoxic against TS/A cancer cell lines, but remained non-cytotoxic toward the HBL100 cell line (healthy cell lines). PTA-Me+ and PTA were measurably cytotoxic to normal cells as well.56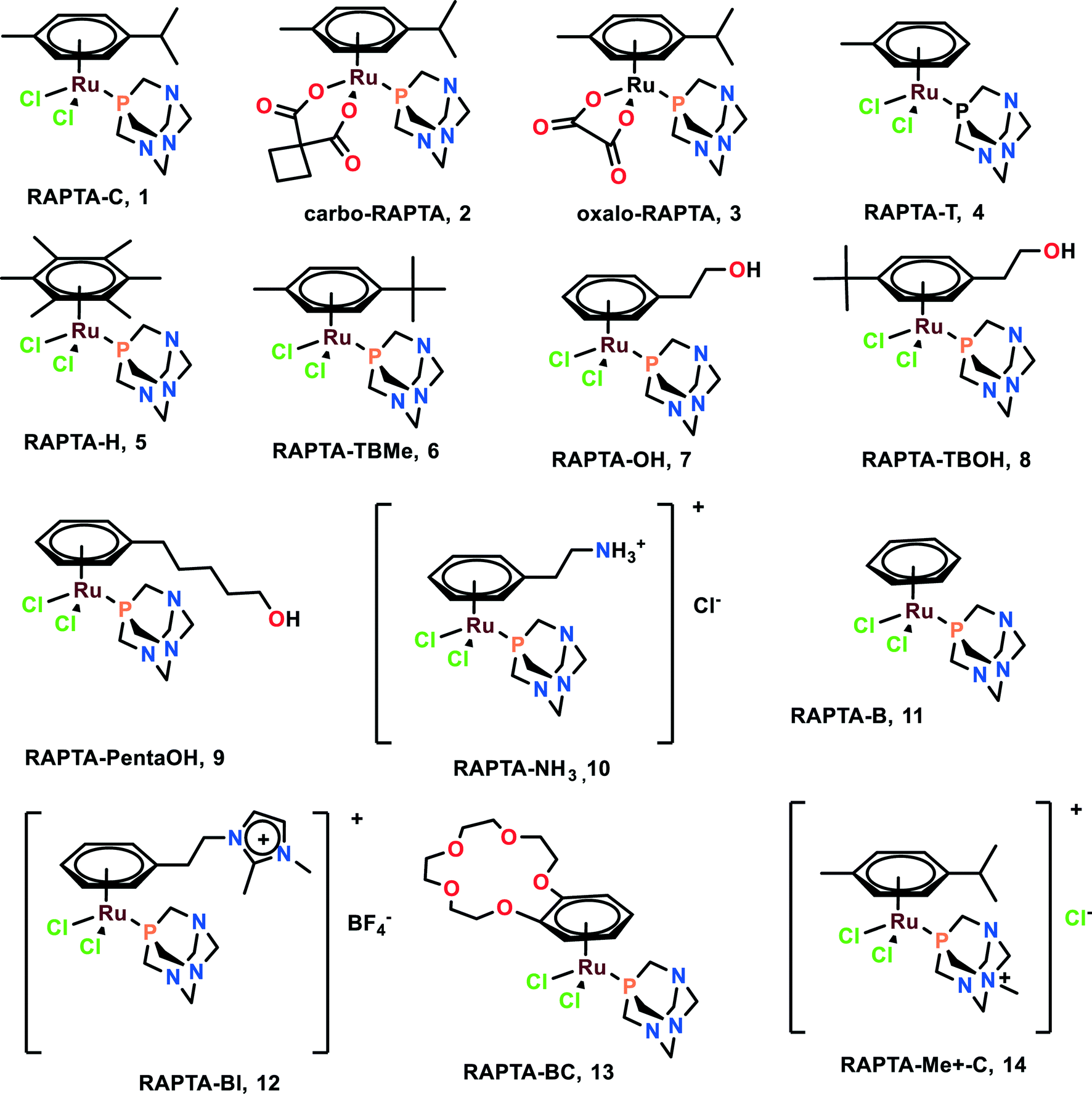 | ||
| Fig. 2 Chemical structures of RAPTA complexes referred to in this article. | ||
RAPTA-C, when working synergistically with erlotinib (EGFR inhibitor), strongly inhibited the cell viability of human endothelial cells (ECRF24 and HUVEC) and ovarian carcinoma (A2780 and A2780cisR). This synergistic combination also led to the better penetration of the complexes within the cells and also functioned as a potent anti-angiogenic agent in vivo.52 Since EGFR plays a significant role in tumour progression and metastasis,57 this combination may be used as an effective anti-metastatic agent, although further investigations are required for application in this regard.
RM175
RM175, [(η6 -C6H5Ph)Ru(N,Nen)Cl]+ (en = 1,2-ethylenediamine), as the hexafluorophosphate salt was among the first compounds to be tested for anticancer activities by the Sadler group (Fig. 3).58,59 The arene substituent facilitates the entry of the complex within the cells due to its hydrophobic nature and on aquation of the complex, it binds to the N7 guanine of DNA. The arene ring extension also enables the hydrophobic interaction of RM175 and DNA through intercalation between the base pairs.60,61 Several studies have been carried out to determine the mechanism related to the anti-metastatic nature of this complex, thus discovering new potential cellular targets. Besides binding to DNA, it was found to inhibit MMP-2 (metalloproteinase-2).60 This MMP-2 plays a crucial role in the metastasis and invasion of the cells, through the suppression of the adaptive immune system, degradation of the extracellular matrix (ECM) and modulating various pathways, which facilitate tumour invasion and progression.62,63 This governs the tumour microenvironment and also modulates cell growth and angiogenesis through various signaling pathways.63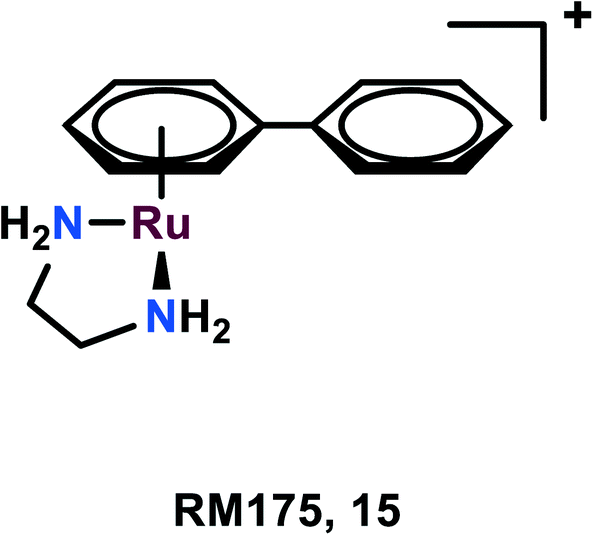 | ||
| Fig. 3 The chemical structure of RM175 complexes referred to in this article. | ||
To determine the mechanism related to the inhibitory activities of RM175, many proteins have been explored including fibronectin, collagen, and poly-L-lysine. As fibronectin and collagen are the components of the extracellular matrix and poly-L-lysine attracts the cells through electrostatic interaction, these proteins might interact with malignant cells in vivo. The MDA-MB-231 cell line when treated with RM175 tends to become more resistant to the detachment from the poly-L-lysine and fibronectin, depending upon which the cells were being grown. RM175 was found to be selective towards the invasive cell line as compared to non-invasive and non-tumourigenic cells. RM175 was found to inhibit the cellular contact-induced movement (haptotaxis) in HBL100 and MDA-MB-231 cells, which prevents metastasis formation produced by stimulus.60 Furthermore, it was found to show significant metastasis reduction in MCa mammary carcinoma in comparison with the primary tumour when tested in vivo, which further adds to the aforementioned explainations.60
ONCO4417
Substituting the RM175 counter chloride ion with the hexafluorophosphate PF6 anion leads to the formation of ONCO4417, which has also shown anti-neoplastic activities and G2/M phase cell cycle arrest. This complex has been found to have similar efficacy to cisplatin in various cell lines like ovarian, lung, esophageal, pancreatic, melanoma, and colorectal. However, it has also shown efficient activity against cisplatin-resistant cell lines like A2780cisR, which suggests this complex does not share any cross-resistance mechanism.64,65 These complexes were found to efficiently inhibit tumour metastasis.26Other significant complexes and their mechanisms
It was also found that by the incorporation of the Ru(arene) fragment, the complex exhibited enhanced anti-migratory properties. These complexes have shown increased inhibition of the wound closure in a wound-healing assay, which was comparable to the recognized anti-metastatic agent sunitinib.33,66,67 It was also reported that some Ru(II)–arene NSAIDs and tetrazole complexes have also shown anti-migratory activities through inhibiting wound closure in wound healing assays.68,69 There are also some Ru(II) diimine complexes that target mitochondria, which have shown anticancer and antimetastasis activities through signaling mediated by reactive oxygen species (ROS).70 Ru(II)phenanthroimidazole derivative complexes were also synthesized, which stabilized the c-Myc G-quadruplex DNA, thus downregulating the activity of c-Myc and causing anti-proliferative activities.71 Another Ru(II)–arene complex, (cym)Ru(5-bromo-8-hydroxyquinoline) was found to inhibit cell invasion and migration, both in the monolayer as well as in the spheroid.72 The novel Ru(II) imidazole–mesalazine Schiff base complexes were synthesized, which showed the unique property of the growth inhibition of 3D colon cancer stem cells without enhancing the stemness of the cells, thus maintaining tight regulation of gene expression.73Ruthplatins
Novel ruthplatins (16–19), which are bimetallic anticancer drugs, were designed and reported to treat tumour metastasis and drug resistance (Fig. 4). Ruthplatins are water-soluble Pt(IV)–Ru(II) heterodinuclear complexes whose activities were evaluated on both 2D and 3D tumour models.74 These were also found to have noteworthy cytotoxic activities against cisplatin-resistant cells A2780cisR and A549cisR. These complexes were also found to be active against MCF-7, MDA-MB-231 (breast cancer cells), HeLa (cervical cancer cells), and HL60 (leukemia cancer cells). However, they remain less cytotoxic towards MRC-5 normal lung fibroblasts in comparison to cisplatin. Similarly, these ruthplatins were found to be significantly active in the 3D spheroid model of MCF-7, where the number of dead cells was significantly higher in the case of ruthplatins at lower concentrations and in less time in comparison with cisplatin. This indicates the effectiveness of ruthplatins in eradicating the tumour cells in the 3D environment. The cytotoxicity of these complexes can be attributed to the S-phase cell cycle arrest. The wound-healing assay determined that ruthplatins prevent the migration ability of the cells, as only 23% and 46% wound closure occurred over the time interval of 12 h and 24 h, in comparison with sunitinib (positive control).66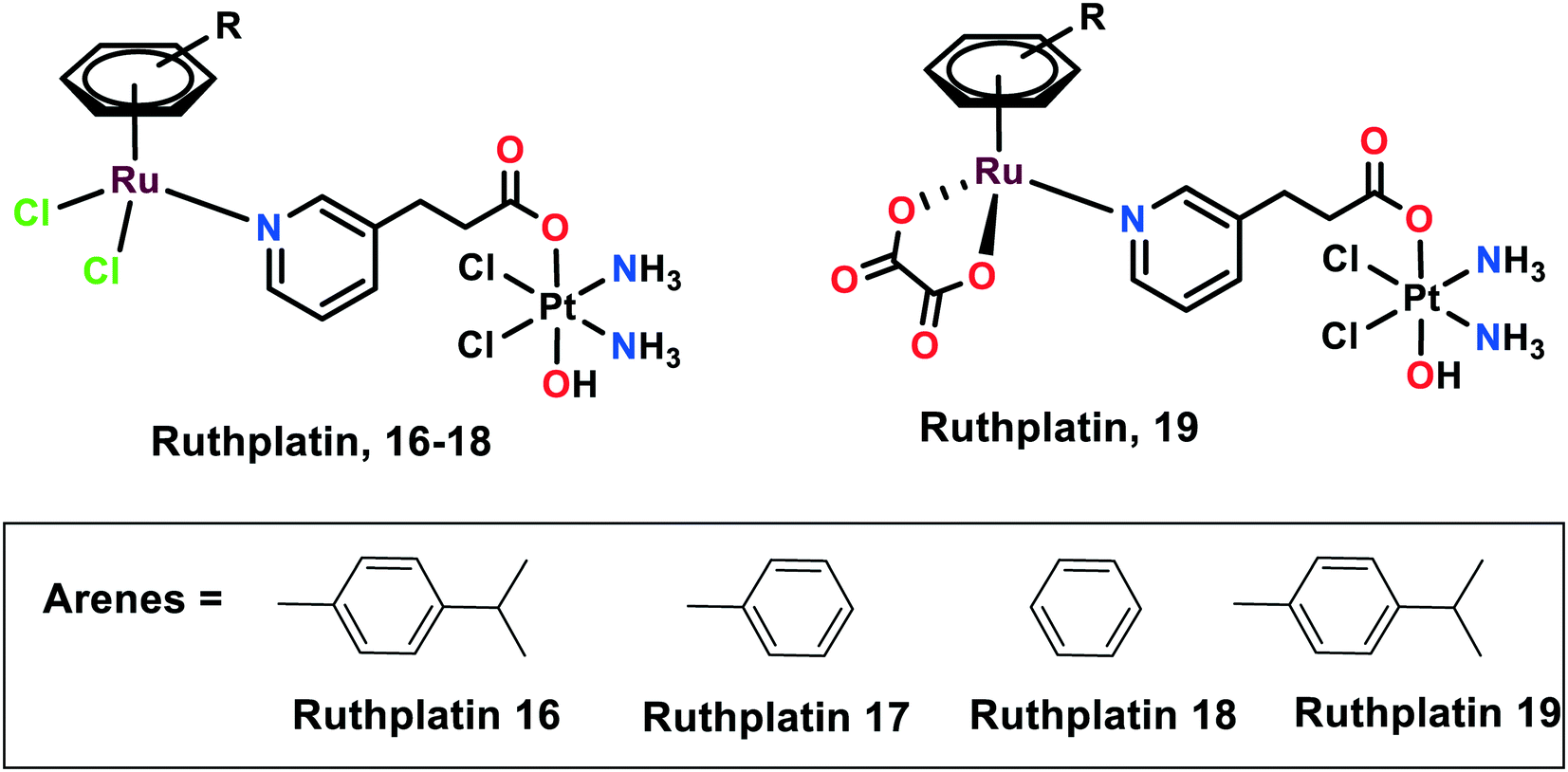 | ||
| Fig. 4 The chemical structures of ruthplatin complexes referred to in this article. | ||
Organoruthenium complexes with 8-hydroxyquinolines
Two novel organoruthenium 8-hydroxyquinolinato complexes (20 and 21) were synthesized. The cytotoxicity of these complexes was evaluated on A549 (lung adenocarcinoma), MG-63 (human osteosarcoma), and MCF-7 cells, and cisplatin was used as the reference (Fig. 5). These complexes showed higher selectivity than cisplatin. This higher selectivity performance can be attributed to redox balance disruption leading to G2/M phase cell cycle arrest, thus causing apoptosis through the mitochondrial pathway.75 On performing a clonogenic assay to estimate the cellular reproductive potential, it was found that both compounds inhibited colony formation in a dose-dependent manner but complex 21 showed a higher inhibition effect on colony formation than complex 20. The migration and invasion assays revealed that both the complexes inhibited migration on MG-63 and MCF-7 but only complex 21 inhibited migration in A549 cells. Complex 21, however, showed significant inhibition of the invasive activity of the cancer cells in the order of MG-63 > MCF-7 > A549. On performing experiments on multicellular spheroids of MG-63, MCF-7, and A549, it was found that both complexes showed anticancer activities on spheroids; however, complex 20 showed enhanced anticancer activity as compared to complex 21 and cisplatin. The results were in contrast to the 2D models since complex 21 was active in 2D models and complex 20 was active in 3D models. The probable reason for this contrast may be the involvement of several factors like hypoxic conditions, redox potential, bioavailability, and cellular uptake, which play a major role in defining the anticancer activities of such kinds of drugs.76 Thus, the combined analysis of complexes on both 2D and 3D models gives a reliable assessment of the anticancer potential activities of the novel complexes.72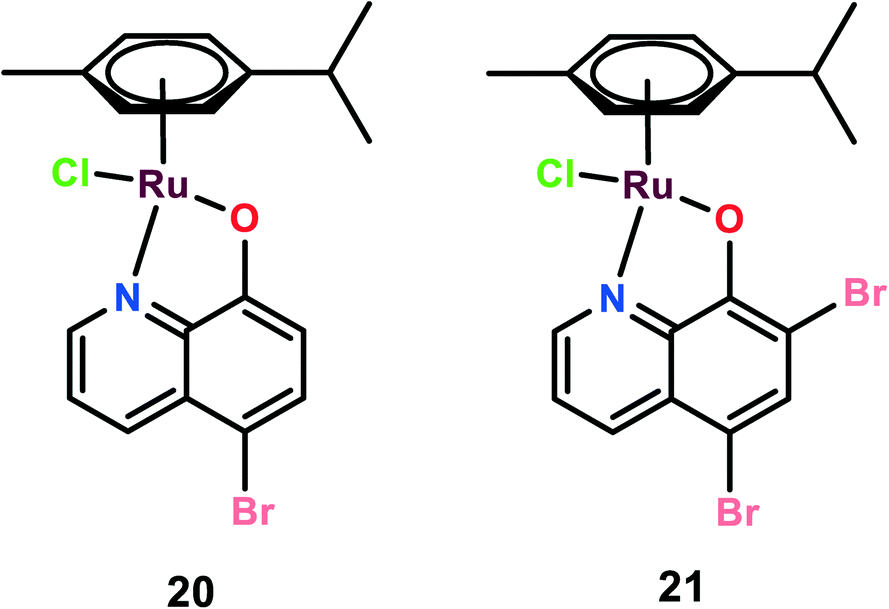 | ||
| Fig. 5 The chemical structures of Ru(II) hydroxyquinolines complexes referred to in this article. | ||
Ruthenium(II)–arene complexes with acylthiourea as ligands
The half-sandwich Ru(II) complexes with acylthiourea ligands of the general type [Ru(η6-p-cymene)(PPh3)-(S)Cl]PF6 (22m–27m) and [Ru(η6-p-cymene)(PPh3)(S–O)]PF6 (22b–27b) where S/S–O = N′,N′-disubstituted acylthiourea were synthesized and characterized (Fig. 6). The activities of the complexes were evaluated on five cell lines, viz. DU145, A549, MCF-10A, MDA-MB-231, and MRC-5.77 The complexes are highly selective towards breast cancer cell lines in comparison to cisplatin, although they were found to be cytotoxic towards DU145, A549 as well; thus, 26b, 27b and their monodentate analog 26m, 27m were chosen for further studies. It is interesting to note that monodentate ligand-containing complexes were cytotoxic towards DU145 and A549, however, bidentate ligand-containing complexes were active against MDA-MB-231. It is also worth indicating that the complexes with R1 as thiophene were cytotoxic to all the cell lines. The length of the R2 chain affected the cytotoxicity of the complexes; the increase in the alkane chain length of R2 enhanced the cytotoxicity of the complexes. R2 with ethyl group was the most cytotoxic among all the complexes. The probable reason for increased chain length affecting the cytotoxicity is because it enhances the lipophilicity of the complexes, thus further enhancing the cellular uptake of the complexes. These complexes were found to significantly inhibit the colony formation in MDA-MB-231 cells. Also, the complexes inhibited the migration of the cells when evaluated by both wound healing and Boyden chamber migration assays. It was found that these complexes significantly inhibited the wound closure when compared with the control and these results were supported by the Boyden chamber assay where the number of migrated cells was significantly inhibited upon treatment with the complexes.77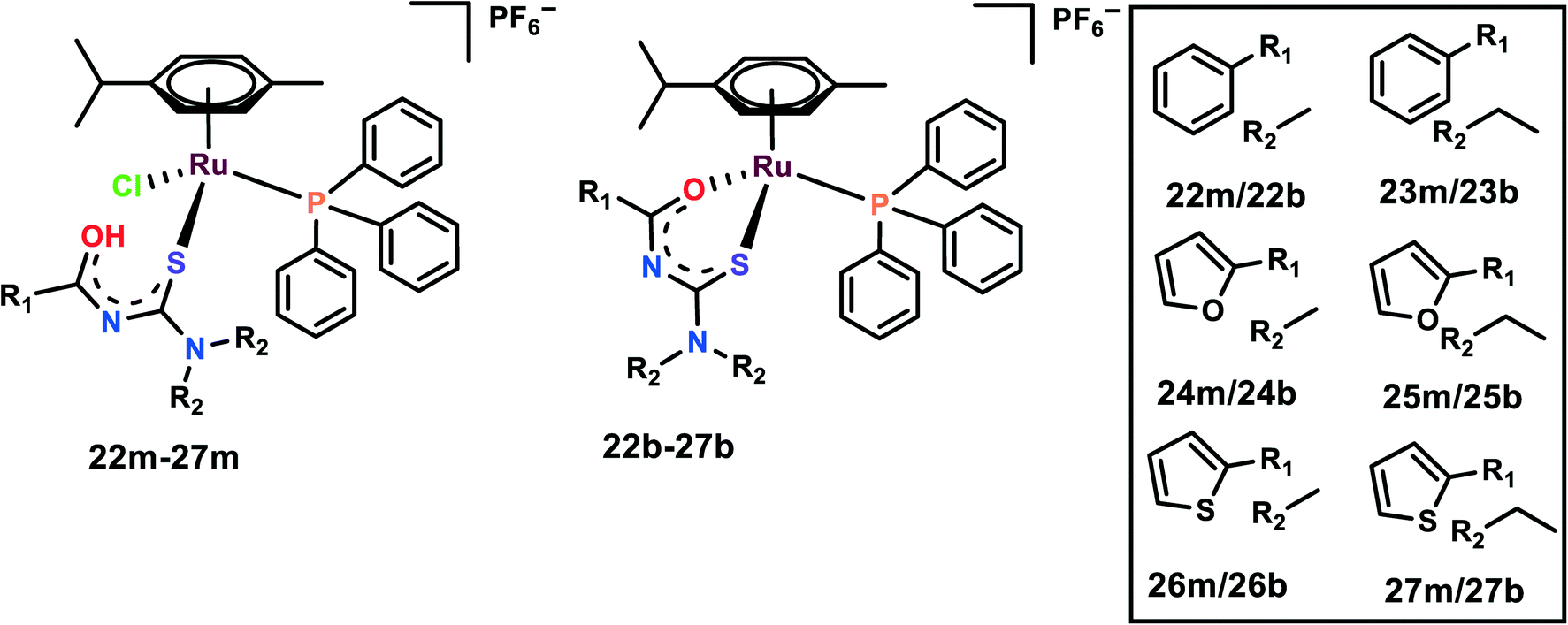 | ||
| Fig. 6 Chemical structures of Ru(II) acylthiourea complexes referred to in this article. | ||
Picolinate Ru(II) complexes
Five complexes were synthesized with the general structural formula [(η6-p-cymene)Ru(L)Cl2], (L: 3-acetylpyridine (28), 2-amino-5-chloropyridine (29)), and [(η6-p-cymene)Ru(HL)Cl], (HL: 2,3-pyridinedicarboxylic acid (30), 2,4-pyridinedicarboxylic acid (31)), and [(η6-p-cymene)RuCl(picolinic acid)]·H2O (32) (Fig. 7).78 These complexes were tested on various cancer cell lines such as B16, MDA-MB-361, HeLa, LS-174, and MDA-MB-453, and exhibited low anti-proliferative activities with IC50 > 200 μM, except for complex 32 with the IC50 value of 81.97 μM.79,80 Despite the low level of cytotoxicity and genotoxicity, few complexes show anti-metastatic potential.46,81,82 Similar results were obtained when these complexes were used to treat endothelial cell lines (model system for angiogenesis) EA.hy 926, and MS1 cell lines. On the cell cycle analysis of all the complexes on EA.hy 926 and HeLa cells, it was observed that only complex 32 showed a decrease in the cell percentage in the G1 phase and a slight increase in the S phase on HeLa cells with no apoptotic effects. On determining the cellular distribution of Ru(II) complexes within the HeLa through ICP-OES, it was found that all the complexes were internalized within the cells. However, it was complex 32 that was found in higher concentration in DNA fraction, thus emphasizing its efficient DNA binding nature. None of the other complexes showed any indication of DNA binding, which may be attributed to the ligands' structural differences that may affect the complexes–DNA interactions. To determine the anti-metastatic potential, various experiments were carried out. On performing gelatin zymography it was observed that after 24 h treatment, complex 32 displayed inhibitory activity on the secretion of MMP-2 and MMP-9 in HeLa and EA.hy 926, respectively. These MMPs proteins are considered to play a significant role in invasion and metastasis as they dissolve the basement membrane and degrade the extracellular matrix (ECM). Among the various MMPs proteins, MMP-2 (capable of degrading gelatin) and MMP-9 are considered to be very significant in the invasion and metastasis processes.83 On performing the matrigel invasion assay it was found that complexes 29 and complex 32 reduced the number of invasive cells by 36% and 20% on treatment with 100 μM and 41 μM, respectively. The pro-adhesive properties of the complexes were evaluated on HeLa cells grown on a plastic substrate (uncoated bottom of plates) showing resistance to the trypsin treatment when compared with the control. It was observed that on 100 μM treatment with complexes 29 and 30, the cells showed almost three times more adhesion as compared to the control, which also correlates with the matrigel invasion assay. On testing the migration inhibition ability of the complexes through a wound healing assay, it was found that only complex 32 showed a significant inhibitory effect on the migration of the cell. To investigate the effects of these complexes on angiogenesis, a tube formation experiment was performed. With the treatment of endothelial cells MS1 with appropriate media on matrigel, these cells started to reorganize, show projections, form cell to cell contacts and form polygon-like structures.84 The treatment of these cells with complexes reduced the ability of endothelial cells to differentiate and extend into tube-like structures. Complexes 28, 31 and 32 showed some potential inhibitory effects on tube formation on endothelial cells, but complex 32 showed the highest inhibitory effect among all the tested complexes.78 To further investigate the DNA-repair-dependent cellular response due to cytotoxic DNA lesions caused by the complexes–DNA interactions, the mRNA and protein expression levels of ERCC1 and MSH1 were elucidated. Through quantitative real-time PCR (RQ-PCR) the mRNA expression level of ERCC1 was determined after the treatment of HeLa cells for 24 h. ERCC1 was found to play a significant role in cell response to other types of DNA lesions and its repair function expands beyond the NER (nucleotide excision repair). The NER plays a notable role in the repair of DNA-platinum adducts.85 Complex 32 was found to upregulate the expression level of ERCC1, whereas other complexes showed no or down-regulated expression level. However, after 24 h of treatment of complex 32 on HeLa cells, the expression level of ERCC1 was found to be down regulated, thus this effect of complex 32 has to be further investigated. Moreover the protein expression level of MSH2 was further investigated. MSH2 plays a crucial role in mismatch repair system since it has a role in correction mismatches that occur in the DNA during DNA replication.86 Only complex 32 was able to decrease the protein level of MSH2; the other complexes seemed to have no effect on the protein level of MSH2. The upregulation of ERCC1 and modulation of the MSH2 protein level suggest the involvement of a DNA-repair-dependent response.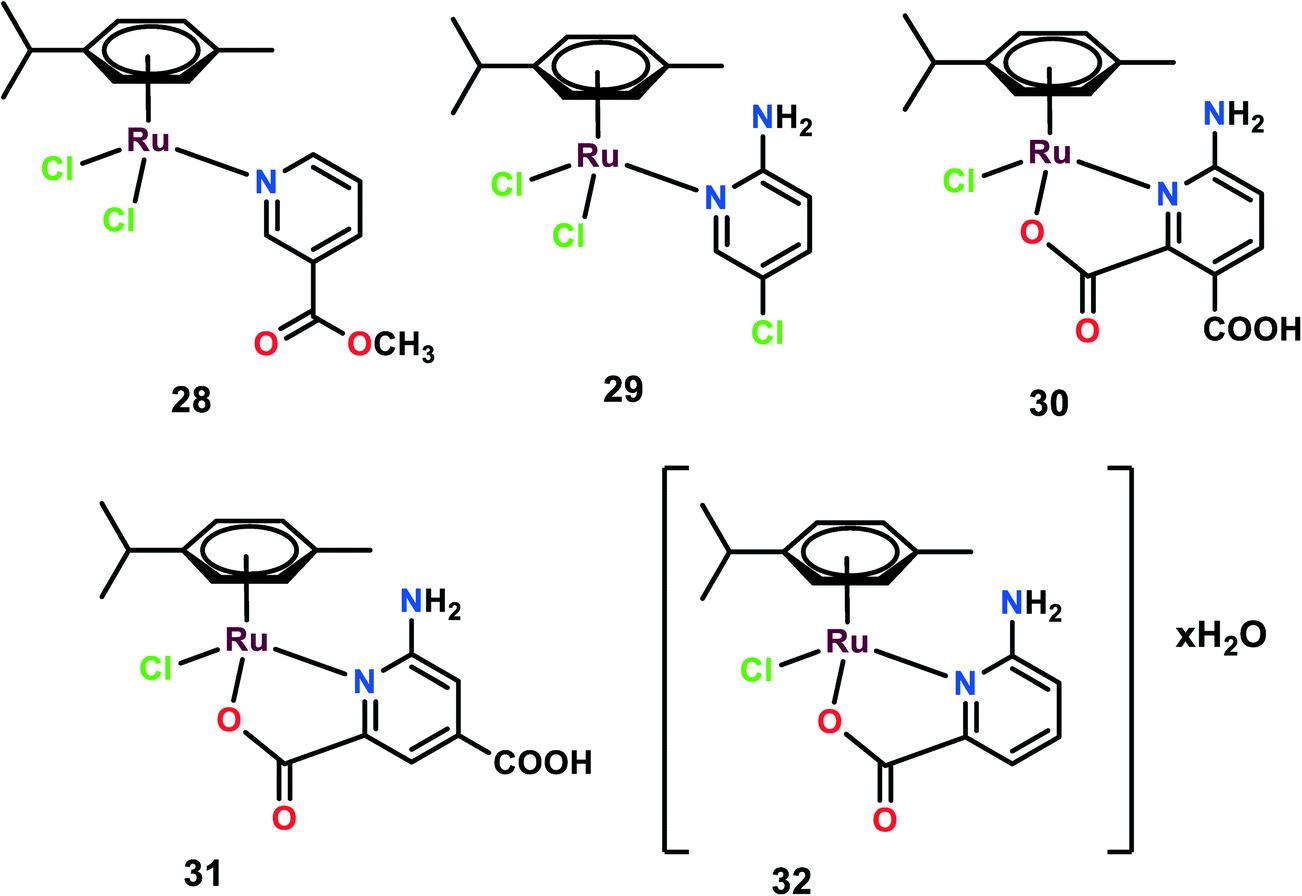 | ||
| Fig. 7 Chemical structures of Ru(II) picolinate complexes referred to in this article. | ||
DAPTA complexes
The anti-metastatic studies of RAPTA-C, RAPTA-T and their analogues [Ru(η6-p-cymene)Cl2(DAPTA)] (DAPTA = (3,7-diacetyl-1,3,7-triaza-5-hosphabicyclo[3.3.1]nonane)) (DAPTA-C) (33) and [Ru(η6-toluene)Cl2(DAPTA)] (DAPTA-T) (34) were studied for their endothelial cell (EC) function through in vitro bio-assays (Fig. 8).87 These complexes were determined to inhibit the EC growth. All the complexes inhibited the viability and growth of HUVEC (primary) and ECRF24 (immortalized) EC cells after 72 h incubation. The ED50 values were found to be 300–500 μg mL−1 (∼500–1000 μM). To further confirm the selectivity of these complexes towards EC, the complexes were tested against colorectal cancer cells (LS174T). The ED values were similar to that of EC for RAPTA-C, RAPTA-T, and DAPTA-T, but were less prominent in DAPTA-C. The apoptotic effects of complexes on cells were evaluated through flow cytometry, which depicted that only RAPTA-T can show apoptosis of about 24%, whereas anginex (angiogenesis inhibitor, which acts through an apoptosis-inducing mechanism) showed 17% of apoptotic cells. The other complexes did not show any significant changes in the apoptotic cells.88 To further investigate the effects of complexes on EC function, a migration assay was performed on ECRF24 cells, which indicated that RAPTA-T inhibited the migration of the cells about >90% at 1 mg mL−1 in a dose-dependent manner. The migration assay was assessed at 6 h along with the determination of the cell proliferation. The cell proliferation was found to not be affected, thus the probable reason for the anti-migratory effect might be anti-angiogenic activity. To further assess the in vitro anti-migratory ability of the complexes, the EC sprout assay (bFGF driven) was performed on HUVEC cells. It was observed that RAPTA and DAPTA-C inhibited endothelial sprout formation in a dose-dependent manner upon 16 h of treatment of cells with the complexes, whereas DAPTA-T showed no effect on the endothelial sprouting. The anti-angiogenic effects of the complexes were further explored in vivo, using the CAM (chicken embryo chorioallantoic membrane) assay. Both DAPTA-C and RAPTA-T were found to suppress neovascularization, decrease the number of branches and form avascular zones in a dose-dependent manner. The distortion in the organization of the capillary plexus was also observed in the treatment of cells with the DAPTA-C and RAPTA-T. At higher concentrations, the RAPTA-C was found to show significant effects in demolishing the capillary bed area on the treated region as compared to DAPTA-C. These complexes were explored for the in vivo model of PDT (photodynamic therapy)-induced angiogenesis. This model allowed the monitoring of the development of the vascular network for two days after the treatment, especially concerning regrowth and vascular occlusion, as described previously.89,90 The treatment with the complexes RAPTA-T and DAPTA-C immediately after PDT showed distorted newly formed vessels, unorganized capillary plexus, and inhibited vascular growth; these effects were more prominent with increasing the concentration of the complexes.90 DAPTA-C showed a moderate effect in comparison with RAPTA-C with substantial inhibition of angiogenesis and inhibition of the reperfusion of pre-existing vessels when administered in the same concentration.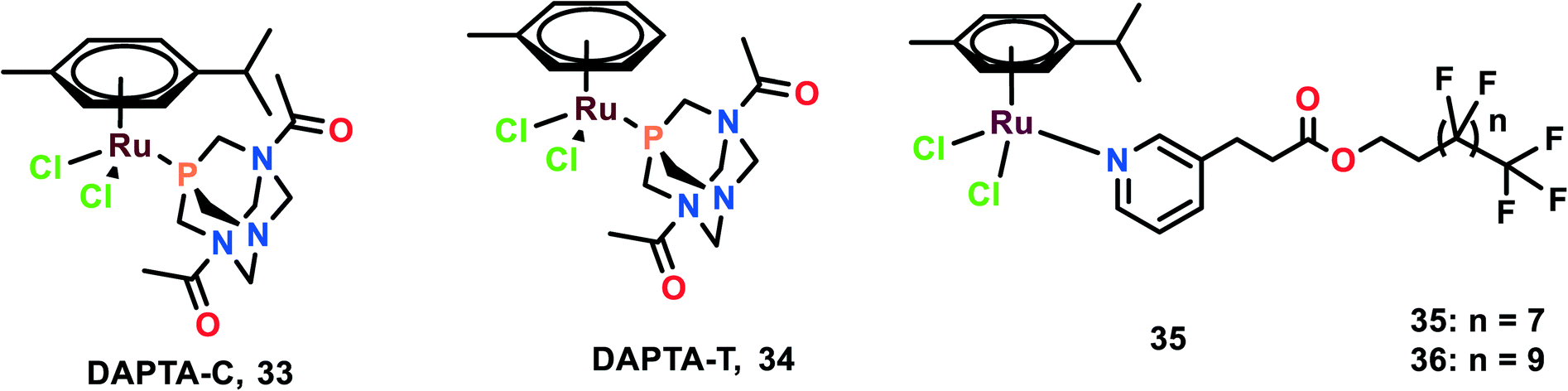 | ||
| Fig. 8 Chemical structures of DAPTA complexes and RAPTA with perfluorinated chain complex referred to in this article. | ||
Similar experiments were carried out using two bifunctional ruthenium(II)–p-cymene complexes with perfluorinated side-chains attached through pyridine ligands (35 and 36). The effects of these complexes on endothelial cell lines (HUVEC and ECRF24) along with noncancerous human embryonic kidney HEK293 and numerous cancerous cell lines (SW480, and LS174T) were also investigated.91
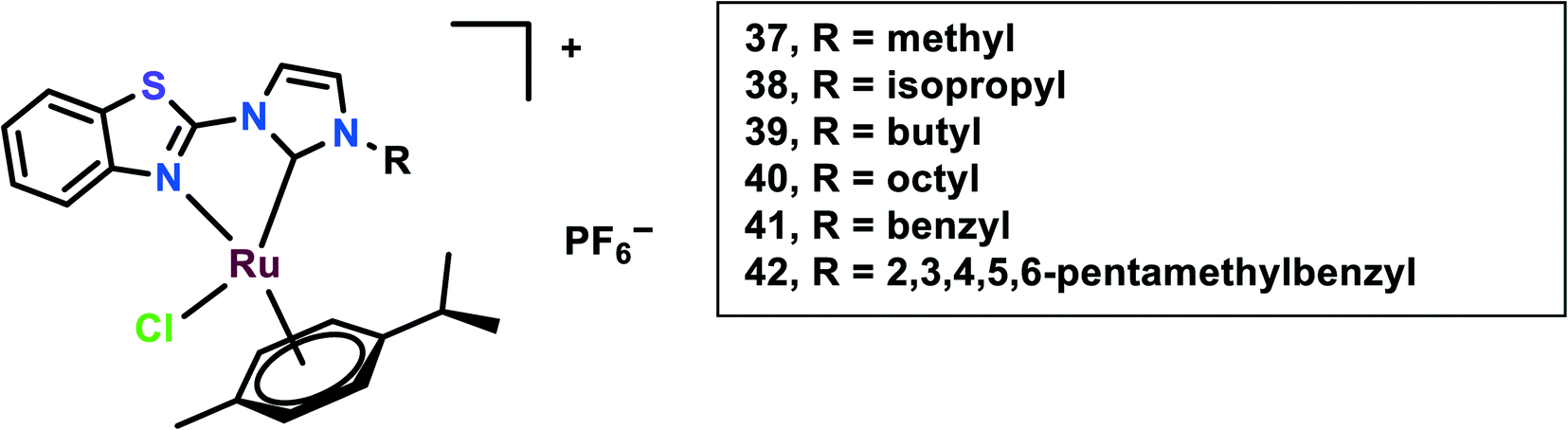
NHC-coordinated Ru(II)–arene complexes
In this study, N-heterocyclic carbine (NHC) ligands coordinated with the Ru(II) complexes were synthesized. Benzothiazole-functionalized Ru(II) NHC complexes (37–42) having different substituents were taken into account (Fig. 9).92 The in vitro cytotoxicity of these complexes was evaluated on six cancer cell lines, viz. HCT-116 (colon cancer) and LoVo, A549 and HT-29, A2780 cells and HeLa. Ru37–Ru39 were found to be inactive against all the cell lines, however, Ru40 had significant cytotoxicity against HT-29 and A2780 cells with IC50 values of 8.51 ± 0.69 and 2.74 ± 0.15 μM, respectively. This difference in cytotoxicity might be because of an increase in the length of alkyl substituents. The peripheral substituent groups of Ru41–Ru42 modulated the cytotoxic potency of the complexes; however, Ru42 showed significantly higher cytotoxicity in all the cell lines but was even more effective in A2780 cells. Overall the complexes showed in vitro cytotoxicity in the order of Ru42 > Ru40 > Ru41 > Ru37, Ru38, Ru39. The Ru40–Ru42 were found to be more lipophilic and had significantly higher accumulation in the A2780 cell line. To evaluate the anti-proliferation activity, the EdU (5-ethynyl-20-deoxyuridine) incorporation assay was performed. This showed that the Ru40 and Ru42 dramatically reduced cell proliferation on increasing the concentration, up to 15% and 16%, respectively; thus, these complexes work in a dose-dependent manner when compared with cisplatin. Also through the migration assay, it was found that the wound closure ratio in the A2780 control cells (33%) were significantly reduced to 11.7% and 8.7% on treatment with Ru40 and Ru42, respectively. These complexes (Ru40 and Ru42) were found to cause increases in ROS through DCFDA staining, mitochondrial dysfunction through JC-1 staining, and cell cycle arrest at the G2/M phase through FACS analysis, thus causing apoptosis. The in vivo systemic tolerance of these complexes was studied using compounds dissolved in DMSO/saline (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution and intraperitoneally injecting them into healthy ICR mice (n = 8 in each group) at different doses (10 and 20 mmol kg−1). This treatment showed that the mice were sufficiently tolerant toward the treatment with the complexes since most of the mice did not show a reduction in weight in comparison with cisplatin, which showed a more than 20% weight reduction on drug treatment within 15 days. The anti-metastatic effects of the compounds were further studied in vivo using Balb/c nude mice bearing A2780 cell-derived ovarian cancer. Each mouse in the treatment groups (n = 8 in each group) was intraperitoneally injected with 200 mL of A2780 cells suspension (5 × 107 cells per mL in PBS). From the next day, drugs were intraperitoneally administered in 200 mL of DMSO/saline (1:1, v/v) solution. During the experiment, the body weights of the mice were recorded and at the end of the study the ovaries and tumours were collected for analysis. Drug administration was reduced to three times on days 0, 3, and 6, to avoid potential toxicity. Ru40 and Ru42 drug administration at the dose of 20 mmol kg−1 yielded mean tumour growth inhibition (TGI) rates of 65.2%, and 68.7%, respectively, on comparison with cisplatin treatment with a TGI rate of 36.7%. The ovaries in the Ru(II) complex-treated mice had normal morphologies, however, the DMSO/saline-treated mice had cancer cells migrate to the ovaries and uterus. This indicated the anti-metastasis effect of the complexes.
:1, v/v) solution and intraperitoneally injecting them into healthy ICR mice (n = 8 in each group) at different doses (10 and 20 mmol kg−1). This treatment showed that the mice were sufficiently tolerant toward the treatment with the complexes since most of the mice did not show a reduction in weight in comparison with cisplatin, which showed a more than 20% weight reduction on drug treatment within 15 days. The anti-metastatic effects of the compounds were further studied in vivo using Balb/c nude mice bearing A2780 cell-derived ovarian cancer. Each mouse in the treatment groups (n = 8 in each group) was intraperitoneally injected with 200 mL of A2780 cells suspension (5 × 107 cells per mL in PBS). From the next day, drugs were intraperitoneally administered in 200 mL of DMSO/saline (1:1, v/v) solution. During the experiment, the body weights of the mice were recorded and at the end of the study the ovaries and tumours were collected for analysis. Drug administration was reduced to three times on days 0, 3, and 6, to avoid potential toxicity. Ru40 and Ru42 drug administration at the dose of 20 mmol kg−1 yielded mean tumour growth inhibition (TGI) rates of 65.2%, and 68.7%, respectively, on comparison with cisplatin treatment with a TGI rate of 36.7%. The ovaries in the Ru(II) complex-treated mice had normal morphologies, however, the DMSO/saline-treated mice had cancer cells migrate to the ovaries and uterus. This indicated the anti-metastasis effect of the complexes.
 | ||
| Fig. 9 Chemical structures of Ru(II) NHC coordinated complexes referred to in this article. | ||
Selectivity of Ru(II)–arene complexes
Despite the significant pharmacological properties of Ru(II)–arene complexes, it is equally important to pay attention to increasing the efficacy and selectivity of these complexes. Chemists have begun exploiting various characteristics of tumour environments or targeting strategies,93 hence, many complexes are synthesized that get activated by the differences between tumours and normal cells, like hypoxia and acidic conditions, which are prevalent in tumour cells.94,95 Ru(II)–Arene complexes have been synthesized with perfluorinated phosphine ligands, which show modest thermo-responsive activation; however, these complexes were further modified for enhanced selective cytotoxicity.96 It has also been reported that Ru(II)–arene complexes containing lipophilic chains via the modification of a PTA, isonicotinic ester ligands, or imidazole lead to increased antiproliferative activity, presumably due to increased cell uptake but not necessarily to increased cancer cell selectivity.97–99 The nature of lipophilic chains (i.e., alkyl or perfluoroalkyl) and their length have a significant impact on the cytotoxicity and selectivity of the Ru(II)–arene complexes.87,93,96 It has also been observed that the electron-donating or withdrawing groups attached to the ligand, and the aquation of the complexes also have a significant effect on the selectivity and cytotoxicity of the ruthenium complexes.71,100–102 The attachment of Ru(II)–arene complexes to macromolecules, which leads to selective permeation and retention of complexes in the tumour vasculature can also play a part.103,104 There are several examples that include macromolecular systems containing Ru(II)–arene complexes, multinuclear Ru(II)–arene metallacages, metallacycles, and copolymer and polymer conjugates of RAPTA complexes for increased selectivity.104–109 Thus, the attached ligands or macromolecules play a crucial role in modulating the activity and efficacy of ruthenium complexes (Fig. 10).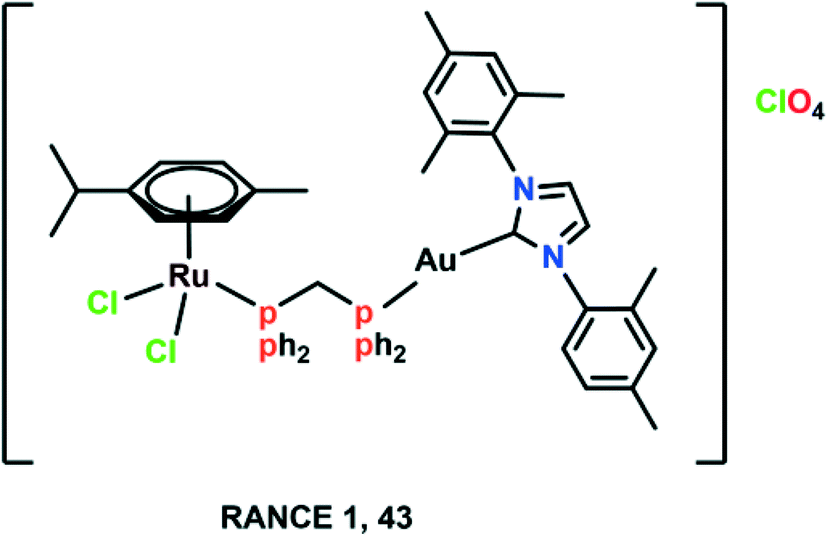 | ||
| Fig. 10 The chemical structure of the heterometallic Ru(II)–arene gold complex (RANCE 1) referred to in this article. | ||
Significance and techniques used for determining anti-metastatic properties
General mechanism of action
A large number of Ru(II)–arene complexes have been synthesized, which exhibit anti-tumour properties, having different targets and mechanisms of action. These properties majorly depend on the associated ligands, and the availability of uncoordinated sites in ruthenium complexes.12,110 The mechanisms of action of ruthenium complexes as anticancer agents generally include mitochondria, death receptors, the endoplasmic reticulum (ER) and DNA-mediated pathways.111 Ru(II)–Arene complexes have been shown to target mitochondria, thus immediately disrupting the mitochondrial membrane potential. This mitochondrial membrane disruption leads to the release of cytochrome c and activates cascade pathways, thus causing apoptosis.68 Another mechanism involves the binding of the Fas ligand and the TNF-related apoptosis-inducing ligand (TRAIL) to Fas, and DR4/5 leads to the recruitment of FADD (Fas-associated death domain), thus further activating the caspase pathway.26,69 The endoplasmic reticulum plays a notable role in protein synthesis, protein folding and protein transportation. However, due to external stimuli, when the ER comes under stress, it begins to accumulate the misfolded proteins, which leads to the ER-associated degradation (ERAD) and unfolded protein response (UPR). Thus, in such conditions, when ER homeostasis cannot be maintained, it activates the programmed cell death pathway.112 Along with these mechanisms, DNA remains one of the major targets for Ru(II)–arene complexes, owing to their covalent and noncovalent interactions with DNA.100,113,114 The binding of the complexes to DNA leads to transcription inhibition or disrupted DNA, which leads to the activation of programmed cell death or irreversible apoptosis.111 Some Ru(II)–arene complexes have also been found to block metastasis by inhibiting angiogenesis, invasion and proliferation processes, by targeting microtubules, the c-Myc gene, MMPs, P53 protein, VEGF and EGF pathways.29,71,73,111,115–117 A heterometallic Ru(II)–arene gold complex (43) was found to be significantly more cytotoxic as compared to the activity of individual metals on the Caki-1 (human renal carcinoma cell line) cell line. They were found to inhibit migration, invasion and angiogenesis by inhibiting the MMPs secretion, decreasing the expression of VEGF, IL (interleukins) and cathepsins.118 Some Ru(II)–arene complexes can also target cancer stem cells without affecting cancer cell stemness in 3D colon cancer stem cell spheroids, thus leading to intact regulation of cell gene expression (Fig. 11).29,73,119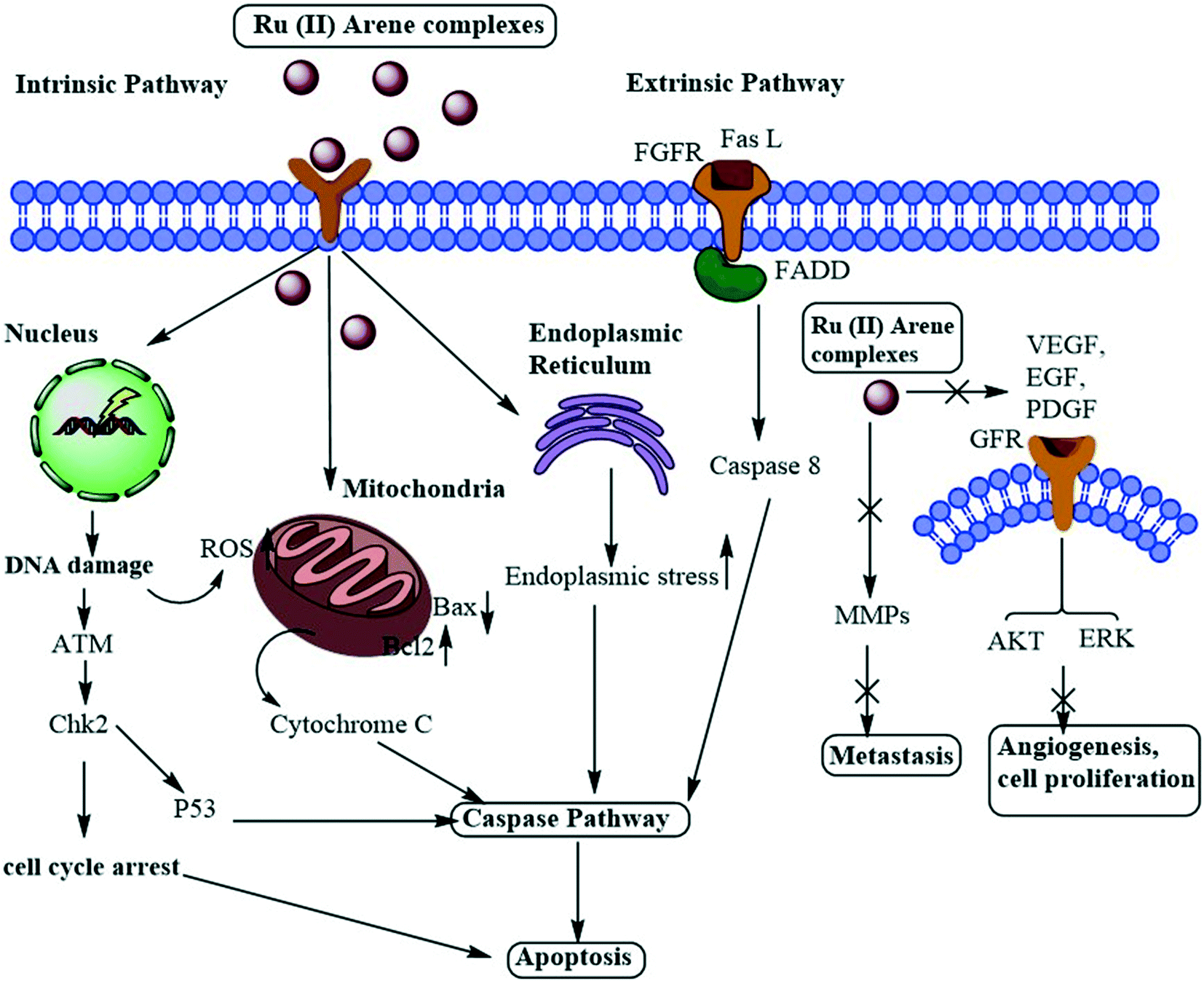 | ||
| Fig. 11 Schematic representation of the general mechanism of action of Ru(II)–arene complexes as anti-metastatic agents. | ||
Metastasis
Metastasis is generally the cause of cancer-related mortality and morbidity. It is described as the spreading of primary cells to the surrounding tissues and then to distant organs.120 Metastasis involves a series of sequential and interrelated events that must be completed to successfully form metastatic tumours. For the metastasis cascade to occur, the primary cells must dissociate from the primary location, penetrate the lymphatic and circulatory system, dodge the immune system, and extravasate to the new capillary bed, and invade and proliferate in the distant organ.121–123 The following functions are required to complete the sequence of events for metastasis: interaction with the local environment, migration, invasion, and angiogenesis-inducing ability.121Angiogenesis
As the primary tumour grows, it requires a blood supply that can provide nutrients and metabolites through diffusion exchange. Angiogenesis is the process by which blood supply is established to the tumour cells, which is actually the growth of the cells from existing vasculature.124 In 1970, Judah Folkman found that if the tumour spheroid size increased beyond 1–2 mm in diameter, at this stage, the diffusion of nutrients and metabolites becomes rate limiting, and thus the formation of new blood vessels is required for the continuous growth of the tumour.125 These neovessels provide nutrients to the tumours, as well as an escape route for the tumour cells to enter the circulation. There are two types of angiogenesis: sprouting angiogenesis involves sprouts of endothelial cells growing towards the angiogenic stimulus, and may add blood vessels to the tissue portions previously devoid of the blood vessels; intussusceptive angiogenesis involves the existing vessels being split as a result of the invasion to form transvascular tissue pillars that can expand.126,127 Hence, the inhibition of angiogenesis can be a potential mode for tumour metastasis control. Also, it has been indicated earlier that primary tumours with higher vascular density have a higher incidence of metastasis than poorly vascular tumours; thus, the vascular density might be a prognostic indicator of metastatic potential.128 The following experiments were designed to determine the tendency for angiogenesis in tumours.Adhesion
In healthy tissues, homeostasis is intensely dependent on the cell-to-cell adhesion and cell-to-extracellular matrix (ECM) interaction. Normal epithelial cells retain their tissue structure by the cell-to-cell adhesion and cell-to-ECM interactions. Also, the two hallmarks of cancer, which are anchorage-independent growth and cell-to-cell adhesion loss, are both dependent on cell adhesion.130 The complex adhesion mechanisms involve adherens and tight junctions, and desmosomes regulate the interepithelial cellular interaction. The malignant and transformed cells may display the loss of adherens junctions, which results in the detachment of the cells from the epithelial cells.131 All through this process, the adherens junctions mediated by E cadherins (important for cell–cell adhesion) are lost.132 Thus, the adhesion assay helps to determine the interaction of cells with other cells and the ECM.Migration and invasion
As mentioned earlier, cancer cell migration and invasion and their entrance into the blood or lymphatic tissues is necessary for metastasis to take place.135 Migration and invasion allow the cells to change positions within the tissues. The neoplastic cells take the route of the blood or lymphatic system to disseminate into circulation and reach distant organs.135 In migration, the cells need to modify their stiffness and shape to interact with the surrounding tissues. The different cell modifications include filamentous actin, different structural and signaling proteins, leading to dynamic interaction with the ECM substrate. Thus, analyzing the cell extension can also be a method for determining cell motility.Once the cells can cross the basement membrane and ECM, the process of invasion has been instigated. Now, the cells can intravasate into the lymphatic or circulatory system and travel to distant organs. This process involves several proteins for the degradation of the basement membrane and ECM along with some structural and signaling proteins to regulate cell motility and migration.122 Evaluating the effects of complexes on migration and invasion also provides an idea about the anti-metastatic properties of the complexes.
Gene expression analysis and western blotting
The transformation of normal cells into metastatic cells requires major phenotypic changes, such as changes in cell surface receptors, growth factor, cytoskeleton function, signal transduction, and proteolytic enzyme production. It also leads to changes in cell interaction with ECM or the microenvironment. The transformed cells can disrupt cell–cell adhesion properties and they can modify the interaction with the microenvironment in favor of their proliferation, survival, and dissemination.121 The tumour environment, especially the extracellular matrix, plays a crucial part in cancer progression. The matrix metalloproteinases (MMP) are responsible for remodeling ECM and are present in almost all cancers.140,141 The transcription of MMPs is generally low and is firmly regulated. Thus, it has been found that increased levels of MMPS can be co-related with the progression of cancer, cancer cell proliferation, and tumour size.142–144 Several adherens, like cadherins, integrins, and proteoglycans, also play a significant role in metastasis.121 Thus, analyzing the effect of complexes on these host tissue interactions, proteins, and RNA levels may give us some insight into the anti-metastatic activities of the complexes.:AcOH:H2O (2.5:1:6.5) and then destained in 4% MeOH with 8% AcOH. To increase the sensitivity, the gels were further destained in a solution of 1% Triton X-100 (1–2 h). The activity bands were observed as a clear transparent band against the blue background of Coomassie Blue.144
Prospects and problems
In recent years, researchers have explored the development of multifunctional ruthenium(II) complexes combined with other therapies, including PDT, radiosensitization, targeted therapy, and photothermal therapies.61,146–149 Using these combination therapies, the efficacy and selectivity of the complexes may be improved. However, despite the development of various ruthenium complexes, their aquation, instability, and low cytotoxicity remain major concerns. These issues can be resolved by using bionanomaterials, which increase the permeability, selectivity and retention of the complexes. These nano-delivery vehicles can effectively carry a large amount of drugs, thus increasing the drug accumulation. These carriers also protect the drug from degradation, thus enhancing the retention of the drugs in the body.111,150 Recently, dinuclear trithiolato-bridged arene Ru complexes, which have been encapsulated in apoferritin (AFt) nanocages, were investigated. These complexes were highly cytotoxic towards mortalized murine fibroblast BALB/c-3T3 transformed with SV40 virus (SVT2) and human epidermoid carcinoma A431 malignant cells, but showed moderate selectivity towards normal BALB/c-3T3 cells.150,151 Many nano-ruthenium complexes have been developed but not much investigation has been done on nano Ru(II)–arene complexes so far.152–154 Thus, exploring the formation and activity of nano Ru(II)–arene complexes would be of great interest and importance.Generally, 2D models are used to study the anti-metastatic activities in the case of Ru(II)–arene complexes, which do not provide a clear picture of all the steps and interactions taking place in metastatic cancer. 3D models do give further insightful information about the steps involved in the metastasis like adhesion, migration, and sprout formation, but still cannot give accurate information about the metastatic processes in vivo. Thus for further experiments, tumour formation in the mouse models, either spontaneous or in experimental in vivo models, may give a better understanding of the effect of complexes in vivo.39 Gene expression profiling, although a costly method, can be used to determine the changes in the number of gene expression levels occurring after treatment. Although every method has some shortcomings, in vivo studies may still be helpful in the detection of proper targets and pathways for complexes. Also, flow cytometric studies can be used for the determination of the changes in the levels of protein expression on treatment; this method is comparatively fast and quantitative for the detection of protein levels, thus will help gain further information about the metastatic protein changes occurring due to the treatment of the complexes. Thus, by doing further extensive studies and involving various techniques, significant information might be obtained about the metastatic target.
Authors contribution
SM did conceptualization and proof reading, writing of the manuscript and editing was done by CS, chemical structure drawn and editing done by SS.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
We are grateful to IIT Indore for providing necessary facilities. C. S. thanks the Council of Scientific and Industrial Research (CSIR) for her fellowship. S. S. thanks MHRD for his fellowship.References
- M. J. Thun, J. O. DeLancey, M. M. Center, A. Jemal and E. M. Ward, Carcinogenesis, 2010, 31, 100–110 CrossRef CAS PubMed.
- B. Kar, N. Roy, S. Pete, P. Moharana and P. Paira, Inorg. Chim. Acta, 2020, 512, 119858 CrossRef CAS.
- M. Lu and X. Zhan, EPMA J., 2018, 9, 77–102 CrossRef.
- T. Gianferrara, I. Bratsos and E. Alessio, Dalton Trans., 2009, 7588–7598 RSC.
- J. Matthey, Ruthenium in Medicine, Platinum Met. Rev., 2001, 45(2), 62 Search PubMed.
- T. Boulikas and M. Vougiouka, Oncol. Rep., 2003, 10, 1663–1682 CAS.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS.
- A.-M. Florea and D. Büsselberg, Cancers, 2011, 3, 1351–1371 CrossRef CAS.
- T. Lazarević, A. Rilak and Ž. D. Bugarčić, Eur. J. Med. Chem., 2017, 142, 8–31 CrossRef.
- C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201–3209 RSC.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- S. Strasser, E. Pump, R. C. Fischer and C. Slugovc, Monatsh. Chem., 2015, 146, 1143–1151 CrossRef CAS.
- S. Y. Lee, C. Y. Kim and T.-G. Nam, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS PubMed.
- N. Muhammad and Z. Guo, Curr. Opin. Chem. Biol., 2014, 19, 144–153 CrossRef CAS PubMed.
- G. R. Jadhav, S. Sinha, M. Chhabra and P. Paira, Bioorg. Med. Chem. Lett., 2016, 26, 2695–2700 CrossRef CAS.
- K. C. Gatter, G. Brown, I. S. Trowbridge, R. E. Woolston and D. Y. Mason, J. Clin. Pathol., 1983, 36, 539–545 CrossRef CAS PubMed.
- W. Zhang, F. Zhang, Y.-L. Wang, B. Song, R. Zhang and J. Yuan, Inorg. Chem., 2017, 56, 1309–1318 CrossRef CAS.
- G. Gupta, P. Kumari, J. Y. Ryu, J. Lee, S. M. Mobin and C. Y. Lee, Inorg. Chem., 2019, 58, 8587–8595 CrossRef CAS PubMed.
- K.-H. Leung, H.-Z. He, D. S.-H. Chan, W.-C. Fu, C.-H. Leung and D.-L. Ma, Sens. Actuators, B, 2013, 177, 487–492 CrossRef CAS.
- D.-L. Ma, M. Hiu-Tung Kwan, D. Shiu-Hin Chan, P. Lee, H. Yang, V. Pui-Yan Ma, L.-P. Bai, Z.-H. Jiang and C.-H. Leung, Analyst, 2011, 136, 2692–2696 RSC.
- W. Wang, K. Vellaisamy, G. Li, C. Wu, C.-N. Ko, C.-H. Leung and D.-L. Ma, Anal. Chem., 2017, 89, 11679–11684 CrossRef CAS.
- W. Wang, L. Lu, K.-J. Wu, J. Liu, C.-H. Leung, C.-Y. Wong and D.-L. Ma, Sens. Actuators, B, 2019, 288, 392–398 CrossRef CAS.
- X. Zhen, R. Qu, W. Chen, W. Wu and X. Jiang, Biomater. Sci., 2021, 9, 285–300 RSC.
- K. Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS PubMed.
- D. A. Smithen, H. Yin, M. H. R. Beh, M. Hetu, T. S. Cameron, S. A. McFarland and A. Thompson, Inorg. Chem., 2017, 56, 4121–4132 CrossRef CAS.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS PubMed.
- N. Y. S. Lam, D. Truong, H. Burmeister, M. V. Babak, H. U. Holtkamp, S. Movassaghi, D. M. Ayine-Tora, A. Zafar, M. Kubanik, L. Oehninger, T. Söhnel, J. Reynisson, S. M. F. Jamieson, C. Gaiddon, I. Ott and C. G. Hartinger, Inorg. Chem., 2018, 57, 14427–14434 CrossRef CAS PubMed.
- S. Parveen, Appl. Organomet. Chem., 2020, 34, e5687 CrossRef CAS.
- P. Moharana, D. Ghosh and P. Paira, Inorg. Chem. Commun., 2021, 124, 108364 CrossRef CAS.
- M. V. Babak and W. H. Ang, Metal ions in life sciences, 2018, p. 18 Search PubMed.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- A. Sigel, H. Sigel, E. Freisinger and R. K. O. Sigel, Metallo-Drugs: Development and Action of Anticancer Agents, Walter de Gruyter GmbH & Co KG, 2018 Search PubMed.
- A. K. Singh, D. S. Pandey, Q. Xu and P. Braunstein, Coord. Chem. Rev., 2014, 270–271, 31–56 CrossRef CAS.
- M. V. Babak, D. Plażuk, S. M. Meier, H. J. Arabshahi, J. Reynisson, B. Rychlik, A. Błauż, K. Szulc, M. Hanif, S. Strobl, A. Roller, B. K. Keppler and C. G. Hartinger, Chem. – Eur. J., 2015, 21, 5110–5117 CrossRef CAS.
- G. P. Gupta and J. Massagué, Cell, 2006, 127, 679–695 CrossRef CAS.
- M. E. Menezes, S. K. Das, I. Minn, L. Emdad, X.-Y. Wang, D. Sarkar, M. G. Pomper and P. B. Fisher, Adv. Cancer Res., 2016, 132, 1–44 CrossRef CAS.
- A. Gandalovičová, D. Rosel, M. Fernandes, P. Veselý, P. Heneberg, V. Čermák, L. Petruželka, S. Kumar, V. Sanz-Moreno and J. Brábek, Trends Cancer, 2017, 3, 391–406 CrossRef.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 2017, 1549–1560 CrossRef CAS.
- A. Bergamo and G. Sava, Chem. Soc. Rev., 2015, 44, 8818–8835 RSC.
- G. Bertini, H. B. Gray, P. H. Gray, J. S. Valentine, E. I. Stiefel and P. E. Stiefel, Biological Inorganic Chemistry: Structure and Reactivity, University Science Books, 2007 Search PubMed.
- T. Pieper, K. Borsky and B. K. Keppler, in Metallopharmaceuticals I: DNA Interactions, ed. M. J. Clarke and P. J. Sadler, Springer, Berlin, Heidelberg, 1999, pp. 171–199 Search PubMed.
- L. Morbidelli, S. Donnini, S. Filippi, L. Messori, F. Piccioli, P. Orioli, G. Sava and M. Ziche, Br. J. Cancer, 2003, 88, 1484–1491 CrossRef CAS PubMed.
- A. Vacca, M. Bruno, A. Boccarelli, M. Coluccia, D. Ribatti, A. Bergamo, S. Garbisa, L. Sartor and G. Sava, Br. J. Cancer, 2002, 86, 993–998 CrossRef CAS PubMed.
- S. Pacor, S. Zorzet, M. Cocchietto, M. Bacac, M. Vadori, C. Turrin, B. Gava, A. Castellarin and G. Sava, J. Pharmacol. Exp. Ther., 2004, 310, 737–744 CrossRef CAS PubMed.
- B. Gava, S. Zorzet, P. Spessotto, M. Cocchietto and G. Sava, J. Pharmacol. Exp. Ther., 2006, 317, 284–291 CrossRef CAS PubMed.
- M. I. Webb and C. J. Walsby, Metallomics, 2013, 5, 1624 CrossRef CAS PubMed.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. DiGiovine, G. Pezzoni, L. Sartor and S. Garbisa, Clin. Cancer Res., 2003, 9, 1898–1905 CAS.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem. – Eur. J., 2011, 17, 3562–3566 CrossRef CAS.
- R. H. Berndsen, A. Weiss, U. K. Abdul, T. J. Wong, P. Meraldi, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Sci. Rep., 2017, 7, 43005 CrossRef CAS PubMed.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- S. Chatterjee, S. Kundu, A. Bhattacharyya, C. G. Hartinger and P. J. Dyson, J. Biol. Inorg. Chem., 2008, 13, 1149 CrossRef CAS.
- A. Casini, C. Gabbiani, F. Sorrentino, M. P. Rigobello, A. Bindoli, T. J. Geldbach, A. Marrone, N. Re, C. G. Hartinger, P. J. Dyson and L. Messori, J. Med. Chem., 2008, 51, 6773–6781 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS.
- T. Sasaki, K. Hiroki and Y. Yamashita, BioMed Res. Int., 2013 DOI:10.1155/2013/546318.
- R. E. Morris, R. E. Aird, P. del Socorro Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- A. Bergamo, A. Masi, A. F. A. Peacock, A. Habtemariam, P. J. Sadler and G. Sava, J. Inorg. Biochem., 2010, 104, 79–86 CrossRef CAS.
- R. Carter, A. Westhorpe, M. Romero, A. Habtemariam, C. Gallevo, Y. Bark, N. Menezes, P. Sadler and R. Sharma, Sci. Rep., 2016, 6, 20596 CrossRef CAS PubMed.
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS PubMed.
- H. Nagase, R. Visse and G. Murphy, Cardiovasc. Res., 2006, 69, 562–573 CrossRef CAS PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- R. Foster, D. Cole, S. Mead, P. Sadler and K. Grimshaw, Cancer Res., 2009, 69, 889–889 Search PubMed.
- L. Ma, R. Ma, Z. Wang, S.-M. Yiu and G. Zhu, Chem. Commun., 2016, 52, 10735–10738 RSC.
- Y. Benabdelouahab, L. Muñoz-Moreno, M. Frik, I. de la Cueva-Alique, M. A. El Amrani, M. Contel, A. M. Bajo, T. Cuenca and E. Royo, Eur. J. Inorg. Chem., 2015, 2015, 2295–2307 CrossRef CAS PubMed.
- C. Sonkar, N. Malviya, R. Ranjan, S. Pakhira and S. Mukhopadhyay, ACS Appl. Bio Mater., 2020, 3, 4600–4612 CrossRef CAS.
- C. Sonkar, N. Malviya, N. Sinha, A. Mukherjee, S. Pakhira and S. Mukhopadhyay, BioMetals, 2021, 1–18 Search PubMed.
- Z. Xu, D. Kong, X. He, L. Guo, X. Ge, X. Liu, H. Zhang, J. Li, Y. Yang and Z. Liu, Inorg. Chem. Front., 2018, 5, 2100–2105 RSC.
- C. Fan, Q. Wu, T. Chen, Y. Zhang, W. Zheng, Q. Wang and W. Mei, MedChemComm, 2014, 5, 597–602 RSC.
- M. C. Ruiz, J. Kljun, I. Turel, A. L. Di Virgilio and I. E. León, Metallomics, 2019, 11, 666–675 CrossRef CAS PubMed.
- S. Acharya, S. Ghosh, M. Maji, A. R. U. Parambil, S. Singh and A. Mukherjee, Chem. Commun., 2020, 56, 5421–5424 RSC.
- L. Ma, R. Ma, Z. Wang, S.-M. Yiu and G. Zhu, Chem. Commun., 2016, 52, 10735–10738 RSC.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- E. Schreiber-Brynzak, E. Klapproth, C. Unger, I. Lichtscheidl-Schultz, S. Göschl, S. Schweighofer, R. Trondl, H. Dolznig, M. A. Jakupec and B. K. Keppler, Invest. New Drugs, 2015, 33, 835–847 CrossRef CAS PubMed.
- B. N. Cunha, L. Luna-Dulcey, A. M. Plutin, R. G. Silveira, J. Honorato, R. R. Cairo, T. D. de Oliveira, M. R. Cominetti, E. E. Castellano and A. A. Batista, Inorg. Chem., 2020, 59, 5072–5085 CrossRef CAS PubMed.
- N. Gligorijević, S. Aranđelović, L. Filipović, K. Jakovljević, R. Janković, S. Grgurić-Šipka, I. Ivanović, S. Radulović and Ž. L. Tešić, J. Inorg. Biochem., 2012, 108, 53–61 CrossRef PubMed.
- S. Nikolić, I. Ćirić, A. Roller, V. Lukeš, V. B. Arion and S. Grgurić-Šipka, New J. Chem., 2017, 41, 6857–6865 RSC.
- I. Ivanovic, S. Grguric-Sipka, N. Gligorijevic, S. Radulovic, A. Roller, Z. Tesic and B. Keppler, J. Serb. Chem. Soc., 2011, 76, 53–61 CrossRef CAS.
- E. S. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS PubMed.
- M. Galanski, V. Arion, M. Jakupec and B. Keppler, Curr. Pharm. Des., 2003, 9, 2078–2089 CrossRef CAS PubMed.
- J. Cathcart, A. Pulkoski-Gross and J. Cao, Genes Dis., 2015, 2, 26–34 CrossRef PubMed.
- H.-W. Cheng, Y.-F. Chen, J.-M. Wong, C.-W. Weng, H.-Y. Chen, S.-L. Yu, H.-W. Chen, A. Yuan and J. J. W. Chen, J. Exp. Clin. Cancer Res., 2017, 36, 27 CrossRef PubMed.
- A.-L. Lu, X. Li, Y. Gu, P. M. Wright and D.-Y. Chang, Cell Biochem. Biophys., 2001, 35, 141–170 CrossRef CAS PubMed.
- G.-M. Li, Cell Res., 2008, 18, 85–98 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnières, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- A. W. Griffioen, S. D.-W. van der Schaft, A. F. Barendsz-Janson, A. Cox, H. A. S. Boudier, H. F. Hillen and K. H. Mayo, Biochem. J., 2001, 354, 233–242 CrossRef CAS.
- P. Nowak-Sliwinska, J.-P. Ballini, G. Wagnières and H. van den Bergh, Microvasc. Res., 2010, 79, 21–28 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, J. R. van Beijnum, M. van Berkel, H. van den Bergh and A. W. Griffioen, Angiogenesis, 2010, 13, 281–292 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, C. M. Clavel, E. Păunescu, M. T. te Winkel, A. W. Griffioen and P. J. Dyson, Mol. Pharmaceutics, 2015, 12, 3089–3096 CrossRef CAS PubMed.
- C. Chen, C. Xu, T. Li, S. Lu, F. Luo and H. Wang, Eur. J. Med. Chem., 2020, 203, 112605 CrossRef CAS PubMed.
- C. M. Clavel, E. Păunescu, P. Nowak-Sliwinska, A. W. Griffioen, R. Scopelliti and P. J. Dyson, J. Med. Chem., 2015, 58, 3356–3365 CrossRef CAS PubMed.
- F. Y. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernandez, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- S. Balaji, M. K. Mohamed Subarkhan, R. Ramesh, H. Wang and D. Semeril, Organometallics, 2020, 39, 1366–1375 CrossRef CAS.
- C. M. Clavel, E. Păunescu, P. Nowak-Sliwinska and P. J. Dyson, Chem. Sci., 2014, 5, 1097 RSC.
- A. K. Renfrew, L. Juillerat-Jeanneret and P. J. Dyson, J. Organomet. Chem., 2011, 3, 772–779 CrossRef.
- I. Dragutan, V. Dragutan and A. Demonceau, Molecules, 2015, 20, 17244–17274 CrossRef CAS PubMed.
- G. Suess-Fink, F.-A. Khan, L. Juillerat-Jeanneret, P. J. Dyson and A. K. Renfrew, J. Cluster Sci., 2013, 3, 313–324 Search PubMed.
- J. Zhao, X. Zhang, H. Liu, Z. Xiong, M. Li and T. Chen, J. Organomet. Chem., 2019, 898, 120869 CrossRef CAS.
- H. Huang, P. Zhang, Y. Chen, L. Ji and H. Chao, Dalton Trans., 2015, 44, 15602–15610 RSC.
- Q. Wu, C. Fan, T. Chen, C. Liu, W. Mei, S. Chen, B. Wang, Y. Chen and W. Zheng, Eur. J. Med. Chem., 2013, 63, 57–63 CrossRef CAS PubMed.
- M. Sweta, P. J. Jay, A. J. Domb and K. Neeraj, Curr. Pharm. Des., 2006, 12, 4785–4796 CrossRef PubMed.
- P. Govender, N. C. Antonels, J. Mattsson, A. K. Renfrew, P. J. Dyson, J. R. Moss, B. Therrien and G. S. Smith, J. Organomet. Chem., 2009, 694, 3470–3476 CrossRef CAS.
- P. Govender, A. K. Renfrew, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2011, 40, 1158–1167 RSC.
- P. Govender, L. C. Sudding, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2013, 42, 1267–1277 RSC.
- M. A. Furrer, F. Schmitt, M. Wiederkehr, L. Juillerat-Jeanneret and B. Therrien, Dalton Trans., 2012, 41, 7201–7211 RSC.
- B. M. Blunden, H. Lu and M. H. Stenzel, Biomacromolecules, 2013, 14, 4177–4188 CrossRef CAS PubMed.
- B. M. Blunden, D. S. Thomas and M. H. Stenzel, Polym. Chem., 2012, 3, 2964 RSC.
- S. Kapitza, M. Pongratz, M. A. Jakupec, P. Heffeter, W. Berger, L. Lackinger, B. K. Keppler and B. Marian, J. Cancer Res. Clin. Oncol., 2005, 131, 101–110 CrossRef CAS PubMed.
- J. Liu, H. Lai, Z. Xiong, B. Chen and T. Chen, Chem. Commun., 2019, 55, 9904–9914 RSC.
- J. Boelens, S. Lust, F. Offner, M. E. Bracke and B. W. Vanhoecke, In Vivo, 2007, 21, 215–226 CAS.
- B. Pragti, K. Kundu, C. Sonkar, R. Ganguly and S. Mukhopadhyay, Polyhedron, 2021, 207, 115379 CrossRef CAS.
- C. Tan, S. Lai, S. Wu, S. Hu, L. Zhou, Y. Chen, M. Wang, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, J. Med. Chem., 2010, 53, 7613–7624 CrossRef CAS PubMed.
- S. Acharya, M. Maji, M. P. Chakraborty, I. Bhattacharya, R. Das, A. Gupta and A. Mukherjee, Inorg. Chem., 2021, 60, 3418–3430 CrossRef CAS PubMed.
- J. Du, E. Zhang, Y. Zhao, W. Zheng, Y. Zhang, Y. Lin, Z. Wang, Q. Luo, K. Wu and F. Wang, Metallomics, 2015, 7, 1573–1583 CrossRef CAS PubMed.
- B. Biersack, M. Zoldakova, K. Effenberger and R. Schobert, Eur. J. Med. Chem., 2010, 45, 1972–1975 CrossRef CAS PubMed.
- B. T. Elie, Y. Pechenyy, F. Uddin and M. Contel, J. Biol. Inorg. Chem., 2018, 23, 399–411 CrossRef CAS PubMed.
- I. Marech, C. Leporini, M. Ammendola, M. Porcelli, C. D. Gadaleta, E. Russo, G. De Sarro and G. Ranieri, Cancer Lett., 2016, 380, 216–226 CrossRef CAS PubMed.
- D. Tarin, Semin. Cancer Biol., 2011, 21, 72–82 CrossRef CAS PubMed.
- M. Bacac and I. Stamenkovic, Annu. Rev. Pathol.: Mech. Dis., 2008, 3, 221–247 CrossRef CAS PubMed.
- M. J. Duffy, P. M. McGowan and W. M. Gallagher, J. Pathol., 2008, 214, 283–293 CrossRef CAS PubMed.
- T. N. Seyfried and L. C. Huysentruyt, Crit. Rev. Oncog., 2013, 18, 43–73 CrossRef PubMed.
- T. H. Adair and J.-P. Montani, Overview of Angiogenesis, Morgan & Claypool Life Sciences, 2010 Search PubMed.
- L. M. Sherwood, E. E. Parris and J. Folkman, N. Engl. J. Med., 1971, 285, 1182–1186 CrossRef PubMed.
- P. H. Burri and M. R. Tarek, Anat. Rec., 1990, 228, 35–45 CrossRef CAS PubMed.
- J. H. Caduff, L. C. Fischer and P. H. Burri, Anat. Rec., 1986, 216, 154–164 CrossRef CAS PubMed.
- B. R. Zetter, Annu. Rev. Med., 1998, 49, 407–424 CrossRef CAS PubMed.
- K. Yamamoto, T. Takahashi, T. Asahara, N. Ohura, T. Sokabe, A. Kamiya and J. Ando, J. Appl. Physiol., 2003, 95, 2081–2088 CrossRef PubMed.
- M. Janiszewska, M. C. Primi and T. Izard, J. Biol. Chem., 2020, 295, 2495–2505 CrossRef CAS PubMed.
- M. Perez-Moreno, C. Jamora and E. Fuchs, Cell, 2003, 112, 535–548 CrossRef CAS PubMed.
- E. Cerami, J. Gao, U. Dogrusoz, B. E. Gross, S. O. Sumer, B. A. Aksoy, A. Jacobsen, C. J. Byrne, M. L. Heuer, E. Larsson, Y. Antipin, B. Reva, A. P. Goldberg, C. Sander and N. Schultz, Cancer Discovery, 2012, 2, 401–404 CrossRef PubMed.
- I. Gurgul, O. Mazuryk, M. Łomzik, P. C. Gros, D. Rutkowska-Zbik and M. Brindell, Metallomics, 2020, 12, 784–793 CrossRef CAS PubMed.
- C. Pelillo, H. Mollica, J. A. Eble, J. Grosche, L. Herzog, B. Codan, G. Sava and A. Bergamo, J. Inorg. Biochem., 2016, 160, 225–235 CrossRef CAS PubMed.
- A. F. Chambers, A. C. Groom and I. C. MacDonald, Nat. Rev. Cancer, 2002, 2, 563–572 CrossRef CAS PubMed.
- A. Zamora, S. A. Pérez, V. Rodríguez, C. Janiak, G. S. Yellol and J. Ruiz, J. Med. Chem., 2015, 58, 1320–1336 CrossRef CAS PubMed.
- X.-D. Gu, L.-L. Xu, H. Zhao, J.-Z. Gu and X.-H. Xie, Braz. J. Med. Biol. Res., 2017, 50, 32–39 Search PubMed.
- M. Chen, X. Huang, J. Lai, L. Ma and T. Chen, Chin. Chem. Lett., 2021, 32, 158–161 CrossRef CAS.
- N. Malviya, C. Sonkar, R. Ganguly, D. Bhattacherjee, K. P. Bhabak and S. Mukhopadhyay, ACS Appl. Mater. Interfaces, 2019, 11, 47606–47618 CrossRef CAS PubMed.
- M. Bloomston, E. E. Zervos and A. S. Rosemurgy, Ann. Surg. Oncol., 2002, 9, 668–674 CrossRef PubMed.
- M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS PubMed.
- S.-C. Liu, S.-F. Yang, K.-T. Yeh, C.-M. Yeh, H.-L. Chiou, C.-Y. Lee, M.-C. Chou and Y.-S. Hsieh, Clin. Chim. Acta, 2006, 371, 92–96 CrossRef CAS PubMed.
- J. Ueda, M. Kajita, N. Suenaga, K. Fujii and M. Seiki, Oncogene, 2003, 22, 8716–8722 CrossRef CAS PubMed.
- S. S. Lakka, M. Rajan, C. Gondi, N. Yanamandra, N. Chandrasekar, S. L. Jasti, Y. Adachi, K. Siddique, M. Gujrati, W. Olivero, D. H. Dinh, G. Kouraklis, A. P. Kyritsis and J. S. Rao, Oncogene, 2002, 21, 8011–8019 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- P. Zhang, H. Huang, J. Huang, H. Chen, J. Wang, K. Qiu, D. Zhao, L. Ji and H. Chao, ACS Appl. Mater. Interfaces, 2015, 7, 23278–23290 CrossRef CAS PubMed.
- Z. Deng, L. Yu, W. Cao, W. Zheng and T. Chen, Chem. Commun., 2015, 51, 2637–2640 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- G. Petruk, D. M. Monti, G. Ferraro, A. Pica, L. D'Elia, F. Pane, A. Amoresano, J. Furrer, K. Kowalski and A. Merlino, ChemMedChem, 2019, 14, 594–602 CrossRef CAS PubMed.
- A. Koceva-Chyła, K. Matczak, P. Hikisz, K. Durka, K. Kochel, G. Süss-Fink, J. Furrer and K. Kowalski, ChemMedChem, 2016, 11, 2171–2187 CrossRef PubMed.
- P. Thangavel, B. Viswanath and S. Kim, Int. J. Nanomed., 2017, 12, 2749–2758 CrossRef CAS PubMed.
- D. Sun, Y. Liu, Q. Yu, X. Qin, L. Yang, Y. Zhou, L. Chen and J. Liu, Biomaterials, 2014, 35, 1572–1583 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, R. C. Huxford-Phillips and W. Lin, Chem. Soc. Rev., 2012, 41, 2673–2685 RSC.
| This journal is © The Royal Society of Chemistry 2022 |