
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRepurposing the HCV NS3–4A protease drug boceprevir as COVID-19 therapeutics†‡
Rick
Oerlemans§
a,
Angel Jonathan
Ruiz-Moreno§
 abc,
Yingying
Cong
d,
Nilima
Dinesh Kumar
de,
Marco A.
Velasco-Velazquez
bc,
Constantinos G.
Neochoritis
f,
Jolanda
Smith
e,
Fulvio
Reggiori
d,
Matthew R.
Groves
a and
Alexander
Dömling
*af
abc,
Yingying
Cong
d,
Nilima
Dinesh Kumar
de,
Marco A.
Velasco-Velazquez
bc,
Constantinos G.
Neochoritis
f,
Jolanda
Smith
e,
Fulvio
Reggiori
d,
Matthew R.
Groves
a and
Alexander
Dömling
*af
aDepartment of Drug Design, University of Groningen, The Netherlands. E-mail: a.s.s.domling@rug.nl
bDepartamento de Farmacología y Unidad Periférica de Investigación en Biomedicina Traslacional, Facultad de Medicina, Universidad Nacional Autónoma de México (UNAM), Ciudad de México, Mexico
cPrograma de Doctorado en Ciencias Biomédicas, UNAM, Ciudad de México, Mexico
dDepartment of Biomedical Sciences of Cells and Systems, University of Groningen, University Medical Center Groningen, The Netherlands
eDepartment of Medical Microbiology and Infection Prevention, University of Groningen, University Medical Center Groningen, Groningen 9700 RB, The Netherlands
fTelesis Pharma, Groningen, The Netherlands
First published on 21st December 2020
Abstract
The rapid growth of COVID-19 cases is causing an increasing death toll and also paralyzing the world economy. De novo drug discovery takes years to move from idea and/or pre-clinic to market, and it is not a short-term solution for the current SARS-CoV-2 pandemic. Drug repurposing is perhaps the only short-term solution, while vaccination is a middle-term solution. Here, we describe the discovery path of the HCV NS3–4A protease inhibitors boceprevir and telaprevir as SARS-CoV-2 main protease (3CLpro) inhibitors. Based on our hypothesis that α-ketoamide drugs can covalently bind to the active site cysteine of the SARS-CoV-2 3CLpro, we performed docking studies, enzyme inhibition and co-crystal structure analyses and finally established that boceprevir, but not telaprevir, inhibits replication of SARS-CoV-2 and mouse hepatitis virus (MHV), another coronavirus, in cell culture. Based on our studies, the HCV drug boceprevir deserves further attention as a repurposed drug for COVID-19 and potentially other coronaviral infections as well.
Introduction
Since emerging in Wuhan, China, in December 2019, the coronavirus (CoV) disease 2019 (COVID-19) epidemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has progressed rapidly into a pandemic.2 COVID-19 is characterized by fever, cough, fatigue, shortness of breath, pneumonia, and other respiratory tract symptoms, and in many cases progresses to death.2–4 As of August 30, 2020, there have been more than 25 million confirmed cases, and more than 830.000 deaths reported worldwide. Moreover, COVID-19 is making an immense negative impact on the world's economy and has become a huge societal burden.5SARS-CoV2 is an enveloped, non-segmented, positive-sense RNA virus that is part of the order Nidovirales, in the CoV virus family, which is broadly distributed in humans and other mammals.6,7 SARS-CoV-2 is classified into the beta-CoV genera. Recent studies highlighted that SARS-CoV-2 genes share >80% nucleotide identity and >89% nucleotide similarity with SARS-CoV genes.8–10 Upon cell entry, two polypeptides, pp1a and pp1ab, are produced by the host translation machinery directly from the CoV genome.11,12 These two polypeptides self-cleave proteolytically into 11 and 16 individual non-structural (nsp) proteins, respectively, that are essential for viral replication.13 CoV encode either two or three proteases that are involved in the self-cleavage of pp1a and pp1ab. They are the papain-like protease (PLpro), present with nsp3, and the 3C-like proteinase (3CLpro) or Mpro, localized in nsp5.14 Most CoV encode two PLpros within nsp3, except the gamma-CoV, SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV-2.14,15 Importantly, 3CLpro plays a critical role in CoV replication and unlike structural/accessory protein-encoding genes, displays a considerable similarity between members of CoV, in particular beta-CoV.11,12 Therefore, it is a promising target for the discovery and the development of a pan-anti-CoV inhibitor.7,16
We investigated FDA-approved drugs with electrophile warheads for their potential to inhibit the SARS-CoV-2 proteases (Fig. 1A). Both proteases are cysteine proteases. The great majority of cysteine protease inhibitors function by a covalent mechanism where the nucleophilic sulfhydryl forms an (ir)reversible bond with an electrophilic warhead (α-ketoamide, for instance) of the inhibitor (Fig. 1B). Such covalent inhibitors have several advantages, including an increased ligand efficiency, overcoming competition with native ligands and less recurrent dosing due to sustained duration of action.17 Thus, we focused our attention on subgroups of drugs, e.g. electrophiles such as α-ketoamides and nitriles, which potentially can undergo a covalent modification of the active site cysteine of SARS-CoV-2 proteases. Our first finding was that nitrile containing gliptins are potential SARS-CoV-2 protease inhibitors.16 Here, we report that α-ketoamide hepatitis C virus (HCV) protease inhibitors are also inhibitors of SARS-CoV-2 3CLpro (Fig. 1C). In particular, we present the finding of boceprevir and telaprevir as 3CLpro SARS-CoV-2 inhibitors, suggested by docking studies, supported by biochemical and co-crystal structure analyses, and finally provide evidence that boceprevir, but not telaprevir, inhibits viral replication in cellular assays assessing the replication of mouse hepatitis virus (MHV), also a beta-CoV, and SARS-CoV-2.
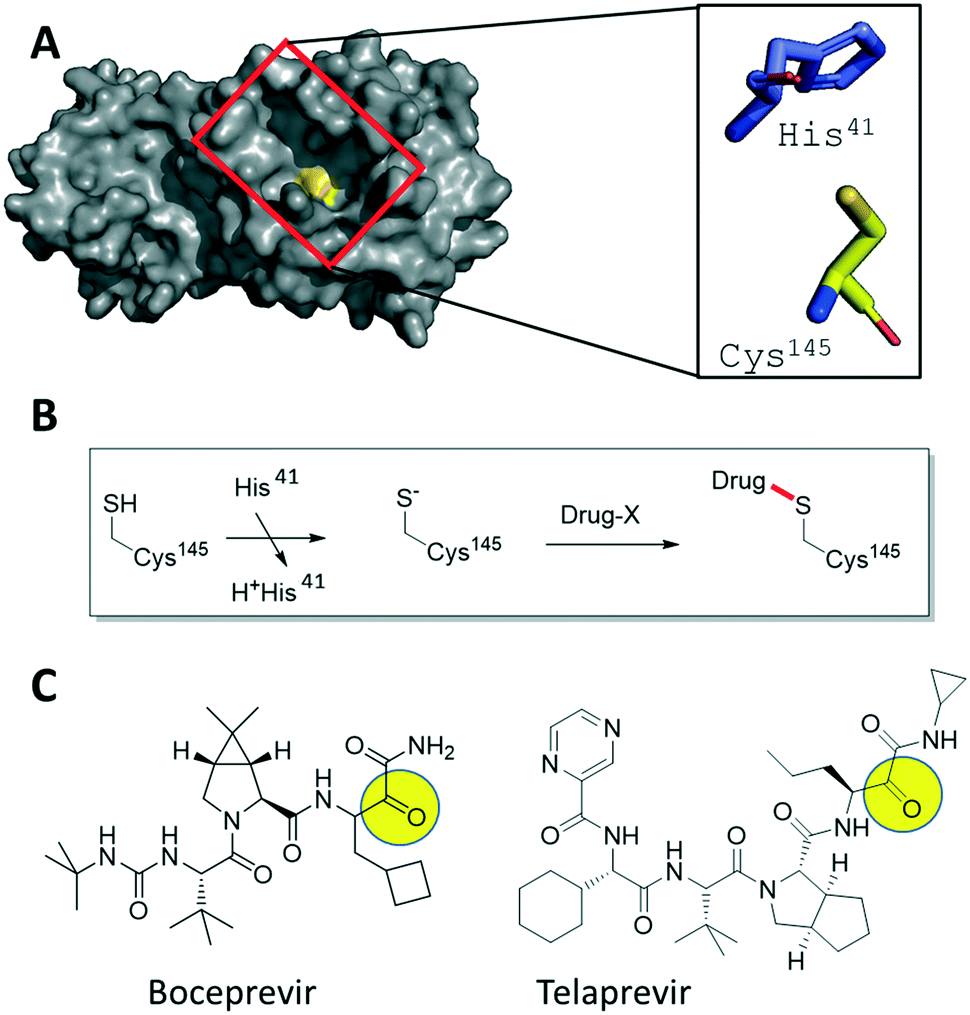 | ||
| Fig. 1 Hypothesis-driven illustration of α-ketoamide HCV NS3–4A protease inhibitors as covalent SARS-CoV-2 3CLpro inhibitors. A) Crystal structure of SARS-CoV-2 3CLpro (PDB ID 6LU7, grey surface presentation) highlighting the active site (red box) and the catalytic Cys145 (yellow). The catalytic dyad His41 and Cys145 are shown as stick presentation. B) Schematic mechanism of the addition of an electrophilic drug onto the active site Cys145 involving the His41 base. C) 2D structures of the marketed HCV drugs boceprevir and telaprevir, highlighting with a yellow circle the electrophilic α-ketoamide groups. | ||
Methods
Chemicals and reagents
Boceprevir, telaprevir and bafilomycin A1 were obtained from Advanced Chemblocks Inc, Combi-Blocks and Sigma-Aldrich, respectively, and dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich) at concentration of 100 mM, 100 mM and 200 μM, respectively. Boceprevir and telaprevir were stored at 4 °C, and bafilomycin at −20 °C.Molecular docking
The methodology for covalent docking was performed as reported.16 Briefly, the crystal structure of SARS-CoV-2 3CLpro (PDB ID 6LU7) was used as a receptor, and boceprevir and telaprevir as ligands. Protein was prepared by removing co-crystallized waters, solvent molecules, and adding charges and hydrogens using Chimera 1.14.18 Isomeric SMILES codes for ligands were retrieved from PubChem and prepared for docking by setting the absolute stereo flags, adding explicit hydrogens and tautomeric states at pH 7.4. 3D coordinates were generated with Standardized 19.20.0 (http://www.chemaxon.com). For covalent docking, the sulfur atom of the reactive Cys145 of 3CLpro was used as the linker to form the covalent bond. For each ligand, 50 runs of genetic algorithm for the conformational search were performed and each pose was evaluated employing the PLP Chemscore scoring function using the GOLD software from the Cambridge Crystallography Data Center (CCDC).19Cloning, protein expression and purification
An E. coli codon-optimized gene encoding SARS-CoV-2 3CLpro was purchased from Eurofins Genomics (Fig. S1‡). The gene was subcloned into the pET-28a(+) vector using NcoI and XhoI restriction enzymes (NEB). Initial tests showed that the resulting C-terminal 6His-tag was reducing enzymatic activity so in order to be able to obtain a native C terminus, PCR was performed with specific primers GCA GGT CTC GAG AGG CCC CTG AAA CGT AAC GCC GC (5′ → 3′) and CGC AAG CCC ATG GCG GC (5′ → 3′) to introduce a human Rhinovirus 3C protease (HRV 3Cpro) compatible cleavage site on the C terminus (SGVTFQGP). The PCR product was then digested and cloned into the pET-28a(+) vector using NcoI and XhoI.The resulting pET-28a-3CLpro plasmid was transformed into competent BL21 Star (DE3) E. coli strain and a single colony was used to inoculate 20 ml of Luria–Bertani (LB) medium, supplemented with 50 μg ml−1 kanamycin and 35 μg ml−1 chloramphenicol, before growth at 37 °C. After 16 h, the preculture was added to 2 l of LB medium (supplemented with 50 μg ml−1 kanamycin and 35 μg ml−1 chloramphenicol) and incubated at 37 °C in a shaking incubator (180 rpm). Expression of the fusion protein was induced by addition of 0.5 mM isopropyl-1-β;-D-thiogalactopyranoside (abcr chemicals) when the culture OD600 reached 0.6. At this point, the culture was transferred to an 18 °C shaking incubator (180 rpm) and after an overnight incubation, the bacteria were harvested by centrifugation at 4000g for 20 min. Pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 10 mM imidazole, 20 μg ml−1 DNAse 1, 0.4 mg ml−1 lysozyme), lysed by sonication (50% amplitude, 2 s on/15 s off, for 5 min) and then clarified by centrifugation at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 45 min. The supernatant was loaded on a 5 ml HiTrap HP column (GE Healthcare), washed with 5 column volumes of washing buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 25 mM imidazole) and subsequently eluted in the elution buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 250 mM imidazole). His-tagged HRV 3Cpro was added to the eluted fraction (1:10 w/w) and this mixture was dialyzed overnight at 4 °C against 50 mM Tris-HCl, 1 mM TCEP, 300 mM NaCl, to remove the imidazole and cleave the C-terminal 6xHis tag. Reverse nickel-NTA purification was then performed to elute untagged SARS-CoV-2 3CLpro, which was then further purified by size-exclusion chromatography (SEC) using a HiLoad 16/60 S200 (GE Healthcare) equilibrated with the SEC buffer (20 mM HEPES, pH 7.5, 50 mM NaCl). Finally, the purified SARS-CoV-2 3CLpro was concentrated to 5 mg ml−1 in a Vivaspin centrifugal concentrator (molecular weight cut-off (MWCO) 10 kDa, Sartorius).
000g for 45 min. The supernatant was loaded on a 5 ml HiTrap HP column (GE Healthcare), washed with 5 column volumes of washing buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 25 mM imidazole) and subsequently eluted in the elution buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 250 mM imidazole). His-tagged HRV 3Cpro was added to the eluted fraction (1:10 w/w) and this mixture was dialyzed overnight at 4 °C against 50 mM Tris-HCl, 1 mM TCEP, 300 mM NaCl, to remove the imidazole and cleave the C-terminal 6xHis tag. Reverse nickel-NTA purification was then performed to elute untagged SARS-CoV-2 3CLpro, which was then further purified by size-exclusion chromatography (SEC) using a HiLoad 16/60 S200 (GE Healthcare) equilibrated with the SEC buffer (20 mM HEPES, pH 7.5, 50 mM NaCl). Finally, the purified SARS-CoV-2 3CLpro was concentrated to 5 mg ml−1 in a Vivaspin centrifugal concentrator (molecular weight cut-off (MWCO) 10 kDa, Sartorius).
Crystallization and structure determination
Initial apo seed crystals were obtained by the sitting drop vapour diffusion method at 18 °C with drops consisting of 0.5 μl protein solution (5 mg ml−1) and 0.5 μl reservoir solution (0.2 M potassium sodium tartrate tetrahydrate, 0.2 M lithium acetate, 20% PEG 3350). These initial SARS-CoV-2 3CLpro crystals were used to make a seed stock using the Glass Seed Bead™ kit (Hampton). To obtain co-crystals of SARS-CoV-2 3CLpro with inhibitors, a pre-incubation co-crystallization protocol was followed. Briefly, protein at 0.5 mg ml−1 was incubated with 300 μM compound at 4 °C overnight. After centrifugation at 20000g for 10 min to remove aggregates and precipitates, the protein solution was concentrated in a Vivaspin centrifugal concentrator (MWCO 10 kDa, Sartorius) to 5 mg ml−1 and an additional 300 μM compound was added and left to incubate at 4 °C for 3 h. After centrifugation, the pre-incubated protein solutions were used to obtain co-crystals by the sitting drop vapour diffusion method.20 For boceprevir the best crystals were grown in drops containing 1.5 μl pre-incubated protein solution (5 mg ml−1), 1 μl reservoir solution (100 mM MES pH 6.75, 16% (w/v) PEG 6000, 5% (v/v) DMSO, 300 μM boceprevir) and 0.5 μl seed stock (diluted 1:500). For telaprevir, the best crystals were grown in drops containing 1.5 μl pre-incubated protein solution (5 mg ml−1), 1 μl reservoir solution (100 mM MES pH 6.75, 18% (w/v) PEG 6000, 5% (v/v) DMSO, 300 μM telaprevir) and 0.5 μl seed stock (diluted 1:500). The cryo-protectant solution consisted of reservoir solution supplemented with 25% (v/v) glycerol.
X-ray diffraction data was collected on beamline P11 of the Deutsches Elektronen-Synchrotron (DESY) (Hamburg, Germany) at 100 K. Data integration and scaling was performed using XDS21 and Aimless22 from the CCP4 software suite.23 The DIMPLE software in CCP4 (ref. 23) was used to obtain the solved structures, utilizing a previously solved SARS-CoV-2 3CLpro structure (ligand stripped) as the reference model (PDB: 6LU7). The DIMPLE output models were then subjected to iterative cycles of model building with COOT and refinement with REFMAC.24,25 The structures were deposited into the PDB (6ZRT and 6ZRU for telaprevir and boceprevir, respectively). Data collection and refinement statistics can be found in Table S1.‡ Similar structures were deposited by other groups (6XQS, 7C7P, 6WNP) during the course of this project.
3CLpro enzymatic assay
The in vitro 3CLpro enzymatic assay to assess compound inhibition was setup following a published protocol for SARS-CoV 3CLpro,26 with slight modifications. Briefly, a continuous kinetic FRET assay was used to measure SARS-CoV-2 3CLpro activity, against the substrate 2-aminobenzoyl-SVTLQSG-Tyr (NO2)-R (Genscript). The cleavage of the FRET substrate was monitored by measuring the increase in fluorescence in a FLUOstar Omega platereader (BMG labtech) at excitation and emission wavelengths of 330 nm and 420 nm, respectively. Total volume for the assay was 200 μl, containing 250 nM SARS-CoV-2 3CLpro and 100 μM substrate in the assay buffer (100 mM potassium phosphate, pH 8, 3% DMSO). The reaction was monitored for 15 min immediately after adding the FRET substrate. Initial rates were calculated via linear regression, using the first 2 min of the reaction. To determine the inhibitory activity of the compounds, the enzyme was incubated with 12 different concentrations of compound (20 nM – 400 μM telaprevir, 5 nM – 100 μM boceprevir) for 1 h at 25 °C, followed by initial rate determination. Dose response curves and IC50 values were obtained using the Prism8 software by plotting the initial rates against inhibitor concentration and performing non-linear regression. The initial rates were normalized to controls, control without enzyme (100% inhibition) and control without compound (0% inhibition). All measurements were performed in triplicate.Cell culture and the cytotoxic assay
The African green monkey Vero E6 cell line (ATCC CRL-1586), kindly provided by Gorben Pijlman (Wageningen University, Wageningen, the Netherlands) and the mouse LR7 cell line were maintained in Dulbecco's minimal essential medium (DMEM) (Gibco), high glucose supplemented with 10% fetal bovine serum (FBS) (Lonza), 100 U ml−1 penicillin, 100 U ml−1 and streptomycin. Cells were mycoplasma negative and maintained at 37 °C under 5% CO2.For the cytotoxic assay, LR7 or Vero E6 cells were seeded in 96-well plates at a density of 1 × 104 cells per well and cultured in DMEM containing 10% FBS at 37 °C under 5% CO2 for 24 h, followed by addition of serial dilutions (0–500 μM) of the tested drugs. Cells were allowed to grow for 8 h at 37 °C and proliferation was analysed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Briefly, the medium was removed before to add 100 μl of 0.5 mg ml−1 MTT solution (Sigma-Aldrich) and incubate cells at 37 °C for 4 h. Then 100 μl of DMSO were added and incubated for 30 min to solubilize the formazan crystals. Absorbance of each well at OD490 was measured using a GloMax-Multi Detection System (Promoega) and cell survival percentage was calculated as OD490 of the sample/OD490 of the control.
Virus stocks and titration
MHV strain A59 was propagated in LR7 cells in DMEM and the virus titer was determined on LR7 cells and the culture infectious dose (TCID50) units per ml of supernatant were calculated according to the Reed–Muench method.27 The SARS-CoV-2 strain/NL/2020 was obtained from European Virus archive global (EVAg-010V-03903). For virus production, Vero E6 cells were infected at a multiplicity of infection (MOI) of 1 and 48 h post infection (p.i.) supernatants containing progeny virions were harvested, centrifuged, aliquoted and stored at −80 °C. The virus was passaged twice after receiving it from EVAg and passage 2 virus was used for subsequent experiments. Titration was performed using plaque assay on Vero E6 cells. Briefly, Vero E6 cells were seeded at a density of 1.3 × 105/ml in 12-well plates. Next day, cells were infected for 2 h with 10-fold serial dilutions of samples following which, cells were overlaid with 1:1 mixture of 2% agarose (Lonza) and 2X MEM medium (Gibco). On day 3, plaques were fixed using 10% formaldehyde (Alfa Aesar) and stained using crystal violet (Sigma-Aldrich). Titers were calculated and reported as plaque-forming units (PFU) per ml.
Antiviral assays
For MHV infections, LR7 cells were seeded at a density of 1 × 105 cells per well in 12-well plates with a 10 mm diameter coverslip in it in DMEM containing 10% FBS, before to remove the medium and infect cells with MHV in DMEM at MOI 1. The inoculum was removed after 1 h, cells washed twice with 1x PBS (137 mM NaCl, 10 mM phosphate pH 7.4, 2.7 mM KCl) and fresh DMEM medium containing 2% FBS added. The tested drugs were added after another 1 h in the new medium and the cells were collected at 8 h p.i. Specifically, the cover slips were removed and fixed with 4% paraformaldehyde. After permeabilization using 0.2% Triton X-100 and subsequent blocking with PBS buffer containing 1% fetal calf serum, viral non-structural protein (nsp)2 and nsp3 were detected using the anti-nsp2/nsp3 antiserum, a kind gift from Susan Baker,28 followed by incubation with secondary antibody conjugated to Alexa-488 (Life Technologies). Fluorescence signals were captured with a Leica sp8 confocal microscope (Leica) and the nsp2/nsp3-positive cells were counted as infected cells. The cells in the 12 wells plates, in contrast, were harvested in 100 μl of 2× sample buffer (65.8 mM Tris–HCl, pH 6.8, 26.3% glycerol, 2.1% SDS and 0.01% bromophenol blue) for 30 min on ice, sonicated for 1 min and boiled. Equal protein amounts were separated by SDS-PAGE and after western blot, proteins were detected using specific antibodies against MHV N protein (a kind gift from Stuart Siddell, University of Bristol, UK29) and β-actin (Merck Millipore), and with secondary antibody conjugated to Alexa-488 (Life Technologies). Fluorescence signals were analyzed Odyssey imaging system (LI-COR) and signal intensities were normalized and quantified using the ImageJ software (NIH).For SARS-CoV-2 infections, Vero E6 cells were seeded at a density of 6 × 104/well in 24 well plates in DMEM containing 10% FBS. Next day, plates were transferred to a Biosafety level 3 (BSL3) facility and replaced with 200 μl of DMEM medium containing 2% FBS and the virus inoculum (MOI 1). Following 2 h adsorption at 37 °C, virus inoculum was removed, after which cells were washed twice and replaced with DMEM media containing 10% FBS in combination with increasing concentrations of compounds or the equivalent volumes of DMSO as the control. Supernatants were harvested at 8 h p.i. and titrated using plaque assay and data were normalized to the non-treated control. In contrast, cells were washed with ice-cold PBS and lysed with TRIzol reagent (Sigma) according to the manufacturer's instructions. First-strand cDNA was synthesized by using Moloney murine leukemia virus (M-MLV) reverse transcriptase and oligo (dT) (both from Invitrogen). Real-time PCR was performed using an CFX connect Thermocycler (Bio-Rad). The expression levels of SARS-CoV-2 nsp3 gene (forward primer: 5′-GCCTATACAGTTGAACTCGGT; reverse primer: 5′-CAATGCCCAGTGGTGTAAGT) were normalized to that of GAPDH (forward primer: 5′-AGCCACATCGCTCAGACAC; reverse primer: 5′-GCCCAATACGACCAAATCC) according to the comparative cycle threshold method used for quantification as recommended by the manufacture's protocol.
Statistical analyses
Statistical significances were evaluated using the two-tailed heteroscedastic t-test before calculating the p-values. Individual data points from each independent experiment were used for the calculation of the significance.Results and discussion
Molecular docking of α-ketoamides onto 3CLpro
Based on the understanding of the molecular mode-of-action of drugs on their receptors, we initially computationally investigated approved drugs for their potential covalent interaction with the proteases of SARS-CoV-2, as a repurposing approach.30 Drug families that attracted our attention were nitrile-containing gliptins and α-ketoamide bearing HCV NS3–4A protease inhibitors.16 Approved HCV NS3–4A protease inhibitors work through different mechanisms. N-Acylsulfonamides, e.g., danoprevir, inhibit the protease through a non-covalent mechanism, whereas α-ketoamides, e.g., boceprevir, forms a covalent bond with the active site serine. Therefore, only α-ketoamide HCV NS3–4A protease inhibitors were considered in our study as they interact covalently with the SARS-CoV-2 3CLpro. Computational docking of several HCV inhibitors, i.e., boceprevir, telaprevir and narlaprevir, into SARS-CoV-2 3CLpro indeed showed promising electrostatic and shape complementarity for the formation of a covalent bond between the active site Cys145 and the α-ketoamide group of these molecules and an overall complementary fit into the enlarged binding pocket. Amongst the three docked HCV NS3–4A protease inhibitors, boceprevir showed the highest ligand efficiency and was predicted to be superior to telaprevir and narlaprevir (Table S2‡). However, we could not further examine narlaprevir further because we did not have access to this molecule. None of the HCV NS3–4A protease inhibitors, however, docked in the second SARS-CoV-2 protease PLpro with appreciable affinity.Fig. 2 shows the best docking poses of boceprevir and telaprevir. Interestingly, our docking results showed that the covalent bond and the α-ketoamide moiety could be stabilized by the formation of hydrogen bonds with the His41 and Gly143. These results encouraged us to perform further studies to assess the use of boceprevir and telaprevir as potential inhibitors of SARS-CoV-2 3CLpro.
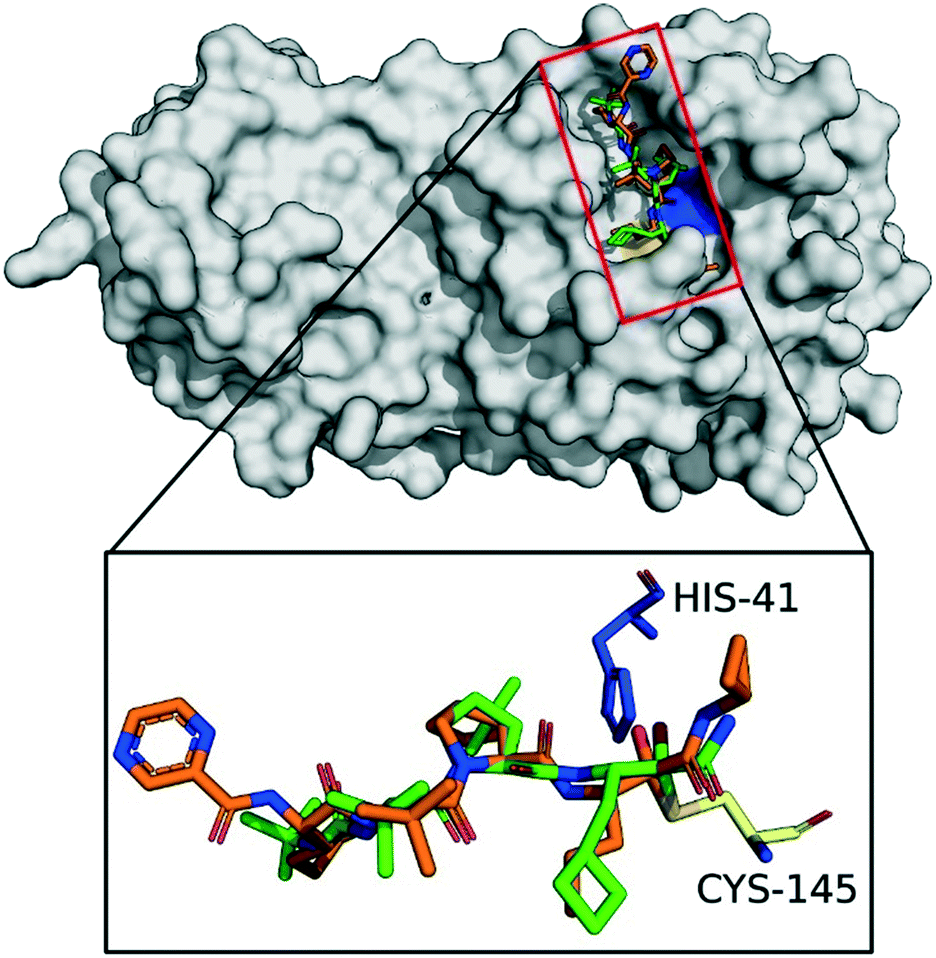 | ||
| Fig. 2 Docking of boceprevir and telaprevir onto SARS-CoV-2 3CLpro. The binding site of SARS-CoV-2 3CLpro (gray surface) with computationally docked boceprevir (green sticks) and telaprevir (orange sticks). The amino acids of the catalytic dyad are highlighted: Cys145 in yellow and His41 in blue sticks. | ||
In vitro 3CLpro enzymatic inhibition by boceprevir and telaprevir
Encouraged by the promising docking results, we determined whether boceprevir and telaprevir could inhibit the enzymatic activity of 3CLpro. For this, we took advantage of an established fluorescence resonance energy transfer (FRET) assay.26 SARS-CoV-2 3CLpro was recombinantly expressed and purified, and using the FRET substrate 2-aminobenzoyl-SVTLQSG-Tyr(NO2)-R, the cleavage of this reporter peptide by 3CLpro was monitored. The IC50 of each compound was determined as described in Methods.Boceprevir showed promising inhibition of 3CLpro with an IC50 of 1.59 μM (Fig. 3A). Telaprevir, in contrast, showed weak inhibition of cleavage activity, reaching 100% inhibition at 200 μM with an IC50 of 55.72 μM (Fig. 3B). These results, in agreement with the in silico predictions, indicate that boceprevir is a better inhibitor of 3CLpro.
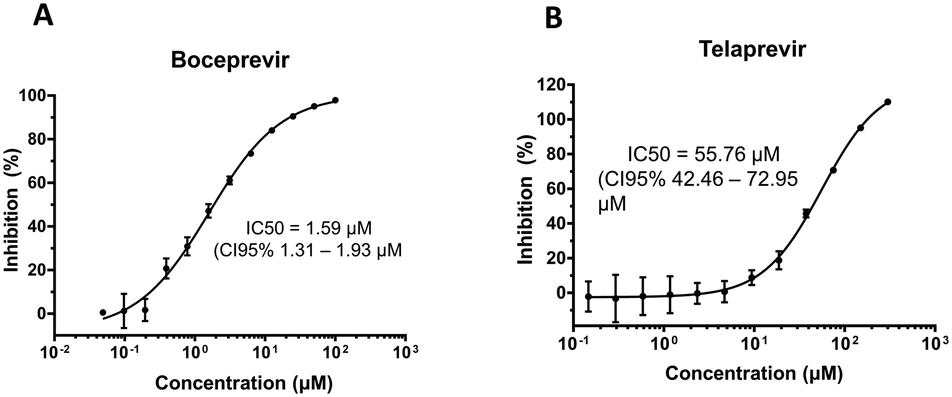 | ||
| Fig. 3 Boceprevir and telaprevir are 3CLpro inhibitors. Inhibition of cleavage activity of SARS-CoV-2 3CLpro in the presence of increasing concentrations of A) boceprevir and B) telaprevir. | ||
Crystal structures of SARS-CoV-2 3CLpro with boceprevir and telaprevir
In order to better understand how boceprevir and telaprevir inhibit the SARS-CoV-2 3CLpro and why their inhibitory activities differ, we determined the co-crystal structures of these drugs with 3CLpro. The crystal structure of SARS-CoV-2 3CLpro in complex with boceprevir was solved to 2.1 Å in space group C2 (Fig. S2‡ -PDB ID 6ZRU-). The asymmetric unit consists of a single monomer of SARS-CoV-2 3CLpro, but the active dimer is formed by a second molecule of SARS-CoV-2 3CLpro, which is related by crystallographic symmetry (Fig. S2B‡). Each monomer consists of three domains, domain I (residues 8–99), domain II (residues 100–183) and domain III (201–303), with the substrate-binding site, including the Cys145–His41 catalytic dyad, being located in a cleft between domains I and II.31,32 The substrate-binding site is made up of four conserved subsites: S1′, S1, S2 and S4.The electron density map clearly shows boceprevir bound in the 3CLpro active site (Fig. S2A‡) where the carbonyl of the electrophilic α-ketoamide forms a 1.8 Å covalent bond with the sulphur of the catalytic Cys145, forming a S,O-acetale. The oxygen of the α-ketoamide forms important hydrogen bonds with the main chain amides of Cys145 and Gly143, occupying the oxyanion hole, and the hydroxyl group, resulting from the covalent addition to the α-ketoamide, forms a hydrogen bond with the sidechain of His41, which all stabilize the conformation. The cyclobutylmethyl group of boceprevir is positioned shallowly into the S1 pocket, angling up and away. The main chains of His164 and Glu166 form hydrogen bonds with the amide bonds on the main chain of boceprevir. The dimethyl-3-aza bicycle moiety inserts deeply into the S2 pocket, making extensive hydrophobic contacts with His41, Met49, Met165, Asp187, Arg188 and Gln189 (Fig. 4A and B). The tert-butyl group is relatively solvent exposed, displaying only minor hydrophobic interactions with the sidechain of Glu166. Finally, the tert-butyl urea group orients deep into the S4 pocket, with the urea group being stabilized by several hydrogen bonds with the main chain oxygen of Glu166 and the tert-butyl undergoing hydrophobic interactions with the sidechains of Met165, Gln192, Leu167 and Pro168.
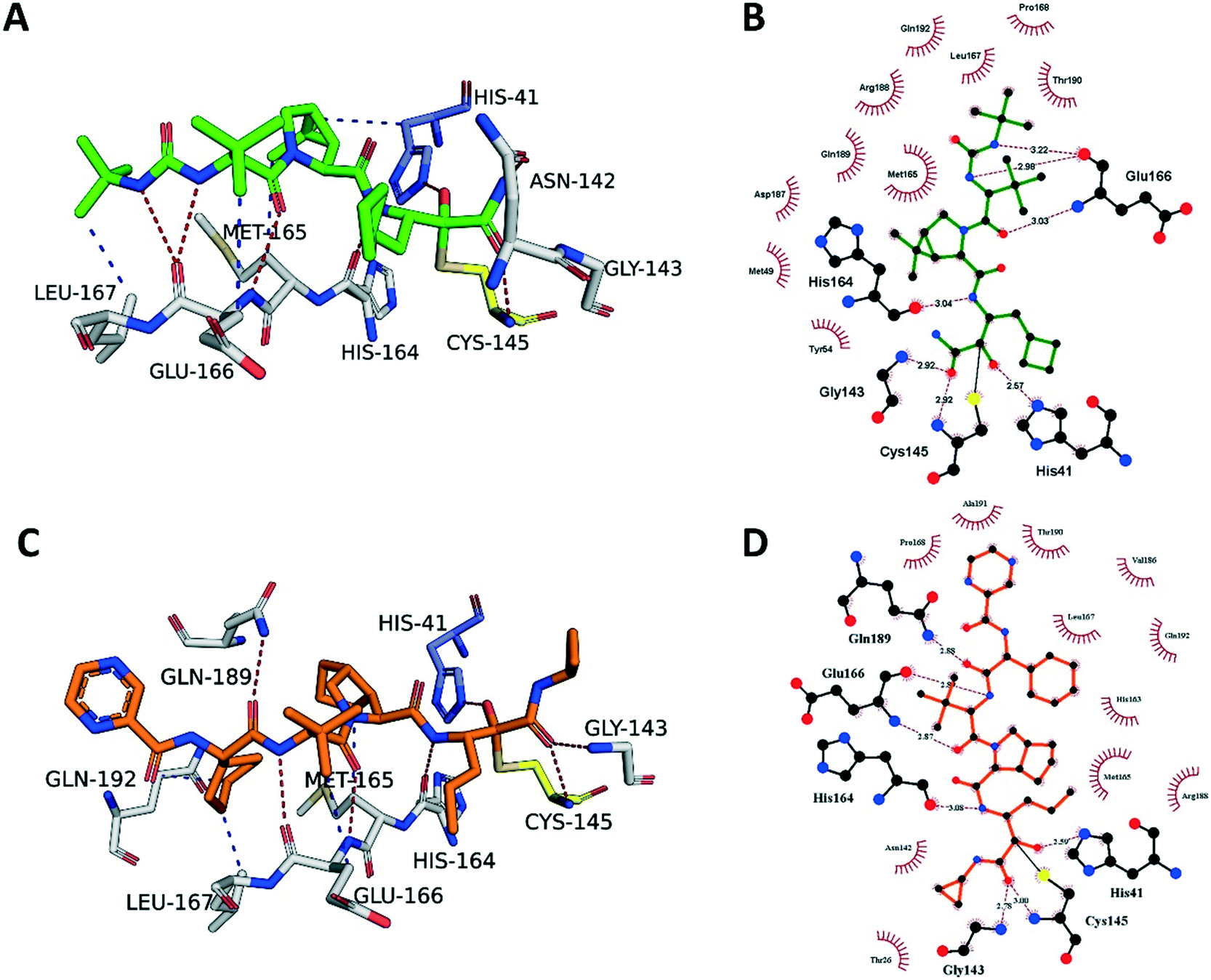 | ||
| Fig. 4 Boceprevir and telaprevir binding to SARS-CoV-2 3CLpro. A) The boceprevir (green sticks) and C) telaprevir (orange sticks) binding sites showing the interactions with the key residues (white sticks). Hydrogen bonds are shown as blue lines, hydrophobic interactions as gray dashed lines. B) Schematic diagram of boceprevir–3CLpro and D) telaprevir–3CLpro interactions, made using Ligplot.1 Green dashed lines represent hydrogen bonds, red curved lines indicate hydrophobic interactions. | ||
We compared the best pose from the virtual docking of boceprevir with our co-crystal structure PDB: 6ZRU, resulting in a root mean square deviation (RMSD) over all atoms of 1.47 Å, exhibiting almost perfect matching of the α-ketoamide warhead and sulfur atom comprising the covalent bond.
The crystal structure of SARS-CoV-2 3CLpro in complex with telaprevir was solved at 2.1 Å resolution in space group C2 (Fig. S3‡ -PDB ID 6ZRT-). The electron density map clearly shows telaprevir bound in the 3CLpro active site (Fig. S3A‡) and it is very similar to the boceprevir–3CLpro complex (Fig. S2A‡). The α-ketoamide from telaprevir forms a covalent adduct in the same orientation as boceprevir and is stabilized by the same hydrogen bonds with Cys145, Gly143 and His41. However, telaprevir contains a cyclopropyl substituent on the ketoamide nitrogen, potentially providing steric and conformational hindrance for the α-ketoamide to orient itself in the S1′ pocket, making covalent adduct formation less effective. The propyl moiety of telaprevir protrudes deeper into the S1 site compared to the cyclobutylmethyl group of boceprevir, displacing an ordered water molecule present in the S1 site of the 3CLpro–boceprevir complex that is absent in the telaprevir complex (Fig. S2A and S3A‡). Similarly to boceprevir, the main chain amides of telaprevir form hydrogen bonds with the backbone atoms of His614, Glu166 and Gln189.
The bicyclic moiety of telaprevir orients into the S2 pocket, making hydrophobic contacts with Arg188, Gln192 and Met165 (Fig. 4C and D). However, the penetration into the pocket is not as deep as the dimethyl-3-aza bicycle of boceprevir and the hydrophobic contacts are not as extensive. The tert-butyl group of telaprevir takes the same orientation as the corresponding group of boceprevir and is relatively solvent exposed, making only minor hydrophobic contacts with the sidechain of Glu166. The last difference is at the S4 pocket, where telaprevir has a cyclohexyl group, displaying hydrophobic interactions with Met165, Leu167 and Gln192, oriented deeply into the S4 pocket, followed by a relatively solvent exposed pyrazinamide moiety extending out of the active site, making only minor van der Waals interactions with Pro168, Thr190 and Ala191 (Fig. 4C and D). In contrast, boceprevir has the tert-butyl urea group that is fully located in the binding site.
We compared our virtual docking against our co-crystal structure PDB: 6ZRT. The best docked pose of telaprevir exhibited a RMSD value of 1.22 Å over all atoms against the crystal pose. The docking showed the highest deviation on the 2-pyrazine carboxamide moiety, indicating flexibility of this functional group when binding to 3CLpro, in line with the relatively high solvent exposure observed in the crystal structure. On the other hand, the orientation of the α-ketoamide warhead of the docked pose was nearly identical to the crystal pose(s), including the stabilizing hydrogen bonds with Cys145, His41, His164 and Gly143 (Fig. 4C and D).
As of now, apart from our released crystal-structures, other published and not published 3CLpro structures containing boceprevir33–35 and telaprevir34,36–38 are available. The other available boceprevir–3CLpro complex structures show highly similar binding poses to ours (Fig. S4A‡). Interestingly, the high-resolution structures (6WNP, 7K40, 7C6S and 7BRP) show a well-ordered water molecule coordinating an interaction between the amide group of the α-ketoamide and the backbone oxygen of Thr26 through hydrogen bonds. This water molecule is not present in the released 3CLpro – telaprevir structures, with the cyclopropyl group likely displacing it (Fig. S4B‡). Additionally, an overlay of available telaprevir structures shows conformational flexibility of the propyl moiety that is located in the S1 pocket, seemingly displacing the ordered water molecule in some structures (6ZRT, 7K6D) but not in others (7K6E, 7C7P and 6XQS). The pyrazinamide moiety of telaprevir also shows multiple conformations among the crystal structures.
Boceprevir and telaprevir in beta-CoV infections
Next, we tested whether boceprevir and telaprevir inhibited beta-CoV replication in cells. To this aim, we first perform a cytotoxic assay in the Vero E6 and LR7 cell lines to determine the maximal non-toxic concentration of these compounds that could be used in cells. Using the MTT method, we found out that the maximal non-toxic concentration of these drugs in these cell lines was 72.5 μM for both (Fig. 5A). To minimize the chance of possible side effects we decided to use a concentration of 40 μM of boceprevir and telaprevir for further cell studies. | ||
| Fig. 5 Boceprevir and telaprevir in beta-CoV infections. A) The cytotoxicity of boceprevir and telaprevir in LR7 and Vero E6 cell lines. Cells were treated with the indicated doses of boceprevir and telaprevir for 6 h. Cell viability was subsequently measured using the MTT assay. All the MTT values were normalized to the 0.1% DMSO-only treatment, which represents 100% cell viability. B) LR7 cells were infected with MHV at MOI 1 for 2 h before adding 40 μM boceprevir, 40 μM telaprevir or 0.1% DMSO (ctrl) for another 6 h. Controls were cells not exposed to MHV (mock) or incubated with both MHV and 400 nM bafilomycin A1 (BafA1), which blocks virus cell entry. Proteins were separated by SDS-PAGE and western blot membranes probed with an antibody against either MHV N protein or GAPDH (top part). The N protein expression in each sample was quantified and normalized to the GAPDH signal. Results are expressed relative to the ctrl (low part). C) Cells treated as in panel B were processed for immunofluorescence using antibodies against MHV nsp2 and nsp3 proteins, and DAPI staining. The number of infected cells was subsequently determined. D) LR7 cells were infected with MHV at MOI 1 for 2 h before adding the indicated concentrations of boceprevir for another 6 h. Cells treated with DMSO only (ctrl), BafA1-treated cells and cells not inoculated with MHV (mock) were used as controls. N protein levels were then examined as in panel B. E) Cells were treated as in panel D before statistically evaluating the percentage of infected cells by immunofluorescence as in panel C. F) Vero E6 cells were inoculated with SARS-CoV-2 at MOI 1, and treated with 40 μM boceprevir at the same time or after 2 h. At 8 h p.i., cell supernatants were collected and the number of produced infectious viral particles was determined using a plaque assay. Cells treated with DMSO only (ctrl), BafA1-treated cells and cells not inoculated with SARS-CoV-2 (mock) were used as controls. Results are expressed relative to the ctrl. G) Vero E6 cells were infected with SARS-CoV-2 at MOI 1 for 2 h before adding the indicated concentrations of boceprevir for another 6 h. Cells were lyses and the replication of SARS-CoV-2 was measured by assessing the expression levels of the mRNA encoding for nsp3 by RT-PCR and normalizing to those encoding for GAPDH. Cells treated with DMSO only (ctrl), BafA1-treated cells and cells not inoculated with SARS-CoV-2 (mock) were used as controls. Results are expressed relative to the ctrl. H) Culture supernatants of the samples analysed in panel G were examined by plaque assay as in panel F. Results are expressed relative to the ctrl. All data are represented as mean ± standard deviation of at least three independent experiments. Student t test was used to evaluate statistical differences and a p value ≤ 0.05 was considered significant with *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001. | ||
As a first analysis, we explored the antiviral effect of the two compounds in LR7 cells infected with mouse hepatitis virus (MHV), a model beta-CoV. We opted to add boceprevir and telaprevir 2 h p.i. The reason behind this choice was twofold. First, a specific inhibitor of 3CLpro should be able to block the viral replication, thus after virus cell entry has occurred. Second, a positive outcome of this approach would indicate that the compound can also be used to treat infected cells, something relevant from a therapeutic point of view. Thus, LR7 cells were infected with MHV at MOI 1 as described in the Methods section, and 40 μM boceprevir or 40 μM telaprevir were added 2 h p.i. The incubation was continued for an extra 6 h before processing the cells for either western blot (WB) with anti-MHV N protein antibodies, to assess viral protein production, or immunofluorescence (IF) with anti-nsp2/nsp3 antibodies, to determine the number of infected cells. As a positive control, we used bafilomycin A1, an inhibitor of the lysosomal H+-ATPase that increases the pH in the compartments of the endolysosomal system, blocking the cell entry of multiple viruses including CoV.39–41 This drug was therefore added at the same time as the virus inoculum and as expected, showed a strong reduction of MHV replication in both WB and IF readouts (Fig. 5B and C). The same assays also revealed that 40 μM telaprevir has no antiviral effect, but rather promoted the MHV infection (Fig. 5B and C). In contrast and seemingly in line with the results of the in vitro 3CLpro enzymatic assay, treatment with 40 μM boceprevir showed a significant inhibition of both MHV N protein production and the percentage of infected cells, to an extent similar to bafilomycin A1 treatment.
Next, we investigated whether the antiviral effect of boceprevir is dose-dependent, which would confirm its specificity. As shown in Fig. 5D and E, LR7 cells infected with MHV were treated with boceprevir at concentrations from 4 nM to 40 μM, with a serial dilution factor of 10. Interestingly, MHV N protein production was decreased by boceprevir in a dose-dependent manner, with a reduction of approximately 90% at a concentration of 40 μM and still of 40% at 4 nM, in comparison to the DMSO-treated infected cells (Fig. 5D). The dose-dependent inhibition of MHV replication by boceprevir was confirmed by determining the percentage of infected cells. This number was reduced by approximately 75% in cells subjected to 40 μM boceprevir and still by approximately 25% in those incubated with 40 nM, in comparison to the mock-treated infected cells (Fig. 5E). Altogether, the results show that boceprevir can efficiently inhibit MHV infection.
Next, we turned to SARS-CoV-2. Vero E6 cells were infected with SARS-CoV-2 at MOI and treated with 40 μM boceprevir at 2 h p.i. At 8 h p.i., cell culture supernatants were collected to titrate the progeny virus by plaque assay. We also added 40 μM boceprevir at the time of virus infection, to explore whether this could further enhance the observed effects of this compound. As shown in Fig. 5F and G, 40 μM boceprevir added at 2 h p.i. reduced the virus egression by approximately 50%, while its addition at the same time as the inoculum decreased virus progeny of approximately 75%.
While preparing our manuscript, an article appeared also showing that boceprevir can inhibit SARS-CoV-2 infection, and in this study the antiviral effects were started to be observed at a concentration of 1 μM.42 Thus, we infected Vero E6 cells with SARS-CoV-2 and at 2 h p.i., added boceprevir at concentrations ranging from 0.1 to 100 μM, with a serial dilution factor of 10. At 8 p.i., cell culture supernatants were collected for the plaque assay while cells were lysed to extract the RNA and quantify viral replication by real-time PCR. This latter assay showed that 100 μM boceprevir effectively inhibited viral gene expression while 10 μM showed a decrease of approximately 50% (Fig. 5F and G). A lower concentration of boceprevir had no antiviral effect. Using the plaque assay, we only could observe a significant inhibition of SARS-CoV-2 egression only with the 100 μM boceprevir (Fig. 5H). Taken altogether, our results show that boceprevir has an evident antiviral effect against beta-CoV at concentrations ranging from 40–100 μM, but the working concentrations might be varying between different beta-CoV.
Discussion
We described the structural basis of the interaction of the HCV NS3–4A protease inhibitors boceprevir and telaprevir with the SARS-CoV-2 3CLpro and established that α-ketoamide HCV NS3–4A protease inhibitors can inhibit SARS-CoV-2 3CLpro. This is surprising since the two enzymes are evolutionary unrelated. The HCV NS3–4A is a serine protease depending on a catalytic triade, while SARS-CoV-2 is a cysteine protease with a catalytic dyad. Moreover, the substrate specificity of the two proteases is different and thus the shape and the electrostatic of the substrate pockets. We showed the added value of using computational docking in the identification of these drugs that could potentially be repurposed, displaying highly similar docking poses to the crystal structures. We showed that the HCV NS3–4A protease inhibitors boceprevir (and to a lesser extent telaprevir) are inhibitors of the SARS-CoV-2 3CLpro in vitro, with boceprevir also showing potent inhibition of viral replication in vivo. As seen in the co-crystal structures, there are differences in subsite binding that could explain the increased inhibitory activity of boceprevir compared to telaprevir. Though the mode of inhibition of these two compounds with respect to the covalent adduct formation with the catalytic Cys145 is the same, the divergence mainly lies in the interactions with the S1′, S2 and S4 subpockets (Fig. 4, 6 and S4‡). Since covalent complex formation is dependent on the formation of an initial non-covalent precomplex, these distinctions could explain the difference in inhibitory potency. Boceprevir binds deeper into the S2 pocket and makes more extensive hydrophobic interactions than telaprevir. Additionally, in recently released, higher resolution structures of boceprevir, a well-ordered water molecule is shown to coordinate an interaction with the amide group of the α-ketoamide and the backbone oxygen of Thr26 through hydrogen bonds31,35,43 (Fig. S4A‡), potentially stabilizing the covalent conformation. This interaction is absent in the telaprevir structures due to the cyclopropyl moiety occupying that space (Fig. S4B‡). Finally, telaprevir shows higher conformational flexibility in the S1 pocket and in the location of its pyrazinamide group. The less favourable interactions, coupled with higher degrees of conformational flexibility could explain the lower inhibitory potency of telaprevir in comparison to boceprevir. | ||
| Fig. 6 Alignments of binding modes of boceprevir and telaprevir. The SARS-CoV-2 3CLpro (gray surface) in complex with boceprevir (green sticks) and telaprevir (orange sticks). The residues od the catalytic dyad as shows in yellow surface for Cys145, and blue surface for His41. | ||
Another α-ketoamide HCV NS3–4A protease inhibitor, narlaprevir also showed promising docking results, however, it could not be tested in vitro or in cell culture since it was not accessible to us. While preparing this manuscript, another research group published the discovery of the HCV NS3–4A protease inhibitor boceprevir as a 3CLpro inhibitor of SARS-CoV-2.30 Although our study confirmed in gross their findings, it also reached some different conclusions. In particular we describe the co-crystal structure of boceprevir and 3CLpro, leading to a sound basis of understanding of the molecular interaction of the drug with the receptor. Our measured enzyme inhibition and cellular viral replication are higher than the ones reported in the previous study. The differences might be explained by a different time-point of addition of boceprevir, and other details such a culture medium, and virus stock. Additionally, we found that not only boceprevir but also the FDA-approved telaprevir from the same drug class of HCV NS3–4A protease inhibitors, inhibits the 3CLpro in vitro. We describe here for the first time the molecular basis of the 3CLpro inhibition by telaprevir. The anti-SARS-CoV-2 mode-of-action of both drugs rely on the presence of an α-ketoamide moiety. Thus, we propose that the approved α-ketoamide HCV NS3–4A protease inhibitors boceprevir, telaprevir and possibly also other members of this family (e.g., narlaprevir) are promising COVID-19 repurposing candidates. While, the potency of the herein proposed repurposed drugs in an in vivo setting are unknown, they deserve further attention as potential treatments for COVID-19 patients. Boceprevir, in particular, is safe for humans and is on the market since 2011 for the treatment of HCV infections and showed a limited number of side effects. Moreover, pharmacokinetics and pharmacodynamics in humans are well known.44 The human plasma exposure of boceprevir was measured as Cmax 1.72 μg ml−1, whereas Cmin 0.08 μg ml−1, which is in the similar range as the herein reported cellular viral replication inhibition of 40 μM (21 μg ml−1).45 Another potential application of this drug is based on the good potency for MHV, showing that it may represent an effective pan-anti-CoV inhibitor. It is well established that CoV infect farm animals. Thus, these compounds could be relevant for the cattle industry and possibly for future CoV epidemics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research in the lab of F. R. was supported by NWO Corona: Fast-track data (440.20.015), ZonMW TOP (91217002), ALW Open Programme (ALWOP.310), Marie Skłodowska-Curie Cofund (713660) and Marie Skłodowska Curie ETN (765912) grants, and by Emergent Ventures (Mercatus Center, George Mason University, USA), and by the Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT IV200121) of UNAM. AJRM would like to acknowledge scholarship CONACYT number 584534.Notes and references
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS.
- N. Chen, M. Zhou, X. Dong, J. Qu, F. Gong, Y. Han, Y. Qiu, J. Wang, Y. Liu, Y. Wei, J. A. Xia, T. Yu, X. Zhang and L. Zhang, Lancet, 2020, 395, 507–513 CrossRef CAS.
- D. Wang, B. Hu, C. Hu, F. Zhu, X. Liu, J. Zhang, B. Wang, H. Xiang, Z. Cheng, Y. Xiong, Y. Zhao, Y. Li, X. Wang and Z. Peng, JAMA, J. Am. Med. Assoc., 2020, 323, 1061 CrossRef CAS.
- Y. Yang, Q.-B. Lu, M.-J. Liu, Y.-X. Wang, A.-R. Zhang, N. Jalali, N. E. Dean, I. Longini, M. E. Halloran, B. Xu, X.-A. Zhang, L.-P. Wang, W. Liu and L.-Q. Fang, medRxiv, 2020 DOI:10.1101/2020.02.10.20021675.
- I. Chakraborty and P. Maity, Sci. Total Environ., 2020, 728, 138882 CrossRef CAS.
- C. Huang, Y. Wang, X. Li, L. Ren, J. Zhao, Y. Hu, L. Zhang, G. Fan, J. Xu, X. Gu, Z. Cheng, T. Yu, J. Xia, Y. Wei, W. Wu, X. Xie, W. Yin, H. Li, M. Liu, Y. Xiao, H. Gao, L. Guo, J. Xie, G. Wang, R. Jiang, Z. Gao, Q. Jin, J. Wang and B. Cao, Lancet, 2020, 395, 497–506 CrossRef CAS.
- M. T. ul Qamar, S. M. Alqahtani, M. A. Alamri and L.-L. Chen, J. Pharm. Anal., 2020, 10(4), 313–319 CrossRef.
- P. Zhou, X. L. Yang and X. G. Wang, et al. , Nature, 2020, 579, 270–273 CrossRef CAS.
- N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi, R. Lu, P. Niu, F. Zhan, X. Ma, D. Wang, W. Xu, G. Wu, G. F. Gao and W. Tan, N. Engl. J. Med., 2020, 382, 727–733 CrossRef CAS.
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes and Y.-Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS.
- S. Perlman and J. Netland, Nat. Rev. Microbiol., 2009, 7, 439–450 CrossRef CAS.
- A. R. Fehr and S. Perlman, in Coronaviruses: Methods and Protocols, ed. H. J. Maier, E. Bickerton and P. Britton, Springer New York, New York, NY, 2015, DOI:10.1007/978-1-4939-2438-7_1, pp. 1–23.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS.
- A. M. Mielech, Y. Chen, A. D. Mesecar and S. C. Baker, Virus Res., 2014, 194, 184–190 CrossRef CAS.
- D. Shin, R. Mukherjee, D. Grewe, D. Bojkova, K. Baek, A. Bhattacharya, L. Schulz, M. Widera, A. R. Mehdipour, G. Tascher, P. P. Geurink, A. Wilhelm, G. J. van der Heden van Noort, H. Ovaa, S. Müller, K. P. Knobeloch, K. Rajalingam, B. A. Schulman, J. Cinatl, G. Hummer, S. Ciesek and I. Dikic, Nature, 2020, 587(7835), 657–662 CrossRef.
- M. Groves, A. Domling, A. J. R. Moreno, A. R. Romero, C. Neochoritis and M. Velasco-Velázquez, 2020, DOI: DOI:10.26434/chemrxiv.12110760.v1.
- A. Tuley and W. Fast, Biochemistry, 2018, 57, 3326–3337 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
- M. A. Dessau and Y. Modis, J. Visualized Exp., 2011, e2285, DOI:10.3791/2285.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204–1214 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- P. Skubák, G. N. Murshudov and N. S. Pannu, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2196–2201 CrossRef.
- J. E. Blanchard, N. H. Elowe, C. Huitema, P. D. Fortin, J. D. Cechetto, L. D. Eltis and E. D. Brown, Chem. Biol., 2004, 11, 1445–1453 CrossRef CAS.
- S. Biacchesi, M. H. Skiadopoulos, L. Yang, B. R. Murphy, P. L. Collins and U. J. Buchholz, J. Virol. Methods, 2005, 128, 192–197 CrossRef CAS.
- J. J. Schiller, A. Kanjanahaluethai and S. C. Baker, Virology, 1998, 242, 288–302 CrossRef CAS.
- B. Schwarz, E. Routledge and S. G. Siddell, J. Virol., 1990, 64, 4784–4791 CrossRef CAS.
- A. Dömling and L. Gao, Chem, 2020, 6, 1283–1295 Search PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS.
- L. Fu, F. Ye, Y. Feng, F. Yu, Q. Wang, Y. Wu, C. Zhao, H. Sun, B. Huang, P. Niu, H. Song, Y. Shi, X. Li, W. Tan, J. Qi and G. F. Gao, Nat. Commun., 2020, 11, 4417 CrossRef CAS.
- D. W. Kneller, S. Galanie, G. Phillips, H. M. O'Neill, L. Coates and A. Kovalevsky, Structure, 2020, 28(12), 1313–1320 CrossRef CAS.
- RCSB PDB - 7K40: Crystal Structure of SARS-CoV-2 Main Protease (3CLpro/Mpro) in Complex with Covalent Inhibitor Boceprevir at 1.35 A Resolution, https://www.rcsb.org/structure/7K40.
- RCSB PDB - 7K6D: SARS-CoV-2 Main Protease Co-Crystal Structure with Telaprevir Determined from Crystals Grown with 40 nL Acoustically Ejected Mpro Droplets at 1.48 A Resolution (Cryo-protected), https://www.rcsb.org/structure/7K6D.
- RCSB PDB - 7K6E: SARS-CoV-2 Main Protease Co-Crystal Structure with Telaprevir Determined from Crystals Grown with 40 nL Acoustically Ejected Mpro Droplets at 1.63 A Resolution (Direct Vitrification), https://www.rcsb.org/structure/7K6E.
- RCSB PDB - 7C7P: Crystal structure of the SARS-CoV-2 main protease in complex with Telaprevir, https://www.rcsb.org/structure/7C7P.
- T. C. Nash and M. J. Buchmeier, Virology, 1997, 233, 1–8 CrossRef CAS.
- S. Matsuyama, M. Ujike, S. Morikawa, M. Tashiro and F. Taguchi, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12543–12547 CrossRef CAS.
- G. Simmons, D. N. Gosalia, A. J. Rennekamp, J. D. Reeves, S. L. Diamond and P. Bates, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11876–11881 CrossRef CAS.
- C. Ma, M. D. Sacco, B. Hurst, J. A. Townsend, Y. Hu, T. Szeto, X. Zhang, B. Tarbet, M. T. Marty, Y. Chen and J. Wang, Cell Res., 2020, 30, 678–692 CrossRef CAS.
- RCSB PDB - 6WNP: X-ray Structure of SARS-CoV-2 main protease bound to Boceprevir at 1.45 A.
- H. Shankar, K. Bichoupan and D. T. Dieterich, Expert Opin. Drug Metab. Toxicol., 2013, 9, 1647–1657 CrossRef CAS.
- M. Johnson, J. Borland, S. Chen, P. Savina, B. Wynne and S. Piscitelli, Br. J. Clin. Pharmacol., 2014, 78, 1043–1049 CrossRef CAS.
Footnotes |
| † Available crystal structures of boceprevir–3CLpro and telaprevir–3CLpro complexes are available at PDB under codes 6ZRU and 6ZRT respectively. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00367k |
| § Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |