
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCollaborative virtual screening to elaborate an imidazo[1,2-a]pyridine hit series for visceral leishmaniasis†
Yuichiro
Akao
a,
Stacie
Canan‡
b,
Yafeng
Cao
c,
Kevin
Condroski§
b,
Ola
Engkvist
 d,
Sachiko
Itono¶
a,
Rina
Kaki
e,
Chiaki
Kimura
e,
Thierry
Kogej
d,
Kazuya
Nagaoka
f,
Akira
Naito
e,
Hiromi
Nakai
e,
Garry
Pairaudeau
g,
Constantin
Radu||
h,
Ieuan
Roberts
g,
Mitsuyuki
Shimada**
a,
David
Shum
h,
Nao-aki
Watanabe
f,
Huanxu
Xie
c,
Shuji
Yonezawa
e,
Osamu
Yoshida
e,
Ryu
Yoshida
e,
Charles
Mowbray
i and
Benjamin
Perry
*i
d,
Sachiko
Itono¶
a,
Rina
Kaki
e,
Chiaki
Kimura
e,
Thierry
Kogej
d,
Kazuya
Nagaoka
f,
Akira
Naito
e,
Hiromi
Nakai
e,
Garry
Pairaudeau
g,
Constantin
Radu||
h,
Ieuan
Roberts
g,
Mitsuyuki
Shimada**
a,
David
Shum
h,
Nao-aki
Watanabe
f,
Huanxu
Xie
c,
Shuji
Yonezawa
e,
Osamu
Yoshida
e,
Ryu
Yoshida
e,
Charles
Mowbray
i and
Benjamin
Perry
*i
aTakeda Pharmaceutical Company Limited, 26-1 Muraoka-Higashi 2-chrome, Fujisawa, Kanagawa 251-8555, Japan
bCelgene Corporation, Celgene Global Health, 10300 Campus Point Drive, San Diego, California 92121, USA
cWuXi AppTec Company Ltd., 666 Gaoxin Road, East Lake High-Tech Development Zone, Wuhan 430075, People's Republic of China
dAstraZeneca Discovery Sciences, R&D, AstraZeneca, Gothenburg, Sweden
eShionogi & Co., Ltd, 3-1-1, Futaba-cho, Toyonaka-shi, Osaka, Japan
fEisai Co., Ltd, 1-3,Tokodai 5-chome, Tsukuba, Ibaraki 300-2635, Japan
gAstraZeneca, Discovery Sciences, R&D, AstraZeneca, Cambridge, UK
hInstitut Pasteur Korea, 16, Daewangpangyo-ro 712 beon-gil, Bundang-gu, Seongnam-si, Gyeonggi-do, 13488 Republic of Korea
iDrugs for Neglected Diseases initiative, 15 Chemin Louis Dunant, Geneva 1202, Switzerland. E-mail: bperry@dndi.org
First published on 21st January 2021
Abstract
An innovative pre-competitive virtual screening collaboration was engaged to validate and subsequently explore an imidazo[1,2-a]pyridine screening hit for visceral leishmaniasis. In silico probing of five proprietary pharmaceutical company libraries enabled rapid expansion of the hit chemotype, alleviating initial concerns about the core chemical structure while simultaneously improving antiparasitic activity and selectivity index relative to the background cell line. Subsequent hit optimization informed by the structure–activity relationship enabled by this virtual screening allowed thorough investigation of the pharmacophore, opening avenues for further improvement and optimization of the chemical series.
Introduction
Leishmaniasis is generally considered to be one of the most neglected tropical diseases and has strong links with poverty.1–3 It is a complex vector-borne disease caused by more than 20 species of the protozoan genus Leishmania with symptoms ranging from localized skin ulcers to a lethal systemic disease. Of the four leishmaniasis syndromes, visceral leishmaniasis (VL) represents the greatest burden to global health, with data from 2015 suggesting it is responsible for almost 1.4 million DALYs (disability adjusted life-years) lost, representing almost 6% of all DALYs caused by diseases on the World Health Organization′s list of neglected tropical diseases (NTDs).4 Caused by infection with L. donovani and L. infantum parasites transmitted by phlebotomine sand flies, visceral leishmaniasis manifests in patients as swelling of spleen and liver, renal dysfunction and, if left untreated, death. Current estimates suggest an annual new infection rate for VL of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 90000 cases, with most cases found in eastern Africa, Brazil and India.2,5–8
000 to 90000 cases, with most cases found in eastern Africa, Brazil and India.2,5–8
Although treatments for VL exist, these medications are rarely fit for purpose, with major issues linked to resistance, geographic variability in efficacy, toxicity, availability and affordability, and often long and painful treatment periods requiring many weeks of hospitalization.9–13 As a result, the past decade has seen increased efforts to develop new treatments for Leishmania related syndromes such as VL, with a primary focus on delivering pre-clinical candidates with improved profiles (broader geographic utility, reduced or no toxicity, oral dosing, up to 10 day treatment period).5 Target product profiles for these next generation VL treatments have been published and discussed widely.14,15
A combination of target-based and phenotypic-based drug discovery approaches have been applied extensively to identify new chemical matter for anti-leishmanial discovery projects, as well as the kinetoplastid field in general.9,16–18 Despite some excellent progress in target-based approaches, these often suffer from poor translation into efficacy in in vitro infection models,19 demonstrating the importance not only of a relevant mechanism of action but also the correct physicochemical properties to access the parasites once intracellular infection has taken hold. The phenotypic approach bypasses this issue by engaging cell-infection assays as the primary screen, however, these approaches often suffer from a combination of a low hit rate and identification of a high percentage of unsuitable chemical matter, with many hit lists comprising compounds rich in PAINS,20 toxicophores and intractable chemistries. Nevertheless, these approaches have yielded an interesting pipeline of novel potential treatments for VL, some of which have progressed to the early stages of clinical or preclinical evaluation.21 These include CRK12 inhibitor GSK3186899,22 the Leishmania proteasome inhibitors GSK3494245 and LXE-408,23,24 as well as DNDI-6174, DNDI-6148 and DNDI-0690, compounds identified via phenotypic drug discovery efforts.25
Despite this promising situation, the reality of clinical-level attrition in drug discovery R&D dictates that if the global health community are to ensure delivery of new, improved treatments to VL patients, further new chemical entities (NCEs) capable of tackling Leishmania infections still need to be identified and developed.26,27 Alongside the traditional target and phenotypic approaches mentioned above, recent reports have highlighted a number of efforts to identify novel anti-leishmanial NCEs via repurposing of existing drugs and clinical stage compounds,28 combined scaffold and parasite hopping,29 the screening of natural products,30 as well as investigation into host-mediated treatments for Leishmania infections.31
We report herein on early discovery efforts around an anti-leishmanial chemotype derived from the output of a kinetoplastid high throughput screening (HTS) campaign. Since 2012 we (DNDi) have tested over 2.2 million compounds in high throughput high content screening against kinetoplastid parasites including Leishmania donovani and Trypanosoma cruzi, the causative agent of Chagas disease,32 searching for novel and attractive small molecules as starting points for drug discovery campaigns.†† Seeking to improve the efficiency and throughput of the triaging of results from these phenotypic screening efforts led to the creation of the NTD drug discovery booster, a pre-competitive virtual screening model comprising key players from pharmaceutical R&D.33
NTD drug discovery booster
In the traditional approach to initial exploration of chemical space around a new HTS hit, commercial compound catalogues are probed for similar molecules and small sets of closely designed analogues are synthesized for testing.34 As an alternative to this, we engaged an innovative in silico approach, termed the NTD drug discovery booster (“Booster”). The Booster process engages collaborative rounds of virtual ligand-based screening across multiple large, proprietary pharmaceutical company databases to identify and test both close analogues to the hit, as well as structurally differentiated compounds fitting the same base pharmacophore. The hit structure (“Seed”) is shared with scientists at each partner pharmaceutical company, in silico similarity searches are performed probing the proprietary collections of each partner, and up to 100 prospective “similar” molecules per partner are screened against the parasite(s) of interest. The definition of “similar” is independently determined by each partner, and could be focused on both SAR annotation as well as more general pharmacophore-led exploration, and could involve the use of both commercial and proprietary computational approaches. Sharing the results of these experimental screens with the providing partners, as well as anonymized sharing of the structure of the most improved molecule from the entire collaborative screening endeavor (“Improved Hit”), allows a second, more refined iteration of in silico screening to be performed, again providing approximately 100 molecules per partner. Repetition of this iterative screening/sharing process continues until improvements cease to be made, or until SAR around a chemotype emerges.The use of both orthogonal in silico techniques as well as orthogonally differentiated libraries, previously demonstrated to have minimal overlap,35 maximizes both the richness of the SAR explored around the initial seed phenotype, and increases the chances of identifying an interesting change of chemotype with similar or improved anti-parasitic activity, i.e. a scaffold hop. Furthermore, these newly identified scaffold hops can be subsequently mined using the same Booster approach, thus maximizing the value of the hits coming from HTS.
Results and discussion
Hit identification
Compound 1 was identified as a hit with anti-trypanosomal activity from a high throughout screen against Trypanosoma cruzi. Follow up confirmation of activity validated both the anti-trypanosomal activity of 1 in a T. cruzi intracellular infection assay (U2OS as host cell) as well as demonstrating the compound's efficacy against Leishmania donovani in an intracellular infection assay (THP1 as host cell) (Fig. 1). As mentioned previously, we are wary of the high hit rate of intractable, non-drug-like and/or PAINS-like compounds in the kinetoplastid literature, and our initial analysis of the 1 chemotype flagged up the 2-pyridyl 3 amino substituted imidazo[1,2-a]pyridine core as a PAINS motif.20 Further investigation of the literature around 1 identified earlier reports about a related structure with moderate potency against the related leishmania parasite L. major, one of the causative parasitic agents of cutaneous leishmaniasis.36 In this report the chemotype did not contain the 2-pyridyl motif nor the amino substituent at the 3-position of the imidazo[1,2-a]pyridine, but rather a 2,3 biaryl substitution pattern. In this instance the structure is, therefore, not classified as a PAINS motif, however the reported SAR demonstrated potencies around an order of magnitude lower than those observed for 1, despite the authors using an axenic promastigote assay which circumvents the additional challenge for compounds to access intracellular parasites. In addition, we found examples of launched drugs which contain the imidazo[1,2-a]pyridine core from 1, including the so-called Z-drugs zolpidem and alpidem, as well as reports of structures similar to 1 confirmed as hits in other infectious disease screens such as Mycobacterium tuberculosis and vaccina virus.37,38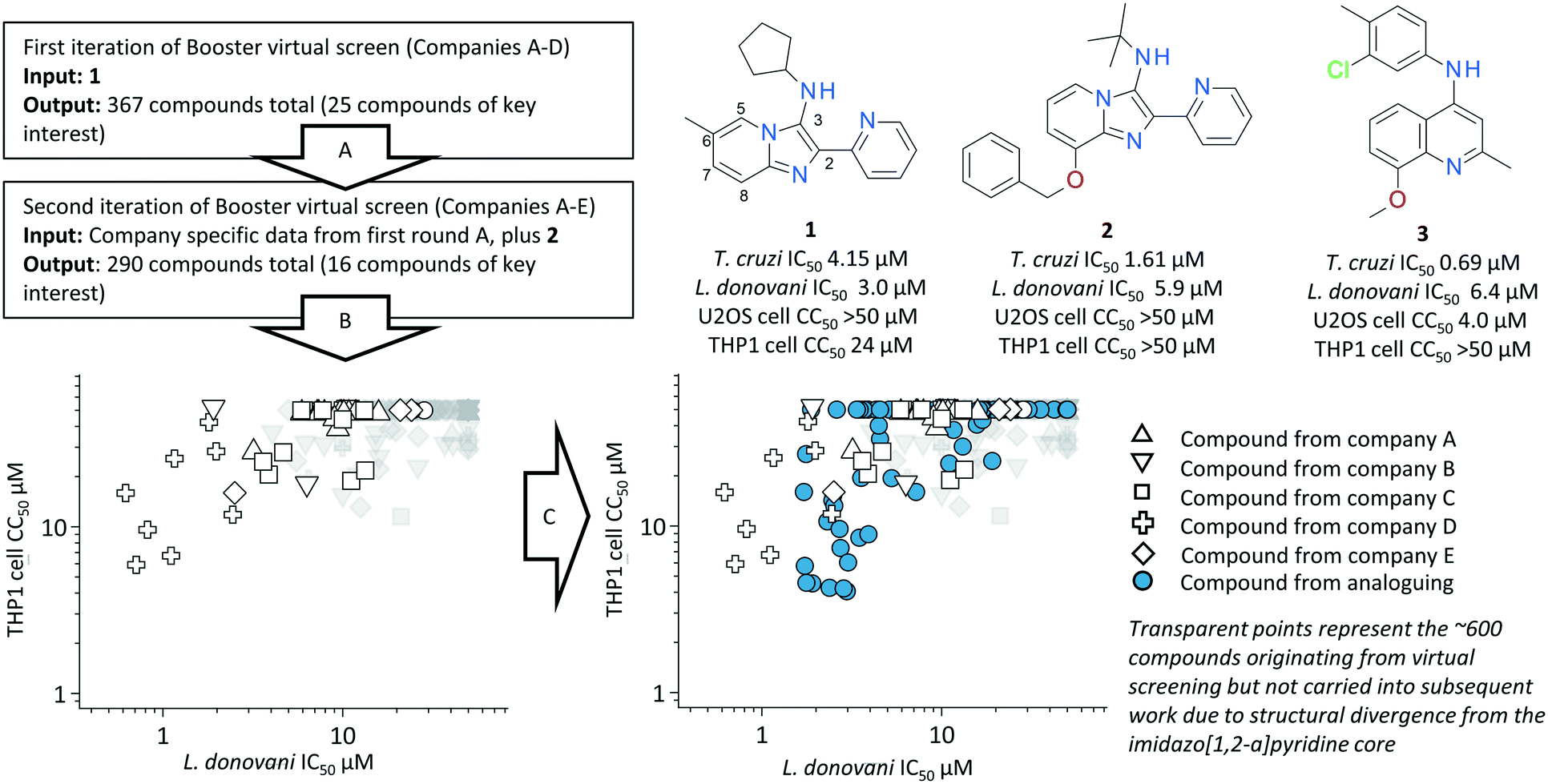 | ||
| Fig. 1 Overview of Booster in silico screening process; first (A) and second (B) round of virtual screening result in data set of compounds with annotated SAR around 1; data set is further annotated via expansive analoguing (C). | ||
Combining the above findings with the nature of the assay used to identify the anti-leishmanial effect of the compound (high content imaging with parasite count) and the fact that the compound demonstrated anti-parasitic activity in more than one assay and against multiple related kinetoplastid parasites, we decided to cautiously investigate further. We were aware from the literature of the potential for a metal-chelation-driven pharmacological effect due to the bidentate heteroaryl-heteroaryl system in the core, indeed it could be hypothesized that this is the source of PAINS-like activity in other assays.39 Therefore, a key step for further investigation of 1 was to gain confidence via SAR and orthogonal assays that the observed anti-parasitic activity was unlikely to be driven by metal chelation, and that the PAINS flag could be legitimately discounted. The two key read-outs we targeted to achieve this were a) the ability of the chemotype to achieve a broader selectivity index for Leishmania donovani relative to the background host cell, and b) clear evidence that altering the core away from the PAINS-like chemotype maintained anti-parasitic activity, as suggested by the earlier paper from Marhadour et al.36
Hit elaboration and validation via collaborative in silico screening
Compound 1 was submitted as a “seed” to five partners of the NTD Booster consortium: AstraZeneca plc, Celgene Corporation, Eisai Co., Ltd, Shionogi & Co., Ltd, and Takeda Pharmaceutical Company Ltd. Since the pre-competitive mechanism of the Booster project requires anonymity and a high degree of confidentiality on in silico methods and library composition between partners, identities of the partners are presented as blinded in this study as “Companies A–E”.‡‡The partners performed two successive iterations of ligand-based virtual screening against their proprietary compound collections: cycle A and cycle B (see Fig. 1). Each distinct library was probed using a partner-specific in silico similarity search approach as described in Table 1 (see ESI† for detailed description). The compounds identified via these virtual screening efforts were investigated in a high content cell-based L. donovani infection assay using THP1 host cells. Assay read-out from this high content approach demonstrated the compound's efficacy at total parasite clearance as well as host cell cytotoxicity, yielding an anti-parasitic IC50, a host cell CC50 and a selectivity index (SI) between the two read-outs (SI = host cell IC50/parasite IC50; see ESI† for more details).
| Company and computational approach | Number of compounds (actives)a | |
|---|---|---|
| On scaffoldb | Scaffold hopc | |
| a Actives defined as compounds with SI > 5 against one or both parasites. b Compounds with a 2-substituted imidazo[1,2-a]pyridine core. c Compounds without a 2-substituted imidazo[1,2-a]pyridine core. | ||
| A- ECFP4 (ref. 40) similarity (initial Tanimoto cut-off 0.7 descending incrementally until sufficient compounds had been identified) | 45 (12) | 15 (3) |
| B- Series of substructure-based queries prioritized with Tanimoto similarity caluculated by use of Morgan fingerprint approach. Final selection cherry picked by eye | 69 (10) | 121 (18) |
| C- ECFP4 (ref. 40) similarity (Tanimoto cut-off 0.6) and in-house fingerprint search (Tanimoto cut-off 0.7). Subsequent in-house “quality”, commercial availability, and IP filters | 85 (11) | 16 (0) |
| D- Daylight and ChemAxon fingerprint similarity ranking,41,42 followed by Openeye pharmacophore alignment43,44 | 58 (12) | 56 (4) |
| E- The top scoring 150 compounds were selected by Tanimoto similarity calculation using the FCFP4 fingerprint, followed by refinement to 96 compounds based on maximized diversity | 106 (11) | 86 (9) |
| All companies | 363 (56) | 294 (34) |
Compounds with a SI > 5 were considered as demonstrating legitimate anti-parasitic activity. Results of this process are shown in Fig. 1. The first sequence of virtual screening (Fig. 1A) identified a total of 367 compounds from 4 partners, of which 25 demonstrated at least some degree of potency against L. donovani and shared the core imidazo[1,2-a]pyridine motif. Interestingly a new batch of compound 1, which displayed similar potency – cytotoxicity profile to the batch from the original HTS, appeared within this set (Fig. 2A). Compound 2 also appeared within this set, which demonstrated the potential of a new vector for exploration at the 8-position of the imidazo[1,2-a]pyridine core, as well as demonstrating efficacy with no background cell cytotoxicity. Parasitology data on each company's selection of compounds was fed back to the respective partner organizations. Parasitology data and the structure of compound 2 was shared with all partners and a second round of virtual screening was then completed using this elaborated parasitology data (Fig. 1B). The second round of in silico screening yielded a further 290 compounds from 5 participating organizations, of which 16 contained the imidazo[1,2-a]pyridine core and demonstrated some level of efficacy against L. donovani. Retrospective analysis of these two rounds of in silico screening demonstrated the power and versatility of this approach to hit to lead elaboration. Of the 657 compounds identified via in silico screening, almost 45% of the compounds represented a scaffold hop, defined in this instance as a compound not containing a 2-substituted imidazo[1,2-a]pyridine as the core. The hit rate of actives within this set of scaffold hops was only marginally lower than for those “on-scaffold” compounds which contained the original chemotype (11.5% compared to 15.4%) – yet these more structurally ambitious scaffold hop compounds would arguably not be included in a traditional hit to lead campaign due to the inherent increase in synthetic complexity of scoping multiple chemotypes. Compound 3 is a key example of such a scaffold hop originating from this approach, demonstrating significant potencies against both T. cruzi and L. donovani whilst representing a significant deviation away from the imidazo[1,2-a]pyridine core.§§
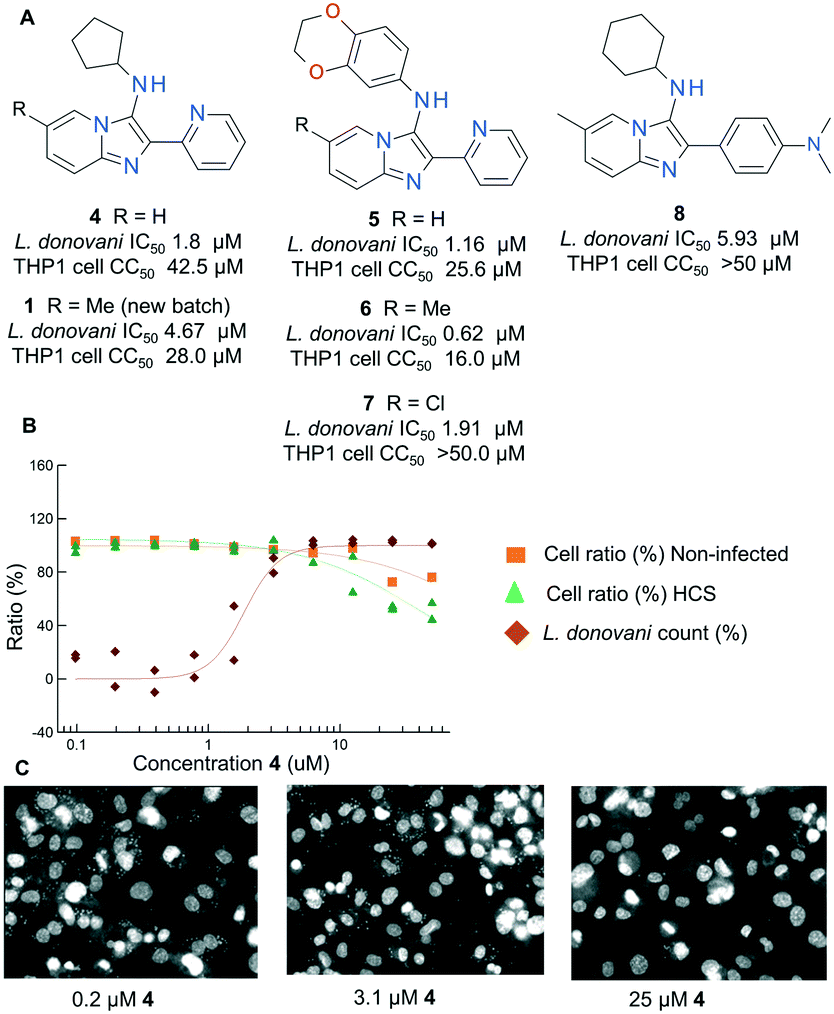 | ||
| Fig. 2 A) Diverse compounds identified from the booster giving confidence in the viability of the chemical series; B) dose response curves for 4; C) high content images of L. donovani infected THP1 cells treated with various concentrations of 4. | ||
Combining the parasitology data for the key active compounds containing the original imidazo[1,2-a]pyridine core structure of 1, we collated data to form a hit series and move forward into detailed investigation and targeted analoguing as detailed below. An initial survey of the data generated by these rounds of virtual screening resulted in a number of initial findings: firstly, there appeared to be at least 5 vectors around the core which tolerated changes whilst retaining anti-leishmanial activity (positions 2, 3, 6, 7 and 8). Secondly, although certain structural changes in the chemotype induced significant cytotoxicity against the host THP1 cell line, we identified key examples which demonstrate acceptable differentiation between anti-parasitic activity and cell based cytotoxicity, with the most selective compounds achieving a selectivity index (SI) of >20 (4, 5, 6, 7). Analysis of the anti-parasitic activity and cell line cytotoxicity curves and high content images clearly demonstrated concentration points for most compounds where a full antiparasitic effect was observed with no cytotoxic observation on the background cell line (see Fig. 2B and C). Thirdly, we were able to identify compounds which achieved an antiparasitic effect with no background cell cytotoxicity whilst removing the 2-pyridyl functionality at the 2 position of the core (8), removing some suspicions about a metal-chelation driven SAR. These findings convinced us that the cytotoxicity observed for the series of compounds was not uniquely responsible for the observed anti-parasitic effect, and that the series merited further investigation. Finally, we also observed that the anti-leishmanial SAR around the scaffold was potentially non-additive, with the impact of certain functionality changes being influenced by substitution patterns elsewhere in the structure (Fig. 2A). This appeared particularly true for substitutions of the 6- and 7-position of the core imidazo[1,2-a]pyridine, as demonstrated by compounds 4–6, where the impact of a 6-methyl group improves potency when the 3-position is aryl substituted, but negatively impacts potency when the 3-position is alkylated. We therefore decided that rather than follow a rigid systematic investigation of each area of the molecule as an SAR follow-up strategy, we would use an empirical approach combining both single- and multi-dimensional SAR exploration. We made subtle variations around the 6- and 7-position substitution at the same time as exploring the 2- and 3-position. This was intended to give as broad a coverage of the chemotype as possible in a smaller number of compounds, and to avoid missing key improvements masked by the hypothesized non-additive SAR.
Synthesis and analoging
Analoging was driven using the representative synthetic pathway shown in Scheme 1.¶¶ Various substituted 2-aminopyridines A were converted into 2-aryl imidazo[1,2-a]pyridines Bvia iodine-mediated condensation with arylmethylketones. Subsequent regioselective bromination of the 3-position of the imidazo[1,2-a]pyridines yielded 3-bromoimidazo[1,2-a]pyridines C, which were then converted into representative final products Dvia palladium mediated coupling with various primary and secondary amines. In some instances, representative final compounds D were obtained directly in one step from aminopyridines Avia tosic acid-mediated three-component cyclization with isonitriles and aryl aldehydes. One such example of D, 2-(pyridin-2-yl)-N-(2,4,4-trimethylpentan-2-yl)imidazo[1,2-a]pyridin-3-amine (E), was cleaved under acidic conditions to give 3-aminoimidazo[1,2-a]pyridine F, and subsequently converted into representative final compounds Gvia palladium mediated coupling with various aryl bromides. Further exploitation of intermediate F was achieved via amidation followed by borane mediated reduction of the amide to give N-alkylated representative final compounds K.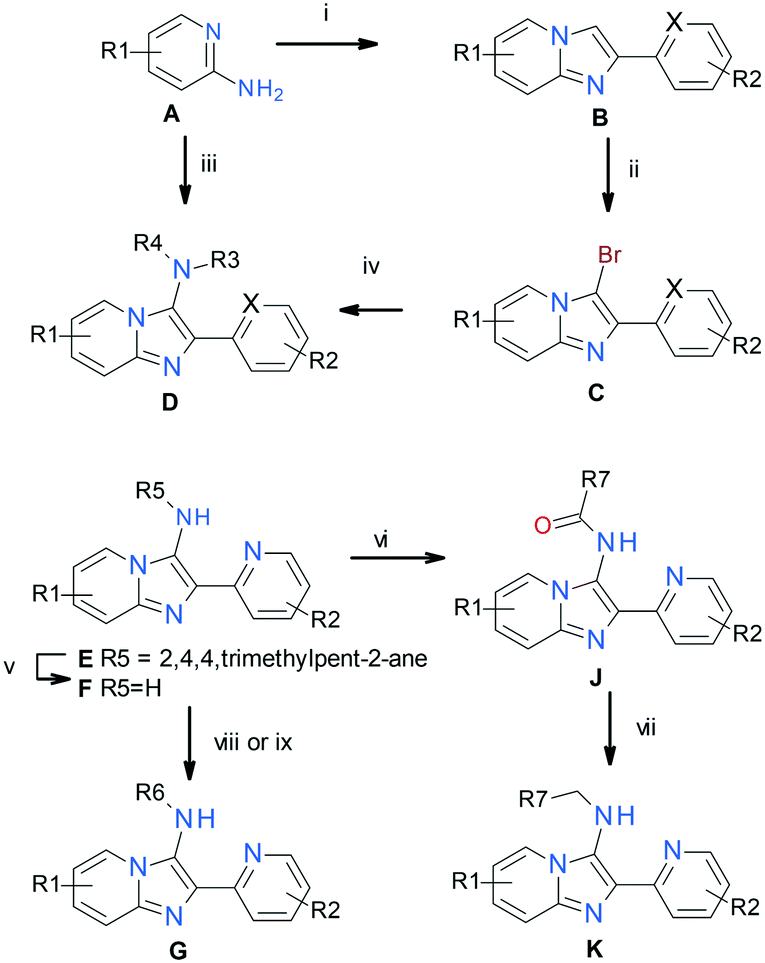 | ||
| Scheme 1 i) ArCOCH3, I2, NaOH, H2O, 110 °C; ii) NBS, MeCN, 20 °C; iii) R3-NC, TsOH, ArCHO, MeOH, 70 °C; iv) R3R4NH, t-BuONa, Brettphos-Pd-G3, t-amylalcohol, 90 °C; v) HCl, MeOH, 20 °C; vi) R7COOH, EDCI, pyridine, 25 °C; vii) BH3SMe2, THF, 60 °C; viii) ArX, t-BuONa, Pd2(dba)3, XantPhos, PhMe, 110 °C; ix) ix) Ti(i-PrO)4, NaBH3CN, dioxane, 60 °C | ||
Structure activity relationship (SAR)
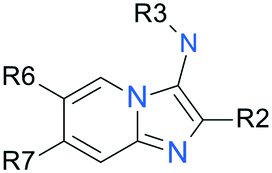
SAR elaboration of the core chemotype was focused on three distinct regions (Tables 2–4).|||| Scanning of the 3-position of the imidazo[1,2-a]pyridine core revealed some initial SAR of this position (Table 2), suggesting that compounds with both amino alkyl and amino aryl moieties at this position could demonstrate anti-parasitic activity. Although small lipophilic alkyl groups demonstrated moderate anti-parasitic activities and selectivity indices (9, 12), presence of both basic (10, 15) and non-basic (13, 16) heteroatoms appear detrimental to anti-parasitic activity. Conversely, presence of additional polar heteroatoms or halogens in the case of aniline-type substitution in the 3-position seemed to enhance potency and in certain cases selectivity over host cell cytotoxicity (5, 21), although, as previously mentioned, this selectivity seemed to partly depend on the nature of additional substitution at positions 6 and 7 on the imidazo[1,2-a]pyridine core (22–25, 29–30). A combination of alkyl and aryl substituents on the 3-position amine appeared to result in an interesting combination of both potency and selectivity over background cytotoxicity (20). It became clear that there were extensive opportunities to modulate physicochemical properties of the compound at the 3-position of the chemotype.|
|
||||||
|---|---|---|---|---|---|---|
| Compound | R3 | R6 | R7 | IC50a μM | CC50b μM | Clintc |
| a L. donovani parasite clearance in infected THP1 cells. b THP1 cytotoxicity. c Mouse microsomes, μL min−1 (mg.pr)−1. | ||||||
| 4 | N-Cyclopentylamino- | H | H | 1.80 | 42.5 | |
| 5 | N-(2,3-Dihydro-1,4-benzodioxin-6-yl)-amino | H | H | 1.16 | 25.6 | |
| 9 | N-2,2,2-Trifluoroethylamino- | H | H | 10.2 | >50 | |
| 10 | N-2-(N′,N'Dimethyl)-ethyl-1,2 diamino- | H | H | >50 | >50 | |
| 11 | N-2-Methyl-2-hydroxy-propylamino- | H | H | 30.4 | >50 | 171 |
| 12 | N-Cyclopropylmethylamino- | H | H | 3.57 | 19.4 | |
| 13 | N-Oxetan-2-ylmethylamino- | H | H | >50 | >50 | |
| 14 | N-Tetrahydrofuran-3-ylamino- | H | H | 29.4 | >50 | |
| 15 | N-Pyrolidin-3-ylamino- | H | H | >50 | >50 | |
| 16 | N-Morpholino- | H | H | >50 | >50 | |
| 17 | N-Benzylamino- | H | H | 9.34 | >50 | |
| 18 | N-Phenylamino- | H | H | 1.11 | 6.69 | |
| 19 | N-4-Fluorophenylamino- | Me | H | 2.71 | 9.61 | |
| 20 | N-(N-Methyl)-4-fluorphenylamino- | H | H | 3.9 | >50 | >1000 |
| 21 | N-3-Fluorophenylamino- | H | H | 3.70 | >50 | 242 |
| 22 | N-2-Fluorophenylamino- | H | OMe | 1.73 | 5.75 | 133 |
| 23 | N-3,5-Difluorophenylamino- | H | OMe | 1.91 | 4.3 | 62 |
| 24 | N-2,5-Dichlorophenylamino- | H | OMe | 3.91 | 8.93 | |
| 25 | N-5-Fluoropyridin-2-ylamino- | H | OMe | 1.71 | 16.0 | 81 |
| 26 | N-Pyridin-2-ylaminoamino- | H | Me | 4.53 | >50 | 130 |
| 27 | 4-Trifluoromethoxyphenylamino- | H | H | 3.49 | >50 | 223 |
| 28 | N-4-Cyananophenylamino- | H | H | 7.76 | >50 | |
| 29 | N-4-Methoxyphenylamino- | Me | H | 2.31 | 10.7 | |
| 30 | N-3-Methoxyphenylamino- | Me | H | 2.53 | 13.2 | |
| 31 | N-2-Methoxyphenylamino- | H | H | 2.61 | >50 | 326 |
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R3 | R2 | R6 | R7 | IC50a μM | CC50b μM | Clintc |
| a L. donovani parasite clearance in infected THP1 cells. b THP1 cytotoxicity. c Mouse microsomes, μL min−1 (mg.pr)−1. d Value for library batch (value for resynthesized batch). | |||||||
| 4 | Cyclopentyl | Pyrid-2-yl | H | H | 1.80 | 42.5 | |
| 32 | 5-Methoxypyrid-2-yl | H | H | 5.26 | 19.3 | ||
| 33 | 4-Methoxypyrid-2-yl | H | H | 1.76 | 27.1 | 932 | |
| 34 | Pyrid-3-yl | H | H | >50 | >50 | ||
| 35 | 2-Methylpyrid-4-yl | H | H | 42.2 | >50 | ||
| 36 | 3,4-Dimethoxyphenyl | H | H | 16.2 | >50 | ||
| 37 | 4-(Dimethylamino)phenyl | H | Me | 7.37 (3.2)d | >50 (29.3)d | >1000 | |
| 38 | H | H | 7.9 | >50 | |||
| 39 | 4-Methoxyphenyl | H | Me | 10.2 | >50 | ||
| 40 | 2-Fluorophenyl | H | H | 25.9 | >50 | ||
| 41 | 2-Fluoro-4-methoxyphenyl | H | H | 22.0 | >50 | ||
| 42 | Thiazol-2-yl | H | H | >50 | >50 | ||
| 43 | N-Methylimidazo2-yl | H | H | >50 | >50 | ||
| 8 | Cyclohexyl | 4-N,N-Dimethylaminophenyl | Me | H | 5.93 | >50 | |
| 44 | 4-Methoxyphenyl | H | Me | 6.51 | >50 | ||
| 45 | 4-Ethoxyphenyl | H | H | 9.4 | >50 | ||
| 46 | 4-Tertbutylphenyl | H | H | 13.2 | >50 | ||
| 47 | 4-Fluorophenyl | 5-Methoxypyrid-2-yl | H | H | 2.5 | 15.4 | 126 |
| 48 | 4-Methoxypyrid-2-yl | H | H | 2.85 | 4.22 | >1000 | |
| 49 | 5-Chloropyrid-2-yl | H | H | 11.5 | >50 | ||
| 50 | N-Methylimidazo-2-yl | H | H | >50 | >50 | ||
| 51 | 3-Fluorophenyl | N-Methylimidazo-4-yl | H | OMe | 4.46 | 40 | <12 |
| 52 | Thiazol-4-yl | H | OMe | 15.7 | 40.6 | ||
| 53 | N-Methylpyrazol-3-yl | H | OMe | 36.2 | >50 | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Compound | R3 | R6 | R7 | IC50a μM | CC50b μM | Clintc |
| a L. donovani parasite clearance in infected THP1 cells. b THP1 cytotoxicity. c Mouse microsomes, μL min−1 (mg.pr)−1. d Library batch (resynthesized batch). e Library batch (initial HTS batch). | ||||||
| 19 | N-4-Fluorophenylamino- | Me | H | 2.71 | 9.61 | |
| 54 | CN | H | >50 | >50 | ||
| 55 | CF3 | H | 35.4 | >50 | ||
| 56 | MeO | H | 2.38 | 4.26 | 923 | |
| 57 | Cl | H | >50 | >50 | ||
| 58 | H | CN | >50 | >50 | 146 | |
| 59 | H | CF3 | 11.0 | 23.7 | ||
| 60 | H | MeO | 1.77 | 4.56 | 117 | |
| 61 | H | Me | 0.71 (2.45)d | 5.9 (11.8)d | 355 | |
| 21 | N-3-Fluorophenylamino | H | H | 3.7 | >50 | |
| 67 | H | Me | 2.47 | 14.24 | ||
| 62 | CH2OMe | H | 7.19 | 16.0 | ||
| 63 | CH2NMe2 | H | 5.12 | >50 | ||
| 64 | H | CH2OMe | 2.74 | 7.37 | 92 | |
| 65 | H | CH2NMe2 | 4.54 | 33.3 | ||
| 66 | H | OH | 16.87 | 43.3 | ||
| 1 | N-Cyclopentylamino- | Me | H | 4.67 (2.9)e | 28.0 (24.0)e | |
| 4 | H | H | 1.80 | 42.5 | ||
| 68 | H | Me | 1.88 | >50 | >1000 | |
| 5 | N-(2,3-Dihydro-1,4-benzodioxin-6-yl)-amino- | H | H | 1.16 | 25.6 | |
| 6 | Me | H | 0.62 (1.98)d | 16.0 (28.3)d | 411 | |
| 7 | Cl | H | 1.91 (6.33)d | >50 (17.4)d | >1000 | |
| 17 | N-Benzylamino- | H | H | 9.34 | >50 | |
| 69 | Me | H | 3.37 | >50 | >1000 | |
| 18 | N-Phenylamino- | H | H | 1.11 | 6.69 | |
| 70 | H | Me | 0.82 | 9.59 | ||
| 10 | N-2-(N′,N'Dimethyl)-ethyl1,2 diamino- | H | H | >50 | >50 | |
| 16 | N-Morpholino | H | H | >50 | >50 | |
| 71 | H | OMe | 17.9 | >50 | 137 | |
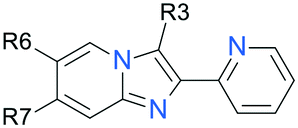
Investigation of the 2-position of the core (Table 3) revealed that small substitutions such as chloro and methoxy on the 4- and 5-position 2-pyridyl were tolerated (32, 33, 42–47) and could be exploited to tune microsomal stability. Moving the position of the pyridyl nitrogen to the 3 or 4 position removed anti-leishmanial activity (34, 35), however replacement of the 2-pyridyl with 5-membered nitrogen-containing heteroaromatic rings was tolerated in certain instances (51), and this change also had a major impact on the metabolic stability of the compound. Interestingly, replacement of the pyridyl with electron enriched phenyls such as 4-anilino (37, 38) and 4-alkoxy substituted (44, 45) retained moderate activity and generally removed any evidence of cytotoxicity, suggesting that the anti-parasitic efficacy of the chemotype is likely not driven by a bidentate metal chelation effect. This observation of multiple non-2-pyridyl analogues further supported our earlier hypothesis regarding the redundancy of the PAINS flag for the initial hit 1.
Broader substitution of the 6- and 7-positions of the core imidazo[1,2-a]pyridine revealed a clear negative effect of withdrawing electron density from the core heterocycle, with cyano (54, 58) and trifluoromethyl (55, 59) substituents negatively impacting the antiparasitic effect (Table 4). Attempts to probe further away from the core negatively impacted potency and/or cytotoxicity (62–65), however incorporation of heteroatoms in this position could be used to improve microsomal stability (64). Scanning the impact of 6- and 7-substitution relative to the functionality at position 3 reconfirmed the previous observation that SAR appears non-additive, with changes having negative or positive impact on potency and cytotoxicity depending on the nature of the 3-position substitution.
ADME and pharmacokinetic profiling
To complete the initial scoping of this chemical series, we profiled a selection of compounds in preliminary in vitro solubility and metabolic stability assays (Table 5). In general compounds appeared poorly soluble at pH 7.4, although this could be optimized through incorporation of additional polarity in the 3-position of the core (25). Kinetic solubility at pH 2.0 was consistently favourable, and the compounds generally demonstrated rapid intrinsic clearance in both human and mouse microsomes, with the intrinsic clearance of both species tracking when moderate improvements were observed in microsomal stability for compounds 25 and 57, which again suggested that ADME property optimization was strongly tied to the presence of polar heteroatoms in the 3-position of the imidazo[1,2-a]pyridine. Compiling the broader SAR around intrinsic microsomal mouse clearance in vitro (Tables 2–5) shows that improvement was generally achievable through the introduction of additional polarity, for example via incorporation of additional nitrogen in the pendant aryl groups at positions 2- and 3- of the core, or through ether groups at the 7-position of the core scaffold. To understand the translation of these in vitro ADME parameters into in vivo exposure, compound 23 was profiled in mouse pharmacokinetic studies at 1 mg kg−1 i.v. and 50 mg kg−1 p.o. (Fig. 3). Despite moderate in vitro Clint and in vivo bioavailability, 23 demonstrated high clearance in the mouse, resulting in relatively low overall exposure in terms of CMax and time of exposure after oral dosing, with no compound observed in the plasma after 24 hours. Such exposure relative to the anti-parasitic activity of the compound was considered insufficient to progress the compound to an in vivo infection model of Leishmania.| Compound | Clint μL min−1 (mg.pr)−1 | Kinetic solubility μg mL−1 | ||
|---|---|---|---|---|
| Mouse | Human | pH 7.4 | pH 2.0 | |
| 6 | 411 | 95 | 8 | 72 |
| 7 | >1000 | >100 | <0.5 | >76 |
| 25 | 81 | 35 | >76 | >76 |
| 37 | >1000 | >100 | <0.5 | 67 |
| 61 | 354 | >100 | <0.5 | 73 |
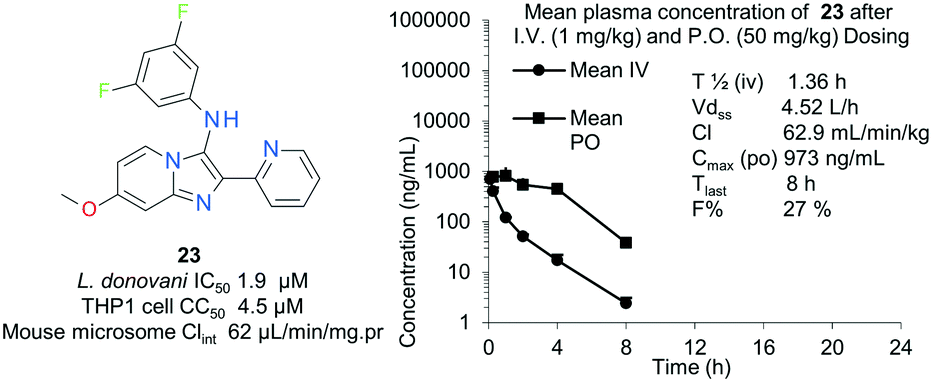 | ||
| Fig. 3 Pharmacokinetic profiling of 23 in mouse. | ||
Conclusions
Work continues to optimize the imidazo[1,2-a]pyridine series informed by the SAR generated by the in-silico Booster process and subsequent informed design. The primary focus for this future work will be further improvement of the in vitro ADME parameters in order to achieve prolonged exposure in vivo in line with a profile capable of demonstrating efficacy in an infection model of Leishmania. This may, however, be complicated by the clearly non-additive anti-parasitic and cytotoxicity SAR, although certain key findings from the SAR identified so far should enable improvement of anti-parasitic activity, microsomal stability and widening of the selectivity index between anti-parasitic effect and cell cytotoxicity. The overall approach of using the drug discovery booster as a multi-party in silico screening approach to complete the first phase of hit-to-lead and hit validation allowed us to rapidly confirm and validate the activity and relevance of the series as well as inform and guide the initial SAR exploration. Further analysis of the impact and utility of the Booster approach, which has to date been applied to over 20 different hit series,33 will be the subject of future communication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge John Cuff, Leelapavan Tadoori, Jean Robert Ioset, Tatsuro Kuzuki, Daisuke Imoto, Midori Morioka, Dominique Junod and Jean Pierre Paccaud for their help in implementation and logistics of the Booster project, Chen Min and Jiajia Zhang for ADME-PK studies, Louise Burrows for help in manuscript preparation. We acknowledge the generous support of Global Health Innovative Technology Fund (GHIT) for financial support of this project, and our collaboration partners at Abbvie Inc, Astellas Pharma Inc and Merck KGaA for participation in the Booster project outside of this particular series. The Drugs for Neglected Diseases initiative (DNDi) is grateful to its donors, public and private, who have provided funding to DNDi since its inception in 2003. A full list of DNDi's donors can be found at http://www.dndi.org/donors/donors/.Notes and references
- WHO, Visceral leishmaniasis, https://www.who.int/leishmaniasis/visceral_leishmaniasis/en/, (accessed April 17, 2020) Search PubMed.
- S. Burza, S. L. Croft and M. Boelaert, Lancet, 2018, 392, 951–970 CrossRef.
- J. Alvar, S. Yactayo and C. Bern, Trends Parasitol., 2006, 22, 552–557 CrossRef.
- N. J. Kassebaum, et al. , Lancet, 2016, 388, 1603–1658 CrossRef.
- J. Alvar, I. D. Vélez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. de Boer, PLoS One, 2012, 7, 1–6 CrossRef.
- CDC - Leishmaniasis - Epidemiology & Risk Factors, https://www.cdc.gov/parasites/leishmaniasis/epi.html, (accessed April 17, 2020) Search PubMed.
- WHO, Epidemiological situation, https://www.who.int/leishmaniasis/burden/en/, (accessed June 10, 2020) Search PubMed.
- GHO, By category, Number of cases of visceral leishmaniasis reported - Data by country, https://apps.who.int/gho/data/node.main.NTDLEISHVNUM?lang=en, (accessed June 10, 2020) Search PubMed.
- J. N. Sangshetti, F. A. Kalam Khan, A. A. Kulkarni, R. Arote and R. H. Patil, RSC Adv., 2015, 5, 32376–32415 RSC.
- F. Chappuis, S. Sundar, A. Hailu, H. Ghalib, S. Rijal, R. W. Peeling, J. Alvar and M. Boelaert, Nat. Rev. Microbiol., 2007, 5, 873–882 CrossRef CAS.
- J. Delgado, J. Macías, J. A. Pineda, J. E. Corzo, P. González-Moreno, R. de La Rosa, A. Sánchez-Quijano, M. Leal and E. Lissen, Am. J. Trop. Med. Hyg., 1999, 61, 766–769 CrossRef CAS.
- H. C. Maltezou, J. Biomed. Biotechnol., 2010, 2010, 617521 CrossRef.
- B. Monge-Maillo and R. López-Vélez, Drugs, 2013, 73, 1863–1888 CrossRef CAS.
- K. Katsuno, J. N. Burrows, K. Duncan, R. H. van Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. T. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS.
- Target Product Profile – Visceral Leishmaniasis – DNDi, https://www.dndi.org/diseases-projects/leishmaniasis/tpp-vl/, (accessed April 17, 2020) Search PubMed.
- A. S. Nagle, S. Khare, A. B. Kumar, F. Supek, A. Buchynskyy, C. J. N. Mathison, N. K. Chennamaneni, N. Pendem, F. S. Buckner, M. H. Gelb and V. Molteni, Chem. Rev., 2014, 114, 11305–11347 CrossRef CAS.
- S. M. Lee, M. S. Kim, F. Hayat and D. Shin, Molecules, 2019, 24 CAS.
- I. H. Gilbert, J. Med. Chem., 2013, 56, 7719–7726 CrossRef CAS.
- J. A. Hutton, V. Goncalves, J. A. Brannigan, D. Paape, M. H. Wright, T. M. Waugh, S. M. Roberts, A. S. Bell, A. J. Wilkinson, D. F. Smith, R. J. Leatherbarrow and E. W. Tate, J. Med. Chem., 2014, 57, 8664–8670 CrossRef CAS.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS.
- Portfolio – DNDi, https://www.dndi.org/diseases-projects/portfolio/, (accessed April 17, 2020) Search PubMed.
- M. G. Thomas, M. de Rycker, M. Ajakane, S. Albrecht, A. I. Álvarez-Pedraglio, M. Boesche, S. Brand, L. Campbell, J. Cantizani-Perez, L. A. T. Cleghorn, R. C. B. Copley, S. D. Crouch, A. Daugan, G. Drewes, S. Ferrer, S. Ghidelli-Disse, S. Gonzalez, S. L. Gresham, A. P. Hill, S. J. Hindley, R. M. Lowe, C. J. Mackenzie, L. Maclean, S. Manthri, F. Martin, J. Miguel-Siles, V. L. Nguyen, S. Norval, M. Osuna-Cabello, A. Woodland, S. Patterson, I. Pena, M. T. Quesada-Campos, I. H. Reid, C. Revill, J. Riley, J. R. Ruiz-Gomez, Y. Shishikura, F. R. C. Simeons, A. Smith, V. C. Smith, D. Spinks, L. Stojanovski, J. Thomas, S. Thompson, T. Underwood, D. W. Gray, J. M. Fiandor, I. H. Gilbert, P. G. Wyatt, K. D. Read and T. J. Miles, J. Med. Chem., 2019, 62, 1180–1202 CrossRef CAS.
- S. Wyllie, S. Brand, M. Thomas, M. de Rycker, C. wa Chung, I. Pena, R. P. Bingham, J. A. Bueren-Calabuig, J. Cantizani, D. Cebrian, P. D. Craggs, L. Ferguson, P. Goswami, J. Hobrath, J. Howe, L. Jeacock, E. J. Ko, J. Korczynska, L. MacLean, S. Manthri, M. S. Martinez, L. Mata-Cantero, S. Moniz, A. Nühs, M. Osuna-Cabello, E. Pinto, J. Riley, S. Robinson, P. Rowland, F. R. C. Simeons, Y. Shishikura, D. Spinks, L. Stojanovski, J. Thomas, S. Thompson, E. V. Gaza, R. J. Wall, F. Zuccotto, D. Horn, M. A. J. Ferguson, A. H. Fairlamb, J. M. Fiandor, J. Martin, D. W. Gray, T. J. Miles, I. H. Gilbert, K. D. Read, M. Marco and P. G. Wyatt, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9318–9323 CrossRef CAS.
- S. Khare, A. S. Nagle, A. Biggart, Y. H. Lai, F. Liang, L. C. Davis, S. W. Barnes, C. J. N. Mathison, E. Myburgh, M. Y. Gao, J. R. Gillespie, X. Liu, J. L. Tan, M. Stinson, I. C. Rivera, J. Ballard, V. Yeh, T. Groessl, G. Federe, H. X. Y. Koh, J. D. Venable, B. Bursulaya, M. Shapiro, P. K. Mishra, G. Spraggon, A. Brock, J. C. Mottram, F. S. Buckner, S. P. S. Rao, B. G. Wen, J. R. Walker, T. Tuntland, V. Molteni, R. J. Glynne and F. Supek, Nature, 2016, 537, 229–233 CrossRef CAS.
- M. van den Kerkhof, D. Mabille, E. Chatelain, C. E. Mowbray, S. Braillard, S. Hendrickx, L. Maes and G. Caljon, Int. J. Parasitol.: Drugs Drug Resist., 2018, 8, 81–86 CAS.
- M. J. Waring, J. Arrowsmith, A. R. Leach, P. D. Leeson, S. Mandrell, R. M. Owen, G. Pairaudeau, W. D. Pennie, S. D. Pickett, J. Wang, O. Wallace and A. Weir, Nat. Rev. Drug Discovery, 2015, 14, 475–486 CrossRef CAS.
- M. Hay, D. W. Thomas, J. L. Craighead, C. Economides and J. Rosenthal, Nat. Biotechnol., 2014, 32, 40–51 CrossRef CAS.
- R. L. Charlton, B. Rossi-Bergmann, P. W. Denny and P. G. Steel, Parasitology, 2018, 145, 219–236 CrossRef.
- B. Singh, J. A. Bernatchez, L. I. McCall, C. M. Calvet, J. Ackermann, J. M. Souza, D. Thomas, E. M. Silva, K. A. Bachovchin, D. M. Klug, H. B. Jalani, S. Bag, M. J. Buskes, S. E. Leed, N. E. Roncal, E. C. Penn, J. Erath, A. Rodriguez, R. J. Sciotti, R. F. Campbell, J. McKerrow, J. L. Siqueira-Neto, L. Ferrins and M. P. Pollastri, ACS Med. Chem. Lett., 2020, 11, 249–257 CrossRef CAS.
- M. Álvarez-Bardón, Y. Pérez-Pertejo, C. Ordóñez, D. Sepúlveda-Crespo, N. M. Carballeira, B. L. Tekwani, S. Murugesan, M. Martinez-Valladares, C. García-Estrada, R. M. Reguera and R. Balaña-Fouce, Mar. Drugs, 2020, 18, 187 CrossRef.
- R. Balaña-Fouce, M. Y. Pérez Pertejo, B. Domínguez-Asenjo, C. Gutiérrez-Corbo and R. M. Reguera, Drug Discovery Today, 2019, 24, 1209–1216 CrossRef.
- WHO, Chagas disease, https://www.who.int/chagas/home_more/en/, (accessed June 12, 2020) Search PubMed.
- NTD – DNDi, https://www.dndi.org/diseases-projects/open-innovation/drug-discovery-booster/, (accessed April 17, 2020) Search PubMed.
- J. P. Hughes, S. S. Rees, S. B. Kalindjian and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249 CrossRef CAS.
- T. Kogej, N. Blomberg, P. J. Greasley, S. Mundt, M. J. Vainio, J. Schamberger, G. Schmidt and J. Hüser, Drug Discovery Today, 2013, 18, 1014–1024 CrossRef.
- S. Marhadour, P. Marchand, F. Pagniez, M. A. Bazin, C. Picot, O. Lozach, S. Ruchaud, M. Antoine, L. Meijer, N. Rachidi and P. le Pape, Eur. J. Med. Chem., 2012, 58, 543–556 CrossRef CAS.
- M. Nuth, L. Huang, Y. L. Saw, N. Schormann, D. Chattopadhyay and R. P. Ricciardi, J. Med. Chem., 2011, 54, 3260–3267 CrossRef CAS.
- P. A. Mak, S. P. S. Rao, M. Ping Tan, X. Lin, J. Chyba, J. Tay, S. H. Ng, B. H. Tan, J. Cherian, J. Duraiswamy, P. Bifani, V. Lim, B. H. Lee, N. Ling Ma, D. Beer, P. Thayalan, K. Kuhen, A. Chatterjee, F. Supek, R. Glynne, J. Zheng, H. I. Boshoff, C. E. Barry, T. Dick, K. Pethe and L. R. Camacho, ACS Chem. Biol., 2012, 7, 1190–1197 CrossRef CAS.
- J. Dam, Z. Ismail, T. Kurebwa, N. Gangat, L. Harmse, H. M. Marques, A. Lemmerer, M. L. Bode and C. B. de Koning, Eur. J. Med. Chem., 2017, 126, 353–368 CrossRef CAS.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS.
- daylight, https://www.daylight.com/ Search PubMed.
- Chemaxon, http://www.chemaxon.com/ Search PubMed.
- Openeye, https://www.eyesopen.com/ Search PubMed.
- P. C. D. Hawkins, A. G. Skillman and A. Nicholls, J. Med. Chem., 2007, 50, 74–82 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic chemistry, in silico screening, and assay experimental details, including analytical data for all compounds. See DOI: 10.1039/d0md00353k |
| ‡ Current address: Elgia Therapeutics, Inc., San Diego, CA, USA. |
| § Current address: Loxo Oncology at Lilly, Boulder CO, USA. |
| ¶ Current address: Axcelead Drug Discovery Partners, Inc., Kanagawa, Japan. |
| || Current address: Molecular Devices, NYC, NY, USA. |
| ** Current address: The University of Tokyo, Tokyo, Japan. |
| †† Compound libraries for HTS efforts at DNDi were sourced from a combination of commercial and proprietary collections. |
| ‡‡ Assignment of blinded names A–E is random. Specific contributions of each partner were, and remain, anonymous within the collaboration. |
| §§ A full data set containing the compounds synthesized during this effort can be found in the supporting information and has been made fully available online: https://dndi.org/research-development/portfolio/drug-discovery-booster/ |
| ¶¶ Full synthetic details for all compounds in ESI.† |
| |||| 3 was followed up further by subsequent rounds of in silico screening via this consortium approach, results to be communicated in future correspondence. |
| This journal is © The Royal Society of Chemistry 2021 |