
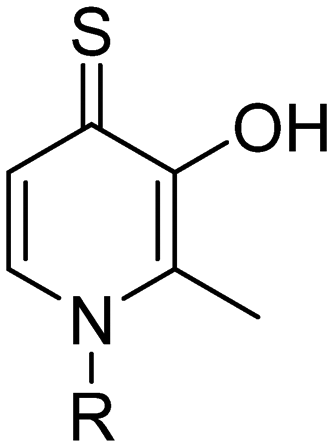
Identification of 3-hydroxy-1,2-dimethylpyridine-4(1H)-thione as a metal-binding motif for the inhibition of botulinum neurotoxin A†
Lucy
Lin
 a,
Lewis D.
Turner
a,
Peter
Šilhár‡
a,
Sabine
Pellett
b,
Eric A.
Johnson
b and
Kim D.
Janda
*a
a,
Lewis D.
Turner
a,
Peter
Šilhár‡
a,
Sabine
Pellett
b,
Eric A.
Johnson
b and
Kim D.
Janda
*a
aDepartment of Chemistry and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA 92037, USA. E-mail: kdjanda@scripps.edu
bDepartment of Bacteriology, University of Wisconsin, 1550 Linden Drive, Madison, Wisconsin 53706, USA
First published on 12th November 2020
Abstract
Botulinum neurotoxin serotype A (BoNT/A) is an important therapeutic target owing to its extremely potent nature, but also has potential use as a biowarfare agent. Currently, no therapeutic exists to reverse the long-lasting paralysis caused by BoNT/A. Herein, we describe the identification of 3-hydroxy-1,2-dimethylpyridine-4(1H)-thione (3,4-HOPTO) as a metal binding warhead for the inhibition of BoNT/A1. An initial screen of 96 metal binding fragments identified three derivatives containing the 3,4-HOPTO scaffold to inhibit the BoNT/A1 light chain (LC) at >95% at 1 mM. Additional screening of a 3,4-HOPTO sub-library identified structure–activity relationships (SARs) between N-substituted 3,4-HOPTO derivatives and the BoNT/A1 LC. Subsequent synthesis was conducted to improve on inhibitory potency – achieving low μM biochemical IC50 values. Representative compounds were evaluated in a cellular-based assay and showed promising μM activity.
Introduction
Botulinum neurotoxins (BoNTs) have gained significant attention in recent years as a potential therapeutic for chronic pain management and other nerve-related diseases.1,2 BoNT serotype A in particular is incredibly potent and its effects are long-lasting, only requiring dosing every few months. However, the off-target effects of BoNT treatments have become a concern; adverse effects include iatrogenic botulism, issues swallowing and breathing, over-all muscle weakness, double vision, blurred vision, and loss of bladder control.3 The characteristics that lend to BoNT/A's potential as a therapeutic also contribute to its danger; BoNT/A is the most potent neurotoxin known to man, with an estimated human LD50 of 1–2 ng kg−1.4 Intoxication with BoNT/A results in botulism, a disease characterized by flaccid paralysis leading to death by asphyxiation. Due to its ease of production and lethality, its potential use as a biowarfare agent has warranted its classification by the CDC as a tier 1 category A biological agent alongside anthrax and Ebola.5Currently, the standard treatment for botulism is an equine-derived antitoxin. The antitoxin must be administered within 72 hours of intoxication to maximize its effect, as it cannot enter the neuronal compartment to neutralize internalized BoNT/A, where the toxin remains active for several months.6–8 Additional limitations to this therapeutic strategy include adverse reactions to horse serum and high costs. Furthermore, even with the aid of antibody treatment, patients may spend weeks to months in the hospital, often requiring mechanical nutrition and ventilation;4 cost of treatment ranges from $55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–350000 per patient.9,10 The high medical costs and burden on the healthcare system of several patients requiring mechanical ventilation has become painfully apparent in the current COVID-19 pandemic. Thus, there is an unmet need for a treatment strategy that is capable of arresting the activity of BoNT/A after motor neurons have been intoxicated.
000–350000 per patient.9,10 The high medical costs and burden on the healthcare system of several patients requiring mechanical ventilation has become painfully apparent in the current COVID-19 pandemic. Thus, there is an unmet need for a treatment strategy that is capable of arresting the activity of BoNT/A after motor neurons have been intoxicated.
BoNT/A is a 150 kD metalloprotease composed of a heavy chain (HC, 100 kD), which mediates specific neuronal cell association, endocytosis, and toxin translocation into cells, and a light chain (LC, 50 kD) which possesses the catalytic domain, the source of BoNT/A's proteolytic activity.11–13 Once translocated into neurons, the LC is free to cleave its substrate, synaptosomal nerve-associated protein-25 (SNAP-25). SNAP-25 is a SNAP receptor (SNARE) protein required for vesicle fusion to the cellular membrane.14 In the absence of fully intact SNAP-25, motor-neurons are unable to release acetylcholine, resulting in the flaccid muscle paralysis characteristic of botulism.14 Post-intoxication treatments for botulism must disrupt the action of the BoNT/A LC inside the neuronal cells. Thus, an ideal strategy to address this issue would be the development of a cell-permeable small molecule inhibitor.8 However, despite significant effort, no small molecule inhibitors of BoNT/A have seen clinical success.
Recently, our group reported the use of a bifunctional strategy which leverages the potency of a metal binding group (MBG) active site inhibitor tethered to a cysteine-reactive warhead to covalently inhibit enzymatic activity of the BoNT/A1 LC.15 While the proof-of concept inhibitors were successful, the potential off-target effects resulting from both the highly reactive warhead and the hydroxamic acid moiety remain a concern.16–18 Thus, there is a continued need for the discovery of MBGs that are capable of inhibiting the BoNT/A LC enzymatic activity, which can then be adapted for use in the bifunctional approach. Herein, we describe the identification and development of 3-hydroxy-1,2-dimethylpyridine-4(1H)-thione (3,4-HOPTO) as an active site inhibitor of BoNT/A1.
Results and discussion
To begin, we employed a fragment-based screening approach, selecting the Cohen fragment library (CFL-1) as it contains a diverse selection of MBGs.19 Screening was performed using a robust fluorescence resonance energy transfer (FRET) assay that has been used extensively to evaluate small-molecule inhibitors of the BoNT/A1 LC (see ESI† 1.2).20 A total of 96 fragments were evaluated against the BoNT/A1 LC at a concentration of 1 mM (Fig. S1†). Six of the fragments (Fig. 1), reduced BoNT/A1 LC activity by over 95% at the tested concentration. While 1 and 2 showed excellent activity, we concluded that these were unsuitable candidates due to several potential liabilities, including cell permeability, lack of growth vectors, and solubility. Compound 3 contains the quinolinol core which has previously been described.21–23 Therefore, we decided to focus our efforts on compounds 4–6, all of which contained the 3,4-HOPTO MBG, which has been shown to inhibit other metalloproteases.19,24–26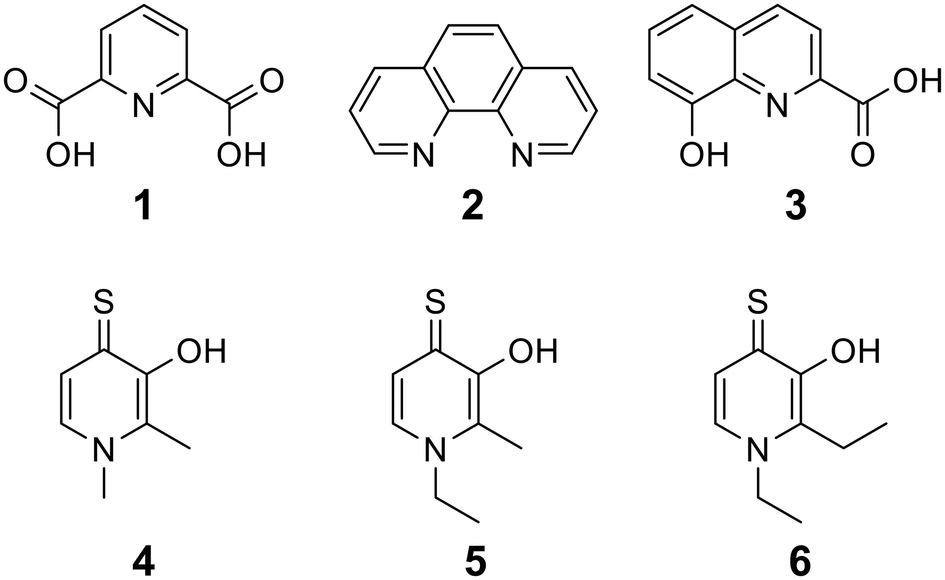 | ||
| Fig. 1 Hits obtained from Cohen's library (CFL-1.1), screened at 1 mM against the BoNT/A1 LC. | ||
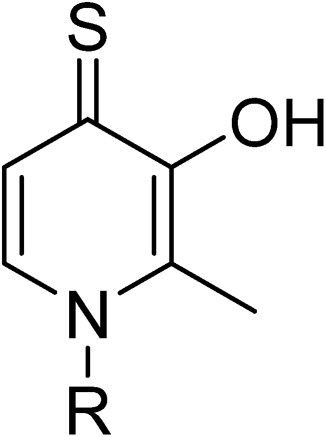
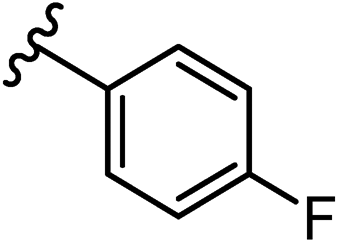
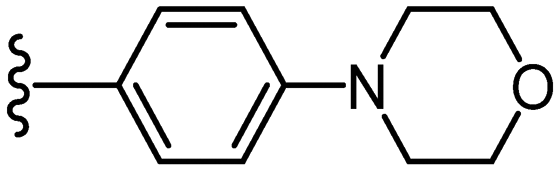
An additional screen of a 3,4-HOPTO-based library (eCFL-1)19 containing 18 compounds substituted on the nitrogen was carried out (Table 1). In general, phenyl derivatives that were directly attached to the nitrogen (7–14) were either inactive or had poor potency, with the exception of compound 7. The mono-para-substituted lipophilic ethoxy group appears to be better placed within the active site when compared to smaller (8 and 9) and larger (10 and 11) para-substituted compounds, suggesting a ‘medium’-sized substituent is most desired when there is a constrained vector for the phenyl group. Bulkier di-substituted compounds (12–14) were not well tolerated, outlining potential steric and orientation constraints of the active site in this region. An increase in flexibility introduced by the insertion of a one- or two-carbon linker between the phenyl ring and the nitrogen atom saw an increase in potency (15–24), supporting this notion. This is best demonstrated through the increase in potency between compounds 8 and 15. We hypothesize that the additional flexibility and length afforded by the linker may facilitate more favourable hydrophobic binding interactions, most notably π-stacking with aromatic residues in the BoNT/A1 LC active site. Moreover, there is little difference in inhibitory potency for compounds bearing the same substituent in different substitution patterns (i.e.16 and 17, 22 and 23), implying more steric freedom. The most influential factor determining changes in potency appears to be steric bulk, demonstrated by the negligible difference in potencies between compounds 18 and 19 where the pyridyl nitrogen has minimal effects on binding. Compounds 20–23 contain an ethylene linker and exhibit similar potencies to their methylene counterparts and are equipotent with compound 24 which possesses a large naphthalene group. This apparent tolerance for steric bulk in this region is consistent with previous work, which established the plasticity of the BoNT/A1 LC.27,28 An X-ray crystal structure (PDB ID: 4HEV) of an adamantane-based hydroxamate inhibitor in complex with the BoNT/A1 LC shows the bulky adamantane group occupying a hydrophobic pocket.27 In direct contrast to this, an X-ray crystal structure (PDB ID: 2IMB) of L-arginine hydroxamate bound in the LC shows the active site to be significantly more polar.29 The variability in the active site conformations means that larger, more bulky hydrophobic inhibitors can be tolerated, while maintaining the same configuration of the zinc ion and catalytic residues.


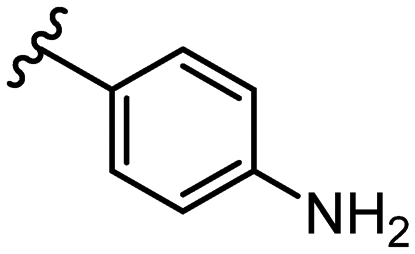
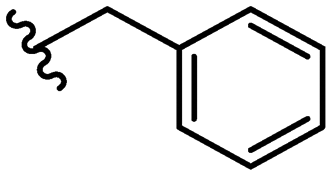
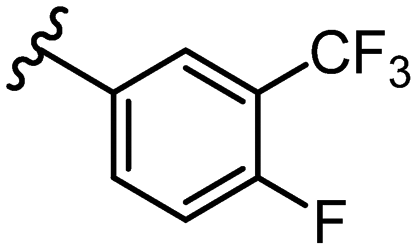
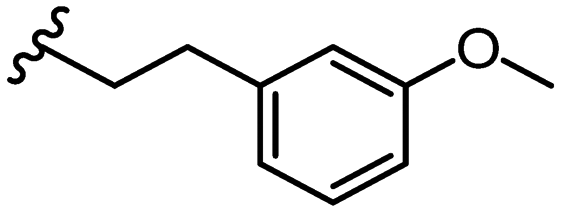


SAR library expansion
To further probe SARs, we synthesized derivatives of the 3,4-HOPTO scaffold by condensation of primary amine precursors with thiomaltol (26)30 using various conditions (Scheme 1), as described in literature.19,25 A total of 19 compounds were synthesized and their IC50 values for the BoNT/A1 LC obtained (Table 2).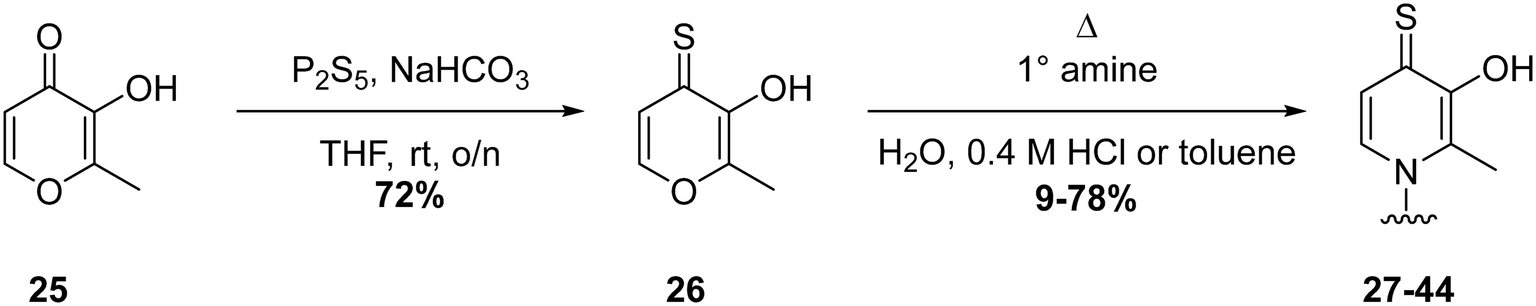 | ||
| Scheme 1 General synthesis of 3,4-HOPTO derivatives 27–44. Full synthetic details provided in ESI.† | ||
|
|
|||||
|---|---|---|---|---|---|
| Compound | R group | IC50a (μM) | Compound | R group | IC50a (μM) |
| a IC50 values are reported as mean ± SD (n = 3). | |||||
| 27 |
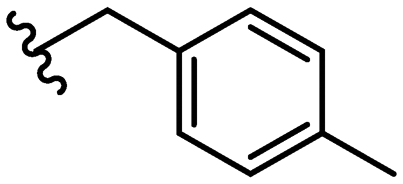
|
5.0 ± 1.0 | 35 |
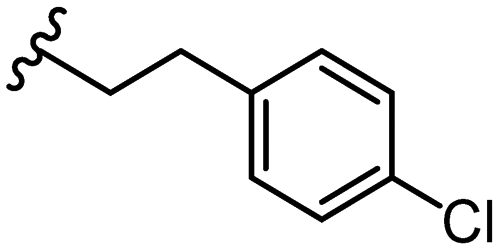
|
1.9 ± 0.2 |
| 28 |
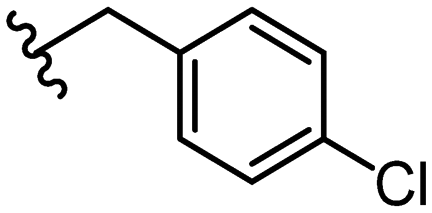
|
11.3 ± 3.4 | 36 |

|
1.7 ± 0.4 |
| 29 |
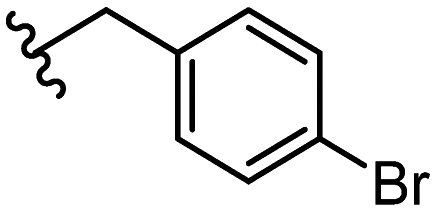
|
2.2 ± 0.8 | 37 |

|
2.8 ± 1.1 |
| 30 |

|
31.9 ± 1.7 | 38 |
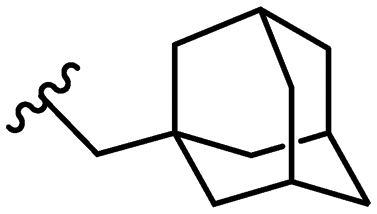
|
4.9 ± 2.6 |
| 31 |

|
>50 | 39 |
|
>50 |
| 32 |
|
3.0 ± 0.5 | 40 |
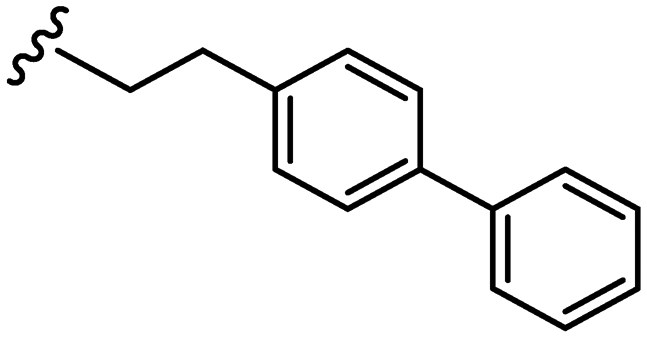
|
1.8 ± 0.1 |
| 33 |
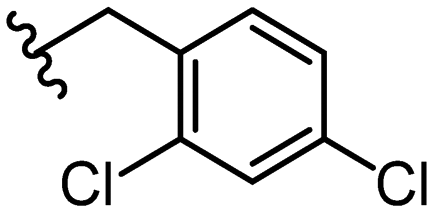
|
>50 | 41 |
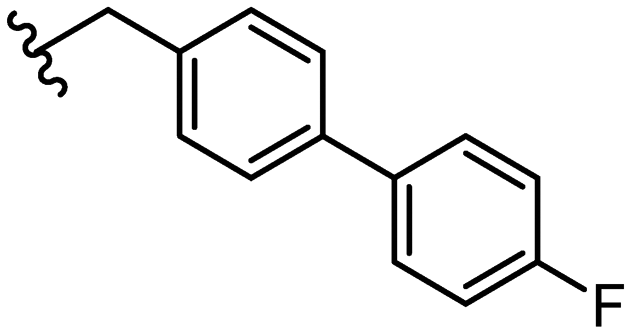
|
1.4 ± 0.1 |
| 34 |
|
1.6 ± 0.2 | 42 |
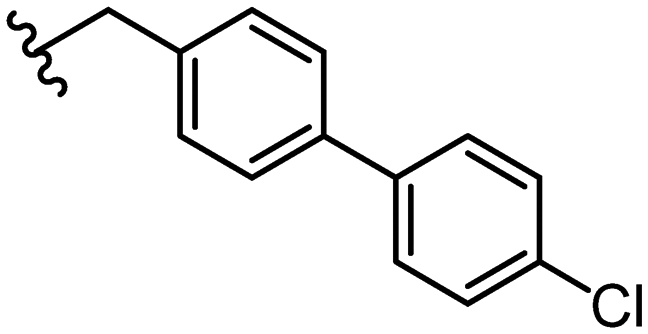
|
1.3 ± 0.3 |
| 21 |
|
3.2 ± 0.7 | 43 |
|
1.8 ± 0.4 |
| 44 |
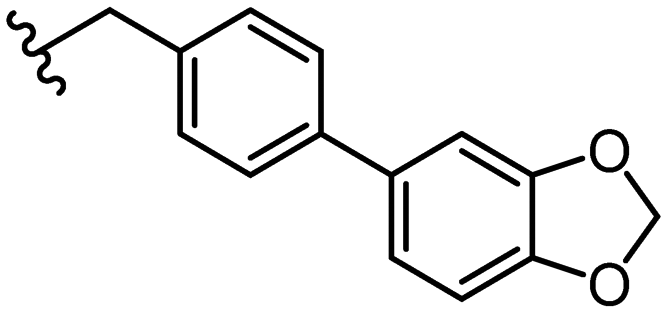
|
1.4 ± 0.5 | |||
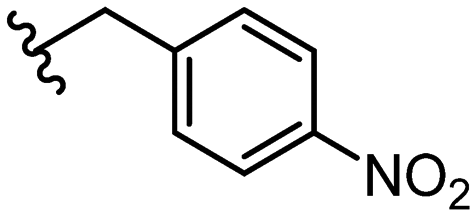
Due to the favourable binding of para-substituted lipophilic phenyl derivatives (7 and 21), we decided to investigate the SAR in this region further (27–32). Lipophilic substituents (27–29) performed better than less lipophilic substituents (30 and 31). Interestingly, chloro derivative 28 was significantly less potent than both methyl (27) and bromo derivatives (29), suggesting a stronger role for electrostatic interactions than observed in earlier data. Compounds 30 and 31 exhibit significantly lower potencies than their lipophilic counterparts, which reflects previous results (9) (Table 1). Yet, 32 possesses a nitro group and shows comparable potency to compounds with lipophilic substituents. This strengthens the hypothesis that there may be an electrostatic component to binding in this region of the active site. The addition of a second –Cl to the ring (33) results in a complete loss of potency. However, extension of the methylene linker to an ethylene linker results in inhibitory activity being regained, with other ethylene-based compounds (21, 35–37) exhibiting equipotency. The additional flexibility and length likely allows the aromatic rings to engage more favourably with hydrophobic aromatic residues (Ile161, Phe194, and Phe369) within the P1 pocket of the active site.29 Past success with adamantane derivatives of BoNT/A1 LC active site inhibitors led us to synthesize 38 and 39. In this instance, the ethylene linker (39) resulted in a complete loss in activity while the methylene derivative (38) remained relatively potent. We thus concluded that the length and flexibility of this appendage has an optimum range, and that the pi-interactions of the aryl groups are likely important. Compounds 40–44 possess linear bi-aryl systems, all of which show equipotent single-digit μM activity and overall flat SAR.
Mechanism of inhibition
In the absence of crystallographic evidence, we sought to verify the mechanism of inhibition of this series of compounds by other means. The SNAPtide assay employed in this inhibitor discovery campaign makes use of a truncated substrate based only on the active-site binding sequence of SNAP-25, which does not interact with either of the two exo-sites that are necessary for SNAP-25 substrate binding.31–33 Furthermore, the 3,4-HOPTO scaffold is a MBG, designed to target the active-site zinc. These two pieces of evidence suggest that the 3,4-HOPTO series of inhibitors are active-site inhibitors. In order to probe the mechanism of inhibition, compound 40 was evaluated in a competition assay against 2,4-dichlorocinnamic hydroxamate (DCHA), a well-characterized active-site inhibitor with a zinc-chelation mechanism that has been confirmed through X-ray crystallography.29,34 Compounds 40 and DCHA were simultaneously incubated at various concentrations with the BoNT/A1 LC in the SNAPtide assay. A two-inhibitor binding model (Fig. 2A and B) was used to fit the inhibition curves (Fig. 2C) to determine how the presence of DCHA impacts the BoNT/A1 LC inhibition profile of compound 40. When the inhibitors are mutually exclusive, i.e. only one of the two can be bound at one time, α is equal to 0. Conversely, when the inhibitors are non-mutually exclusive, α is unconstrained. The extra-sum-of-squares F-test was used to compare the two cases, finding that the mutually exclusive model was preferred (Table S1†). This indicates that it is very unlikely that 40 can inhibit the BoNT/A1 LC at the same time as DCHA. From this evidence, in tandem with the presence of an MBG and its inhibitory activity in the SNAPtide assay, we can conclude that the mechanism of BoNT/A1 LC inhibition by 3,4-HOPTO compounds is most likely competitive active-site inhibition. | ||
| Fig. 2 Competition assay between 40 and DCHA. A) An enzyme model for substrate catalysis in the presence of two inhibitors. B) Equation derived from the two-inhibitor model. C) Mutually exclusive model curve fitting. Each data set represents a different concentration of DCHA, as indicated by the legend. | ||
Target specificity
Target specificity is a major concern for all small molecules that employ an MBG mechanism of inhibition. The 3,4-HOPTO scaffold itself (26) and related structures (7e, 12e, Fig. S1†) have been shown to be relatively promiscuous for several matrix metalloproteinases (MMPs).24 Compound 41 was thus screened on a panel of MMPs to evaluate its target selectivity (Fig. 3). Gratifyingly, we found that, with the exception of MMP-9, the compound was at least two-fold more selective for BoNT/A LC than the other enzymes. MMP-9 is implicated in regulation, inflammation, and restructuring of the extracellular matrix, and is a prolific target for the treatment of cancers and inflammatory disease.35–37 The complexity of the protease web makes it difficult to predict the outcome of MMP-9 inhibition, though off-target effects associated with long-term dosing of broad-spectrum MMP inhibitors include musculoskeletal syndrome and gastrointestinal disorders.38,39 However, the ultimate goal of our inhibitor discovery campaign is irreversible inhibition of the BoNT/A1 LC to alleviate symptoms of botulism, which is non-regenerative and should therefore only require a short dosing regimen. Additionally, the eventual addition of a proximity-based Cys-reactive warhead should impart an additional degree of selectivity.15 Thus, we expect that for our specific application, off-target effects associated with MMP-9 inhibition by 40 will be minimal.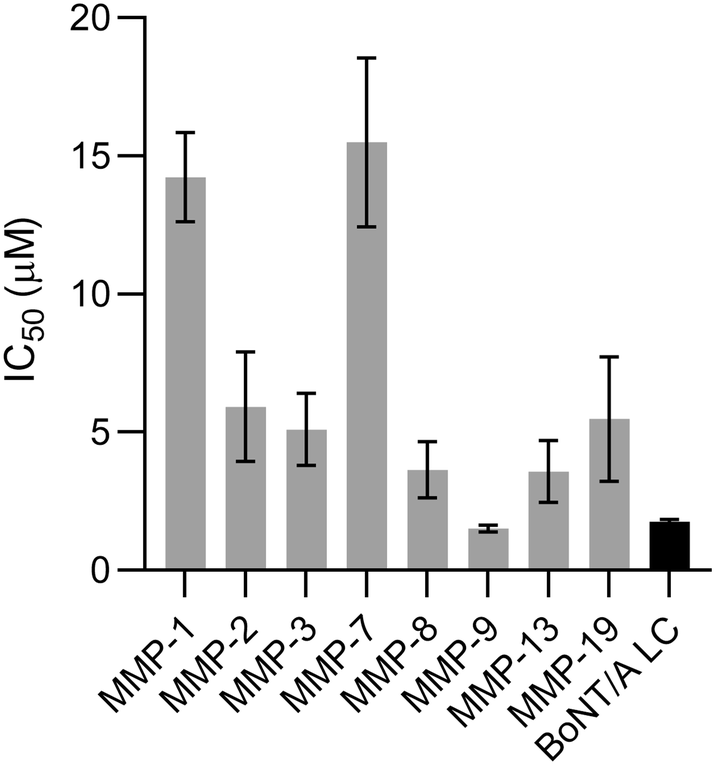 | ||
| Fig. 3 IC50 data of 40 when tested against various MMPs. IC50 values are reported as mean ± SD (n = 3) and can be found in Table S2.† | ||
Cellular evaluation
The promising inhibitory potency of the synthesized 3,4-HOPTO derivatives prompted evaluation in a cell-based assay which measures protection of SNAP-25 cleavage from the BoNT/A1 LC in the native intracellular compartment. Representative compounds 32, 21, 40–44 were selected based on the BoNT/A1 LC inhibition data presented in Table 2. Compounds were evaluated on human-induced pluripotent stem cell (hIPSC) derived neurons that had been pre-exposed to BoNT/A1, and SNAP-25 cleavage quantified by Western blot Table 3.40|
|
|||||||
|---|---|---|---|---|---|---|---|
| Cpd no. | R group | BoNT/A LC IC50a (μM) | hIPSC IC50a (μM) | Cpd no. | R group | BoNT/A LC IC50a (μM) | hIPSC IC50a (μM) |
| a IC50 values are reported as mean ± SD (n = 3). b SD not calculated due to no variation in fit value. | |||||||
| 32 |
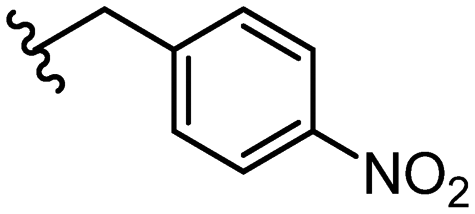
|
3.1 ± 0.5 | 100 ± 9 | 42 |
|
1.3 ± 0.3 | 31 ± 3 |
| 34 |

|
1.6 ± 0.2 | ∼200 | 43 |
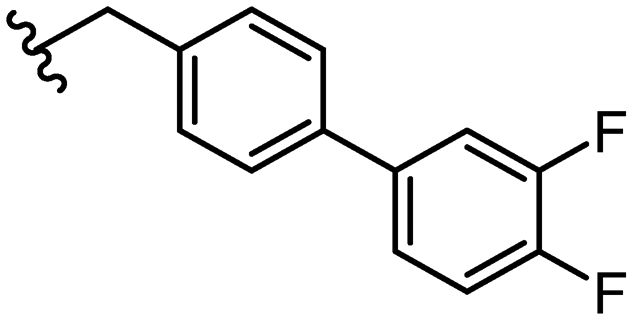
|
1.8 ± 0.4 | 39 ± 0b |
| 40 |

|
1.8 ± 0.1 | 13 ± 2 | 44 |
|
1.4 ± 0.5 | 51 ± 4 |
| 41 |
|
1.4 ± 0.1 | 33 ± 6 | ||||

Unsurprisingly, an overall decrease in potency was observed between enzyme- and cell-based IC50 values. In particular, poor cell-permeation due to the charged nature of the nitro group of 32 is likely a crucial factor in its poor inhibitory potency, although the lack of SNAP-25 protection afforded by 34 is unexplained. As seen in the enzyme IC50 data, there is a limited SAR trend between compounds 34 and 40–44. Interestingly, compound 40 exhibits the best protection against SNAP-25 cleavage, between 2- to 3-fold better than the other biphenyl derivatives 41–44 and only suffered a 7-fold reduction in cell potency compared to the enzyme assay. Despite possessing similar enzyme-potencies, 40 and 32, have drastically different cell potencies. It is therefore likely that any putative cell permeability issues are not related to the 3,4-HOPTO scaffold itself, and that potency in the cell assay may be tuned by improving physicochemical properties.
Conclusion
In summary, 3,4-HOPTO derivatives were identified as BoNT/A1 LC inhibitors through a screen of 96 metal binding fragments. A subsequent screen of a 3,4-HOPTO sub-library outlined SARs based on N-substitution of the 3,4-HOPTO core. Synthesis of 18 additional compounds identified several derivatives exhibiting low micromolar activity against the BoNT/A1 LC, with active-site mechanism of inhibition being confirmed through competition studies with DCHA. Compound 40 showed reasonable selectivity for BoNT/A1 LC over the majority of matrix metalloproteinases. Several inhibitors exhibited modest cellular activity, indicating good cell permeability, lipophilicity, and aqueous solubility. Overall, the 3,4-HOPTO scaffold is a compelling candidate for further development as a bifunctional covalent inhibitor of BoNT/A1 LC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Seth M. Cohen at University of California San Diego for the use of the 3,4-HOPTO sub-library and William H. Tepp at University of Wisconsin-Madison for producing the purified BoNT/A1 used in the cell-based assays. Research was supported by the National Institutes of Health grants R01 AI119564 (to KDJ), the Natural Sciences and Engineering Research Council of Canada (NSERC) grant PGSD3-502274 (to LL), and the Skaggs Institute for Chemical Biology (to LL). This is manuscript #30042 from the Scripps Research Institute.References
- A. Cocco and A. Albanese, Toxicon, 2018, 147, 77–83 CrossRef CAS.
- E. J. Schantz and E. A. Johnson, Microbiol. Rev., 1992, 56, 80 CrossRef CAS.
- E. Yiannakopoulou, Pharmacology, 2015, 95, 65–69 CrossRef CAS.
- S. S. Arnon, R. Schechter, T. V. Inglesby, D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. D. Fine, J. Hauer, M. Layton, S. Lillibridge, M. T. Osterholm, T. O'Toole, G. Parker, T. M. Perl, P. K. Russell, D. L. Swerdlow and K. Tonat, JAMA, 2001, 285, 1059–1070 CrossRef CAS.
- C. F. D. C. A. P. (CDC), Botulism Annual Summary, 2016, Center for Disease Control and Prevention (CDC), Atlanta, Georgia, 2017 Search PubMed.
- C. O. Tacket, W. X. Shandera, J. M. Mann, N. T. Hargrett and P. A. Blake, Am. J. Med., 1984, 76, 794–798 CrossRef CAS.
- L. Simpson, Toxicon, 2013, 68, 40–59 CrossRef CAS.
- F. H. Al-Saleem, Z. Nasser, R. M. Olson, L. Cao and L. L. Simpson, J. Pharmacol. Exp. Ther., 2011, 338, 503 CrossRef CAS.
- R. St John, B. Finlay and C. Blair, Can. J. Infect. Dis. Med. Microbiol., 2001, 12, 275–284 Search PubMed.
- L. M. Wein and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9984 CrossRef CAS.
- R. Pellizzari, O. Rossetto, G. Schiavo and C. Montecucco, Philos. Trans. R. Soc., B, 1999, 354, 259–268 CrossRef CAS.
- G. Schiavo, M. Matteoli and C. Montecucco, Physiol. Rev., 2000, 80, 717–766 CrossRef CAS.
- L. K. Koriazova and M. Montal, Nat. Struct. Biol., 2002, 10, 13 CrossRef.
- J. Rizo and T. C. Südhof, Nat. Rev. Neurosci., 2002, 3, 641 CrossRef CAS.
- L. Lin, M. E. Olson, T. Sugane, L. D. Turner, M. A. Tararina, A. L. Nielsen, E. K. Kurbanov, S. Pellett, E. A. Johnson, S. M. Cohen, K. N. Allen and K. D. Janda, J. Med. Chem., 2020, 63, 11100–11120 CrossRef CAS.
- S. Kitamura, K. Sugihara and K. Tatsumi, Biochem. Mol. Biol. Int., 1994, 34, 1197–1203 CAS.
- L. Du, D. G. Musson and A. Q. Wang, J. Pharm. Biomed. Anal., 2006, 42, 556–564 CrossRef CAS.
- P. Hermant, D. Bosc, C. Piveteau, R. Gealageas, B. Lam, C. Ronco, M. Roignant, H. Tolojanahary, L. Jean, P.-Y. Renard, M. Lemdani, M. Bourotte, A. Herledan, C. Bedart, A. Biela, F. Leroux, B. Deprez and R. Deprez-Poulain, J. Med. Chem., 2017, 60, 9067–9089 CrossRef CAS.
- A. Agrawal, S. L. Johnson, J. A. Jacobsen, M. T. Miller, L.-H. Chen, M. Pellecchia and S. M. Cohen, ChemMedChem, 2010, 5, 195–199 CrossRef CAS.
- L. Lin, M. E. Olson, L. M. Eubanks and K. D. Janda, Acc. Chem. Res., 2019, 52, 2322–2331 CrossRef CAS.
- P. T. Bremer, M. Adler, C. H. Phung, A. K. Singh and K. D. Janda, J. Med. Chem., 2017, 60, 338–348 CrossRef CAS.
- D. Caglič, M. C. Krutein, K. M. Bompiani, D. J. Barlow, G. Benoni, J. C. Pelletier, A. B. Reitz, L. L. Lairson, K. L. Houseknecht, G. R. Smith and T. J. Dickerson, J. Med. Chem., 2014, 57, 669–676 CrossRef.
- V. Roxas-Duncan, I. Enyedy, V. A. Montgomery, V. S. Eccard, M. A. Carrington, H. Lai, N. Gul, D. C. Yang and L. A. Smith, Antimicrob. Agents Chemother., 2009, 53, 3478–3486 CrossRef CAS.
- J. A. Jacobsen, J. L. Fullagar, M. T. Miller and S. M. Cohen, J. Med. Chem., 2011, 54, 591–602 CrossRef CAS.
- A. L. Garner, A. K. Struss, J. L. Fullagar, A. Agrawal, A. Y. Moreno, S. M. Cohen and K. D. Janda, ACS Med. Chem. Lett., 2012, 3, 668–672 CrossRef CAS.
- J. A. Lewis, J. Mongan, J. A. McCammon and S. M. Cohen, ChemMedChem, 2006, 1, 694–697 CrossRef CAS.
- P. Šilhár, N. R. Silvaggi, S. Pellett, K. Čapková, E. A. Johnson, K. N. Allen and K. D. Janda, Bioorg. Med. Chem., 2013, 21, 1344–1348 CrossRef.
- N. R. Silvaggi, D. Wilson, S. Tzipori and K. N. Allen, Biochemistry, 2008, 47, 5736–5745 CrossRef CAS.
- N. R. Silvaggi, G. E. Boldt, M. S. Hixon, J. P. Kennedy, S. Tzipori, K. D. Janda and K. N. Allen, Chem. Biol., 2007, 14, 533–542 CrossRef CAS.
- G. R. Flematti, E. D. Goddard-Borger, D. J. Merritt, E. L. Ghisalberti, K. W. Dixon and R. D. Trengove, J. Agric. Food Chem., 2007, 55, 2189–2194 CrossRef CAS.
- J. J. Schmidt and R. G. Stafford, US Pat., US201202754A1, 2012 Search PubMed.
- M. A. Breidenbach and A. T. Brunger, Nature, 2004, 432, 925–929 CrossRef CAS.
- S. Chen, J.-J. P. Kim and J. T. Barbieri, J. Biol. Chem., 2007, 282, 9621–9627 CrossRef CAS.
- G. E. Boldt, J. P. Kennedy and K. D. Janda, Org. Lett., 2006, 8, 1729–1732 CrossRef CAS.
- G. V. Halade, Y. F. Jin and M. L. Lindsey, Pharmacol. Ther., 2013, 139, 32–40 CrossRef CAS.
- A. Yabluchanskiy, Y. Ma, R. P. Iyer, M. E. Hall and M. L. Lindsey, Physiology, 2013, 28, 391–403 CrossRef CAS.
- S. Mondal, N. Adhikari, S. Banerjee, S. A. Amin and T. Jha, Eur. J. Med. Chem., 2020, 194, 112260 CrossRef CAS.
- B. G. Fields, Cells, 2019, 8, 984 CrossRef.
- M. A. Rudek, J. Venitz and W. D. Figg, Pharmacotherapy, 2002, 22, 705–720 CrossRef CAS.
- R. C. M. Whitemarsh, M. J. Strathman, L. G. Chase, C. Stankewicz, W. H. Tepp, E. A. Johnson and S. Pellett, Toxicol. Sci., 2012, 126, 426–435 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00320d |
| ‡ Present address: 217 Kameničany, 01854 Slovakia. |
| This journal is © The Royal Society of Chemistry 2021 |