
Towards a RIOK2 chemical probe: cellular potency improvement of a selective 2-(acylamino)pyridine series†
Christopher B.
Wang
 ab,
Álvaro
Lorente-Macías
c,
Carrow
Wells
ab,
Julie E.
Pickett
ab,
Alfredo
Picado
ab,
William J.
Zuercher
abd and
Alison D.
Axtman
*ab
ab,
Álvaro
Lorente-Macías
c,
Carrow
Wells
ab,
Julie E.
Pickett
ab,
Alfredo
Picado
ab,
William J.
Zuercher
abd and
Alison D.
Axtman
*ab
aStructural Genomics Consortium, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: alison.axtman@unc.edu
bDivision of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
cDepartamento de Química Farmacéutica y Orgánica, University of Granada, Granada, 18071, Spain
dLineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
First published on 11th November 2020
Abstract
RIOK2 is an understudied kinase associated with a variety of human cancers including non-small cell lung cancer and glioblastoma. No potent, selective, and cell-active chemical probe currently exists for RIOK2. Such a reagent would expedite re-search into the biological functions of RIOK2 and validate it as a therapeutic target. Herein, we describe the synthesis of naphthyl–pyridine based compounds that have improved cellular activity while maintaining selectivity for RIOK2. While our compounds do not represent RIOK2 chemical probes, they are the best available tool molecules to begin to characterize RIOK2 function in vitro.
Introduction
Right open reading frame kinase 2 (RIOK2) is an atypical kinase involved in maturation of the small ribosome subunit (40S) and cell cycle progression.1 While it contains a kinase motif and demonstrates the ability to hydrolyze ATP in vitro, RIOK2 and its associated RIO kinase family (RIOK1/3) differ from typical eukaryotic protein kinases due to a lack of conserved substrate binding domains and activation loop motifs.2,3 This difference, combined with a lack of a known substrate has led to the suggestion that RIOK2 serves as an ATPase rather than a kinase.4 Regardless, RIOK2 acts as a necessary factor in the maturation and export pathway of ribosomal RNA through interactions with the nuclear export protein CRM1 and the RNA-binding protein NOB1.5 Its function in ribosome biogenesis is also postulated to involve the recycling of the necessary trans-factor proteins involved in 40S-ribosome maturation, with in vitro studies showing a dependence on catalytic activity for proper functioning.4 RIOK2 also coordinates with PLK1 in regulating mitotic progression and with mTOR complexes in increasing oncogenic AKT signaling; demonstrating the dual role of RIOK2 in both cell cycle regulation and ribosome biogenesis.6,7 While RIOK2 has been implicated in these cellular pathways, a full understanding of these and other biological functions mediated by this understudied kinase remain to be fully elucidated.2,8 The lack of characterization around RIOK2 made it eligible for inclusion on the illuminating the druggable genome (IDG) list of dark kinases.9Clinically, RIOK2 is overexpressed in multiple cancers that carry poor prognoses, including non-small cell lung cancer (NSCLC) and glioblastoma (GB). The overexpression of RIOK2 in these cancers is associated with increased mortality, poor clinical outcomes, and lymph node metastasis.10,11 The increased expression of RIOK2 in these cancers may be functionally relevant as knockdown of RIOK2 via shRNA and miRNA was found to decrease both viability and migration for both glioblastoma and NSCLC tumor cells.10,12 A selective RIOK2 chemical probe would enable further characterization of the effects of RIOK2 inhibition and explore its potential as a therapeutic target in cancer and other indications.
Although a handful of potent RIOK2 inhibitors have been identified by high-throughput screening (HTS) campaigns, none have been selective for RIOK2.13,14 However, a recent publication identified naphthyl–pyridine–amide compound 1 as having submicromolar biochemical binding affinity for RIOK2 (Kd = 160 nM). 1 bound only 11 other kinases with a Kd < 3 μM out of a panel of 456 kinase binding assays.15 In contrast, sunitinib and midostaurin, two additional published inhibitors with high affinity for RIOK2, were reported in the same assay to bind more than 190 kinases with a Kd < 3 μM (Fig. S1†). Thus, inhibitor 1 is considered the most selective RIOK2 inhibitor reported to date.15
To determine the utility of 1 as a chemical tool to characterize RIOK2 biological function, the cellular penetrance and RIOK2 engagement in cells by inhibitor 1 was explored via a bioluminescence resonance energy transfer assay that employs nano-luciferase (NanoBRET).16 Our RIOK2 NanoBRET assay represents the first and only available cell-based assay to directly assess RIOK2 affinity. Briefly, HEK293 cells were transfected with a vector containing RIOK2 fused to a nanoluciferase (NLuc) and then incubated with a cell-permeable fluorescent energy transfer tracer capable of binding to RIOK2. By increasing the concentration of test compounds, dose-dependent displacement of the tracer from the RIOK2–NLuc fusion could be observed, allowing for the determination of an IC50 value from the quantifiable changes in BRET signal. We found that the potent biochemical binding affinity of 1 was not recapitulated in this cellular milieu (IC50 = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 nM) (Fig. 1). With the goal of increasing the cellular potency of 1, we designed analogues to further explore SAR resulting from changes to the naphthyl–pyridine–amide scaffold.
600 nM) (Fig. 1). With the goal of increasing the cellular potency of 1, we designed analogues to further explore SAR resulting from changes to the naphthyl–pyridine–amide scaffold.
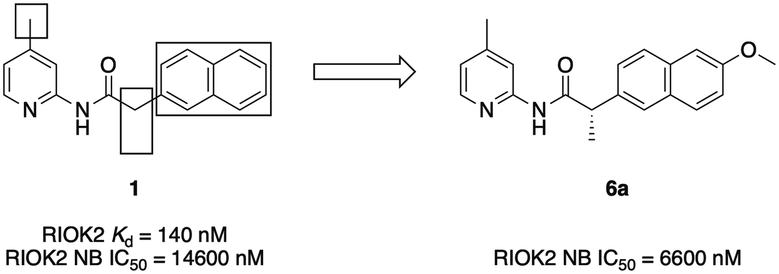 | ||
| Fig. 1 Structure of inhibitor 1 with areas of SAR exploration boxed alongside lead analogue 6a. | ||
The poor cellular potency of 1 in the NanoBRET assay relative to its in vitro binding affinity could be attributed to several different potential factors. These include low cellular permeability in general or specifically to the cellular location of RIOK2 within HEK293 cells, competition with endogenous ATP levels, or even partial cellular degradation via hydrolysis of the amide. We carried out the NanoBRET assay in permeabilized cells (treated with digitonin) as well as in ATP-depleted cells (treated with rotenone), neither of which impacted the observed cellular IC50 value, providing evidence that general cellular permeation and ATP competition were likely not the basis for the poor cellular activity observed (Fig. S2†).14 Instead, we postulated that the discrepancy between the in vitro binding and NanoBRET potencies might be an inherent difference between the DiscoverX binding and NanoBRET cellular assays and not specific to the compounds tested.14 To support this possibility, several other reported RIOK2 inhibitors built upon different scaffolds with potent biochemical Kd/IC50 values, including promiscuous compounds, failed to manifest IC50 values <1 μM in the RIOK2 NanoBRET assay (data not shown). Based on our result with 1, all analogues moving forward were tested at a concentration up to 30 μM in the RIOK2 NanoBRET assay.
Study design
Analogues were designed based upon SAR developed through analysis of the published library as well as through structure-guided design using a published crystal structure of RIOK2 (PDB code 6HK6).17 Analysis of the human RIOK2 structure co-crystalized with a derivative of 1 (PDB 6HK6) led to three observations: i) the pyridine ring and amide nitrogen form hydrogen bonds to the main chain portion of I191, ii) the para position substituent on the pyridine ring faces a small hydrophobic pocket close to the I245 side chain, and iii) hydrophobic interactions exist between the naphthyl ring and the adjacent side chains I111 and M101 (Fig. 2).17 It is important to note that when this structure was solved, RIOK2 crystallized as a homodimer in eight of ten molecules within the asymmetric unit, allowing for interactions between the co-crystalized ligand and residues from an adjacent RIOK2 unit (H200 or K45). While a recent report suggests RIOK2 may exist as an inactive homodimer, the residues known to bind ATP are blocked when two RIOK2 monomers assemble.18 Based on the precluded ATP site in the homodimeric RIOK2 structure, the monomeric RIOK2 structure should be used to guide analogue design. Accordingly, we did not use these interactions between RIOK2 monomers when designing compounds herein.17 To maintain the key hydrogen bonds to the kinase hinge region, we did not alter the core pyridine–amide when substituting various other portions of the scaffold of 1 (Fig. 1). The observation that the binding pocket immediately adjacent to the pyridine was limited in volume led to the choice of attaching only small substituents to the pyridine ring. Furthermore, the depth of this side pocket was explored through extending the chain connecting the pyridine and naphthyl portions of the molecule. A variety of substituents that differ in size, hydrogen-bonding potential and electronics were attached at the 4-position of the pyridine based upon the observation that this position has the potential to interact with the pocket and might tune the activity. Finally, based upon the largely hydrophobic nature of the portion of the pocket that accommodates the naphthyl ring, hydrophobicity was considered an asset when considering replacements for this core.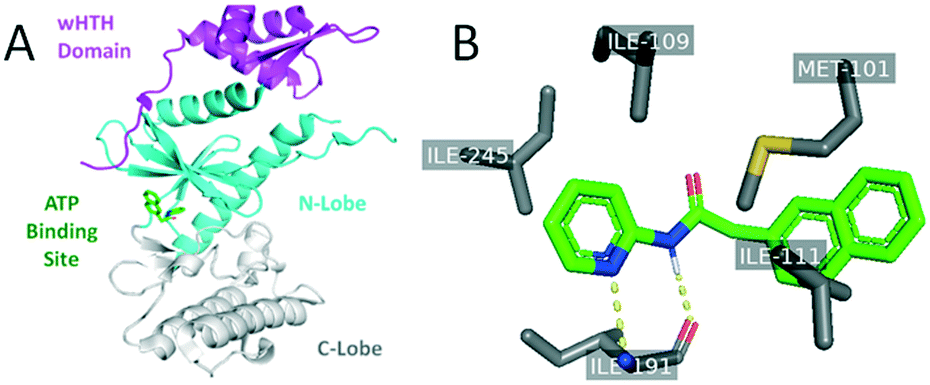 | ||
| Fig. 2 Published co-crystal structure of RIOK2 in complex with an analogue of 1 (PDB: 6HK6). Panel A is color-coded to identify the RIOK2 wHTH domain (purple), N-lobe (cyan), and C-lobe (grey). Panel B shows specific protein–ligand interactions identified between RIOK2 and its co-crystallized ligand. | ||
Analogues were prepared using two different routes. Scheme 2 was only employed when compound preparation was found to be refractory to Scheme 1 conditions. Compounds 1 and 6–14 were prepared via HATU-mediated amide coupling (Scheme 1). Analogues 2, 3, 4, and 5 were synthesized through conversion of the starting acid to the corresponding acid chloride, followed by amide formation (Scheme 2). These conditions were found to be quite robust, as yields were generally high (>70%).
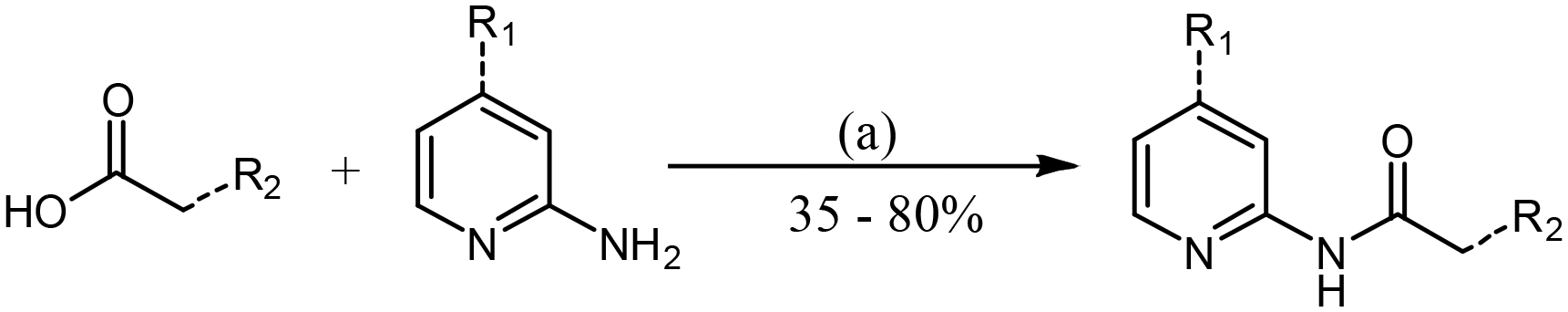 | ||
| Scheme 1 General procedure for the synthesis of 2-amide–pyridines via HATU amide coupling. Reagent and conditions: (a) HATU, DIPEA, DMF, RT, 12 h. | ||
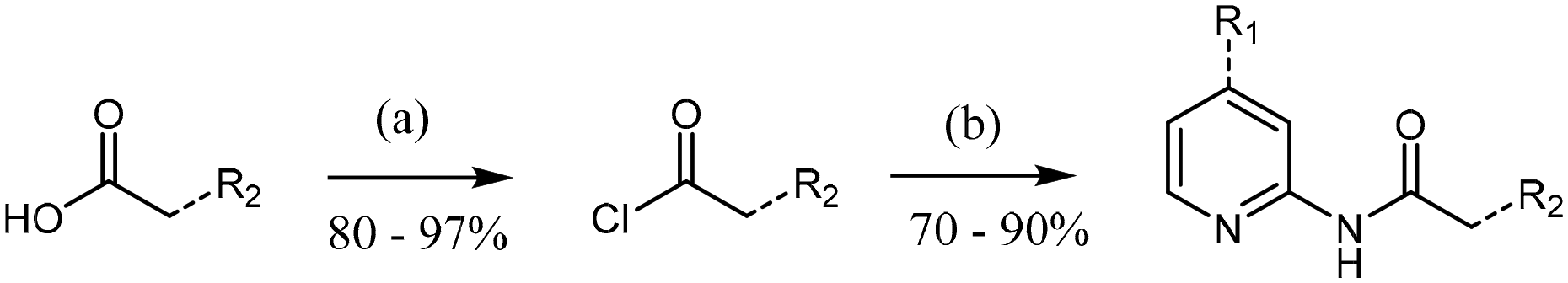 | ||
| Scheme 2 General procedure for the synthesis of 2-amide-pyridines via an acyl chloride intermediate. Reagent and conditions: (a) SOCl2, CH2Cl2, 0 °C, 3 h; (b) aminopyridine, CH2Cl2, RT, 12 h. | ||
Results and discussion
Several compounds demonstrated improved potency relative to the parent 1. The addition of a trifluoromethyl (2), methoxy (3), or chloro (5) group on the 4-position of the pyridine ring [R1] increased activity, while ethyl (10) and bromo (4) substituents did not. We propose that the electron-withdrawing effects of these substituents increase the acidity of the amide hydrogen and promote its ability to form a hydrogen bond with the main chain carbonyl of I191. The enhanced potency of 3, which bears an electron-donating methoxy group on the pyridine, is hypothesized to result from both increasing the basicity of the pyridine ring and potentially promoting the formation of a hydrogen bond with an additional main-chain amide. The similar activity of the 4-bromo (4) and 4-ethyl (10) pyridines to parent compound 1 suggests that incorporation of a larger group, especially one that cannot participate in hydrogen bonding, does not improve affinity.Variation of the linker length (12) or the inclusion of heteroatoms in the linker (13) resulted in loss of RIOK2 affinity. Furthermore, replacing the naphthalene ring [R2] with a benzodioxane (7), benzodioxole (8), or dimethyl–phenyl (11) ring caused a loss in activity. We hypothesized that these structural changes could abolish interactions with the M101 and I111 side chains of RIOK2, which favor a methylene linker and hydrophobic bicyclic ring structure. Hydrophobic residues, including M101 and I111, at the entrance to the ATP-binding site make key interactions with the naphthalene ring, which are proposed to be lost when changing the core in 7, 8, and 11 to less hydrophobic systems. Interactions with these residues are not only important for binding affinity, but also are proposed to contribute to the selectivity of the naphthalene-based analogues for RIOK2 over its RIOK family members since M101 and I111 are found in RIOK2 but not in RIOK1 or RIOK3 (I101/H111).17 It is also possible that extension of the linker length results in insertion of the pyridine ring into the binding pocket that cannot be accommodated, resulting in steric clash and decreased binding potency. In contrast, ring substitution with a biphenyl analogue (14) maintained activity. While a prior publication demonstrated that a 4-methyl biphenyl analogue demonstrated three-fold lower binding affinity than its naphthyl counterpart, analogue 14 had similar potency to its naphthyl complement 3. We propose that this could be due to the 4-methoxy on the pyridine ring causing a change in the binding mode, resulting in favorable interactions between the biphenyl ring and RIOK2 hydrophobic residues. Due to ease of synthesis, further development of analogues containing this biphenyl ring offer a promising alternative scaffold to the naphthyl ring.
The addition of both an alpha-methyl adjacent to the carbonyl [R3] and 6-methoxy on the naphthyl ring [R2] (6) results in a significant potency gain versus the parent compound. There is no potency difference between the (S) and (R) alpha-methyl enantiomers 6a and 6b, respectively. There is no additive effect observed when a 4-methoxy is appended to the pyridine ring [R1] of the (S) alpha-methyl/6-methoxy analogue (9). The improved activity that results from addition of both an alpha-methyl and 6-methoxy (3) is likely due to the alpha-methyl rather than the 6-methoxy substitution. Assuming a similar binding pose to 1, the 6-methoxy on the naphthalene ring of 6 would project towards solvent. No difference in potency was observed between the racemic 6, (S) 6a and (R) 6b alpha-methyl analogues. We propose that the same interaction that is made by the racemic alpha-methyl analogue is maintained by the enantiomers, possibly due to free rotation of this bond. Furthermore, the lack of additional potency observed from the 4-methoxy-pyridine (S)-alpha-methyl enantiomer (9) led us to hypothesize that 9 may bind in a different pose than 3.
To assess if substitutions of the core scaffold of 1 affected selectivity against RIOK2, activity of analogues with improved cellular potency were tested against off-target kinases known to bind 1. RIOK2 itself was not included in this study as no RIOK2 enzymatic assay is currently offered by a commercial vendor. Efforts to collaboratively develop an activity assay using full-length RIOK2 protein have not yet been successful, partially since RIOK2 substrates have not yet been identified. The DiscoverX RIOK2 binding assay is the source for the majority of literature data generated for this understudied kinase. While 1 was shown to have high selectivity for RIOK2 in the DiscoverX assay, this activity was not fully replicated in an orthogonal enzymatic kinase inhibition assay at Eurofins (Table 2). This finding demonstrates the importance of evaluating compounds using different assay formats to minimize the contributions from assay-specific factors. While the DiscoverX assay relies on active site-directed competition to assess binding in a heterogeneous format, the Eurofins assay measures incorporation of radiolabeled ATP into a peptide substrate to assess enzymatic activity homogenously. Furthermore, the DiscoverX platform is ATP-free, while all radiometric enzymatic assays were carried out at the Km of ATP. Finally, different protein constructs of the same kinase are used in each respective assay.
|
|
||||
|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | RIOK2 NanoBRET IC50 values (μM) |
| a IC50 values >10 μM are results obtained from n = 1 determination. b IC50 values are results obtained from n = 3 determinations. c IC50 values are results obtained from n = 4 determinations. | ||||
| 1 | CH3 | 2-Naphthalene | H | 14.6a |
| 2 | CF3 | 2-Naphthalene | H | 7.5 ± 0.62c |
| 3 | OCH3 | 2-Naphthalene | H | 5.2 ± 0.43c |
| 4 | Br | 2-Naphthalene | H | 13.4a |
| 5 | Cl | 2-Naphthalene | H | 5.0 ± 0.79c |
| 6 | CH3 | 2-[6-Methoxy-naphthalene] | CH3 (rac.) | 5.2 ± 1.03c |
| 6a | CH3 | 2-[6-Methoxy-naphthalene] | CH3 (S) | 6.6 ± 0.48b |
| 6b | CH3 | 2-[6-Methoxy-naphthalene] | CH3 (R) | 7.6 ± 0.35b |
| 7 | CH3 | 6-[1,4-Benzodioxane] | H | >30a |
| 8 | CH3 | 5-[1,3-Benzodioxole] | H | >30a |
| 9 | OCH3 | 2-[6-Methoxy-naphthalene] | CH3 (S) | 5.3 ± 0.86b |
| 10 | CH2CH3 | 2-Naphthalene | H | 14.5a |
| 11 | OCH3 | 2-[3,4-Dimethyl-phenyl] | H | 28a |
| 12 | CH3 | CH2-[2-Naphthalene] | H | >30a |
| 13 | CH3 | 2-Naphthol | H | >30a |
| 14 | OCH3 | 2-(4-Biphenyl) | H | 7.6 ± 0.79b |

| Off-target kinase | Binding affinities Kd (nM) | Enzymatic kinase activity assay values IC50 (nM) |
|---|---|---|
| All IC50 values are results obtained from n = 2 determinations. N.D. not determined. | ||
| RIOK2 | 140 | N.D. |
| MAPK8 | 220 | >10000 |
| MAPK10 | 340 | 4700 |
| GSK3α | 870 | 5200 |
| SNRK | 890 | >10000 |
| HIPK1 | 1100 | >10000 |
We hypothesize that the hydrophobic nature of 1 allows it to bind tightly into hydrophobic grooves in the kinases listed in Table 2. This high affinity binding, however, does not translate to potent inhibition of kinase function. While a complete loss of activity was observed for MAPK8, SNRK, and HIPK1 in the respective enzymatic assays, a 14-fold and 6-fold decrease was seen with MAPK10 and GSK3α, respectively. The micromolar inhibitory activity of MAPK10 and GSK3α by compound 1 in corresponding enzymatic assays demonstrates that this compound is not exquisitely selective. MAPK10 and GSK3α are two of the kinases that had reported binding affinities within 8-fold of RIOK2 in the original publication.15 Several other off-target kinases reported to have binding affinities within 8-fold of RIOK2 in the original publication (MAPK8, SNRK, and HIPK1), however, were found to be inactive in the corresponding enzymatic assays.15 Data in Table 2 refine the reported selectivity profile of compound 1, confirming that some of the proposed off-target kinases are not inhibited in an enzymatic assay format while others are inhibited in the micromolar potency range. Our aspirational goal for a RIOK2 chemical probe is to deliver a compound with nanomolar RIOK2 inhibitory activity such that there would be a large potency window between RIOK2 and off-target kinases. With respect to off-target kinases, it is important to note that kinases from the RIOK family differ significantly from most typical protein kinases, including MAPK10 and GSK3α, and that these differences in ATP binding site and substrate binding residues can be exploited to improve selectivity. Versus other RIOK2 inhibitors in the literature and included in our introduction (sunitinib and midostaurin), 1 remains the most selective RIOK2-targeting compound and the 2-(acylamino)pyridine scaffold is suggested to be the best choice for chemical optimization toward a RIOK2 chemical probe.
The most potent inhibitors in the RIOK2 NanoBRET assay were tested against the same panel of off-target kinase assays alongside 1 (Table 3). None of the compounds showed any activity with MAPK8, SNRK, and HIPK1 at the concentration range used in the respective enzymatic assays. On the other hand, some substitutions that improved cellular potency for RIOK2 also increased inhibition of MAPK10 and GSK3α. Structural modifications resulted in nuanced changes in selectivity. Relative to 1, the chloride substitution in analogue 5 increased inhibition potency of both MAPK10 and GSK3α, while the methoxy in compound 3 resulted in enhanced inhibition potency of MAPK10 only. Interestingly, the addition of an alpha-methyl and 6-methoxy on the naphthyl ring in compound 6 resulted in more potent inhibition only in the case of the racemic alpha-methyl. The (S) enantiomer (6a) did not increase inhibition of MAPK10 or GSK3α. While RIOK2 does not seem to favor one enantiomer over the racemate, other kinases are sensitive to this stereocenter. Given this ability to tune selectivity via this center of a cell active compound, we propose that the scaffold bearing an alpha-methyl and 6-methoxy holds the greatest potential for further development of a chemical probe for RIOK2. Versus the parent compound (1), analogue 6a maintains a similar selectivity profile against the off-target kinases (Table 3) but demonstrates enhanced cellular affinity for RIOK2 (Table 1).
| Cmpd | MAPK8 | MAPK10 | GSK3α | SNRK | HIPK1 |
|---|---|---|---|---|---|
| All IC50 values are results obtained from n = 2 determinations. | |||||
| 1 | >10000 |
4700 | 5200 | >10000 |
>10000 |
| 3 | >10000 |
1300 | 4800 | >10000 |
>10000 |
| 5 | >10000 |
520 | 860 | >10000 |
>10000 |
| 6 | >10000 |
780 | 1900 | >10000 |
>10000 |
| 6a | >10000 |
5100 | 4500 | >10000 |
>10000 |
Conclusions
We have prepared a small library of naphthyl–amide compounds targeting RIOK2. Several of our analogues out-perform literature reported exemplars in terms of cell-based activity, as demonstrated in our RIOK2 cellular target engagement assay. The RIOK2 NanoBRET assay described is the first and only assay that allows direct assessment of RIOK2 binding in live cells. Our initial library has expanded the RIOK2-specific SAR around this scaffold and informed the design of subsequent analogues to further enhance cellular activity. Analysis of the selectivity of our new compounds encourages the use of this scaffold for development of a chemical probe. We provide cell active RIOK2-targeting compounds that can be further optimized to furnish chemical tools that will facilitate illumination of RIOK2 biology.Experimental section
Chemicals and solvents
All reactions were performed using solvents and reagents purchased from Fisher Chemical, Sigma Aldrich, Combi-Blocks or Enamine.Chromatography
Reactions were monitored by thin-layer chromatography carried out on silica gel plates using ultraviolet light to visualize.Analytical techniques
NMR spectra were recorded on a Varian Inova 400 MHz spectrometer with CDCl3 and CD3CN as the solvent. The 1H NMR chemical shifts are reported as δ values in parts per million (ppm) relative to tetramethylsilane (TMS, δ = 0.00). The residual chloroform signal, CHCl3 (δ = 7.26) was used as reference and for CH3CN signal (quintet, centerline δ = 1.94) was used as reference. 13C NMR chemical shifts are reported as δ values in parts per million (ppm) relative to TMS, the CDCl3 signal (triplet, centerline δ = 77.0) and CD3CN signal (singlet, δ = 118.2) were used as reference. Column chromatography was completed using a Biotage Isolera Prime automated flash purification system with HPLC-grade solvents. Characterization of all compounds via1H and 13C NMR are included in the ESI† section. All compounds were >98% pure by 1H/13C NMR.Procedures for preparation of key compounds 2, 3, 5, 6, 6a, and 14
4-(Trifluoromethyl)pyridin-2-amine (100 mg, 0.617 mmol) and i-Pr2NEt [DIPEA] (1.3 eq.) were suspended in tetrahydrofuran [THF] (4 mL) and stirred at room temperature for 15 min prior to the addition of 2-(naphthalen-2-yl)acetyl chloride (151 mg, 0.740 mmol). The reaction solution was further stirred overnight at room temperature for 12 h. After this period, the reaction mixture was diluted with dichloromethane (DCM) and washed with water and saturated aqueous NaHCO3 solution (2×). The combined organic layers were dried over anhydrous Na2SO4 and then, the solvent was evaporated via reduced pressure to afford a crude reaction mixture. Purification of the crude reaction mixture by column chromatography (silica gel, 0–15% EtOAc/n-hexanes) yielded a white solid (2, 41.7 mg, 20%). 1H NMR (400 MHz, CDCl3) δ 8.55 (dd, J = 1.7, 0.9 Hz, 1H), 8.32 (d, J = 5.2 Hz, 1H), 8.13 (bs, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.87–7.81 (m, 2H), 7.80 (s, 1H), 7.57–7.46 (m, 2H), 7.44 (dd, J = 8.4, 1.8 Hz, 1H), 7.25–7.18 (m, 1H), 3.95 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.88, 152.01, 148.41, 141.01 (d, J = 34.1 Hz), 133.76, 132.90, 130.96, 129.38, 128.69, 127.94, 127.88, 127.14, 126.78, 126.49, 122.68 (d, J = 273.8 Hz), 115.67 (q, J = 3.4 Hz), 110.29 (q, J = 4.1 Hz), 45.23. HRMS-ESI (m/z): [M + H]+ calcd for C18H14F3N2O, 331.1058, found 331.1053.
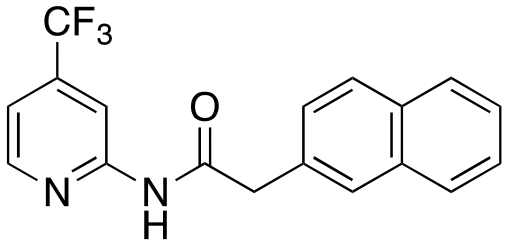
4-Methoxypyridin-2-amine (124 mg, 0.999 mmol) and i-Pr2NEt [DIPEA] (1.3 eq.) were suspended in tetrahydrofuran [THF] (4 mL) and stirred at room temperature for 15 min prior to the addition of 2-(naphthalen-2-yl)acetyl chloride (225 mg, 1.1 mmol). The reaction solution was further stirred overnight at room temperature for 12 h. After this period, the reaction mixture was diluted with dichloromethane (DCM) and washed with water and saturated aqueous NaHCO3 solution (2×). The combined organic layers were dried over anhydrous Na2SO4 and then, the solvent was evaporated via reduced pressure to afford a crude reaction mixture. Purification of the crude reaction mixture by column chromatography (silica gel, 0–20% EtOAc/n-hexanes) yielded a white solid (3, 60.7 mg, 21%). 1H NMR (400 MHz, CDCl3) δ 8.75 (bs, 1H), 7.97 (d, J = 5.9 Hz, 1H), 7.90 (d, J = 2.4 Hz, 1H), 7.87–7.76 (m, 3H), 7.79–7.74 (m, 1H), 7.53–7.43 (m, 2H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 6.56 (dd, J = 5.9, 2.4 Hz, 1H), 3.90 (s, 2H), 3.86 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.03, 167.97, 152.98, 147.59, 133.71, 132.79, 131.42, 129.07, 128.53, 127.86, 127.85, 127.25, 126.55, 126.24, 108.19, 98.77, 55.63, 45.18. HRMS-ESI (m/z): [M + H]+ calcd for C18H17N2O2, 293.1290, found 293.1286.
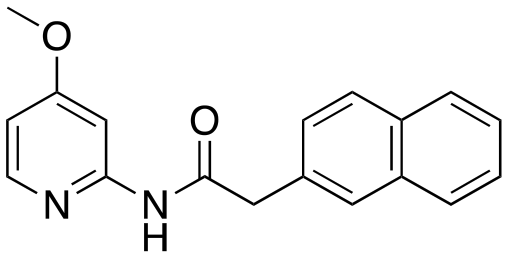
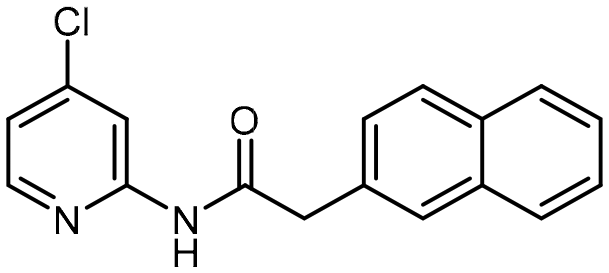
To a solution of 2-naphthaleneacetic acid (248.28 mg, 1.3 mmol) in 3 mL of dry dichloromethane [DCM], thionyl chloride [SOCl2] (5.0 eq.) was added at 0 °C and the mixture was slowly warmed to rt and stirred for 5 h. The solution was removed in a rotary evaporator under reduced pressure and quantitative yield assumed. The crude acid chloride was carried on without further purification. A dry flask containing 4-chloropyridin-2-amine (128.01 mg, 1.0 mmol) and triethylamine [Et3N] (1.2 eq.) was stirred for 5–10 min under a nitrogen atmosphere. Next, anhydrous DCM (4 mL) was added to the reaction flask followed by dropwise addition of 2-(naphthalen-2-yl)acetyl chloride (204.65 mg, 1.2 mmol). The resulting mixture was stirred at rt for 12 h. After this period, the reaction mixture was diluted with DCM and washed with water and saturated aqueous NaHCO3 solution (2×). The combined organic layers were dried over anhydrous Na2SO4 and then the solvent was evaporated via reduced pressure to afford a crude reaction mixture. Purification of the crude reaction mixture by column chromatography (silica gel, 40% EtOAc/n-hexanes) to yield a white solid (5, 166.7 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.35 (d, J = 1.8 Hz, 1H), 8.04 (d, J = 5.5 Hz, 1H), 7.86–7.78 (m, 3H), 7.76 (s, 1H), 7.53–7.46 (m, 2H), 7.41 (dd, J = 8.4, 1.8 Hz, 1H), 7.00 (dd, J = 5.3, 1.9 Hz, 1H), 3.91 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 169.7, 151.9, 147.7, 146.3, 133.5, 132.6, 130.9, 129.0, 128.4, 127.7, 127.7, 126.9, 126.5, 126.2, 120.3, 114.3, 44.9. HRMS-ESI (m/z): [M + H]+ calcd for C17H14ClN2O, 297.0795, found 297.078844.
4-Methylpyridin-2-amine (108.14 mg, 1.0 mmol), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate [HATU] (1.0 eq.), and i-Pr2NEt [DIPEA] (3 eq.) were suspended in N,N-dimethyl-formamide [DMF] (3 mL) and stirred at room temperature for 15 min prior to the addition of 2-(6-methoxynaphthalen-2-yl)propanoic acid (230.26 mg, 1.0 mmol). The reaction solution was further stirred overnight at room temperature for 12 h. After this period, the reaction mixture was diluted with dichloromethane (DCM) and washed with water and saturated aqueous NaHCO3 solution (2×). The combined organic layers were dried over anhydrous Na2SO4 and then, the solvent was evaporated via reduced pressure to afford a crude reaction mixture. Purification of the crude reaction mixture by column chromatography (silica gel, 10–40% EtOAc/n-hexanes) yielded a white solid (6, 240.3 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 8.56 (s, 1H), 8.11 (s, 1H), 7.98 (d, J = 5.1 Hz, 1H), 7.69–7.65 (m, 3H), 7.38 (dd, J = 8.5, 1.8 Hz, 1H), 7.13 (dd, J = 9.0, 2.5 Hz, 1H), 7.09–7.07 (m, 1H), 6.79 (d, J = 5.1 Hz, 1H), 3.89 (s, 3H), 3.84 (q, J = 7.1 Hz, 1H), 2.33 (s, 3H), 1.63 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.9, 157.7, 151.3, 150.1, 146.8, 135.5, 133.8, 129.2, 128.9, 127.6, 126.2, 125.9, 120.8, 119.1, 114.6, 105.5, 55.2, 47.9, 21.3, 18.3. HRMS-ESI (m/z): [M + H]+ calcd for C20H21N2O2, 321.1603, found 321.15926.

4-Methylpyridin-2-amine (108.14 mg, 1.0 mmol), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate [HATU] (1.0 eq.), and i-Pr2NEt [DIPEA] (3 eq.) were suspended in N,N-dimethyl-formamide [DMF] (3 mL) and stirred at room temperature for 15 min prior to the addition of (S)-2-(6-methoxynaphthalen-2-yl)propanoic acid (230.26 mg, 1.0 mmol). The reaction solution was further stirred overnight at room temperature for 12 h. After this period, the reaction mixture was diluted with dichloromethane (DCM) and washed with water and saturated aqueous NaHCO3 solution (2×). The combined organic layers were dried over anhydrous Na2SO4 and then, the solvent was evaporated via reduced pressure to afford a crude reaction mixture. Purification of the crude reaction mixture by column chromatography (silica gel, 10–40% EtOAc/n-hexanes) yielded a white solid (6a, 240.3 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.11 (s, 1H), 7.98 (d, J = 5.1 Hz, 1H), 7.72–7.66 (m, 3H), 7.43–7.35 (m, 1H), 7.14 (dd, J = 9.0, 2.5 Hz, 1H), 7.10–7.08 (m, 1H), 6.80 (dd, J = 5.2, 1.5 Hz, 1H), 3.90 (s, 3H), 3.85 (q, J = 7.1 Hz, 1H), 2.33 (s, 3H), 1.64 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 173.0, 157.7, 151.3, 150.2, 146.6, 135.5, 133.8, 129.2, 128.9, 127.7, 126.2, 125.9, 120.8, 119.1, 114.5, 105.6, 55.2, 48.0, 21.3, 18.3. HRMS-ESI (m/z): [M + H]+ calcd for C20H21N2O2, 321.1603, found 321.15909.
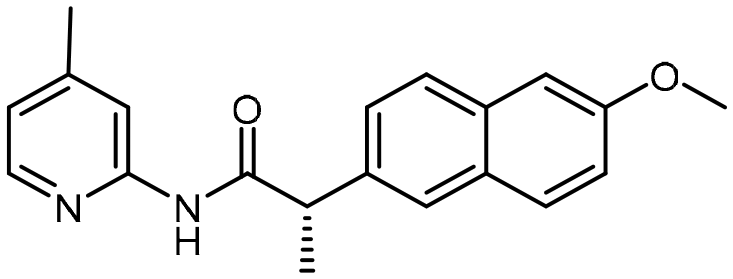
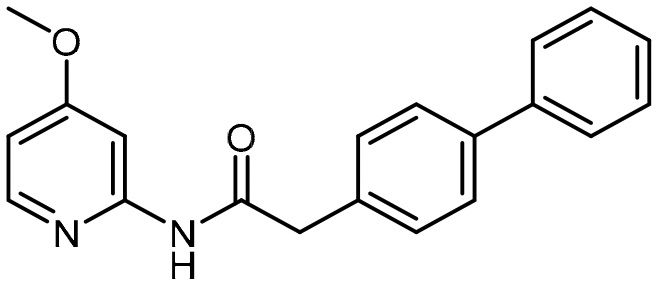
4-Methoxypyridin-2-amine (124 mg, 1.0 mmol), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate [HATU] (1.0 eq.), and i-Pr2NEt [DIPEA] (3 eq.) were suspended in N,N-dimethyl-formamide [DMF] (3 mL) and stirred at room temperature for 15 min prior to the addition of 2-([1,1′-biphenyl]-4-yl)acetic acid (212.08 mg, 1.0 mmol). The reaction solution was further stirred overnight at room temperature for 12 h. After this period, the reaction mixture was diluted with dichloromethane (DCM) and washed with water and saturated aqueous NaHCO3 solution (2×). The combined organic layers were dried over anhydrous Na2SO4 and then, the solvent was evaporated via reduced pressure to afford a crude reaction mixture. Purification of the crude reaction mixture by column chromatography (silica gel, 10–40% EtOAc/n-hexanes) yielded a white solid (14, 254.5 mg, 80%). 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.07 (d, J = 5.8 Hz, 1H), 7.76 (d, J = 2.4 Hz, 1H), 7.65–7.59 (m, 4H), 7.49–7.40 (m, 4H), 7.38–7.34 (m, 1H), 6.64 (dd, J = 5.8, 2.4 Hz, 1H), 3.81 (s, 3H), 3.76 (s, 2H). 13C NMR (101 MHz, CD3CN) δ 170.9, 168.3, 154.4, 149.9, 141.5, 140.5, 135.6, 130.9, 129.8, 128.3, 128.2, 127.8, 107.7, 99.4, 56.1, 44.1. HRMS-ESI (m/z): [M + H]+ calcd for C20H19N2O2, 319.1447, found 319.14348.
Abbreviations used
| AKT | Protein kinase B |
| ATP | Adenosine triphosphate |
| BRET | Bioluminescence resonance energy transfer |
| CRM1 | Chromosomal maintenance 1/exportin 1 |
| GB | Glioblastoma |
| GSK3α | Glycogen synthetase kinase 3-alpha |
| HEK293 | Human embryonic kidney cells |
| HIPK1 | Homeodomain-interacting protein kinase 1 |
| HTS | High throughput screening |
| IDG | Illuminating the druggable genome |
| MAPK10 | Mitogen activated protein kinase 10 |
| MAPK8 | Mitogen activated protein kinase 8 |
| mTOR | Mammalian target of rapamycin |
| NanoBRET | Bioluminescence resonance energy transfer using nanoluciferase |
| NLuc | Nanoluciferase |
| NOB1 | NIN1 (RPN12) binding protein 1 homolog |
| NSCLC | Non-small cell lung cancer |
| PDB | Protein data bank |
| PLK1 | Polo-like kinase 1 |
| RIO | Right open reading frame |
| SAR | Structure activity relationship |
| SNRK | SNF-related serine/threonine-protein kinase |
| wHTH | Winged helix turn helix |
Authors contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. C. B. W. and A. L. M. completed the chemical synthesis, purification, and characterization of compound analogues. C. B. W. and A. P. executed all structural analyses. J. E. P. and C. W. performed the biological NanoBRET assays and graphical determination of IC50 values. C. B. W. and A. D. A. selected compounds and kinases for selectivity screening. W. J. Z. and A. D. A. managed all aspects of the study design to completion and A. D. A. served as the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The construct for the RIOK2 NanoBRET measurements were kindly provided by Promega. We thank the following individuals at the UNC Eshelman School of Pharmacy for their expertise and help in reviewing the manuscript: Q. Zhang, A. Devanathan, D. Carpenter, and C. Lee. We thank the UNC Department of Chemistry Mass Spectrometry Core Laboratory for their assistance with mass spectrometry analyses. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Genentech, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. Research reported in this publication was supported in part by the NC Biotechnology Center Institutional Support Grant 2018-IDG-1030, and by the NIH 1U24DK11604. C. B. W is additionally supported by a Gateway to Research Scholarship provided by the American Foundation for Pharmaceutical Education (AFPE) Foundation (RAMSES Proposal No. 20-0328). A. L. M. acknowledges support from the Spanish MECD (FPU 14/00818).References
- N. LaRonde-LeBlanc and A. Wlodawer, J. Biol. Chem., 2005, 280(45), 37297–37300 CrossRef CAS.
- T. Liu, M. Deng, J. Li, X. Tong, Q. Wei and X. Ye, J. Biol. Chem., 2011, 286(42), 36352–36360 CrossRef CAS.
- N. LaRonde-LeBlanc and A. Wlodawer, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1754(1–2), 14–24 CrossRef CAS.
- S. Ferreira-Cerca, V. Sagar, T. Schäfer, M. Diop, A.-M. Wesseling, H. Lu, E. Chai, E. Hurt and N. LaRonde-LeBlanc, Nat. Struct. Mol. Biol., 2012, 19(12), 1316–1323 CrossRef CAS.
- I. Zemp, T. Wild, M.-F. O'Donohue, F. Wandrey, B. Widmann, P.-E. Gleizes and U. Kutay, J. Biol. Chem., 2009, 185(7), 1167–1180 CAS.
- T. Liu, M. Deng, J. Li, X. Tong, Q. Wei and X. Ye, J. Biol. Chem., 2011, 286(42), 36352–36360 CrossRef CAS.
- R. D. Read, T. R. Fenton, G. G. Gomez, J. Wykosky, S. R. Vandenberg, I. Babic, A. Iwanami, H. Yang, W. K. Cavenee, P. S. Mischel, F. B. Furnari and J. B. Thomas, PLoS Genet., 2013, 9(2), e1003253 CrossRef CAS.
- T. H. Geerlings, A. W. Faber, M. D. Bister, J. C. Vos and H. A. Raué, J. Biol. Chem., 2003, 278(25), 22537–22545 CrossRef CAS.
- D.-T. Nguyen, S. Mathias, C. Bologa, S. Brunak, N. Fernandez, A. Gaulton, A. Hersey, J. Holmes, L. J. Jensen, A. Karlsson, G. Liu, A. Ma'ayan, G. Mandava, S. Mani, S. Mehta, J. Overington, J. Patel, A. D. Rouillard, S. Schürer, T. Sheils, A. Simeonov, L. A. Sklar, N. Southall, O. Ursu, D. Vidovic, A. Waller, J. Yang, A. Jadhav, T. I. Oprea and R. Guha, Nucleic Acids Res., 2017, 45(D1), D995–D1002 CrossRef CAS.
- R. D. Read, T. R. Fenton, G. G. Gomez, J. Wykosky, S. R. Vandenberg, I. Babic, A. Iwanami, H. Yang, W. K. Cavenee, P. S. Mischel, F. B. Furnari and J. B. Thomas, PLoS Genet., 2013, 9(2), e1003253 CrossRef CAS.
- K. Liu, H.-L. Chen, S. Wang, M.-M. Gu, X.-M. Chen, S.-L. Zhang, K.-J. Yu and Q.-S. You, Sci. Rep., 2016, 6(1), 28666 CrossRef CAS.
- K. Liu, H. Chen, Q. You, Q. Ye, F. Wang, S. Wang, S. Zhang, K. Yu, W. Li and M. Gu, Int. J. Oncol., 2018, 53(1), 257–265 CAS.
- O. Fedorov, B. Marsden, V. Pogacic, P. Rellos, S. Müller, A. N. Bullock, J. Schwaller, M. Sundström and S. Knapp, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(51), 20523–20528 CrossRef CAS.
- M. I. Davis, J. P. Hunt, S. Herrgard, P. Ciceri, L. M. Wodicka, G. Pallares, M. Hocker, D. K. Treiber and P. P. Zarrinkar, Nat. Biotechnol., 2011, 29(11), 1046–1051 CrossRef CAS.
- T. Varin, A. G. Godfrey, T. Masquelin, C. A. Nicolaou, D. A. Evans and M. Vieth, Biochim. Biophys. Acta, Proteins Proteomics, 2015, 1854(10), 1630–1636 CrossRef CAS.
- J. D. Vasta, C. R. Corona, J. Wilkinson, C. A. Zimprich, J. R. Hartnett, M. R. Ingold, K. Zimmerman, T. Machleidt, T. A. Kirkland, K. G. Huwiler, R. F. Ohana, M. Slater, P. Otto, M. Cong, C. I. Wells, B.-T. Berger, T. Hanke, C. Glas, K. Ding, D. H. Drewry, K. V. M. Huber, T. M. Willson, S. Knapp, S. Müller, P. L. Meisenheimer, F. Fan, K. V. Wood and M. B. Robers, Cell Chem. Biol., 2018, 25(2), 206–214 CrossRef CAS .e11.
- J. Wang, T. Varin, M. Vieth and J. M. Elkins, Open Biol., 2019, 9(4), 190037 CrossRef CAS.
- F. Maurice, N. Pérébaskine, S. Thore and S. Fribourg, RNA Biol., 2019, 16(11), 1633–1642 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Figures that summarize previously reported data, biological assay methods and curves, and spectral characterization data. See DOI: 10.1039/d0md00292e |
| This journal is © The Royal Society of Chemistry 2021 |