
Identification of P218 as a potent inhibitor of Mycobacterium ulcerans DHFR†
Gustavo P.
Riboldi
 ab,
Rachael
Zigweid
c,
Peter J.
Myler
cd,
Stephen J.
Mayclin
ef,
Rafael M.
Couñago
*ab and
Bart L.
Staker
*c
ab,
Rachael
Zigweid
c,
Peter J.
Myler
cd,
Stephen J.
Mayclin
ef,
Rafael M.
Couñago
*ab and
Bart L.
Staker
*c
aCentro de Química Medicinal (CQMED), Centro de Biologia Molecular e Engenharia Genética (CBMEG), Universidade Estadual de Campinas (UNICAMP), Campinas, SP 13083-875, Brazil. E-mail: rafael.counago@unicamp.br
bStructural Genomics Consortium, Departamento de Genética e Evolução, Instituto de Biologia, UNICAMP, Campinas, SP 13083-886, Brazil
cCenter for Infectious Disease Research, Seattle Children's Research Institute, Seattle, Washington 98109, USA. E-mail: bart.staker@seattlechildrens.org
dDepartment of Pediatrics, University of Washington, Seattle, Washington 91895, USA
eSeattle Structural Genomics Center for Infectious Disease (SSGCID), Seattle, Washington 98109, USA
fUCB, Bainbridge Island, Washington 98110, USA
First published on 22nd October 2020
Abstract
Mycobacterium ulcerans is the causative agent of Buruli ulcer, a debilitating chronic disease that mainly affects the skin. Current treatments for Buruli ulcer are efficacious, but rely on the use of antibiotics with severe side effects. The enzyme dihydrofolate reductase (DHFR) plays a critical role in the de novo biosynthesis of folate species and is a validated target for several antimicrobials. Here we describe the biochemical and structural characterization of M. ulcerans DHFR and identified P218, a safe antifolate compound in clinical evaluation for malaria, as a potent inhibitor of this enzyme. We expect our results to advance M. ulcerans DHFR as a target for future structure-based drug discovery campaigns.
Introduction
Buruli ulcer is a necrotizing skin disease caused by Mycobacterium ulcerans infection. Skin ulcers are a result of the host immune response to mycolactone, an immunosuppressive polyketide-derived macrolide cytotoxin secreted by M. ulcerans.1–3 Buruli ulcer is considered a major public health problem in endemic areas,4 which include humid rural tropical zones prone to seasonal flooding and/or linked to low-lying wetland areas.5,6 Current treatment – oral (rifampin) and injectable (streptomycin) antibiotics – is effective but requires daily antibiotic injections for 8 weeks and is associated with long-term hearing loss.7 Thus, a major goal of Buruli ulcer research is finding an all-oral therapeutic strategy that shortens the duration of treatment and avoids severe side-effects.8–11Unlike mammals, microbes are unable to obtain folate from the environment and rely on de novo production of reduced folate species for the biosynthesis of critical cellular components, including methionine, glycine, serine, N-formylmethionyl-tRNA, purines, and thymidine.12,13 Thus, folate metabolism is a vulnerable pathway in microbes and antifolate compounds have found widespread use in the clinic. Two enzymes in the microbial folate pathway are validated targets for antifolate compounds: dihydropteroate synthase (DHPS), and dihydrofolate reductase (DHFR). Compounds inhibiting these enzymes are effective against pathogenic Mycobacterium species, including M. ulcerans,14,15M. leprae,16–19 the causative agent of leprosy, and M. tuberculosis,13,20–22 the causative agent of tuberculosis. In the clinic, dapsone, (DDS; diaminodiphenyl sulfone) a sulfone antibiotic targeting DHPS, is used with rifampicin and clofazimine to treat leprosy and p-aminosalicylic acid (PAS), a prodrug targeting DHFR,23 is a second-line treatment for tuberculosis. Nevertheless, the treatment of Buruli ulcer or other mycobacterial infections with antifolates remains incompletely explored.
As in other mycobacteria, M. ulcerans (Mul)DHFR is encoded by a single gene (dfrA or folA).24 The MulDHFR amino acid sequence is 74, 28, and 32% identical to M. tuberculosis (Mtb), Staphylococcus aureus (Sau) and Escherichia coli (Eco)DHFR, respectively. While trimethoprim (TMP), a diaminopyrimidine antibiotic used against both Gram-positive and Gram-negative bacteria, is a potent inhibitor of Eco- and SauDHFR,25 TMP is ineffective against M. tuberculosis and M. ulcerans. However, combining dapsone with epiroprim, a different diaminopyrimidine antibiotic that targets DHFR, is effective in culture against M. ulcerans.15,17,26
Despite differences in amino acid sequence, bacterial and human DHFR enzymes have a conserved structural architecture consisting of two distinct sub-domains. The adenosine binding subdomain provides the binding site for the NADPH cofactor adenosine moiety. The major (or “loop”) sub-domain encompasses most of the protein and has three loop regions – M20, F-G, and G-H; important for substrate binding and enzyme catalysis.27 Amino acid differences in the “loop” subdomain are thought to be responsible for determining inhibitor selectivity towards DHFR in different organisms. Mutations to residues in this subdomain are associated with the emergence of bacterial strains resistant to anti-folate antibiotics, mainly in Gram-negative bacteria.28–30
Focused medicinal chemistry efforts have produced potent and cell-permeable DHFR inhibitors that are efficacious against M. tuberculosis.20–22 To enable similar programs for Buruli ulcer, we report here the biochemical and structural characterization of MulDHFR and the identification of P218 – a DHFR inhibitor undergoing clinical studies as a therapy for malaria – as a potent MulDHFR inhibitor.31 We expect our work to contribute to the development of future therapeutic strategies for Buruli ulcer based on anti-folates.
Experimental
Gene cloning and recombinant protein production
Cloning, expression and purification were conducted as part of the Seattle Structural Genomics Center for Infectious Disease (SSGCID)32,33 following standard protocols described previously.34–36 Prokaryotic expression vectors containing either the wild-type (WT) or the cysteine-89 variant mutant (C89S) of M. ulcerans DHFR were obtained from the Seattle Structural Genomics Center for Infectious Disease (clone ID MyulA.01062.a.B1.GE39104 and MyulA.01062.a.B11.GE42658, respectively; http://www.SSGCID.org). The C89S variant was created by performing site-directed mutagenesis on the WT using a Quick-Change Lightning kit, based upon the UCLA MBI-SERp Server Identification of high surface-entropy residues. Primers were designed using the Quick-Change Primer Design Program available online at http://www.agilent.com/genomics/qcpd.The gene was cloned into the ligation independent cloning (LIC) expression vector pBG1861 encoding a non-cleavable, N-terminal 6xHis fusion tag (amino acid sequence: MAHHHHHH).34 Plasmid DNA was transformed into chemically competent E. coli BL21(DE3)R3 Rosetta cells. The plasmids containing MulDHFR and MulDHFR-C89S were expression tested and 2 litres of culture were grown using auto-induction media37 in a LEX Bioreactor (Epiphyte Three Inc.) as previously described.38
MulDHFR and MulDHFR-C89S were purified in a two-step protocol consisting of a Ni2+-affinity chromatography (IMAC) step and size-exclusion chromatography (SEC). All chromatography runs were performed on an ÄKTApurifier 10 (GE) using automated IMAC and SEC programs according to previously described procedures.36 Bacterial pellets-thawed in a 42 °C water bath and vortexed gently were resuspended in 180 ml lysis buffer containing 20 mM HEPES pH 7.2–7.4, 300 mM NaCl, 5% (v/v) glycerol, 0.5% CHAPS, 30 mM imidazole, 21 mM MgCl2, 1 mM TCEP, and 2 protease inhibitor tablets and lysed by sonication for 15 minutes (5 s on, 10 s off, 70% amplitude, on ice). After sonication, the crude lysate was clarified with 2 μl of benzonase and incubated while mixing at room temperature for 45 minutes. The lysate was then clarified by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rev min−1 for 1 h at 4 °C using a Sorvall centrifuge (Thermo Scientific). The supernatant was filtered through a 0.45 μM syringe filter, then passed over a Ni-NTA His-Trap FF 5 ml column (GE Healthcare) which was pre-equilibrated with loading buffer composed of 20 mM HEPES pH 7.0, 300 mM NaCl, 5% (v/v) glycerol, and 30 mM Imidazole. The column was washed with 20 column volumes (CV) of loading buffer and was eluted with 10 CVs of loading buffer plus 0.5 M imidazole. Peak fractions, as determined by UV at 280 nm, were pooled and concentrated to 5 ml with a 3 K Pall filter. A SEC column (Superdex 75, GE) was equilibrated with running buffer composed of 300 mM NaCl, 20 mM HEPES pH 7.0, 5% (v/v) glycerol, and 1 mM TCEP. The peak fractions were collected and analysed for the presence of the protein of interest using SDS-PAGE. The peak fractions were pooled and concentrated using an Amicon purification system (Millipore). Aliquots of 110 μl were flash-frozen in a dry ice and ethanol bath and stored at −80 °C until use. The protein was quantitated by UV-light (extinction coefficient of 37470 M−1 cm−1) using a Nanodrop spectrophotometer (Thermo). The molecular mass of the purified protein was confirmed by intact mass, LC-MS.
000 rev min−1 for 1 h at 4 °C using a Sorvall centrifuge (Thermo Scientific). The supernatant was filtered through a 0.45 μM syringe filter, then passed over a Ni-NTA His-Trap FF 5 ml column (GE Healthcare) which was pre-equilibrated with loading buffer composed of 20 mM HEPES pH 7.0, 300 mM NaCl, 5% (v/v) glycerol, and 30 mM Imidazole. The column was washed with 20 column volumes (CV) of loading buffer and was eluted with 10 CVs of loading buffer plus 0.5 M imidazole. Peak fractions, as determined by UV at 280 nm, were pooled and concentrated to 5 ml with a 3 K Pall filter. A SEC column (Superdex 75, GE) was equilibrated with running buffer composed of 300 mM NaCl, 20 mM HEPES pH 7.0, 5% (v/v) glycerol, and 1 mM TCEP. The peak fractions were collected and analysed for the presence of the protein of interest using SDS-PAGE. The peak fractions were pooled and concentrated using an Amicon purification system (Millipore). Aliquots of 110 μl were flash-frozen in a dry ice and ethanol bath and stored at −80 °C until use. The protein was quantitated by UV-light (extinction coefficient of 37470 M−1 cm−1) using a Nanodrop spectrophotometer (Thermo). The molecular mass of the purified protein was confirmed by intact mass, LC-MS.
Enzyme assays
Dihydrofolate reductase enzyme activity was assessed by dihydronicotinamide-adenine dinucleotide phosphate (NADPH) reduction followed by tetrahydrofolate (THF) formation. Enzyme assays were performed in 384-well, black, non-binding plates (Corning). Assay mixture consisted of reaction buffer (50 mM PIPES, pH 7.3, 0.02% Tween-20), NADPH (400 μM), BSA (1.5 mg mL−1) and DTT (6 mM). The reaction was started by the addition of the enzyme substrate 7,8-dihydrofolate (diHF) (300 μM, final concentration). Final volume for the reaction mixture was 30 μL. NADPH fluorescence intensity (FI) was followed (excitation 340 nm/emission 445 nm) for 30 minutes at 25 °C using a ClarioSTAR® (BMG Labtech) plate reader. The acquired data was analysed using the MARS software (BMG Labtech). Enzyme titration experiments were performed by serial dilution of either wild-type or C89S M. ulcerans DHFR in assay reaction buffer. Experiments to determine MulDHFR and MulDHFR-C89S kinetic parameters (KM, Kcat) were performed by serial dilution of diHF at fixed enzyme concentrations (10.0 nM). Half-maximal inhibitory concentrations (IC50) were assessed by performing enzyme activity assays in the presence of increasing inhibitor concentrations, serially diluted in dimethyl sulfoxide (DMSO, Sigma-Aldrich). Compounds in 100% DMSO were transferred to the reaction plate using an automated liquid handler robot (Felix, Analytik Jena AG), and incubated for 30 min, at 25 °C, before the reaction was started by the addition of diHF. The final DMSO concentration in the assay mixture was 0.3 or 0.07%, depending on the pin tool used to transfer the compounds to the assay plate. Ki values were calculated assuming competitive inhibition and used the Cheng–Prusoff relationship:39Ki = IC50/(1 + ([diHF]/KM,diHF)), where [diHF] is the diHF concentration used in the IC50 assay (300 μM). Graphical plots, enzyme kinetics, inhibitory pattern estimation and statistical analysis were performed in Prism-Graphpad 8.0. Values shown in graphical plots are the average of three data points + standard deviations collected during a single experiment. All experiments were performed at least three times.Protein crystallization and structure determination
For crystallization, NADPH and P218 were added to purified MulDHFR-C89S (12.5 mg ml−1) at 6-fold molar excess. Sitting drops were set at 1:1 ratio composed of 400 nL of the protein-P218–NADPH mixture to 400 nL reservoir solution. Crystallization experiments were performed at 14 °C. The best-diffracting crystals grew from the Morpheus crystallization screen (Molecular Dimensions) in a solution containing 12.5% (w/v) PEG 1000, 12.5% (w/v) PEG 3350, 12.5% (v/v) MPD, 0.03 M diethylene glycol, 0.03 M triethylene glycol, 0.03 M tetraethylene glycol, 0.03 M pentaethylene glycol and 0.1 M MOPS/HEPES-Na pH 7.5. Because of the makeup of the mother liquor, crystals were vitrified in liquid nitrogen with no additional cryo-protection. Diffraction data were collected at LS-CAT at the Advanced Photon Source Life Sciences Collaborative Access Team (APS LS-CAT) beamline 21-ID-D equipped with a Dectris Eiger 9M detector at a wavelength of 0.8666 Å. Data sets were reduced with the XDS package.40 Molecular replacement (MR) was performed with Molrep41 from the CCP4 package,42 using the M. tuberculosis structure as template (PDB ID 1DF7).43 Manual model building was performed using Coot,44 the structure was refined in reciprocal space with Phenix.45 NADPH and P218 atoms were refined with full (1.0) occupancy. Structure validation was performed using MolProbity.46 Structure factors and coordinates have been deposited in the PDB with the PDB ID 6UWW.
Results & discussion
Recombinant protein production
To the best of our knowledge, recombinant production of M. ulcerans DHFR has not been reported before. We successfully produced recombinant wild-type MulDHFR in E. coli cells with an N-terminal poly-histidine tag to facilitate protein purification. The protein was purified from the clarified cell lysate using a combination of immobilized metal ion affinity chromatography (IMAC) and size exclusion chromatography. The N-terminal poly-histidine tag was not removed. Using LC-MS, we verified that the purified protein had the expected molecular weight (19082.7 Da). This recombinant protein was used in biochemical assays and protein crystallization attempts.
Unfortunately, initial attempts to obtain crystals using recombinant wild-type MulDHFR were not successful (see below). To maximize chances of obtaining crystals we created a small series of mutant proteins with single or double residue changes hypothesized to alter crystallization properties.47 We targeted non-conserved cysteine or charged residues predicted to be surface exposed based on comparisons to the M. avium (Mav-DHFR) or M. tuberculosis structures43 (overall sequence identity levels of 69 and 74% to MulDHFR, respectively). We then produced a mutant version of the enzyme in which MulDHFR cysteine 89 was replaced with a serine (MulDHFR-C89S). A sequence comparison to M. avium identified MulDHFR-E96 as a non-conserved and surface exposed residue. We mutated this residue to alanine in order to match the sequence of the M. avium DHFR, which was previously solved at high resolution. We also constructed the double mutant MulDHFR-C89S/E96A. Production of mutant proteins followed the same protocol used for the wild-type enzyme and the identity of the purified protein was verified by LC-MS.
Biochemical characterization of recombinant MulDHFR
To determine that both recombinant proteins were enzymatically active, we followed the DHFR reaction by monitoring the oxidation of NADPH into NADP catalysed by MulDHFR in the presence of 7,8-dihydrofolate (diHF). Using increasing amounts of the purified enzyme, we observed a dose-dependent reduction in NADPH fluorescence, indicating the recombinant MulDHFR was active. These enzyme titration curves were performed with an excess amount of cofactor NADPH (400 μM), and showed a linear relationship between the observed DHFR reaction rate and the final enzyme concentration used in the assay (ranging from 0.4 to 24 nM). Based on these results, we chose to use 10 nM of MulDHFR in all subsequent biochemical experiments. Further, our data showed that both wild-type and C89S MulDHFR have similar enzymatic activities (Fig. 1A).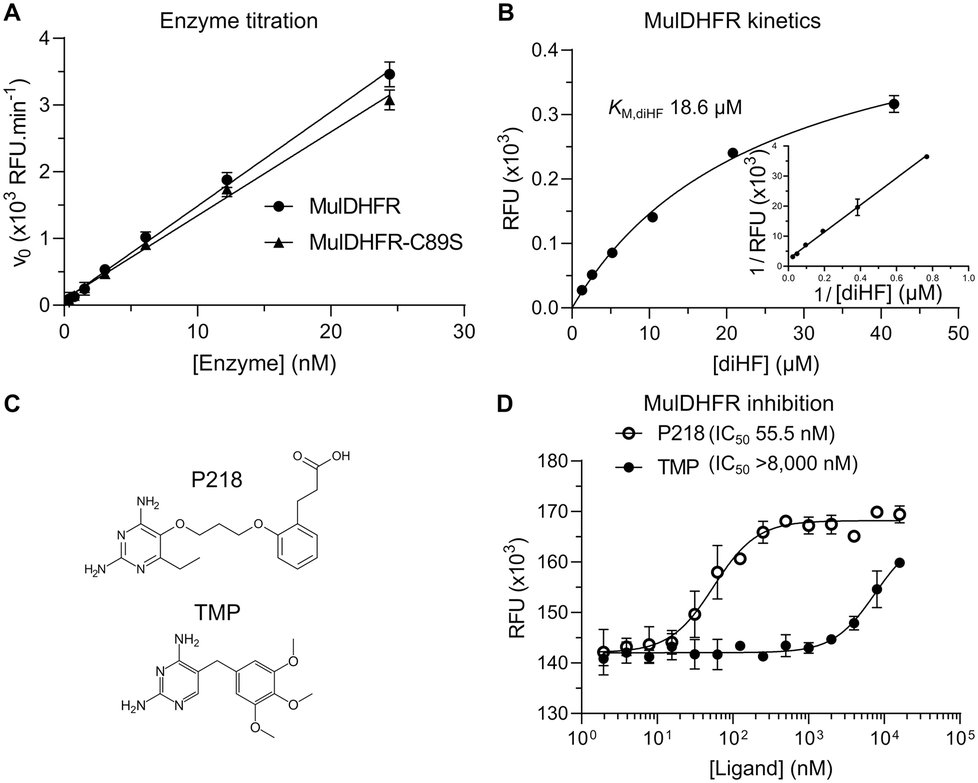 | ||
| Fig. 1 MulDHFR and MulDHFR-C89S have similar enzymatic properties and can be inhibited by P218. (A) Linear relationship between increasing concentrations of wild-type (circles) or C89S mutant (triangles) MulDHFR and the reaction rates (initial velocities – v0). (B) Hyperbolic relationship between substrate concentration and the rate (initial velocity – normalized v0) of the DHFR-catalysed reaction for the wild-type enzyme. The inset shows the Lineweaver–Burk plot of the same kinetic data. The value of the Michaelis–Menten constant (KM – defined as the concentration of substrate diHF needed to reach the reaction's half-maximal velocity) was obtained from the x-axis intercept in Lineweaver–Burk plot. (C) Chemical structures of P218 (top) and trimethoprim (TMP, bottom). (D) Enzyme inhibition of wild-type MulDHFR by TMP (filled symbols) and P218 (empty symbols). The half-maximal inhibitory concentration (IC50) for each compound is shown in parenthesis. Data shown are mean + SD of triplicates. | ||
Determining the enzyme kinetic parameters for MulDHFR can help future drug discovery campaigns. Establishing the enzyme Vmax and KM for its substrate diHF (KM,diHF) allows inhibitor constants (Ki) for diHF-competing ligands to be directly estimated from half-maximal inhibitory concentrations values (IC50). Ki values can then be used to compare the potency of the same compound for different enzymes. Further, determining how ligands impact an enzyme KM and Vmax can help establish their enzyme inhibition mechanisms. The kinetic parameters for the M. tuberculosis DHFR enzyme have already been established and were found to be comparable to those obtained for the enzymes from pathogenic Gram-positive and Gram-negative organisms, such as EcoDHFR and SauDHFR25,48,49 (Table 1). To determine the enzyme kinetic parameters for wild-type MulDHFR, we first obtained initial velocity rates (v0) from reaction progress curves at increasing concentrations of the enzyme substrate diHF. Fitting these data to Michaelis–Menten kinetics allowed us to estimate a KM value for the substrate diHF (KM,diHF) of 12.3 + 4.9 μM, and a Vmax value for the reaction of 123.7 nM s−1 ng (Fig. 1B). Based on these values, we calculated a Kcat value of 6.2 s−1 for wild-type MulDHFR. Similar values were obtained for MulDHFR-C89S, further suggesting that the C89S point mutation did not interfere with the protein's enzymatic activity (Table 1). Our data indicated that the kinetic parameters of MulDHFR are comparable to those previously established for the enzyme from M. tuberculosis, E. coli and S. aureus (Table 1).
Enzyme inhibition studies identify P218 as a potent MulDHFR inhibitor
While DHFR inhibitors, such as TMP (Fig. 1C, bottom), have found wide-use against Gram-positive and Gram-negative bacteria in the clinic,50 a similar therapeutic strategy for mycobacterial infections has not been fully explored. Efforts to develop new antibiotics against mycobacterial infections has led to the exploration of anti-folate compounds targeting DHFR as treatment options. TMP bactericide activity is potentiated by sulfonamide-based antibiotics that target dihydropteroate synthase (DHPS), an enzyme upstream of DHFR in the folate pathway.51 Sulfonamides can reduce DHFR substrate diHF, removing it from competing with TMP for the DHFR folate-binding site.50 TMP is a potent inhibitor of EcoDHFR (Ki ∼ 0.2 nM)25 and SauDHFR (Ki ∼ 4.4 nM)49 but is only weakly active against MtbDHFR (Ki ∼ 1.5 μM).48 To ascertain if TMP could inhibit MulDHFR, we obtained initial velocity (v0) values for reactions performed at increasing concentrations of TMP. From this data we estimated the half-maximal inhibitory concentration of TMP for MulDHFR (IC50,TMP > 8000 nM) and MulDHFR-C89S (IC50,TMP > 5000 nM) (Fig. 1D and S1†), indicating that TMP is a weak inhibitor of MulDHFR and helping rationalize the reported lack of activity of this compound towards M. ulcerans.15P218 is a derivative of WR99210, a diaminopyridine compound originally developed for the Plasmodium enzyme and shown active against Mycobacteria52,53 (Fig. 1C, top). Results from phase I clinical studies (registered at http://ClinicalTrials.gov with the identifier NCT02885506) showed that P218 has a promising safety profile in humans, although the compound may need further development to improve its short half-life.31 IC50 values of P218 for MulDHFR were obtained as described above for TMP and our data confirmed that P218 is a potent inhibitor of MulDHFR (IC50,TMP = 55.5 nM). Using the formalism of Cheng–Prusoff,39 and the obtained IC50,P218 and KM,diHF values, we computed an equilibrium inhibition constant (Ki,P218) of ∼3.2 nM for P218 against MulDHFR activity. We performed similar experiments for MulDHFR-C89S and obtained similar IC50 values for the mutant enzyme (IC50,TMP = 32.8 nM), showing P218 has similar potency for both the wild-type and the mutant enzyme used for the structural studies described below.
The crystal structure of MulDHFR-C89S bound to cofactor NADPH and inhibitor P218
Currently, there are no MulDHFR structures available. To better understand P218 binding to MulDHFR, and to provide a starting point for future structure-based drug design programs for this enzyme, we obtained the co-crystal structure of MulDHFR-C89S bound to P218 and cofactor NADPH to a resolution of 0.9 Å (Fig. 2). As we could not obtain co-crystals of P218 bound to the wild-type protein, all structural work described below was performed with the MulDHFR-C89S mutant. In the light of our biochemical analysis above, we are confident the C89S mutation caused minimum, if any, disturbance to the native enzyme structure. MulDHFR-C89S crystals were obtained in the presence of excess cofactor NADPH and inhibitor P218. We used the structure of MtbDHFR (PDB ID 1DF7) as a search model in molecular replacement to solve the crystallographic phase problem (Table S1†). No density was observed for the first two N-terminal residues in MulDHFR-C89S, likely due to disorder, and the final protein model consisted of residues 3–165 (Fig. 2). Both the ligand P218 and the cofactor NADPH could be placed unambiguously into the electron density unfilled by the protein model (OMIT map shown for P218 in Fig. 2). MulDHFR-C89S crystal structure is similar to that of its counterpart from M. tuberculosis (root mean square deviation, r.m.s.d. = 0.45 Å for 135 equivalent Cα atoms), as expected from the high identity levels between these two proteins (74%) (Fig. S2†).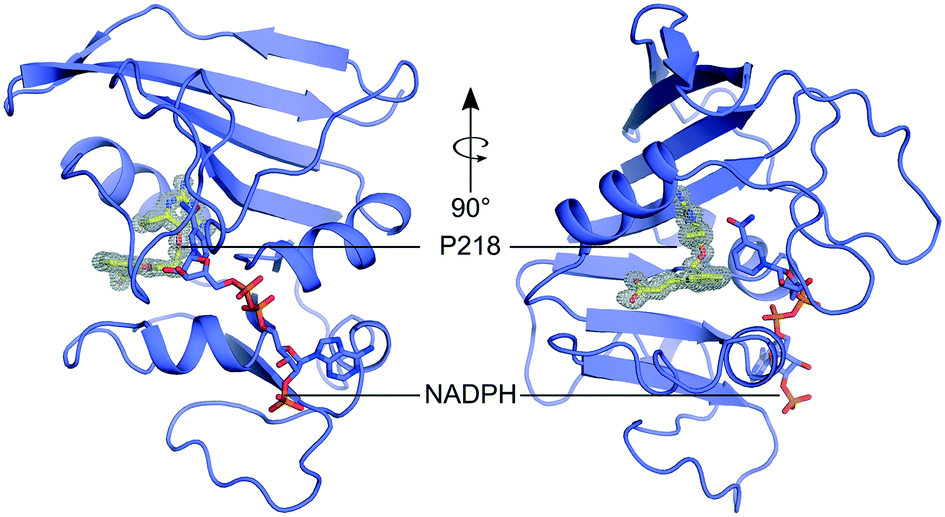 | ||
| Fig. 2 The structure of MulDHFR-C89S bound to P218 and NADPH. Cartoon representation of the MulDHFR-C89S structure. P218 and NADPH molecules are shown as stick. P218 is covered by a polder OMIT map54 (grey mesh represents the mFobs-DFmodel OMIT difference density contoured at 3.0σ). | ||
Our co-crystal structure offered a detailed view of the binding of P218 to MulDHFR-C89S (Fig. 3). The ligand 2,4-diaminopyrimidine (DAP) moiety anchored P218 deep into the enzyme active site, sandwiched between the aromatic side chain of Phe33 and cofactor NADPH amide group. Further, the amino groups in P218 DAP make hydrogen bonds to side chain atoms from residues Asp29 and Tyr106, and to main chain atoms from residues Ile7 and Ile100 in MulDHFR-C89S. P218 five-atom linker confers the ligand enough flexibility for its benzyl group to point away from Phe41 side chain while allowing the ligand α-carboxylate group to interact via a bidentate hydrogen bond to Arg62. Structurally equivalent arginine residues to MulDHFR Arg62 are conserved amongst DHFR enzymes from different organisms, as this residue interacts directly with the substrate dihydrofolate α-carboxylate group.27,55
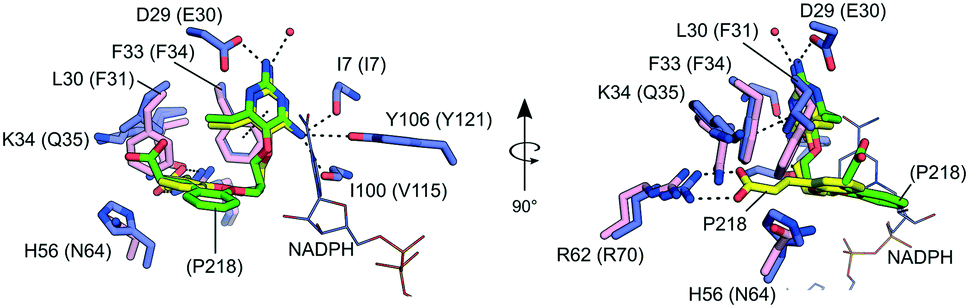 | ||
| Fig. 3 Details of P218 binding to MulDHFR and HsaDHFR. Amino acid residues within the folate-binding site of MulDHFR-C89S (blue sticks) and HsaDHFR (pink sticks). Residues/ligands in parenthesis are for the human enzyme. The ligand P218 is shown as sticks in yellow, for the MulDHFR co-structure, or green, for the HsaDHFR co-structure. The cofactor NADPH as seen in the MulDHFR co-structure is also shown (lines). Dashed lines indicate possible hydrogen bonds between the ligand and atoms from the protein or the solvent (shown as a red sphere) in MulDHFR-C89S crystals. | ||
The sequence and structural conservation between human and M. ulcerans DHFR enzymes may raise concerns about the safety of using anti-folates as a therapeutic strategy to treat Buruli ulcer. However, P218 is ∼1000-fold more active against MulDHFR, as compared to the human enzyme (HsaDHFR Ki,P218 ∼ 2.8 μM).56 The co-structure of P218 bound to HsaDHFR revealed that the ligand α-carboxylate group pointed away from the enzyme dihydrofolate-interacting arginine residue (Arg70 in HsaDHFR). In the human enzyme, residues Phe31 (Leu30 in MulDHFR) and Gln35 (Lys34 in MulDHFR) sterically interfere with P218 binding, and prevent the compound α-carboxylate group from reaching the conserved arginine residue (Fig. 3). The resulting difference in ligand binding mode is thought to be responsible for P218 low activity towards the human enzyme.
Conclusion
P218 is a safe DHFR inhibitor already under clinical investigation for malaria31,56 and here we have identified it as a potent inhibitor of the M. ulcerans DHFR enzyme. Our structural and biochemical characterization of M. ulcerans DHFR and its interaction with P218 offers an opportunity to further develop P218 as a therapeutic strategy against Buruli ulcer. To reduce the chances of emerging resistance to anti-folates, we suggest P218 be used in combination with other antimicrobial agents currently used for Buruli ulcer treatment, especially those that can be administered orally and do not have severe side effects, such as rifampin.7–9Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Brazilian agencies FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) (2013/50724-5 and 2014/50897-0) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) (465651/2014-3). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome [106169/ZZ14/Z]. This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272201700059C. We thank the staff of the Life Sciences Core Facility (LaCTAD) at UNICAMP for the Genomics and Proteomics analysis.Notes and references
- F. S. Sarfo, R. Phillips, M. Wansbrough-Jones and R. E. Simmonds, Cell. Microbiol., 2016, 18, 17 CrossRef CAS.
- K. M. George, D. Chatterjee, G. Gunawardana, D. Welty, J. Hayman, R. Lee and P. L. C. Small, Science, 1999, 5403, 854 CrossRef.
- D. S. Walsh, F. Portaels and W. M. Meyers, Trans. R. Soc. Trop. Med. Hyg., 2008, 102, 969 CrossRef.
- WHO Meeting on Buruli ulcer and other skin NTDs - Final Report, 2019.
- T. Wagner, M. E. Benbow, T. O. Brenden, J. Qi and R. C. Johnson, Int. J. Health Geogr., 2008, 7, 25 CrossRef.
- M. Debacker, F. Portaels, J. Aguiar, C. Steunou, C. Zinsou, W. Meyers and M. Dramaix, Emerging Infect. Dis., 2006, 12, 1325 CrossRef.
- S. Klis, Y. Stienstra, R. O. Phillips, K. M. Abass, W. Tuah and T. S. van der Werf, PLoS Neglected Trop. Dis., 2014, 8, e2739 CrossRef.
- P. J. Converse, D. V. Almeida, R. Tasneen, V. Saini, S. Tyagi, N. C. Ammerman, S. Y. Li, N. M. Anders, M. A. Rudek, J. H. Grosset and E. L. Nuermberger, PLoS Neglected Trop. Dis., 2018, 12, e0006728 CrossRef.
- N. Scherr, R. Bieri, S. S. Thomas, A. Chauffour, N. P. Kalia, P. Schneide, M. T. Ruf, A. Lamelas, M. S. S. Manimekalai, G. Grüber, N. Ishii, K. Suzuki, M. Tanner, G. C. Moraski, M. J. Miller, M. Witschel, V. Jarlier, G. Pluschke and K. Pethe, Nat. Commun., 2018, 9, 5370 CrossRef CAS.
- T. F. Omansen, T. S. van der Werf and R. O. Phillips, in Buruli Ulcer: Mycobacterium Ulcerans Disease, 2019, p. 203 Search PubMed.
- W. A. Nienhuis, Y. Stienstra, W. Thompson, P. C. Awuah, K. M. Abass, W. Tuah, N. Y. Awua-Boateng, E. O. Ampadu, V. Siegmund, J. P. Schouten, O. Adjei, G. Bretzel and T. S. van der Werf, Lancet, 2010, 375, 664 CrossRef CAS.
- Y. Minato, J. M. Thiede, S. L. Kordus, E. J. McKlveen, B. J. Turman and A. D. Baughn, Antimicrob. Agents Chemother., 2015, 59, 5097 CrossRef CAS.
- M. R. Nixon, K. W. Saionz, M. S. Koo, M. J. Szymonifka, H. Jung, J. P. Roberts, M. Nandakumar, A. Kumar, R. Liao, T. Rustad, J. C. Sacchettini, K. Y. Rhee, J. S. Freundlich and D. R. Sherman, Chem. Biol., 2014, 21, 819 CrossRef CAS.
- S. R. Pattyn and J. van Ermengem, Int. J. Lepr. Other Mycobact. Dis., 1968, 36, 427 CAS.
- A. M. Dhople, J. Antimicrob. Chemother., 2001, 47, 93 CrossRef CAS.
- A. M. Dhople, Int. J. Antimicrob. Agents, 2002, 19, 71 CrossRef CAS.
- A. M. Dhople, Int. J. Antimicrob. Agents, 1999, 12, 319 CrossRef CAS.
- A. M. Dhople, Arzneim. Forsch., 1999, 49, 267 CAS.
- A. M. Dhople, I. Ortega, J. K. Seydel and G. D. Gardner, Indian J. Lepr., 1990, 62, 76 CAS.
- A. Kumar, A. Guardia, G. Colmenarejo, E. Pérez, R. R. Gonzalez, P. Torres, D. Calvo, R. M. Gómez, F. Ortega, E. Jiménez, R. C. Gabarro, J. Rullás, L. Ballell and D. R. Sherman, ACS Infect. Dis., 2016, 1, 604 CrossRef.
- B. Hajian, E. Scocchera, S. Keshipeddy, N. G-Dayanandan, C. Shoen, J. Krucinska, S. Reeve, M. Cynamon, A. C. Anderson and D. L. Wright, PLoS One, 2016, 11, e0161740 CrossRef.
- B. Hajian, E. Scocchera, C. Shoen, J. Krucinska, K. Viswanathan, N. G-Dayanandan, H. Erlandsen, A. Estrada, K. Mikušová, J. Korduláková, M. Cynamon and D. Wright, Cell Chem. Biol., 2019, 26, 781 CrossRef CAS.
- J. Zheng, E. J. Rubin, P. Bifani, V. Mathys, V. Lim, M. Au, J. Jang, J. Nam, T. Dick, J. R. Walker, K. Pethe and L. R. Camacho, J. Biol. Chem., 2013, 388, 23447 CrossRef.
- T. P. Stinear, T. Seemann, S. Pidot, W. Frigui, G. Reysset, T. Garnier, G. Meurice, D. Simon, C. Bouchier, L. Ma, M. Tichit, J. L. Porter, J. Ryan, P. D. R. Johnson, J. K. Davies, G. A. Jenkin, P. L. C. Small, L. M. Jones, F. Tekaia, F. Laval, M. Daffé, J. Parkhill and S. T. Cole, Genome Res., 2007, 17, 192 CrossRef CAS.
- M. Cammarata, R. Thyer, M. Lombardo, A. Anderson, D. Wright, A. Ellington and J. S. Brodbelt, Chem. Sci., 2017, 8, 4062 RSC.
- H. H. Locher, H. Schlunegger, P. G. Hartman, P. Angehrn and R. L. Then, Antimicrob. Agents Chemother., 1996, 40, 1376 CrossRef CAS.
- M. R. Sawaya and J. Kraut, Biochemistry, 1997, 36, 586 CrossRef CAS.
- R. De Groot, M. Sluijter, A. D. De Bruyn, J. Campos, W. H. F. Goessens, A. L. Smith and P. W. M. Hermans, Antimicrob. Agents Chemother., 1996, 40, 2131 CrossRef CAS.
- G. E. Dale, C. Broger, A. D'Arcy, P. G. Hartman, R. DeHoogt, S. Jolidon, I. Kompis, A. M. Labhardt, H. Langen, H. Locher, M. G. P. Page, D. Stüber, R. L. Then, B. Wipf and C. Oefner, J. Mol. Biol., 1997, 266, 23 CrossRef CAS.
- A. Pikis, J. A. Donkersloot, W. J. Rodriguez and J. M. Keith, J. Infect. Dis., 1998, 178, 700 CAS.
- M. F. Chughlay, E. Rossignol, C. Donini, M. El Gaaloul, U. Lorch, S. Coates, G. Langdon, T. Hammond, J. Möhrle and S. Chalon, Br. J. Clin. Pharmacol., 2020, 86, 1113 CrossRef CAS.
- P. Myler, R. Stacy, L. Stewart, B. Staker, W. Van Voorhis, G. Varani and G. Buchko, Infect. Disord.: Drug Targets, 2009, 9, 493 CAS.
- R. Stacy, D. W. Begley, I. Phan, B. L. Staker, W. C. Van Voorhis, G. Varani, G. W. Buchko, L. J. Stewart and P. J. Myler, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 979 CrossRef CAS.
- R. Choi, A. Kelley, D. Leibly, S. Nakazawa Hewitt, A. Napuli and W. Van Voorhis, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 998 CrossRef CAS.
- D. A. Serbzhinskiy, M. C. Clifton, B. Sankaran, B. L. Staker, T. E. Edwards and P. J. Myler, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 594 CrossRef CAS.
- C. M. Bryan, J. Bhandari, A. J. Napuli, D. J. Leibly, R. Choi, A. Kelley, W. C. Van Voorhis, T. E. Edwards and L. J. Stewart, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 1010 CrossRef CAS.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207 CrossRef CAS.
- M. Vedadi, J. Lew, J. Artz, M. Amani, Y. Zhao, A. Dong, G. A. Wasney, M. Gao, T. Hills, S. Brokx, W. Qiu, S. Sharma, A. Diassiti, Z. Alam, M. Melone, A. Mulichak, A. Wernimont, J. Bray, P. Loppnau, O. Plotnikova, K. Newberry, E. Sundararajan, S. Houston, J. Walker, W. Tempel, A. Bochkarev, I. Kozieradzki, A. Edwards, C. Arrowsmith, D. Roos, K. Kain and R. Hui, Mol. Biochem. Parasitol., 2007, 151, 100 CrossRef CAS.
- C. Yung-Chi and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099 CrossRef.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125 CrossRef CAS.
- A. Vagin and A. Teplyakov, J. Appl. Crystallogr., 1997, 30, 1022 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235 CrossRef CAS.
- R. Li, R. Sirawaraporn, P. Chitnumsub, W. Sirawaraporn, J. Wooden, F. Athappilly, S. Turley and W. G. J. Hol, J. Mol. Biol., 2000, 295, 307 CrossRef CAS.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486 CrossRef CAS.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213 CrossRef CAS.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12 CrossRef CAS.
- Z. S. Derewenda, Structure, 2004, 12, 529 CrossRef CAS.
- E. L. White, L. J. Ross, A. Cunningham and V. Escuyer, FEMS Microbiol. Lett., 2004, 232, 101 CrossRef CAS.
- C. R. Bourne, E. W. Barrow, R. A. Bunce, P. C. Bourne, K. D. Berlin and W. W. Barrow, Antimicrob. Agents Chemother., 2010, 54, 3825 CrossRef CAS.
- A. Wróbel, K. Arciszewska, D. Maliszewski and D. Drozdowska, J. Antibiot., 2020, 73, 5 CrossRef.
- O. Sköld, Vet. Res., 2001, 32, 261 CrossRef.
- S. C. C. Meyer, S. K. Majumder and M. H. Cynamon, Antimicrob. Agents Chemother., 1995, 39, 1862 CrossRef CAS.
- A. Kumar, M. Zhang, L. Zhu, R. P. Liao, C. Mutai, S. Hafsat, D. R. Sherman and M. W. Wang, PLoS One, 2012, 7, e39961 CrossRef CAS.
- D. Liebschner, P. V. Afonine, N. W. Moriarty, B. K. Poon, O. V. Sobolev, T. C. Terwilliger and P. D. Adams, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 148 CrossRef CAS.
- J. A. Ribeiro, S. M. Chavez-Pacheco, G. S. De Oliveira, C. Dos Santos Silva, J. H. P. Giudice, G. A. Libreros-Zúñiga and M. V. B. Dias, Acta Crystallogr., Sect. D: Struct. Biol., 2019, 75, 682 CrossRef CAS.
- Y. Yuthavong, B. Tarnchompoo, T. Vilaivan, P. Chitnumsub, S. Kamchonwongpaisan, S. A. Charman, D. N. McLennan, K. L. White, L. Vivas, E. Bongard, C. Thongphanchang, S. Taweechai, J. Vanichtanankul, R. Rattanajak, U. Arwon, P. Fantauzzi, J. Yuvaniyama, W. N. Charman and D. Matthews, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16823 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00303d |
| This journal is © The Royal Society of Chemistry 2021 |