DOI:
10.1039/D0MD00259C
(Research Article)
RSC Med. Chem., 2021,
12, 95-102
Pharmacophore-based screening of diamidine small molecule inhibitors for protein arginine methyltransferases†
Received
23rd July 2020
, Accepted 13th September 2020
First published on 30th September 2020
Abstract
Protein arginine methyltransferases (PRMTs) are essential epigenetic and post-translational regulators in eukaryotic organisms. Dysregulation of PRMTs is intimately related to multiple types of human diseases, particularly cancer. Based on the previously reported PRMT1 inhibitors bearing the diamidine pharmacophore, we performed virtual screening to identify additional amidine-associated structural analogs. Subsequent enzymatic tests and characterization led to the discovery of a top lead K313 (2-(4-((4-carbamimidoylphenyl)amino)phenyl)-1H-indole-6-carboximidamide), which possessed low-micromolar potency with biochemical IC50 of 2.6 μM for human PRMT1. Limited selectivity was observed over some other PRMT isoforms such as CARM1 and PRMT7. Molecular modeling and inhibition pattern studies suggest that K313 is a nonclassic noncompetitive inhibitor to PRMT1. K313 significantly inhibited cell proliferation and reduced the arginine asymmetric dimethylation level in the leukaemia cancer cells.
Introduction
Epigenetics refers to the study of functional changes in the genome without altering the underlying nucleotide sequence.1,2 Epigenetic alterations, especially DNA methylation and histone modifications, play critical regulatory roles in eukaryotic biology. Aberrant epigenetic regulation is causative to various disease states, including diabetes, inflammation, and cancer.3 Protein arginine methyltransferases (PRMTs) modulate chromatin remodeling by depositing methyl marks on specific arginine residues of nucleosomal histones.4 Major PRMTs also have a broad spectrum of non-histone substrates that are pertinent to numerous signaling pathways.5 Nine PRMT members have been found in mammalian cells that transfer the methyl group from S-adenosylmethionine (SAM or AdoMet) to replace a hydrogen atom on the ω-nitrogen of arginine guanidine group of protein substrates, generating three types of methylated states.6,7 Type I PRMTs (PRMT1, 2, 3, 4, 6 and 8) are responsible for the formation of monomethylarginine (MMA) and asymmetric dimethylarginine (ADMA); type II PRMTs (contributed predominantly by PRMT5 and marginally by PRMT9) catalyze the formation of MMA and symmetric dimethylarginine (SDMA); and type III enzyme (PRMT7) only produces MMA.8 NDUFAF7 and METTL23 are two additional PRMTs reported recently,9,10 but their methyltransferase activity remains to be characterized and validated. PRMT1 is predominant in type I PRMTs that accounts for more than 50% of ADMA formation in mouse embryonic fibroblasts as demonstrated by conditional knockout experiments.11 PRMTs are involved in critical biological processes, including cell death, cell cycle progression, and RNA processing.12 Although biological roles of PRMTs are not fully understood, overexpression and dysregulation of PRMTs are commonly observed in cancers, cardiovascular disorders, and many other pathological conditions.13–15
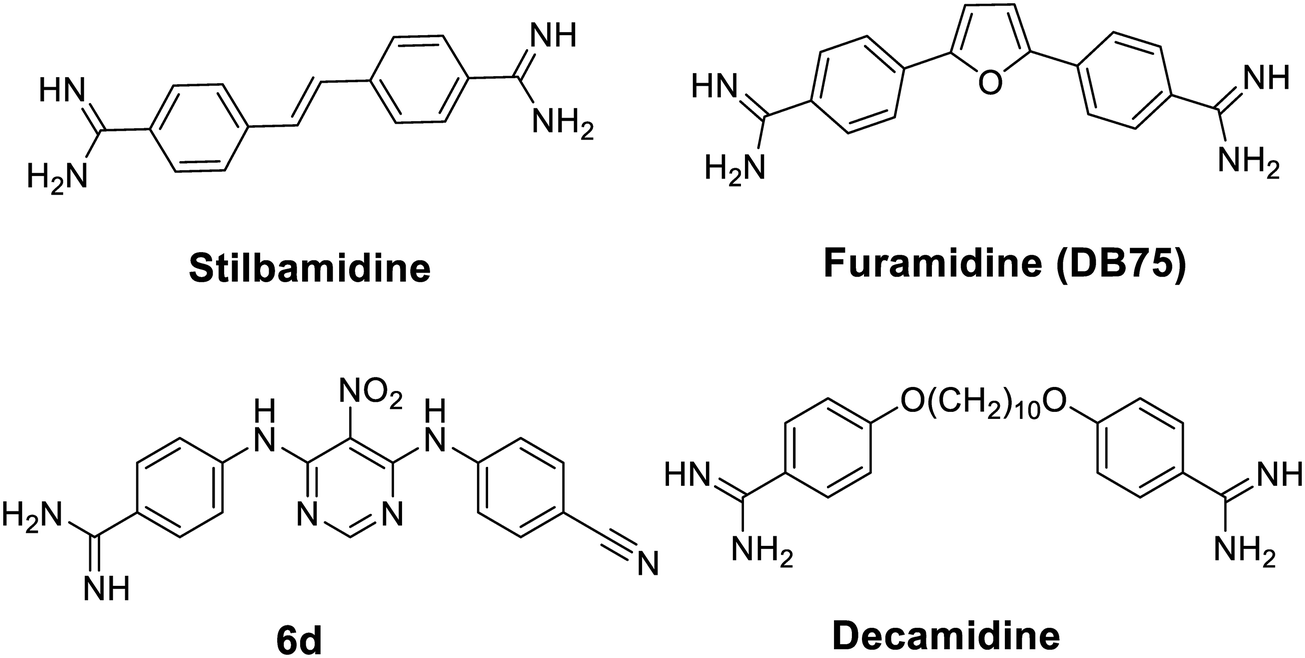
An increasing number of inhibitors have been reported to target various histone modifying enzymes, some of which have been approved by the FDA to treat hematological cancer (e.g. histone deacetylases inhibitors vorinostat, belinostat, and romidepsin) and many more are in the clinical trials.16 Small molecule inhibitors of PRMTs have potentials to be therapeutic agents targeting diseases modulated by aberrant PRMT activities.17 Selective PRMT inhibitors are also powerful chemical tools to elucidate biological functions of PRMT isoforms.18 Thus far, quite a few PRMT small molecule inhibitors have been reported.19–21 Notably, a diamidine compound stilbamidine (Fig. 1), was first reported by Jung and coworkers to show inhibitory activity against PRMT1.22 Diamidine compounds such as stilbamidine,23 pentamidine,24,25 DAPI26,27 and furamidine28 are previously known as DNA binders and antiparasitic agents. We hypothesize that the amidine moiety, which is rigidly planar, highly basic and contains two terminal nitrogen atoms, can potentially mimic the side chain guanidino group of the arginine substrate, thus serving the basis of designing small molecule inhibitors to target PRMT enzymes. From a biochemical screening on diamidine-structure focused library, we identified furamidine (Fig. 1) as a PRMT1-targeting lead inhibitor.29 The IC50 value of furamidine was 9.4 μM against PRMT1, better than stilbamidine (IC50 15.2 μM). Importantly, the potency of furamidine for PRMT1 is 18-fold, >40-fold, and 30-fold higher against PRMT5, CARM1, and PRMT6, respectively. Kinetic characterization and molecular docking suggested that furamidine primarily acted as a substrate-competitive inhibitor. Furamidine significantly reduced cell growth and ADMA level in leukemia and glioblastoma cell lines.29,30 It was further used as a chemical probe to study biological functions of PRMT1 in Smad signaling, in alternative RNA splicing, and in conferring TKI resistance of non-small cell lung cancer.31–33 We further designed and synthesized a series of diamidine compounds bearing varied lengths of central methylene linker, which led to the discovery of decamidine (Fig. 1), that displayed a 2-fold increase in potency for PRMT1 than furamidine, while pentamidine only showed IC50 of 81 μM.34 Compound 6d (Fig. 1), which contains one amidine group, was also reported as a PRMT inhibitor.35 These series of studies demonstrated that diamidine structure is an bona fide pharmacophore for PRMT inhibitor development. In this work, we carried out a ligand-based pharmacophore screening with furamidine as a start to identify improved diamidine leads for PRMT1 inhibition.
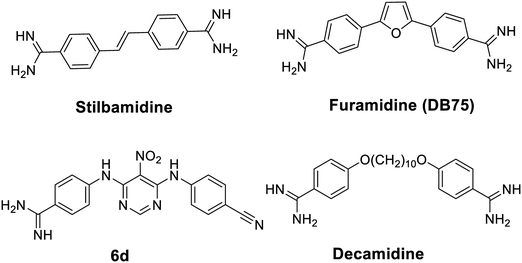 |
| | Fig. 1 Structures of the reported amidine-containing inhibitors for PRMT1. | |
Results and discussion
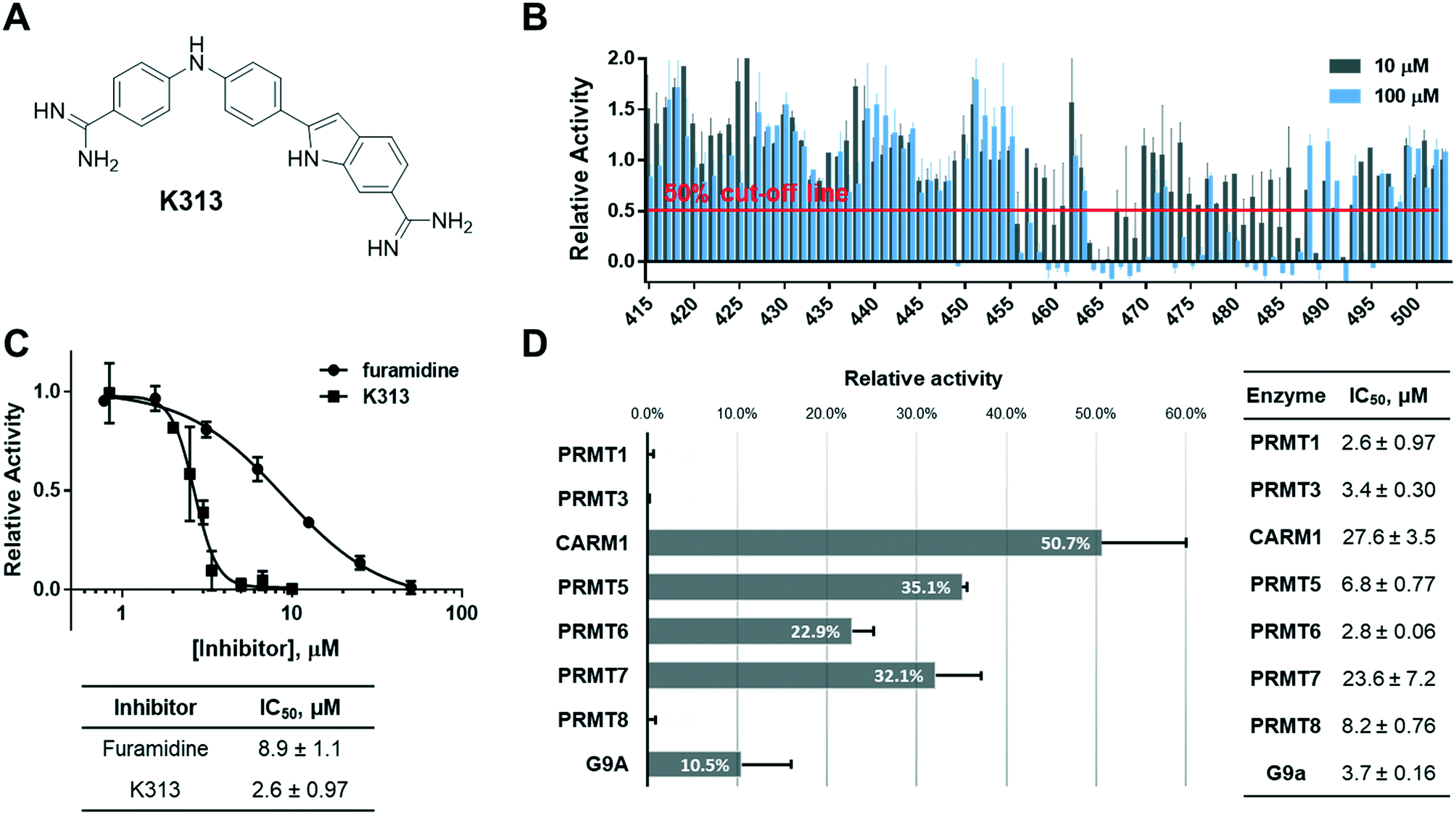
Because furamidine is thus far the most potent and selective diamidine inhibitor for PRMT1, we set out to conduct a ligand-based virtual screening with furamidine as the ligand model to further expand the diversity of diamidines for PRMT inhibition (ESI† Fig. S1). Both charge and shape similarity filtrations were applied to search the National Cancer Institute (NCI) Diversity Set (a total of 260![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 071 compounds).36 Each compound was expanded into a set of 50 three-dimensional conformations using Omega 2.5.1.4 of OpenEye software.37 The three-dimensional shape comparison between DB75 and the molecules in the NCI Diversity Set was carried out using ROCS 3.2.1.4.38 The top 1000 ranked compounds from the shape-based screen were then assessed for similarity to DB75 using EON 2.2.0.5 (ref. 39) to calculate an electrostatic Tanimoto (ET) score, which is a measure of the electrostatic similarity between two small molecules. A total of 830 compounds, representing the merging of the top 500 hits from the two screens, were subsequently selected. 406 of these top 830 compounds were obtained from the Development Therapeutics Program (DTP) depository of the NCI. These 406 top hits (numbered from K1 to K406) were subjected to biochemical screening by using the radiometric scintillation proximity assay (SPA)40 at single concentration of 10 μM each against recombinant PRMT1 (exemplary for type I PRMTs) and PRMT5 (exemplary for type II PRMTs) (ESI† Fig. S2). The inhibitory activity test yielded 33 hits that inhibited PRMT1 activity by more than 50% at 10 μM, which suggested their improved potency as compared to furamidine (Fig. 1) whose IC50 is 8.9 μM in this assay (Fig. 2C). Further concentration-dependent tests found that compound K313, (2-(4-((4-carbamimidoylphenyl)amino)phenyl)-1H-indole-6-carboximidamide) (Fig. 2A), is the best lead among the 33 hits, with an IC50 of 2.6 ± 0.97 μM for PRMT1 (Fig. 2C). To obtain more structural diamidine analogs for PRMT1 inhibition, we conducted a second round of virtual screening search on the NCI Diversity Set by using the structure of K313 as the filtration template. With a similar prioritization strategy in the furamidine-based virtual screening, we were able to obtain 89 more compounds from the DTP depository (numbered from K415 to K503) and screened them at 10 μM and 100 μM against PRMT1 in SPA measurement. In this second screening, 14 compounds were able to inhibit PRMT1 activity by more than 50% at 10 μM (Fig. 2B). K313 and K465 possessed the best potency against PRMT1 (K313 IC50 = 2.6 ± 0.97 μM, Fig. 2C; K465 IC50 = 2.4 ± 1.0 μM, Fig. 4A).
071 compounds).36 Each compound was expanded into a set of 50 three-dimensional conformations using Omega 2.5.1.4 of OpenEye software.37 The three-dimensional shape comparison between DB75 and the molecules in the NCI Diversity Set was carried out using ROCS 3.2.1.4.38 The top 1000 ranked compounds from the shape-based screen were then assessed for similarity to DB75 using EON 2.2.0.5 (ref. 39) to calculate an electrostatic Tanimoto (ET) score, which is a measure of the electrostatic similarity between two small molecules. A total of 830 compounds, representing the merging of the top 500 hits from the two screens, were subsequently selected. 406 of these top 830 compounds were obtained from the Development Therapeutics Program (DTP) depository of the NCI. These 406 top hits (numbered from K1 to K406) were subjected to biochemical screening by using the radiometric scintillation proximity assay (SPA)40 at single concentration of 10 μM each against recombinant PRMT1 (exemplary for type I PRMTs) and PRMT5 (exemplary for type II PRMTs) (ESI† Fig. S2). The inhibitory activity test yielded 33 hits that inhibited PRMT1 activity by more than 50% at 10 μM, which suggested their improved potency as compared to furamidine (Fig. 1) whose IC50 is 8.9 μM in this assay (Fig. 2C). Further concentration-dependent tests found that compound K313, (2-(4-((4-carbamimidoylphenyl)amino)phenyl)-1H-indole-6-carboximidamide) (Fig. 2A), is the best lead among the 33 hits, with an IC50 of 2.6 ± 0.97 μM for PRMT1 (Fig. 2C). To obtain more structural diamidine analogs for PRMT1 inhibition, we conducted a second round of virtual screening search on the NCI Diversity Set by using the structure of K313 as the filtration template. With a similar prioritization strategy in the furamidine-based virtual screening, we were able to obtain 89 more compounds from the DTP depository (numbered from K415 to K503) and screened them at 10 μM and 100 μM against PRMT1 in SPA measurement. In this second screening, 14 compounds were able to inhibit PRMT1 activity by more than 50% at 10 μM (Fig. 2B). K313 and K465 possessed the best potency against PRMT1 (K313 IC50 = 2.6 ± 0.97 μM, Fig. 2C; K465 IC50 = 2.4 ± 1.0 μM, Fig. 4A).
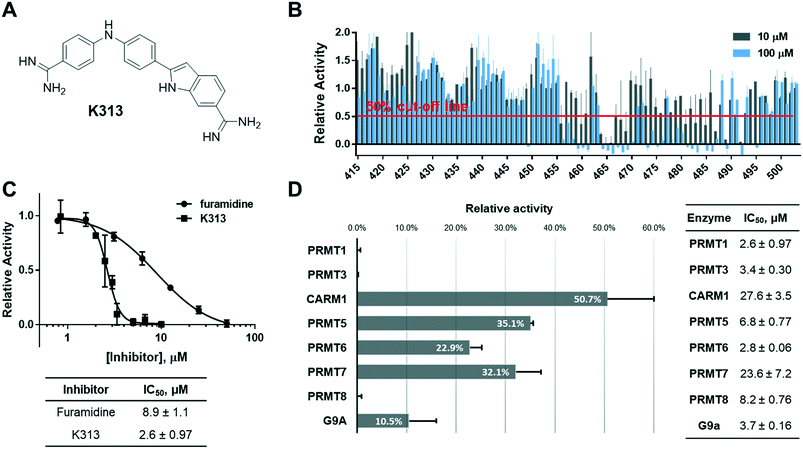 |
| | Fig. 2 Identification of compound K313. (A) Structure of K313. (B) Two-concentration (10 μM and 100 μM) screening result of the K313-derived library, n = 2. (C) IC50 curves and values of K313 and furamidine, n = 2 for each compound shown. (D) Inhibitory activity of K313 at 10 μM against a panel of methyltransferases and the corresponding IC50 values. | |
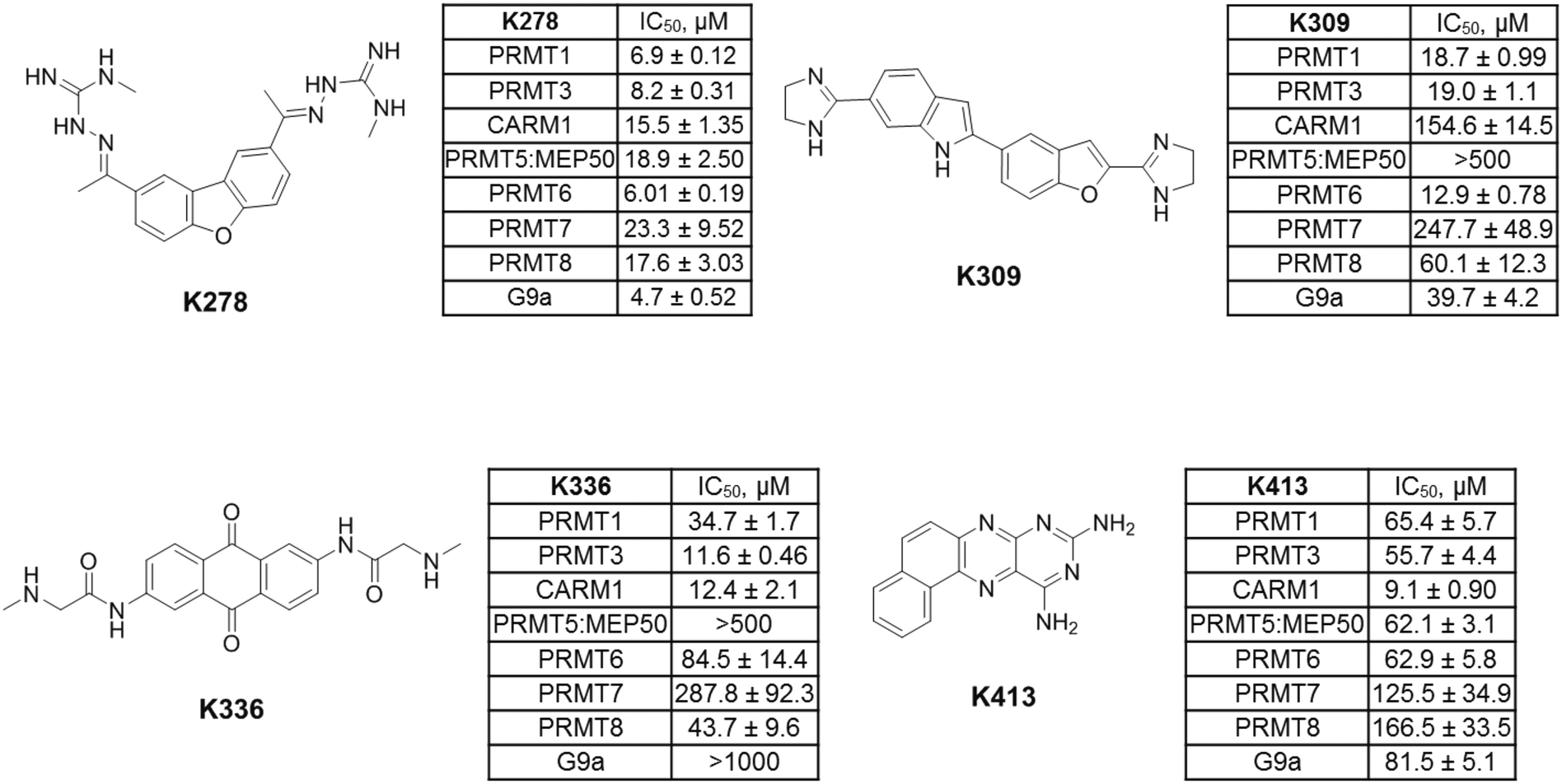
Next, the inhibitory activity of K313 against a panel of methyltransferases containing seven PRMT isoforms (PRMT1, PRMT3, CARM1, PRMT5:MEP50 complex, PRMT6, PRMT7, and PRMT8) and a lysine methyltransferase (G9a) was determined. Compound K313 was serially diluted and added to the enzyme, cofactor and substrate mixture, and the changes in the amount of methylated substrates was quantified by SPA. For each enzyme, the reaction was kept at initial rate conditions that the reaction yield is controlled under 10%, and the substrate concentrations are close to their Km. Details of the substrates and reaction conditions are summarized in Table S1.† The selectivity profile at a single-point concentration of 10 μM proved that K313 is a potent inhibitor for tested PRMTs in the panel, with strongest activity against PRMT1 but also significantly inhibited PRMT3 and PRMT8 (Fig. 2D). The selectivity of the other hits representing different chemical scaffold was also measured (Fig. 3), among which K413 was the most potent against CARM1, K336 was more potent against PRMT3 and CARM1, and K278 and K309 were more potent for type-I members PRMT1, 3, and 6. In general, these positively charged, heteroaromatic ring-containing structures tend to inhibit type-I PRMT members stronger than PRMT5. Fine tuning of these hits by altering their substructures or adding extra functional groups could further improve potency and selectivity.
 |
| | Fig. 3 Selectivity profiles of selected hits from the furamidine-derived library against a panel of protein methyltransferases. The final concentrations of the enzyme, [3H]-SAM, and histone protein or peptide are summarized in Table S1.† | |
We performed a structure–activity relationship (SAR) analysis with the top experimental hits. These compounds were categorized into three series according to their frame structures and summarized in Fig. 4. In series 1 (Fig. 4A–D), compounds share two indene-like cores connected by a two-carbon linker with symmetrical feature, in which K465 showed the best IC50 at 2.4 ± 1.0 μM in this series. The substitution of the 5-membered ring can affect the activity of the compounds. Having a hydrogen bond donor NH in the 5-membered ring (position X1, X2, X3 and X4) offered improved potency than having a carbon or oxygen in the same position, as seen in many structures of Fig. 4A and B. The additional methyl group on the double-bond linker of K457 did not bring a significant change on the potency, comparing to K467 (Fig. 4A). Changing the two amidine groups to dihydroimidazole rings decreased potency, comparing K473 to K464. The flexible linkers are not favored, comparing K461 to K465, and K458 to K469 (Fig. 4D). However, the cis conformation showed less significant effect (Fig. 4C). It is possible that the flexible linkers induce additional entropy penalty for inhibitor–enzyme binding. In series 2 (Fig. 4E–H), a benzamidine moiety is connected at the para-position with an indene-like core. Similar to series 1, dihydroimidazole rings are less favored than amidine groups, comparing K480 to K476. For compounds in Fig. 4G and H, NH substitution at X1 position of the 5-membered ring provides better potencies than carbon or oxygen. Replacement of the ethylene linker with an amide bond lowers the potency, as reflected by compounds in Fig. 4F and H. Lastly, in series 3 (Fig. 4I), inhibitors bearing different lengths of the linkers are listed. The shorter linker (K309 and K489) is more preferred than the four-carbon diene linker (K475 and K481). This likely reflects the size limitation of the binding pocket in PRMT1.
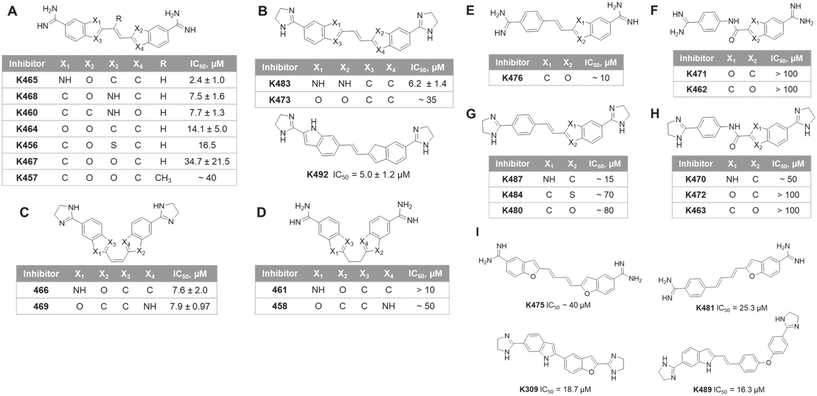 |
| | Fig. 4 Structure of the K313 analogs and their corresponding IC50 values of PRMT1 inhibition. The compounds are categorized in three series by the structure features. (A) to (D), series 1, compounds with symmetrical features that two indene-like cores connected by a two-carbon linker. (E) to (H), series 2, compounds with a benzamidine moiety is connected at the para-position with an indene-like core. (I), series 3, compounds with various linker lengths. | |
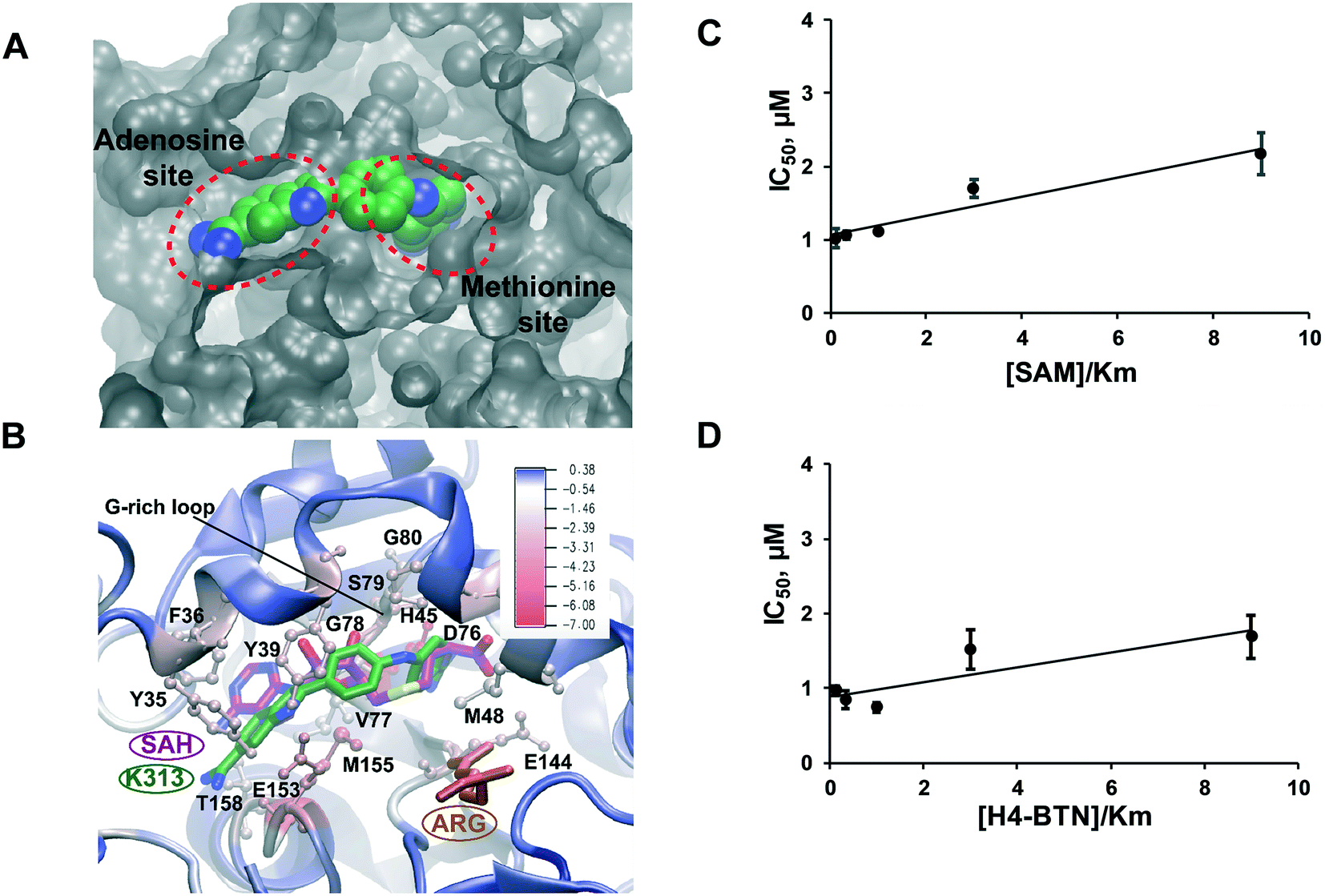
To explore the molecular mechanism of K313 in PRMT1 inhibition, molecular docking was performed using a hPRMT1 homology model as the target with AutoDock4.41 The region including the SAM-binding site and substrate arginine site was covered with a grid for docking. Subsequently, 50 ns molecular dynamics (MD) simulation was conducted for the predicted PRMT1–K313 complex from the docking. Following MD, the binding energy for K313 was evaluated using molecular mechanics/Poisson–Boltzmann solvent-accessible surface area (MM–PBSA) calculations.42 The substituents on the linker of K313 significantly increased its binding affinity. The estimated binding energy of K313 (−37.90 kcal mol−1) was less than that of furamidine (DB75, −28.9 kcal mol−1),29 which is in agreement with the experimental IC50. To elucidate the detailed atomic interactions responsible for stabilizing K313 in the PRMT1, the binding free energy was decomposed individual contributions per residue level of the PRMT1. As shown in Fig. 5A and B, the diamidine moiety of K313 inserts into both the SAM adenosine binding site and methionine binding site, which differs with the previously reported binding mode of furamidine.29 In our previous molecular modeling analysis, furamidine was inserted into the substrate binding site and only partially overlapped with the cofactor binding site. The two amidine groups engaged with the glutamate residues via hydrogen bonding. Experimentally, it was primary competitive with the peptide while was non-competitive for the cofactor.29 In the K313–PRMT1 model, the interaction of the amidine groups with the acidic residues has remained (Fig. 5B). However, comparing to furamidine, K313 possesses longer and more hydrophobic backbone between the two amidine groups (Fig. 2A). One amidine group of K313 deeply inserts into the cofactor methionine pocket, interacts with the Gly-rich loop of PRMT1 (residues G78 and G80) via hydrogen bonds with the side chain of D76 and the backbone carbonyl group of S79; and the second amidine group and adenosine moiety of K313 is embedded by several hydrophobic residues (Val77, Try35, Phe36, and Tyr39, YFXY motif) that are responsible for hydrophobic interactions (π–π interactions). To further examine its inhibition mode, we measured the IC50 of K313 at various concentrations (0.3125–4.5 μM) of the substrate peptide or cofactor, while keeping the other component at a fixed concentration close to the Km value (i.e. 0.5 μM) (ESI† Fig. S3). A slight increase in the IC50 values were observed in both cases (Fig. 5C and D), which is the pattern of non-classic non-competitive inhibition. The structural difference between K313 and furamidine might explain their different inhibition mode to PRMT1. The longer backbone of K313 allows deeper placement in the cofactor pocket, which also provides additional hydrophobic interactions that might account for its stronger binding with PRMT1 than furamidine. The molecular modeling in this study and previous studies29,34 showed the importance of the amidine groups for this type of PRMT inhibitors. It is also reflected in the SAR of compounds in Fig. 4 that replacing diamidine with dihydroimidazole rings lowered the inhibitor's potency. Meanwhile, the lengths and hydrophobicity of the linker between the two amidine groups may play a role in tuning the inhibitor potency, as we have seen in the SAR of compounds in Fig. 4 that the amide bond linker was not favored, along with our previous study on decamidine34 that certain linker lengths are more preferred.
 |
| | Fig. 5 Mode of action of K313. (A) The modeled binding pocket of K313 bound to PRMT1 and (B) ligand–residue binding energies from MM/PBSA energy decomposition. The protein (in cartoon representation) is colored according to the residue contribution values in the free energy decomposition from red (negative) to blue (positive). K313, SAH and substrate ARG are colored green, purple and purple, respectively. (C) and (D) K313 is a noncompetitive inhibitor of PRMT1. IC50 data were obtained at varying concentrations of cofactor SAM or substrate peptide H4-BTN. Km value of 0.5 μM was used for both SAM and H4-BTN (n = 2). | |
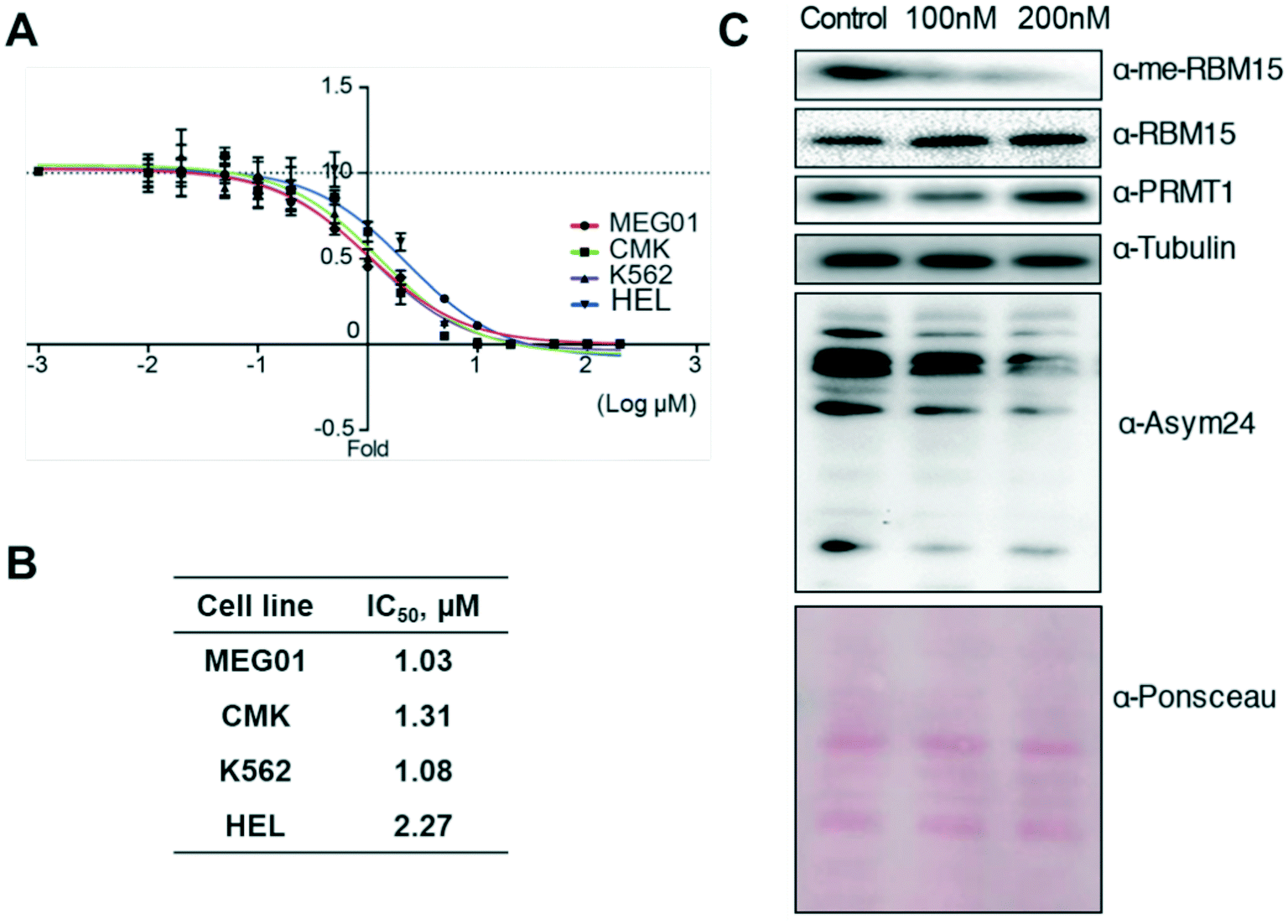
Major PRMTs have been reported to play oncogenic roles in regulating cancer biology.43 PRMT1 plays critical roles to regulate alternative RNA splicing in leukaemia through methylation of its substrate RBM15.31 Herein, we assessed the anti-cancer activity of K313 in several leukaemia cell lines. The cell growth inhibition IC50 values of K313 ranged from 1.03 μM to 2.27 μM after 72-hour treatment in MEG01, CMK, K562 and HEL cell lines, which are in close range of its biochemical potency (Fig. 6A and B). Next, the methylation level of RBM15 and total methylation level of the cells were evaluated by using immunoblotting. At 100 nM and 200 nM of K313 treatment in MEG01 cells, the methylation level of RBM15 was significantly reduced in a dose-dependent manner. A decrease in signal intensity was also observed in the pan-asymmetric methylation (α-Asym24, Fig. 6C). These data coincide with the previous observations that the diamidine inhibitors exhibit effective anti-neoplastic performance in cancer cells which is well in line with their inhibitory activity to reduce arginine asymmetric methylation level in substrate proteins.29–31,33
 |
| | Fig. 6 K313 inhibits proliferation of leukemia cell lines via blocking PRMT1 activity. (A and B) K313 inhibited leukemic cell growth. (A) K313 was diluted serially and added to the MEG01, CMK, K562, and HEL cell cultures and cell viability was measured after 72 h treatment. The same amount of DMSO was added to the control. (B) The sensitivity curves were plotted to obtain IC50 values. (C) Treatment of K313 decreased the arginine methylation level MEG01 cells. MEG01 cells were cultured with DMSO control or K313 for 24 h, and the cell extract samples were resolved by SDS-PAGE. PRMT1 activity was measured by the methylation level of substrate protein RBM15 by anti-methyl-RBM15 antibody and the total asymmetric arginine methylation level (α-Asym24 antibody). Equal loading was confirmed by Ponceau S staining. | |
Conclusion
In this work, we conducted a pharmacophore-based virtual screening in combination with biochemical assays for PRMT inhibitor development, which resulted in the identification of a variety of diamidine-bearing compounds that inhibited PRMT1 activity at single-digit micromolar level. In particular, we discovered the diamidine compound K313 as a new potent PRMT1 inhibitor that shows strong anti-cancer activity in leukemia cells. Together with our previous work,29,34 these results have further demonstrated the importance of the diamidine pharmacophore in designing PRMT isoform selective inhibitors. Diamidine compounds exemplified by furamidine (DB75) and K313 provide lead structures that can be potentially further optimized into therapeutic agents for cancer treatment and chemical probes for epigenetic research.
Experimental section
Materials and reagents
PRMT5:MEP50 complex was purchased from Reaction Biology Corp (part number: HMT-22-148). PRMT1 other proteins were obtained from bacterial expression as described below. Cofactor [3H]-SAM was purchased from Perkin Elmer (part number: NET155V001MC). Histone H3.3 was purchased from New England Biolabs (part number: M2507S). Biotinylated histone H4 peptide was synthesized in lab, method described in ESI.†
Protein expression and purification of recombinant methyltransferases
Recombinant His-tagged human PRMT1, PRMT3, PRMT8 and G9a were expressed in E. coli. In brief, the corresponding pET28b plasmid was transformed into BL21(DE3) (Stratagene) by heat shock method. Transformed bacteria were incubated in LB media at 37 °C for growth and then at 16 °C for protein expression with 0.3 mM IPTG induction. Cells were harvested by centrifuge and lysed by microfluidics cell disrupter. The supernatant containing target protein was loaded onto the Ni-charged His6x-tag binding resin (Novagen) in equilibrium buffer (25 mM Na-HEPES, pH 7.0, 300 mM NaCl, 1 mM PMSF, 10% glycerol (v/v) and 30 mM imidazole). Beads were washed thoroughly by washing buffer (25 mM Na-HEPES, pH 7.0, 300 mM NaCl, 1 mM PMSF, 10% glycerol (v/v) and 70 mM imidazole), and protein was eluted with elution buffer (25 mM Na-HEPES, pH 7.0, 300 mM NaCl, 1 mM PMSF, 100 mM EDTA, 10% glycerol (v/v) and 200 mM imidazole). Recombinant GST tagged CARM1 and PRMT7 on pGEX2T or 4T plasmid were expressed in E. coli. The supernatant containing target protein was loaded onto the Glutathione Sepharose 4B resin (GE Healthcare) in column buffer (25 mM Na-HEPES, pH 7.0, 250 mM NaCl, 1 mm EDTA, 1 mM PMSF, and 0.1% (v/v) Triton x100). Beads were washed thoroughly by the column, and protein was eluted with elution buffer (50 mM Na-HEPES, pH 7.0, 10 mM reduced glutathione and 1 mM PMSF). All the eluted protein solutions were dialyzed into buffer containing 25 mM Na-HEPES, pH 7.0, 300 mM NaCl, 10% glycerol (v/v) and 1 mM DTT. Protein purity were checked by 12% SDS-PAGE, and concentration was determined by Bradford assay.
Biochemical assays
The biochemical screening and inhibition mode studies were performed using scintillation proximity assay (SPA) method on 96-well plates. The selectivity profiling on a panel of methyltransferases was also performed using SPA except for CARM1, which was performed using the radioactive filter plate assay (FPA) with histone H3.3 protein as the substrate. Both SPA and FPA measures the signal from the methylated histone substrates, which carries [3H] labeled methyl group from [3H]-SAM. Detailed description can be found in the ESI.†
K313 identity and purity
1H and 13C NMR spectra were recorded on a Varian Mercury plus 400 MHz spectrometer (ESI† Fig. S4). 1H NMR (400 MHz, DMSO-d6) δ 7.85–7.80 (m, 3H), 7.69 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.4 Hz, 1H), 7.35 (dd, J = 8.3 Hz, 1.5 Hz, 1H), 7.22 (d, J = 8.7 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 6.86 (s, 1H); 13C NMR (400 MHz, DMSO-d6) δ 166.87, 164.73, 149.04, 142.72, 141.84, 136.45, 133.48, 130.44, 127.40, 125.23, 120.33, 120.07, 119.59, 119.29, 117.13, 115.27, 112.25, 98.67. Mass spectra were recorded on a Bruker Esquire 3000 plus ion trap mass spectrometer equipped with an ESI (electrospray) ion source. Target compound K313 was >95% purity, as determined by high-performance liquid chromatography (HPLC). HPLC was performed on a Shimadzu LC-20AT instrument using an Aeris peptide XB-C18 column (4.6 mm × 250 mm, 3.6 μm).
Abbreviations
| ADMA | Asymmetric dimethylarginine |
| HTS | High throughput screening |
| MMA | Monomethylarginine |
| PRMTs | Protein arginine methyltransferases |
| SAM |
S-Adenosyl methionine |
| SDMA | Symmetric dimethylarginine |
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgements
This work was funded by NIH grant GM126154 to Y. G. Z., NIH grant GM110387 and NSF grant MCB-2027902 to I. I., and NIH grants R21CA202390 and R011DK110574 to X. Z. We thank the Proteomics and Mass Spectrometry Facility and the Chemical Sciences Magnetic Resonance Facility of the University of Georgia for their assistance in collecting the MS and NMR data. Computational resources were provided in part by an allocation from the National Science Foundation XSEDE program CHE110042.
References
- A. Bird, Nature, 2007, 447, 396–398 CrossRef CAS.
-
Y. G. Zheng, in Epigenetic Technological Applications, ed. Y. G. Zheng, Academic Press, San Diego, 2015, pp. 1–18 Search PubMed.
- G. Egger, G. Liang, A. Aparicio and P. A. Jones, Nature, 2004, 429, 457 CrossRef CAS.
- A. Di Lorenzo and M. T. Bedford, FEBS Lett., 2011, 585, 2024–2031 CrossRef CAS.
- M. T. Bedford and S. G. Clarke, Mol. Cell, 2009, 33, 1–13 CrossRef CAS.
- R. S. Blanc and S. Richard, Mol. Cell, 2017, 65, 8–24 CrossRef CAS.
- M. D. Fulton, T. Brown and Y. G. Zheng, Chem. Rec., 2018, 18, 1792–1807 CrossRef CAS.
- J. Fuhrmann, K. W. Clancy and P. R. Thompson, Chem. Rev., 2015, 115, 5413–5461 CrossRef CAS.
- V. F. Rhein, J. Carroll, S. Ding, I. M. Fearnley and J. E. Walker, J. Biol. Chem., 2013, 288, 33016–33026 CrossRef CAS.
- Y. Hatanaka, T. Tsusaka, N. Shimizu, K. Morita, T. Suzuki, S. Machida, M. Satoh, A. Honda, M. Hirose, S. Kamimura, N. Ogonuki, T. Nakamura, K. Inoue, Y. Hosoi, N. Dohmae, T. Nakano, H. Kurumizaka, K. Matsumoto, Y. Shinkai and A. Ogura, Cell Rep., 2017, 20, 2756–2765 CrossRef CAS.
- S. Dhar, V. Vemulapalli, A. N. Patananan, G. L. Huang, A. Di Lorenzo, S. Richard, M. J. Comb, A. Guo, S. G. Clarke and M. T. Bedford, Sci. Rep., 2013, 3, 1311 CrossRef.
- M. T. Bedford and S. Richard, Mol. Cell, 2005, 18, 263–272 CrossRef CAS.
- Y. Yang and M. T. Bedford, Nat. Rev. Cancer, 2013, 13, 37–50 CrossRef CAS.
- E. Guccione and S. Richard, Nat. Rev. Mol. Cell Biol., 2019, 20, 642–657 CrossRef CAS.
- R. M. Baldwin, A. Morettin and J. Cote, World J. Biol. Chem., 2014, 5, 115–129 Search PubMed.
- S. X. Pfister and A. Ashworth, Nat. Rev. Drug Discovery, 2017, 16, 241–263 CrossRef CAS.
- R. A. Copeland, M. E. Solomon and V. M. Richon, Nat. Rev. Drug Discovery, 2009, 8, 724–732 CrossRef CAS.
- M. K. Luo, ACS Chem. Biol., 2012, 7, 443–463 CrossRef CAS.
- H. Hu, K. Qian, M. C. Ho and Y. G. Zheng, Expert Opin. Invest. Drugs, 2016, 25, 335–358 CrossRef CAS.
- H. U. Kaniskan, K. D. Konze and J. Jin, J. Med. Chem., 2015, 58, 1596–1629 CrossRef CAS.
- E. Smith, W. Zhou, P. Shindiapina, S. Sif, C. Li and R. A. Baiocchi, Expert Opin. Ther. Targets, 2018, 1–19 Search PubMed.
- A. Spannhoff, R. Heinke, I. Bauer, P. Trojer, E. Metzger, R. Gust, R. Schüle, G. Brosch, W. Sippl and M. Jung, J. Med. Chem., 2007, 50, 2319–2325 CrossRef CAS.
- N. Gresh and B. Pullman, Mol. Pharmaceutics, 1984, 25, 452–458 CAS.
- K. J. Edwards, T. C. Jenkins and S. Neidle, Biochemistry, 1992, 31, 7104–7109 CrossRef CAS.
- M. Cory, R. R. Tidwell and T. A. Fairley, J. Med. Chem., 1992, 35, 431–438 CrossRef CAS.
- J. Portugal and M. J. Waring, Biochim. Biophys. Acta, Gene Struct. Expression, 1988, 949, 158–168 CrossRef CAS.
- A. A. Farahat, A. Kumar, M. Say, A. E.-D. M. Barghash, F. E. Goda, H. M. Eisa, T. Wenzler, R. Brun, Y. Liu, L. Mickelson, W. D. Wilson and D. W. Boykin, Bioorg. Med. Chem., 2010, 18, 557–566 CrossRef CAS.
- J. B. Chaires, J. Ren, D. Hamelberg, A. Kumar, V. Pandya, D. W. Boykin and W. D. Wilson, J. Med. Chem., 2004, 47, 5729–5742 CrossRef CAS.
- L. Yan, C. Yan, K. Qian, H. Su, S. A. Kofsky-Wofford, W. C. Lee, X. Zhao, M. C. Ho, I. Ivanov and Y. G. Zheng, J. Med. Chem., 2014, 57, 2611–2622 CrossRef CAS.
- S. F. Samuel, A. J. Marsden, S. Deepak, F. Rivero, J. Greenman and P. Beltran-Alvarez, Proteomes, 2018, 6, 44 CrossRef CAS.
- L. Zhang, N. T. Tran, H. Su, R. Wang, Y. Lu, H. Tang, S. Aoyagi, A. Guo, A. Khodadadi-Jamayran, D. Zhou, K. Qian, T. Hricik, J. Cote, X. Han, W. Zhou, S. Laha, O. Abdel-Wahab, R. L. Levine, G. Raffel, Y. Liu, D. Chen, H. Li, T. Townes, H. Wang, H. Deng, Y. G. Zheng, C. Leslie, M. Luo and X. Zhao, eLife, 2015, 4, e07938 CrossRef.
- J. Xu, A. H. Wang, J. Oses-Prieto, K. Makhijani, Y. Katsuno, M. Pei, L. Yan, Y. G. Zheng, A. Burlingame, K. Bruckner and R. Derynck, Mol. Cell, 2013, 51, 5–19 CrossRef CAS.
- T. Iderzorig, J. Kellen, C. Osude, S. Singh, J. A. Woodman, C. Garcia and N. Puri, Biochem. Biophys. Res. Commun., 2018, 496, 770–777 CrossRef CAS.
- J. Zhang, K. Qian, C. Yan, M. He, B. A. Jassim, I. Ivanov and Y. G. Zheng, MedChemComm, 2017, 8, 440–444 RSC.
- X.-R. Yu, Y. Tang, W.-J. Wang, S. Ji, S. Ma, L. Zhong, C.-H. Zhang, J. Yang, X.-A. Wu, Z.-Y. Fu, L.-L. Li and S.-Y. Yang, Bioorg. Med. Chem. Lett., 2015, 25(22), 5449–5453 CrossRef CAS.
- J. H. Voigt, B. Bienfait, S. Wang and M. C. Nicklaus, J. Chem. Inf. Comput. Sci., 2001, 41, 702–712 CrossRef CAS.
- P. C. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Chem. Inf. Model., 2010, 50, 572–584 CrossRef CAS.
- P. C. Hawkins, A. G. Skillman and A. Nicholls, J. Med. Chem., 2007, 50, 74–82 CrossRef CAS.
- G. Tresadern, D. Bemporad and T. Howe, J. Mol. Graphics Modell., 2009, 27, 860–870 CrossRef CAS.
- J. Wu, N. Xie, Y. Feng and Y. G. Zheng, J. Biomol. Screening, 2012, 17, 237–244 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
- J. M. Wang, P. Morin, W. Wang and P. A. Kollman, J. Am. Chem. Soc., 2001, 123, 5221–5230 CrossRef CAS.
- Y. Yang and M. T. Bedford, Nat. Rev. Cancer, 2013, 13, 37–50 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures and information for virtual screening, selectivity panel screening, molecular modeling, cellular assays, and NMR spectra are included in the supporting information. See DOI: 10.1039/d0md00259c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Chunli
Yan
b,
Hairui
Su
c,
Tran
Dang
a,
Bo
Zhou
a,
Zhenyu
Wang
b,
Xinyang
Zhao
c,
Ivaylo
Ivanov
b,
Meng-Chiao
Ho
d and
Y. George
Zheng
a,
Chunli
Yan
b,
Hairui
Su
c,
Tran
Dang
a,
Bo
Zhou
a,
Zhenyu
Wang
b,
Xinyang
Zhao
c,
Ivaylo
Ivanov
b,
Meng-Chiao
Ho
d and
Y. George
Zheng