
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Levulinic acid-based bioplasticizers: a facile approach to enhance the thermal and mechanical properties of polyhydroxyalkanoates†
Alessandro
Sinisi
 a,
Micaela
Degli Esposti
*ab,
Simona
Braccini
c,
Federica
Chiellini
c,
Susana
Guzman-Puyol
d,
José Alejandro
Heredia-Guerrero
d,
Davide
Morselli
*ab and
Paola
Fabbri
ab
a,
Micaela
Degli Esposti
*ab,
Simona
Braccini
c,
Federica
Chiellini
c,
Susana
Guzman-Puyol
d,
José Alejandro
Heredia-Guerrero
d,
Davide
Morselli
*ab and
Paola
Fabbri
ab
aDepartment of Civil, Chemical, Environmental and Materials Engineering (DICAM), Università di Bologna, Via Terracini 28, Bologna 40131, Italy. E-mail: davide.morselli6@unibo.it; micaela.degliesposti@unibo.it
bNational Interuniversity Consortium of Materials Science and Technology (INSTM), Via Giusti 9, Firenze 50121, Italy
cDepartment of Chemistry and Industrial Chemistry, Università di Pisa, Via G. Moruzzi 13, 56124 Pisa, Italy
dInstituto de Hortofruticultura Subtropical y Mediterránea “La Mayora”, Universidad de Málaga-Consejo Superior de Investigaciones Científicas (IHSM, UMA-CSIC), Av. Louis Pasteur, 49, 29010 Málaga, Spain
First published on 4th November 2021
Abstract
Plasticizers are the most used polymer additives world-wide. Nowadays, conventional plasticizers (e.g. phthalates) do not meet the requirements in terms of renewability, biodegradability and cytotoxicity that have become necessary, especially if they are compounded with biopolymers. In this study, novel bioplasticizers are synthesized from levulinic acid via a protecting-group-free three-step process. After FT-IR and NMR characterization of the synthesized molecules, their plasticization effect has been tested with poly(3-hydroxybutyrate) (PHB) as a model semicrystalline biopolyester characterized by a narrow processing window, slow re-crystallization and high brittleness, which limit its processability and diffusion. The proposed bioplasticizers show remarkable miscibility with PHB and low leaching. The bioplasticizers also show a remarkable plasticization effect in terms of reducing the glass transition and melting temperatures (17 °C and 8 °C, respectively), which are comparable with the performance of the best commercially available green plasticizers. Furthermore, flexibility and crystallinity are positively affected, leading to an overall reduction in the typical brittleness of PHB. The observed effects result in an expansion of the temperature range in which PHB can be processed without thermal degradation. Moreover, the incorporation of the levulinic acid-based additives does not significantly affect the typical biodegradability and biocompatibility of PHB, showing their promising features as bioplasticizers for both environmental and biomedical applications.
Introduction
The growing environmental awareness concerning the depletion of fossil reserves, greenhouse gas emissions and waste management has been increasingly dissuading companies from investing in traditional plastics.1 As a consequence, there is a rapidly growing research interest toward more sustainable polymers2 based on renewable feedstocks, which biodegrade at the end of their life cycle without leaching out any harmful pollutants.3 In addition to the important environmental benefits, the physical properties of the emerging biopolymers should also be comparable with those of conventional plastics in order to represent a realistic alternative. In this sense, compounding polymers with suitable additives is a simple and effective approach to overcome poor mechanical properties, limited thermal stability and/or manufacturing problems. Nonetheless, while the development of new biopolymers has been rapidly increasing in the last decade, the related range of additives has not been growing that fast.4 This has resulted in an urgent need of developing a new generation of sustainable “bioplasticizers” that can improve the processability of the biopolymers and tune their properties and, at the same time, preserve their renewability and/or cytocompatibility and/or biodegradability.5Polyhydroxyalkanoates (PHAs) are a very promising class of fully biobased thermoplastic polyesters produced by different bacterial strains as intracellular energy storage compounds.6 PHAs are well-known for their high biodegradability upon exposure to soil, composting sites or water bodies (sea, lake, etc.).7 These biopolyesters have been recently proposed as materials for biomedical applications thanks to their excellent biocompatibility and bioresorbability.8,9 Moreover, they have found applications as materials for food-packaging10 and as substrates for green electronics and piezoelectric devices.11–15 Poly(3-hydroxybutyrate) (PHB) is the most widespread member of the PHA family. PHB is a semicrystalline isotactic stereoregular polyester, which can reach a crystallinity degree of up to 80%16 which makes this material particularly stiff and brittle.17 In addition, PHB suffers from secondary crystallization, which slowly occurs during the lifetime of the material, thus further affecting its properties.18 Moreover, the PHB processability is strongly limited by its thermal-degradation temperature (approx. 200 °C) which occurs a few tens degree above its melting temperature (typically 175 °C).19 All these disadvantages can be easily overcome by compounding PHB with a plasticizer, to induce a decrease in glass transition (Tg) and melting temperatures (Tm), elastic modulus and melt viscosity.20,21 However, not only the plasticization performance is important for a plasticizer suitable for bioplymer, as PHB. In particular, an innovative plasticizer (bioplasticizer) does not have to alter the biodegradability, biocompatibility and bioresorbability typical of these biopolymers.22,23 Traditional petroleum-based plasticizers such as phthalates, trimellitates, and dicarboxylates have shown high plasticization efficiency; however, they also have very severe toxicity issues24–27 and a high environmental impact28,29 due to their leaching.30 This led the USA,31 Canada32 and the European Union,33,34 among others, to strictly regulate the use of petroleum-based plasticizers, such as phthalates in the manufacturing of children's toys, biomedical devices and materials for food-packaging, and incentivized the development of safer and greener alternatives. In this context, bio-based alternatives,35 typically used in poly(vinyl chloride) (PVC), have been investigated as potential PHB plasticizers. Among them, starch derivatives such as starch-adipate and grafted starch-urethane were evaluated in terms of changes in the mechanical and thermal properties.36 Epoxidized linseed and soybean vegetable oils (EVOs), as well as citrate derivatives,20,35,39–41 have been also intensively studied.37–39 Although EVOs are cheap raw materials extracted from biomass (plants, fruits, seeds or wood), their epoxidation process usually requires harsh conditions and harmful reagents and solvents such as carboxylic acids, peroxides and benzene42,43 that definitly affect the sustainablility of these plasticizers synthesis. Furthermore, EVOs cannot ensure the long-lasting properties of the plastic compound since they have shown a high leaching rate especially under UV irradiation.44
Currently, citrate derivatives find extensive use as phthalate alternatives in medical grade plastics.45 Although citrate esters are believed to be much less harmful than phthalates, most of the available toxicity data are related to acetyltributyl citrate (ATBC),46 and only limited information is available regarding other citrates.47–49
Among the biobased feedstocks used for producing sustainable alternatives to phthalates, levulinic acid (LA) has been increasingly drawing attention due to its renewability, chemical versatility, and relatively low price.50 It is indeed not by chance that LA has been classified as one of the most promising biomass derived building blocks by both the USA51 and European Union52 and it is considered a key intermediate in the synthesis of several value-added products.53 Recently, Xuan and co-workers developed a family of linear and branched LA-based esters and tested their performance in poly(lactic acid) (PLA), evaluating the effect of structural variations on their plasticizing efficiency and migration properties.54 Almost simultaneously, our group designed a new class of biobased asymmetric ketal–diester derivatives of LA, synthesized by means of a selective protecting-group-free route. These molecules exhibited a very high plasticization effect towards PVC, showing performances comparable to the ones of commercially available phthalates even at lower concentrations.55 This was a preliminary study on a model commodity plastic that is the most compounded material worldwide.
The aim of this work is to develop LA-based ketal–esters capable of inducing the plasticization effect in semicrystalline polyesters such as PHB, which represents one of the most promising bio-based materials of the upcoming future. The herein proposed ketal–ester bioadditives efficiently decrease the glass transition temperature, melting temperature and stiffness of PHB with only 20 phr (per hundred of resin) of the additive, showing a plasticization performance comparable with that of the so-called “green plasticizers”. In addition, the synthesized bioplasticizers do not alter the cytotoxicity and biodegradability typical of PHB, envisioning their potential use in the manufacturing of plastic biomedical devices and food packaging materials.
Results and discussion
Five LA-based ketal–ester plasticizers have been synthesized by modifying the three-step protecting-group-free route (Scheme 1) previously developed by our group, using only materials that can be obtained from renewable resources.55 As shown in Scheme 1, the prepared molecules are characterized by the same central structure (ketal–ester) and different side chains: myristoyl (C14), stearyl (C18), isovaleryl (C5), phenyl (C6) and benzyl (C7). The synthesized ketal–diesters have been coded as KE-myr, KE-stear, KE-isoval, KE-phen and KE-benz according to the side chain present in them.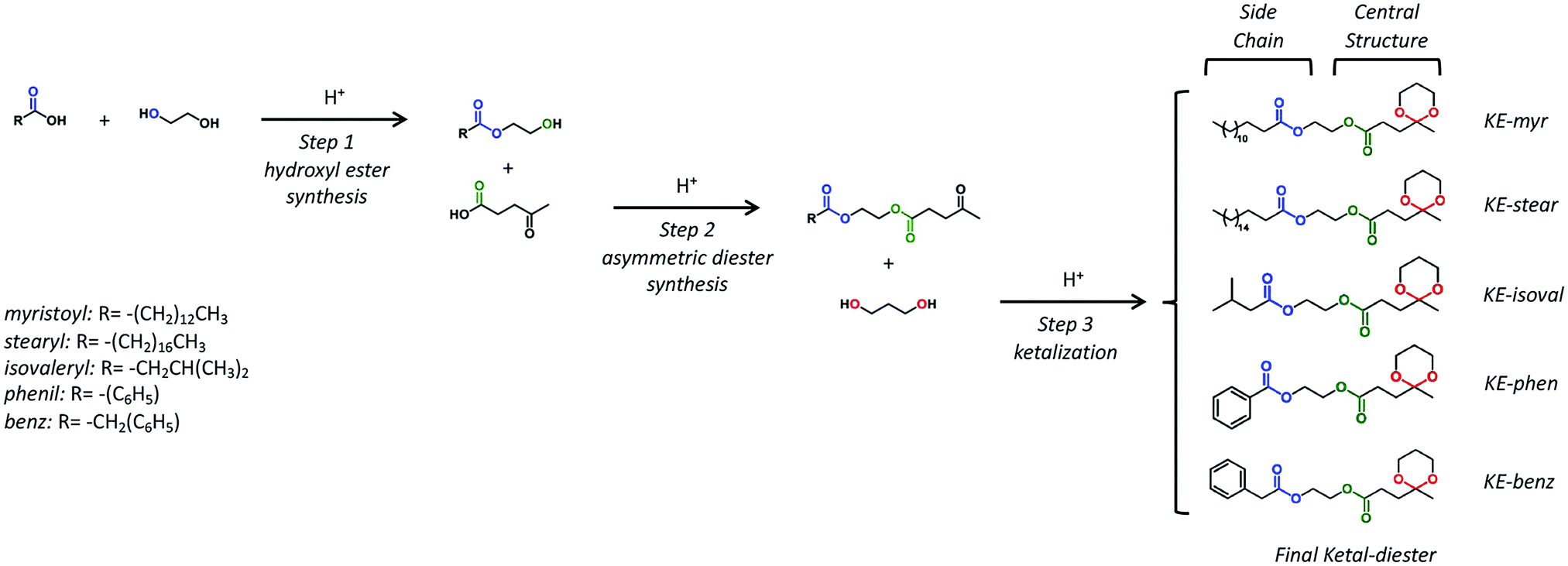 | ||
| Scheme 1 The three-step protecting-group-free synthetic route to obtain asymmetrical ketal–diester bioplasticizers. | ||
The chemical structure evolution of the intermediates and their corresponding final additives have been studied by FT-IR analysis on the products obtained in each step. The example of KE-myr is used as the representative and reported in Fig. S1 (ESI†). The structure of the final ketal–esters has been determined by 1D nuclear magnetic resonance (NMR). In the 1H-NMR spectra shown in Fig. 1 (assignments in the ESI†), the ketal-ring formation is confirmed by the three multiplets at approx. 3.90, 1.80, and 1.53 ppm corresponding to the methylene protons (C, E) and to the diastereotopic protons (D) of the six-membered ketal ring, respectively. Moreover, protons (G, F) and (A) of the levulinate moiety are observed as triplets at approximately 2.50 and 2.00 ppm and as a singlet at 1.40 ppm, respectively. The 1H-NMR results are then further supported by 13C-NMR investigations and related assignments reported in Fig. S2 in the ESI.†
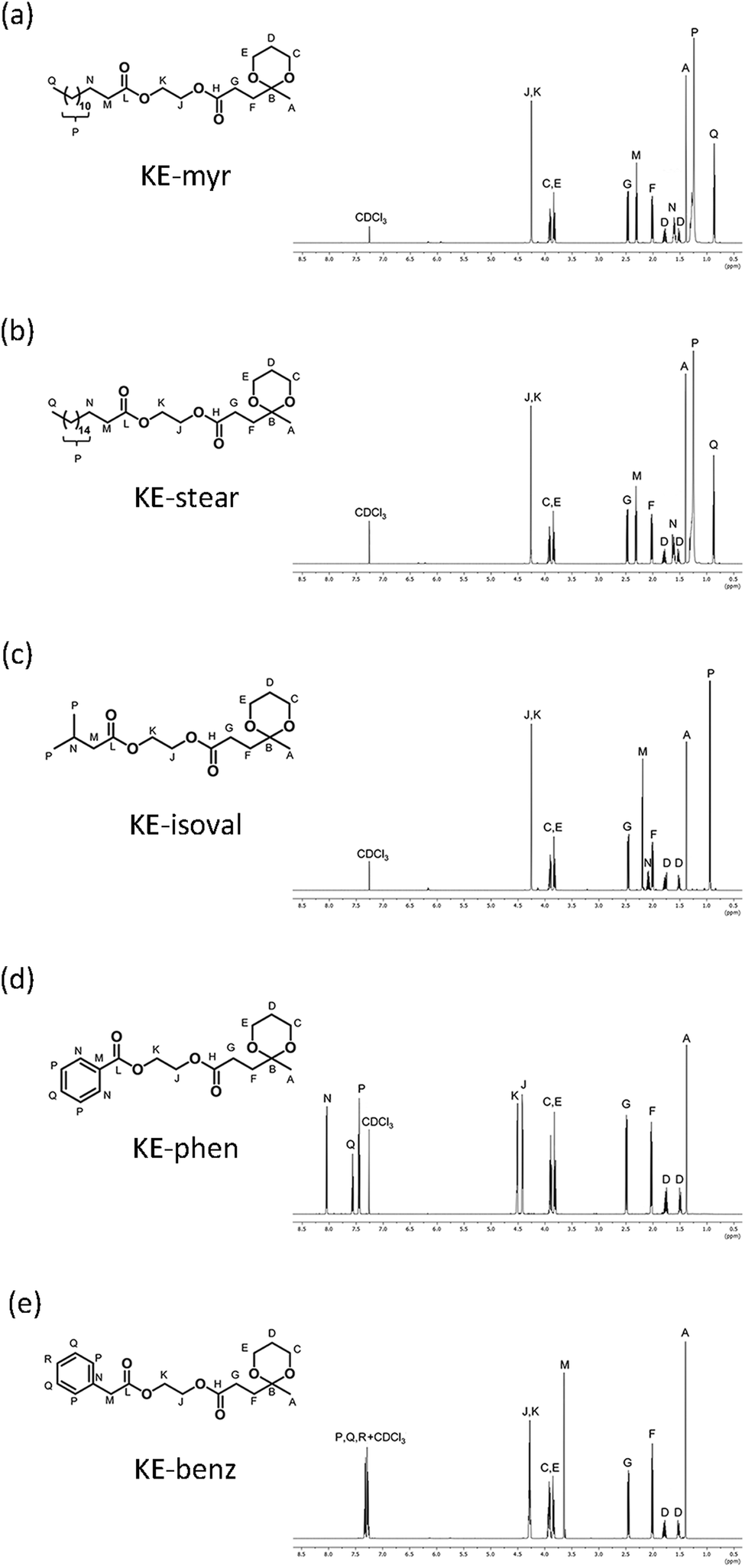 | ||
| Fig. 1 1H-NMR spectra and the corresponding proton assignments of the final ketal–esters: (a) KE-myr, (b) KE-stear, (c) KE-isoval, (d) KE-phen and (e) KE-benz. | ||
Neat PHB and PHB compounded films, with 10 and 20 phr (per hundred of resin) of the plasticizer, have been prepared by solvent casting for the following characterization studies (photographs of the films are provided in Fig. S3, ESI†).
High miscibility of the plasticizer in the desired polymer is the first parameter to consider in order to have an effective plasticization.56 This has been evaluated through FESEM investigations on the cross sections of the neat and compounded PHB films with 20 phr additives. As clearly shown in Fig. 2, all samples present only one homogeneous phase even at high magnification (in the insets), confirming the mutual miscibility of the components. Generally, compatibility arises thanks to the presence of intermolecular attractive forces between the additive molecules and polymeric chains.57 This is the reason why polar moieties have been introduced during the initial design of the plasticizers.58 Specifically, the excellent compatibility between PHB and the ketal–diesters can be ascribed to the carbonyl–carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯CO) intermolecular non-covalent interactions, where the lone pairs (n) of the oxygen atoms of the carbonyl groups are delocalized over the antibonding orbitals (π*) of the nearby carbonyl CO bonds (n → π*).59,60 Thanks to these interactions, both polymer and plasticizer CO groups are polarized, resulting in a sequential chain of O⋯C n → π* interactions, which improve the mutual attraction and result in high miscibility as also similarly observed in other polyesters such as poly(lactic acid).61
O⋯CO) intermolecular non-covalent interactions, where the lone pairs (n) of the oxygen atoms of the carbonyl groups are delocalized over the antibonding orbitals (π*) of the nearby carbonyl CO bonds (n → π*).59,60 Thanks to these interactions, both polymer and plasticizer CO groups are polarized, resulting in a sequential chain of O⋯C n → π* interactions, which improve the mutual attraction and result in high miscibility as also similarly observed in other polyesters such as poly(lactic acid).61
 | ||
| Fig. 2 Cross-section FESEM images (secondary electrons) of neat PHB and plasticized films. The insets show high-resolution images. | ||
The intermolecular interactions between PHB and additives not only lead to a homogeneous material, but also lower the plasticizer migration, preventing its leaching from the polymeric compound. Limited leaching is a crucial requirement for an additive, ensuring long-lasting properties of the compound and avoiding environmental contamination or human exposure. The potential leaching has been evaluated by extraction tests (24 h) both in water and n-hexane in order to simulate the hydrophilic and lipophilic environments. As shown in Fig. 3, the extractable fraction (f) in water is remarkably low, with a value less than 1.6% for all additives considering the extracted fraction of neat PHB. As expected, when n-hexane is used, the average weight loss is higher due to the high miscibility of the synthesized additives in this solvent, which forces their migration. Nevertheless, f is less than 3.5% for all plasticizers, taking into account the small extracted fraction observed for neat PHB (Fig. 3). This has to be considered a promising result since it is comparable with the value obtained under similar conditions for the so-called “non-migrating plasticizers”.62
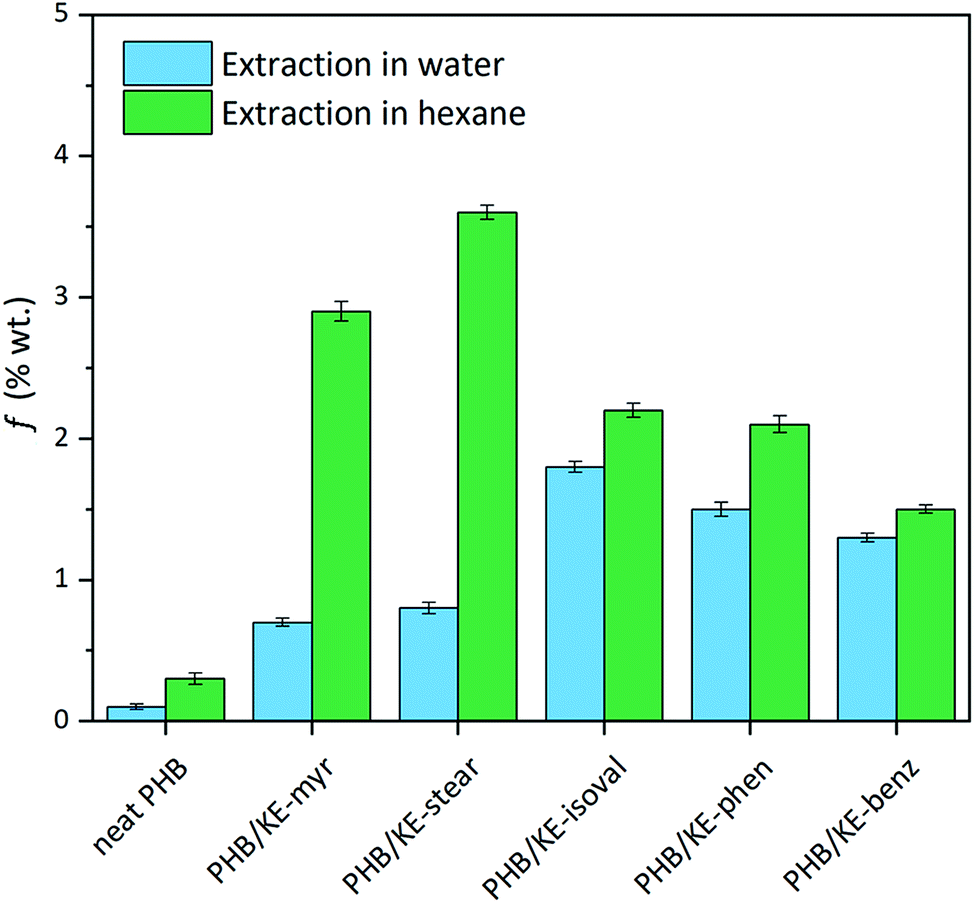 | ||
| Fig. 3 Extractable fraction (f) of neat and plasticized PHB films calculated using eqn (1) after 24 h of extraction test in water (light blue) and n-hexane (green). | ||
The role of a plasticizer is to separate the neighboring polymer chains by breaking up some of the attractive inter-chain interactions.58 The smaller number of cohesion points among the macromolecular chains results in an enhanced polymer molecular mobility, which leads to a decrease in the polymer Tg, since less energy is required to overcome the forces that keep the polymeric chains closely bonded.63,64 The plasticizing performance of the proposed molecules has been evaluated in terms of Tg reduction (Table S1, ESI†) as extrapolated from the differential scanning calorimetry (DSC) thermograms (in Fig. S4, ESI†) of neat and compounded PHB films. As shown in Fig. 4a, the plasticizer KE-stear and KE-isolval showed a comparable Tg decreasing effect of approx. −5 °C at 10 phr of additive content, and −8 °C at a concentration of 20 phr. The additives KE-myr, KE-phen, and KE-benz are shown to be more effective leading to Tg reduction of approx. −10 and −17 °C by increasing the additive content from 10 to 20 phr, respectively (Fig. 4a). According to the free volume theory, plasticizers intercalate and diffuse into the polymer, increasing the internal space between the macromolecules. Based on the side chain, the proposed ketal–diesters can be divided into aliphatic (KE-myr, KE-stear and KE-isoval) and aromatic (KE-phen and KE-benz) chain bearing derivatives. The largest experimentally observed decrease of Tg is induced by KE-myr (C14 linear side chain). Although a branched plasticizer is typically more efficient than the corresponding linear one,58 KE-isoval showed a slightly worse plasticizing effect than KE-myr. Probably, the isopropyl side chain of KE-isoval is not sufficiently branched to induce a free volume comparable with the one provided by the long linear aliphatic compounds. On the other hand, aromatic side chain additives such as KE-phen and KE-benz due to the steric hindrance largely affect the Tg of PHB (Fig. 4a).
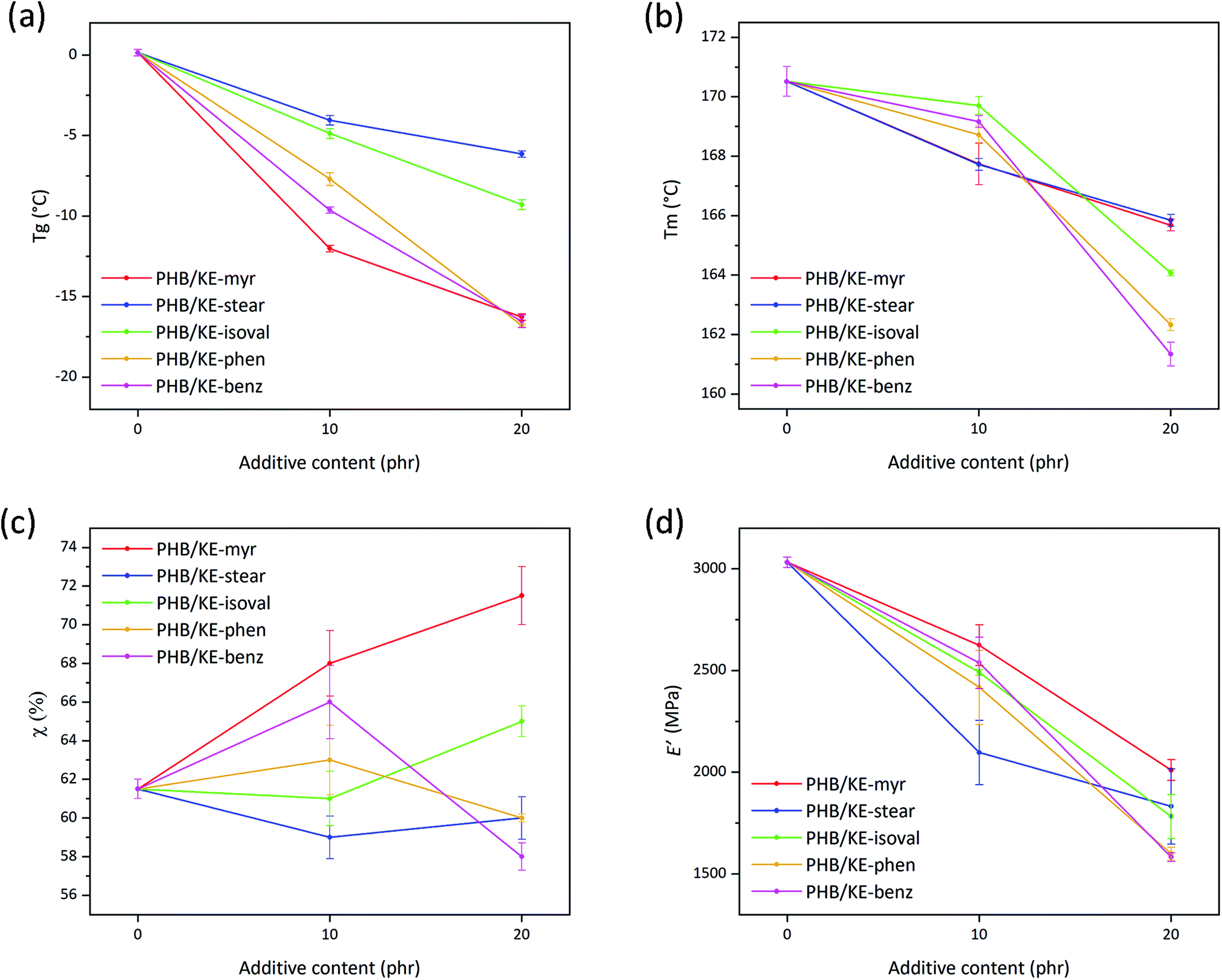 | ||
| Fig. 4 (a) Glass transition temperature (Tg) and (b) melting temperature (Tm) determined by DSC for neat and plasticized PHB films as a function of the additive content. (c) Crystallinity degree (χ) calculated using eqn (2) from the DSC curves for neat and plasticized PHB films as a function of the additive content. (d) Storage modulus (E′) values (extrapolated at 25 °C) as a function of the additive content for neat and plasticized PHB films. | ||
Importantly, the overall plasticization performance of the presented ketal–diesters is comparable with the effect of different citrate esters, which currently represent one of the most common commercially available “green” alternatives to phthalates.65 Chaos and co-workers, who studied the plasticizing effect of tributyl citrate on PHB, observed a Tg reduction of 16 °C with an additive concentration of 20 wt% (approx. 17 phr).20 This Tg reduction is basically the same result obtained using the herein proposed plasticizers KE-myr, KE-phen and KE-benz loaded at 20 phr (Fig. 4a). On the other hand, a very low Tg reduction (approx. 3 °C) was observed elsewhere66 by using the pomace extract from grape waste, confirming that the herein proposed ketal–esters are very promising plasticizers and can compete with the commercially available products.
PHB is a semicrystalline polymer, thus characterized by both Tg and Tm. Typically, plasticizers have a more important effect on the amorphous part, but in this case, it has been found that the additives have also a lowering effect on the Tm of the polymer (Table S1, ESI†). The addition of the plasticizers leads to an overall decrease in the Tm of PHB as shown in Fig. 4b. Specifically, at 10 phr, KE-myr and KE-stear are more effective than KE-isoval, KE-phen and KE-benz, lowering the Tm from 171 °C to approximately 168 °C. Conversely, at higher contents, KE-isoval, KE-phen and KE-benz perform better than KE-myr and KE-stear, resulting in further decreased Tm values (164, 162, and 161 °C, respectively). The polymer melting is only related to the crystallite features (such as crystallites size, morphology and stability), which mainly affect the DSC endothermic peak shape and the Tm value. Typically, neat PHB is characterized by a double melting peak, whose small shoulder is attributed to lower molecular weight species, polymorphism, and different crystallite sizes.67 As observed in the thermograms shown in Fig. S4 (ESI†), all plasticizers actually induce changes in the melting peak of PHB. In particular, the typical double-peak shape undergoes a broadening effect and shifts toward lower temperatures regardless of the additive contents.
Considering the narrow temperature range (approx. 20 °C) where PHB can be processed without thermal degradation,68 the observed decreases of Tg and Tm are remarkable results for enhancing its processability in the molten state.
The observed effect on the Tm allows to suppose a possible effect on the crystallinity degree of PHB. Melting enthalpy (ΔHm) values (listed in Table S2, ESI†), obtained from the DSC curves (Fig. S4, ESI†), were used to calculate the degree of crystallinity (by eqn (2)) of the samples as shown in Fig. 4c and listed in Table S2 (ESI†). Generally, an increase in crystallinity is consistent with the plasticization effect. In fact, the increase of free volume and molecular mobility allows the macromolecular chains to rearrange in new configurations resulting in further nucleation.20,69 However, the observed crystallinity decrease (Fig. 4c) caused by KE-phen and KE-benz at 20 phr may be rationalized considering the chemical structure of the additives. Both plasticizers are characterized by polarizable aromatic rings, which can form many points of mutual attraction along the polymer chains. This may prevent the chains from lining up and compactly packing into crystals. On the other hand, additives such as KE-myr, KE-stear, and KE-isoval that bear aliphatic non-polar side chains do not introduce additional cohesive links along the macromolecular chains, allowing them to move and rearrange into ordered structures.
The previously discussed higher chain molecular mobility affects not only the biopolyester thermal behavior, but also the mechanical properties, making the samples more flexible already at room temperature.70 As described by Marcilla and Beltran,58 when a plasticizer is added, the amorphous portion of the polymer swells due to the increase in free volume. Consequently, the mobility of the chains is enhanced as well as the overall flexibility of the material. Storage modulus (E′) values at 25 °C, extrapolated from the results of the dynamic thermal mechanical analysis (the DMTA curves are shown in Fig. S5, ESI†), are a suitable parameter to evaluate the effect of the proposed plasticizers on the viscoelastic properties of PHB. As shown in Fig. 4d, the plasticizers led to an overall decrease of E′ for both tested contents, confirming their effective plasticization (values also listed in Table S1, ESI†). The E′ at 25 °C is constantly under 2000 MPa for all additives loaded at 20 phr. This value is significantly lower than the E′ reported by Audic et al. using epoxidized vegetable oils added at a similar content (in the range 3000–3500 MPa).71 Considering that epoxidized vegetable oils are one of the best green plasticizers, the presented ketal–esters can be considered a very promising alternative to reduce the brittleness of PHB at room temperature.
Interestingly, among the plasticized samples, a correlation between E′ and crystallinity can be noticed. In detail, at 10 phr, KE-myr and KE-benz, which induce the highest degrees of crystallinity (68% and 66%, respectively), also show the highest moduli compared to the other additives (2625 and 2538 MPa, respectively). On the other hand, KE-stear, which is the only additive able to decrease the PHB crystallinity degree at 10 phr, results in the lowest value of E′ (2097 MPa). KE-phen and KE-benz show different performances depending on their content, increasing the crystallinity of PHB to 63% and 66% at 10 phr, while at 20 phr they decrease it to 60% and 58%, respectively. As a result, the two aromatic additives at 20 phr lead to the lowest observed E′ values (1600 and 1583 MPa, respectively). Conversely, KE-myr induces the highest degree of crystallinity, showing therefore the lowest E′ reduction at 20 phr (2011 MPa). This behavior may be explained taking into account the so-called rigid amorphous fraction (RAF), which is a metastable nanophase at the interface between the crystallites and the surrounding phase in semicrystalline polymers.72 This fraction is constituted of amorphous chain portions, the mobility of which is hindered by the neighboring crystalline phase.73 As reported by Di Lorenzo et al., full mobilization of the RAF of PHB takes place at a temperature around 70 °C. At 25 °C (temperature at which E′ has been extrapolated), RAF is still vitrified, acting as a further tensed and rigid domain within the polymer.18 Consequently, the higher values of E′ found for the samples with higher degree of crystallinity may be due to the higher amount of RAF related to the crystalline phase.
Knowing the unique biocompatibility and resorbability of PHB in living systems, cytocompatibility tests have been performed to ascertain whether the synthesized ketal–diesters can affect this property. For this, the mouse embryo fibroblast Balb/3T3 clone A31 cell line was directly incubated with different dilutions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:4) of the neat and compounded PHB sample extracts (extraction time 24 h). According to the international standard ISO 10993-5:2009, employed for the tests, a material shall be considered non-cytotoxic if the cell viability is ≥70% of the control. As shown in Fig. 5, the results highlight that all extracts diluted at 1:4 are fully cytocompatible. In addition, very promising results were obtained from the extracts at a dilution ratio of 1:2 (data summarized in Table S3, ESI†). Indeed, only the extract containing the KE-isoval additive leads to a cell viability value close to 40% of the control. It is noteworthy that KE-isoval is also the plasticizer that showed the highest values of leaching in water. Probably it hydrolyses yielding an acidic compound, which slightly decreases the pH of the extract and inhibits the cell growth. However, it is important to underline that the 1:4 ratio represents the most realistic dilution envisioning the potential application of the proposed compounded material as medical grade components, tubes, blood bags and scaffolds for tissue regeneration.
:2 and 1:4) of the neat and compounded PHB sample extracts (extraction time 24 h). According to the international standard ISO 10993-5:2009, employed for the tests, a material shall be considered non-cytotoxic if the cell viability is ≥70% of the control. As shown in Fig. 5, the results highlight that all extracts diluted at 1:4 are fully cytocompatible. In addition, very promising results were obtained from the extracts at a dilution ratio of 1:2 (data summarized in Table S3, ESI†). Indeed, only the extract containing the KE-isoval additive leads to a cell viability value close to 40% of the control. It is noteworthy that KE-isoval is also the plasticizer that showed the highest values of leaching in water. Probably it hydrolyses yielding an acidic compound, which slightly decreases the pH of the extract and inhibits the cell growth. However, it is important to underline that the 1:4 ratio represents the most realistic dilution envisioning the potential application of the proposed compounded material as medical grade components, tubes, blood bags and scaffolds for tissue regeneration.
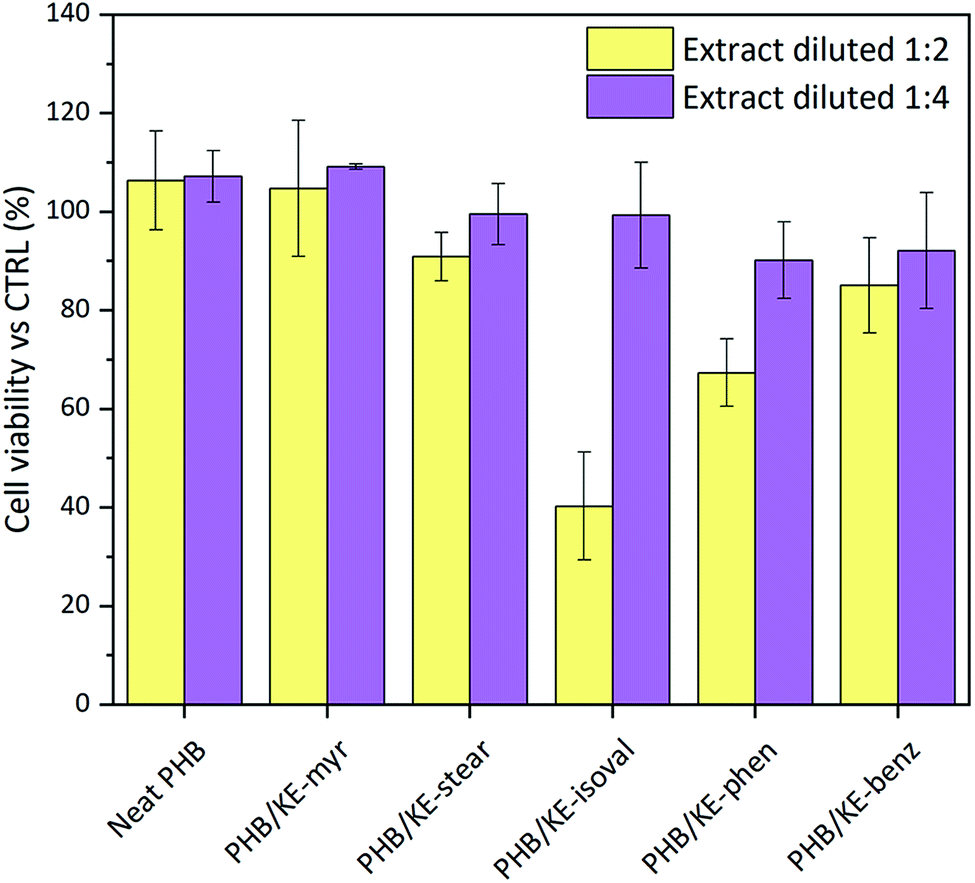 | ||
| Fig. 5 Evaluation of the viability of mouse embryo fibroblasts Balb/3T3 clone A31 cells after 24 h of incubation. In yellow: extract diluted at 1:2, and in purple: extract diluted at 1:4. | ||
The biodegradability of neat PHB and compounded with KE additives (20 phr) was assessed by immersing the samples in seawater for 30 days. Fig. 6a reports the typical biological oxygen demand (BOD) values registered every day for the entire period of the analysis for each sample. All samples started biodegrading after 4 days and after a period of rising a plateau was reached at different days depending on the sample: 10 days for KE-phen, 15 days for KE-isoval, KE-benz, and KE-stear, 17 days for KE-myr, and 21 days for neat PHB. The final BOD values (or BOD30) were also dependent on the type of additive used (if any), inset of Fig. 6b. Thus, these values ranged from ∼3.8 mg O2 L−1 for KE-myr to ∼9.0 mg O2 L−1 for KE-phen. Neat PHB showed an intermediate behavior with a value of ∼6.3 mg O2 L−1. In the explanation of this tendency can participate many factors such as the sample's crystallinity, the interactions between the additive and the polymer matrix, and the different nature of the ester bond of the additives.74–77 To locate the biodegradability potential of the KE samples, the final BOD values of KE-myr, KE-phen, and neat PHB were compared to those of other common biopolymers such as polylactide (PLA), cotton, lignin, xylan, and amorphous cellulose (Fig. 6b).78–80 Interestingly, the values are similar to those of cotton and lignin and much higher than those of PLA that shows almost no biodegradation in seawater. The weight loss after the immersion in seawater for 30 days was also measured (Fig. 6c). The inset of Fig. 6c displays the weight losses (by eqn (3)) of all the KE samples and neat PHB. As observed, KE-myr, neat PHB, and KE-stear showed weight losses less than 15%, while for KE-benz, KE-isoval, and KE-phen the weight losses were more than 22%. In general, these values are between the weight losses described under similar conditions for cotton and amorphous cellulose (Fig. 6c).78–80 Finally, the effect on the samples’ morphology of the biodegradability in seawater was characterized by SEM (Fig. 6d). KE-myr and KE-phen, which are the samples with the lowest and highest biodegradability, respectively, have been selected to show such effects. Before the immersion in seawater, both samples presented very similar rough surfaces. However, after 30 days, some differences were noticed. KE-myr showed little changes with the appearance of small pores, while in the case of KE-phen such pores were accompanied by some higher scratches typical of surface erosion mechanisms.74
 | ||
| Fig. 6 (a) Typical BOD curves for the KE samples and neat PHB as a function of the immersion time in seawater. (b) BOD30 values of KE-myr, KE-phen, and neat PHB in comparison to other biopolymers. The inset shows the BOD30 values of all the KE samples and neat PHB. (c) Weight losses (using eqn (3)) after immersion for 30 days in seawater of KE-myr, KE-phen, and neat PHB in comparison to other biopolymers. The inset shows the weight losses after immersion for 30 days in seawater of all the KE samples and neat PHB. (d) The SEM top-view images of KE-myr and KE-phen samples before and after BOD tests. Scale bar: 25 μm. | ||
Conclusion
Novel bioplasticizers for enhancing the limiting mechanical and thermal properties of polyhydroxyalkanoates are herein presented. The levulinic acid-based ketal–ester bioplasticizers were synthesized, fully characterized and tested as plasticizing additives in PHB. In particular, we found that with only 20 phr of plasticizer that bears benzyl side chains, the glass transition and melting temperatures of PHB can be reduced by 17 °C and 8 °C, respectively. Also the storage modulus is affected, showing a reduction of approx. 50% with respect to the neat PHB, resulting in a flexible polymer at room temperature. It is noteworthy that the observed plasticization efficiency is comparable with that of the commercial green additives on the market. In addition, the proposed plasticizers do not significantly affect both cytocompatibility and biodegradability typical of this family of biopolyesters, which represents an important step forward in the polymer additive field. The presented results support the use of ketal–esters as very promising sustainable plasticizers for several other biopolyesters as innovative alternatives to the polluting and toxic conventional additives on the market.Experimental section
Materials
Commercially available reagents and solvents were used as received without further purification. Levulinic acid (98.0%), ethylene glycol (anhydrous, 99.8%), 1,3-propanediol (98.0%), myristic acid (≥98.0%), stearic acid (95.0%), benzoic acid (99.5%), p-toluenesulfonic acid monohydrate (PTSA, ≥98.5%), sulfuric acid (H2SO4, 96%), sodium sulfate (Na2SO4, anhydrous, ≥99.0%), sodium chloride (NaCl, ≥99.5%), filter agent (Celite® 545), diethyl ether (≥99.0%), chloroform (CHCl3, HPLC grade), deuterated chloroform (CDCl3, 99.8 atom% D, contains 0.03% v/v TMS), dichloromethane (≥99.9%), ethyl acetate (≥99.5%), n-hexane (≥95.0%), water (HPLC grade), methanol (≥99.8%), tetrahydrofuran (THF, HPLC grade) and toluene (HPLC grade) were purchased from Sigma-Aldrich. Isopentanoic acid (99.0%), phenylacetic acid (99.0%) and sodium carbonate (Na2CO3, ≥99.0%) were supplied by Merck. Analytical thin layer chromatography (TLC) was performed using pre-coated aluminium-backed plates (Merck Kieselgel 60 F254) and visualized using a solution of potassium permanganate (KMnO4, 0.06 M). For column chromatography, silica gel (MN Kieselgel 60, 0.063–0.2 mm, 70–230 mesh, Macherey-Nagel) was used. For cytocompatibility evaluation, the mouse embryo fibroblast Balb/3T3 clone A31 cell line from American Type Culture Collection (ATCC CCL-163) was selected. Cells were propagated as indicated by the supplier using Dulbecco's modified eagle medium (DMEM) (Sigma-Aldrich) supplemented with 4 mM of L-glutamine (Sigma-Aldrich), 1% of penicillin:streptomycin solution (10000 U mL−1: 10 mg mL−1; Sigma-Aldrich), 10% of calf serum (Sigma-Aldrich) and antimycotic (complete DMEM). Viability and proliferation were investigated using WST-8 tetrazolium salt reagent (Microtech).
Custom grade PHB (Sigma-Aldrich, Italy) was carefully purified as described elsewhere.81 In brief, as received PHB was solubilized in warm CHCl3, filtered through Celite and precipitated in a large excess of cold methanol, in order to completely remove the fermentation residues. Number average molecular weight (Mn) of 71600 g mol−1 and weight average molecular weight (Mw) of 245100 g mol−1 (all data in Table S4, ESI†) were determined after the purification step by gel permeation chromatography (GPC) (chromatogram in Fig. S6a, experimental details in the ESI†). The purified PHB was characterized by 1H-nuclear magnetic resonance (NMR) (experimental details in the ESI†) in order to confirm the absence of any possible additive that can affect the plasticization effect. 1H-NMR spectra and signal assignments of PHB are shown in Fig. S6b (ESI†).
Synthesis of ketal–diester derivatives of levulinic acid
Ketal–diesters were synthesized by modifying the selective three-step protecting-group-free route previously developed.55 Briefly, a solvent-free reaction between ethylene glycol and selected carboxylic acids led selectively to different 2-hydroxyethyl esters which, in the second step, reacted with the carboxylic moiety of levulinic acid giving the corresponding asymmetric diesters. In the last step, the acetalization of the remaining ketone group with 1,3-propanediol resulted in the target cyclic ketal–diester compounds, bearing side chains with distinctive chemical features (Scheme 1).PHB films preparation
In order to evaluate the plasticizing effect, the synthesized ketal–esters were added to a PHB/CHCl3 solution (12.5 mg mL−1) and cast in a Petri dish for obtaining films (approx. 70 μm thick). The additives were added to the polymer solution at 10 and 20 per hundred of resin (phr). The solutions were dried overnight at room temperature in an aspiration hood. A neat PHB film was also prepared as a reference in order to observe the variations in the properties due to the addition of the herein synthesized ketal–esters (such as thermal behavior, crystallinity degree and dynamic mechanical properties). As shown in Fig. S3 (ESI†), the addition of the plasticizers does not affect the aspect of the neat PHB film, which is partially transparent due to its semicrystalline natire.Characterization studies
To confirm the chemical structure of the synthesized intermediates and final additives, FT-IR and 1D NMR experiments were carried out (FT-IR results and experimental details are reported in the ESI†). 1H-NMR and 13C-NMR spectra were recorded at room temperature using a Varian Mercury 400 spectrometer at 400 MHz (nominal frequency: 399.92 MHz) for 1H and at 150 MHz (nominal frequency: 150.80 MHz) for 13C. Relaxation delays of 1 s and pulses at 45 degrees were employed. 13C-NMR spectra were acquired in the 1H broad-band decoupled mode. CDCl3 containing 0.03 vol% of TMS as internal reference was used as solvent for the analysis. Chemical shifts (δ) are reported in ppm relative to residual solvent signals (CHCl3, 7.26 ppm for 1H-NMR; CHCl3, 77.16 ppm for 13C-NMR). The following abbreviations are used to indicate the multiplicity in NMR spectra: s, singlet; d, doublet; t, triplet; m, multiplet; br s, broad signal. All the spectra were processed using VnmrJ software (Varian, Inc.).The miscibility of the levulinic acid derivatives in PHB was evaluated in the cross-sections of the compounded PHB films and compared to that of neat PHB film. The cross-sections were prepared by cryo-fracturing in liquid nitrogen and coated with gold (thickness 10 nm) by the electrodeposition method to impart electrical conduction. The so-prepared cross-sections were investigated by field emission scanning electron microscopy (FESEM) using a Nova NanoSEM 450 electron microscope (FEI Company-Bruker Corporation), applying an accelerating voltage of 5 kV.
The migration resistance of the plasticizers was evaluated by extraction tests in either deionized water or n-hexane, following the standard method ASTM D1239-14. Samples of approximately 300 mg of both neat and plasticized PHB films (20 phr additive content) were placed in a closed vessel containing 20 mL of the given extracting solvent for 24 hours at room temperature under gentle magnetic stirring. Afterwards, the samples were dried for 24 hours at 40 °C under vacuum and re-weighed. The weight loss corresponds to the extractable fraction (f) and it was calculated according to the following equation:
| f (%) = [(W1 − W2)/W1] × 100 | (1) |
The thermal behavior of the prepared films was evaluated by differential scanning calorimetry (DSC, Q10, TA Instruments), fitted with a standard DSC cell and equipped with a discovery refrigerated cooling system (RCS90, TA Instruments). Samples of approx. 10 mg were placed in aluminum pans and subjected to three heating cycles from −60 °C to +195 °C (hold for 5 min) at a heating rate of 10 °C min−1. The cooling rate was 10 °C min−1 during the first cooling scan and as fast as possible (quenching) during the second cooling scan. The DSC cell was purged with dry nitrogen at 50 mL min−1. The system was calibrated both in temperature and enthalpy with indium standard. The DCS curves were processed using TA Universal Analysis 2000 software (TA Instruments) in order to extrapolate the peak melting temperature (Tm) and the enthalpy of melting (ΔHm) from the area of the endothermic melting signal of the second heating scans, in order to remove the influence of material processing. The degree of crystallinity (χ) was calculated according to the following equation:
| χ (%) = [ΔHm/(ΔHm,0·wPHB)] × 100 | (2) |
Dynamic mechanical thermal analysis (DMTA, Q800, TA Instrument) was carried out in triplicate in order to measure the temperature-dependent viscoelastic properties of the material, particularly storage modulus (E′). Rectangular-shaped samples (15 × 10 × 0.07 mm) were tested in the film tension mode at a constant frequency of 1 Hz under strain control fixed at 0.4%. The heating cycle was from −60 to 60 °C at a rate of 3 °C min−1 and under nitrogen gas flow. DMTA curves were processed using TA Universal Analysis 2000 Version 4.5 A Build 4.5.0.5 software (TA Instruments).
The cytocompatibility of the PHB films containing the synthesized plasticizers was evaluated using biological tests based on international standards BS EN ISO 10993-5:2009 and BS EN ISO 10993-12:2012. Both neat and plasticized PHB films were cut using a puncher (5 mm diameter) and sterilized under UV light for 45 min. After this, each sample was placed in a centrifuge tube containing a certain amount of complete growth medium (extraction ratio 0.2 g mL−1) and incubated at 37 °C and 5% CO2 for 24 h to allow the formation of the extracts. Direct contact assays were then performed in triplicate by directly incubating the cells (Balb/3T3 clone A31) with different dilutions of the prepared extracts. Briefly, cells were seeded in 96-well culture plates at a concentration of 3 × 103 per well and incubated at 37 °C in a 5% CO2-enriched atmosphere and allowed to proliferate for 24 h. Cells were then incubated with diluted sample extracts (dilution ratios of 1:2 and 1:4) for 24 h. Cells incubated with the complete growth medium were used as control (CTRL). Finally, cells were incubated for 4 h at 37 °C and 5% CO2 with the WST-8 reagent at 1:10 dilution in order to evaluate their viability and proliferation. Measurements of formazan dye absorbance were carried out using a microplate reader (Biorad) at 450 nm. The assays were performed in triplicate, and the average values were expressed as a percentage of the control.
Biodegradability was assessed on neat and plasticized PHB samples using a standard biochemical oxygen demand (BOD) test by measuring the oxygen consumption during the biodegradation process in seawater. For this, carefully weighed samples (approx. 200 mg) were finely ground and immersed in amber bottles with 164 mL of seawater from the Malaga (Spain) area shoreline. The oxygen consumed during biodegradation was recorded for 30 days at 20 °C in the dark by using OxyTop®-i measuring heads (WTW). Seawater with no sample was used as the reference. Measurements were carried out in triplicate and the results were averaged to obtain the main value.
After BOD tests, the remaining samples were collected, washed with distilled water, dried for 24 h under vacuum, and weighed. The weight loss was calculated as follows:
| Weight loss (%) = (ms − mb)/ms × 100 | (3) |
The morphology of the films was characterized by SEM before and after the BOD tests in seawater by using a JEOL JSM-6490LV microscope working in the high vacuum mode, with an acceleration voltage of 5 kV. Samples were previously coated with ∼30 nm of gold using a JEOL ION SPUTTER JFC 1100.
Author contributions
Alessandro Sinisi: investigation and writing – original draft. Micaela Degli Esposti: visualization and supervision. Simona Braccini: investigation. Federica Chiellini: writing-original draft. Susana Guzman-Puyol: investigation and writing – original draft. José Alejandro Heredia-Guerrero: investigation and writing-original draft. Davide Morselli: conceptualization, supervision, visualization, writing – original draft, and writing – review & editing. Paola Fabbri: conceptualization and funding acquisition.Funding
M. D. E., D. M., P. F. received funding from the Italian Ministry of Education, University and Research (MIUR) under the program “Research Projects of National Interest (PRIN)”. Project title: “Development and promotion of the Levulinic acid and Carboxylate platforms by the formulation of novel and advanced PHA-based biomaterials and their exploitation for 3D printed green-electronics applications” (VISION); grant number: 2017FWC3WC. J. A. H.-G. was supported by the Spanish “Ministerio de Ciencia, Innovación y Universidades” project RYC2018-025079-I/AEI/10.13039/501100011033 (co-financed by the European Social Fund, ESF) and by the Spanish Research Council (CSIC) project 202040E003.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to Dr Alessandra Petroli (UNIBO) for the technical support on NMR investigations. Dr Mauro Zapparoli (CIGS – UNIMORE) and Dr Francesca Bisi (UNIMORE) are also acknowledged for the technical support on SEM and DMTA analyses, respectively.References
- P. B. Smith and G. F. Payne, ACS Symposium Series, 2011, vol. 1063, pp. 1–10 Search PubMed.
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed.
- M. Rahman and C. S. Brazel, Prog. Polym. Sci., 2004, 29, 1223–1248 CrossRef CAS.
- J. P. Harmon and R. Otter, ACS Sustainable Chem. Eng., 2018, 6, 2078–2085 CrossRef CAS.
- Z. Li, J. Yang and X. J. Loh, NPG Asia Mater., 2016, 8, 1–20 CrossRef CAS.
- C. R. Álvarez-Chávez, S. Edwards, R. Moure-Eraso and K. Geiser, J. Cleaner Prod., 2012, 23, 47–56 CrossRef.
- D. Puppi and F. Chiellini, Appl. Mater. Today, 2020, 20, 100700 CrossRef.
- L. Brunetti, M. Degli Esposti, D. Morselli, A. R. Boccaccini, P. Fabbri and L. Liverani, Mater. Lett., 2020, 278, 128389 CrossRef CAS.
- D. Z. Bucci, L. B. B. Tavares and I. Sell, Polym. Test., 2007, 26, 908–915 CrossRef CAS.
- L. Valentini, P. Fabbri, M. Messori, M. Degli Esposti and S. Bittolo Bon, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 596–602 CrossRef CAS.
- P. Cataldi, P. Steiner, T. Raine, K. Lin, C. Kocabas, R. J. Young, M. Bissett, I. A. Kinloch and D. G. Papageorgiou, ACS Appl. Polym. Mater., 2020, 2, 3525–3534 CrossRef CAS.
- S. Bittolo Bon, I. Chiesa, D. Morselli, M. Degli Esposti, P. Fabbri, C. De Maria, T. Foggi Viligiardi, A. Morabito, G. Giorgi and L. Valentini, Mater. Des., 2021, 201, 109492 CrossRef CAS.
- S. Bittolo Bon, I. Chiesa, M. Degli Esposti, D. Morselli, P. Fabbri, C. De Maria, A. Morabito, R. Coletta, M. Calamai, F. S. Pavone, R. Tonin, A. Morrone, G. Giorgi and L. Valentini, ACS Appl. Mater. Interfaces, 2021, 13, 21007–21017 CrossRef PubMed.
- S. Bittolo Bon, L. Valentini, M. Degli Esposti, D. Morselli, P. Fabbri, V. Palazzi, P. Mezzanotte and L. Roselli, J. Appl. Polym. Sci., 2021, 138, e49726 CrossRef.
- J. K. Hobbs, T. J. McMaster, M. J. Miles and P. J. Barham, Polymer, 1996, 37, 3241–3246 CrossRef CAS.
- P. Anbukarasu, D. Sauvageau and A. Elias, Sci. Rep., 2015, 5, 1–14 Search PubMed.
- G. J. M. de Koning, A. H. C. Scheeren, P. J. Lemstra, M. Peeters and H. Reynaers, Polymer, 1994, 35, 4598–4605 CrossRef CAS.
- I. Janigová, I. Lacík and I. Chodák, Polym. Degrad. Stab., 2002, 77, 35–41 CrossRef.
- A. Chaos, A. Sangroniz, A. Gonzalez, M. Iriarte, J. R. Sarasua, J. del Río and A. Etxeberria, Polym. Int., 2019, 68, 125–133 CrossRef CAS.
- A. Kovalcik, L. Sangroniz, M. Kalina, K. Skopalova, P. Humpolíček, M. Omastova, N. Mundigler and A. J. Müller, Int. J. Biol. Macromol., 2020, 161, 364–376 CrossRef CAS PubMed.
- H. C. Erythropel, M. Maric and D. G. Cooper, Chemosphere, 2012, 86, 759–766 CrossRef CAS PubMed.
- R. Jamarani, H. C. Erythropel, J. A. Nicell, R. L. Leask and M. Marić, Polymers, 2018, 10, 834 CrossRef PubMed.
- R. U. Halden, Annu. Rev. Public Health, 2010, 31, 179–194 CrossRef PubMed.
- H. Frederiksen, N. E. Skakkebæk and A. M. Andersson, Mol. Nutr. Food Res., 2007, 51, 899–911 CrossRef CAS PubMed.
- C. Yang, S. A. Harris, L. M. Jantunen, J. Kvasnicka, L. V. Nguyen and M. L. Diamond, Environ. Sci. Technol., 2020, 54, 8186–8197 CrossRef CAS PubMed.
- S. Benjamin, E. Masai, N. Kamimura, K. Takahashi, R. C. Anderson and P. A. Faisal, J. Hazard. Mater., 2017, 340, 360–383 CrossRef CAS PubMed.
- D. W. Gao and Z. D. Wen, Sci. Total Environ, 2016, 541, 986–1001 CrossRef CAS PubMed.
- A. Lenoir, R. Boulay, A. Dejean, A. Touchard and V. Cuvillier-Hot, Environ. Sci. Pollut. Res., 2016, 23, 16865–16872 CrossRef CAS PubMed.
- X.-F. Wei, E. Linde and M. S. Hedenqvist, npj Mater. Degrad., 2019, 3, 18 CrossRef.
- United States Congress, Public Law 110–314—Aug. 14, 2008, Consumer Product Safety Improvement Act, https://www.congress.gov/110/plaws/publ314/PLAW-110publ314.pdf, visited in July 2021.
- Phthalates Regulations, https://laws-lois.justice.gc.ca/PDF/SOR-2016-188.pdf, visited in July 2021.
- European Commision, COMMISSION REGULATION (EU) 2018/2005, https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32018R2005&from=EN, visited in July 2021.
- Eurpean Commission, Commission Staff Working Document (SWD) on endocrine disruptors, https://ec.europa.eu/environment/pdf/chemicals/2020/10/SWD_on_Endocrines_disruptors.pdf, visited in July 2021.
- P. Jia, H. Xia, K. Tang and Y. Zhou, Polymers, 2018, 10, 1303 CrossRef PubMed.
- L. H. Innocentini-Mei, J. R. Bartoli and R. C. Baltieri, Macromol. Symp., 2003, 197, 77–88 CrossRef CAS.
- D. M. Panaitescu, C. A. Nicolae, A. N. Frone, I. Chiulan, P. O. Stanescu, C. Draghici, M. Iorga and M. Mihailescu, J. Appl. Polym. Sci., 2017, 134, 1–14 CrossRef.
- D. Garcia-Garcia, J. M. Ferri, N. Montanes, J. Lopez-Martinez and R. Balart, Polym. Int., 2016, 65, 1157–1164 CrossRef CAS.
- J. S. Choi and W. H. Park, Polym. Test., 2004, 23, 455–460 CrossRef CAS.
- M. G. A. Vieira, M. A. Da Silva, L. O. Dos Santos and M. M. Beppu, Eur. Polym. J., 2011, 47, 254–263 CrossRef CAS.
- R. C. Baltieri, L. H. I. Mei and J. Bartoli, Macromol. Symp., 2003, 197, 33–44 CrossRef CAS.
- S. P. Carlson and K. D. Chang, J. Am. Oil Chem. Soc., 1985, 62, 934–939 CrossRef.
- N. Rao, S. Kaujalgikar, B. I. Chaudhary, S. Bhide, S. Morye and S. Agashe, US Pat., US985036B2, 2017.
- M. Bocqué, C. Voirin, V. Lapinte, S. Caillol and J. J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 11–33 CrossRef.
- F. Chiellini, M. Ferri, A. Morelli, L. Dipaola and G. Latini, Prog. Polym. Sci., 2013, 38, 1067–1088 CrossRef CAS.
- A. Takeshita, J. Igarashi-migitaka, K. Nishiyama, H. Takahashi, Y. Takeuchi and N. Koibuchi, Toxicol. Sci., 2011, 123, 460–470 CrossRef CAS PubMed.
- Y. Xu, S. H. Park, K. N. Yoon, S. J. Park and M. C. Gye, Environ. Res., 2019, 172, 675–683 CrossRef CAS PubMed.
- T. T. Bui, G. Giovanoulis, A. P. Cousins, J. Magnér, I. T. Cousins and C. A. de Wit, Sci. Total Environ., 2016, 541, 451–467 CrossRef CAS PubMed.
- Y. Xu and M. C. Gye, Chemosphere, 2018, 204, 523–534 CrossRef CAS PubMed.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- T. Werpy and G. R. Petersen, Top Value Added Chemicals From Biomass: Volume I: Results of Screening for Potential Candidates from Sugars and Synthesis Gas, https://www.nrel.gov/docs/fy04osti/35523.pdf, visited in July 2021.
- L. Nattrass, C. Biggs, A. Bauen, C. Parisi, E. Rodríguez-Cerezo and M. Gómez-Barbero, The EU bio-based industry: Results from a survey, 2016 DOI:10.2791/806858.
- F. D. Pileidis and M. M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed.
- W. Xuan, M. Hakkarainen and K. Odelius, ACS Sustainable Chem. Eng., 2019, 7, 12552–12562 CAS.
- A. Sinisi, M. Degli Esposti, M. Toselli, D. Morselli and P. Fabbri, ACS Sustainable Chem. Eng., 2019, 7, 13920–13931 CrossRef.
- P. H. Daniels, J. Vinyl Addit. Technol., 2009, 15, 219–223 CrossRef CAS.
- V. V. Senichev and V. Y. Tereshatov, Handbook of Plasticizers, William Andrew Publishing, Norwich, NY, 3rd edn, 2017, pp. 135–164 Search PubMed.
- A. Marcilla and M. Beltrán, Handbook of Plasticizers, ChemTec Publishing, Ontario, Canada, 3rd edn, 2017, pp. 119–134 Search PubMed.
- A. Rahim, P. Saha, K. K. Jha, N. Sukumar and B. K. Sarma, Nat. Commun., 2017, 8, 1–12 CrossRef CAS PubMed.
- S. K. Singh, K. K. Mishra, N. Sharma and A. Das, Angew. Chem., Int. Ed., 2016, 55, 7801–7805 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, Chem. Commun., 2013, 49, 7699–7701 RSC.
- J. Yuan and B. Cheng, Sci. Rep., 2017, 7, 9277 CrossRef PubMed.
- A. El-Hadi, R. Schnabel, E. Straube, G. Müller and S. Henning, Polym. Test., 2002, 21, 665–674 CrossRef CAS.
- P. H. Daniels and A. Cabrera, J. Vinyl Addit. Technol., 2015, 21, 7–11 CrossRef CAS.
- T. Mekonnen, P. Mussone, H. Khalil and D. Bressler, J. Mater. Chem. A, 2013, 1, 13379–13398 RSC.
- P. Persico, V. Ambrogi, A. Baroni, G. Santagata, C. Carfagna, M. Malinconico and P. Cerruti, Int. J. Biol. Macromol., 2012, 51, 1151–1158 CrossRef CAS PubMed.
- L. M. W. K. Gunaratne and R. A. Shanks, Eur. Polym. J., 2005, 41, 2980–2988 CrossRef CAS.
- G. Foli, M. Degli Esposti, D. Morselli and P. Fabbri, Macromol. Rapid Commun., 2020, 41, 1900660 CrossRef CAS PubMed.
- R. S. Kurusu, C. A. Siliki, É. David, N. R. Demarquette, C. Gauthier and J. M. Chenal, Ind. Crops Prod., 2015, 72, 166–174 CrossRef CAS.
- W. George, Handbook of Plasticizers, ChemTec Publishing, Ontario, Canada, 3rd edn, 2017, pp. 1–6 Search PubMed.
- J. L. Audic, L. Lemiègre and Y. M. Corre, J. Appl. Polym. Sci., 2014, 131, 1–7 CrossRef.
- M. L. Di Lorenzo, M. Gazzano and M. C. Righetti, Macromolecules, 2012, 45, 5684–5691 CrossRef CAS.
- I. Chodak, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, pp. 451–477 Search PubMed.
- H. Tsuji and K. Suzuyoshi, Polym. Degrad. Stab., 2002, 75, 347–355 CrossRef CAS.
- R. Pantani and A. Sorrentino, Polym. Degrad. Stab., 2013, 98, 1089–1096 CrossRef CAS.
- M. Rutkowska, M. Jastrzębska and H. Janik, React. Funct. Polym., 1998, 38, 27–30 CrossRef CAS.
- M. Rutkowska, K. Krasowska, A. Heimowska, M. Smiechowska and H. Janik, Iran. Polym. J., 2000, 9, 221–227 CAS.
- G. Scoponi, S. Guzman-Puyol, G. Caputo, L. Ceseracciu, A. Athanassiou and J. A. Heredia-Guerrero, Polymer, 2020, 193, 122371 CrossRef CAS.
- P. Cataldi, M. Cassinelli, J. A. Heredia-Guerrero, S. Guzman-Puyol, S. Naderizadeh, A. Athanassiou and M. Caironi, Adv. Funct. Mater., 2020, 30, 1907301 CrossRef CAS.
- G. Tedeschi, S. Guzman-Puyol, L. Ceseracciu, U. C. Paul, P. Picone, M. Di Carlo, A. Athanassiou and J. A. Heredia-Guerrero, Biomacromolecules, 2020, 21, 910–920 CrossRef CAS PubMed.
- M. Degli Esposti, F. Chiellini, F. Bondioli, D. Morselli and P. Fabbri, Mater. Sci. Eng., C, 2019, 100, 286–296 CrossRef CAS PubMed.
- P. J. Barham, A. Keller, E. L. Otun and P. A. Holmes, J. Mater. Sci., 1984, 19, 2781–2794 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, more characterizations such as FTIR spectra of the intermediates, 13C-NMR spectra of the final ketal–esters, photographs of samples, DSC thermograms, DMTA curves, and full characterization of the purified PHB used in this work are reported. See DOI: 10.1039/d1ma00833a |
| This journal is © The Royal Society of Chemistry 2021 |