
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic reduction of CO2 by halide perovskites: recent advances and future perspectives
Muhammad Ali
Raza
a,
Feng
Li
 a,
Meidan
Que
*b,
Liangliang
Zhu
*a and
Xi
Chen
*c
a,
Meidan
Que
*b,
Liangliang
Zhu
*a and
Xi
Chen
*c
aSchool of Chemical Engineering, Northwest University, Xi’an 710069, China. E-mail: zhu.liangliang@nwu.edu.cn
bCollege of Materials Science and Engineering, Xi’an University of Architecture and Technology, Xi’an 710055, China. E-mail: mdque@xauat.edu.cn
cEarth Engineering Center, Center for Advanced Materials for Energy and Environment, Department of Earth and Environmental Engineering, Columbia University, New York, NY10027, USA. E-mail: xichen@columbia.edu
First published on 5th October 2021
Abstract
Photocatalytic CO2 reduction to generate energy-rich fuels through solar energy provides an attractive route to alleviate the global energy crisis and environmental concerns. Searching for various photocatalysts with high catalytic activity and selectivity for the transformation of CO2 is the key strategy to accomplish this goal. Halide perovskite nanomaterials, with the advancements of facile synthesis, excellent light-harvesting, efficient exciton generation, long carrier diffusion length, and abundant surface sites, have great potential in solar energy conversion. In this review, the fundamental photocatalytic mechanism for CO2 reduction is firstly highlighted and the impact of the structural properties of halide perovskites is presented. Then, recent advancements of the reaction medium, halide perovskites (e.g., ABX3 structure and A2B′B′′X6 structure), and their composites (e.g., oxide, sulfide, carbide, metal–organic framework, noble metal, and carbon derivative) as photocatalysts are described and analyzed. Finally, potential research approaches and future perspectives for the improvement of halide perovskite-based photocatalysts toward efficient CO2 reduction are briefly discussed. This review sheds light on the upcoming innovative photocatalysts for CO2 reduction into high value-added products.
 Muhammad Ali Raza | Muhammad Ali Raza received his M.S. in Environmental Engineering from Northwestern Polytechnical University in 2017. Now, he is pursuing the PhD degree at Northwest University, China. His research direction is photocatalytic CO2 reduction. |
 Feng Li | Feng Li is a graduate student at Northwest University, Xi'an, China. His research interests focus on materials and processes for carbon capture and utilization. |
 Meidan Que | Meidan Que is currently working as an associate professor at Xi'an University of Architecture and Technology in China. She got her PhD at Xi'an Jiaotong University in 2018 working on optical materials for perovskite solar cells. From Sep. 2017 to Sep. 2018, she studied as a visiting scholar at Brown University. Currently, her research interests focus on semiconductor quantum dots for photovoltaic systems and nanocomposites for photocatalyst applications. |
 Liangliang Zhu | Liangliang Zhu is working as an associate professor at the School of Chemical Engineering of Northwest University, Xi'an, China. He received his PhD degree from Xi'an Jiaotong University, Xi'an, China, in 2018. His research interest is in multi-scale materials for carbon capture and utilization, and flexible structures. |
 Xi Chen | Xi Chen is a professor at the Department of Earth and Environment Engineering at Columbia University. He is the Director of the Earth Engineering Center, and the Center for Advanced Materials for Energy and Environment at Columbia University. He received his PhD from Harvard University in 2001 and joined Columbia in 2003. He has received numerous awards including the Presidential Early Career Award for Scientists and Engineers (PECASE), ASME Sia Nemat-Nasser Early Career Award, and the SES Young Investigator Medal. |
1. Introduction
Tremendous natural environmental changes have made people around the world increase crisis awareness when we are facing unprecedented nature. Great attention has been paid to the depletion of natural resources and the impact of the emission of greenhouse gases on the environment due to the burning of fossil fuels.1–4 Solar-driven carbon dioxide (CO2) conversion is an encouraging strategy to alleviate the negative effect of greenhouse gases,5,6 which uses artificial photosynthesis and photocatalysis of solar energy for carbon feedbacks and is also a straightforward and flexible pathway of CO2 valorization.7,8Since the landmark breakthrough in 1972, the pioneering work on ultraviolet (UV) driven photocatalytic production of hydrogen by TiO29 has spurred enormous interest and motivated decades of scientific research and progress, which are of particular importance for energy transformation (e.g., reduction of CO2 and water splitting), chemical transformations, and the decontamination of organic pollutants.10 During recent years, the rapid increase in the CO2 concentration in the environment has caused widespread concern.11 Extensive efforts have been devoted to stabilizing and controlling the concentration of CO2 in the environment, leading to CO2 capture and renewable energy production. In particular, the state-of-the-art photocatalytic reduction of CO2 has been considered as one of the most promising ways.12
From the perspective of long-term development, it is essential to explore cost-efficient photocatalysts with outstanding properties and performances. An ideal photocatalytic material should possess wide-range and superior light absorption, effective charge separation, excellent stability and proper redox ability.13 Upon illumination, the photogenerated charge carriers are produced in the conduction band (CB) and valence band (VB) in semiconductors, respectively. When the energy structure of these semiconductors straddles the redox potentials, their photogenerated charge carriers would involve the surface redox reaction. However, most pure semiconductors (g-C3N4,14 TiO2,15 and Cu2O16) as photocatalysts are far from the ideal state yet, because of various limitations, e.g., quick recombination of photo-generated charge carriers, large bandgap, and poor photoreduction potential. Hence, there is still a huge demand to explore low-cost, stable, and effective artificial photocatalysts for CO2 fixation.
Recently, halide perovskites have drawn enormous attention by virtue of their excellent photoelectric properties, for example, long charge carrier diffusion length, adjustable bandgap, high absorption coefficient, and excellent external quantum efficiency.17–21 Due to these impressive characteristics and attractive properties, halide perovskites have been extensively employed in the fields of photoelectric conversion,22 reduction reaction of CO2, and hydrogen evolution reaction.23–27 These successful applications also encourage more applications in other traditional fields.28 Recent developments in the synthesis methods (e.g., hot-injection, ligand-assisted reprecipitation scheme, ultrasonic method, solvothermal method, microwave assisted method, and ball milling)29–35 have promoted halide perovskites marching to practical usage.36 Through controlling the production method, reaction temperature, capping ligand, and additional reaction conditions, halide perovskites with variable chemical components (e.g., inorganic, organic, and inorganic hybrid), dimensions, sizes, and morphologies can be obtained.37–41 These efforts are expected to bring new opportunities for halide perovskites in the application of photocatalysis.
Halide perovskites have recently attracted great attention towards the reduction of CO2 into high value-added products, from basic and theoretical research to applied science.42–52 Shown in Fig. 1 is a timeline of the development of halide perovskites as photocatalysts for CO2 reduction.
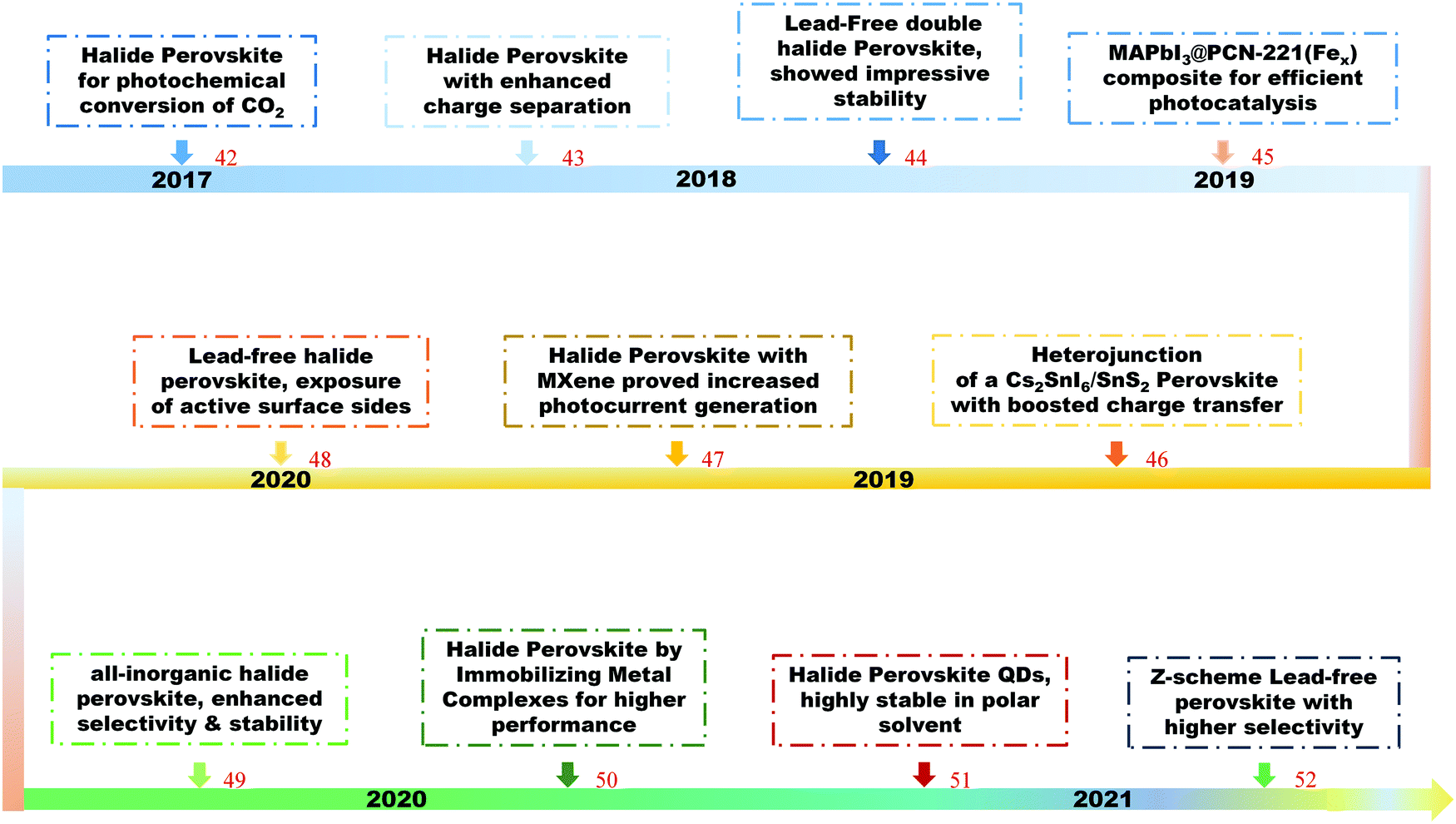 | ||
| Fig. 1 The development of halide perovskites as photocatalysts for CO2 reduction. | ||
Up to now, there have been some review works of halide perovskites as photocatalysts focusing on the stability issues, or modification strategies through structural engineering and interfacial modulations.53–57 However, a timely and comprehensive review is still in demand concerning the photoreduction mechanism, reductants and products, the structure and photocatalytic performances, and the exploration efforts on halide perovskites and their composites for further improvements. In this review, we start with the basic mechanism of the photoreduction of CO2, and the reductants and products. After that, the fundamental photoelectrical properties of halide perovskites are described collectively, including their crystal structure, defect tolerance, toxicity, luminescence properties, and photostability issues. Insights into the molecular calculations of halide based perovskites concerning their unique electronic structure and physio-chemical properties are also included. Then, we summarize the existing exploration efforts on halide perovskites and their composites in tuning the selectivity of photocatalytic reduction of CO2 as well as increasing the active sites. Finally, we present the outlook for upcoming directions and the great potential of halide perovskites toward photocatalytic CO2 reduction.
2. Photocatalytic mechanism for CO2 reduction
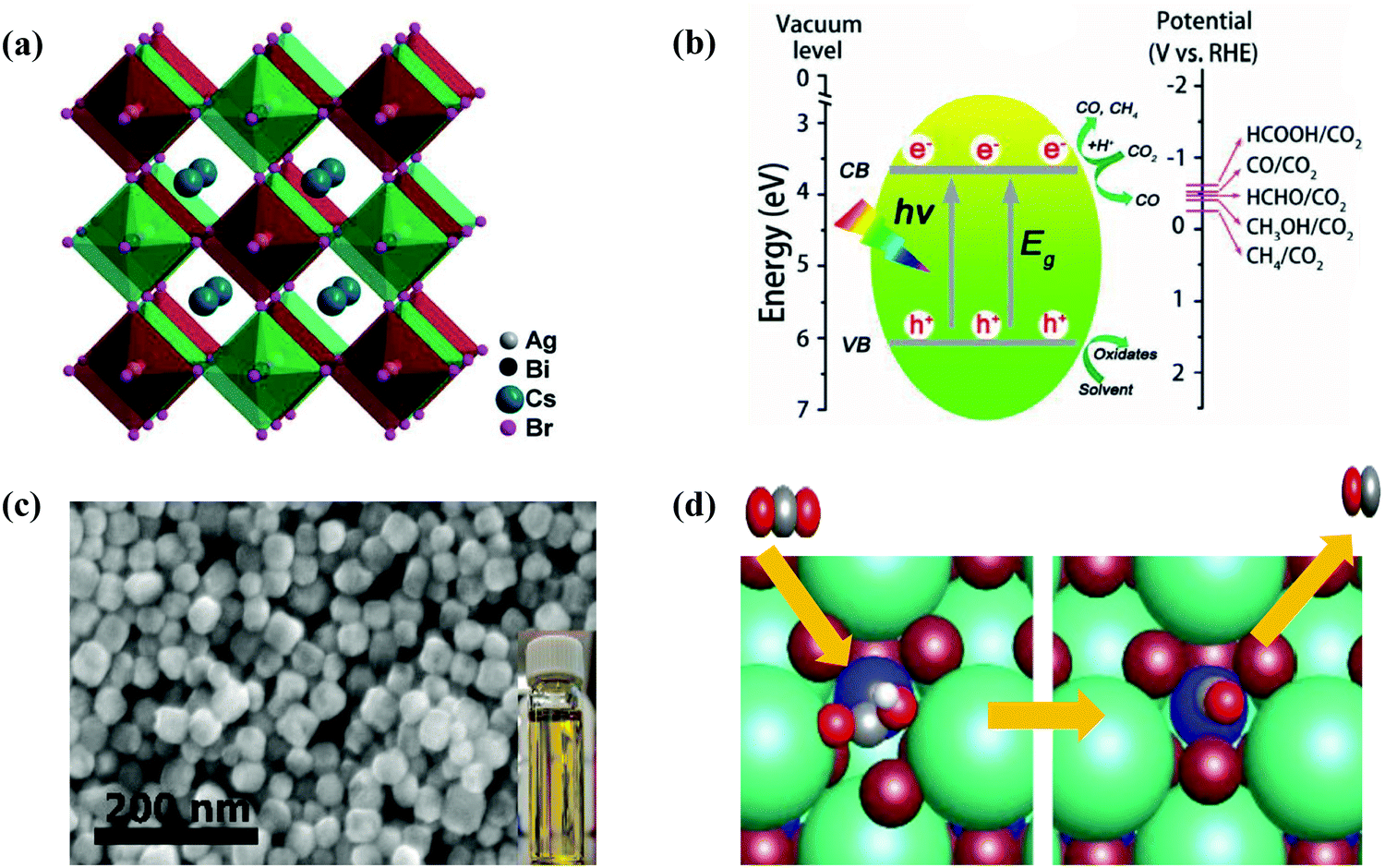
In the process of photocatalysis, the abundantly available solar energy is transformed into electrical/chemical/thermal energies through semiconductor materials.13,58–61 In general, the photocatalysis process for CO2 reduction mainly consists of three critical synergistic steps, as illustrated in Fig. 2. In the first step, photon absorption occurs to create electron–hole pairs. During this catalytic reaction, incident light induces the transition of electrons from the valence band (VB) to the conduction band (CB), leaving an equal number of holes in the VB. A suitable band structure of the photocatalyst is required to reduce CO2 or oxidize water. However, in the second step, a large number of fractions of photogenerated electron–hole pairs may be consumed by recombination. The recombination is dominant over charge separation or vice versa, depending on the relative time scale among the rate of recombination and photogenerated carriers. This issue can be resolved via a very complex interaction among dimension, surface properties, material crystallinity, and several other structural factors. The design of balanced active sites was utilized to facilitate charge separation. The final step is the CO2 adsorption. This redox reaction involves the transfer of electrons from the photocatalyst to CO2 molecule. In general, larger surface area of the photocatalyst can lead to higher rate of CO2 adsorption thanks to more active sites.62–64 The photocatalytic activity is generally associated with two main conditions: (i) the bandgap energy (Eg) of the photocatalyst should be lower than the energy of the incident light (hv; the absorption energy). (ii) The reduction potential of the reacting species should be positioned between the CB and VB values of the photocatalyst material. The former condition specifies a narrower bandgap that can help the effective utilization of incident photons. Conversely, the latter condition reveals that a higher value of CB potential and a lower value of VB potential are thermodynamically advantageous toward the reduction as well as oxidation reactions of the reacting species, respectively. Therefore, it is inevitable to seek the balance point owing to the existence of the noticeable inconsistency as discussed in the above conditions. However, this is quite a challenging task to obtain both broad light absorption and robust redox capability simultaneously for pure component semiconductor materials. Moreover, the photogenerated electrons in the CB can easily recombine with holes, or be trapped in the defect state, or easily come back to the VB for a pure-component material, which would decrease the efficiency of solar energy utilization.65 Hence, designing a suitable heterogeneous photocatalyst is an effective approach to overcome the above-mentioned issues. Generally, there are four vital rules played by cocatalysts: (i) enhancing the separation and transfer of charges, (ii) boosting the performance, and the selectivity of photoreduction of CO2, (iii) improving the durability of photocatalysts in a peculiar environment (e.g. high humidity and high temperature), and (iv) suppressing the side reactions (e.g., H2 reduction).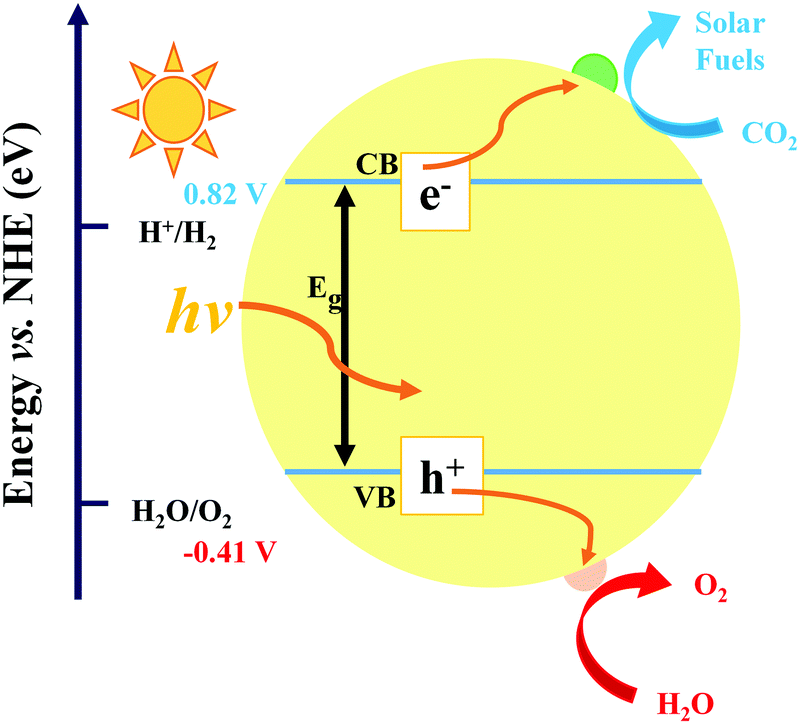 | ||
| Fig. 2 Schematic diagram of the photocatalytic CO2 reduction mechanism. | ||
Photocatalytic reduction of CO2 with a semiconductor-based cocatalyst is affected by many aspects, for example, photocatalyst loading, particle size, structure, composition, dispersion, crystal facets, alloy phase, morphology, and valence states. In a photocatalyst system, the maximum photocatalytic performance is accomplished at an optimum loading of cocatalysts. An additional loading of the cocatalyst may lead to deterioration of catalytic activity owing to the following factors. (i) Too much cocatalyst can hinder the incident light and reduce the light absorption ability, hence decreasing the photogenerated electron–hole pairs. (ii) Excessive cocatalyst percentage could in turn shield the active sites on the surface of the photocatalyst, therefore lessening the contact with H2O and CO2 molecules. (iii) Excessive loading may cause the irregular dispersion and formation of bigger particles, consequently reducing the catalytic performance.
A closed gas circulation reactor is usually used for photoreduction of CO2, where the responded CO2 decreased in the existence of semiconductors under continuous illumination.66–71 Since the Gibbs free-energy (ΔG) value for the reduction reaction of CO2 to produce carbon monoxide (CO) is ∼258.95 kJ mol−1, the bandgap of no less than 1.35 eV is needed. Then rapid separation and transfer of the photogenerated charge carriers to the particular catalytic active sites of the photocatalysts triggers the electrochemical reaction, resulting in chemical fuel. For photocatalytic reduction of CO2 in the existence of water, the CB must be negatively paralleled to the reduction potential of CO2 (−1.07 V vs. normal hydrogen electrode (NHE)) for CO2 reduction to CO at pH = 0. Simultaneously, the VB should be larger than the oxidation potential of water (1.23 V vs. NHE at pH 0) to procedure O2. Fig. 3 displays the schematic representation of the bandgap and band position of reported typical halide perovskite materials and CO2 reduction potential.
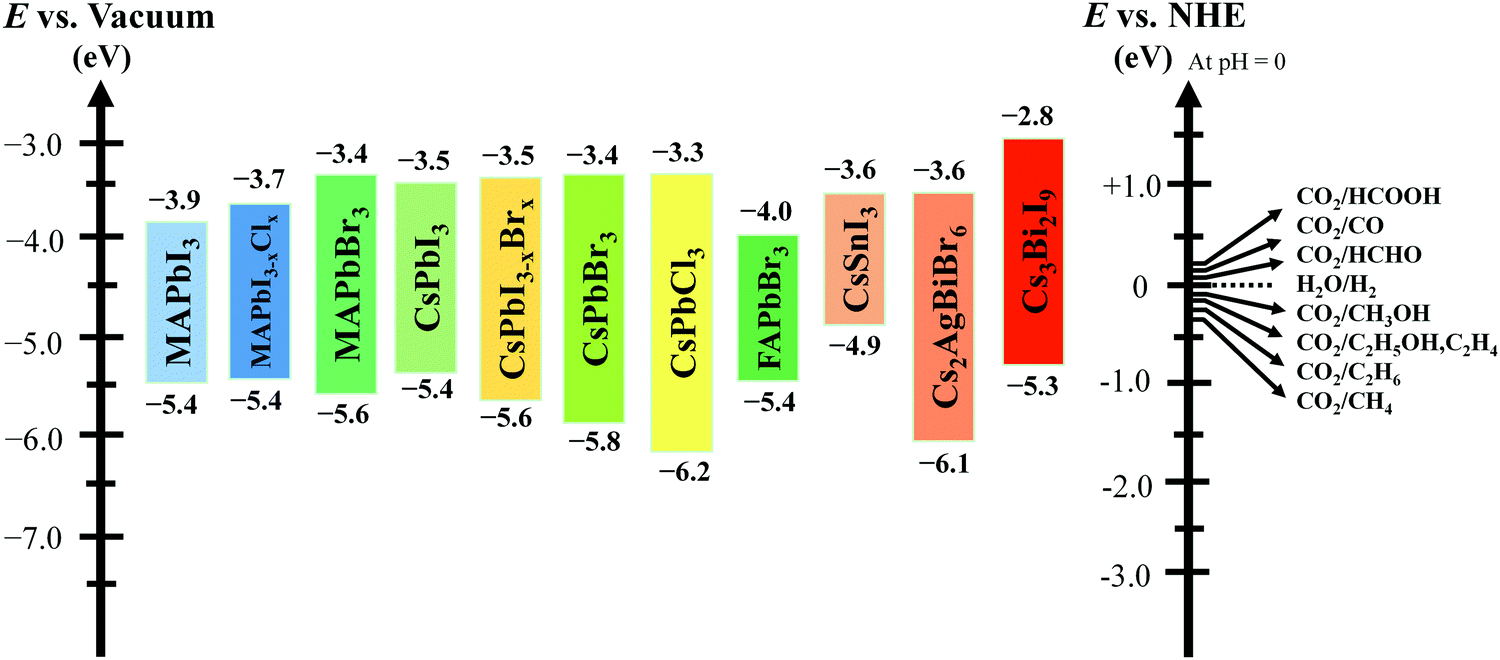 | ||
| Fig. 3 Conduction and valence band position of halide-based perovskite materials with relative CO2 reduction potential. | ||
Photocatalytic CO2 reduction reaction is a high energy-driven process, because of the carbon-based compound that has a thermodynamically stable and high bond energy of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (750 kJ mol−1).72,73 Therefore, the photocatalytic reduction reaction of CO2 also needs prior activation. The process of mass transfer of reactants, CO2 adsorption and creation of active sites for CO2 reduction can be improved by increasing the surface area of a photocatalyst. A large number of nanostructured materials for the photocatalyst were developed to achieve larger surface areas, including zero dimensional (0D) nanoparticles,74 one dimensional (1D) nanowires/nanotubes,75 two dimensional (2D) nanosheets,76,77 and three dimensional (3D) hierarchical nanostructures.78 Furthermore, various porous and hollow materials were extensively studied for CO2 conversion because of their increased surface area and porosity. The adsorption and activation of CO2 can also be facilitated by tuning the surface defects of photocatalysts, which can be quite helpful in improving the reactivity and photochemical properties of the photocatalytic processes.79,80 Strongly negative reduction potential is required for the activation of CO2, which occurs on the surface of photocatalysts by the chemisorption method to convert a linear molecule to bend carbonate anion radical (CO2˙−) via mono- or bi-dentate coordination. Hence, this reactive carbonate anion is the critical factor for various photoreduction of CO2. To create chains of hydrocarbons, more reactive CO2˙− species can promote reactions via two different ways (carbene and formaldehyde), as illustrated in Table 1.81 However, the obtained products are methane (CH4) and the formation of some additional products (C1–C3 hydrocarbons) in the photocatalytic reduction of CO2. Hori et al. suggested the conversion tracking of CO2 to CO by reactive CO2˙−, and subsequently the reaction with a proton.82 For the photocatalytic CO2 reduction reaction, the selectivity of product formation is still challenging and relies on several factors, such as reaction intermediate, and active sites of the photocatalysts. The release of protons initiates these reactions in the presence of water vapor, H2, or in both cases. These above-mentioned protons form different reduction products after reaction with CO2 in the existence of photogenerated electrons. The efficiency of the photoreduction process of CO2 can be shown in terms of product formation rate and the conversion rate of CO2.83 CO2 reduction starts with the formation of new carbon bonds by the cleavage of the OCO bond. Photocatalytic reduction of CO2 is a proton-assisted multielectron reduction process that comprises various intermediate steps as shown in Table 1, for obtaining high-value products (e.g., CO, formic acid, methane, and methanol).84,85
O (750 kJ mol−1).72,73 Therefore, the photocatalytic reduction reaction of CO2 also needs prior activation. The process of mass transfer of reactants, CO2 adsorption and creation of active sites for CO2 reduction can be improved by increasing the surface area of a photocatalyst. A large number of nanostructured materials for the photocatalyst were developed to achieve larger surface areas, including zero dimensional (0D) nanoparticles,74 one dimensional (1D) nanowires/nanotubes,75 two dimensional (2D) nanosheets,76,77 and three dimensional (3D) hierarchical nanostructures.78 Furthermore, various porous and hollow materials were extensively studied for CO2 conversion because of their increased surface area and porosity. The adsorption and activation of CO2 can also be facilitated by tuning the surface defects of photocatalysts, which can be quite helpful in improving the reactivity and photochemical properties of the photocatalytic processes.79,80 Strongly negative reduction potential is required for the activation of CO2, which occurs on the surface of photocatalysts by the chemisorption method to convert a linear molecule to bend carbonate anion radical (CO2˙−) via mono- or bi-dentate coordination. Hence, this reactive carbonate anion is the critical factor for various photoreduction of CO2. To create chains of hydrocarbons, more reactive CO2˙− species can promote reactions via two different ways (carbene and formaldehyde), as illustrated in Table 1.81 However, the obtained products are methane (CH4) and the formation of some additional products (C1–C3 hydrocarbons) in the photocatalytic reduction of CO2. Hori et al. suggested the conversion tracking of CO2 to CO by reactive CO2˙−, and subsequently the reaction with a proton.82 For the photocatalytic CO2 reduction reaction, the selectivity of product formation is still challenging and relies on several factors, such as reaction intermediate, and active sites of the photocatalysts. The release of protons initiates these reactions in the presence of water vapor, H2, or in both cases. These above-mentioned protons form different reduction products after reaction with CO2 in the existence of photogenerated electrons. The efficiency of the photoreduction process of CO2 can be shown in terms of product formation rate and the conversion rate of CO2.83 CO2 reduction starts with the formation of new carbon bonds by the cleavage of the OCO bond. Photocatalytic reduction of CO2 is a proton-assisted multielectron reduction process that comprises various intermediate steps as shown in Table 1, for obtaining high-value products (e.g., CO, formic acid, methane, and methanol).84,85
| Equation | Product | E 0 (V) |
|---|---|---|
| CO2 + 1e− = CO2˙− | Carbonate anion radical | −1.90 |
| CO2 + 2H+ + 2e− = HCOOH (aq) | Formic acid | −0.61 |
| CO2 + 2H+ + 2e− = CO (g) + H2O | Carbon monoxide | −0.53 |
| CO2 + 4H+ + 4e− = HCHO (aq) + H2O | Formaldehyde | −0.48 |
| CO2 + 6H+ + 6e− = CH3OH (aq) + H2O | Methanol | −0.38 |
| CO2 + 8H+ + 8e− = CH4 (g) + H2O | Methane | −0.24 |
3. Crystal structure property of halide perovskites
3.1. ABX3 structure
Halide perovskites employed by photocatalytic CO2 reduction facilitate band alignment with the CB position and hinder the photogenerated charge carriers from undergoing recombination. For achieving the higher productivity and selectivity of halide perovskite nanocrystals (NCs) for photoreduction of CO2, fundamental understandings of the relationships between the structure (chemical composition as well as morphology) and CO2 selectivity need to be determined.89 In this section, the impact stemming from halide perovskites from the unit cell will be reviewed. The unit cell is the simplest and smallest volume of a material. The properties of materials derive from special characteristics of the unit cell, e.g., the bond angle, bond length, and symmetry. Moreover, the diffusion lengths and lifetimes of charge carriers in optoelectronic materials are more affected by crystal boundaries and crystal defects.One of the main chemical formulas of halide perovskites can be symbolized as ABX3, where A and B represent two different kinds of cations and X represents a halide anion (i.e., Cl, Br, and I). Cation B restricts at the median of the octahedron body composed of six halide anions (Fig. 4a).90–92 Conventionally, A and B are cations with 12- and 6-fold coordination with X anions at the corner in ABX3 halide perovskites (Fig. 4a), respectively.93–95 Recently, various high-performing perovskite materials were developed with A being formamidinium (FA), methylammonium (MA), or Cs; B being Pb, Bi, or Sn; and X being Cl, Br or I.93–106
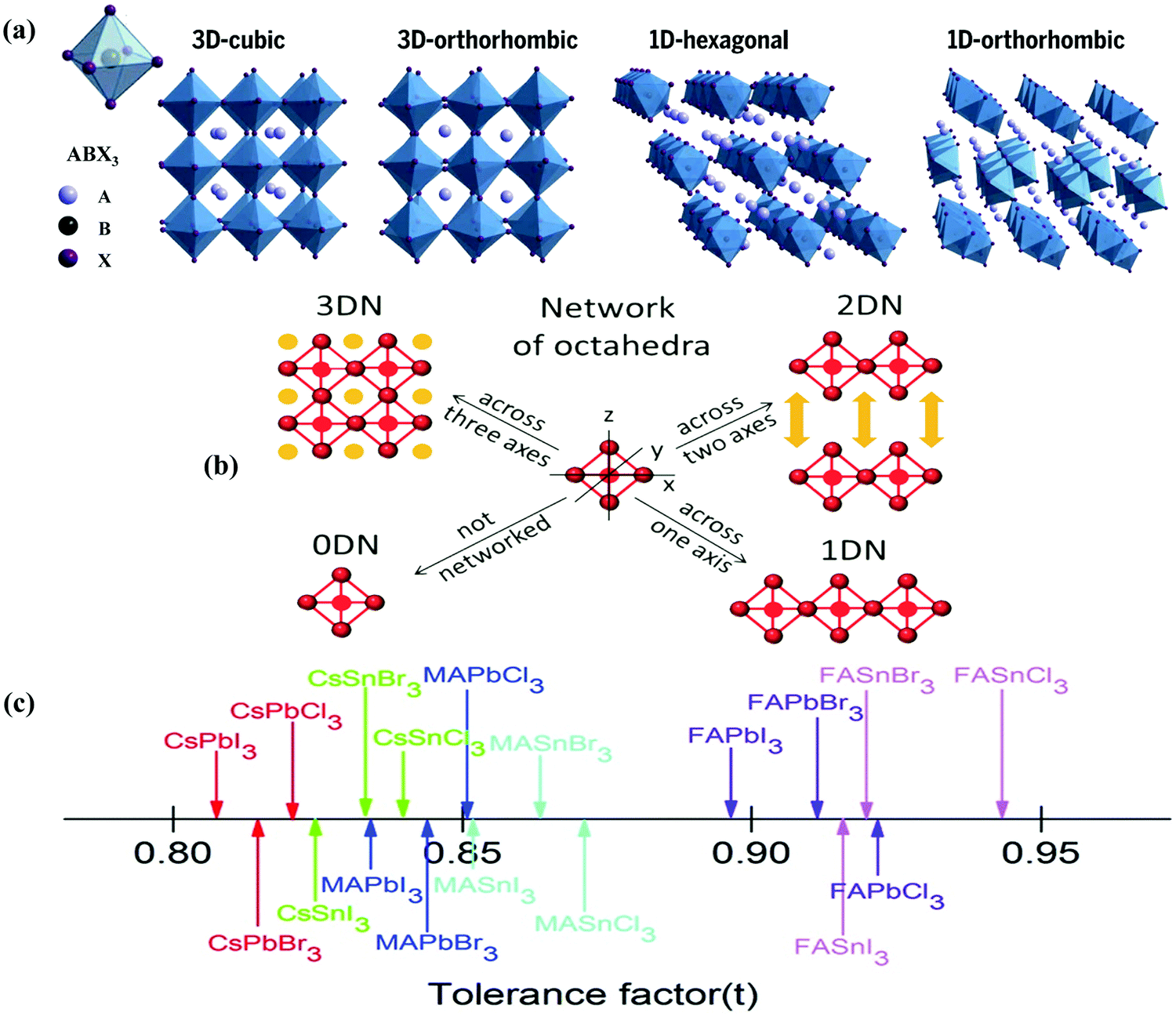 | ||
| Fig. 4 Structures and physical properties of halide perovskites. (a) Crystal structure of 3D-cubic, 3D-orthorhombic, 1D-hexagonal, and 1D-orthorhombic phases. From ref. 92, reprinted with permission from AAAS. (b) Schematic structural diagrams of the change network of octahedra. Reproduced with permission from ref. 107 at https://pubs.acs.org/doi/10.1021/acsenergylett.6b00705; further permissions are directed to the ACS. (c) Tolerance factor of ABX3 halide perovskites. Republished with permission of the Royal Society of Chemistry, from ref. 111; permission conveyed through Copyright Clearance Center, Inc. | ||
When A site is employed with higher organic cations, low-dimensional halide perovskite structures may form, where the inorganic network connectivity has degenerated to 2D sheets, 1D chains, or 0D clusters (Fig. 4b).107 Conversely, the crystal symmetry will reduce and the cubic structure is distorted. In bulk lead halide perovskites, three polymorphs are usually noticed: cubic, tetragonal, and orthorhombic phases in the order of decreasing symmetry (Fig. 4a).92 At higher temperatures, the more stable phase is the cubic phase, and the temperature of phase transitions is clearly defined. For NCs, surface effects may regulate the relative stabilities of several polymorphs, which gains limited attention. All as-synthesized lead halide perovskite NCs crystallize into three-dimensional (3D) phases as follows: MAPbI3 NC is tetragonal; FAPbBr3, FAPbI3, and MAPbBr3 NCs are pseudocubic; and CsPbI3 and CsPbBr3 NCs are orthorhombic at room temperature.92 One of the most significant features of halide perovskites is their higher tolerance for defects. The nature of halide perovskites is defect-tolerant generally owing to their electronic band structure, where the VB maximum is mostly composed of an anti-bonding orbital,108–110 and the CB minimum becomes stabilized via the strongest spin–orbit coupling. Such defect tolerance behavior involves the conservation of a pristine Eg upon the formation of typical defects, due to their defect energy levels residing entirely in either the VB or CB, rather than within the Eg itself. The large hollow between octahedra (A-site) is preoccupied through one or a combination of three big cations (CH(NH2)2+, CH3NH3+, or Cs+), yielding generally the structure of ABX3. The tolerance factor calculated by 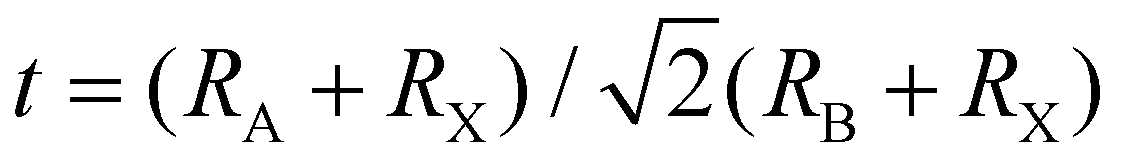 , where RA, RB, and RX are the ionic radii of the corresponding ions, should be near 1 to keep a higher symmetry cubical structure of the perovskite. Fig. 4c presents the tolerance factors for the best widespread Pb or Sn halide perovskites. Due to the larger numbers of Pb or Sn atoms occupying the B sites of halide perovskites, the A site must be larger enough to placate the tolerance factor.111
, where RA, RB, and RX are the ionic radii of the corresponding ions, should be near 1 to keep a higher symmetry cubical structure of the perovskite. Fig. 4c presents the tolerance factors for the best widespread Pb or Sn halide perovskites. Due to the larger numbers of Pb or Sn atoms occupying the B sites of halide perovskites, the A site must be larger enough to placate the tolerance factor.111
3.2. Luminescence properties
Halide perovskite NCs have excellent luminescence without advance electronic surface passivation. Protesescu et al. presented nearly ideal photoluminescence efficiency from colloidal CsPbBr3 NCs.32 The composition of lead halide perovskite NCs can be appropriately adjusted through cation or anion exchange, as shown in Fig. 5a.38 The photoluminescence spectra of lead halide perovskite NCs span the whole visible light range and their peak positions are tunable through modifying the composition (Fig. 5b), size, and shape.92 Higher photoluminescence quantum yield with tunable emission, low costs, and simple synthesis of halide perovskite NCs make them appealing in practical applications. However, the toxicity of lead, sensitivity to the atmosphere condition (humidity, oxygen, daylight, and temperature), and limited yield still hamper further photocatalysis application of lead-based halide perovskite NCs. Halide perovskites have great photophysical properties, including high absorption coefficients, low trap densities, long charge diffusion lengths, and broadly tunable bandgaps. These properties allow for light-harvesting from ultra-visible to the near inferred spectral range.46,112 Besides, the band positions of halide perovskites mostly fulfill the thermodynamic requirements for photocatalytic reduction of CO2.113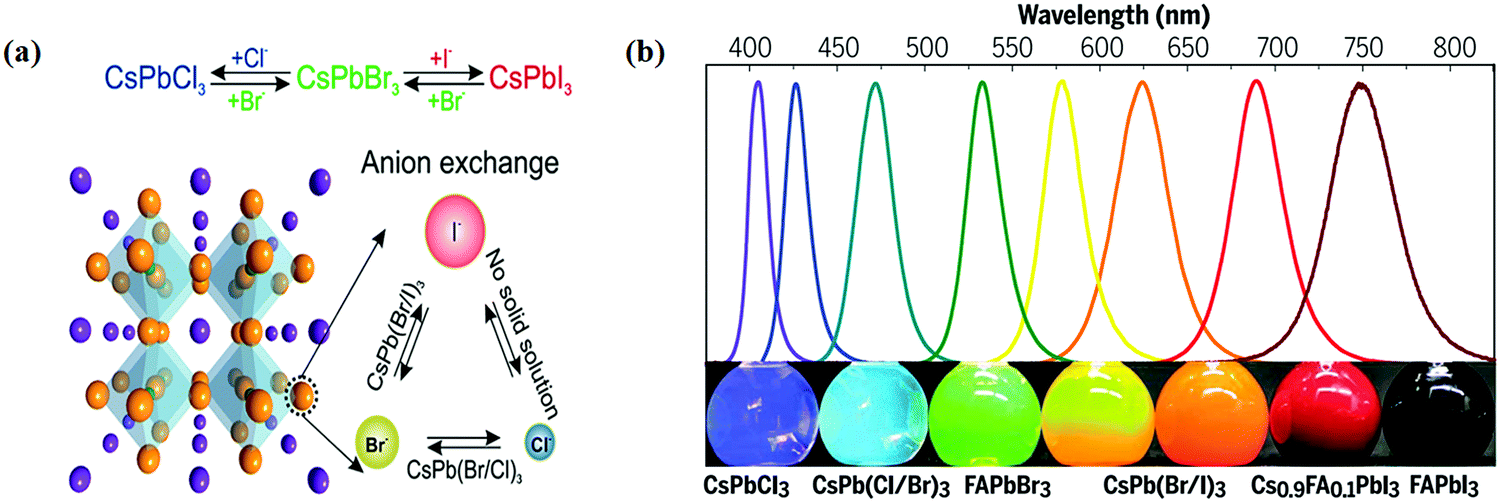 | ||
| Fig. 5 (a) Schematic of the halide anion-exchange process. Reproduced with permission from ref. 38 at https://pubs.acs.org/doi/abs/10.1021/acs.nanolett.5b02404; further permissions are directed to the ACS. (b) Photoluminescence spectra and the corresponding photographs (under varied daylight and UV excitation) of colloid composition-tuned APbX3 NCs. From ref. 92, reprinted with permission from AAAS. | ||
3.3. Other structures
The lead halide perovskite-based NCs with a 3D APbX3 crystal structure and composition have become the focus of much research interest up to now. However, the inherent toxicity and overall reactivity of these halide perovskites have also motivated the scientists to investigate them in various research directions. Firstly, the structural instability and higher ionicity of LHP NCs could usually be taken as a positive aspect, as the APbX3 lattice could be easily rationalized into other phases. This has motivated the researchers to investigate NCs more extensively by exploring their composition and structural characteristics that are defined as “perovskite-related structures”, for example Cs4PbX6 and CsPb2X5 (often called zero-dimensional (0D) and two dimensional (2D) structures, respectively), whereas the characterization of the 3D APbX3 structure was observed to have corner-sharing [PbX6]4− octahedra with the A+ cations filling the holes formed by four neighboring PbX64− octahedra ensuing in a cubic or pseudo-cubic structure (Fig. 6a). The PbX64− octahedra in A4PbX6 structures are dissociated in all extents and the halide ions are no longer shared between them (Fig. 6b).114,115 Layered perovskites have recently been under a lot of scrutinization process. Similar to that of layered double hydroxides containing alternating Cs+ and [Pb2X5]− polyhedron layers, the CsPb2X5 has emerged as a 2D version of lead halide perovskite materials with a tetragonal phase (Fig. 6c).116,117 Another type that contains an alternating layer of corner-sharing [PbX6]4− octahedra and bulky cations is known as the 2D perovskite A2PbX4 phase (Fig. 6d).118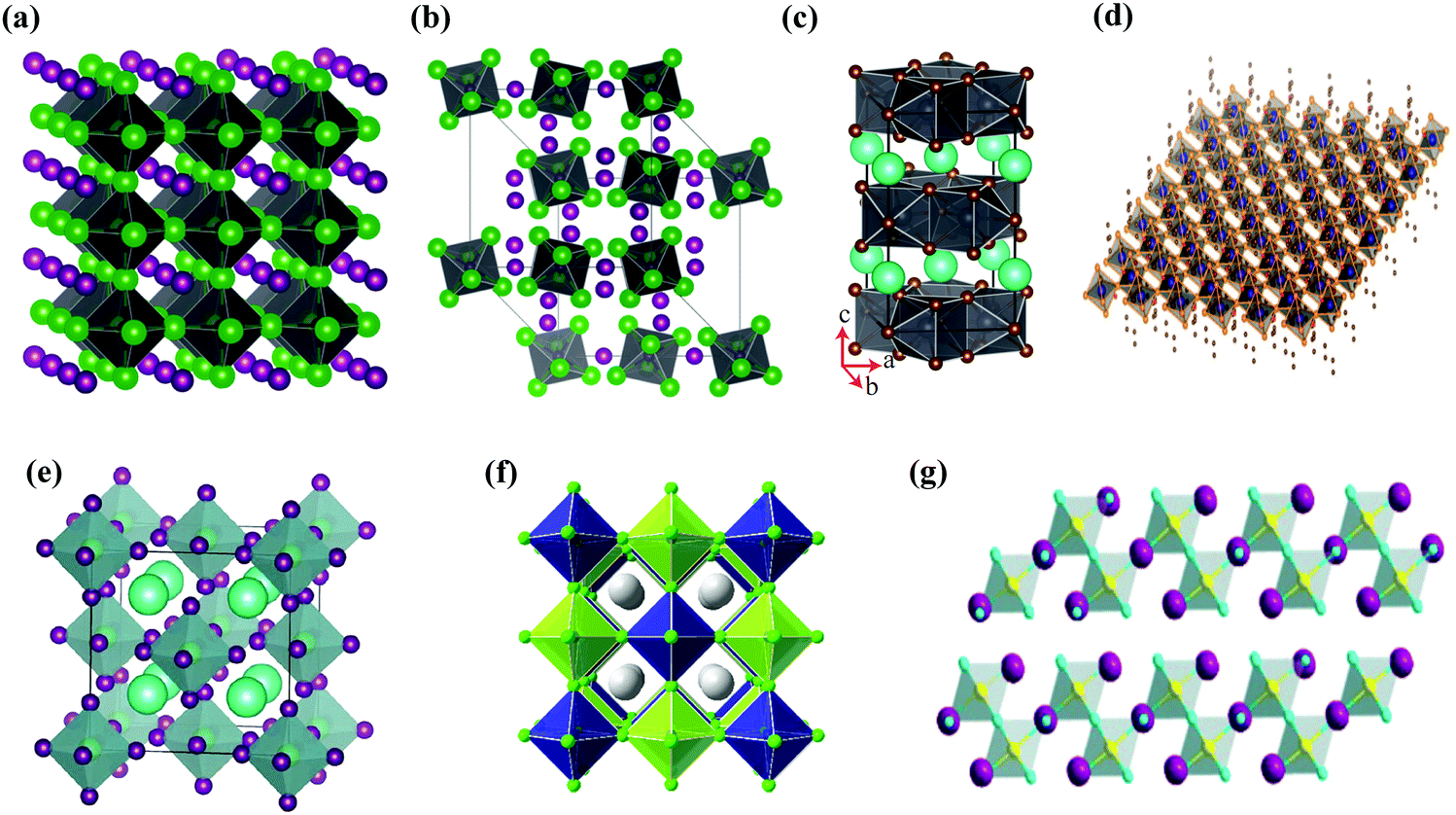 | ||
| Fig. 6 Schematic illustration of different halide structures: (a) pseudocubic ABX3 (3D) and (b) A4BX6 (reproduced with permission from ref. 115 at https://pubs.acs.org/doi/abs/10.1021/acs.nanolett.6b05262; further permissions are directed to the ACS); (c) AB2X5 (2D), reprinted with permission from ref. 116, Copyright 2017 by the American Physical Society; (d) A2BX4, from ref. 118, reprinted with permission from AAAS. (e) A2BX6 (0D), reproduced from ref. 120 under the Creative Commons CC BY license (https://creativecommons.org/licenses/by/4.0/); (f) A2B+B3+X6 (3D), reprinted with permission from ref. 123, Copyright 2016 American Chemical Society; (g) A3B2X9 (2D), reprinted with permission from ref. 121, Copyright 2017 American Chemical Society. | ||
In APbX3 NC systems, the lead toxicity and its bioaccumulation in the ecosystem are known as key drawbacks, which in turn motivate the researchers to find alternative materials with similar optoelectronic characteristics, such as Cs2SnI6 NCs.119–121 To date, there has been very little success. Cs2SnI6 crystallizes in the face-centered cubic structure. Four [SnI6]2− octahedra at the corners and face centers and eight Cs+ cations at the tetragonal interstitials make up the unit cell (Fig. 6e). A Cs2SnI6 structure is a perovskite derivative that is made by removing half of the Sn atoms at regular intervals from each center of the [SnI6] octahedron.120 Because of this, the structure is also known as a “vacancy ordered double perovskite.” Two primary techniques are now being explored in the search for lead-free metal halide compounds: a “simple” substitution of Pb2+ cations with other less toxic divalent metal ions from the same group IV, such as Sn or Ge, and a “complex” substitution of Pb2+ cations with other less toxic divalent metal ions from the same group IV,122 such as Sn or Ge or a substitution of one monovalent M+ and one trivalent M3+ cation for every two divalent Pb2+ ions (i.e., 2Pb2+ → B+ + B3+), resulting in quaternary A2B+B3+X6 systems also known as “double perovskites” as shown in Fig. 6f.123 Other transition or post-transition metals, like Fe3+ and Bi3+, were used to examine the diversity of halide compounds associated with LHPs.124–127 Cs3M2X9 (M = Fe3+ and Bi3+) crystallizes in the hexagonal space group P63/mmc. This is made up of isolated clusters, each of which is made up of two face-sharing octahedra and has the M2Br93− formula, with Cs+ acting as a bridging ion between the clusters. Antimony-based halide compounds, on the other hand, form in a layered shape with each Sb2Br93− cluster sharing corners with three octahedra (Fig. 6g).121
4. Performance of photocatalytic CO2 reduction
4.1. Reaction medium
The solvent plays a critical role in photocatalytic reactions. Halide perovskites are unstable in a polar solvent; therefore, extensive research studies have been accomplished to find a suitable medium for photocatalytic reduction of CO2. Low-polar ethyl acetate was utilized as the solvent due to the higher solubility of CO2, which guarantees durable stability for CsPbBr3 NCs.42 Thereafter, various groups reported the boost in the selectivity of CO2 conversion (99%) with the suppression of production of H2, with the addition of a small amount of water (<50 μl) in ethyl acetate medium.45,100,101,113 However, employing acetonitrile/water (0.3 vol%) mixture showed photocatalytic reduction of CO2 with a high conversion rate (149 μmol g−1 h−1) of CO2 to CO, compared to the ethyl acetate/water (0.3 vol%) mixture upon light irradiation using CsPbBr3 NCs combined with porous g-C3N4. Moreover, enhanced selectivity as well as productivity of the photocatalytic reduction of CO2 was obtained with a high content of diluted water (1.2 vol%).128Owing to several polarities, dielectric constant, and CO2 solubility, the selection of solvents can exert a higher effect on the reaction rate and selectivity. The pairing of a robust co-catalyst is the key factor for enhancing the performance of photocatalysis. Meanwhile, it can not only separate the photogenerated electrons by the creation of a Schottky junction within the co-catalyst and photocatalyst to hinder the charge recombination, but also lower the kinetic bottlenecks in the activation of CO2.129,130 In the present case, the best nonaqueous solvent was ethyl acetate, which resulted in the reduction reaction of CO2 with an electron yield rate of 2.74 μmol g−1 h−1 along with 95.2% selectivity. Furthermore, a photo-deposition of Pt co-catalyst boosts the electron yield rate to 5.66 μmol g−1 h−1.131 Numerous additional solvents such as toluene, benzene, etc. were also explored for photocatalytic reduction of CO2 as a photocatalysis medium in halide perovskite NCs.101 Besides, owing to reducing the formation of less active CO2, higher hydration products and selectivity, solid-vapor CO2 reduction systems also remain very attractive. Mu et al. utilized hexafluorobutyl methacrylate to protect cobalt-doped CsPbBr3/Cs4PbBr6 NCs for the photoreduction of CO2 in the aqueous medium. They adopted pure water as a solvent, where water was used as a sacrificial reductant and electron source. The content of 2% Co on the surface of the NCs (2%Co@CsPbBr3/Cs4PbBr6) was employed to enhance the activity for photocatalytic reduction of CO2.93 They proposed that Co2+ acted as an active center to promote CO2 absorption and accrued two electrons from CsPbBr3/Cs4PbBr6; on account of that, the Co2+ center formed a Co-CO2 complex and reacted with H+ to facilitate the formation of CO.
4.2. Halide perovskites
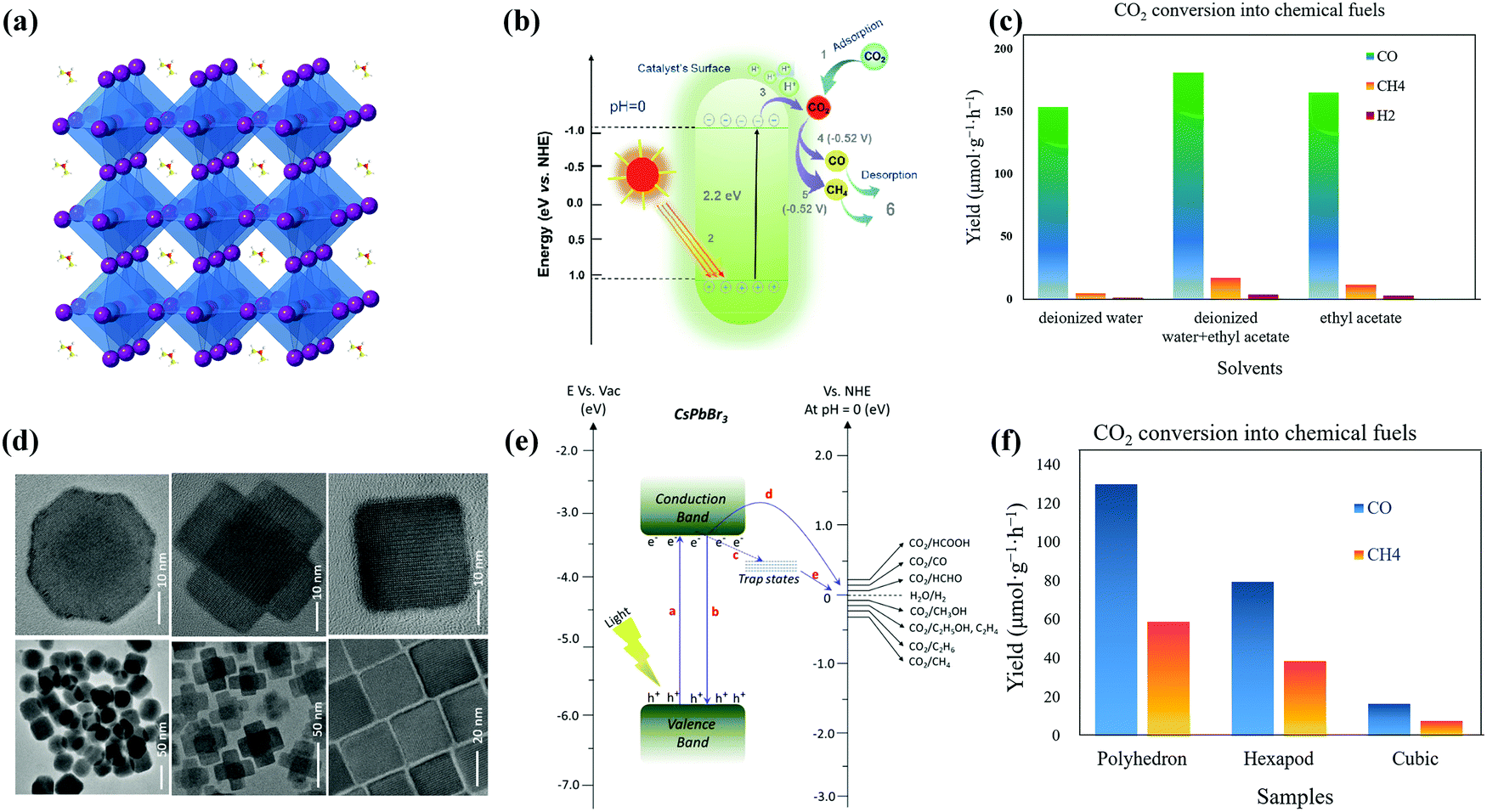 | ||
| Fig. 7 (a) Crystal structure of the cubic FAPbBr3 perovskite and (b) schematic band diagram of photocatalytic CO2 reduction into chemical fuels for FAPbBr3 QDs. Reprinted from ref. 51, Copyright 2021, with permission from Elsevier. (c) CO2 reduction reaction by FAPbBr3 QDs in DI, DI/EA, and EA systems (data from ref. 51). (d) TEM and HRTEM images of non-cubes, hexapods, and cube-shaped nanostructures. (e) Schematic representation of the conduction and valence band position of the CsPbBr3 catalyst with the relative potential of CO2 reduction. (d and e) Reprinted with permission from ref. 136, Copyright 2020 American Chemical Society. (f) Formation of CO and CH4 from CO2 reduction reactions using noncube, polyhedron, or hexapod photocatalysts (data from ref. 136). | ||
Several approaches have been reported and applied to deal with the instability issues of MHPs. Examples include (i) replacing organic cations MA or FA with inorganic cations (e.g., Cs+);144,145 (ii) creating heterostructures to spatially separate photogenerated excitons;99 and (iii) encapsulating halide perovskites to minimize direct contact with highly polar environments.45,93 In this context, the incident light absorption range for metallic complexes can be extended by coupling with halide perovskite NCs. Meanwhile, these metallic complexes are expected to be responsible for catalytic sites designed for halide perovskite NCs and capture excited electrons from halide perovskite NCs for selective photoreduction of CO2. Lead-free inorganic halide double perovskites were based on antimony or bismuth and noble metals.33 Most halide double perovskites have been described to have a cubic face-centered structure, which has the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and a lattice parameter ranging from approximately 10 to 12 Å.123,146 This new type of perovskite consists of B'Cl6 and B''Cl6 octahedra alternating in a rock-salt face-centered cubic structure. George Volonakis et al.147 used first-principles calculations to observe the patterns in the electronic and optical properties in the whole family of double halide perovskites, A2B′B′′X6 with A = Cs; B′′ = Bi and Sb; B′ = Cu, Ag, and Au; and X = Cl, Br, and I. Their calculations showed highly tunable carrier effective masses as well as optical gaps across the visible-light range of the electromagnetic spectra. Moving away from lead-based metal halide perovskites toward lead-free halide perovskites, Zhou et al. proved the photocatalytic reduction of CO2 over double halide perovskite Cs2AgBiBr6 NCs in the presence of light in ethyl acetate.44,148 The crystal structure of cubic Cs2AgBiBr6 NCs as well as constant advancement of CH4 and CO on double halide perovskite Cs2AgBiBr6 NCs in the presence of solar light radiation is illustrated in Fig. 8(a and b). Lu et al. reported that substitution of unsaturated oleic acid with saturated octanoic acid was hazardous to synthesize pure Cs3Sb2Br9 NCs without the CsBr contamination as shown in Fig. 8c.48 Cs3Sb2Br9 NCs showed much higher efficiency of photocatalytic reduction reaction of CO2, compared to Pb-based CsPbBr3 NCs. For photocatalytic reduction of CO2, a bound state is observed for COOH* on the surface of Cs3Sb2Br9 NCs in which a Br ion is partially replaced to allow for Sb–C bond formation as shown in Fig. 8d.48 Dai et al. reported that the mesoporous silica-supported Cs3Bi2Br9 NCs were developed. They observed that the Cs3Bi2Br9 revealed outstanding photocatalytic properties in selective aromatic as well as non-aromatic C−H bond activation in the presence of sunlight.149 Moreover, several environmentally friendly MHPs, such as Cs2InAgCl6,150 CsSnI3,151 Cs3Sb2I9,152 and Cs3Sb2Br9,121 have been successfully produced. These metal-based HPs showed great optoelectronic properties for photocatalytic applications.
m and a lattice parameter ranging from approximately 10 to 12 Å.123,146 This new type of perovskite consists of B'Cl6 and B''Cl6 octahedra alternating in a rock-salt face-centered cubic structure. George Volonakis et al.147 used first-principles calculations to observe the patterns in the electronic and optical properties in the whole family of double halide perovskites, A2B′B′′X6 with A = Cs; B′′ = Bi and Sb; B′ = Cu, Ag, and Au; and X = Cl, Br, and I. Their calculations showed highly tunable carrier effective masses as well as optical gaps across the visible-light range of the electromagnetic spectra. Moving away from lead-based metal halide perovskites toward lead-free halide perovskites, Zhou et al. proved the photocatalytic reduction of CO2 over double halide perovskite Cs2AgBiBr6 NCs in the presence of light in ethyl acetate.44,148 The crystal structure of cubic Cs2AgBiBr6 NCs as well as constant advancement of CH4 and CO on double halide perovskite Cs2AgBiBr6 NCs in the presence of solar light radiation is illustrated in Fig. 8(a and b). Lu et al. reported that substitution of unsaturated oleic acid with saturated octanoic acid was hazardous to synthesize pure Cs3Sb2Br9 NCs without the CsBr contamination as shown in Fig. 8c.48 Cs3Sb2Br9 NCs showed much higher efficiency of photocatalytic reduction reaction of CO2, compared to Pb-based CsPbBr3 NCs. For photocatalytic reduction of CO2, a bound state is observed for COOH* on the surface of Cs3Sb2Br9 NCs in which a Br ion is partially replaced to allow for Sb–C bond formation as shown in Fig. 8d.48 Dai et al. reported that the mesoporous silica-supported Cs3Bi2Br9 NCs were developed. They observed that the Cs3Bi2Br9 revealed outstanding photocatalytic properties in selective aromatic as well as non-aromatic C−H bond activation in the presence of sunlight.149 Moreover, several environmentally friendly MHPs, such as Cs2InAgCl6,150 CsSnI3,151 Cs3Sb2I9,152 and Cs3Sb2Br9,121 have been successfully produced. These metal-based HPs showed great optoelectronic properties for photocatalytic applications.
 | ||
| Fig. 8 (a) Crystal structure of cubic Cs2AgBiBr6. (b) Schematic diagram of the CO2 photoreduction on the surface of Cs2AgBiBr6 NCs. (a and b) Reproduced with permission from ref. 44, Copyright 2018 Wiley. (c) SEM image of Cs3Sb2Br9 NCs. The inset shows the yellow color and high transparency of the solution. (d) CO2 reduction mechanism by Cs3Sb2Br9 NCs. (c and d) Republished (adapted) with permission from the Royal Society of Chemistry from ref. 48; permission conveyed through Copyright Clearance Center, Inc. | ||
4.3. Semiconductors with halide perovskites
 | ||
| Fig. 9 (a) Band structure of the TiO2/CsPbBr3 composite photocatalyst. (b) Photocatalytic activities of CO2 reduction for TiO2, CsPbBr3 QDs, and TiO2/CsPbBr3 composite, respectively. (a and b) Reproduced from ref. 153, under the Creative Commons CC BY license (http://creativecommons.org/licenses/by/4.0/). (c) A schematic illustration of CO2 fixation and conversion in TiO2-encapsulated CsPbBr3 NCs, and (d) photocatalytic CO2 reduction test results. (c and d) Reproduced with permission from ref. 43, Copyright 2018 Wiley. | ||
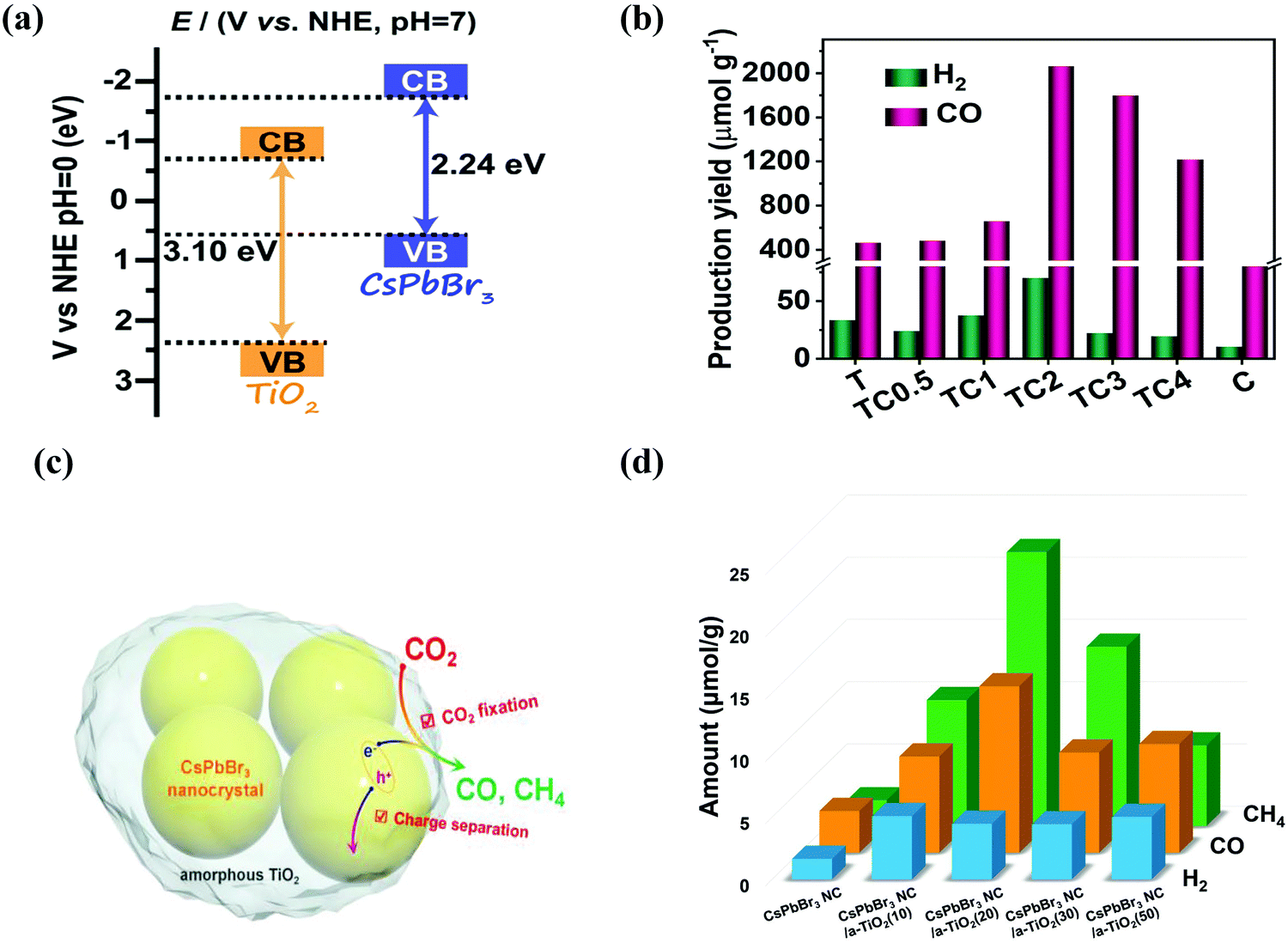
When developing the stronger reduction potential of CsPbBr3 to produce a Z-scheme heterojunction photocatalyst, it is best to search for a suitable oxidative semiconductor. Moreover, the oxidative semiconductor has the following properties: (i) strong interactions with CsPbBr3; (ii) visible-light response; (iii) high catalytic activity for the oxidation reaction; and (iv) compatible band structure with CsPbBr3. Bismuth tungsten oxide (Bi2WO6) is a unique oxide with visible-light response,154 which contains perovskite-type [WO4]2− layers and alternating [Bi2O2]2+.155,156 For oxidative reactions, Bi2WO6 also has an oxygen-deficient surface that provides abundant active sites, and it is preferred as an ideal photocatalyst for O2 evolution.157,158 Notably, the band structures among CsPbBr3 and Bi2WO6 were well-matched, and in principle, they can also be incorporated into a Z-scheme heterojunction. Wang et al. developed a 0D/2D heterojunction of CsPbBr3 QDs/Bi2WO6 nanosheet (CPB/BWO) photocatalyst for photocatalytic reduction of CO2.159 The larger surface area of 2D Bi2WO6 effectively decorated 0D CsPbBr3 on its surface. The close contact between Bi2WO6 and CsPbBr3 permitted an excellent interface for the charge transfer and separation.160,161 The advanced CPB/BWO heterojunction photocatalyst exhibited higher activities for photocatalytic reduction of CO2 as compared to the Bi2WO6 nanosheets and pristine CsPbBr3 QDs (Fig. 10a–c), respectively.159 The separated holes and electrons in CsPbBr3 QDs and Bi2WO6 nanosheets can be proficiently used for photocatalytic reduction of CO2. The yield of CPB/BWO is 503 μmol g−1, which is 9.5 fold that of CsPbBr3.
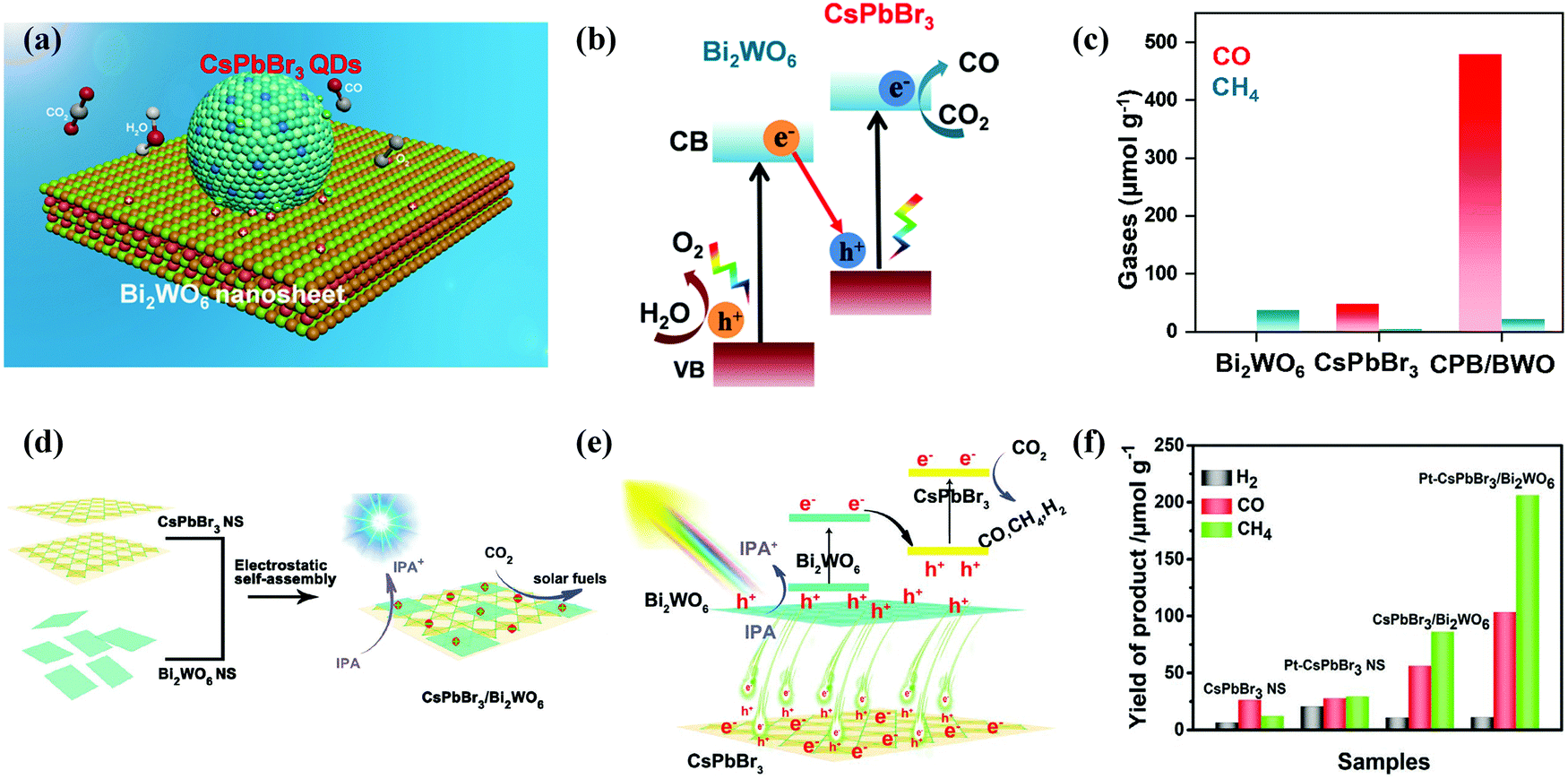 | ||
| Fig. 10 (a) Z-Scheme photocatalytic diagram of the 0D/2D CsPbBr3/Bi2WO6 composite for CO2 reduction. (b) Schematic illustration of Z-scheme of the energy band structure of the CsPbBr3/Bi2WO6 composite. (c) Gas yields in the ethyl acetate/water system over various catalysts. (a–c) Reprinted with permission from ref. 159, Copyright 2020 American Chemical Society. (d) Schematic diagram of the 2D/2D CsPbBr3/Bi2WO6 heterojunction for CO2 reduction. (e) Schematic diagram of the energy band structure of the 2D/2D CsPbBr3/Bi2WO6 heterojunction. (f) Photocatalytic activity of different samples. (d–f) Reproduced with permission from ref. 167. Copyright 2020 Wiley. | ||
Besides, a 2D/2D heterojunction is considered as the most promising approach to manipulate the photoreduction efficiency of semiconductor catalysts. Recently, a lot of efforts have been devoted to preparing 2D/2D heterostructures.162,163 This is because the large surface area of 2D ultrathin nanosheets, rich active sites, special electronic structure, and short charge transport distance are useful for the catalytic reaction.164–166 Recently, Jiang et al. designed a lead halide perovskite-based 2D/2D direct Z scheme heterojunction by assembling ultrathin Bi2WO6 nanosheets on the surface of CsPbBr3 nanosheets through an electrostatic self-assembly process (Fig. 10d–f).167 Bi2WO6 has been identified as one of the best visible-light oxidation photocatalysts, which have higher oxidation ability, low manufacturing costs and controlled synthesis.168,169 The CsPbBr3/Bi2WO6 heterostructure provided higher photocatalytic performance for CO2 reduction activity. The yield of the 2D/2D CsPbBr3/Bi2WO6 hybrid was 153.0 μmol g−1 (10.9 μmol g−1 for H2, 56.4 μmol g−1 for CO, and 86.0 μmol g−1 for CH4).167
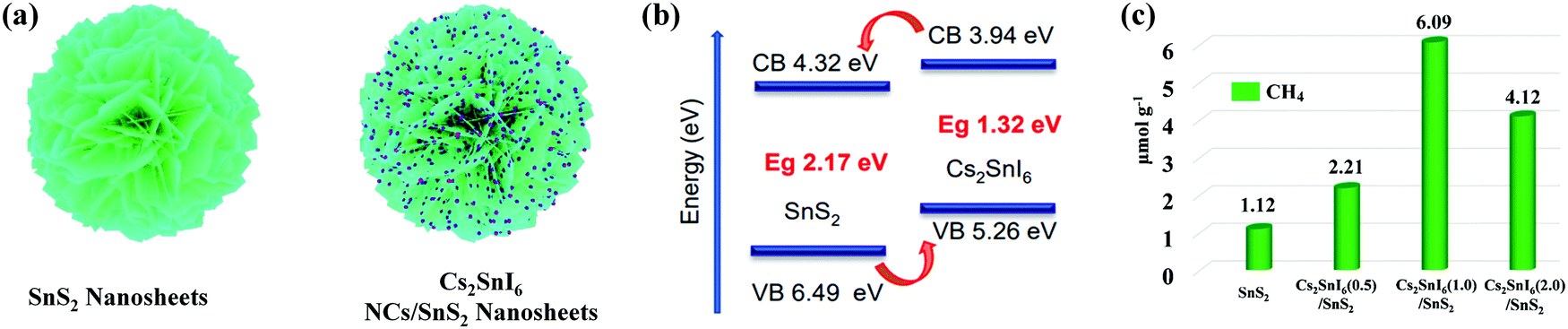 | ||
| Fig. 11 (a) Schematic illustration of the synthesis of the Cs2SnI6/SnS2 heterostructure. (b) Diagram of the energy band structure of Cs2SnI6 and SnS2. (c) Comparison of photocatalytic CO2 reduction activities of SnS2, Cs2SnI6(0.5)/SnS2, Cs2SnI6(1.0)/SnS2, and Cs2SnI6(2.0)/SnS2. Reprinted with permission from ref. 46, Copyright 2019 American Chemical Society. | ||
Fig. 12a–c shows the representation of the CsPbBr3 QD/GO composite photocatalyst and its corresponding TEM image.42 It was noted that, in ethyl acetate, the pure CsPbBr3 QDs catalyzed the photocatalytic reduction of CO2 with an electron consumption rate of 23.75 μmol g−1 h−1 over 99.2% selectivity. Through the combination of highly conducting materials, such as GO, which has charge extraction abilities, the charge recombination was suppressed.181 The CsPbBr3 NCs/GO composite enhanced the electron consumption rate from 23.75 to 29.82 μmol g−1 h−1 with 99.15% selectivity.
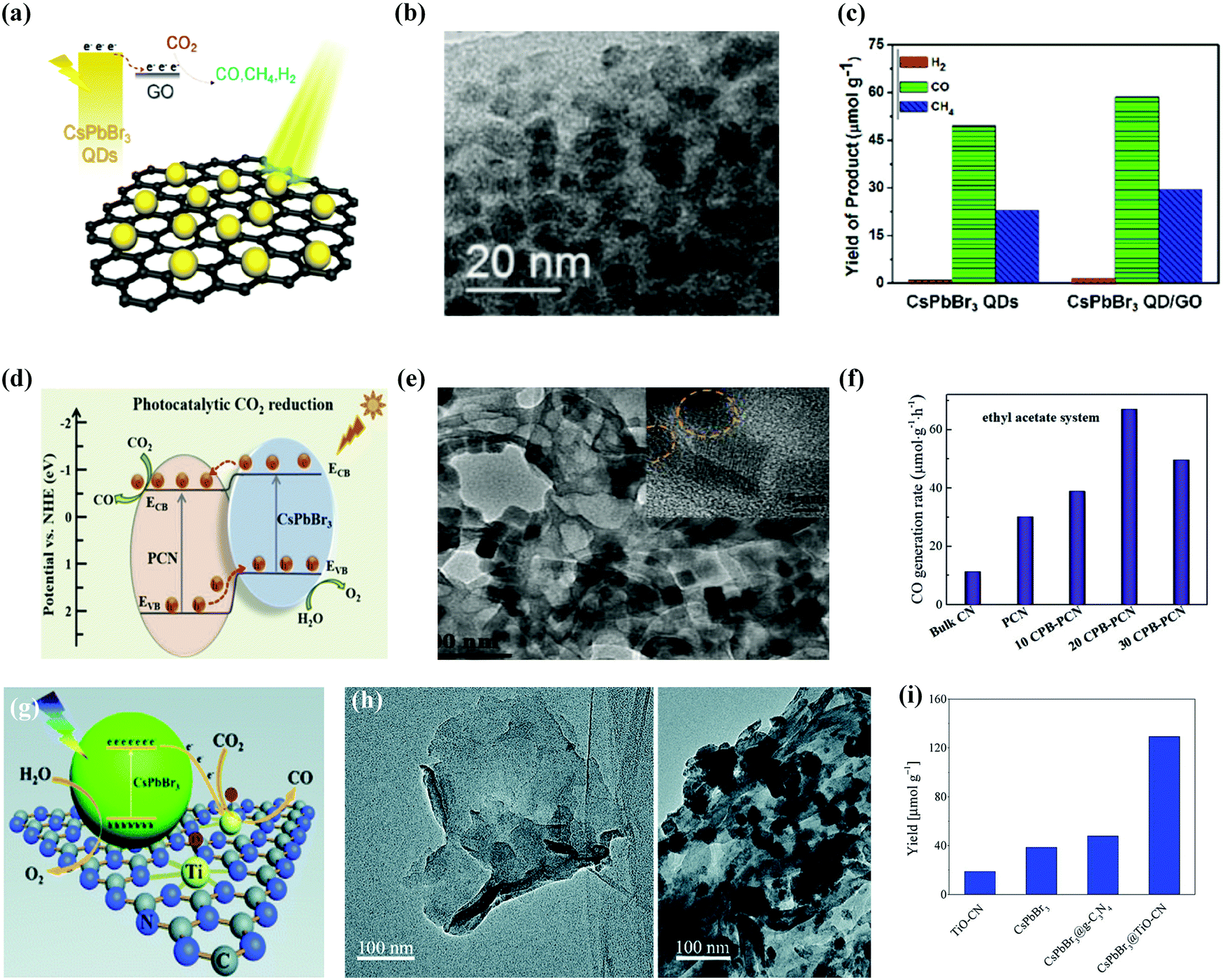 | ||
| Fig. 12 Heterostructure of CsPbBr3/GO composite photocatalysts for enhanced CO2 reduction. (a) Schematic model, (b) TEM image, and (c) histogram of different products in the presence and absence of GO with CsPbBr3 QDs. (a–c) Reprinted with permission from ref. 42, Copyright 2017 American Chemical Society. (d) Amino-assisted anchoring of CsPbBr3 QDs on porous g-C3N4 for photocatalytic CO2 reduction. (e) TEM image of CsPbBr3-PCN. (f) Generation of CO in acetonitrile/water and ethyl acetate/water systems over various photocatalysts. (d–f) Reproduced with permission from ref. 182, Copyright 2018 Wiley. (g) Photocatalytic CO2 reduction by CsPbBr3 nanocrystals with graphitic carbon nitride nanosheets containing titanium-oxide composite. (h) TEM images of TiO-CN without and with CsPbBr3 nanocrystals. (i) The yield of CO generation from photocatalytic CO2 reduction reactions with TiO-CN, CsPbBr3, CsPbBr3@g-C3N4, and CsPbBr3@TiO-CN. (g–i) Republished with permission of the Royal Society of Chemistry, from ref. 98; permission conveyed through Copyright Clearance Center, Inc. | ||
Ou et al. described that anchoring of CsPbBr3 QDs over porous g-C3N4 (PCN) also improved the performance of photocatalytic reduction of CO2 in acetonitrile/water or ethyl acetate/water solution.182 With 20 wt% CsPbBr3 QDs (CPB) on NHx-rich porous g-C3N4 nanosheets (CPB-PCN), CsPbBr3 QDs were linked with PCN by a N-Br bond which boosted the photogenerated charge carrier separation, as shown in Fig. 12d–f.182 The above-mentioned strategy helped to achieve the maximum rate of CO formation in the acetonitrile/water mixture compared with bare CsPbBr3 QDs.183
Based on the above-mentioned composite structure, Guo et al. functionalized CsPbBr3 NCs with graphitic carbon nitride, which had a titanium-oxide species (TiO-CN) through N–Br and O–Br bonding, and developed an effective catalyst system for photocatalytic reduction of CO2 utilizing water as the source of electrons.98 The introduction of TiO-CN could improve the number of catalytic active sites, along with a rapid interfacial charge separation between CsPbBr3 and TiO-CN owing to their promising energy-offsets as well as chemical bonding performances (Fig. 12g–i). They also reported that the CsPbBr3@TiO-CN composite increased the separation of photogenerated charges and improved the number of catalytic active sites that led to the formation of CO after 10 hours of irradiation, which was 3 times higher than that of pure CsPbBr3.98
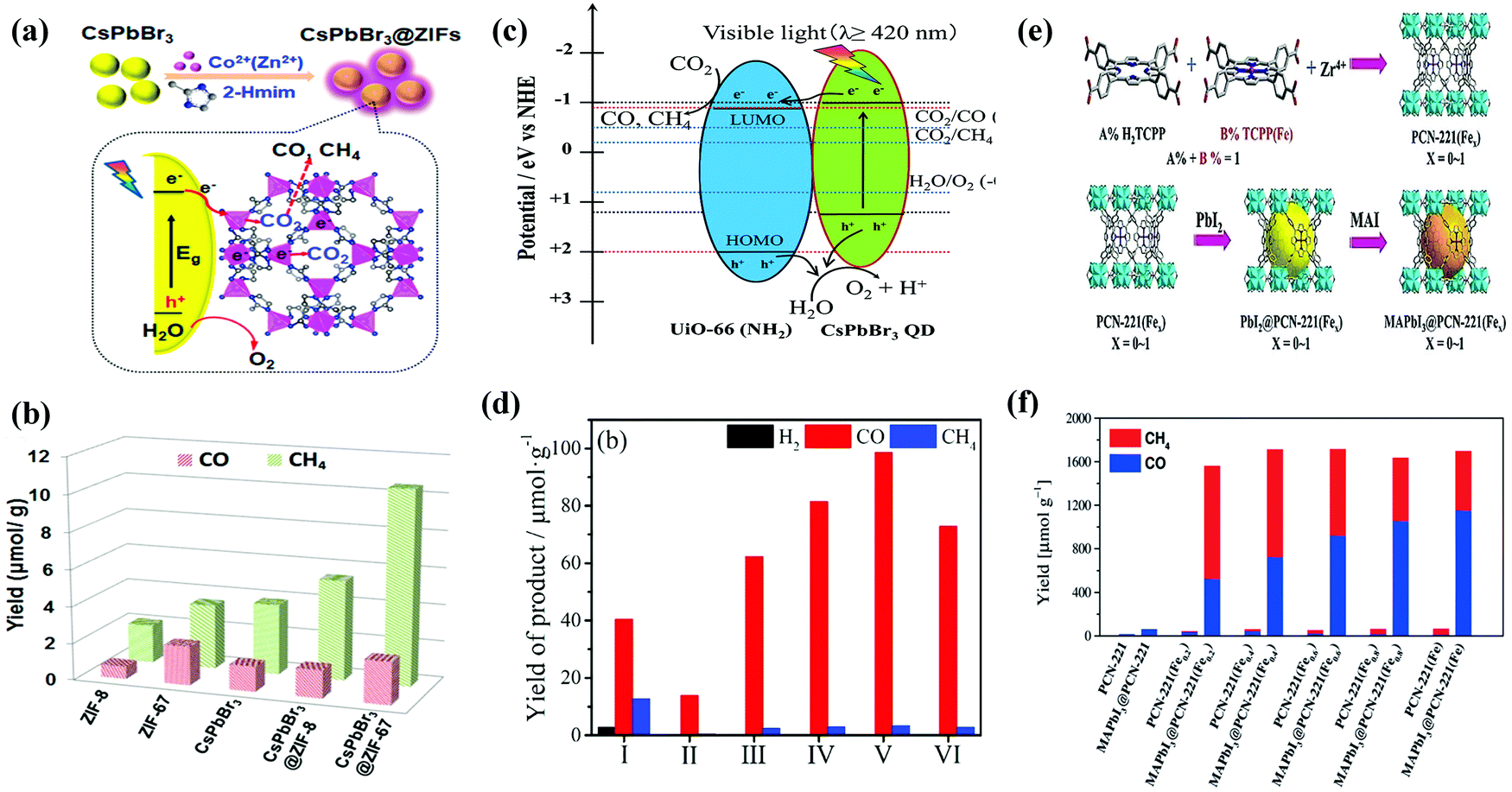 | ||
| Fig. 13 (a) Schematic illustration of the fabrication process and CO2 photoreduction process of CsPbBr3/ZIFs. (b) The product yield after 3 h of photocatalytic reaction. (a and b) Reprinted with permission from ref. 102, Copyright 2018 American Chemical Society. (c) Schematic illustration of a possible mechanism of photocatalytic CO2 reduction. (d) Photocatalytic CO2 reduction performance for CsPbBr3 QDs and x%-CsPbBr3 QDs/UiO-66(NH2) nanocomposite. (c and d) Reprinted from ref. 113, Copyright (2019), with permission from Elsevier. (e) Schematic representation for the synthesis of PCN-221(Fex) MOF and MAPbI3@PCN221(Fex) composite photocatalysts for stable and product selective CO2 reduction. (f) Histogram of product formation using PCN-221(Fex) and MAPbI3@PCN-221(Fex). (e and f) Reproduced with permission from ref. 45, Copyright 2019 Wiley. | ||
4.4. Conductor with halide perovskites
Xiao and co-workers examined the energy conversion in the Ag-CsPbBr3 system as well as dynamics of plasmon-induced hot electrons, which showed a significant hot-electron transfer efficiency near 50%.195 The studies about plasmon-exciton interaction for the hybrid method of plasmonic Au NPs over halide perovskite NCs are still at a nascent development.
The CsPbBr3–Au nanocomposite was used as a photocatalyst for artificial photoreduction of CO2 by two different types of light sources (λ > 420 nm and λ > 580 nm). Based on the Kelvin probe force microscopy (KPFM) results and the spectroscopic characterization, two-dimensional interaction processes for the CsPbBr3–Au nanocomposite are shown in Fig. 14.194 Even though there is a significant difference between the Fermi level (EF) of CsPbBr3 and the work function (WF) of Au, energy level alignment can be obtained with their close contact (Fig. 14a and b). Consequently, photogenerated electrons would mostly accumulate in the CB of CsPbBr3, which raises its EF negatively, and thermodynamically supports the electrons to transfer into the Au.196,197 The electrons accumulated in Au consequently contribute to the photoreduction reaction of CO2, as demonstrated in Fig. 14c. Only the Au nanoparticle-based photo-responsive absorption occurred under the light condition of λ >580 nm, while CsPbBr3 based photo-responsive absorption started at wavelength λ >530 nm. To initiate CO2 reduction more efficiently, the LSPR-induced hot electrons encompassing higher energy potential in plasmonic Au nanoparticles could be further transferred rapidly to CsPbBr3. Hence, this kind of plasmon-exciton exchange process makes the CsPbBr3–Au nanocomposite work well over the longer wavelength in turn as shown in Fig. 14d.
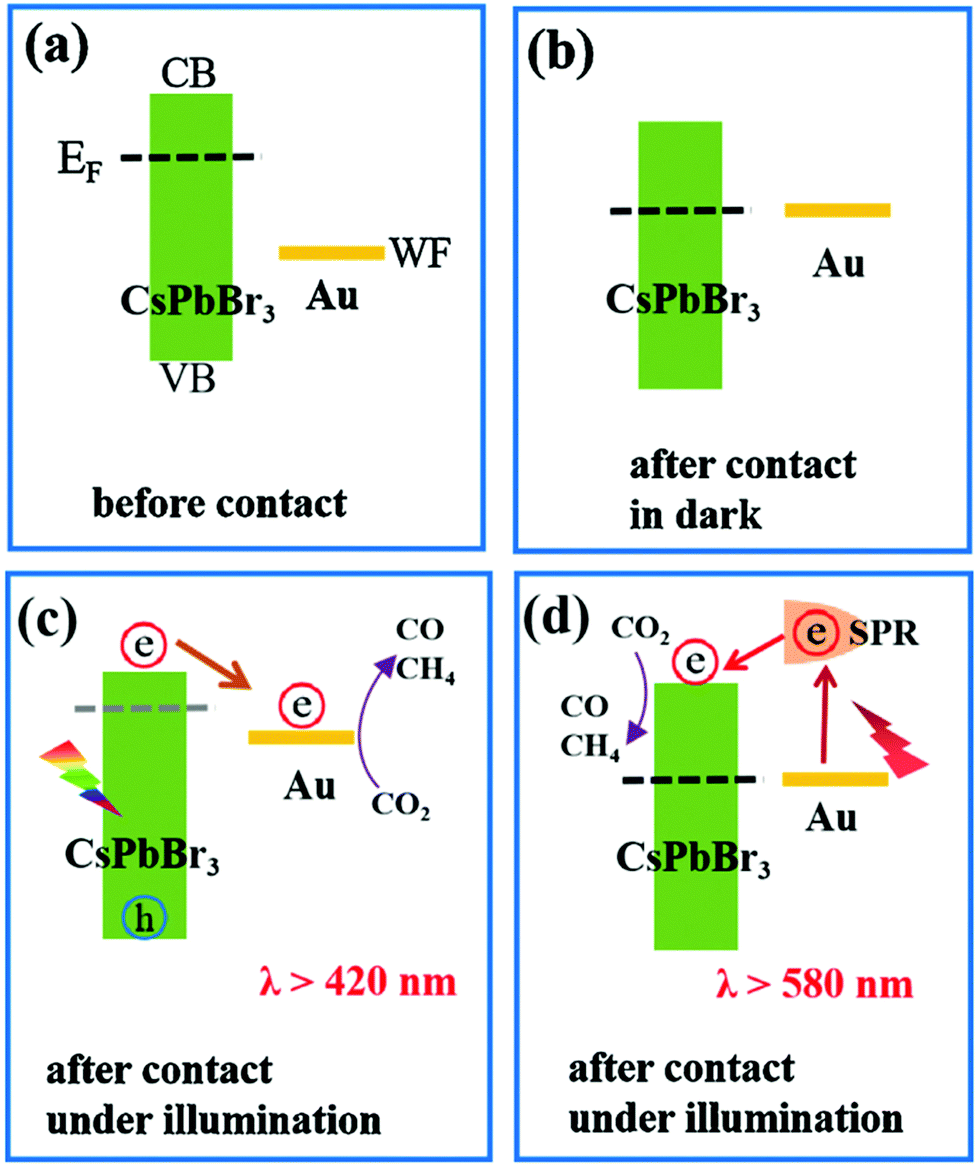 | ||
| Fig. 14 (a) Band structure of CsPbBr3 nanocrystals and Au nanoparticles before contact. (b) Fermi level alignment after their close contact in the dark. Schematic diagram of two excitation wavelength-dependent charge separation mechanisms in the CsPbBr3–Au nanocomposite under light irradiation with (c) λ >420 nm or (d) λ >580 nm. (a–d) Reprinted from ref. 194, Copyright 2021, with permission from Elsevier. | ||
Xu et al. described an innovative 0D CsPbBr3 nanocrystal (CsPbBr3 NC)/2D Pd nanosheet (Pd-NS) composite photocatalyst for the effective and stable photocatalytic reduction of CO2 under sunlight in the existence of water vapor.100 The Pd-NS not only played an important role as an electron collector to rapidly separate the electron–hole pairs in CsPbBr3 NC and controlled the unwanted radiative charge recombination, but also modified the kinetics of catalytic reduction of CO2 reaction. Therefore, the addition of Pd-NS increased the consumption rate of photoelectrons from 9.85 μmol g−1 h−1 to 33.7 μmol g−1 h−1 under visible light (>420 nm);100 especially, the incorporation of a 2D metallic semiconductor not only formed a Schottky junction at the interface to increase the electron transfer from the semiconductor to the metal, but also presented photocatalytic reaction sites to promote the transfer of electrons and increased the resulting chemical reactions,164,198 as presented in Fig. 15a–c.
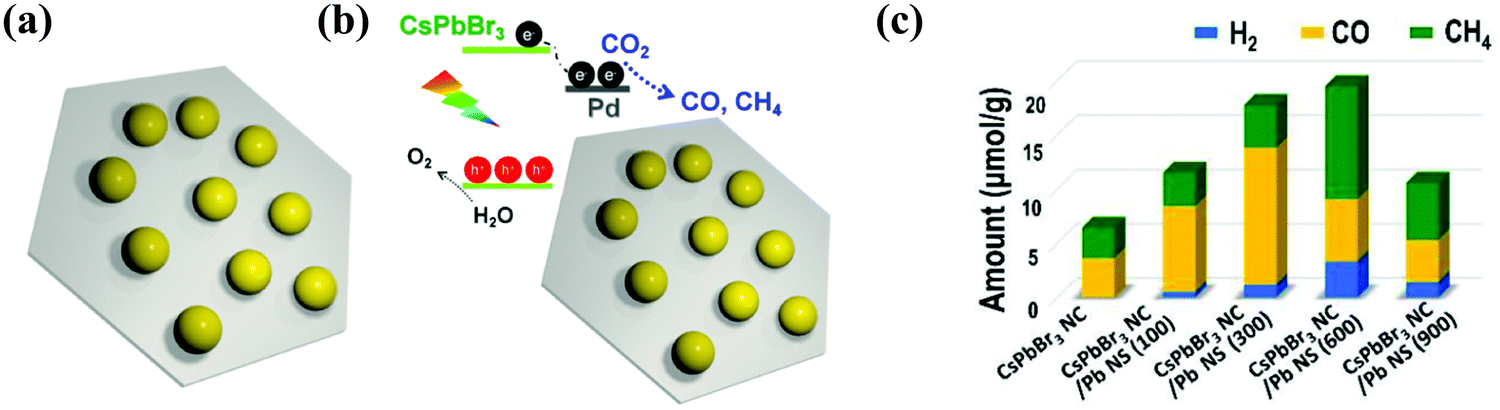 | ||
| Fig. 15 (a) Diagram of the CsPbBr3 NC/Pd-NC composite. (b) The sketch of the composite materials (CsPbBr3 NC)/(Pd-NC) and their band alignments. (c) Photocatalytic CO2 reduction performance. (a–c) Reprinted (adapted) with permission from ref. 100, Copyright 2018 American Chemical Society. | ||
Pan and co-workers demonstrated that the incorporation of MXene to CsPbBr3 (CsPbBr3 NCs/MXene-20) increased the yields of CO and CH4 linearly. The maximum CH4 and CO yield rate was 7.25 and 26.32 μmol g−1 h−1, respectively (Fig. 16a and b),47 which was superior or at least comparable to that of the MXene free CsPbBr3 NCs (<4.42 μmol g−1 h−1) or other state-of-the-art CsPbBr3 NC-based heterostructure photocatalysts.42,43,102,112,135,183 Que and co-workers successfully synthesized a FAPbBr3 QD/MXene nanosheet heterostructure via a facile process. Under visible-light irradiation, the as-prepared FAPbBr3 QD/MXene composite exhibited a remarkable photocatalytic activity in the presence of water. To stimulate the quick separation of excitons, the nanosheets of MXene played a role as an electron acceptor and facilitated a large number of active sites. Optimized performance with a higher electron consumption rate of 717.18 μmol g−1 h−1 was achieved via the FAPbBr3 QD/MXene composite (Fig. 16c and d), which showed a 2.08 times better performance as compared to the pure FAPbBr3 QDs.210
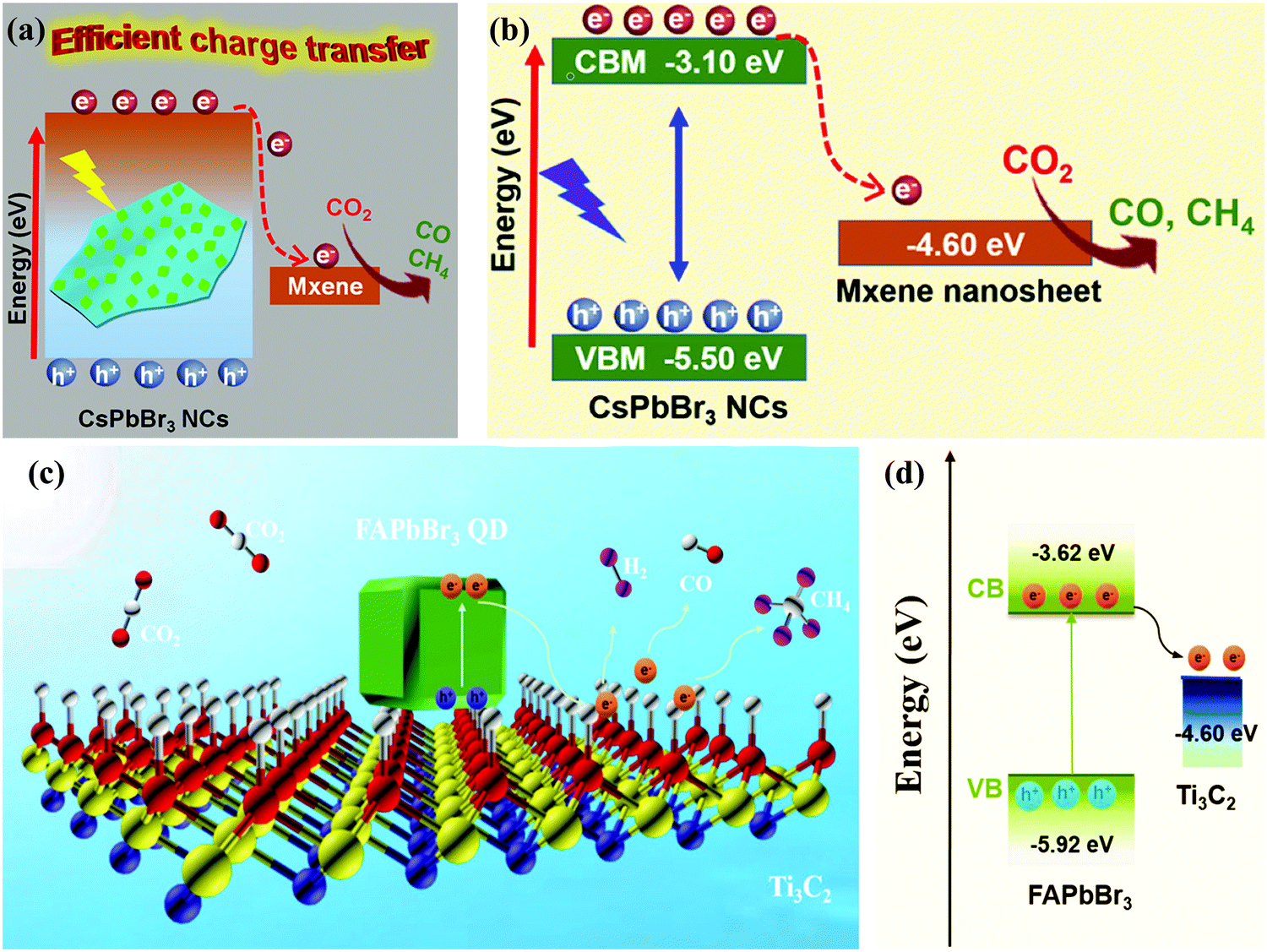 | ||
| Fig. 16 (a) Schematic illustration and (b) the relative energy diagram of the CsPbBr3 NC/MXene heterostructures suitable for photocatalytic CO2 reduction. (a and b) Reprinted with permission from ref. 47, Copyright 2019 American Chemical Society. (c) Schematic diagram of the potential mechanism of photocatalytic CO2 reduction in the FAPbBr3/MXene composite. (d) Energy diagram of the band structure of FAPbBr3/MXene. (c and d) Reprinted with permission from ref. 210, Copyright 2021 American Chemical Society. | ||
5. Molecular computation
Halide based perovskites have been extensively studied through theoretical/computational analysis concerning their unique electronic structure and physio-chemical properties. Their photo-conversion efficiency greatly depends on the balance electron, bandgap, absorption coefficient, hole mobility and intrinsic recombination rates.211–213 Changes (element or molecular ion) in the structure of halide perovskites could significantly alter the optoelectronic properties. In many cases, replacing cations or anions in HPs may cause different Coulomb interaction which results in the shifting of conduction and valence band positions according to their Fermi level energy.214 Bandgap is considered as one of the critical properties of a photocatalyst. To extend the light absorption to the visible region, the bandgap required for photocatalytic reaction is ∼2.00 eV. However, the position of the conduction band minimum (CBM) should be lower than −0.52 eV (vs. NHE PH = 0, the energy barriers of reducing CO2 to CO) for the CO2 reduction reaction,215,216 which is thermodynamically more feasible for the reduction reaction.Liu and co-workers217 conducted density functional theory (DFT) calculations of lead-free perovskites to investigate the valves of band gaps of CsGeI3, MAGeI3, and FAGeI3 using exchange–correlation functionals. Some commonly used exchange–correlation functionals are Generalized Gradient Approximation (GGA) and Perdew–Burke–Ernzerhof (PBE) which were found to be consistent with experimental values. The same group also performed hybrid functional Heyd–Scuseria–Ernzerhof-06 (HSE06) calculations to obtain more precise values of band gaps, which were very close to experimental values. The optimized structures, calculated band structure, and density of states (DOS) of MAGeI3 are shown in Fig. 17a–c. HSE06 and PBE functionals were used to calculate the bandgap of ABX3 HPs, with and without spin orbit coupling (SOC) calculations.218 Tang et al. elucidated the catalytic ability of pure CsPbBr3 perovskite and doped CsPbBr3 perovskites with different metals (Co, Fe, Ni, etc.) by density functional theory calculations.139 It was shown that methane is formed with higher selectivity and the smaller band gap between Co-doped and Fe-doped perovskites improved the efficiency of the photocatalyst as shown in Fig. 17f. The adsorption free energy of intermediates was found to be much lower than that of pure perovskite (Fig. 17f). Besides, the calculations also showed that the Co and Fe doped catalyst can enhance the catalytic activity of CsPbBr3 towards CO2 reduction. When dopants (Co or Fe) were introduced into CsPbBr3, the band-edge states were created by Co-d and Fe-d orbitals as they contributed more actively to the CB. The formed band-edge states reduced the bandgap from 2.32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV to 1.79 and 1.71eV for Co-doped for Fe-doped cases, respectively. The reduced bandgap not only improved light absorption in the visible spectrum but also promoted effective charge separation and transfer efficiency.139,219 Density of states was calculated as shown in Fig. 17d and e for the Co-doped and the Fe-doped cases respectively. The electron-donating ability of Co and Fe is greater than that of Pb, leading to improved reduction performance of doped HPs. Besides, calculation results showed that the CO double bond becomes weaker for the Co-doped and Fe-doped structures and CO2* is activated to produce HCOO*.139
eV to 1.79 and 1.71eV for Co-doped for Fe-doped cases, respectively. The reduced bandgap not only improved light absorption in the visible spectrum but also promoted effective charge separation and transfer efficiency.139,219 Density of states was calculated as shown in Fig. 17d and e for the Co-doped and the Fe-doped cases respectively. The electron-donating ability of Co and Fe is greater than that of Pb, leading to improved reduction performance of doped HPs. Besides, calculation results showed that the CO double bond becomes weaker for the Co-doped and Fe-doped structures and CO2* is activated to produce HCOO*.139
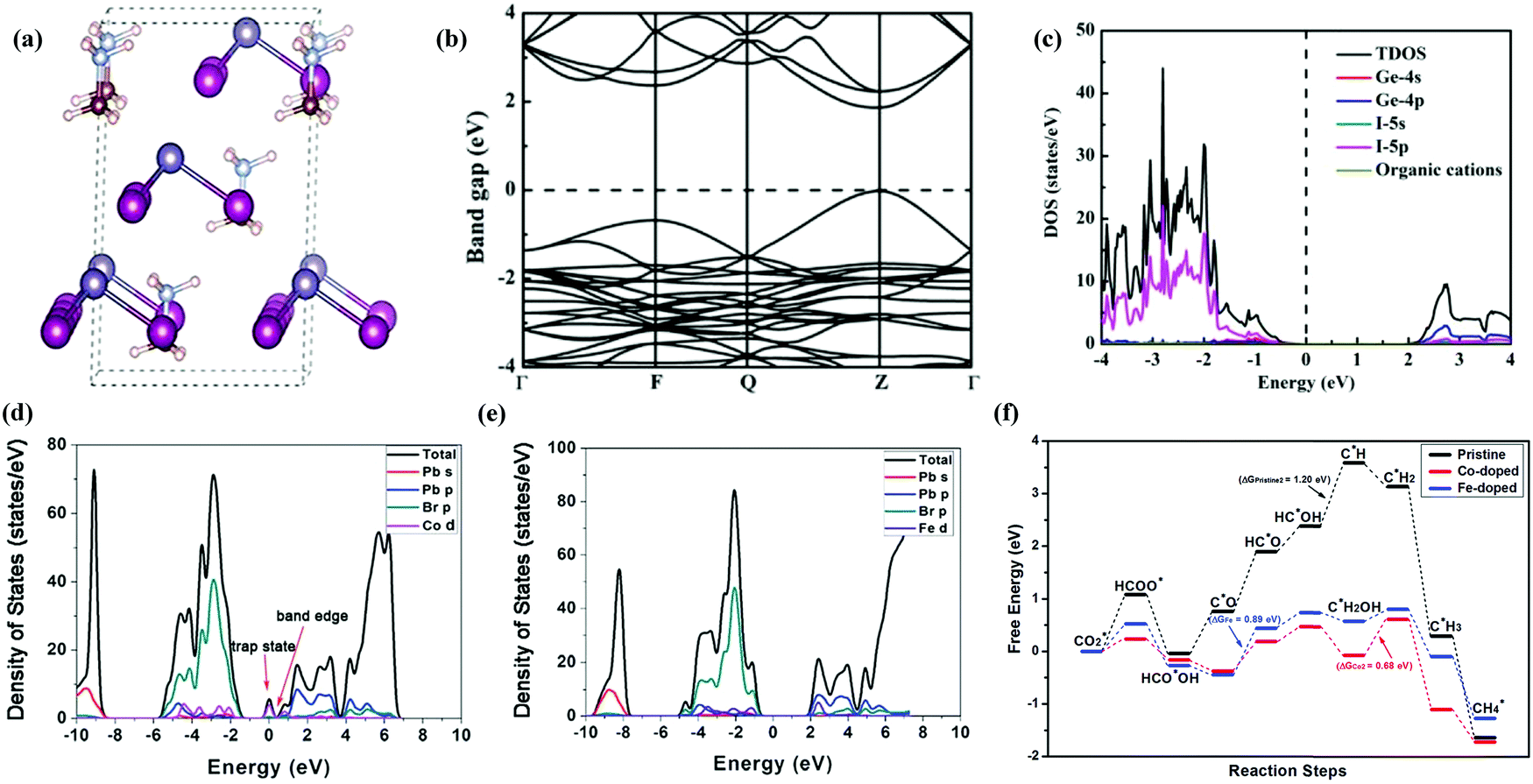 | ||
| Fig. 17 (a) Optimized structure, (b) calculated band structure, and (c) DOS of MAGeI3. Reprinted with permission from ref. 217, Copyright 2019 American Chemical Society. (d) Calculated density of state diagrams of Co-doped CsPbBr3, (e) calculated density of state diagrams of Fe-doped CsPbBr3, and (f) free-energy diagrams of the most favored paths of CO2 reduction by pristine, Co-doped and Fe-doped CsPbBr3. Republished with permission of the Royal Society of Chemistry, from ref. 139; permission conveyed through Copyright Clearance Center, Inc. | ||
6. Conclusion and future outlook
In this review, we summarize the recent progress achieved in exploring perovskite nanomaterials in terms of photocatalysts for CO2 reduction. Improvement methods in this field are discussed systematically including utilizing newly emerging halide perovskite nanomaterials along with their modifications in terms of structural engineering, interfacial modulation through the formation of the heterostructure, metal ion doping, surface modification, encapsulation with several types of co-catalysts, or using conducting substrates. Table 2 displays the performance of recent perovskite-based photocatalysts. Significant advances have been made to the conversion of CO2 into useful energy-bearing hydrocarbons by many attractive approaches, while significant research effort is still needed for improvement in the field to accelerate practical implementation. Moreover, some fundamental studies are highly required to resolve the bottleneck problems, including the kinetic problem in multi-electron reduction processes, water oxidation reaction, poor reduction selectivity, and low quantum yield of the photocatalysts. These issues should be extensively examined and studied for future improvements to be employed on an industrial scale.| Photocatalyst | Medium | Light source | Products | Yield (μmol g−1 h−1) | Highest efficiency (%) | Stability (h) | Ref. |
|---|---|---|---|---|---|---|---|
| CsPbBr3 QDs | Ethyl acetate/water | 300 W Xe lamp (AM 1.5G) | CO + CH4 + H2 | 4.3 + 1.5 + 0.1 | — | >8 | 135 |
| CsPbBr3@g-C3N4 | Ethyl acetate | 450 W Xe lamp AM 1.5G | CO + CH4 | 2.1 + 22.8 | — | >12 | 220 |
| CsPb(Br0.5/Cl0.5)3 NCs | Ethyl acetate | 300 W Xe lamp (AM 1.5G) | CO + CH4 | 85.2 + 12.0 | — | >9 | 97 |
| CsPbBr3 QD/GO | Ethyl acetate | 150 mW cm−2 (AM 1.5G) | CO + CH4 + H2 | 4.9 + 2.5 + 0.1 | 0.025 | >12 | 42 |
| CsPbBr3@TiO-CN | Ethyl acetate/water | 300 W Xe lamp (≥400 nm) | CO | 12.9 | — | >10 | 98 |
| CsPbBr3 NCs/MXene | Ethyl acetate | 300 W Xe lamp (≥420 nm) | CO + CH4 | 26.3 + 7.3 | — | >9 | 47 |
| CsPbBr3 NCs/BZNW/MRGO | (CO2 + water vapor) | 150 W Xe lamp (AM 1.5G) | CO + CH4 | 0.9 + 6.3 | — | >3 | 99 |
| CsPbBr3 NCs/Pd NS | (CO2 + water vapor) | 150 W Xe lamp (≥420 nm) | CO + CH4 + H2 | 1.9 + 3.5 + 1.1 | 0.035 | >3 | 100 |
| CsPbBr3-Re(600) | Toluene/isopropanol | 150 W Xe lamp (≥420 nm) | CO + H2 | 104.4 + 5.6 | — | >3 | 101 |
| CsPbBr3 NCs/a-TiO2 | Ethyl acetate/isopropanol | 150 W Xe lamp (AM 1.5G) | CO + CH4 + H2 | 3.9 + 6.7 + 1.5 | — | >3 | 43 |
| CsPbBr3 NCs@ZIF-67 | (CO2 + water vapor) | 150 mW cm−2 (AM 1.5G) | CO + CH4 | 2.1 + 3.5 | 0.035 | >3 | 102 |
| CsPbBr3 NCs@ZIF-8 | (CO2 + water vapor) | 100 W Xe lamp (AM 1.5G) | CO + CH4 | 0.7 + 2.0 | — | >3 | 102 |
| CsPbBr3 QDs/UiO66(NH2) | Ethyl acetate/water | 300 W Xe lamp (≥420 nm) | CO + CH4 | 8.2 + 0.3 | — | >12 | 113 |
| MAPbI3@PCN221(Fex) | Ethyl acetate/water | 300 W Xe lamp (≥400 nm) 100 mW cm−2 | CO + CH4 | 4.2 + 13.0 | — | 25 | 45 |
| Fe: CsPbBr3 NCs | Ethyl acetate/water | 450 W Xe lamp 150 mW cm−2 | CO + CH4 | 3.2 + 6.1 | — | >3 | 221 |
| FAPbBr3/Bi2WO6 | Benzyl alcohol | 150 W Xe lamp AM 1.5G 100 mW cm−2 | CO + Benzaldehyde | 170.0 + 250.0 | — | — | 222 |
| FAPbBr3 QDs | Ethyl acetate/water | 300 W Xe lamp 100 mW cm−2 | CO + CH4 + H2 | 181.3 + 16.9 + 2.37 | — | >3 | 51 |
| CsPbBr3 QDs/Bi2WO6 | Ethyl acetate/water | 100 mW cm−2 > 400 nm | CO + CH4 | Totally 50.3 | — | >10 | 159 |
| Cs2SnI6/SnS2 NCs | (CH3OH + CO2 + water vapor) | 150 mW cm−2 (≥400 nm) | CH4 | 6.1 | — | >3 | 46 |
| Cs3Sb2Br9 | Octadecene | 300 W Xe lamp 100 mW cm−2 | CO | 127.2 | — | >6 | 48 |
| α-Fe2O3/Amine-RGO/CsPbBr3 | (CO2 + water vapor) | 150 W Xe lamp AM 1.5G, >420 nm | CO + CH4 + H2 | 2.4 + 9.5 + 0.3 | — | >15 | 223 |
| CsPbBr3-Ni(tpy) | Ethyl acetate/water | 300 W Xe lamp >400 nm 100 mW cm−2 | CO + CH4 | 431.0 + 48.8 | 0.23 | >4 | 50 |
| Cs2AgBiBr6 NCs | Ethyl acetate | 150 mW cm−2 (AM 1.5G) | CO + CH4 | 2.4 + 1.6 | 0.028 | >6 | 44 |
| Cs3Bi2I9 | (CO2 + water vapor) | 80.38 μW cm−2 (AM 1.5G) | CO + CH4 | 7.7 + 1.5 | — | >10 | 224 |
| Co2%@CsPbBr3/Cs4PbBr6 | Water | 300 W Xe lamp 100 mW cm−2 | CO + CH4 | 12.0 + 1.8 | — | >20 | 225 |
| Mn/CsPb(Br/Cl)3 | Ethyl acetate | 300 W Xe-lamp with AM 1.5 filter | CO, CH4 | 213, 9.1 | — | 9 | 226 |
| Co-CsPbBr3/Cs4PbBr6 | Acetonitrile/water/Methanol | 300 W Xe lamp 100 mW cm−2 | CO | 122 | — | 15 | 227 |
| Pt/CsPbBr3 | Ethyl acetate | 150 W Xe-lamp with 380 nm cut o filter | CO | 5.6 | 0.012 | 30 | 131 |
| Ni and Mn-doped CsPbCl3 NCs | (CO2 + water vapor) | 300 W Xe-lamp with AM 1.5 filter | NiCO MnCO |
169.37, 152.49 | — | 6 | 228 |
| Cs4PbBr6/rGO | Ethyl acetate/water | 300 W Xe-lamp with 420 nm filter (light intensity, 100 mW cm−2) | CO | 11.4 | — | 60 | 229 |
| Cu-RGO-CsPbBr3 | (CO2/water vapor) | Xe-lamp irradiation with a 400 nm filter | CH4 | 12.7 | (1.1 ± 0.15) | 12 | 49 |
| TiO2/CsPbBr3 | Acetonitrile/water | 300 W Xe-arc lamp | CO | 9.02 | — | 16 | 153 |
| Cs2AgBiBr6@g-C3N4 | Ethyl acetate/Methanol | Xe-lamp (80 mW cm−2 light intensity) | CO, CH4 | 2 | — | 12 | 52 |
Promising research directions in halide perovskite-based photocatalysts for future exploration are forecasted including: (i) application of halide perovskites with various modulation strategies,43,150,230,231 such as employing interface engineering, fabricating 3D hierarchical architectures, exploiting ultrathin 2D nanosheets, using the surface fluorination effects, preparing surfaces over layers, creating surface vacancies, etc. and (ii) accomplishing higher light-harvesting ability and enhancing charge separation efficiency via rational design of a novel halide perovskite-based heterostructure.47,232–234 (iii) It's well known that solvent/electrolyte plays a significant role in any catalytic reaction. Since the halide perovskites are unstable in polar solvents, a wide range of research efforts have been devoted towards the exploration of a more suitable medium for efficient photocatalytic CO2 reduction. Owing to the high solubility of CO2, the use of low-polar ethyl acetate was preferred as a better solvent.42 (iv) The utilization of co-catalysts in the photocatalytic process has been proved to be an environmentally benign method with high performance.56 Some efforts could be devoted towards the development of an innovative noble-metal-free co-catalyst. (v) Despite attracting enormous research interest in the efficient photoreduction of CO2, the use of halide perovskites is still restricted on the industrial scale owing to their low scale (μmolrange). For artificial photosynthesis, the up to date solar-to-fuel efficiency value is ∼1.1%,49 which is ∼10 times that of natural photosynthesis,235 but is still very low for industrial applications. Given that solar energy conversion efficiency is >20% in solar photovoltaics, the value required for the feasible artificial photosynthesis should be 10% or above. The commercialization of CO2 photoreduction reaction is currently obstructed by the poor stability, low selectivity and high cost. One promising way to address the challenge is combining the halide perovskite with other efficient semiconductors for improving charge separation capacity and optical behaviors, so as to enhance the catalytic efficiency and stability. Therefore, the development of long-lasting, cost-effective, and potentially scalable halide perovskites is highly required for a sustainable future. (vi) Doping with proper metal ions and altering the reaction system for obtaining multiple doped halide perovskites should also be proposed for the possible improvement of the photocatalytic performance and their stability for reduction reaction of CO2. (vii) Another issue which should be addressed is the toxic nature of halide perovskites that hinders their commercial applications. In order to get environmentally friendly halide perovskites, substitution of lead (Pb) with other transition metals such as Sn, Sb and Bi is necessary. However, these transition metal substituted HPs have relatively poor stability as compared to Pb-based halide perovskites. Any further development of Pb-free HPs should address the limitations related to stability. (viii) In recent years, there has been an increasing amount of literature on photocatalytic reduction of CO2 which focuses mainly on higher conversion rate of CO2. The typical products of this photocatalytic reaction are single carbon atom compounds such as methane, carbon monoxide, formic acid, etc. However, current research in photocatalytic CO2 reduction by halide perovskites has emphasized the productivity as well as selectivity of compounds with two or more carbons (C2+), for example, ethanol, ethane, acetic acid, etc.87 Further research is required to investigate the productivity and selectivity of these C2+ products via photocatalytic reduction of CO2 by halide perovskites to valuable products such as alkenes, aromatic hydrocarbons and alcohols, rather than less valuable products. These challenges we are facing could provide countless opportunities for further improvement in photocatalytic activity. We hope that the readers can get more new insights into the recent advancements and future directions toward the energy conversion applications of halide perovskites in the field of photocatalysis.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (12002271, 11872302, and 62004155), Scientific Research Project of Shaanxi Provincial Department of Education (20JK0714), Xi’an Science and Technology Plan Project, China (2019220914SYS024CG046), and Earth Engineering Center and Center for Advanced Materials for Energy and Environment at Columbia University.References
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1701784 CrossRef PubMed.
- H. He, M. Zhong, D. Konkolewicz, K. Yacatto, T. Rappold, G. Sugar, N. E. David, J. Gelb, N. Kotwal and A. Merkle, Adv. Funct. Mater., 2013, 23, 4720–4728 CrossRef CAS.
- Y. He, L. Zhang, B. Teng and M. Fan, Environ. Sci. Technol., 2015, 49, 649–656 CrossRef CAS PubMed.
- W. Zhang, A. R. Mohamed and W.-J. Ong, Angew. Chem., 2020, 59, 22894–22915 CrossRef CAS PubMed.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu and Y. Yan, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CrossRef CAS.
- P. V. Kamat, ACS Energy Lett., 2017, 2, 1586–1587 CrossRef CAS.
- Ş. Neaţu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef PubMed.
- T. Masson-Delmotte, P. Zhai, H. Pörtner, D. Roberts, J. Skea, P. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan and R. Pidcock, World Meteorological Organization, Geneva, Tech. Rep, 2018 Search PubMed.
- E. Karamian and S. Sharifnia, J. CO2 Util., 2016, 16, 194–203 CrossRef CAS.
- H. Zhou, Y. Qu, T. Zeid and X. Duan, Energy Environ. Sci., 2012, 5, 6732–6743 RSC.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- M. Ge, Q. Li, C. Cao, J. Huang, S. Li, S. Zhang, Z. Chen, K. Zhang, S. S. Al-Deyab and Y. Lai, Adv. Sci., 2017, 4, 1600152 CrossRef PubMed.
- K. Li, A. D. Handoko, M. Khraisheh and J. Tang, Nanoscale, 2014, 6, 9767–9773 RSC.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- Z. Shi, S. Li, Y. Li, H. Ji, X. Li, D. Wu, T. Xu, Y. Chen, Y. Tian, Y. Zhang, C. Shan and G. Du, ACS Nano, 2018, 12, 1462–1472 CrossRef CAS PubMed.
- H. Zai, C. Zhu, H. Xie, Y. Zhao, C. Shi, Z. Chen, X. Ke, M. Sui, C. Chen, J. Hu, Q. Zhang, Y. Gao, H. Zhou, Y. Li and Q. Chen, ACS Energy Lett., 2018, 3, 30–38 CrossRef CAS.
- F. Cao, D. Yu, X. Li, Y. Zhu, Z. Sun, Y. Shen, Y. Wu, Y. Wei and H. Zeng, J. Mater. Chem. C, 2017, 5, 7441–7445 RSC.
- Q. Chen, H. Zhou, Y. Fang, A. Z. Stieg, T.-B. Song, H.-H. Wang, X. Xu, Y. Liu, S. Lu, J. You, P. Sun, J. McKay, M. S. Goorsky and Y. Yang, Nat. Commun., 2015, 6, 7269 CrossRef CAS PubMed.
- K. Chen, X. Deng, G. Dodekatos and H. Tüysüz, J. Am. Chem. Soc., 2017, 139, 12267–12273 CrossRef CAS PubMed.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M. H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef PubMed.
- G. Do Park, C. W. Lee and K. T. Nam, Curr. Opin. Electrochem., 2018, 11, 98–104 CrossRef.
- X. Song, G. Wei, J. Sun, C. Peng, J. Yin, X. Zhang, Y. Jiang and H. Fei, Nat. Catal., 2020, 3, 1027–1033 CrossRef CAS.
- L.-F. Gao, W.-J. Luo, Y.-F. Yao and Z.-G. Zou, Chem. Commun., 2018, 54, 11459–11462 RSC.
- A. Guerrero and J. Bisquert, Curr. Opin. Electrochem., 2017, 2, 144–147 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- E. A. Tsiwah, Y. Ding, Z. Li, Z. Zhao, M. Wang, C. Hu, X. Liu, C. Sun, X. Zhao and Y. Xie, CrystEngComm, 2017, 19, 7041–7049 RSC.
- D. M. Jang, D. H. Kim, K. Park, J. Park, J. W. Lee and J. K. Song, J. Mater. Chem. C, 2016, 4, 10625–10629 RSC.
- Z. Long, H. Ren, J. Sun, J. Ouyang and N. Na, Chem. Commun., 2017, 53, 9914–9917 RSC.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- F. Zhang, H. Zhong, C. Chen, X. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, O. Nazarenko, D. N. Dirin and M. V. Kovalenko, ACS Appl. Nano Mater., 2018, 1, 1300–1308 CrossRef CAS PubMed.
- J. Wang, J. Liu, Z. Du and Z. Li, J. Energy Chem., 2021, 54, 770–785 CrossRef.
- C. Zhang, B. Wang, W. Li, S. Huang, L. Kong, Z. Li and L. Li, Nat. Commun., 2017, 8, 1138 CrossRef PubMed.
- P. Fu, Q. Shan, Y. Shang, J. Song, H. Zeng, Z. Ning and J. Gong, Sci. Bull., 2017, 62, 369–380 CrossRef CAS.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- D. Yang, M. Cao, Q. Zhong, P. Li, X. Zhang and Q. Zhang, J. Mater. Chem. C, 2019, 7, 757–789 RSC.
- Y. Chen, M. He, J. Peng, Y. Sun and Z. Liang, Adv. Sci., 2016, 3, 1500392 CrossRef PubMed.
- S. Bonabi Naghadeh, B. Luo, G. Abdelmageed, Y.-C. Pu, C. Zhang and J. Z. Zhang, J. Phys. Chem. C, 2018, 122, 15799–15818 CrossRef CAS.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- Y. F. Xu, X. D. Wang, J. F. Liao, B. X. Chen, H. Y. Chen and D. B. Kuang, Adv. Mater. Interfaces, 2018, 5, 1801015 CrossRef.
- L. Zhou, Y. F. Xu, B. X. Chen, D. B. Kuang and C. Y. Su, Small, 2018, 14, 1703762 CrossRef PubMed.
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- X.-D. Wang, Y.-H. Huang, J.-F. Liao, Y. Jiang, L. Zhou, X.-Y. Zhang, H.-Y. Chen and D.-B. Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS PubMed.
- A. Pan, X. Ma, S. Huang, Y. Wu, M. Jia, Y. Shi, Y. Liu, P. Wangyang, L. He and Y. Liu, J. Phys. Chem. Lett., 2019, 10, 6590–6597 CrossRef CAS PubMed.
- C. Lu, D. S. Itanze, A. G. Aragon, X. Ma, H. Li, K. B. Ucer, C. Hewitt, D. L. Carroll, R. T. Williams and Y. Qiu, Nanoscale, 2020, 12, 2987–2991 RSC.
- S. Kumar, M. Regue, M. A. Isaacs, E. Freeman and S. Eslava, ACS Appl. Energy Mater., 2020, 3, 4509–4522 CrossRef CAS.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- M. Que, Y. Zhao, L. Pan, Y. Yang, Z. He, H. Yuan, J. Chen and G. Zhu, Mater. Lett., 2021, 282, 128695 CrossRef CAS.
- Y. Wang, H. Huang, Z. Zhang, C. Wang, Y. Yang, Q. Li and D. Xu, Appl. Catal., B, 2021, 282, 119570 CrossRef CAS.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- J. Luo, W. Zhang, H. Yang, Q. Fan, F. Xiong, S. Liu, D.-S. Li and B. Liu, EcoMat, 2021, 3, e12079 CrossRef CAS.
- C. B. Hiragond, N. S. Powar and S.-I. In, Nanomaterials, 2020, 10, 2569 CrossRef CAS PubMed.
- S. Shyamal and N. Pradhan, J. Phys. Chem. Lett., 2020, 11, 6921–6934 CrossRef CAS PubMed.
- Y. Xu, M. Cao and S. Huang, Nano Res., 2021, 14, 3773–3794 CrossRef CAS.
- L. Li and M. Wang, Advanced Nanomatericals for Solar Photocatalysis, in Advanced Catalytic Materials – Photocatalysis and Other Current Trends, ed. L. E. Norena and J.-A. Wang, IntechOpen, 2016 Search PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- F. Wang, Q. Li and D. Xu, Adv. Energy Mater., 2017, 7, 1700529 CrossRef.
- L. Ran, J. Hou, S. Cao, Z. Li, Y. Zhang, Y. Wu, B. Zhang, P. Zhai and L. Sun, Sol. RRL, 2020, 4, 2070045 CrossRef.
- J. M. Elward and A. Chakraborty, J. Chem. Theory Comput., 2013, 9, 4351–4359 CrossRef CAS PubMed.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef CAS PubMed.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed.
- H. Du, Y.-N. Liu, C.-C. Shen and A.-W. Xu, Chin. J. Catal., 2017, 38, 1295–1306 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- Z. Li, J. Feng, S. Yan and Z. Zou, Nano Today, 2015, 10, 468–486 CrossRef CAS.
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- J. H. Zejian Wang, S.-F. Ng, W. Liu, J. Huang, P. Chen and W.-J. Ong, Acta Phys. - Chim. Sin, 2021, 37, 2011033 Search PubMed.
- J. Y. Choi, C. K. Lim, B. Park, M. Kim, A. Jamal and H. Song, J. Mater. Chem. A, 2019, 7, 15068–15072 RSC.
- Z. He, J. Zhang, X. Li, S. Guan, M. Dai and S. Wang, Small, 2020, 16, 2005051 CrossRef CAS PubMed.
- W. Chen, B. Han, C. Tian, X. Liu, S. Liang, H. Deng and Z. Lin, Appl. Catal., B, 2019, 244, 996–1003 CrossRef CAS.
- Z. Sun, Q. Fan, M. Zhang, S. Liu, H. Tao and J. Texter, Adv. Sci., 2019, 6, 1901084 CrossRef CAS PubMed.
- Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao and P. K. Wong, Adv. Mater., 2018, 30, 1706108 CrossRef PubMed.
- Y. Wang, S. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2019, 58, 17236–17240 CrossRef CAS PubMed.
- H. Shen, T. Peppel, J. Strunk and Z. Sun, Solar RRL, 2020, 4, 1900546 CrossRef CAS.
- P. Kar, S. Farsinezhad, N. Mahdi, Y. Zhang, U. Obuekwe, H. Sharma, J. Shen, N. Semagina and K. Shankar, Nano Res., 2016, 9, 3478–3493 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610–7627 CrossRef CAS PubMed.
- R. Shi, G. I. Waterhouse and T. Zhang, Sol. RRL, 2017, 1, 1700126 CrossRef.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- S. Wang, B. Y. Guan and X. W. D. Lou, J. Am. Chem. Soc., 2018, 140, 5037–5040 CrossRef CAS PubMed.
- Z. Fan, K. Sun and J. Wang, J. Mater. Chem. A, 2015, 3, 18809–18828 RSC.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- Y. F. Mu, W. Zhang, X. X. Guo, G. X. Dong, M. Zhang and T. B. Lu, ChemSusChem, 2019, 12, 4769–4774 CrossRef CAS PubMed.
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS PubMed.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- D. Weber, Z. Naturforsch. B, 1978, 33, 1443–1445 CrossRef.
- S.-H. Guo, J. Zhou, X. Zhao, C.-Y. Sun, S.-Q. You, X.-L. Wang and Z.-M. Su, J. Catal., 2019, 369, 201–208 CrossRef CAS.
- X.-X. Guo, S.-F. Tang, Y.-F. Mu, L.-Y. Wu, G.-X. Dong and M. Zhang, RSC Adv., 2019, 9, 34342–34348 RSC.
- Y. Jiang, J.-F. Liao, Y.-F. Xu, H.-Y. Chen, X.-D. Wang and D.-B. Kuang, J. Mater. Chem. A, 2019, 7, 13762–13769 RSC.
- Y.-F. Xu, M.-Z. Yang, H.-Y. Chen, J.-F. Liao, X.-D. Wang and D.-B. Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS.
- Z.-C. Kong, H.-H. Zhang, J.-F. Liao, Y.-J. Dong, Y. Jiang, H.-Y. Chen and D.-B. Kuang, Sol. RRL, 2020, 4, 1900365 CrossRef CAS.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- J. Chen, J. Yin, X. Zheng, H. Ait Ahsaine, Y. Zhou, C. Dong, O. F. Mohammed, K. Takanabe and O. M. Bakr, ACS Energy Lett., 2019, 4, 1279–1286 CrossRef CAS.
- Q. Wang, L. Tao, X. Jiang, M. Wang and Y. Shen, Appl. Surf. Sci., 2019, 465, 607–613 CrossRef CAS.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S.-M. Chang and E. W.-G. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed.
- M.-Z. Yang, Y.-F. Xu, J.-F. Liao, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, J. Mater. Chem. A, 2019, 7, 5409–5415 RSC.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- V. K. Ravi, G. B. Markad and A. Nag, ACS Energy Lett., 2016, 1, 665–671 CrossRef CAS.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8515 CrossRef CAS PubMed.
- A. Filippetti and A. Mattoni, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 125203 CrossRef.
- Z. Zhu, Q. Sun, Z. Zhang, J. Dai, G. Xing, S. Li, X. Huang and W. Huang, J. Mater. Chem. C, 2018, 6, 10121–10137 RSC.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger and K. Katsiev, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- S. Wan, M. Ou, Q. Zhong and X. Wang, Chem. Eng. J., 2019, 358, 1287–1295 CrossRef CAS.
- Q. A. Akkerman, A. L. Abdelhady and L. Manna, J. Phys. Chem. Lett., 2018, 9, 2326–2337 CrossRef CAS PubMed.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- F. Iyikanat, E. Sari and H. Sahin, Phys. Rev. B, 2017, 96, 155442 CrossRef.
- K. A. Huynh, D. L. T. Nguyen, V.-H. Nguyen, D.-V. N. Vo, Q. T. Trinh, T. P. Nguyen, S. Y. Kim and Q. V. Le, J. Chem. Technol. Biotechnol., 2020, 95, 2579–2596 CAS.
- L. Dou, A. B. Wong, Y. Yu, M. Lai, N. Kornienko, S. W. Eaton, A. Fu, C. G. Bischak, J. Ma, T. Ding, N. S. Ginsberg, L. W. Wang, A. P. Alivisatos and P. Yang, Science, 2015, 349, 1518–1521 CrossRef CAS PubMed.
- Y. Fu, H. Zhu, C. C. Stoumpos, Q. Ding, J. Wang, M. G. Kanatzidis, X. Zhu and S. Jin, ACS Nano, 2016, 10, 7963–7972 CrossRef CAS PubMed.
- M. Faizan, K. C. Bhamu, G. Murtaza, X. He, N. Kulhari, M. M. Al-Anazy and S. H. Khan, Sci. Rep., 2021, 11, 6965 CrossRef PubMed.
- J. Zhang, Y. Yang, H. Deng, U. Farooq, X. Yang, J. Khan, J. Tang and H. Song, ACS Nano, 2017, 11, 9294–9302 CrossRef CAS PubMed.
- T. C. Jellicoe, J. M. Richter, H. F. J. Glass, M. Tabachnyk, R. Brady, S. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Böhm, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS PubMed.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- F. Wei, F. Brivio, Y. Wu, S. Sun, P. D. Bristowe and A. K. Cheetham, J. Mater. Chem. C, 2018, 6, 3573–3577 RSC.
- Y. Zhang, J. Yin, M. R. Parida, G. H. Ahmed, J. Pan, O. M. Bakr, J.-L. Brédas and O. F. Mohammed, J. Phys. Chem. Lett., 2017, 8, 3173–3177 CrossRef CAS PubMed.
- B. Yang, J. Chen, F. Hong, X. Mao, K. Zheng, S. Yang, Y. Li, T. Pullerits, W. Deng and K. Han, Angew. Chem., Int. Ed., 2017, 56, 12471–12475 CrossRef CAS PubMed.
- Y. Hu, S. Zhang, X. Miao, L. Su, F. Bai, T. Qiu, J. Liu and G. Yuan, Adv. Mater. Interfaces, 2017, 4, 1700131 CrossRef.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570 CrossRef CAS PubMed.
- W. Tu, Y. Xu, J. Wang, B. Zhang, T. Zhou, S. Yin, S. Wu, C. Li, Y. Huang and Y. Zhou, ACS Sustainable Chem. Eng., 2017, 5, 7260–7268 CrossRef CAS.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- Y.-X. Chen, Y.-F. Xu, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, Sustainable Energy Fuels, 2020, 4, 2249–2255 RSC.
- S. Thapa, K. Bhardwaj, S. Basel, S. Pradhan, C. J. Eling, A. M. Adawi, J.-S. G. Bouillard, G. J. Stasiuk, P. Reiss and A. Pariyar, Nanoscale Adv., 2019, 1, 3388–3391 RSC.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Bertolotti, N. Masciocchi, A. Guagliardi and M. V. Kovalenko, J. Am. Chem. Soc., 2016, 138, 14202–14205 CrossRef CAS PubMed.
- I. Lignos, L. Protesescu, D. B. R. Emiroglu, R. Maceiczyk, S. Schneider, M. V. Kovalenko and A. J. deMello, Nano Lett., 2018, 18, 1246–1252 CrossRef CAS PubMed.
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Chem. – Eur. J., 2017, 23, 9481–9485 CrossRef CAS PubMed.
- S. Shyamal, S. K. Dutta, T. Das, S. Sen, S. Chakraborty and N. Pradhan, J. Phys. Chem. Lett., 2020, 11, 3608–3614 CrossRef CAS PubMed.
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, J. Phys. Chem. Lett., 2014, 5, 1035–1039 CrossRef CAS PubMed.
- Y. Fu, H. Zhu, J. Chen, M. P. Hautzinger, X.-Y. Zhu and S. Jin, Nat. Rev. Mater., 2019, 4, 169–188 CrossRef CAS.
- C. Tang, C. Chen, W. Xu and L. Xu, J. Mater. Chem. A, 2019, 7, 6911–6919 RSC.
- G. Abdelmageed, L. Jewell, K. Hellier, L. Seymour, B. Luo, F. Bridges, J. Z. Zhang and S. Carter, Appl. Phys. Lett., 2016, 109, 233905 CrossRef.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- Y. Kim, E. Yassitepe, O. Voznyy, R. Comin, G. Walters, X. Gong, P. Kanjanaboos, A. F. Nogueira and E. H. Sargent, ACS Appl. Mater. Interfaces, 2015, 7, 25007–25013 CrossRef CAS PubMed.
- S. You, S. Guo, X. Zhao, M. Sun, C. Sun, Z. Su and X. Wang, Dalton Trans., 2019, 48, 14115–14121 RSC.
- Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780 CAS.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- G. Volonakis, M. Filip, A.-A. Haghighirad, N. Sakai, B. Wenger, H. Snaith and F. Giustino, J. Phys. Chem. Lett., 2016, 7, 1254–1259 CrossRef CAS PubMed.
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS PubMed.
- Y. Dai, C. Poidevin, C. Ochoa-Hernández, A. A. Auer and H. Tüysüz, Angew. Chem., Int. Ed., 2020, 59, 5788–5796 CrossRef CAS PubMed.
- G. Volonakis, A. A. Haghighirad, R. L. Milot, W. H. Sio, M. R. Filip, B. Wenger, M. B. Johnston, L. M. Herz, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2017, 8, 772–778 CrossRef CAS PubMed.
- I. Chung, J.-H. Song, J. Im, J. Androulakis, C. D. Malliakas, H. Li, A. J. Freeman, J. T. Kenney and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 8579–8587 CrossRef CAS PubMed.
- J. Pal, S. Manna, A. Mondal, S. Das, K. V. Adarsh and A. Nag, Angew. Chem., Int. Ed., 2017, 56, 14187–14191 CrossRef CAS PubMed.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed.
- X.-F. Cao, L. Zhang, X.-T. Chen and Z.-L. Xue, CrystEngComm, 2011, 13, 306–311 RSC.
- H. Huang, R. Cao, S. Yu, K. Xu, W. Hao, Y. Wang, F. Dong, T. Zhang and Y. Zhang, Appl. Catal., B, 2017, 219, 526–537 CrossRef CAS.
- C. Zhang and Y. Zhu, Chem. Mater., 2005, 17, 3537–3545 CrossRef CAS.
- Y. Zhou, Y. Zhang, M. Lin, J. Long, Z. Zhang, H. Lin, J. C.-S. Wu and X. Wang, Nat. Commun., 2015, 6, 8340 CrossRef PubMed.
- J. Hu, D. Chen, Z. Mo, N. Li, Q. Xu, H. Li, J. He, H. Xu and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 2073–2077 CrossRef CAS PubMed.
- J. Wang, J. Wang, N. Li, X. Du, J. Ma, C. He and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 31477–31485 CrossRef CAS PubMed.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- D. Deng, K. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- J. Li, Y. Zhao, M. Xia, H. An, H. Bai, J. Wei, B. Yang and G. Yang, Appl. Catal., B, 2020, 261, 118244 CrossRef CAS.
- Z. Xing, J. Hu, M. Ma, H. Lin, Y. An, Z. Liu, Y. Zhang, J. Li and S. Yang, J. Am. Chem. Soc., 2019, 141, 19715–19727 CrossRef CAS PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han and G.-H. Nam, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- Y. Zhao, S. Zhang, R. Shi, G. I. Waterhouse, J. Tang and T. Zhang, Mater. Today, 2020, 34, 78–91 CrossRef CAS.
- W. Wang, W. Zhao, H. Zhang, X. Dou and H. Shi, Chin. J. Catal., 2021, 42, 97–106 CrossRef CAS.
- Y. Jiang, H. Y. Chen, J. Y. Li, J. F. Liao, H. H. Zhang, X. D. Wang and D. B. Kuang, Adv. Funct. Mater., 2020, 2004293 CrossRef CAS.
- N. Zhang, R. Ciriminna, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014, 43, 5276–5287 RSC.
- H. Huang, C. Zhou, X. Jiao, H. Yuan, J. Zhao, C. He, J. Hofkens, M. B. Roeffaers, J. Long and J. A. Steele, ACS Catal., 2019, 10, 1439–1443 CrossRef.
- P. Schulz, D. Cahen and A. Kahn, Chem. Rev., 2019, 119, 3349–3417 CrossRef CAS PubMed.
- G. Kapil, T. Ohta, T. Koyanagi, M. Vigneshwaran, Y. Zhang, Y. Ogomi, S. S. Pandey, K. Yoshino, Q. Shen and T. Toyoda, J. Phys. Chem. C, 2017, 121, 13092–13100 CrossRef CAS.
- H. S. Rao, W. G. Li, B. X. Chen, D. B. Kuang and C. Y. Su, Adv. Mater., 2017, 29, 1602639 CrossRef PubMed.
- Z. Chen, Q. Dong, Y. Liu, C. Bao, Y. Fang, Y. Lin, S. Tang, Q. Wang, X. Xiao and Y. Bai, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- Y. Liu, J. Sun, Z. Yang, D. Yang, X. Ren, H. Xu, Z. Yang and S. Liu, Adv. Opt. Mater., 2016, 4, 1829–1837 CrossRef CAS.
- A. Kaltzoglou, M. Antoniadou, A. G. Kontos, C. C. Stoumpos, D. Perganti, E. Siranidi, V. Raptis, K. Trohidou, V. Psycharis and M. G. Kanatzidis, J. Phys. Chem. C, 2016, 120, 11777–11785 CrossRef CAS.
- B. Lee, C. C. Stoumpos, N. Zhou, F. Hao, C. Malliakas, C.-Y. Yeh, T. J. Marks, M. G. Kanatzidis and R. P. Chang, J. Am. Chem. Soc., 2014, 136, 15379–15385 CrossRef CAS PubMed.
- X. Jiao, X. Li, X. Jin, Y. Sun, J. Xu, L. Liang, H. Ju, J. Zhu, Y. Pan, W. Yan, Y. Lin and Y. Xie, J. Am. Chem. Soc., 2017, 139, 18044–18051 CrossRef CAS PubMed.
- M. M. Kandy, Sustainable Energy Fuels, 2020, 4, 469–484 RSC.
- S. Zhang, Z. He, S. Xu, X. Li, J. Zhang, X. Zhan, M. Dai and S. Wang, Solar RRL, 2021, 5, 2100233 CrossRef CAS.
- J. Low, B. Cheng, J. Yu and M. Jaroniec, Energy Storage Mater., 2016, 3, 24–35 CrossRef.
- S. Kaniyankandy, S. Rawalekar and H. N. Ghosh, J. Phys. Chem. C, 2012, 116, 16271–16275 CrossRef CAS.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., 2018, 130, 13758–13762 CrossRef.
- C. Li, Y. Du, D. Wang, S. Yin, W. Tu, Z. Chen, M. Kraft, G. Chen and R. Xu, Adv. Funct. Mater., 2017, 27, 1604328 CrossRef.
- C. Cao, D.-D. Ma, J.-F. Gu, X. Xie, G. Zeng, X. Li, S.-G. Han, Q.-L. Zhu, X.-T. Wu and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 15014–15020 CrossRef CAS PubMed.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 126, 1052–1056 CrossRef.
- J. Qin, S. Wang and X. Wang, Appl. Catal., B, 2017, 209, 476–482 CrossRef CAS.
- S. Yan, S. Ouyang, H. Xu, M. Zhao, X. Zhang and J. Ye, J. Mater. Chem. A, 2016, 4, 15126–15133 RSC.
- L. Shen, S. Liang, W. Wu, R. Liang and L. Wu, J. Mater. Chem. A, 2013, 1, 11473–11482 RSC.
- G. Gao, Q. Xi, H. Zhou, Y. Zhao, C. Wu, L. Wang, P. Guo and J. Xu, Nanoscale, 2017, 9, 12032–12038 RSC.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- A. A. Zhumekenov, M. I. Saidaminov, M. A. Haque, E. Alarousu, S. P. Sarmah, B. Murali, I. Dursun, X.-H. Miao, A. L. Abdelhady and T. Wu, ACS Energy Lett., 2016, 1, 32–37 CrossRef CAS.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- J.-F. Liao, Y.-T. Cai, J.-Y. Li, Y. Jiang, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, J. Energy Chem., 2021, 53, 309–315 CrossRef.
- X. Huang, H. Li, C. Zhang, S. Tan, Z. Chen, L. Chen, Z. Lu, X. Wang and M. Xiao, Nat. Commun., 2019, 10, 1163 CrossRef PubMed.
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS PubMed.
- F. Liu, Y. Zhang, C. Ding, T. Toyoda, Y. Ogomi, T. S. Ripolles, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, J. Phys. Chem. Lett., 2018, 9, 294–297 CrossRef CAS PubMed.
- Z. Fan, X. Huang, C. Tan and H. Zhang, Chem. Sci., 2015, 6, 95–111 RSC.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4207 CrossRef.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- S. Zhang, Z. He, X. Li, J. Zhang, Q. Zang and S. Wang, Nanoscale Adv., 2020, 2, 3610–3623 RSC.
- R. Meng, J. Huang, Y. Feng, L. Zu, C. Peng, L. Zheng, L. Zheng, Z. Chen, G. Liu and B. Chen, Adv. Energy Mater., 2018, 8, 1801514 CrossRef.
- H. Tao, Q. Fan, T. Ma, S. Liu, H. Gysling, J. Texter, F. Guo and Z. Sun, Prog. Mater. Sci., 2020, 111, 100637 CrossRef CAS.
- M. Arif, G. Yasin, M. Shakeel, M. A. Mushtaq, W. Ye, X. Fang, S. Ji and D. Yan, J. Energy Chem., 2021, 58, 237–246 CrossRef.
- T. P. Nguyen, D. M. Tuan Nguyen, D. L. Tran, H. K. Le, D.-V. N. Vo, S. S. Lam, R. S. Varma, M. Shokouhimehr, C. C. Nguyen and Q. V. Le, Mol. Catal., 2020, 486, 110850 CrossRef CAS.
- Q. Xue, H. Zhang, M. Zhu, Z. Pei, H. Li, Z. Wang, Y. Huang, Y. Huang, Q. Deng and J. Zhou, Adv. Mater., 2017, 29, 1604847 CrossRef PubMed.
- L. Ding, Y. Wei, L. Li, T. Zhang, H. Wang, J. Xue, L.-X. Ding, S. Wang, J. Caro and Y. Gogotsi, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- M. Alhabeb, K. Maleski, T. S. Mathis, A. Sarycheva, C. B. Hatter, S. Uzun, A. Levitt and Y. Gogotsi, Angew. Chem., Int. Ed., 2018, 130, 5542–5546 CrossRef.
- C. B. Hiragond, N. S. Powar and S.-I. In, Nanomaterials, 2020, 10, 2569 CrossRef CAS PubMed.
- M. Que, Y. Zhao, Y. Yang, L. Pan, W. Lei, W. Cai, H. Yuan, J. Chen and G. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 6180–6187 CrossRef CAS PubMed.
- X. Mao, L. Sun, T. Wu, T. Chu, W. Deng and K. Han, J. Phys. Chem. C, 2018, 122, 7670–7675 CrossRef CAS.
- W.-J. Yin, J.-H. Yang, J. Kang, Y. Yan and S.-H. Wei, J. Mater. Chem. A, 2015, 3, 8926–8942 RSC.
- Y. Park and B. Park, Results Phys., 2018, 11, 302–305 CrossRef.
- M. Irfan, S. Azam, S. Hussain, S. A. Khan, M. Sohail, M. Makhdoom, Z. Ali, I. V. Kityk, S. Muhammad and A. G. Al-Sehemi, J. Alloys Compd., 2018, 766, 536–545 CrossRef CAS.
- P. Kanhere and Z. Chen, Molecules, 2014, 19, 19995–20022 CrossRef PubMed.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- D. Liu, Q. Li, H. Jing and K. Wu, J. Phys. Chem. C, 2019, 123, 3795–3800 CrossRef CAS.
- S. T. A. G. Melissen, F. Labat, P. Sautet and T. Le Bahers, Phys. Chem. Chem. Phys., 2015, 17, 2199–2209 RSC.
- X. Liu, J. Fan and C. Huang, Front. Nanotechnol., 2021, 3, 695490 CrossRef.
- S. You, S. Guo, X. Zhao, M. Sun, C. Sun, Z. Su and X. Wang, Dalton Trans., 2019, 48, 14115–14121 RSC.
- S. Shyamal, S. K. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 7965–7969 CrossRef CAS PubMed.
- H. Huang, J. Zhao, Y. Du, C. Zhou, M. Zhang, Z. Wang, Y. Weng, J. Long, J. Hofkens and J. A. Steele, ACS Nano, 2020, 14, 16689–16697 CrossRef CAS PubMed.
- Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780 CAS.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S.-M. Chang and E. W.-G. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed.
- Y.-F. Mu, W. Zhang, X.-X. Guo, G.-X. Dong, M. Zhang and T.-B. Lu, ChemSusChem, 2019, 12, 4769–4774 CrossRef CAS PubMed.
- Y.-W. Liu, S.-H. Guo, S.-Q. You, C.-Y. Sun, X.-L. Wang, L. Zhao and Z.-M. Su, Nanotechnology, 2020, 31, 215605 CrossRef CAS PubMed.
- G.-X. Dong, W. Zhang, Y.-F. Mu, K. Su, M. Zhang and T.-B. Lu, Chem. Commun., 2020, 56, 4664–4667 RSC.
- J. Zhu, Y. Zhu, J. Huang, L. Hou, J. Shen and C. Li, Nanoscale, 2020, 12, 11842–11846 RSC.
- X. Wang, K. Li, J. He, J. Yang, F. Dong, W. Mai and M. Zhu, Nano Energy, 2020, 78, 105388 CrossRef CAS.
- Z. Wang, H. Kim and H. N. Alshareef, Adv. Mater., 2018, 30, 1706656 CrossRef PubMed.
- J. Song, L. Xu, J. Li, J. Xue, Y. Dong, X. Li and H. Zeng, Adv. Mater., 2016, 28, 4861–4869 CrossRef CAS PubMed.
- H. Huang, J. Li, Y. Yi, J. Wang, Y. Kang, P. K. Chu, H. Ong and X.-F. Yu, Chem. Commun., 2018, 54, 2365–2368 RSC.
- R. Begum, M. R. Parida, A. L. Abdelhady, B. Murali, N. M. Alyami, G. H. Ahmed, M. N. Hedhili, O. M. Bakr and O. F. Mohammed, J. Am. Chem. Soc., 2017, 139, 731–737 CrossRef CAS PubMed.
- W. Deng, H. Huang, H. Jin, W. Li, X. Chu, D. Xiong, W. Yan, F. Chun, M. Xie and C. Luo, Adv. Opt. Mater., 2019, 7, 1801521 CrossRef.
- Y. Shiraishi, T. Hagi, M. Matsumoto, S. Tanaka, S. Ichikawa and T. Hirai, Commun. Chem., 2020, 3, 169 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |