
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlood apheresis technologies – a critical review on challenges towards efficient blood separation and treatment
Christine Jurene O.
Bacal
 *ab,
James W.
Maina
b,
Harshal H.
Nandurkar
c,
Maryam
Khaleel
def,
Rosanne
Guijt
g,
Yung
Chang
h,
Karen M.
Dwyer
a and
Ludovic F.
Dumée
*def
*ab,
James W.
Maina
b,
Harshal H.
Nandurkar
c,
Maryam
Khaleel
def,
Rosanne
Guijt
g,
Yung
Chang
h,
Karen M.
Dwyer
a and
Ludovic F.
Dumée
*def
aDeakin University, Geelong, School of Medicine, Waurn Ponds, 3216, Victoria, Australia. E-mail: cbacal@deakin.edu.au
bDeakin University, Geelong, Institute for Frontier Materials, Waurn Ponds, 3216, Victoria, Australia
cAlfred Health, Australian Centre for Blood Diseases, Monash University, Melbourne, 3004, Victoria, Australia
dDepartment of Chemical Engineering, Khalifa University, Abu Dhabi, United Arab Emirates. E-mail: ludovic.dumee@ku.ac.ae
eCenter for Membrane and Advanced Water Technology, Khalifa University, Abu Dhabi, United Arab Emirates
fResearch and Innovation Center on CO2 and Hydrogen, Khalifa University, Abu Dhabi, United Arab Emirates
gCentre for Rural and Regional Futures, Deakin University, Geelong, VIC 3220, Australia
hR&D Center for Membrane Technology and Department of Chemical Engineering, Chung Yuan Christian University, Chung-Li, 32023, Taiwan
First published on 18th October 2021
Abstract
Blood apheresis technologies are crucial during blood donation and toxin removal. Current purification methods such as leukocytapheresis, erythrocytapheresis, thrombocytapheresis and plasmapheresis primarily rely on centrifugation and membrane filtration to separate blood components. Although established and scaled-up, challenges related to the selectivity and long-term impact of these techniques on blood cells and their constituents remain. Shear generated during extraction may stress blood cells leading to cell damage and initiate the complement cascade causing platelet activation or triggering inflammatory responses. Emerging technologies, including microfluidics and selective adsorption present viable alternatives supporting more selective extraction pathways but extensively rely on novel manufacturing or material developments. This review critically assesses recent progress across apheresis technologies relating to advances in surface science and materials engineering to address operating challenges towards selective removal or extraction of blood components in traditional apheresis technologies such as centrifugation and membrane filtration.
 Christine Jurene O. Bacal | Christine Jurene O. Bacal finished her undergraduate degree in Chemistry in the University of the Philippines – Diliman and took up her Masters in the same university. During her Masters, she specialized in bioprospecting of local fungal endophytes for carbohydrate-active enzymes (CAZys). Currently, she is a PhD candidate of both the School of Medicine (SoM) and the Institute for Frontier Materials (IFM) at Deakin University, Australia. Her research is now focused on developing a more selective haemodialysis membrane for acetaminophen overdose patients. |
 Karen M. Dwyer | Prof. Dwyer has more than 20 years clinical experience in the diagnosis and management of renal-related disease, including acute and chronic kidney injury, refractory hypertension and renal transplantation. Her research interests focus on purinergic signalling, specifically the role of CD39 and adenosine signalling in health and disease. She has developed small animal models to examine acute and chronic ischaemic induced kidney injury, and renal, liver and islet transplant related injury. Prof. Dwyer also has an interest in clinical research project specifically surrounding the diabetes arising post transplantation. |
 Ludovic F. Dumée | Ludo is an Assistant Professor at Khalifa University, Abu Dhabi, United Arab Emirates. His work is focused on the development of advanced separation materials stemming from the properties of nanoscale materials. His team investigates routes to generate efficient separation membranes and catalysts from 1D and 2D nanomaterials toward the design of isoporous membranes for ultra-selective separation of cells, proteins, and pollutants. He is active in developing innovative separation technologies with lower operating costs and footprint to support Circular Economy strategies. |
1. Introduction
Blood is an essential biological fluid containing several blood cell types, such as red (RBCs), white (WBCs) blood cells and platelets, all contained in plasma as the liquid component enabling the transfer of the blood throughout the body.1 Abnormalities in these blood components can lead to various blood diseases necessitating treatment known as apheresis.Apheresis is an extracorporeal medical procedure that involves removal of blood components temporarily utilizing centrifugal force, size and structural differences of blood components and surface forces in microchannels as basis of separation.2–5 Apheresis has two types based on purpose – therapeutic and diagnostic.6 In therapeutic apheresis, disease-causing blood components were removed in critically ill patients.7,8 Therapeutic apheresis techniques are utilized to treat patients with multiple myeloma,9 thrombotic thrombocytopenic purpura (TTP) (formation of blood clots in small blood vessels),10,11 neurological diseases,12 end-stage renal disease (ESRD)3,13 as well as drug toxicities.14,15 Meanwhile, diagnostic apheresis involves isolation of blood components for disease prognosis16 and is typically applied in cancer research.17 For instance, mononuclear cells (MNC) which are of low abundance (only ∼0.06% of all blood cells), are often separated from the bloodstream using diagnostic apheresis techniques without the need of biopsy (a procedure that involves taking a tissue specimen from the patient for the purpose of diagnosing a disease).16
Apheresis procedures may also be classified based on the types of blood component to be extracted, and include erythrocytapheresis and leukapheresis for respectively RBC and WBC extraction, and thrombocytapheresis and plasmapheresis for the collection of platelets and blood plasma respectively.18 The apheresis procedure remains challenging due to the complexity of blood matrix, the variations in blood composition between donors19 and risks of inducing cell damage.20 Apheresis techniques can affect cell integrity and biological function21,22 due to the shear stress experienced during the extraction processes. In addition, only a limited number of different components can be collected simultaneously within a single apheresis procedure due to long treatment times which can lead to significant risks of blood contamination caused by transfusion-transmitted pathogens such as HIV, hepatitis B and C virus, prions, to name a few.23 The most mature and clinically implemented technologies enabling blood apheresis include centrifugation and membrane filtration.9 Progress in terms of equipment capabilities, separation material biocompatibility and performance has led to increased shelf-life, greater purity of the extracted components as well as more rapid processing of larger blood volumes.24 Challenges related to platelet activation and biofouling during separation however still prevail, limiting the efficacy and utility of the procedure.24 Hence, an assessment of the efficiency of existing and emerging apheresis techniques in relation to ultra-selective blood component extraction is required, while considering the cost-effectiveness in more mature technologies.12 So far, limited studies described key performance characteristics of the existing apheresis technologies25–29 and few comprehensively benchmarked current practices to emerging and alternative technologies, such as microfluidic-based or sorbent-based apheresis.30 The impact of materials used or the operating parameters on the removal efficiency are also seldomly discussed.31 An analysis of the key performance characteristics of each apheresis techniques is therefore required27 and must be tailored to a specific disease indication.32–34
This review discusses current blood apheresis technologies as well as emerging solutions and materials to separate specific components from blood. A comprehensive overview on the gaps remaining across these technologies in terms of extraction rate, separation yield, throughput and potential effects on cells will be presented. The performance and efficiency of these technologies, for both therapeutic and diagnostic apheresis techniques, will be assessed considering the improvements on surface characteristics, percentage of haemolysis and thrombogenicity. Fresh perspectives for further improvement of the technologies in terms of design and operating procedures, and perspectives towards integration of more efficient and biocompatible materials will also be offered.
2. Centrifugal-based apheresis
Differential centrifugation is the method used to separate blood components from whole blood based on their densities upon increasing either applied centrifugal force, spinning duration or number of centrifugal cycles.2 Plasma has the lowest density thus settles last, platelets and leukocytes have intermediate densities and erythrocytes have the highest density among the blood cell components hence settling first compared to other blood cells at the bottom of the tube (schematic diagram on Fig. 1a).2 Centrifugal-based apheresis is applicable during blood transfusions and isolation of blood cells26 and is the most utilized apheresis technique since it is versatile, fast to prime and set-up.27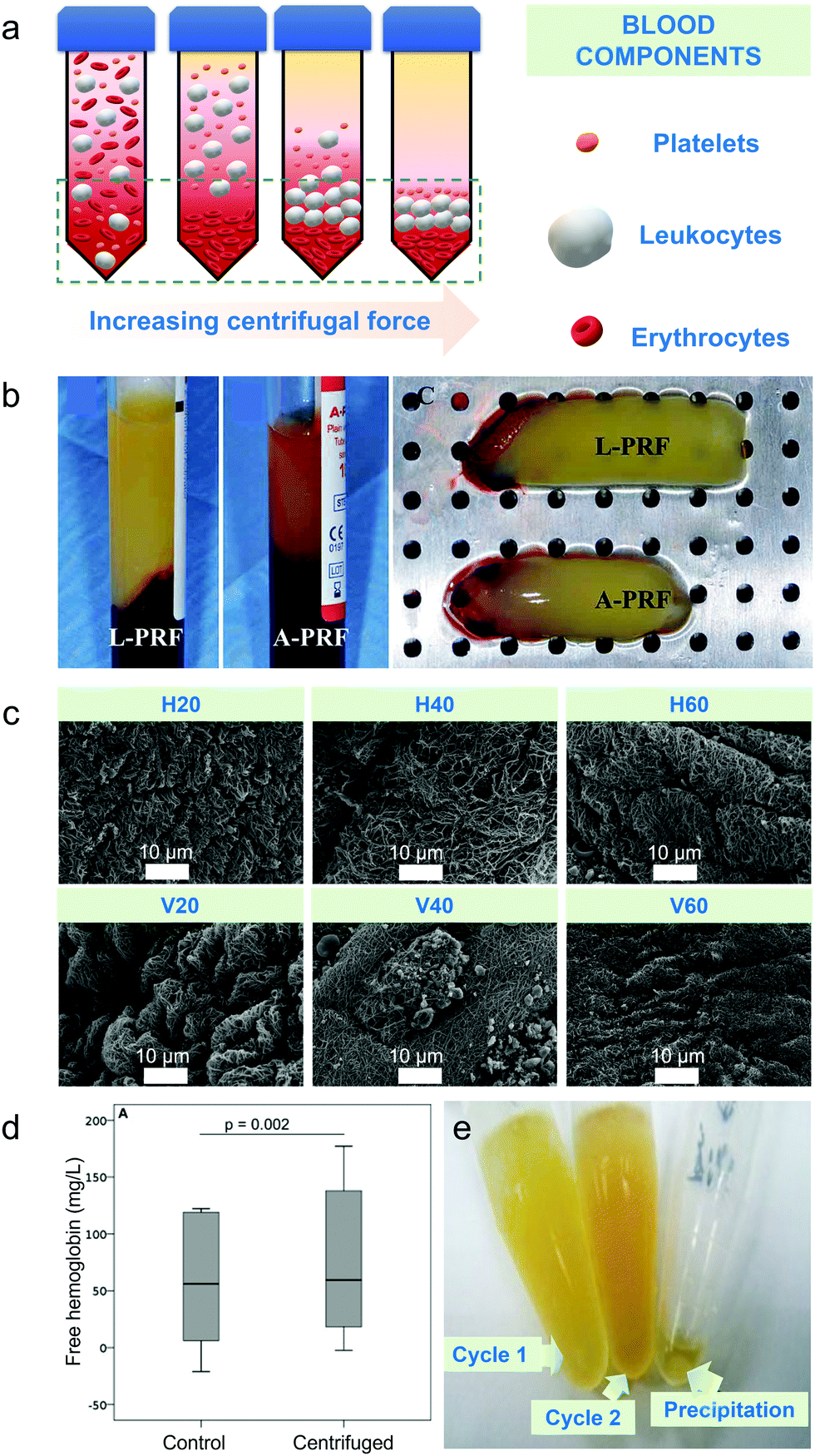 | ||
Fig. 1 Centrifugal-based apheresis. (a) Density-driven separation. Blood components are separated depending on the applied centrifugal force, spinning duration and number of centrifugal cycles. Yellow background represents blood plasma. (b) Impact of centrifugal force on separated blood components during centrifugation.36 Leukocyte- (L-PRF) and platelet-rich fibrin clot (A-PRF) centrifuged at 2700 rpm for 12 min (A) and platelet-rich fibrin clot centrifuged at 1500 rpm for 14 min (B) showed that yellow clot from A-PRF was not fully separated from the red blood cell part due to lower applied centrifugal force. (c) Impact of increasing centrifugal duration on separated blood components. Electron micrographs of fibrin scaffolds at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification showed thicker and less dense filaments upon 20 min centrifugation than 40 and 60 min centrifugation.37 (d) Effect of centrifugation duration on free haemoglobin concentration. Free haemoglobin concentration (g L−1) measured for either 5 or 10 min showed the same result for control and centrifuged sample.21 (e) Impact of number of centrifugation cycles on microRNAs.38 Higher concentration of serum was observed at second centrifugation than the first cycle. Lower detection rates for microRNAs were observed after second centrifugation. 000× magnification showed thicker and less dense filaments upon 20 min centrifugation than 40 and 60 min centrifugation.37 (d) Effect of centrifugation duration on free haemoglobin concentration. Free haemoglobin concentration (g L−1) measured for either 5 or 10 min showed the same result for control and centrifuged sample.21 (e) Impact of number of centrifugation cycles on microRNAs.38 Higher concentration of serum was observed at second centrifugation than the first cycle. Lower detection rates for microRNAs were observed after second centrifugation. | ||
The main challenge of using centrifugation-based apheresis is the occurrence of haemolysis, or rupture of blood cells, caused by shear stress introduced during spinning.35 The extent of haemolysis is dependent on centrifugal operating conditions such as operating mode to be used, centrifugal force, spinning duration and number of centrifugal cycles and will be discussed in this section.
2.1. Operating modes
There are two operating modes in centrifugal-based apheresis, which can either be manual or automatic.2 Manual centrifugation involves pelleting cells in a swinging bucket centrifuge and discarding the supernatant after the centrifugal process.39 Meanwhile, automatic centrifugation involves following a series of pre-set protocols from the centrifuge, depending on application with minimal user intervention.39 Sample recoveries for each operating mode are quantified and compared in the literature using the percentage (%) in recovery, the volume recovered or the number of cells present in the supernatant before and after centrifugation.40 The separation efficiency is, however, a measurement of the amount of blood components extracted or collected within the shortest time possible without compromising the integrity of these components.25,26,41The operation mode must therefore be selected carefully based on the type of cells to be separated to ensure high yields. Overall, in separating blood components for component collection using centrifugation, the yield of the peripheral blood stem cells (PBSC) during leukapheresis should be 2 × 106 CD34 cells or 2 × 108 mononuclear cells (MNCS) per kg body weight of the recipient.41 Also, since packed red blood cells (PRBC) and platelet concentrates (PLTC) contain mostly leukocytes, the number of leukocytes must be <1.2 × 109 if the entire buffy coat, corresponding to blood portion consisting mostly of leukocytes and platelets, is to be discarded.41
The impact of manual or automatic centrifugation on the absolute volume of red blood cells recovered was evaluated and it was observed that using manual centrifugation, ∼201 ± 3 mL per blood unit from a starting volume of 240–300 mL per blood unit and centrifugal speed of 4200 × g42 was recovered yielding a recovery range of 67–84%. When automatic centrifugation was applied, the absolute red blood cell mass volume recovered stood at ∼214 ± 0.6 mL per blood unit from a starting volume of 240–250 mL per blood unit42 giving a higher recovery range of 86–89%. The recoveries of leukocytes and CD34+, a glycoprotein marker of blood stem cells, were also evaluated with both manual and automatic modes. Interestingly, no significant change in WBC count per kg dose recovery (0.9 ± 0.07) was obtained whether manual and automatic centrifugation were used.43 There was also no significant change in the CD34+ count per kg dose recovery (0.9 ± 0.09) when comparing both operation modes43 demonstrating the need to select techniques based on cost-effectiveness. In the same study that compared the recovery of WBCs and CD34+, the separation efficiency was also compared in terms of processing times. Although both centrifugal modes showed no significant differences in recoveries, the processing time for manual centrifugation (35 min) was much longer than that of the automatic centrifugation (28 min) for the same treated blood volume.43 Longer processing times also typically lead to platelet loss and clotting events25,44 thus this factor must also be considered aside from the cost.
The sample purity is also affected by the centrifugal operating mode. The application of manual centrifugation was found to lead to discrepancies in spin speeds while additional user manipulations may lead to large variations in the quality and purity of the separated blood component.45 Automatic centrifugation can produce separated blood components that are more uniform in composition since protocols are set based on the blood component to be separated with minimal user intervention. For instance, the percentage of haematocrit, corresponding to the ratio of red blood cells to whole blood, was compared for both manual and automatic modes of centrifugation. The measured % haematocrit in manual mode was 73 ± 9% compared to 89 ± 7% for the automatic mode, demonstrating that purer red blood cells may be extracted with the latter. The purity variations can be attributed to the fact that manual centrifugation might induce greater mechanical shear stress to the sample due to the longer operating times, which may lead to platelet activation.46 The percentage of haemolysis, or blood cell lysis, corrected for haematocrit for both modes was evaluated and it was found that percentage of haemolysis was greater (0.7 ± 0.20) for manual centrifugation compared to automatic centrifugation (0.4 ± 0.2).42 Automatic centrifugation is therefore currently recommended unless cost prohibitive or additional user intervention is needed.
2.2. Centrifugal force
Centrifugal force, described to be either revolutions per minute (rpm) or relative centrifugal force (rcf) – also known as “g-force” (×g), is the apparent force pulling a blood component of interest away from the centre of rotation, caused by the component's inertia.9,41 Characterization of mechanical properties of blood components is important in diagnosing haematological disorders and optimizing the design of cardiovascular implants and blood circulating devices with respect to blood damage.21 A study discussed the effect of centrifugation on the markers of mechanical damage and RBC membrane.21 An increase in the free haemoglobin yield from 61 mg L−1 to 79 mg L−1 could be attributed to haemolytic damage caused by the forces associated with centrifugation, even at 900 × g.Leukocyte- and platelet-rich fibrin clots (L-PRF) were also separated due to its significance in oral and maxillofacial regenerative therapies via centrifugation.36 It was observed that when centrifugal force applied increased from 1500 rpm to 2700 rpm for 14 and 12 min respectively, the separation between the clot and the red blood cell became more defined regardless of the blood volume used (10 mL for 1500 rpm and 9 mL for 2700 rpm) (Fig. 1b). Cell viability of adipocytes was observed when centrifuged from blood and it was discovered that there was no significant difference in the cell viability in the centrifuged adipocytes.47 After eight accelerations up to 20000 × g for 10 min, the viability remained at around 80–90%.47 An increase in the centrifugal force alone could not be equated to higher purity samples. The level of platelet-rich plasma obtained was 3.2 times that of the concentration of WB baseline when a centrifugal force of 200 × g was used in 5 mL WB for two spins, 10 min each.48 Comparatively, a platelet concentration factor of 3.47 was obtained from an 8.5 mL processed WB when centrifugation was performed at both 130 × g and 250 × g for a period of 15 min when performing a two-spin centrifugation process.49 The centrifugation of WB at 460 × g for 8 min using only one centrifugation spin step led to a platelet concentration factor of 2.67 above baseline.50 When around 5–6 L of blood were processed, effects of centrifugal force on the collection efficiency of mononuclear cells were evaluated using packing factor (PF).51 Packing factor is defined as the exertion of relatively high centrifugal force on the anticoagulated blood entering the spinning channel. Higher platelet drop per litre processed blood was recorded (2.2 ± 0.6%) when PF 4.5 was used instead of PF 4.0 (2.6 ± 0.5%). Reduced platelet loss ultimately led to reduced platelet contamination which improved the quality of the collected cellular component. However, the mechanism on how the decreased centrifugal force affected platelets are not discussed in this study. Hence, centrifugal force must be interlinked to other operating parameters to optimize recovery and purity.
2.3. Spinning duration
The spinning duration refers to the required duration for a centrifugal cycle to end, affecting sample recovery, separation efficiency, purity and characteristics of the extracted cells. Longer centrifugation durations slightly increased the platelet recovery and decreased WBC concentrations in the upper supernatant layer after a first spin step at 100 × g for both 6 min (platelet = 72 ± 3%; WBC = 27 ± 11%) and 10 min (platelet = 79 ± 8%; WBC = 9 ± 4%) intervals.31 Better platelet separation efficiency was also obtained in another study (92%) by applying an acceleration of 1900 × g for 5 min as compared to 1300 × g for 10 min.52 For fibrin scaffolds, less compaction and thicker filaments were observed in electron micrographs when centrifugation duration used was 20 min rather than 40–60 min (Fig. 1c).37 Morphological analysis for fibrin fibre showed that mean fibre diameter decreased after centrifugation from 20 to 60 min on both horizontal (from 358 ± 79 to 205 ± 57 nm, p = 0.04) and vertical (from 311 ± 63 to 138 ± 35 nm, p = 0.037) orientation of the rotors used.In another study however, there is no significant effect in the free haemoglobin (fHb) concentration when the spinning duration was decreased from 7 min (0.07 ± 0.04 g L−1) to 5 min (0.07 ± 0.03 g L−1) at both 3000 × g centrifugal force.53 The same finding was obtained for the free haemoglobin concentration obtained when centrifugal force used was lower at both 900 × g at 5 and 10 min centrifugation (61 mg L−1 for control and 79 mg L−1 for centrifuged samples at both 5 and 10 min) (Fig. 1d)21 Although the difference in duration is small, an increase in fHb concentration however small, can have a significant effect if it is above analyte specific haemolysis cut-off.53 The adverse effects to cells as associated with the spinning duration and centrifugal force was also explored and correlated to adenosine triphosphate (ATP) (organic compound that drives many metabolic processes in cells) release with haemolysis.35 An increase in the concentration of ATP was observed (from 1.4–4.7 nM) as the centrifugation time increased from 1 to 5 min (Fig. 1c) while varying the applied centrifugal force from 900–16000 × g.35 The magnitude of ATP in the packed RBCs obtained using the parameters explored suggests that care must be taken when interpreting ATP transduction data in experiments that utilize centrifugation.
2.4. Number of spins
The number of spins pertains to the number of centrifugal cycles in a run. In most runs, centrifugation is repeated up to two cycles and increasing the number of spins can lead to a higher recovery of separated blood component. Processing of 3.5 mL of blood at 100 × g for 10 min for the first spin and 400 × g for 10 min for the second spin while withdrawing 2/3 of the remnant plasma to obtain platelets was performed.40 Although a high platelet recovery of 70–80% was obtained, approximately 20% of the platelets remained adsorbed in the RBC pellet40 after the second spin. Hence, a recorded 5-fold increase in platelet concentration40 still requires optimization of the number of spins along with centrifugal force and spinning time to increase the yield and quality of the separated blood components.The separation of other components found in blood, such as microRNAs, are critical since they serve as biomarkers for tumour diagnosis and prognosis.38 The content in representative microRNAs was found to decrease after a two-step centrifugation as compared to a single-step centrifugation. The cell count of miR-BART5, a type of microRNA from a virus involved in a variety of human tumours, in the blood plasma was 40 when a two-step centrifugation was applied compared to 33.5 for a single-step centrifugation process, suggesting that the number of viral microRNAs left in the blood decreased.38 The total signal detection rate of microRNAs also decreased (15.3%) when a two-step centrifugation was applied compared to a single-step centrifugation (21.1%) (Fig. 1e).38 For the evaluation of the purity of detected microRNAs, size distribution of the particles from exosomes – carrier of microRNAs with expected particle size of 30–150 nm,54 were assessed by Dynamic Light Scattering (DLS). Particles with mean diameters of 63 nm present after a one-step centrifugation process were found to increase to 170 nm after a two-step centrifugation.38 However, this two-step protocol did not demonstrate if the extracted microRNAs found after both the first and second steps were similar, and determination of the purity for these biomarkers is yet to be confirmed.
Platelet and leukocyte separation were evaluated after a single and two-step process and it was found that platelet concentration only increased by 1.5-fold for single-step compared to 4.7-fold for a two-step process. The leukocyte concentration factor also did not increase for the single-step process but was raised 1.4-fold after the two-step procedure.55
2.5. Conclusions for centrifugal-based apheresis
Although centrifugation has been the most utilized and established method of separating blood components, intrinsic limitations persist. Centrifugation is labour-intensive and expensive, as the whole apheresis process involves elaborate sample handling and storage most especially in industries that supply blood products where high-capacity centrifuges are utilized.20 Variations in separation time, non-reproducibility and low selectivity limit the practical use of traditional centrifugation separation technology.19 The main drawback is often related to strong mechanical shear stress induced on the cells, triggering platelet activation and causing haemolysis which can decrease the utility of the extracted blood component.41,56 Platelet activation primarily affects the quality of the recovered or extracted blood component by overwhelming the size of the component of interest. Consequently, more anticoagulants are required adding complexity with regards to potential long-term adverse effects including tendency to suffer from bleeding. This technique can also lead to platelet loss of up to 50% since platelets have a similar density as blood plasma and are therefore difficult to separate from the main blood matrix.25,44,57,58 Hence, despite the popularity of centrifugation, platelet activation and haemolysis underpin the search for alternative apheresis technologies.3. Membrane-based apheresis
In membrane-based apheresis, blood components are separated based on their differences in size. Membranes are fabricated to generate pore sizes that will sieve the blood component of interest while retaining other relevant components (schematic diagram on Fig. 2a).3 The main application of membrane blood filtration is to remove uremic toxins and excess plasma proteins such as urea, creatinine, immunoglobulins, complement factors and lipoproteins in patients with end-stage renal disease (ERSD) or autoimmune disorders.32–34 It can also be used to filter leukocytes (leukodepletion) to avoid transfusion-related diseases.59 These applications led to the classification of membrane blood filters as leukodepletion filters, plasma separation membranes and haemodialysis membranes.3,60,61 The inherent propensity of the membranes to foul (biofouling),26 corresponding to the formation of a cake-layer composed mostly of biological materials, onto the active part of the membrane however represents a major operating challenge.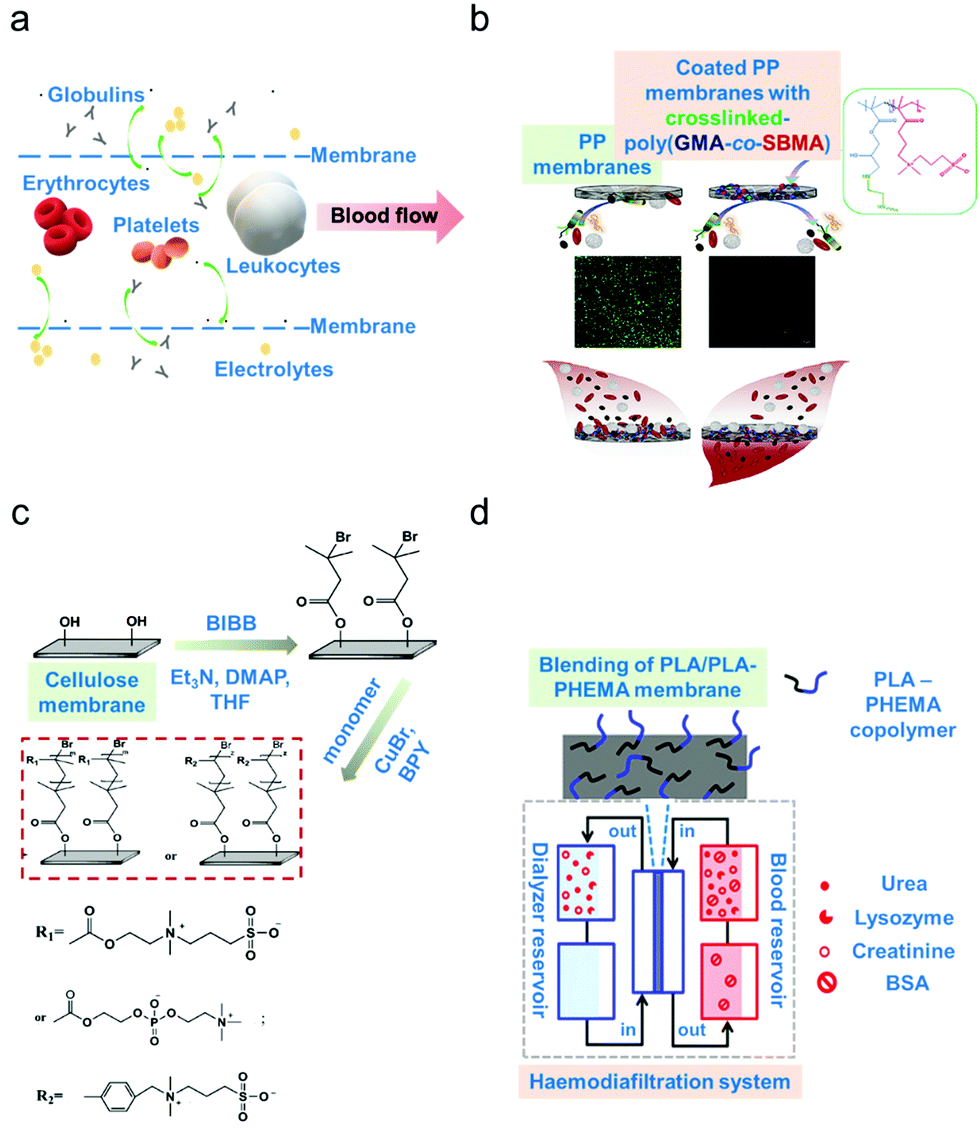 | ||
| Fig. 2 Membrane-based apheresis. (a) Size-driven separation. Smaller-sized blood solutes such as electrolytes and globulins (plasma proteins) pass through the membrane sieves whereas larger blood cells such as the erythrocytes, leukocytes and platelets go along the direction of the blood flow. (b) Example of surface modification via coating. Poly(propylene) (PP) membranes were coated with a copolymer (poly(glycidyl methacrylate)-co-sulfobetaine methacrylate) (poly(GMA-co-SBMA) with zwitterionic properties to minimize membrane fouling.66 (c) Example of surface modification via grafting. Cellulose membranes were modified by grafting zwitterionic polymers via surface-initiated atom transfer radical polymerization (ATRP).67 (d) Example of surface modification via blending. A block copolymer of poly(lactic acid) (PLA)/poly(hydroxyethylmethacrylate) (PHEMA) was blended with the base material which is poly(lactic acid) to form a more biocompatible membrane.68 | ||
Properties of the membrane materials can be modified at will to increase biocompatibility and to improve the selectivity and resistance to biofouling.27,57,62 Various surface modification techniques are employed such as (i) materials blending, in terms of polymer mixing to form composite materials via physical or noncovalent interaction, (ii) surface grafting, whereby a covalent attachment of functional groups on the membrane surface is performed, and (iii) surface coating, whereby a layer covering the membrane surface is added to enhance biocompatibility.63–65 Hence, in this section, potential solutions to address technological gaps in membrane-based apheresis will be discussed including surface modification of the common materials used in membrane fabrication for enhanced biocompatibility. Membrane performance before and after surface modification will be evaluated to determine the suitable techniques that can be applied for improved removal efficiencies.
3.1. Membranes for leukodepletion
Leukodepletion filters extract white blood cells for the purpose of preparing red blood cells (RBCs) and platelets or removing micro-aggregates, clots, and particle debris from stored or salvaged blood components.60 The mechanisms of leukocyte removal could either be driven through cell sieving, based on size or molecular weight cut-off, or cell adhesion, based on the bio-chemical affinity between the membrane and WBCs.60 In the 1980s, Asahi and Pall successively proposed non-woven fabric systems for reducing white blood cells.59,69–71 Surface grafting of leukocyte filters with positively charged polymers was found to enhance the adsorption of electronegatively charged leukocytes.72 In the past 30 years, this concept has been widely used in the development of membrane systems for leukocyte removal processes.69 The currently available commercial leukocyte filters which fulfil the criteria of both the European Council (EC) and the American Association of Blood Banks (AABB) for leukoreduced blood components, as well as newly developed leukocyte filters for enhanced leukodepletion are summarized in Table 1.73| Entry | Material | Surface-modified? | Type of surface modification mainly applied | Blood component recovered | Leukocyte depletion | Recovery |
|---|---|---|---|---|---|---|
| a Abbreviation: ND, no data. | ||||||
| 1 | Nonwoven polyester fibres69,76–78 | Yes | NDa | Erythrocytes | Up to 99.3% | >90% |
| 2 | Nonwoven polyester fibres69,79,83 | Yes | Coating with phosphorylcholine-containing polymers | Platelets | >99% | 93% |
| 3 | Microporous poly(urethane)69,70,77,78 | Yes | Coating with poly(ethylene imine) | Erythrocytes | Up to 99.6% | >90% |
| Platelets | >90% | >90% | ||||
| 4 | Microporous polyurethane85 | Yes | Grafting with Na2SO3, H2SO4, glycine, ammonia, NH(C2H5)2 and NH2C2H4OH | Erythrocytes | ∼13% permeation ratio of erythrocytes | <5% |
| Platelets | ∼25% permeation ratio of platelets | <5% | ||||
| T-Cells | ∼24% permeation ratio of T-cells | ∼20% | ||||
| B-Cells | ∼10% permeation ratio of B-cells | ∼15% | ||||
| CD34+ cells | <1% permeation ratio of CD34+ cells | ∼70% | ||||
| Mononuclear cells | ∼2% permeation ratio of mononuclear cells | <5% | ||||
| 5 | Poly(propylene) membranes86 | Yes | Grafting with poly(DMAEMA-co-PEGMA) | Erythrocytes | 99.6% | 98% |
| 6 | Poly(propylene) membranes66 | Yes | Coating and crosslinking with poly(GMA-co-SBMA) | Erythrocytes | >99.9% | ∼100% |
| 7 | Poly(propylene) nonwovens87 | Yes | Grafting of “LDV” peptidomimetics | Erythrocytes | ∼3.71 log depletion | ∼100% |
| 8 | Melt-blown poly(butylene terephthalate)192 | Yes | Grafting with polar groups (not disclosed) | Erythrocytes | 99.03% | 95.48% |
| 9 | Nonwoven poly(butylene) terephthalate90 | Yes | Grafting of poly(vinyl pyrrolidone) | Erythrocytes | 96% | 92% |
| 10 | Nonwoven poly(butylene) terephthalate91 | Yes | Coating with hydroxyapatite | Erythrocytes | 98.5% | 99.5% |
| 11 | Electrospun poly(ethylene-co-vinyl alcohol)92 | No | ND | Erythrocytes | Up to ∼99% | >90% |
Current commercial leukodepletion filters have removal efficiencies of up to 99% and blood components recovered are at around 90% which satisfies the criteria set by the EC and AABB (Table 1). Membrane surface modification played a key role in enhancing the recovery of leukoreduced blood component yields when these filters are used during leukocyte filtration. Blending, coating or grafting of membrane surfaces affects the surface wettability and charge, thus supporting altered solute/matrix interactions, while also dictating the compatibility of the materials.59 Leukocytes were shown to exhibit a greater adherence to wettable surfaces as modelled by observing changes in the free energy of solid surfaces74 and more hydrophilic filters, characterized by low contact angles (<90°),75 are beneficial to increase the leukocyte removal efficiency.
In one study,83 leukocyte depletion increased from around 55% in unmodified membrane to around 90% upon coating the polyester fibres with phosphorylcholine-containing polymers such as MPC37LMA63 (polymer consisting of 2-methacryloyloxyethylmethacrylate (MPC) and lauryl methacrylate (LMA)) (Table 1, entry 2). This result can be attributed to the reorientation of hydrophobic PC head towards the surface of the base material (polyester) minimizing interfacial energy upon blood contact.84 As a proof of the reorientation phenomenon, the dynamic contact angle of membranes coated with MPC37LMA63 also showed that the initial contact angle was high at around 110° but after a brief immersion in water, the contact angle decreased to around 30°. Upon exposure to an aqueous environment, positively charged PC groups tend to reorient towards the surface minimizing the interaction of the polyester membrane with the blood during coating.
Commercial filters have similar performance with nonwoven polyesters when coated with a hydrophilic polymer, poly(ethylene imine) (PEI).69,70,77,78 A stable coating with a complete coverage of 0.1 mg m−2 based on 14C-labeled PEI mapping of the membrane surface showed that PEI-coated PU leukocyte filters increased the leukocyte depletion efficiency to >95% compared with the unmodified filters with efficiency of ∼80%. PEI enhanced the performance of PU filters by introducing hydrophilic amine groups on the PU membrane surface. X-Ray photoelectron spectroscopy (XPS) results showed an increase in the nitrogen content from 2.4% in unmodified membranes to 3.8% after PEI coating, which verifies that the improved leukocyte removal efficiency is caused by incorporation of a stable PEI coating.
| Entry | Material | Surface-modified? | Type of surface modification applied | Solutes/proteins removed | Flow rate (mL min−1) | Solute clearance (mL min−1) or % removal efficiency | |
|---|---|---|---|---|---|---|---|
| Blood side | Dialysate side | ||||||
| a Abbreviation: ND, no data. b At UF rate = 10 mL min−1. c % decrease in adsorption. d Feed flow rate. e Plasma flow rate. f Ultrafiltration coefficient (mL h−1 m−2 mmHg−1). g Solution flow rate and pure water flow rate, respectively. | |||||||
| 1 | Unmodified cellulose193–196 | No | NDa | Ureab | 10 | 105 | |
| 2 | Cellulose67 | Yes | Grafting with zwitterionic polymers via surface-initiated atom transfer radical polymerization (ATRP) | Plasma proteins | ND | 44%c | |
| 3 | Cellulose acetate99 | Yes | Blending with SiO2 | Urea | 2500d | 99.7% | |
| BSA | 2500d | 1% | |||||
| 4 | Cellulose triacetate29,197–200 | No | ND | Urea | 300 | 500 | 262–287 |
| Creatinine | 300 | 500 | 242–277 | ||||
| Phosphate | 300 | 500 | 227–252 | ||||
| Vitamin B12 | 300 | 500 | 152–202 | ||||
| 5 | Poly(sulfone)201–203 | No | ND | Urea | 300 | 500 | 228–261 |
| Creatinine | 300 | 500 | 200–231 | ||||
| Phosphate | 300 | 500 | 164–210 | ||||
| Vitamin B12 | 300 | 500 | 94–138 | ||||
| 6 | Poly(sulfone)100 | Yes | Blending with Fe2O3 | Urea | 15 | 37.5 | 75.1% |
| Lysozyme | 15 | 37.5 | 35.6% | ||||
| 7 | Poly(sulfone)97 | Yes | Coating with vitamin E | Vitamin B12 | 100 | 10 | ∼100% |
| Creatinine | 100 | 10 | ∼100% | ||||
| Chymotrypsin | 100 | 10 | 82% | ||||
| BSA | 100 | 10 | ∼2% | ||||
| 8 | Poly(sulfone)32,204 | No | ND | Albumin | 40e | 100% | |
| IgG | 40e | 100% | |||||
| IgM | 40e | 100% | |||||
| Lipoprotein-B | 40e | ∼96% | |||||
| 9 | Poly(ether sulfone)145,165,197 | No | ND | Urea | 300 | 500 | 243–293 |
| Creatinine | 300 | 500 | 268–275 | ||||
| Phosphate | 300 | 500 | 254–265 | ||||
| Vitamin B12 | 300 | 500 | 190–206 | ||||
| 10 | Poly(ether sulfone)117 | Yes | Blending with a commercial co-polymer, SlipSkin™ (SS) | Creatinine | 10f | ∼89% | |
| Hippuric acid | 10f | ∼59% | |||||
| Indoxyl sulfate | 10f | ∼100% | |||||
| BSA | 10f | 1.83-Fold increase | |||||
| 11 | Poly(ether sulfone)111 | Yes | Blending with multi-walled carbon nanotube (MWCNT) and poly(vinyl pyrrolidone) | Urea | ND | 125,g 50g | 56.3% |
| Creatinine | ND | 125,g 50g | 55.1% | ||||
| Lysozyme | ND | 125,g 50g | 27.9% | ||||
| 12 | Poly(ether sulfone)101 | Yes | Blending with multi-walled carbon nanotube (MWCNT) grafted-poly(citric) acid | BSA | ND | ND | 63.5% |
| 13 | Poly(ether sulfone)205 | No | ND | Lipopolysaccharides | 15–45e | 50% | |
| TNF-α | 15–45e | 75% | |||||
| C-Reactive protein | 15–45e | 51% | |||||
| Fibrinogen | 15–45e | 51% | |||||
| Total cholesterol | 15–45e | 62% | |||||
| Apolipoprotein B | 15–45e | 59% | |||||
| 14 | Poly(ethylene)58,93,206 | Yes | Coating with ethylene vinyl alcohol copolymer | Total protein | <16–50e | 98% | |
| Albumin | <16–50e | 97% | |||||
| IgG | <16–50e | 93% | |||||
| IgA | <16–50e | 96% | |||||
| IgM | <16–50e | 95% | |||||
| Total cholesterol | <16–50e | 99% | |||||
| 15 | Poly(propylene)207 | Yes | Grafting with poly(2-methoxyethylacrylate) (poly(MEA)) | Total protein | 20e | 100% | |
| Albumin | 20e | 102% | |||||
| Total cholesterol | 20e | 100% | |||||
| Fibrinogen | 20e | 100% | |||||
| Factor VII C | 20e | 99% | |||||
| 16 | Poly(lactic acid)68 | Yes | Blending with grafted poly(lactic acid)-block-poly(2-hydroxyethyl methacrylate) (PLA–PHEMA) copolymers | Urea | ND | 100, 300 | 0.78 |
| Creatinine | ND | 100, 300 | 0.74 | ||||
| Lysozyme | ND | 100, 300 | ∼0.70 | ||||
| BSA | ND | 100, 300 | ∼0.35 | ||||
| 17 | Poly(lactic acid)110 | Yes | Grafting with heparin | Urea | 200 | 500 | 79% |
| Lysozyme | 200 | 500 | 18% | ||||
| BSA | 200 | 500 | <10% | ||||
| 18 | Poly(amide)165 | Yes | Blending with poly(vinylpyrrolidone) and poly(aryl ether sulfone) | Urea | 300 | 500 | 167 |
| Creatinine | 300 | 500 | 146 | ||||
| Phosphate | 300 | 500 | 136 | ||||
| Vitamin B12 | 300 | 500 | 90 | ||||
| 19 | Poly(acrylonitrile)208 | Yes | Blending with sodium methallyl sulfonate | Urea | 300 | 500 | 231–265 |
| Creatinine | 300 | 500 | 204–237 | ||||
| Phosphate | 300 | 500 | 172–207 | ||||
| Vitamin B12 | 300 | 500 | 106–143 | ||||
| 20 | Poly(methyl methacrylate)165,197,209 | No | ND | Urea | 200 | 500 | 184–192 |
| Creatinine | 200 | 500 | 182–191 | ||||
| Phosphate | 200 | 500 | 155–179 | ||||
| Vitamin B12 | 200 | 500 | 106–133 | ||||
| 21 | Poly(methyl methacrylate)210 | No | ND | β2-Microglobulin | ND | ∼64% | |
| 22 | Poly(methyl methacrylate)211 | No | ND | Total plasma | 33e | 50 | |
| Plasma proteins (β-lipoprotein, albumin, IgG, IgM) | 33e | 100 | |||||
| 23 | Poly(urethane)212 | Yes | Coating with 3,4-dichloro-5-hydroxy-2(5H)-furanone incorporated with poly(vinyl pyrrolidone) containing succinimidyl functional groups | Human serum albumin (HSA) | ND | ND | 64–70%c |
| 24 | Nanoporous silicon213 | No | ND | Creatinine | 10 | 10 | ∼109 |
| Urea | 10 | 10 | ∼150 | ||||
| Phosphorus | 10 | 10 | ∼87 | ||||
| β2-Microglobulin | 10 | 10 | ∼27 | ||||
3.2. Membranes for toxin removal
Aside from leukocytes with sizes ranging from 6–10 μm,69 other blood components such as plasma proteins and uremic toxins are removed to achieve haemostasis. Plasma separators are used for the purpose of collecting plasma proteins such as immunoglobulins, albumin and lipopolysaccharides, which are known precursors to endotoxins and cholesterols including low-density lipoprotein (LDL) and high-density lipoprotein (HDL) and to replace unhealthy blood plasma with a fresh and healthy one in patients with autoimmune diseases.9 Plasma components have pore sizes of >100 nm hence the pore size distribution range of membrane plasma separators is typically narrower and finer than that of leukodepletion filters, and in the microfiltration (MF) range of 0.2 to 0.8 μm.93 Haemodialysis membranes, on the other hand, are primarily employed for the removal of low molecular weight uremic toxins (<500 Da) which could be of the size of <10 nm.94,95 Higher molecular weight toxins >60000 Da with a size range >10 nm but <100 nm are removed by haemofiltration membranes, the separation mechanism follows the principle of convection and not diffusion, as with haemodialyzers.3,96 Currently available membranes for toxin removal are presented in Tables 2 and 3.
| Entry | Material | Surface-modified? | Type of surface modification applied | Indicators of anti-thrombogenicity | Indicator of complement system activation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| APTT (s) | PT (s) | PRT (s) | Serum concentration (μg mL−1) | ||||||||
| UM | M | UM | M | UM | M | UM | M | ||||
| a Abbreviation: NT, not tested. | |||||||||||
| 1 | Poly(sulfone)128 | Yes | Grafting with acrylic acid and sulfonated hydroxypropyl chitosan | 36.90 | 179 | 18.10 | 62.10 | 238 | 301 | NTa | NT |
| 2 | Poly(ether imide)129 | Yes | Blending with graphene oxide/poly(vinyl pyrrolidone) nanocomposites | 32 | 75 | 15 | 18 | 325 | 675 | ∼0.04 (C3a), ∼0.0016 (C5a) | ∼0.028 (C3a), 0.0008 (C5a) |
| 3 | Poly(ether imide)214 | Yes | Blending with functionalized multi-walled carbon nanotubes (f-MWCNT)/poly(vinyl pyrrolidone) composites | 42.5 | 84 | 12.5 | 24.2 | NT | NT | NT | NT |
| 4 | Cuprophan98 | Yes | Blending with chitosan interpenetrated with anionic polysaccharides | NT | NT | NT | NT | 600 | 240–480 | 101.5 (iC3b) | 63.2 (iC3b) |
| 5 | Cellulose triacetate215 | No | ND | NT | NT | NT | NT | NT | NT | 1.2 (β-thyroglobulin) | |
| 0.7 (platelet factor 4) | |||||||||||
| 6 | Cellulose acetate99 | Yes | Blending with SiO2 | UM | ∼55% thrombosis | NT | |||||
| M | ∼45% thrombosis | ||||||||||
Plasma separators and haemodialyzers could be made out of cellulose or synthetic polymers such as poly(sulfone), poly(ethylene), poly(lactic acid), to name a few (Table 3). Like in leukodepletion filters, these materials can be modified through the previously specified techniques for a more efficient separation of plasma proteins and uremic toxins. The use of zwitterionic polymers,67 vitamin E97 and chitosan98 are only some of the added materials developed for a more enhanced performance and haemocompatibility. Interestingly, the use of nanoparticles such as silicon dioxide,99 ferric oxide100 and multi-walled carbon nanotubes101 are also explored as anti-fouling agents.
Cellulose acetate (CA) membranes are also used in blood purification to address the problem with the number of –OH group in cellulose membranes that would lead to leukopenia (Table 2, entry 3).103 Here, hydroxyls of the unmodified cellulose were substituted by acetate radicals to minimize hydrophilicity. Although CA membranes were linked to reduced complement activation due to the presence of negative charges on the surface that can adsorb the activated cationic complement peptide,105 there are still a few concerns such as limited mechanical strength and shelf-life.106,107 To address these concerns, a novel method of covalently adding inorganic nanoparticles such as SiO2 with mechanical and thermal stability properties to CA membranes was explored.99 By combining the phase inversion and sol–gel techniques, a CA/SiO2 monophasic hybrid membrane was formed. After optimizing the amount of silica added, it was found out that when 5 wt% silica was added to CA membrane, there was no significant change in contact angle but when 11 wt% and 18 wt% silica were added to the CA membrane respectively, the contact angle decreased from ∼40° to around 14° and 22° respectively. The surface roughness will influence the hydrophilicity but when the average roughness of the hybrid membrane was obtained through atomic force microscopy, it was found that there is not much difference in the nano (1.0–2.5 nm) and the micro (6.0–7.3 nm) range for the CA membranes with 5, 11 and 18 wt% silica added thus surface roughness did not affect the hydrophilicity in this case. With regards to the anti-fouling ability, it was shown that urea is fully permeated with % rejection of ∼0.3% for 11–18% silica–CA hybrid membranes from 2% in unmodified CA membranes whereas BSA is fully rejected in both modified and modified CA at 99% rejection. Hence, this membrane is selective in filtering urea and retaining BSA and plasma proteins with similar size which is highly desired during haemodialysis.
3.2.2.1. Poly(sulfone) and poly(ether sulfone) membranes. Poly(sulfone) (PSf) and poly(ether sulfone) (PES) are the primary materials used in fabricating haemodialysis membranes and plasma filters however, high membrane hydrophobicity causing low removal efficiency of middle-molecular weight toxins such as β2-microglobulin is the current trade-off in using these membrane materials.101 The incorporation of metal oxides is also explored not only in cellulose acetate membranes99 but also in synthetic polymers such as poly(sulfone). Iron(III) oxide (Fe2O3) is blended with poly(sulfone) membranes because aside from its hydrophilicity and nontoxicity, its nanostructure facilitates better water porosity at minimal operating pressure (Table 2, entry 6).100 Although the exact mechanism of how it became a good liquid transport medium is not explained, true enough, PSf/Fe2O3 membranes exhibited more finger-like structures and thinner skin layer than PSf membrane alone. The pore size distribution also shifted to larger mean sizes due to the greater freedom in movement of Fe2O3 near the membrane-moisture interface. The contact angle values decreased from 67° to 55° and water porosity increased from 43% to 67% leading to lesser human serum albumin (HSA) adsorption from 8.23 μg cm−2 to 6.68 μg cm−2 and higher BSA rejection from 91% to 98% in PSf/Fe2O3 membranes. The clearances for the relevant haemodialysis solutes – urea and lysozyme were 75.1% (from 52.3% urea clearance) and 35.6% respectively. Lysozyme clearance was better at 35.6% compared to other studies – 18% using heparin-immobilized poly(lactic acid) (PLA) membrane110 and 28% multi-walled carbon nanotube (MWCNT)-incorporated PES membrane.111 Lysozyme has about the same molecular weight as the common middle-molecular weight β2-microglobulin (∼15 kDa) but the size of lysozyme is 9 nm in length and 1.8 nm in diameter.112 β2-Microglobulin is <200 nm in length and ∼10 nm wide.113 Better representation molecule for middle-molecular weight toxins must be considered in future studies.
Other surface modifications were also applied to PSf membranes such as coating PSf membranes with vitamin E to capture reactive oxygen species in blood that are activated during blood–membrane contact.97 This is not the first time that vitamin E-coated PSf membranes were reported in the literature.114–116 However, this is the first study to demonstrate the mass-transfer characteristic of vitamin E-coated PSf membranes. Filtration experiments on aqueous test solutions of creatinine, vitamin B12, chymotrypsin and albumin were done at flow rates of 100 mL min−1 on the blood side and 10 mL min−1 on the dialysate side. Upon coating PSf membranes with α-tocopherol – the active form of vitamin E, it was discovered that there is no significant difference in the efficiency of small solute removal such as creatinine and vitamin B12 against the uncoated membranes (∼100%). However, significant changes were observed when larger solutes such as chymotrypsin and albumin were filtered. The removal efficiency of chymotrypsin decreased from ∼86% in uncoated membranes to ∼82% in coated membranes during the first 30 min of filtration. Same changes were observed when albumin was filtered wherein the removal efficiency decreased to ∼2% from ∼4% during the first 30 min of filtration. Morphological analysis presented a wedge-shaped structure on the cross-section wherein large pores are filled with white drops that can be related to the successful coating of vitamin E. This result shows that addition of vitamin E influenced the mass transfer properties of the PSf membranes and can make the filter more selective towards small solutes and more discriminative towards middle to high molecular weight proteins such as albumin which is necessary for homeostasis.
Polymer blends were used to generate asymmetric membranes from poly(ether sulfone) (PES) and a co-polymer, SlipSkin™ (SS) with randomly arranged monomeric moieties of hydrophilic NVP (N-vinylpyrrolidone) and hydrophobic BMA (N-butylmethacrylate) (Table 2, entry 10).117 Morphological analyses showed the presence of macrovoids in both PES-SS and PES-PVP membranes while no macrovoids were present across the PES-SS membranes. Enhanced long-term filtration fluxes for both PES-SS (1.3-fold increase) and PES-PVP (5-fold increase) were achieved compared to commercial F8HPS membranes, measured after 24 h of filtration. Interestingly, although the PES-SS membranes exhibited more macrovoids than the PES-PVP membranes, the relative flux for the PES-PVP was higher than that of the PES-SS (2.6-fold higher), which was attributed to leaching of PVP over time thus leading to unstable pore size and surface energy distributions within the materials. ATR-IR spectra confirmed this hypothesis where large variations in the intensity of the carbonyl peaks at ∼1680–1700 cm−1 for PES-PVP membranes were found between the reference sample and after 24 h of filtration. With regards to the anti-fouling ability of the PES-SS membranes, removal of both soluble (creatinine) and protein-bound uremic toxins (hippuric acid, HA and indoxyl sulfate, IS) were examined. The PES-SS membranes has ∼89% better creatinine removal efficiency than commercially available low-flux F8HPS (Fresenius) fibres, which was linked to the presence of a hydrophilic NVP (N-vinylpyrrolidone) block within the SS polymer, into the selective layer and pore surface of the PES-SS hollow fibres. These NVP were anchored by the hydrophobic BMA (N-butylmethacrylate) block of the SS into the membrane polymer matrix leading to less interaction between the membrane and the soluble solute.118,119 Interestingly, there is a low removal efficiency for hippuric acid which is only ∼59% which could be related to the fact that there is a higher protein interaction and adsorption of HA on the surface of the commercial membrane as compared to the PES-SS membrane. Also, HA binds to plasma proteins at a lower degree (∼30%) as compared to IS (∼90%). Hence, no significant difference was recorded for the IS removal for both the commercial and PES-SS membranes.
Adding hydrophilic polymers has been a common surface modification technique for hydrophobic membranes to minimize problems in fouling that will lead to cascade of reactions such as platelet activation and complement system activation. However, hydrophilic polymers tend to swell in water which is eluded in dialysis process as it affects the removal efficiency of membranes.111 Hence, exploration of blending synthetic polymer membranes with nanomaterials that can serve as inorganic fillers to avoid swelling is a growing trend in membrane technology.101 Carbon nanotubes (CNT) have been used as nanofillers for their nanoscale dimensions, which can offer high membrane specific surface area for interactions with contaminants or resources. In addition, both hydrophobic and hydrophilic blood components can interact with this material via π–π stacking and hydrogen bonding with oxygen-rich species (–COOH and –OH) in its chemical structure.111 In the first study that fabricated multi-walled carbon nanotube (MWCNT)/PES membranes, poly(vinyl pyrrolidone) (PVP) was the other additive that was incorporated to avoid MWCNT aggregation while reducing the elution of PVP during filtration (Table 2, entry 11).111 Urea, creatinine and lysozyme were filtered at a rate of 125 mL min−1 at the simulated blood solution side and 50 mL min−1 at the pure water solution side which gave higher removal efficiencies of 56.3% for urea, 55.1% for creatinine and 27.9% for lysozyme than the unmodified membranes. These values are relatively low compared to the performance of other mixed matrix membranes (MMMs) that incorporated inorganic nanofillers (Table 2, entries 3 and 6) and also filtered urea99 (∼97%) and lysozyme100 (∼36%). Possible reasons could be the tendency of PVP to leach after prolonged hours of filtration and the inadequate amount of added MWCNT (0.05–0.2 g) to sustain PVP in the matrix. Hence, another MWCNT/PES MMM membrane was proposed which used MWCNT-grafted poly(citric acid) (PCA) as another additive instead of PVP.101 Hydrophilic poly(citric acid) was grafted with MWCNTs since MWCNTs are more well-dispersed when grafted with polymers and other large organic molecules than grafting with acid alone.120 Addition of solid oxidized (O)-MWCNTs in the PES dope solution increased the solution viscosity from 5121 to 7797 mPa s. However, when the PCA-g-MWCNTs were added, a reduction of viscosity from 7797 mPa s to 6564 mPa s was recorded. This result is an implication of the inverse relationship between the viscosity and the average size or molecular weight distribution of MWCNT. An increase in the molecular weight of the PCA-g-MWCNTs led to greater shear rate thus lesser solution viscosity. As a result, larger voids were formed leading to a lower BSA absorption of 65.3% than the unmodified membrane.
3.3. Conclusions for membrane-based apheresis
Membrane apheresis is one of the most promising blood apheresis technology for its scalability and versatility. From an operational point of view, the focus of membrane apheresis has been directed towards minimizing surface fouling by applying such material modification methods to improve performance and conserve hospital resources by reducing procedure duration.25 This strategy is implemented by ensuring that an increased amount of blood toxins is removed to prevent compromising of the quality and purity of the extracted cells and of the filtered blood that will be returned to the patient at the end of the apheresis procedure. However, only few studies elaborated on the causes for these materials’ enhanced performance, in terms of chemical or surface/interfacial energy changes.74,84 A drive towards improved selectivity in blood apheresis is also required due to the large range and complexity of blood components exhibiting wide size distributions and biochemistries. Efficient speciation of certain components, which are the cause of diseases or of industrial value, may be currently hampered due to the presence of other similarly sized materials. One such example is related to the extraction of IgG and the competitive transfer of albumin during membrane apheresis.61,127 In addition, testing the haemocompatibility of the added material to ensure applicability on extracorporeal blood purification is not yet performed systematically, which should become a good practice with materials scientists to develop relevant materials solutions.128,1294. Adsorption-based apheresis
Sorbent apheresis technologies are developed to typically remove unwanted blood components using separation mechanisms beyond diffusion and convection.4 Specifically, selective sorbent materials for haemoperfusion have been developed since the 1970's for treatment of drug poisoning, to treat immunological diseases and remove uremic toxins.4,130 Some common uremic toxins that must be separated from blood are urea, creatinine and bilirubin. Urea and creatinine are toxic by-products of muscle and protein metabolism and high levels of these toxins implies impaired kidney function.131 Meanwhile, bilirubin is a by-product of red blood cell metabolism and high bilirubin levels indicates impaired liver function.132 Removal of bilirubin is necessary as this toxic metabolite can cause jaundice or condition where skin appears yellow.132 Separation of these toxins using these sorbent technologies is promising since, as opposed to centrifugation or membrane separation, only solutes of interest are retained through physical or chemical adsorption processes (schematic diagram on Fig. 3a).133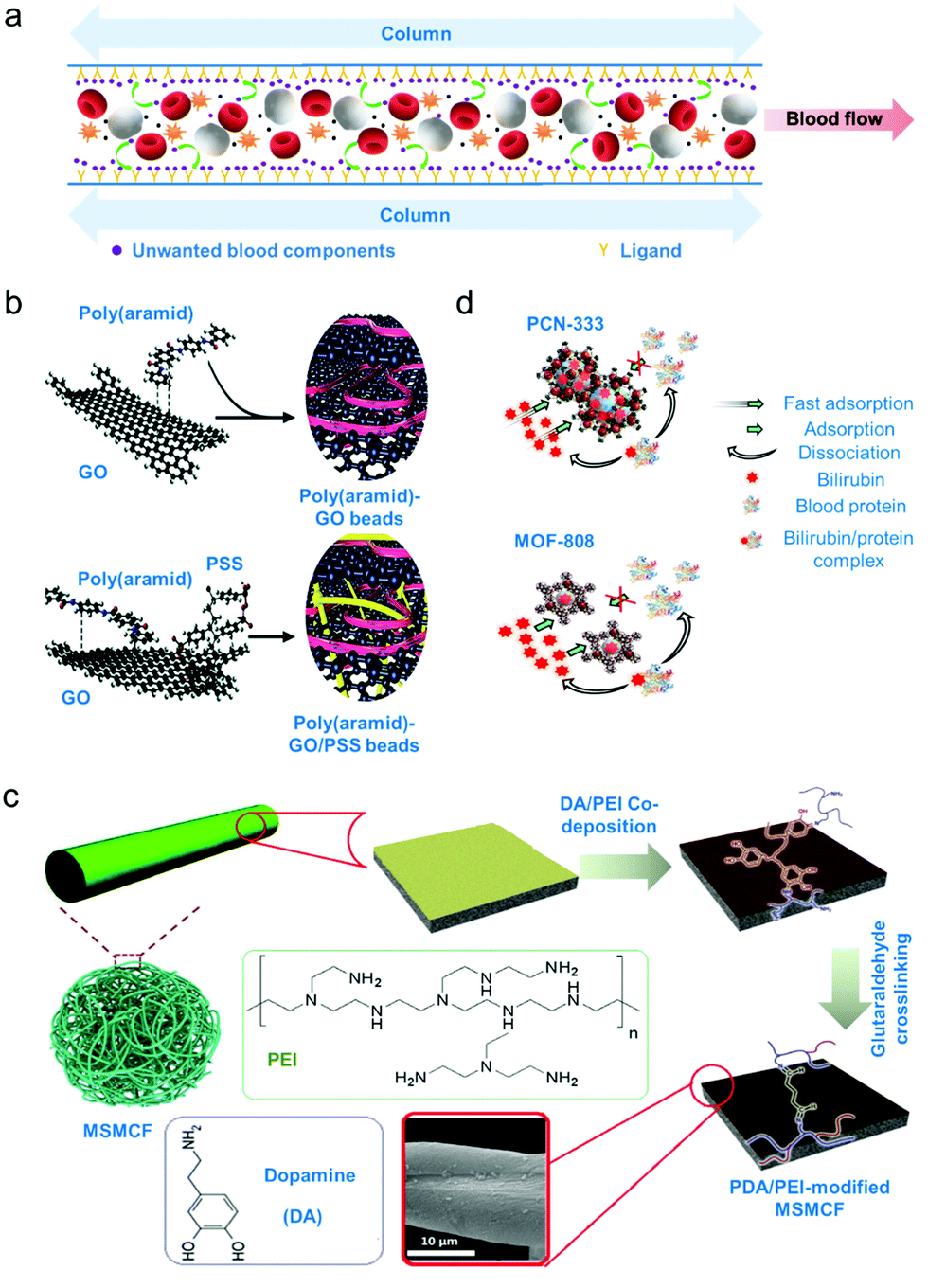 | ||
| Fig. 3 Adsorption-based apheresis. (a) Affinity-based separation. A ligand or sorbent attached to the column surface attracts the blood component of interest by affinity due to charge or shape complementarity. (b) An example of sorbent for direct haemoperfusion. Poly(aramid)–GO/PSS gel beads were crosslinked via π–π interactions.136 (c) An example of sorbent for plasma perfusion. Biocompatibility of sorbents is increased by coating with a hydrophilic polymer. Micrometre-size magnetic carbon fibres (MSMCF) are functionalized with a more stable poly(dopamine)/poly(ethylene imine) (PDA/PEI) coating by crosslinking the magnetically separated MSMCF/PDA/PEI mixture using glutaraldehyde to form PDA/PEI-modified MSMCF.137 (d) Metal–organic frameworks (MOFs) as adsorbent. Mechanism of capture for two cage-based MOFs were shown. High specificity toward bilirubin is demonstrated by dissociation of blood protein from the bilirubin/protein complex after capture.138 | ||
Leaching or release of sorbents over time however, remains a major challenge most especially if the material is not biocompatible, causing adverse effects to the patient such as chills, fever, cutaneous rush, thrombocytopenia and leukopenia.4 Strategies to minimize such adverse reactions or immunological responses from the body such as containing the adsorbents within specifically designed cartridges or modules are critical to allow for proper and safe operation and direct contact with the blood.134 Two solutions have emerged involving (i) separation of the plasma from the blood cells prior to circulation through the sorbent bed or (ii) the coating of the sorbent materials with a secondary bio- or haemocompatible material.135 The applicability and throughput of these strategies will be discussed in this section.
4.1. Sorbents for direct haemoperfusion
In direct perfusion columns, adsorbent cartridges are used to be in direct contact with blood within the column housing.139 In this apheresis mode, the micron-sized sorbent particles provide fast and efficient removal capacity of all the toxins they are designed to capture. However, direct haemoperfusion does not offer control over the fluid balance and the ability to remove other contaminants simultaneously, such as urea.140| Entry | Sorbent | Sorbent modification? | Type of sorbent modification | Pore size (nm) | BET surface area (m2 g−1) | Adsorbed blood component | Adsorption capacity (per gram or %) | Application |
|---|---|---|---|---|---|---|---|---|
| a Abbreviation: ND, no data. | ||||||||
| 1 | Carbon-based sorbent materials140 | No | NDa | 0.8–1 and 5 | 1250 | Creatinine | ∼14 mg | Direct haemoperfusion |
| Indoxyl sulfate (IS) | ∼3.7 mg | |||||||
| Hippuric acid (HA) | ∼13 mg | |||||||
| Interleukin-6 (IL-6) | ∼33 ng | |||||||
| Interleukin-8 (IL-8) | ∼80 ng | |||||||
| 2 | Magnetic carbon fibre137 | Yes | Coated with dopamine-poly(ethylene)imine (PEI) | 35 and 5200 | 146 | Bilirubin | 335 mg | Plasma perfusion |
| 3 | Nanostructured silica materials153 | Yes | Functionalization with 1-hydroxy-2-pyridinone (1,2-HOPO-SAMMS) | ∼6 | 900 | Free and chelated gadolinium (gadopentetate dimeglumine and gadodiamide) | 99% in 1 min (free); 95% in 30 min (gadodiamide) | Direct and plasma perfusion |
| 4 | Cage-based metal organic frameworks (MOFs)133 | No | ND | 1.84–5.5 (cage size) | 2060–4000 | Bilirubin | ∼1003 mg | Plasma perfusion |
| 5 | Kevlar-based composite gel beads142 | Yes | Crosslinking of sodium p-styrenesulfonate (SS) onto Kevlar-graphene oxide (GO) interface | 50000 (gel shell) |
ND | Creatinine | 97% | Direct haemoperfusion |
| Bilirubin | 79% | |||||||
| 6 | Non-ionic microporous poly(styrene adsorbent)156 | No | ND | 4.8 | 795 | Bilirubin | 50% | Plasma perfusion |
| 7 | Heparin-mimetic gel beads145 | Yes | Crosslinking of carrageenan and poly(acrylic acid) networks | 32000–22000 |
ND | Cu2+ | 560 mg | Direct haemoperfusion |
| Creatinine | 15 mg | |||||||
| Low density lipoprotein (LDL) | 18 mg | |||||||
| Bilirubin | 228 mg | |||||||
| 8 | Molecularly-imprinted biomimetic adsorbent157 | No | ND | 3.6 | 210 | α-Amanitin | ∼91–96% | Plasma perfusion |
| β-Amanitin | ∼90–92% | |||||||
| 9 | CCL-25 columns146 | No | ND | ND | ND | CCR-9 expressing cells | 98% | Direct haemoperfusion (leukoreduction) |
| 10 | Solid phase extraction adsorbent158 | No | ND | 2.75 | 183 | Ampicillin sodium | 13.5 mg | Plasma perfusion |
400 s, for the modified samples, and this trend indicated that platelet activation was inhibited. PTT remained at similar concentration for both the control and the modified beads at ∼12 s, which indicated that extrinsic coagulation pathways were not activated. Observing the serum concentrations of the anaphylatoxins C3a and C5a as indicators for complement activation, the serum C3a concentration only increased from 1750 ng mL−1 to 1900 ng mL−1 while the serum C5a concentration only decreased from 38 ng mL−1 to 32 ng mL−1 which is not clinically relevant. Since C5a is a more potent chemoattractant than C3a, K-GO/PSS did not induce inflammatory response from these mediators upon contact of blood to the beads which confirms its haemocompatibility.
4.2. Sorbents for plasma perfusion
Plasma perfusion adsorbents consist of a base material coated with an active functional ligand layer for selective coordination of plasma components.139 During plasma perfusion, the extracted plasma from a plasma separator is flushed through a column containing the adsorbents to prevent damage to the cellular components, known to reduce the adsorption efficiency of the process significantly.1394.3. Conclusions for adsorbent-based apheresis
Adsorbent-based apheresis is a technique that use affinity-based separation enabling specific molecule to capture through electrostatic interactions, hydrogen bonding or shape complementarity. Although adsorption is very efficient and promising overall selectivity, the process may still be improved, and novel surface chemistries must be developed to enable more selective capture mechanisms. The design of specific micro or nano pore distributions may also support performance, but pre-treatment is nevertheless required prior to operation to facilitate the adsorption process. Carbon-based sorbent materials are largely non-selective but represent an excellent scaffold material and their microstructures and pores may be tuned to allow for separation based on size.The haemocompatibility and toxicity of the ligand materials used to build chemical adsorbents remain a challenge by hindering direct blood contact and adding costly cartridge designs. The grafting of antibodies was discontinued for instance due to strong side effects triggering platelet activation.159 Hence, patient safety and disease indications should still be carefully monitored prior to selecting a base material for the sorbent.
5. Microfluidic-based apheresis
Microfluidic-based separation technologies emerged during the late 1970's and gained interest for their ability to separate very efficiently minute blood volumes of approximately up to one nanolitres with flow rates of up to 1 μL min−1.5,28,160 Microfluidics is mainly applied during collection of blood components for novel therapies as part of clinical trials and for isolation of blood components for disease prognosis.161 The main speciation mechanisms rely on size and deformability of blood cells, and can be controlled using the fluidic properties, flow rate and channel geometry (schematic diagram in Fig. 4a).5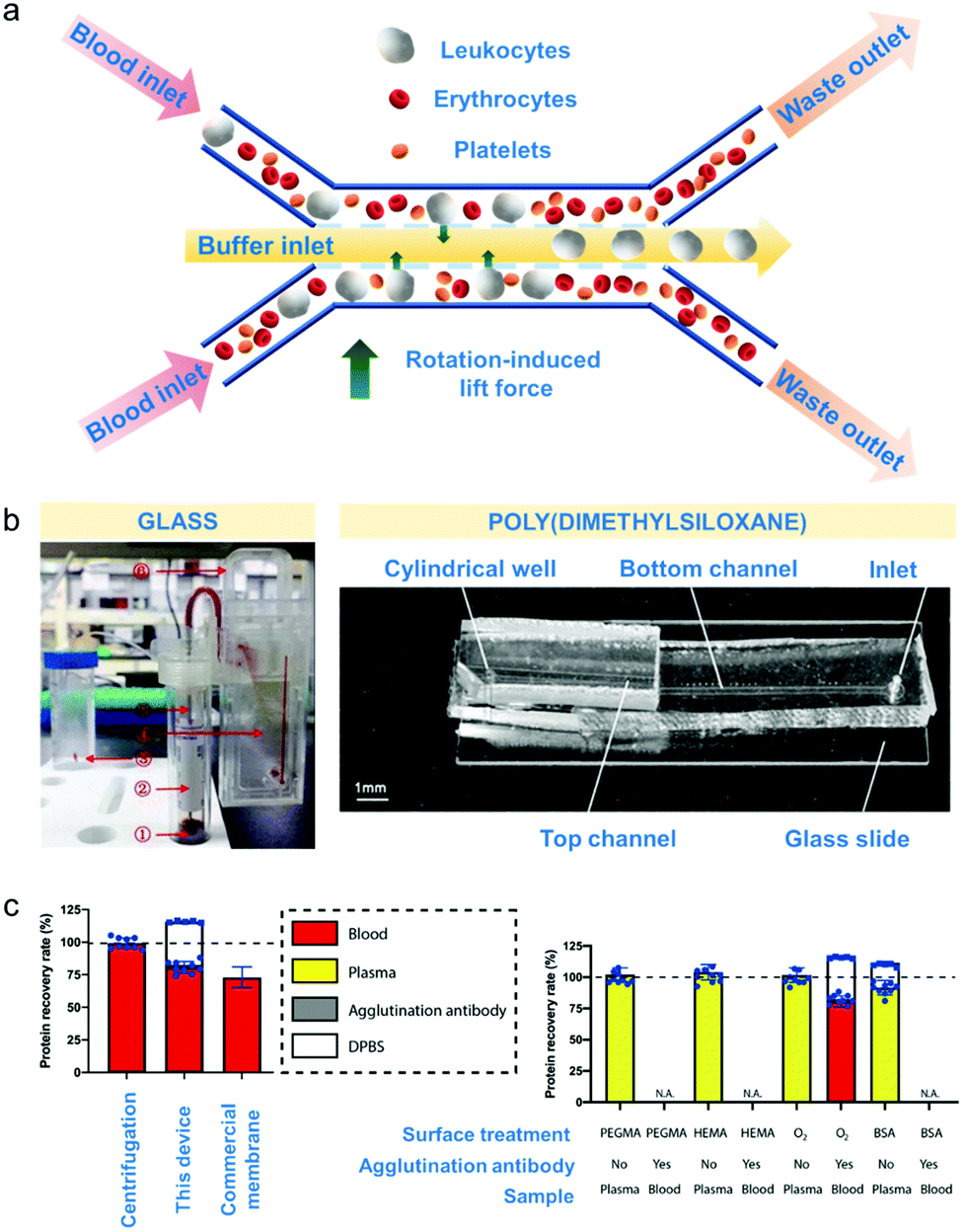 | ||
| Fig. 4 Microfluidic-based apheresis. (a) An example of microfluidic separation.165 As blood sample is introduced, the plasma (liquid component) exerts rotation-induced lift force on the sphere-like blood components such as leukocytes, erythrocytes and platelets which causes size-based lateral migration. (b) Materials used for device fabrication of apheresis chips.145,166 Poly(dimethylsiloxane) or PDMS is the most commonly used but glass is also employed for blood component separation. (c) Comparison of microfluidic apheresis device with already established apheresis techniques (centrifugation and membrane filtration). Total plasma protein recovery (left) is significantly higher than the protein recovery obtained after membrane filtration considering the amount of agglutination antibody protein incorporated with the device Effects of surface treatments such as treating with poly(ethylene glycol)methyl ether methacrylate (PEGMA) solution, hydroxyethylmethacrylate (HEMA) solution, bovine serum albumin (BSA) solution and oxygen plasma (right) show that BSA treatment gave the highest % recovery rate considering also the amount of agglutination antibody incorporated with the device. | ||
Separation by microfluidics technologies can be classified as active, passive or combined as in the case of label-free cell sorting mechanisms.28 In active approaches, external force fields are applied to effect particle motion during blood fractionation.162 Active separation techniques can be based on magnetophoresis, whereby particles move in a magnetic field, acoustophoresis, if particles migrate with soundwaves, and dielectrophoresis, upon non-uniform electric field application.28 Passive techniques do not require external forces to drive the separation and rely on the interaction of phenomena including diffusion, hydrodynamic forces such as inertial and Dean's forces, and the intrinsic physical properties of the cells including size and deformability.28,161,163
The fabrication material employed by most current microfluidic devices developed for diagnostic apheresis is poly(dimethylsiloxane) or PDMS – a transparent and inert polymer that is inexpensive.164 It is commonly used for purposes of blood component separation because aside from it is nontoxic, it entails very simple device fabrication.164 Other materials that can be used are glass and various thermoplastics, which can also be biocompatible but in the prototyping stages fabrication process fabrication can be more complex and have higher infrastructure demands than making PDMS devices by soft lithography.164 Hence, this section will focus on device design principles aside from the fabrication materials used and the surface modification applied towards these materials to increase separation efficiency, throughput and recovery (Table 5).
| Entry no. | Fabrication material | Surface modification for efficient separation | Active/passive | Separation mechanism | Injected sample | Sample volume/throughput (μL min−1) | Blood component separated | Separation efficiency (%) | Recovery (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Abbreviation: ND, no data. | |||||||||
| 1 | Poly(dimethylsiloxane) (PDMS) mixture | Treated with 1% (w/v) aqueous solution of mPEG-silane | Passive167 | Controlled incremental filtration | ∼2–3 mL freshly donated platelet-rich plasma | 1000 | Leukocytes | 99.9 | >80 (platelets) |
| 2 | Poly(dimethylsiloxane) | PDMS bonded to glass slide after oxygen plasma treatment | Passive170 | Microfiltration | 10 μL human blood samples | 1.5 | Leukocytes | 100 | 100 (leukocytes) |
| 3 | Poly(dimethylsiloxane) | PDMS bonded to glass slide after oxygen plasma treatment | Passive171 | Continuous flow diffusion | 1 mL whole blood | 5 | Leukocytes | >97 | ∼50 (erythrocytes) |
| 4 | Poly(dimethylsiloxane) | Glass compounded over a silicon substrate | Passive172 | Crossflow filtration | 100-Fold dilution of blood samples | 10 | Erythrocytes | 85.3 (pillar-type); 93.3 (weir-type) | ∼27 |
| 5 | Poly(dimethylsiloxane) | PDMS bonded to glass slide after oxygen plasma treatment | Passive177 | Hydrodynamic and inertial forces | Human whole blood with 4.3 × 108 cells per mL | Chip 1:15000 |
Erythrocytes | ∼100 | 93.2 (leukocytes) |
| Chip 2:6000 |
|||||||||
| 6 | Poly(dimethylsiloxane) | PDMS bonded to glass slide after oxygen plasma treatment | Passive165 | Size-dependent inertial migration | Spiked cancer cells | 300 | Circulating tumor cells | >99.3 | 83–93 |
| 7 | Glass | ND | Passive145 | Hydrodynamic separation | Spiked samples containing target cells | 200 | Circulating tumor cells | >93 | 99.9 (leukocytes) |
| 8 | Poly(dimethylsiloxane) for magnetic sorter; medical grade cyclic olefin copolymer (for inertial separation array device) | PDMS bonded to glass slide after oxygen plasma treatment | Active17 | Magnetic sorting | 64 mL whole blood spiked with cancer cells | 2800 | Circulating tumor cells | 86 | >86 (CTC) |
| Leukocytes | 99.97 | ||||||||
| Erythrocytes | >99.99 | ||||||||
| Platelets | >99.99 | ||||||||
| 9 | Poly(dimethylsiloxane) | PDMS bonded to glass slide after oxygen plasma treatment | Active185 | Electro-osmotic flow | 0.5 μL fresh human blood and 7.5 μL buffer | NDa | Plasma | 8–26 | NDa |
| 10 | Off-stoichiometry thiolenes | Treatment with PEGMA, HEMA, BSA and oxygen plasma | Passive189 | Capillary flow driven by blood agglutination | 90 μL whole blood | 17 | Plasma | 11 | >82 |
| 11 | Poly(dimethylsiloxane) | Oxygen plasma treatment | Passive166 | Gravity and capillary flow-driven | <10 μL whole blood | NDa | Plasma | 99.9 | <20 |
5.1. Microfluidic devices for leukapheresis
Current leukoreduction retains leukocytes via a membrane filtration process while allowing other blood components to pass with the flow through the pores prior to returning to the body. Membrane fouling, platelet activation and complement system activation are key challenges of membrane apheresis. In order to address this limitation, a high-throughput passive microfluidic device to remove leukocytes from platelet-rich plasma (PRP) in a continuous flow regime using controlled incremental filtration (CIF) was developed (Table 5, entry 1).167 Here, leukocytes were separated from platelets within the PRP by progressively syphoning clarified PRP away from the concentrated leukocyte flow stream. Poly(dimethylsiloxane) (PDMS) was the material used to fabricate the device replicas for separation runs but these replicas were treated with 1% (w/v) aqueous solution of methyl-end capped trimethoxy poly(ethylene glycol) silane (mPEG-silane) to reduce protein adsorption and fouling.168 Uncoated PDMS can lead to higher fibrinogen adsorption causing more immediate platelet activation.169 Using this device, 99.9% of leukocytes were removed based on the filtrate PRP while recovering >80% of the original platelets at volumetric throughputs of ∼1 mL min−1. These results suggest that CIF approaches will enable users in many fields to exploit microfluidic phenomena, even at throughputs nearing the flow rates used for continuous flow centrifugation (∼50 mL min−1).Integrated micro-fabricated filters were used for the separation of leukocytes (Table 5, entries 2 and 3) based on deformability.170,171 Initially, microfilters with gap sizes gradually decreasing from 15 to 3 μm were used.170 The distribution of captured cells was analysed for different flow rates, cell sizes and cell deformability. Changes in fluidic behaviour with variation in flow rates allowed for the collection of leukocytes of different sizes at different flow rates. At high sample flow rate (6 μL min−1), small leukocytes particularly lymphocytes with a size of 7 μm permeated across the smallest microfilter gap size of 3 μm, whereas at lower flow rates (1.5 μL min−1), lymphocytes were captured in a 6 μm microfilter gap size. Although this system offers a low sample throughput compared to the CIF device,167 it does offer a high separation efficiency for WBCs (100%) with regard to other blood components such as cancer cells and RBCs.
Another filter-integrated device based on diffusion in a continuous flow was developed,171 where the geometry of the diffuser was optimized to ensure that the fluid distributes equally across the individual sieve elements. When a flared geometry was used, a separation efficiency of >97% was achieved for leukocytes at a higher sample throughput (5 μL min−1) than previously reported for microfluidic devices (1.5 μL min−1). Hence, the optimised geometry supported greater throughput. The device is versatile since it can also be used for plasma fractionation by reducing the height of filter elements from 2.5 μm to <0.5 μm. Leukocyte and erythrocyte separation however, depends mainly on pressure gradient across filter elements which is a function of operating flow rate and in this case, higher flow rates will permit stronger interaction between the leukocytes and erythrocytes with the filter elements than lower flow rates.
5.2. Microfluidic devices for erythrocytapheresis
Erythrocytes may also be separated from whole blood for blood donation or red cell exchange.41 Two types of passive microfluidic device that are based on crossflow filtration were fabricated to separate RBCs from whole blood (Table 5, entries 4 and 5).172 Microstructures including as micro-pillar arrays and micro-weirs were used in a filtration device. PDMS was still the main fabrication material of choice but in the first chip (Table 5, entry 4),173 glass is compounded with PDMS over a silicon substrate. Considering the microstructures incorporated with the device, the rationale for the material of choice could be the fact that glass compensates for the elasticity of PDMS which may affect accuracy of the fabricated channel dimensions.174 In terms of the mode of membrane filtration, cross flow filtration was used as opposed to dead end filtration to ensure large particles remain suspended while the small particles permeate through the filter; in dead-end filtration large particles may clog or jam the pores.175,176 Consequently, by employing the design principle and fabrication material, separation efficiencies were greater than 95% and 27% for RBC and WBC, respectively.Besides incorporating filters within the microfluidic device, a passive microfluidic chip driven by the inertial migration effect was also developed to separate and enrich leukocytes from whole blood.177 The chip design incorporated 10 switchback curve channels bridged by straight channels for inducing inertial migration effects to concentrate the blood cells while the curved channels were integrated for inducing Dean flow – a secondary cross-sectional force field characterized by the two counter-rotating vortices perpendicular to the direction of the flow for further separation of cells based on their sizes.178 This configuration led to the very high erythrocyte separation efficiency with almost no erythrocyte loss (∼100% capture efficiency) while still recovering ∼93.2% of the leukocytes from whole blood.
5.3. Microfluidic devices for circulating tumour cell (CTC) isolation
Circulating tumour cells (CTCs) are cancer cells that migrate away from primary site and are distributed throughout the body using the bloodstream.145 CTCs are considered attractive biomarkers for cancer diagnosis/prognosis as sampling blood may be less invasive than taking tumour biopsies.17 However, their very low concentration (1–100 CTCs per mL of blood) makes their detection challenging.179 So far, CTCs can be separated based on their physical properties such as size and deformability since CTCs have comparable sizes as other blood cell components.180 Filtration-based separation can address challenges regarding low expression of protein marker for CTCs (epithelial cell adhesion molecule or EpCAM) however, filtration will not be able to extract CTCs from other blood components with similar sizes. Hence, filter-free microfluidics provides an attractive platform for CTC isolation.181 Affinity-based separation can also be done for these blood components.181 The most clinically-validated current commercial technology available to separate EpCAM from whole blood which implements affinity-based separation is the FDA-approved CellSearch® System which contains ferrofluids with EpCAM antibodies. The specificity towards EpCAM clearance is high and ranging between 89 and 93% using this technique.182,183 However, the sensitivity remains low with a very broad result distribution, and on the order of 27 to 70%.182,183 Hence, other approaches are still being explored to separate EpCAM from blood.A passive, multi-flow microfluidic channel was used to isolate CTCs in lung cancer patients (Table 5, entry 6).165 This device was designed for separation based on size-dependent inertial migration. Rotation-induced lift force (FΩ) was the predominant force behind the size-based lateral migration and subsequent CTC isolation. The blood sample and phosphate buffer were introduced using a sample inlet and buffer inlet channel, merging to forming a sandwiched flow configuration in the main channel. The CTCs were collected from the inner outlet whereas other cells outside the size cut-off were collected from the outer outlet. To assess device performance, cancer cells spiked with poly(styrene) particles were used. Consequently, >99% of the spiked samples were isolated with purity of >87% and recovery of >93%. However, red blood cell contamination is an issue during sample preparation since the device isolated CTCs directly from whole blood thus additional cell lysis step for RBCs was required before separation. Samples were also not directly analysed by the instrument, which required immune-phenotyping post speciation.
Meanwhile, a passive, glass wedge-shaped microchip was also used to isolate CTCs from gastric cancer patients (Table 5, entry 7 and Fig. 4b).145 The wedge-shaped chamber provided functionality to the device by gradually decreasing the chamber height which allowed a wide outlet causing blood cells to not clog the channels. In addition, a pillar-array fluid distributor made the blood velocity distribution even across the entire wedge-shaped microchannel which enhanced the anti-clogging effect of the wedge-shaped chamber. RBC would flow and randomly rotate through the micro-chamber when the height of the micro-chamber was larger than the size of RBC but if otherwise, RBC would be oriented flat due to their disk-like shape during sample processing. Interestingly, WBCs would also go through this microchip even the cut-off size of this chip is smaller than the diameter of WBC due to the size-space amplification by the wedge-shaped micro chamber. This design resulted in a capture efficiency of >93% for CTCs when processing the optimal blood volume of 2 mL at 200 μL min−1.
Recognising the potential of magnetic sorting, active microfluidic devices were demonstrated for the isolation of CTCs from whole blood (Table 5, entry 8).17 The magnetic sorter was fabricated by using medical grade cyclic olefin copolymer for the inertial separation array device. PDMS was annealed to a glass slide to avoid further deformations caused by baking or heating during fabrication of the magnetic sorter. Meanwhile, cyclic olefin copolymer (COC) was used for the main chip. COCs are inexpensive polymers derived from cyclic olefin monomers and ethene.184 They are also transparent like PDMS except they have low elasticity and hydrophobic. Protein adsorption could be an issue on materials with hydrophobic surface but in this study, treatments to make the surface of the array device more hydrophilic were not applied. COC has the advantage of being a thermoplastic, hence devices can be made with high throughput by injection moulding. Separation relied solely on the device design rather than the material and occurred when the field gradient was intensified within the sorting channels using channels filled with “soft” (annealed) iron particles to act as magnetic micro-lenses. An increase in the applied magnetic field on inertially focused stream of cells efficiently depleted many leukocytes bound to magnetic beads, yielding 86% for CTCs and separating ∼99% for leukocytes, erythrocytes and platelets respectively. This result was supported by cell counting and phase contrast microscopy experiments. Compared to the previous passive microfluidic devices for blood component separation, the high separation efficiencies here is specific for only one or few blood components.
5.4. Microfluidic devices for plasmapheresis
The presence of important biomarkers and proteins in blood plasma makes plasma a desirable body fluid for clinical analysis.185 In order to achieve efficient separation for point-of-care applications, several different techniques were already applied such as geometrical obstructions,186 cross-flow filtration187 and bifurcation law, referred to as the Zweifach–Fung effect188 or the process wherein shear is applied on cells driving them towards the centre of the channels of the microfluidic device forming a cell-free layer that can be captured at points where these channels divide (bifurcation points).160Instead of hydrodynamic pumps, the electro-osmotic flow (EOF) has also been used to drive the fluid flow for plasmapheresis in micro-channels185 (Table 5, entry 9). The EOF is generated from the walls of the device when an electric field is applied. As the magnitude of the EOF correlates linearly with the electric field strength, fluidic separation mechanisms such as the Zweifach–Fung effect can be realised manipulating the electric field instead of a pump. By employing this separation mechanism, a yield of around 26% of blood plasma was collected from whole blood, with nearly 100% of the blood cells retained in the main channel. It should be noted that the volume of fresh human blood in the micro-channels was only 0.5 μL (diluted with 7.5 μL buffer).
Plasmapheresis was also realised using passive interlocked micropillar scaffolds fabricated from off-stoichiometry thiol-enes (OSTE)189 (Table 5, entry 10 and Fig. 4c). OSTE is a group of polymer synthesized via step polymerization of thiol and -ene groups.190 By offsetting the stoichiometric ratios from 1:1, reactive groups in this polymer could be predicted making it easier for surface functionalization.190 Separation in this device happens when the blood sample clots upon material contact caused by deliberately adding an agglutination antibody to the material. The clots drastically minimize the surface area but allow for passage of the blood plasma through capillary action. When compared with previously established blood separation techniques such as centrifugation and membrane filtration (Fig. 4c, left), protein recovery rate improved from ∼73% using membrane filtration191 to ∼82% using this device. Effects of surface treatments using poly(ethylene glycol)methyl ether methacrylate (PEGMA) solution, hydroxyethylmethacrylate (HEMA) solution, bovine serum albumin (BSA) solution and oxygen plasma was also monitored and results showed that BSA treatment further improved the protein recovery rate to 91.7%. However, it should be accounted for that BSA is a protein itself and the result of measurement of protein recovery using BCA (bicinchoninic) protein assay in this study could be a false positive.
A passive, capillary flow-driven microfluidic device used oxygen plasma to induce a wettability gradient in the cylindrical well fabricated between the hydrophilic top and bottom channels of a PDMS microfluidic device166 (Table 5, entry 11 and Fig. 4b). Upon introduction of blood sample within the hydrophilic device inlet, the difference in the Young-Laplace pressure induced a flow, with capillary action pulling the fluid front to the hydrophobic patch. The increasing contact angle at the hydrophobic patch decreased this force, significantly decreasing the fluid velocity. As the velocity decreased, the cells sedimented due to their higher density while the blood plasma continued to slowly rise, partially facilitated by its lower viscosity. Hence, the combination of gravity and capillarity underpins the plasma separation. When <10 μL of blood sample was injected in the device, 2 μL of plasma was obtained in 15 min with a separation efficiency of 99.9%. It is to be considered, however, that modification must be done for the device to be more applicable in continuous flow mode. The implementation of this approach aimed in this work towards diagnostics still faces challenges and further developments of the geometry of the system must be enabled to control the flow dynamics and facilitate continuous flow operation.
5.5. Conclusions for microfluidic-based apheresis
Microfluidic devices have great potential for use in blood apheresis, offering >99% capture efficiencies for specific blood components. While parallel processing have enabled upscaling of microfluidic processing, microfluidic apheresis is currently more attractive in research since it can operate on very low blood volumes. Plasma separations, in particular, are highly sought after for diagnostic purposes. While some microfluidic approaches have demonstrated volumetric throughputs nearing those of ‘traditional’ approaches (1 mL min−1 for CIF vs. 50 mL min−1 for centrifugation), significant technical advances in parallel processing are required to progress these technologies from research and diagnostics to the collection of blood products at a commercial scale. In terms of material development, other haemocompatible polymers aside from PDMS, COC and OSTE such as poly(methyl methacrylate) (PMMA) must be explored to fully tap the potential of these devices in blood component extraction while considering the polymer properties required for device fabrication. Surface modification such as incorporating zwitterionic polymers (e.g. carboxybetaine or sulfobetaine) or even grafting anticoagulants such as heparin on device channels could be helpful in avoiding fouling as in other apheresis techniques such as membrane filtration.6. Perspectives
Apheresis techniques, which may be utilized for donor blood component collection, removal of disease-provoking elements and overexpressed blood components for blood cell isolation are critical to bio-products separation and pathogen removal. Advantages and limitations for each technique are summarized in Table 6. Centrifugation, membranes, adsorbents and microfluidic devices represent the most established and relevant approaches to this mean and were discussed in detail in this review.| Apheresis technique | Advantages | Limitations |
|---|---|---|
| Centrifugal-based | Most utilized and established | High variabilities in separation time, low reproducibility and selectivity; strong mechanical shear stress inducing platelet activation |
| Membrane-based | Scalable and versatile | Low selectivity especially towards components of same size |
| Adsorbent-based | Highly selective | Low hemocompatibility of ligand materials |
| Microfluidic-based | Highly specific with capture efficiencies >99%; low processing volumes required | Fouling of microfluidic components |
The gaps of current apheresis technologies are primarily linked to yet incomplete or non-cost-effective removal or separation efficiency of bio-products, potential side effects to patients and damage induced to cells during the extraction processes. Alternative processes benefiting from advanced materials with tuned interactions with blood components represent a key avenue to limit biofouling of surfaces and membranes and prevent inopportune cell activation. The scalability of such devices is often limited and implementing simpler fabrication methods could lead to improvements towards more scalable technologies. The impact of blood coagulation proteins for blood apheresis are not prioritized thus preventing the massive application of these technologies in clinical settings.
Hence, future work must be directed on the interfacial chemistries between the blood and the material it interacts with to have a better understanding on the exact mechanism of contact, the functional groups that react and the configuration of the blood molecule that interacted with these functional groups. Future studies on these aspects are especially relevant now when there is an increasing trend towards incorporation of emerging materials whose properties are not yet fully understood, such as inorganic nanofillers incorporated with membrane and sorbents for enhanced mechanical property.
Biocompatible materials such as amphiphilic/zwitterionic polymers can also be explored as coating materials for these devices for enhanced haemocompatibility. Extensive haemocompatibility testing inclusive on the effects of the selected apheresis technology on coagulation proteins must be done as part of material development to aid in the translation of these technologies towards actual apheresis procedures. By comparing the separation efficiencies and percent recoveries of each technique, this review offers a selection of the possible ways of addressing the limitations in blood apheresis giving emphasis on the effects of surface modification methods and material design to enhance separation performance.
Author contributions
Christine Jurene O. Bacal: conceptualization, writing – original draft. Ludovic F. Dumee: conceptualization, supervision, writing – review and editing. Karen M. Dwyer: supervision, writing – review and editing. James W. Maina, Harshal H. Nandurkar, Yung Chang, Maryam T. Khaleel and Rosanne M. Guijt: writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. F. D. acknowledges financial support from the Discovery Early Career Researcher Award (DECRA) from the Australian Research Council (ARC) (180100130) and from Khalifa University of Science and Technology under project RC2-2019-007.References
- J. Hardwick, ISBT Sci. Ser., 2008, 3, 148–176 CrossRef.
- M. El Ekiaby, ISBT Sci. Ser., 2017, 12, 87–90 CrossRef.
- C. Ronco and W. R. Clark, Nat. Rev. Nephrol., 2018, 14, 394–410 CrossRef CAS PubMed.
- W. R. Clark, F. Ferrari, G. La Manna and C. Ronco, Contrib. Nephrol., 2017, 190, 43–57 CrossRef PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- What is Apheresis? https://www.petermac.org/services/treatment/haematological-treatments/apheresis/what-apheresis, (accessed October 13, 2021).
- J. L. Winters, Hematology Am. Soc. Hematol. Educ. Program, 2012, 2012, 7–12 CrossRef PubMed.
- N. Bose and H. Kanzariya, Indian J. Anaesth., 2014, 58, 672–678 CrossRef PubMed.
- M. E. Williams and R. A. Balogun, Clin. J. Am. Soc. Nephrol., 2014, 9, 181–190 CrossRef CAS PubMed.
- C. Klaus, B. Plaimauer, J.-D. Studt, F. Dorner, B. Lämmle, P. M. Mannucci and F. Scheiflinger, Blood, 2004, 103, 4514 CrossRef CAS PubMed.
- E. Sumida, Y. Iwasaki, K. Akiyoshi and S. Kasugai, J. Pharmacol. Sci., 2006, 101, 91–97 CrossRef CAS PubMed.
- N. Worel, B. Mansouri Taleghani and E. Strasser, Transfus. Med. Hemother., 2019, 46, 394–406 CrossRef PubMed.
- R. A. Ward, J. K. Leypoldt, W. R. Clark, C. Ronco, G. J. Mishkin and E. P. Paganini, Semin. Dial., 2001, 14, 160–174 CrossRef CAS PubMed.
- S. Gosselin, D. N. Juurlink, J. T. Kielstein, M. Ghannoum, V. Lavergne, T. D. Nolin and R. S. Hoffman, Clin. Toxicol., 2014, 52, 856–867 CrossRef CAS PubMed.
- A. M. Grunbaum, S. Kazim, M. Ghannoum, M.-A. Kallai-Sanfacon, R. Mangel, E. Villeneuve and S. Gosselin, Clin. Toxicol., 2013, 51, 270–271 Search PubMed.
- A. Urbansky, P. Ohlsson, A. Lenshof, F. Garofalo, S. Scheding and T. Laurell, Sci. Rep., 2017, 7, 17161 CrossRef PubMed.
- A. Mishra, T. D. Dubash, J. F. Edd, M. K. Jewett, S. G. Garre, N. M. Karabacak, D. C. Rabe, B. R. Mutlu, J. R. Walsh, R. Kapur, S. L. Stott, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16839 CrossRef CAS PubMed.
- A. P. Sanchez, R. Cunard and D. M. Ward, J. Clin. Apher., 2013, 28, 20–29 CrossRef PubMed.
- M. Kersaudy-Kerhoas and E. Sollier, Lab Chip, 2013, 13, 3323–3346 RSC.
- S. C. Gifford, B. C. Strachan, H. Xia, E. Vörös, K. Torabian, T. A. Tomasino, G. D. Griffin, B. Lichtiger, F. M. Aung and S. S. Shevkoplyas, PLoS One, 2018, 13, e0190827 CrossRef PubMed.
- L. Wiegmann, D. A. De Zélicourt, O. Speer, A. Muller, J. S. Goede, B. Seifert and V. Kurtcuoglu, Front. Physiol., 2017, 8, 731 CrossRef PubMed.
- S. Fukuda and G. W. Schmid-Schönbein, J. Leukoc. Biol., 2002, 72, 133–139 CAS.
- A. Seltsam, Front. Med., 2017, 4, 219 CrossRef PubMed.
- G. de Sousa and J. Seghatchian, Transfus. Apher. Sci., 2006, 34, 77–83 CrossRef.
- B. Puppe and E. J. Kingdon, Clin. Kidney J., 2014, 7, 201–205 CrossRef CAS PubMed.
- C. Hafer, P. Golla, M. Gericke, G. Eden, G. Beutel, J. J. Schmidt, B. M. W. Schmidt, S. De Reys and J. T. Kielstein, Int. Urol. Nephrol., 2016, 48, 133–138 CrossRef PubMed.
- M. E. Janssens and S. Wakelin, Eur. Oncol. Haematol., 2018, 14, 105–109 CrossRef.
- S. O. Catarino, R. O. Rodrigues, D. Pinho, J. M. Miranda, G. Minas and R. Lima, Micromachines, 2019, 10(9), 593 CrossRef PubMed.
- J. Korsak and Z. Wańkowicz, Blood Purif., 2016, 41, 1–10 CrossRef CAS PubMed.
- D. M. Ward, J. Clin. Apher., 2011, 26, 230–238 CrossRef PubMed.
- A. G. Perez, J. F. Lana, A. A. Rodrigues, A. C. Luzo, W. D. Belangero and M. H. Santana, ISRN Hematol., 2014, 2014, 176060 Search PubMed.
- J. Schwartz, A. Padmanabhan, N. Aqui, R. A. Balogun, L. Connelly-Smith, M. Delaney, N. M. Dunbar, V. Witt, Y. Wu and B. H. Shaz, J. Clin. Apher., 2016, 31, 149–338 Search PubMed.
- J. Schwartz, J. L. Winters, A. Padmanabhan, R. A. Balogun, M. Delaney, M. L. Linenberger, Z. M. Szczepiorkowski, M. E. Williams, Y. Wu and B. H. Shaz, J. Clin. Apher., 2013, 28, 145–284 CrossRef PubMed.
- Z. M. Szczepiorkowski, J. L. Winters, N. Bandarenko, H. C. Kim, M. L. Linenberger, M. B. Marques, R. Sarode, J. Schwartz, R. Weinstein and B. H. Shaz, J. Clin. Apher., 2010, 25, 83–177 CrossRef PubMed.
- J. E. Mancuso, A. Jayaraman and W. D. Ristenpart, PLoS One, 2018, 13, e0203270 CrossRef PubMed.
- D. M. Dohan Ehrenfest, N. R. Pinto, A. Pereda, P. Jiménez, M. D. Corso, B.-S. Kang, M. Nally, N. Lanata, H.-L. Wang and M. Quirynen, Platelets, 2018, 29, 171–184 CrossRef CAS PubMed.
- E. S. Lourenço, G. G. Alves, R. de Lima Barbosa, C. N. Spiegel, R. C. de Mello-Machado, S. Al-Maawi, S. Ghanaati and C. F. de Almeida Barros Mourão, J. Biomed. Mater. Res., Part B, 2021, 109, 60–68 CrossRef PubMed.
- X. H. Zheng, C. Cui, X. X. Zhou, Y. X. Zeng and W. H. Jia, Chin. J. Cancer, 2013, 32, 667–672 CrossRef CAS PubMed.
- M. Lu, D. L. Lezzar, E. Vörös and S. S. Shevkoplyas, J. Blood Med., 2019, 10, 37 CrossRef CAS PubMed.
- R. Dhurat and M. Sukesh, J. Cutan. Aesthet. Surg., 2014, 7, 189–197 CrossRef PubMed.
- D. Basu and R. Kulkarni, Indian J. Anaesth., 2014, 58, 529–537 CrossRef CAS PubMed.
- E. Hussein and A. Enein, Lab. Med., 2014, 45, 238–243 CrossRef PubMed.
- L. Bongo, R. Bhatia and L. S. Lamb, Jr., Blood, 2018, 132, 5693 CrossRef.
- P. Kes, M. E. Janssens, N. Bašić-Jukić and M. Kljak, Transfusion, 2016, 56, 3065–3072 CrossRef CAS PubMed.
- M. Brambilla, L. Rossetti, C. Zara, P. Canzano, P. L. A. Giesen, E. Tremoli and M. Camera, Platelets, 2018, 29, 406–414 CrossRef CAS PubMed.
- H. Fernandez-Munoz, C. Castilla-Llorente, E. M. Plaza, C. Martinez-Millan, I. Heras, P. Iniesta, M. L. Amigo, F. Ferrer-Marin, M. J. Candela, M. L. Lozano, V. Vicente and J. Rivera, Blood Transfus., 2018, 16, 273–278 Search PubMed.
- A. K. Pulsfort, T. P. Wolter and N. Pallua, Ann. Plast. Surg., 2011, 66, 292–295 CrossRef CAS PubMed.
- R. Landesberg, M. Roy and R. S. Glickman, J. Oral Maxillofac. Surg., 2000, 58, 297–300 CrossRef CAS PubMed ; discussion 300–291.
- O. Bausset, L. Giraudo, J. Veran, J. Magalon, J. M. Coudreuse, G. Magalon, C. Dubois, N. Serratrice, F. Dignat-George and F. Sabatier, Biores. Open Access, 2012, 1, 115–123 CrossRef CAS PubMed.
- E. Anitua, J. J. Aguirre, J. Algorta, E. Ayerdi, A. I. Cabezas, G. Orive and I. Andia, J. Biomed. Mater. Res., Part B, 2008, 84, 415–421 CrossRef PubMed.
- M. Punzel, A. Kozlova, A. Quade, A. H. Schmidt and R. Smith, Vox Sang., 2017, 112, 586–594 CrossRef CAS PubMed.
- C. H. Jo, Y. H. Roh, J. E. Kim, S. Shin and K. S. Yoon, J. Oral Implantol., 2013, 39, 525–532 CrossRef PubMed.
- J. Cadamuro, C. Mrazek, A. B. Leichtle, U. Kipman, T. K. Felder, H. Wiedemann, H. Oberkofler, G. M. Fiedler and E. Haschke-Becher, Biochem. Med., 2018, 28, 010704 CrossRef PubMed.
- D. M. Pegtel, K. Cosmopoulos, D. A. Thorley-Lawson, M. A. van Eijndhoven, E. S. Hopmans, J. L. Lindenberg, T. D. de Gruijl, T. Würdinger and J. M. Middeldorp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6328–6333 CrossRef CAS PubMed.
- G. Filardo, E. Kon, M. T. Pereira Ruiz, F. Vaccaro, R. Guitaldi, A. Di Martino, A. Cenacchi, P. M. Fornasari and M. Marcacci, Knee Surg., Sports Traumatol., Arthrosc., 2012, 20, 2082–2091 CrossRef PubMed.
- N. Tynngård, Transfus. Apher. Sci., 2009, 41, 97–104 CrossRef PubMed.
- H. J. Gurland, M. J. Lysaght, W. Samtleben and B. Schmidt, Int. J. Artif. Organs, 1984, 7, 35–38 CrossRef CAS PubMed.
- C. Okafor, D. M. Ward, M. H. Mokrzycki, R. Weinstein, P. Clark and R. A. Balogun, J. Clin. Apher., 2010, 25, 240–249 CrossRef PubMed.
- A. Bruil, T. Beugeling, J. Feijen and W. G. van Aken, Transfus. Med. Rev., 1995, 9, 145–166 CrossRef CAS PubMed.
- A. Sueoka, Ther. Apher., 1998, 2, 78–86 CrossRef CAS PubMed.
- A. Sueoka, Ther. Apher., 1997, 1, 135–146 CrossRef CAS PubMed.
- T. Xiang, C. Cheng and C. Zhao, J. Membr. Sep. Technol., 2014, 3, 162 CrossRef.
- J. F. Hester, Doctor of Philosophy in Polymers Dissertation, Massachusetts Institute of Technology, 1993.
- A. Higuchi, K. Sugiyama, B. O. Yoon, M. Sakurai, M. Hara, M. Sumita, S.-i. Sugawara and T. Shirai, Biomaterials, 2003, 24, 3235–3245 CrossRef CAS PubMed.
- L. Y. Ng, A. W. Mohammad, C. P. Leo and N. Hilal, Desalination, 2013, 308, 15–33 CrossRef CAS.
- C.-C. Lien, P.-J. Chen, A. Venault, S.-H. Tang, Y. Fu, G. V. Dizon, P. Aimar and Y. Chang, J. Membr. Sci., 2019, 584, 148–160 CrossRef CAS.
- P.-S. Liu, Q. Chen, S.-S. Wu, J. Shen and S.-C. Lin, J. Membr. Sci., 2010, 350, 387–394 CrossRef CAS.
- L. Zhu, F. Liu, X. Yu and L. Xue, ACS Appl. Mater. Interfaces, 2015, 7, 17748–17755 CrossRef CAS PubMed.
- S. Singh and A. Kumar, Biotechnol. J., 2009, 4, 1140–1151 CrossRef CAS PubMed.
- A. Bruil, H. A. Oosterom, I. Steneker, B. J. Al, T. Beugeling, W. G. van Aken and J. Feijen, J. Biomed. Mater. Res., 1993, 27, 1253–1268 CrossRef CAS PubMed.
- A. Bruil, W. G. Van Aken, T. Beugeling, J. Feijen, I. Steneker, J. G. Huisman and H. K. Prins, J. Biomed. Mater. Res., 1991, 25, 1459–1480 CrossRef CAS PubMed.
- S. Teraoka, Y. Hayasaka, H. Shoji, M. Mineshima, T. Suzuki, Y. Sasaki, M. Sato, T. Hoshino, K. Era and T. Tojinbara, et al. , ASAIO Trans., 1988, 34, 608–612 CAS.
- R. R. Sharma and N. Marwaha, Asian J. Transfus. Sci., 2010, 4, 3–8 CrossRef CAS PubMed.
- A. W. Neumann, D. R. Absolom, C. J. van Oss and W. Zingg, Cell Biophys., 1979, 1, 79–92 CrossRef CAS PubMed.
- W. A. Zisman, Contact Angle, Wettability, and Adhesion, American Chemical Society, 1964, ch. 1, vol. 43, pp. 1–51 Search PubMed.
- J. Kjeldsen-Kragh and E. Golebiowska, Vox Sang., 2002, 82, 127–130 CrossRef CAS PubMed.
- R. R. Gammon, S. A. Strayer, N. L. Avery and P. D. Mintz, Ann. Clin. Lab. Sci., 2000, 30, 195–199 CAS.
- I. Steneker and J. Biewenga, Transfusion, 1991, 31, 40–46 CrossRef CAS PubMed.
- S. Dzik, Transfus. Med. Rev., 1993, 7, 65–77 CrossRef CAS PubMed.
- S. H. Ye, J. Watanabe and K. Ishihara, J. Biomater. Sci., Polym. Ed., 2004, 15, 981–1001 CrossRef CAS PubMed.
- M.-C. Sin, S.-H. Chen and Y. Chang, Polym. J., 2014, 46, 436–443 CrossRef CAS.
- PuriBlood, http://www.puriblood.com/index.php?lang=en, (accessed March 13, 2021).
- A. L. Lewis, R. N. Freeman, R. P. Redman, L. A. Tolhurst, L. C. Kirkwood, D. M. Grey and T. A. Vick, J. Mater. Sci.: Mater. Med., 2003, 14, 39–45 CrossRef CAS PubMed.
- S. Clarke, M. C. Davies, C. J. Roberts, S. J. Tendler, P. M. Williams, V. O'Byrne, A. L. Lewis and J. Russell, Langmuir, 2000, 16, 5116–5122 CrossRef CAS.
- A. Higuchi, S.-i. Yamamiya, B. O. Yoon, M. Sakurai and M. Hara, J. Biomed. Mater. Res., Part A, 2004, 68A, 34–42 CrossRef CAS PubMed.
- Y.-W. Chen, A. Venault, J.-F. Jhong, H.-T. Ho, C.-C. Liu, R.-H. Lee, G.-H. Hsiue and Y. Chang, J. Membr. Sci., 2017, 537, 209–219 CrossRef CAS.
- E. Gérard, E. Bessy, G. Hénard, T. Verpoort and J. Marchand-Brynaert, J. Biomed. Mater. Res., Part B, 2012, 100, 1513–1523 CrossRef PubMed.
- F. Sadeghi, A. Ajji and P. J. Carreau, J. Membr. Sci., 2007, 292, 62–71 CrossRef CAS.
- F. Sadeghi, A. Ajji and P. J. Carreau, Polym. Eng. Sci., 2007, 47, 1170–1178 CrossRef CAS.
- Y. Cao, J. Liu, R. Zhong, Q. Yu and H. Wang, Artif. Cells, Blood Substitutes, 2012, 40, 317–325 CrossRef CAS PubMed.
- E. J. Kim, G.-D. Yeo, C.-M. Pai and I.-K. Kang, J. Biomed. Mater. Res., Part B, 2009, 90B, 849–856 CrossRef CAS PubMed.
- P. V. Mayuri, A. Bhatt, A. Sabaweeswaran and P. Ramesh, J. Biomater. Sci., Polym. Ed., 2020, 1–18, DOI:10.1080/09205063.2020.1851559.
- R. Hirano, K. Namazuda, J. Suemitsu, T. Harashima and N. Hirata, Transfus. Apher. Sci., 2017, 56, 649–653 CrossRef PubMed.
- R. Vanholder, A. Argilés, U. Baurmeister, P. Brunet, W. Clark, G. Cohen, P. P. De Deyn, R. Deppisch, B. Descamps-Latscha, T. Henle, A. Jorres, Z. A. Massy, M. Rodriguez, B. Stegmayr, P. Stenvinkel and M. L. Wratten, Int. J. Artif. Organs, 2001, 24, 695–725 CrossRef CAS PubMed.
- M. Chmielewski, G. Cohen, A. Wiecek and J. Jesús Carrero, Semin. Nephrol., 2014, 34, 118–134 CrossRef CAS PubMed.
- C. Ronco and G. La, Manna, Contrib. Nephrol., 2017, 190, 124–133 CAS.
- A. C. Yamashita, H. Masaki, E. Kobayashi and T. Sukegawa, Hemodial. Int., 2015, 19, S20–S25 CrossRef PubMed.
- M. M. Amiji, J. Biomater. Sci., Polym. Ed., 1997, 8, 281–298 CrossRef PubMed.
- M. Faria, C. Moreira, T. Eusébio, P. Brogueira and M. N. de Pinho, Cellulose, 2020, 27, 3847–3869 CrossRef CAS.
- N. Said, M. Nidzhom, H. Hasbullah, A. Ismail, P. Goh, M. Hafiz, M. H. Othman, S. Abdullah, S. Kadir, A. Kadir and F. Kamal, Mal. J. Fund. Appl. Sci., 2020, 16, 1–5 CrossRef.
- M. N. Z. Abidin, P. S. Goh, A. F. Ismail, M. H. D. Othman, H. Hasbullah, N. Said, S. H. S. A. Kadir, F. Kamal, M. S. Abdullah and B. C. Ng, Mater. Sci. Eng., C, 2016, 68, 540–550 CrossRef CAS PubMed.
- P. G. Kerr and L. Huang, Nephrology, 2010, 15, 381–385 CrossRef PubMed.
- A. K. Cheung, C. J. Parker, L. A. Wilcox and J. Janatova, Kidney Int., 1990, 37, 1055–1059 CrossRef CAS PubMed.
- Z. Zhang, S. Chen, Y. Chang and S. Jiang, J. Phys. Chem. B, 2006, 110, 10799–10804 CrossRef CAS PubMed.
- R. M. Hakim, D. T. Fearon, J. M. Lazarus and C. S. Perzanowski, Kidney Int., 1984, 26, 194–200 CrossRef CAS PubMed.
- J. C. Hutter, M. J. Kuehnert, R. R. Wallis, A. D. Lucas, S. Sen and W. R. Jarvis, JAMA, 2000, 283, 2128–2134 CrossRef CAS PubMed.
- M. E. Neumann, Nephrol. News Issues, 1996, 10(9), 11 Search PubMed.
- H. D. Humes, W. H. Fissell and K. Tiranathanagul, Kidney Int., 2006, 69, 1115–1119 CrossRef CAS PubMed.
- S. P. Adiga, C. Jin, L. A. Curtiss, N. A. Monteiro-Riviere and R. J. Narayan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 568–581 CAS.
- A. Gao, F. Liu and L. Xue, J. Membr. Sci., 2014, 452, 390–399 CrossRef CAS.
- M. Irfan, A. Idris, N. M. Yusof, N. F. M. Khairuddin and H. Akhmal, J. Membr. Sci., 2014, 467, 73–84 CrossRef CAS.
- J. R. Colvin, Can. J. Chem., 1952, 30, 831–834 CrossRef CAS.
- C. H. Trinh, D. P. Smith, A. P. Kalverda, S. E. V. Phillips and S. E. Radford, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9771–9776 CrossRef CAS PubMed.
- F. Galli, S. Rovidati, L. Chiarantini, G. Campus, F. Canestrari and U. Buoncristiani, Kidney Int., 1998, 54, 580–589 CrossRef CAS PubMed.
- V. Sepe, M. Gregorini, T. Rampino, P. Esposito, R. Coppo, F. Galli and C. Libetta, BMC Nephrol., 2019, 20, 412 CrossRef CAS PubMed.
- M. Kiaii, M. Aritomi, M. Nagase, M. Farah and B. Jung, J. Artif. Organs, 2019, 22, 307–315 CrossRef CAS PubMed.
- O. E. M. ter Beek, D. Pavlenko and D. Stamatialis, J. Membr. Sci., 2020, 604, 118068 CrossRef CAS.
- M. Tijink, J. Janssen, M. Timmer, J. Austen, Y. Aldenhoff, J. Kooman, L. Koole, J. Damoiseaux, R. van Oerle, Y. Henskens and D. Stamatialis, J. Mater. Chem. B, 2013, 1, 6066–6077 RSC.
- Y. Liu, Y. Su, X. Zhao, Y. Li, R. Zhang and Z. Jiang, J. Membr. Sci., 2015, 486, 195–206 CrossRef CAS.
- M. Adeli, E. Mehdipour and M. Bavadi, J. Appl. Polym. Sci., 2010, 116, 2188–2196 CAS.
- T. Xiang, C.-D. Luo, R. Wang, Z.-Y. Han, S.-D. Sun and C.-S. Zhao, J. Membr. Sci., 2015, 476, 234–242 CrossRef CAS.
- L. Wang, J. Wang, X. Gao, Z. Liang, B. Zhu, L. Zhu and Y. Xu, Polym. Chem., 2014, 5, 2836–2842 RSC.
- K. Madhavan Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef CAS PubMed.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- M. M. Flanders, R. Crist and G. M. Rodgers, Clin. Chem., 2003, 49, 169–172 CrossRef CAS PubMed.
- M. Z. Fahmi, M. Wathoniyyah, M. Khasanah, Y. Rahardjo, S. Wafiroh and Abdulloh, RSC Adv., 2018, 8, 931–937 RSC.
- M. Leeman, J. Choi, S. Hansson, M. U. Storm and L. Nilsson, Anal. Bioanal. Chem., 2018, 410, 4867–4873 CrossRef CAS PubMed.
- M.-M. Tu, J.-J. Xu and Y.-R. Qiu, RSC Adv., 2019, 9, 6254–6266 RSC.
- N. J. Kaleekkal, A. Thanigaivelan, M. Durga, R. Girish, D. Rana, P. Soundararajan and D. Mohan, Ind. Eng. Chem. Res., 2015, 54, 7899–7913 CrossRef CAS.
- M. Ghannoum, J. Bouchard, T. D. Nolin, G. Ouellet and D. M. Roberts, Semin. Dial., 2014, 27, 350–361 CrossRef PubMed.
- J. H. Salazar, Lab. Med., 2014, 45, e19–e20 CrossRef.
- L. B. VanWagner and R. M. Green, JAMA, 2015, 313, 516–517 CrossRef CAS PubMed.
- Q. Li, W. Zhao, H. Guo, J. Yang, J. Zhang, M. Liu, T. Xu, Y. Chen and L. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 25546–25556 CrossRef CAS PubMed.
- C. Tetta, R. Bellomo, A. Brendolan, P. Piccinni, A. Digito, M. Dan, M. Irone, G. Lonnemann, D. Moscato, J. Buades, G. La Greca and C. Ronco, Kidney Int., 1999, 56, S15–S19 CrossRef.
- G. La Greca, A. Brendolan, P. M. Ghezzi, R. De Smet, C. Tetta, R. Gervasio and C. Ronco, Int. J. Artif. Organs, 1998, 21, 303–308 CrossRef CAS PubMed.
- K. Cao, C. P. Siepermann, M. Yang, A. M. Waas, N. A. Kotov, M. D. Thouless and E. M. Arruda, Adv. Funct. Mater., 2013, 23, 2072–2080 CrossRef CAS.
- R.-L. Liu, Y. Wang, X.-L. Ge, P. Yu, H.-Q. Liu, M.-C. Wang, W. Lu and Q. Fu, J. Biomater. Sci., Polym. Ed., 2017, 28, 1444–1468 CrossRef CAS PubMed.
- S. E. Cowper, H. S. Robin, S. M. Steinberg, L. D. Su, S. Gupta and P. E. LeBoit, Lancet, 2000, 356, 1000–1001 CrossRef CAS.
- A. Sueoka, Ther. Apher., 1997, 1, 271–283 CrossRef CAS PubMed.
- D. Pavlenko, D. Giasafaki, G. Charalambopoulou, E. van Geffen, K. G. F. Gerritsen, T. Steriotis and D. Stamatialis, Sci. Rep., 2017, 7, 14914 CrossRef CAS PubMed.
- C. A. Howell, S. R. Sandeman, Y. Zheng, S. V. Mikhalovsky, V. G. Nikolaev, L. A. Sakhno and E. A. Snezhkova, Carbon, 2016, 97, 134–146 CrossRef CAS.
- Y. Yang, S. Yin, C. He, X. Wu, J. Yin, J. Zhang, L. Ma, W. Zhao, C. Cheng and C. Zhao, J. Mater. Chem. B, 2020, 8, 1960–1970 RSC.
- R. S. Bedi, D. E. Beving, L. P. Zanello and Y. Yan, Acta Biomater., 2009, 5, 3265–3271 CrossRef CAS PubMed.
- Y. Yu, Chin. Sci. Bull., 2013, 58, 4357–4361 CrossRef CAS.
- X. Song, K. Wang, C.-Q. Tang, W.-W. Yang, W.-F. Zhao and C.-S. Zhao, Biomacromolecules, 2018, 19, 1966–1978 CrossRef CAS PubMed.
- M. Eberhardson, P. Marits, M. Jones, P. Jones, P. Karlen, M. Karlsson, G. Cotton, K. Woznica, B. Maltman, H. Glise and O. Winqvist, Clin. Immunol., 2013, 149, 73–82 CrossRef CAS PubMed.
- J. H. Kang, M. Super, C. W. Yung, R. M. Cooper, K. Domansky, A. R. Graveline, T. Mammoto, J. B. Berthet, H. Tobin, M. J. Cartwright, A. L. Watters, M. Rottman, A. Waterhouse, A. Mammoto, N. Gamini, M. J. Rodas, A. Kole, A. Jiang, T. M. Valentin, A. Diaz, K. Takahashi and D. E. Ingber, Nat. Med., 2014, 20, 1211–1216 CrossRef CAS PubMed.
- J. Ciambella, D. C. Stanier and S. S. Rahatekar, Composites, Part B, 2017, 109, 129–137 CrossRef CAS.
- N. M. Osmond, Adsorpt. Sci. Technol., 2000, 18, 529–539 CrossRef CAS.
- S. Chen, Y. Cao and J. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 349–356 CrossRef CAS PubMed.
- A. Y. Rad, H. Yavuz, M. Kocakulak and A. Denizli, Macromol. Biosci., 2003, 3, 471–476 CrossRef CAS.
- W. Shi, Y. Shen, H. Jiang, C. Song, Y. Ma, J. Mu, B. Yang and D. Ge, J. Membr. Sci., 2010, 349, 333–340 CrossRef CAS.
- W. Yantasee, G. E. Fryxell, G. A. Porter, K. Pattamakomsan, V. Sukwarotwat, W. Chouyyok, V. Koonsiripaiboon, J. Xu and K. N. Raymond, Nanomedicine, 2010, 6, 1–8 CrossRef CAS PubMed.
- Y. Lin, S. K. Fiskum, W. Yantasee, H. Wu, S. V. Mattigod, E. Vorpagel, G. E. Fryxell, K. N. Raymond and J. Xu, Environ. Sci. Technol., 2005, 39, 1332–1337 CrossRef CAS PubMed.
- D. W. Whisenhunt, M. P. Neu, Z. Hou, J. Xu, D. C. Hoffman and K. N. Raymond, Inorg. Chem., 1996, 35, 4128–4136 CrossRef CAS PubMed.
- J. Chen, W. Han, R. Su, J. Chen, W. Zong, Y. Wang, W. Wang, G. Cheng, L. Ou and Y. Yu, Artif. Cells, Nanomed., Biotechnol., 2017, 45, 174–183 CrossRef CAS PubMed.
- L. Tan, R. He, Y. Li, Y. Liang, H. Li and Y. Tang, J. Chromatogr. A, 2016, 1459, 1–8 CrossRef CAS PubMed.
- N. Wu, Z. Luo, Y. Ge, P. Guo, K. Du, W. Tang, W. Du, A. Zeng, C. Chang and Q. Fu, J. Pharm. Anal., 2016, 6, 157–164 CrossRef PubMed.
- T. Agishi, K. Takahashi, T. Yagisawa and K. Ota, ASAIO Trans., 1991, 37, M496–M498 CAS.
- M. E. Warkiani, L. Wu, A. K. Tay and J. Han, Annu. Rev. Biomed. Eng., 2015, 17, 1–34 CrossRef CAS PubMed.
- Z. T. Yu, K. M. Aw Yong and J. Fu, Small, 2014, 10, 1687–1703 CrossRef CAS PubMed.
- A. A. S. Bhagat, H. Bow, H. W. Hou, S. J. Tan, J. Han and C. T. Lim, Med. Biol. Eng. Comput., 2010, 48, 999–1014 CrossRef PubMed.
- H. W. Hou, A. A. S. Bhagat, W. C. Lee, S. Huang, J. Han and C. T. Lim, Micromachines, 2011, 2, 319–343 CrossRef.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- J. Zhou, A. Kulasinghe, A. Bogseth, K. O’Byrne, C. Punyadeera and I. Papautsky, Microsyst. Nanoeng., 2019, 5, 8 CrossRef PubMed.
- M. S. Maria, P. E. Rakesh, T. S. Chandra and A. K. Sen, Sci. Rep., 2017, 7, 43457 CrossRef PubMed.
- H. Xia, B. C. Strachan, S. C. Gifford and S. S. Shevkoplyas, Sci. Rep., 2016, 6, 35943 CrossRef CAS PubMed.
- A. Papra, A. Bernard, D. Juncker, N. B. Larsen, B. Michel and E. Delamarche, Langmuir, 2001, 17, 4090–4095 CrossRef CAS.
- K. M. Kovach, J. R. Capadona, A. S. Gupta and J. A. Potkay, J. Biomed. Mater. Res., Part A, 2014, 102, 4195–4205 Search PubMed.
- A. Mohd Noor, T. Masuda and F. Arai, ROBOMECH J., 2020, 7, 5 CrossRef.
- P. Sethu, A. Sin and M. Toner, Lab Chip, 2006, 6, 83–89 RSC.
- X. Chen, D. F. Cui, C. C. Liu and H. Li, Sens. Actuators, B, 2008, 130, 216–221 CrossRef CAS.
- X. Chen, D. F. Cui, C. C. Liu and H. Li, Sens. Actuators, B, 2008, 130, 216–221 CrossRef CAS.
- H. Jang, M. R. Haq, J. Ju, Y. Kim, S.-M. Kim and J. Lim, Micromachines, 2017, 8, 67 CrossRef.
- P. Bacchin, M. Meireles and P. Aimar, Desalination, 2002, 145, 139–146 CrossRef CAS.
- Y. Lee and M. M. Clark, J. Membr. Sci., 1998, 149, 181–202 CrossRef CAS.
- B. Lombodorj, H. C. Tseng, H. Y. Chang, Y. W. Lu, N. Tumurpurev, C. W. Lee, B. Ganbat, R. G. Wu and F. G. Tseng, Micromachines, 2020, 11(3), 275 CrossRef PubMed.
- S. Ookawara, M. Agrawal, D. Street and K. Ogawa, Chem. Eng. Sci., 2007, 62, 2454–2465 CrossRef CAS.
- P. Li, Z. S. Stratton, M. Dao, J. Ritz and T. J. Huang, Lab Chip, 2013, 13, 602–609 RSC.
- W. J. Allard, J. Matera, M. C. Miller, M. Repollet, M. C. Connelly, C. Rao, A. G. J. Tibbe, J. W. Uhr and L. W. M. M. Terstappen, Clin. Cancer Res., 2004, 10, 6897 CrossRef PubMed.
- Y. Dong, A. M. Skelley, K. D. Merdek, K. M. Sprott, C. Jiang, W. E. Pierceall, J. Lin, M. Stocum, W. P. Carney and D. A. Smirnov, J. Mol. Diagn., 2013, 15, 149–157 CrossRef PubMed.
- B. Hong and Y. Zu, Theranostics, 2013, 3, 377–394 CrossRef CAS PubMed.
- L. Wang, P. Balasubramanian, A. P. Chen, S. Kummar, Y. A. Evrard and R. J. Kinders, Semin. Oncol., 2016, 43, 464–475 CrossRef CAS PubMed.
- P. S. Nunes, P. D. Ohlsson, O. Ordeig and J. P. Kutter, Microfluid. Nanofluid., 2010, 9, 145–161 CrossRef CAS.
- H. Jiang, X. Weng, C. H. Chon, X. Wu and D. Li, J. Micromech. Microeng., 2011, 21, 085019 CrossRef.
- S. Choi, S. Song, C. Choi and J. K. Park, Lab Chip, 2007, 7, 1532–1538 RSC.
- T. Tachi, N. Kaji, M. Tokeshi and Y. Baba, Anal. Chem., 2009, 81, 3194–3198 CrossRef CAS PubMed.
- S. Yang, B. Ji, A. Undar and J. D. Zahn, ASAIO J., 2006, 52, 698–704 CrossRef CAS PubMed.
- W. Guo, J. Hansson and W. van der Wijngaart, Anal. Chem., 2020, 92, 6194–6199 CrossRef CAS PubMed.
- M. Hillmering, G. Pardon, A. Vastesson, O. Supekar, C. F. Carlborg, B. D. Brandner, W. van der Wijngaart and T. Haraldsson, Microsyst. Nanoeng., 2016, 2, 15043 CrossRef CAS PubMed.
- J. Hauser, G. Lenk, J. Hansson, O. Beck, G. Stemme and N. Roxhed, Anal. Chem., 2018, 90, 13393–13399 CrossRef CAS PubMed.
- Z.-Y. Tao, H. Xia, J. Cao and Q. Gao, Malar. J., 2011, 10, 251 CrossRef PubMed.
- C. Ronco, C. Crepaldi, A. Brendolan, L. Bragantini, V. d'Intini, P. Inguaggiato, M. Bonello, B. Krause, R. Deppisch and H. Goehl, Nephrol. Dial. Transplant., 2003, 18, vii10–vii20 CrossRef CAS PubMed.
- R. M. Schaefer, W. Rautenberg, S. Neumann, A. Heidland and W. H. Hörl, Clin. Nephrol., 1986, 26(Suppl 1), S35–S38 Search PubMed.
- L. J. Langsdorf, L. G. Krankel and A. L. Zydney, ASAIO J., 1993, 39, M767–M772 CAS.
- F. Aucella, M. Vigilante, G. Gatta, E. Grandone, D. Colaizzo, M. Margaglione, S. Modoni and C. Stallone, Artif. Organs, 2002, 26, 543–545 CrossRef CAS PubMed.
- N. Meert, S. Eloot, E. Schepers, H.-D. Lemke, A. Dhondt, G. Glorieux, M. Van Landschoot, M.-A. Waterloos and R. Vanholder, Nephrol., Dial., Transplant., 2011, 26, 2624–2630 CrossRef PubMed.
- J. K. Leypoldt, A. K. Cheung, L. Y. Agodoa, J. T. Daugirdas, T. Greene and P. R. Keshaviah, Kidney Int., 1997, 51, 2013–2017 CrossRef CAS PubMed.
- W. R. Clark, R. J. Hamburger and M. J. Lysaght, Kidney Int., 1999, 56, 2005–2015 CrossRef CAS PubMed.
- EXELTRA™ Dialyzer, Single Use, 2003, p. K030974.
- FX high-flux and FX low-flux dialyzers, https://www.freseniusmedicalcare.asia/en/healthcare-professionals/hemodialysis/dialyzers/fx-high-and-low-flux-dialyzers/, (accessed November 2, 2020).
- S. Sibbel, A. Hunt, S. Laplante, W. Beck, M. Gellens and S. M. Brunelli, ASAIO J., 2016, 62, 613–622 CrossRef PubMed.
- Commonly used dialyzer specifications and substitution chart for visiting patients, http://www.bcrenalagency.ca/resource-gallery/Documents/Commonly%20Used%20Dialyzer%20Specifications%20and%20Subs%20Chart.pdf, (accessed September 11, 2019).
- P. Schuff-Werner and B. Holdt, Artif. Organs, 2002, 26, 117–123 CrossRef PubMed.
- W. Samtleben, S. Bengsch, K.-S. Boos and D. Seide, Artif. Organs, 1998, 22, 43–46 CrossRef CAS PubMed.
- N. Hirata, Y. Shizume, J. Shirokaze, J. Suemitsu, H. Yoshida and N. Yamawaki, Ther. Apher. Dial., 2003, 7, 64–68 CrossRef PubMed.
- M. Onishi, K. Shimura, Y. Seita and S. Yamashita, Radiat. Phys. Chem., 1995, 46, 219–223 CrossRef CAS.
- Q. Xia, Y. Yun, Q. Li, Z. Huang and Z. Liang, Des. Monomers Polym., 2016, 20, 201–209 CrossRef PubMed.
- Filtryzer BG Hemodialyzer, https://www.toray-medical.com/en/products/dialysis/detail.html?key=0EB5426D0FD8A757492580D20022E798, (accessed December 30, 2020).
- J. M. Campistol, J. V. Torregrosa, E. Ponz and B. Fenollosa, in Polymethylmethacrylate. A Flexible Membrane for a Tailored Dialysis, ed. C. Ronco, Karger, Basel, 1998, vol. 125, pp. 76–85 Search PubMed.
- I. Iizuka, Z. Yamazaki, Y. Fujimori, T. Takahama, F. Kanai, T. Wada, N. Inoue, T. Sonoda, T. Nogi and M. Kimura, Ther. Apher., 2000, 4, 54–57 CrossRef CAS PubMed.
- D. Xie, L. Howard and R. Almousa, J. Biomater. Appl., 2018, 33, 340–351 CrossRef CAS PubMed.
- S. Kim, B. Feinberg, R. Kant, B. Chui, K. Goldman, J. Park, W. Moses, C. Blaha, Z. Iqbal, C. Chow, N. Wright, W. H. Fissell, A. Zydney and S. Roy, PLoS One, 2016, 11, e0159526 CrossRef PubMed.
- N. J. Kaleekkal, D. Rana and D. Mohan, RSC Adv., 2016, 6, 63156–63170 RSC.
- K. Togo, M. Yamamoto, M. Imai, K. Akiyama and A. C. Yamashita, Ren. Replace. Ther., 2018, 4, 29 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |