
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic polymer-derived single-network inks/bioinks for extrusion-based 3D printing towards bioapplications
Sonu
Kumar

Department of Applied Sciences (Chemistry), Punjab Engineering College (Deemed to be University), Sector 12, Chandigarh, 160012, India. E-mail: sonu.kumar@pec.edu.in
First published on 24th September 2021
Abstract
Three-dimensional (3D) printing, also known as the additive manufacturing technique has revolutionized the field of manufacturing with great impact as compared to the other traditional methods. This technique has shown a steep increase in popularity over the past decade due to its benchmark capabilities of fabricating new and complex 3D constructs, especially towards tissue engineering and regenerative medicine. Among the currently applied 3D printing techniques, extrusion-based 3D printing has garnered particular attention for the employment of ink/bioink materials to enable on-demand personalized fabrication due to its low cost, broad utility for various materials, and ease of controlled printability. However, there is still a lack of diversity in the ink materials with their optimized degradation rate, rheology, and bioactivity for precisely fabricating complex and self-supported cell-laden 3D printed constructs. Therefore, the development of an array of such new materials is a major challenge for synthetic polymer chemists, material scientists and biomedical researchers for widening the future applicability of 3D (bio)printing. This review aims to summarize the recent advances in the rational design and development of ink/bioink materials based on synthetic polymers as single network precursors due to their great opportunity to tune their physicochemical and mechanical properties in order to design and mimic in-human 3D tissue scaffolds with shape retention for both hard and soft tissues.
1. Introduction
The 3D printing-based computer-aided additive manufacturing technique has emerged as a revolutionary manufacturing method and is currently being considered at the forefront of research in material sciences and biomedical engineering.1,2 3D printing is a promising method for the future era of personalized medicine via the layer-by-layer fabrication of patient-specific, customizable medical devices, organs, tissues, and other bio-systems, mimicking their native counterparts with complex and heterogeneous structures.3,4 In the endeavours for such biofabrication, the selection of printing technology, design and materials are critically important for maintaining the structure–property-processing relationships in the 3D printed constructs.5,6 To date, there has been considerable development in the engineering of different types of 3D printing devices7–11 and increasing their printing affordability and reliability such as having tight control over the printing speed, higher resolutions, and multicomponent fabrications.12,13 The development and optimization of the printable ink materials are required to achieve the desired properties including physico-mechanical properties, rheological properties and biofunctionality to obtain self-supporting functional and high-strength products, depending on the applications.14,15 Although there exist different ink materials ranging from polymers to composites and ceramics,16 there is still the need for diversity in 3D printable ink materials. On the other hand, the preparation of 3D printable cell-laden ink materials (i.e. bioinks) has attracted a great deal of interest from researchers in recent years,17 and has been considered as one of the most advanced tools for finding new avenues in tissue engineering,18 regenerative medicine,19 drug delivery,20 cell therapy,21etc. To mimic the biocomplexity and heterogeneity of natural tissues in the 3D printed constructs on various scales, the development of cellular embedded ink materials with “printability” (e.g., with optimized printing process, speed, and resolution are critically important)22,23 is challenging due to the demand for various features depending on the application envisioned as discussed here. Cells are encapsulated within such ink materials, and therefore all the components of the inks such as the functionalized polymer-based main ingredient along with other applied precursors such as catalysts, cross-linker, drugs, bioactive molecules (e.g. peptide sequences,24 growth and differentiation factors, etc.,25,26) should be biocompatible in order to maintain the cell survivability throughout the printing process, as well as in the final printed article. The ink materials should protect the cells and maintain their sensitivity to enable the survival of cells during the printing process for the bio-fabrication, and the desired cellular functions (e.g., cell growth and proliferation, adhesion, and differentiation) should be preserved in the fabricated structures.27 Further, the post-extrusion structural and functional integrity in the product should be maintained by standardizing different parameters such as pressure, temperature, pH, or light to avoid cell resistance.28,29 Finally, the materials should be biodegradable with controlled degradation kinetics and their waste compounds and intermediates should not have any toxic effects.In the effort to develop ink materials for 3D printing with desired mechanical and biological properties, two major categories of material precursors have been adopted: (i) natural polymers (biomaterials), and (ii) synthetic polymers. Natural polymers, such as polysaccharides and proteins, as typically extracted from plants, animals, bacteria, cells, etc., are currently widely applied materials as network precursors in bioink formation due to their greater biocompatible nature and higher cellular proliferation rates in comparison to synthetic polymers.30 Note that in many reports of bioink formulation natural polymers were artificially modified with functional groups, for instance, to enable network crosslinking, however, such artificial biomaterials have been considered in this review in the category of natural polymers only.31,32 Naturally derived polymers are often associated with some disadvantages that can lead to challenges in their utility for biofabrication as follows: (1) batch-to-batch variations, which can lead to complications due to their variable printabilty and the issue of the reproducibility of constructs, and the cellular sensitivity to such variations; (2) tuning the structures, solubility, viscosity and other properties of biopolymers remain challenging since their functionalization is often more difficult; (3) fast biodegradability rate, which is often not suitable. On the other hand, synthetic polymers can overcome these limitations of natural polymers along with the opportunity to be tailored with specific physical, chemical, and biological properties and can further lead to the synthesis of a broad library of new materials for (bio)inks.33 Synthetic polymers also further offer the ability to tune functional properties and rheological behaviours, like the printability and mechanical integrity of the inks, through monomer selection, architecture control, and post-polymerization functionalization opportunities.34,35 Therefore, synthetic polymers have great potential to be exploited as future ink materials of 3D printing for shaping into advanced and highly customisable architectures suitable for medical devices and tissue scaffolds, including both hard and soft tissues.36,37 However, a careful selection of synthetic polymer has to be made in order to maintain biocompatibility and biodegradation issues. For instance, poly(ethylene glycol) (PEG) corresponds is a synthetic polymer that has been approved by the Food and Drug Administration (FDA) and shows good water solubility as well as biocompatibility.38,39 Poly(lactic-co-glycolic) acid (PLGA), polylactic acid (PLA), and polycaprolactone (PCL) are also FDA-approved synthetic polymers for their application in medical devices and are biocompatible and biodegradable in nature.40,41 Therefore, the utilization of synthetic polymers as ink materials can provide advancement in the 3D printing technology with obtaining the highly defined (bio)fabricated constructs with better control size, resolutions, integrity and features as compared to the traditional manufacturing techniques. Gupta et al. exemplified the combination of 3D printing technique with functional plasmonic nanomaterials for exhibiting programmable release from the printed multiplexed array of stimuli-responsive capsules having shells comprised of PLGA polymer within hydrogel matrices.42 In 2019, Camacho and Busari et al. investigated RGDS(biotin)-PCL and RGES(azide)-PCL-based peptide-polymer conjugates for the controlled fabrication of spatially functionalized 3D printed biodegradable scaffolds by using multiple printer heads, which displayed a significant enhancement in NIH 3T3 fibroblast adhesion on the 3D-printed fiber surface of (RGDS(biotin))-PCL.43 Another example of a PCL-based 3D printed scaffold was documented by Rashad et al., where it was subsequently coated with cellulose nanofibrils that significantly improved the hydrophilicity and protein absorption of the PCL scaffold surface, and further enhanced the cellular response (attachment, proliferation, and osteogenic differentiation) of mesenchymal stem cells (hBMSCs) in vitro for the potential application of such 3D printed PCL scaffolds towards bone tissue regeneration in vivo.44 In contrast to the recent burgeoning advancements in the 3D (bio)printing technique, the focus of this review is to highlight the examples and development of the (bio)ink materials based on synthetic polymers as a single-network precursor (or major component) employed for the extrusion-based 3D printing technique towards bioapplications since, to the best of our knowledge, this area of interest has not been previously reviewed. Note, that the formulation of (bio)ink materials based on only natural polymers or combined natural and synthetic polymers is out of the scope of this review article. The existing challenges associated with 3D biofabrication have been also discussed in this article to motivate synthetic polymer chemists to address the current demand for new and advanced (bio)ink materials, which would further help to widen the scope of this unique and advanced technique of extrusion-based 3D printing.
2. The extrusion-based 3D printing technique
Extrusion-based 3D printing is the commercially available and most common rapid prototyping technique due to its affordability, versatility, and compatibility with a wide selection of ink materials usable for biofabrication and tissue regeneration ranging from small vessels to large constructs/organs.45,46 Here, the inks are extruded through a micronozzle of extrusion head and controlled by either an endless screw, pneumatic pressure, or a mechanical piston (Fig. 1A).47,48 The 3D objects are constructed via the blueprints from a computer-aided design (CAD) file by direct-ink-writing in a layer-by-layer fashion, and controlling the xyz stage of the deposition path of the print-heads.49 Extrusion-based 3D printers can be successfully engineered under sterile conditions with nozzle diameters and multiple extrusion heads providing the unique advantage of depositing different types of layers in the final printed article via delivering multiple (bio)ink materials with varying components, cells, and cellular densities (Fig. 1B).8,50 To successfully enable the “printability” of an ink, a suitable viscosity should be achieved, typically by inducing either an external shear force (i.e. shear-thinning property) or pre-crosslinking to enable its extrusion from the nozzle; however as the external mechanical stress is removed post-extrusion, a rapid filament shape retention with efficient recovery of mechanical properties (i.e. self-healing properties) is desired to keep entire and self-supported 3D printed structures with no structural collapse.51 For instance, UV irradiation (or heating) can be applied for such shape retention to the tip of the printing nozzle or directly over the printed structure on the deposition plate. Burdick and coworkers developed a general extrusion-based bioprinting method of “in situ crosslinking” for photo-crosslinkable non-viscous hydrogel inks (such as from 5 wt% methacrylated hyaluronic acid (MeHA)) via introducing light through a photopermeable capillary, which enables their simultaneous extrusion and crosslinking, prior to deposition (Fig. 1C and D).52 The printed constructs depicted the uniform filament formation along with the high viability of the encapsulated cells. Another fascinating approach that has gained significant interest recently is extrusion-based 3D bioprinting using suspension baths, illustrating their ability to suspend and completely encapsulate the extruded filament materials.53 Such secondary support provides the flow restriction of the bioink and triggers the cross-linking (physically or chemically) immediately after the deposition, and enables the printed construct with improved resolution and controlled heterogeneity.54 Feinberg and co-workers presented such an approach as Freeform Reversible Embedding of Suspended Hydrogels (FRESH) printing, where a thermoreversible support bath (CaCl2) was utilized to embed a soft hydrogel (based on alginate) to exemplify the construction of a bioprinted full-sized human heart model.55 Recently, a co-axial extrusion-based 3D bioprinting approach has also been introduced to enable the simultaneous printing of bioink along with cross-linker solutions in a coaxial system to promote gelation during extrusion.56 Khademhosseini, Dentini, and co-workers demonstrated a coaxial needle extrusion system where the internal needle carried the flow of cell-laden bioink materials (based on alginate and gelatin methacroyl) and the external needle was designed for the simultaneous flow of ionic crosslinking solution (CaCl2) to generate the ionically crosslinked hydrogel microfibers at the tip of the dispensing system, which was further secondarily crosslinked covalently via exposure to UV light.57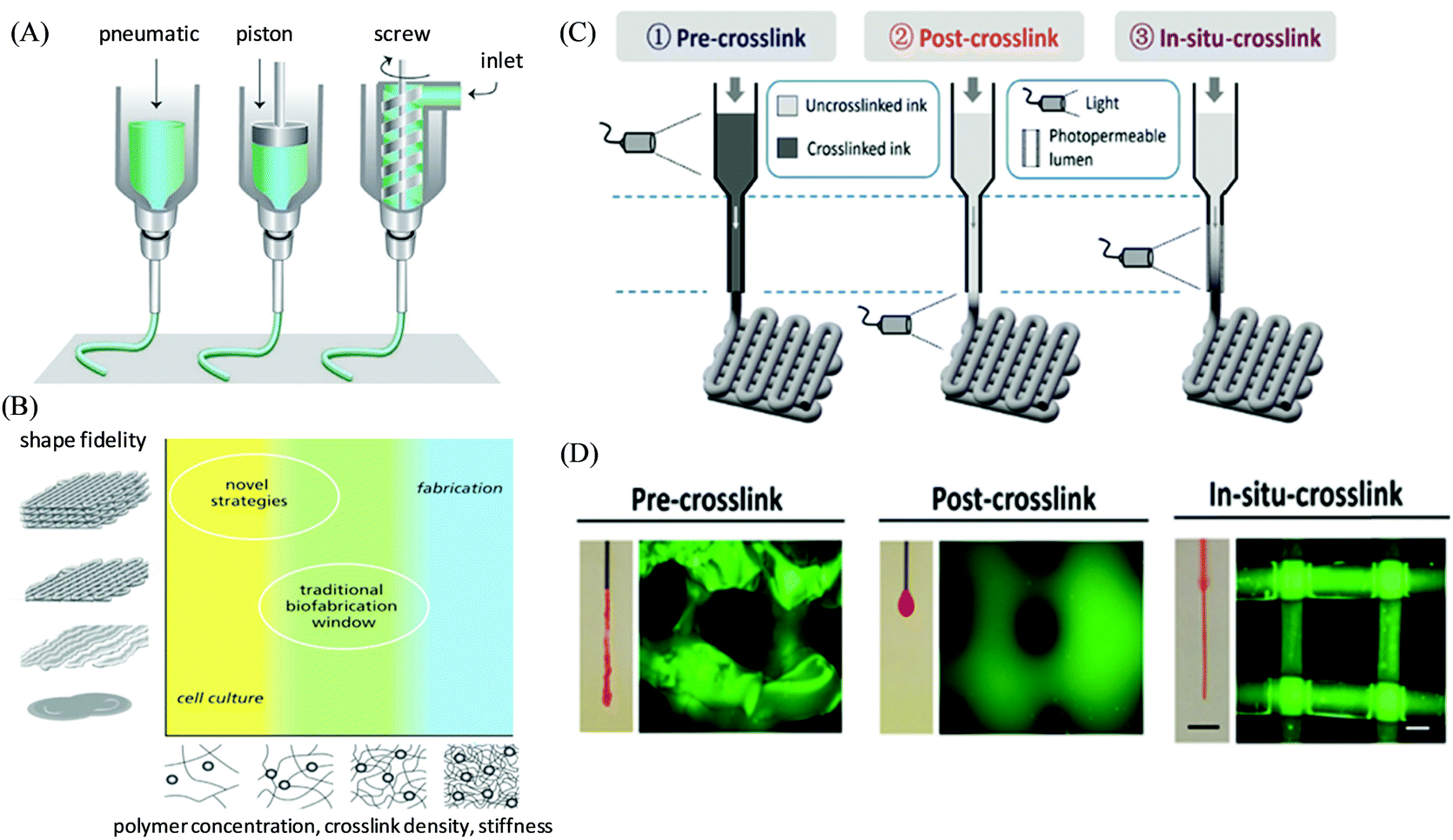 | ||
| Fig. 1 (A) Schematic illustration of the working principle of extrusion-based 3D printing techniques, and (B) the concept of the biofabrication window to improve the printability of polymeric hydrogel bioinks. Adapted with permission from ref. 47. Copyright 2013 John Wiley and Sons. (C) Schematic illustration of different bioprinting approaches for photo-crosslinkable inks via crosslinking before (pre-crosslinking), after (post-crosslinking), or during (in situ crosslinking) extrusion. (D) Representative images of the extruded material from nozzles and the lattice structure of corresponding printed constructs. Reproduced with permission from ref. 52. Copyright 2017 John Wiley and Sons. | ||
3. Hydrogel inks/bioinks based on synthetic polymers as single-network precursors
Hydrogels are highly hydrated three-dimensional polymeric networks and have been exploited for various biomedical applications, such as scaffolds for tissue engineering, and drug delivery.58,59 Hydrogel-based ink materials can provide a perfect soft material for soft tissue engineering since creating a soft tissue is more challenging due to the demand for features like high elasticity, flexibility, viscosity, and inter-layer adhesion.60,61 From a biological point of view, hydrogels having a high water content can be considered as an ideal candidate for cell-laden bioink materials for extrusion-based 3D printing because they can closely simulate the native extracellular matrix (ECM) microenvironments having an aqueous 3D environment, and provide cell survivability with retained rounded morphology and homogeneous encapsulation during the extrusion process, and thereafter promote new tissue formation and functioning in 3D space.18,62 However, the fabrication of hydrogel-based scaffolds or cell-laden constructs with shape fidelity remains challenging, and there is a current demand for optimization of the mechanical properties of the hydrogel materials within a biofabrication window (Fig. 1B).47 On the one hand, the soft hydrogels, which are well suited for cells, are disadvantageous for fabrication processes to achieve the constructs with maintained predesigned structural fidelity. On the other hand, the stiff hydrogels, which can typically provide shape fidelity, can affect the cellular functioning by limiting the migration and proliferation of the cells within the dense polymeric network.In this context, the utilization of synthetic polymers as hydrogel ink materials can tailor the mechanics by maintaining adequate viscoelastic characteristics and integrity,63,64 including self-healing and shear-thinning properties, and also provide opportunities for optimization in its functionality, and degradation kinetics of the printed products.65,66 In general, hydrogel ink formulation requires a specific viscous polymeric solution that can immediately form high networking post-printing either by the physical or chemical cross-linking of polymers.67 Prestwich and coworkers described the formation of extrudable hydrogels with suitable rheological properties for the bioprinting of vessel-like constructs derived from tetrahedral polyethylene glycol tetracrylates via co-crosslinking thiolated hyaluronic acid and gelatin derivatives.68 Joas et al. investigated the hydrogel formulation for extrusion-based 3D printing comprised of the diacrylate of PEG, the anionic and cationic monomers 3-sulfopropyl acrylate and [2-(acryloyloxy)ethyl]trimethylammonium chloride, respectively, via UV irradiation-based photo-polymerization.69 The biodegradable cross-linked hydrogel network (using visible light stimulus) was further illustrated by Xu et al. based on a single-component precursor of diacrylated polycaprolactone-poly(ethylene glycol)-polycaprolactone (PCL–PEG–PCL) triblock copolymer, which was synthesized (theoretical block length 2k–20k–2k) via ring-opening polymerization followed by acryloyl group insertion (Fig. 2).70 The hydrogels were composed of PEG and PCL-based well-known biocompatible synthetic materials and therefore, the hydrogels exhibited support for cellular growth with good survival rates (in vitro) of the encapsulated 3T3 fibroblasts of over 90% from day 1 and day 3 of culture. The hydrogels possessed good mechanical properties with high flexibility and elasticity upon stretching, compressing, and twisting. The viscosities of the obtained hydrogel precursors were optimized to prepare cell/polymer bioink (with PCL–PEG–PCL at 10% of polymer concentration) based on different types of human cells such as human umbilical vein endothelial cells (HUVEC), human aortic smooth muscle cells (SMC), and neonatal human lung fibroblasts (NHLF). 3D bioprinting was performed to create cell-gel constructs upon visible-light exposure, exhibiting their high cellular viability over 83% across all three cell types, within the constructs after 7 days in culture printing. Basic shapes and complex patterns were printed using 20% (w/v) PCL–PEG–PCL solution, and their resolutions were maintained by changing the nozzle diameter (with 18G and 21G needle), indicating the printability of the hydrogels.
 | ||
| Fig. 2 (A) Chemical structure of the acrylated PCL–PEG–PCL triblock copolymer. (B) The preparation of a visible-light-induced cross-linked single-network biodegradable hydrogel having high elasticity for bioprinting with various human cells. (C) Photographs illustrating the mechanical properties of the PCL–PEG–PCL hydrogel upon twisting for four cycles and recovering after release. (D and E) Live/dead cell assay and cellular compatibility, respectively, of different cell types in the printed 10% PCL–PEG–PCL hydrogel carried out immediately after gelation (D0) and after 7 days in culture (D7) (scale bar: 500 μm). (F) Printed sample shapes by using different needle sizes (scale bar: 5 mm). Adapted with permission from ref. 70. Copyright 2018 American Chemical Society (ACS). https://pubs.acs.org/doi/10.1021/acsami.8b01294 (for further permissions related to the material excerpted, readers are directed to the ACS). | ||
3.1 Supramolecular polymers
Supramolecular polymers are derived from a unique combination of supramolecular chemistry and polymer science,71,72 and have appeared as advanced smart materials for diverse applications towards energy harvesting, drug delivery, electronic devices, template synthesis, etc.73,74 Such polymeric chains involve various types of supramolecular interactions based on hydrogen bonding, π–π interactions, metal–ligand coordination, interactions between ions, and host–guest interactions.75,76 Those supramolecular noncovalent interactions offer a dynamic and reversibile nature, providing an opportunity to tune the mechanical and viscoelastic behavior with improved rheological performance of supramolecular polymers,77 which makes them potential candidates for 3D printing-based additive manufacturing to create customizable and dynamic constructs for tissue engineering scaffolds and other biomedical applications.19,78,79 Echalier et al. documented a 3D printable bioink derived from supramolecular peptide-functionalized synthetic polymer-based hydrogels by using soft sol–gel polymerization without using photoactivation or additional organic reagents (Fig. 3).80 Hydrolysis, followed by condensation of the silylated precursors of hybrid bifunctional PEG polymer and integrin ligand (GRGDSP peptide), occurred during the extrusion-based 3D printing process (at room temperature and in physiological buffer with pH 7.2) resulting in a chemically cross-linked network through siloxane (Si–O–Si) bonds. The obtained hybrid PEG-peptide-based 3D scaffolds were then seeded with a mesenchymal stem cell (mMSC) culture, which showed excellent cell viability after 4 days of proliferation live/dead assay on the scaffolds, indicating the compatibility of the hybrid PEG-peptide scaffolds for cell culture.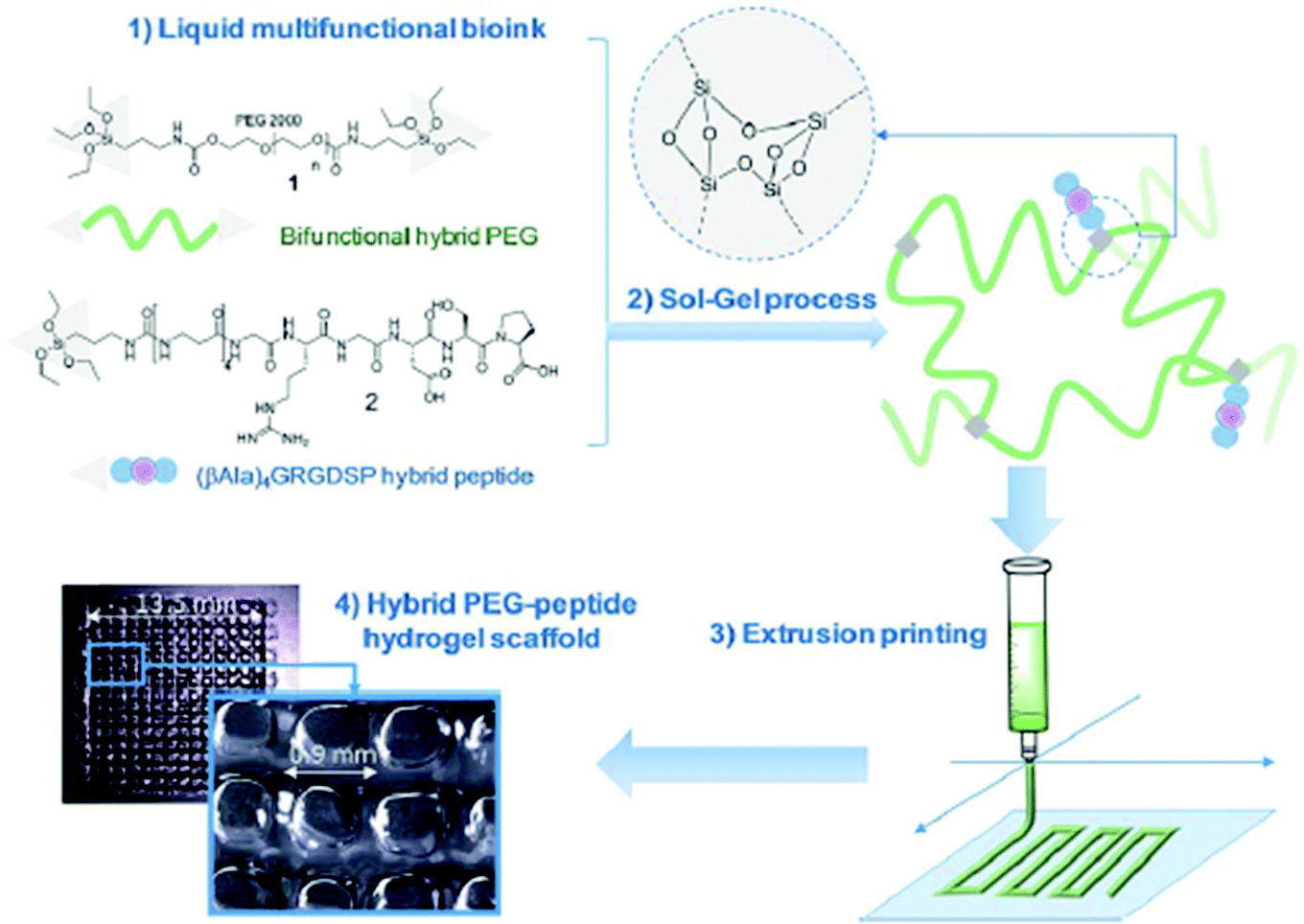 | ||
| Fig. 3 Principle of sol–gel extrusion printing of PEG-peptide-based hybrid hydrogels. Hybrid silylated PEG polymers and hybrid GRGDSP peptides were dissolved in buffer to provide a multicomponent bioink that undergoes condensation during 3D printing to produce hydrogel scaffolds. Reproduced from ref. 80 with permission from The Royal Society of Chemistry. | ||
In another example, Lorson et al. documented thermogelling supramolecular materials as a bioink for 3D bioprinting based on a series of synthetic biocompatible amphiphilic block copolymers comprised of a poly(2-methyl-2-oxazoline) (PMeOx)-based hydrophilic block and poly(2-n-propyl-2-oxazine) (PnPrOzi)-based thermoresponsive segment, yielding PnPrOzin-b-PMeOxn (Fig. 4).81 At concentrations of 20 wt% and above, the copolymers depicted thermogelation between room temperature and 37 °C, and the formed hydrogels exhibited high mechanical strength with G’ values more than 1 kPa, and pronounced isothermal shear thinning behavior with a rapid and complete shear recovery upon the removal of stress. Such rheological properties of the copolymer materials along with their obtained cytocompatibility, enabled their suitability for extrusion-based 3D bioprinting to afford cell-loaded constructs.82 The homogeneous cell distribution was observed within the entire printed constructs generated from NIH 3T3 fibroblasts encapsulated PnPrOzi-b-PMeOx-based bioinks and no negative effects on the cell viability were observed by the dispensing process, with cytocompatiblity (91.5 ± 0.8%) measured 24 h post-printing, which was comparable to the viability of the control cells incorporated in the material but not printed (92.8 ± 1.7%), and untreated cells in the medium (98.9 ± 0.18%).
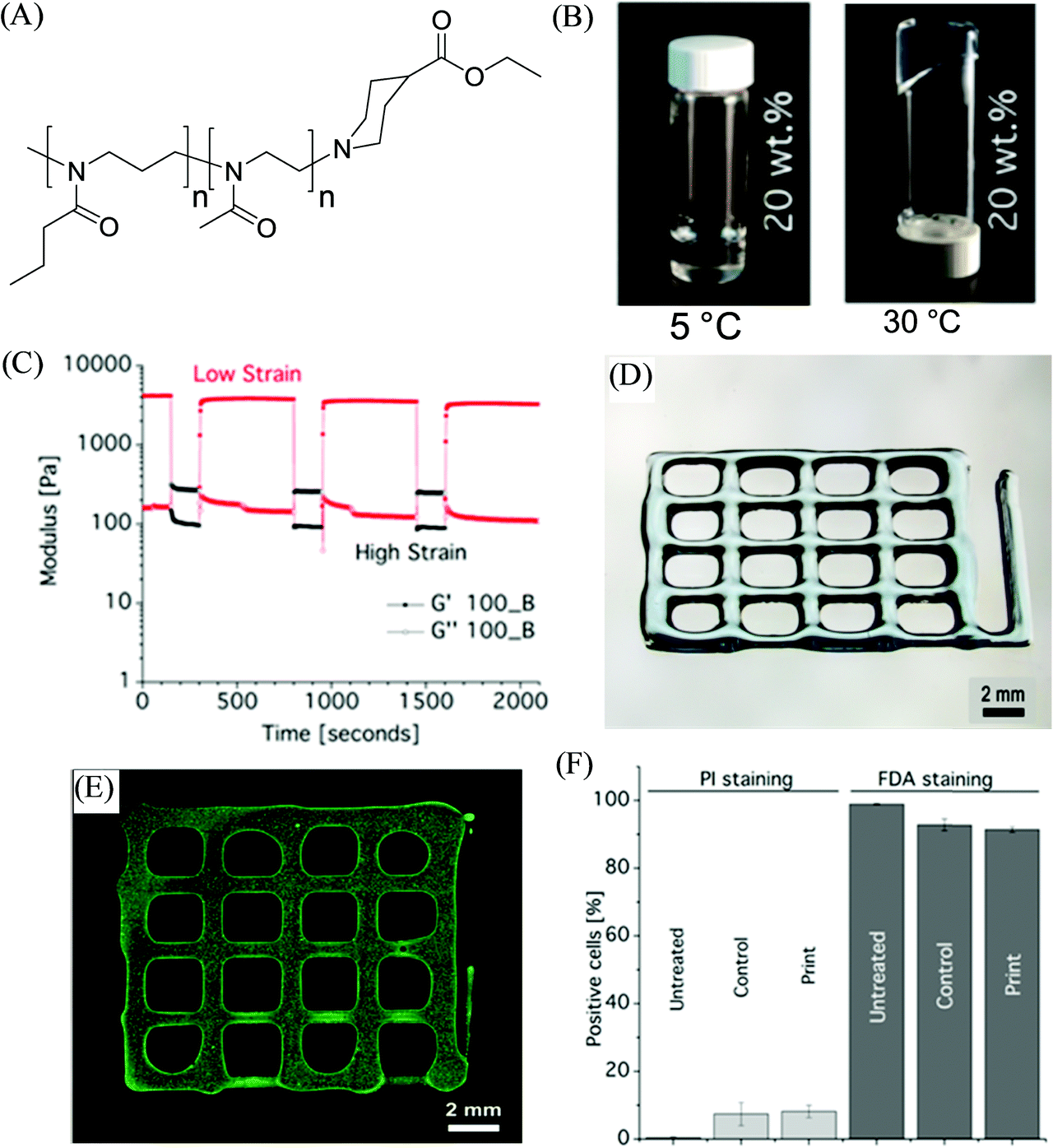 | ||
| Fig. 4 (A) The chemical structure of the investigated diblock copolymer materials PnPrOzin-b-PMeOxn. (B) Photographs of 20 wt% concentration of PnPrOzi100-b-PMeOx100 (100_B) at different temperatures. (C) Shear recovery at 37 °C and 10 rad s−1 of (20 wt%) 100_B (low strain: 0,5%, high strain: 150%). (D) Light microscopy image of hydrogel 3D printed constructs, and (E) cell-loaded printed constructs. (F) FACS analysis results to illustrate the effect of the printing process on the viability of NIH 3T3 fibroblast cells stained with propidium iodide (PI) and fluorescein diacetate (FDA) (“untreated” corresponds to the cells in the medium, “control” refers to the cells redispersed in the bioink but not printed, and “print” refers to the viability of cells after 24 h of printing). Adapted with permission from ref. 81. Copyright 2017 American Chemical Society. | ||
The one-pot fabrication of printable supramolecular polymer-based self-healing hydrogels was demonstrated by Wei et al., derived from guanidinium-based oligopeptide via the dual-enzyme-triggered self-assembly and simultaneous enzyme-initiated polymerization approach (Fig. 5).83 The ink material for such a system was comprised of the mixed solution of monomer poly(ethylene glycol) methacrylate (PEGMA) and NapFFRK-acryloyl (peptidic hydrogelator), and the enzymes horseradish peroxidase (HRP), glucose oxidase (GOx), along with glucose, and acetyl acetone (AcAc), which commenced the two enzyme-mediated redox reaction. The hybrid hydrogel provided an enlarged time window for the in situ viscosity-controlled (of the pre-gel solution) printing into 3D constructs. NIH 3T3 cells-incorporated hybrid hydrogels exhibited a very high survival rate of 99.0 ± 0.8%, and a live/dead assay of the 3D bioprinted construct from such cell-laden bioink materials showed the cell viability of 98.0 ± 2.3%, suggesting the biocompatibilty of the hybrid hydrogels. The preparation of similar hybrid hydrogels by utilizing NapFFK-acryloyl hydrogelators (along with other components GOx, HRP, AcAc, and PEGMA) for their 3D cell printing was further illustrated by Wei et al. in another report.84 Such a supramolecular-polymeric hydrogel exhibited high mechanical strength with reusability and thermal stability along with biodegradability and cellular compatibility, and was successively employed for in situ 3D cellular printing to fabricate a 3D scaffold of NIH-3T3 cells.
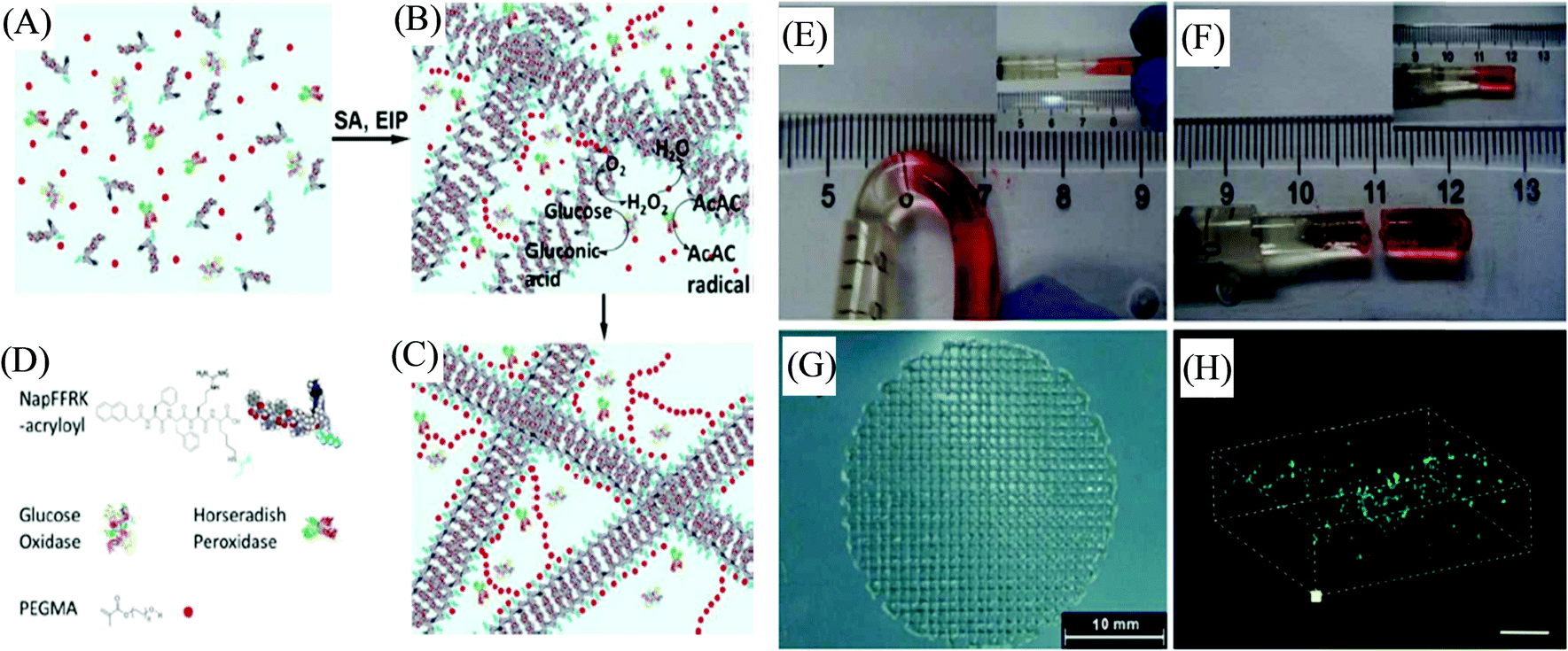 | ||
| Fig. 5 (A) Schematic representation of the pre-gel solution. (B) Dual-enzyme-triggered self-assembly (SA) and simultaneous enzyme-initiated polymer reaction (EIP). (C) The hybrid hydrogel formation with the incorporated dual enzyme. (D) Schematic illustration of the peptidic hydrogelator, enzymes, and PEGMA (AcAc and glucose are omitted). (E) The self-healing of the hybrid hydrogel at the joint by placing two parts together at 3600 s. (F) The joint cannot withstand bending by placing them together at 6000 s. (G) 3D printed constructs. (H) A 3D stack of printed cells in the hybrid hydrogel materials stained with FDA and PI (scale bar represents 300 μm). Reproduced from ref. 83 with permission from The Royal Society of Chemistry. | ||
A self-healable supramolecular polymer material for 3D printing under simplified thermomelting extrusion conditions was described by Wang et al. based on copolymer P(N-acryloyl glycinamide-co-1-vinyl-1,2,4-triazole) cross-linked through dynamic hydrogen bond interactions.85 The prepared hydrogels exhibited thermoplasticity, self-repairability, and reprocessability for various 3D printed constructs over a lower temperature range. By tuning the feed ratios and concentrations of monomer in the copolymer, the effective mechanical properties of the formed supramolecular hydrogels were obtained with high tensile strength up to 1.3 MPa, large stretchability up to more than 1300%, and increased compressibility to 11 MPa at swelling equilibrium state. Moreover, these supramolecular hydrogel biomaterials depicted medicinally desirable antibacterial and anti-inflammatory activities, along with biocompatible properties.
Other notable systems include a 3D printed drug delivery implant developed by Roberts, Hayes, and coworkers based on thermo-responsive supramolecular polyurethane (SPU), able to form a self-assembled polymer network via hydrogen bonding and π–π stacking (Fig. 6).86 The material displayed suitable mechanical properties (self-supporting and stiff, yet flexible) for the hot-melt extrusion-based approach at a relatively low processing temperature (100 °C). To establish the potential application of SPU for the 3D printing of a biomedical device, the biocompatibility of the polymeric films of polyurethane was determined by MTT assay using mouse fibroblasts (L929), which illustrated their non-cytotoxicity with more than 94% of cell viability. A multicomponent synthetic material SPU–PEG–paracetamol was prepared based on SPU co-formulated with PEG (4 wt% and 8 wt%) and incorporating paracetamol drug (16% w/w) to afford 3D printed robust implant constructs that exhibited appropriate mechanical performance, and prolonged drug release over 5 to 8.5 months. Such release rates can be potentially altered by modulating the formulation with varying PEG percentage or molecular weight. In 2019, Binder et al. developed 3D printable inks based on supramolecular polymers as linear and three-arm star biocompatible poly(isobutylene)s exhibiting multiple hydrogen-bonds and their nanocomposites to enable the construction of stable and self-supported structures at room temperature with polymer filament diameters of 200–300 μm.87
 | ||
| Fig. 6 (A) Chemical structure of the dynamic self-assembling supramolecular polyurethane (SPU) materials. (B) Schematic illustration of the SPU–PEG–paracetamol formulation-based 3D printing of the prototype drug-release implant. (C and D) Side-on and end-on view photographs of the 3D printed bar construct from such formulation. Reproduced from ref. 86 with permission from The Royal Society of Chemistry. | ||
3.2 Stimuli-responsive polymers
Stimuli-responsive polymers are smart or intelligent materials that can undergo alterations in their chemical, physical, morphological or mechanical properties upon interactions with their surrounding environment.88,89 In the past decade, these materials have been increasingly applied to a wide range of applications, including controlled and triggered drug delivery, (bio)sensing, actuators, coatings, diagnostics, tissue engineering, and biomedical devices.90,91 Various stimuli have been investigated for such polymers, including pH, temperature, chemicals, light, redox, mechanical forces, biomolecules, electric fields, and magnetic fields. The synergistic integration of stimuli-responsive polymer materials with the 3D printing technique can potentially provide remarkable engineering and materials advancements in the field of additive manufacturing processes to create complex geometries and structures that are not possible via other manufacturing techniques.92,93 For instance, Pluronic F127 has been investigated as a stimuli-responsive ink material for 3D printing, which corresponds to the ABA-type triblock copolymer PEG-PPO-PEG and possesses inverse thermo-gelling properties, and can be readily printed and removed under mild conditions.8 Zhang et al. developed a similar type of hydrogel material with dual stimuli-responsiveness for 3D printing; however such ABA triblock copolymers were comprised of one hydrophilic PEG segment and two hydrophobic poly(isopropyl glycidyl ether) (PiPrGE) segments arranged in a PiPrGE–PEG–PiPrGE configuration, and the corresponding hydrogels were able to exhibit thermoreversibility, as well as a rapid and reversible response to shear forces.94 Hsieh et al. documented bioink materials based on the aqueous dispersions of thermoresponsive biodegradable polyurethane (PU) nanoparticles (comprised of diols of PCL and (L or D,L) PLA of Mn ∼ 2000, mixed in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 M ratio), which formed a hydrogel near 37 °C without using any crosslinker (Fig. 7).95 The hydrogel stiffness was optimized by varying the PU content (25 or 30%) of the dispersion and their neural stem cell (NSC)-laden hydrogel constructs were printed by fused deposition manufacturing equipment. The filaments of the subsequently printed constructs displayed proper shape retention with reasonable swelling (<10% at 72 h), e.g., from a diameter of ∼210 μm at 0 h to ∼220 μm after 72 h for PU2 (30% or 25%) hydrogels. The cell viability of the encapsulated NSCs in the above PU2 hydrogels (∼680–2400 Pa) was found to be greater than 100% at 72 h, indicating excellent proliferation and differentiation. Moreover, the zebrafish embryo neural injury model having the injection with such NSCs (labeled with PKH26 (red fluorescence))-laden 25–30% PU2 hydrogels exhibited their wide dispersion in all brain areas, in particular, for those with 25% PU2 hydrogel, and further promoted the repair of the function of the impaired central nervous system. Murphy et al. illustrated another example of stimuli-responsive ink materials for 3D printing (Fig. 8) based on a photoresponsive polypeptide (with functional photoresponsive crosslinking).96 The hydrogel ink materials, comprised of UV-responsive copolypeptides incorporated with isolucine, glutamic acid, and cysteine (protected with photo-cleavable nitrobenzene groups), and alkyne functionalized 4-arm PEG-propiolate crosslinker, illustrated impressive shear-thinning properties to readily print a range of mechanically stable 3D structures. A catalyst-free nucleophilic thiol–yne “click” reaction was performed upon UV curing to enable crosslinking between the cysteine and propiolate residues, providing crosslinked hydrogel constructs with more than 10 layers exhibiting improved stiffness, high fidelity, and resolution. The biocompatibility of the copolypeptide hydrogel was further investigated with a human dermal fibroblasts cell assay by culturing on the top and within the hydrogel, illustrating almost no cytotoxicity and high viability of cells, respectively.
:1 M ratio), which formed a hydrogel near 37 °C without using any crosslinker (Fig. 7).95 The hydrogel stiffness was optimized by varying the PU content (25 or 30%) of the dispersion and their neural stem cell (NSC)-laden hydrogel constructs were printed by fused deposition manufacturing equipment. The filaments of the subsequently printed constructs displayed proper shape retention with reasonable swelling (<10% at 72 h), e.g., from a diameter of ∼210 μm at 0 h to ∼220 μm after 72 h for PU2 (30% or 25%) hydrogels. The cell viability of the encapsulated NSCs in the above PU2 hydrogels (∼680–2400 Pa) was found to be greater than 100% at 72 h, indicating excellent proliferation and differentiation. Moreover, the zebrafish embryo neural injury model having the injection with such NSCs (labeled with PKH26 (red fluorescence))-laden 25–30% PU2 hydrogels exhibited their wide dispersion in all brain areas, in particular, for those with 25% PU2 hydrogel, and further promoted the repair of the function of the impaired central nervous system. Murphy et al. illustrated another example of stimuli-responsive ink materials for 3D printing (Fig. 8) based on a photoresponsive polypeptide (with functional photoresponsive crosslinking).96 The hydrogel ink materials, comprised of UV-responsive copolypeptides incorporated with isolucine, glutamic acid, and cysteine (protected with photo-cleavable nitrobenzene groups), and alkyne functionalized 4-arm PEG-propiolate crosslinker, illustrated impressive shear-thinning properties to readily print a range of mechanically stable 3D structures. A catalyst-free nucleophilic thiol–yne “click” reaction was performed upon UV curing to enable crosslinking between the cysteine and propiolate residues, providing crosslinked hydrogel constructs with more than 10 layers exhibiting improved stiffness, high fidelity, and resolution. The biocompatibility of the copolypeptide hydrogel was further investigated with a human dermal fibroblasts cell assay by culturing on the top and within the hydrogel, illustrating almost no cytotoxicity and high viability of cells, respectively.
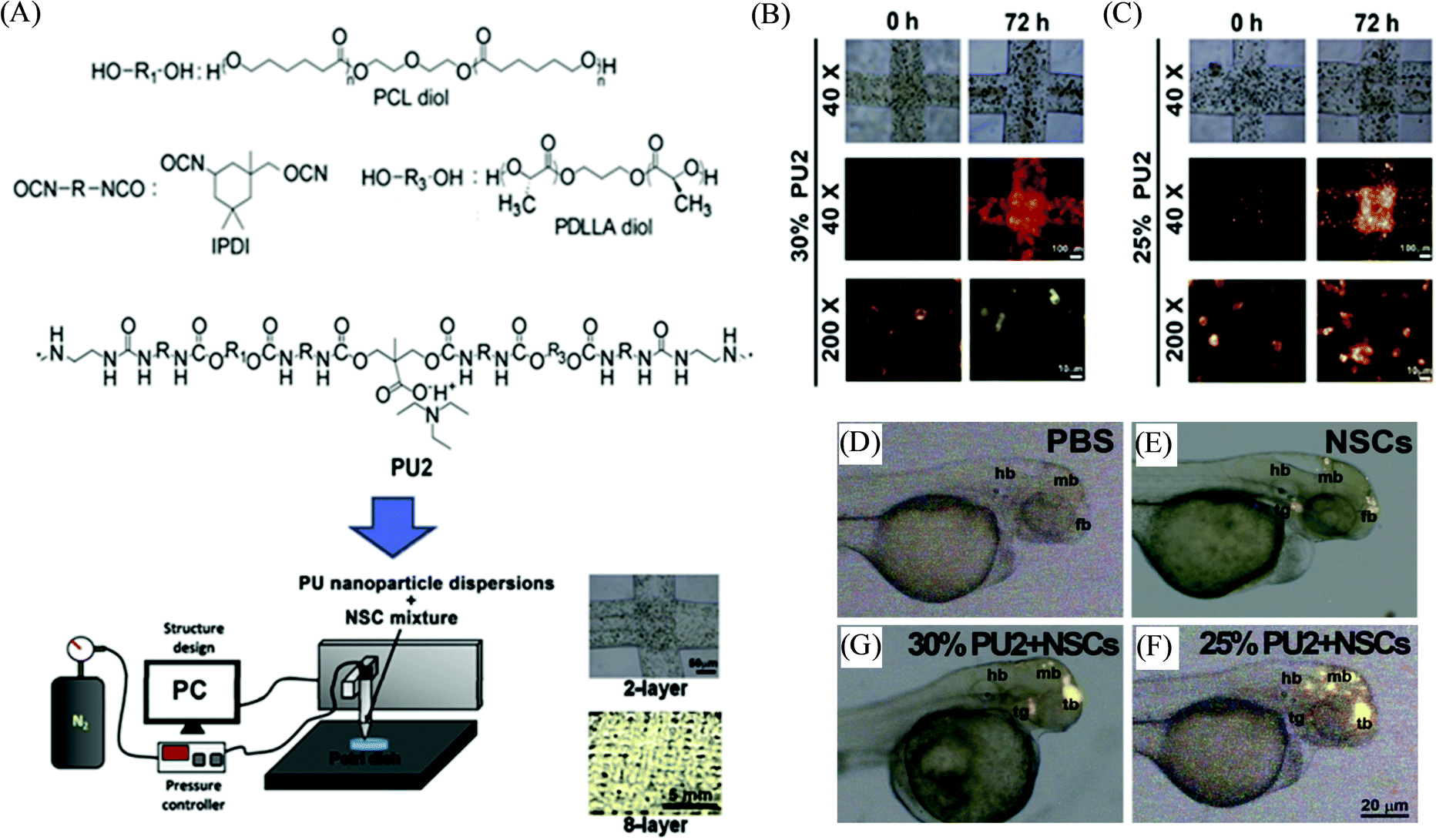 | ||
| Fig. 7 (A) Schematic representation depicting the chemical structure of polyurethane (PU), and the 3D bioprinting process for creating constructs based on the neural stem cell (NSC)-laden PU hydrogel. (B and C) Photographs of NSCs (labeled with PKH26) in the 3D printed stacking fibers at 0 h and 72 h corresponding to 25 and 30% PU2 hydrogels bionks, respectively. (D–G) The distribution of NSCs (labeled with PKH26) in the zebrafish embryos at 48 h post-fertilization, injected with PBS, NSCs, and NSC-laden PU2 hydrogels of 25 and 30%, respectively. Adapted with permission from ref. 95. Copyright 2015 Elsevier. | ||
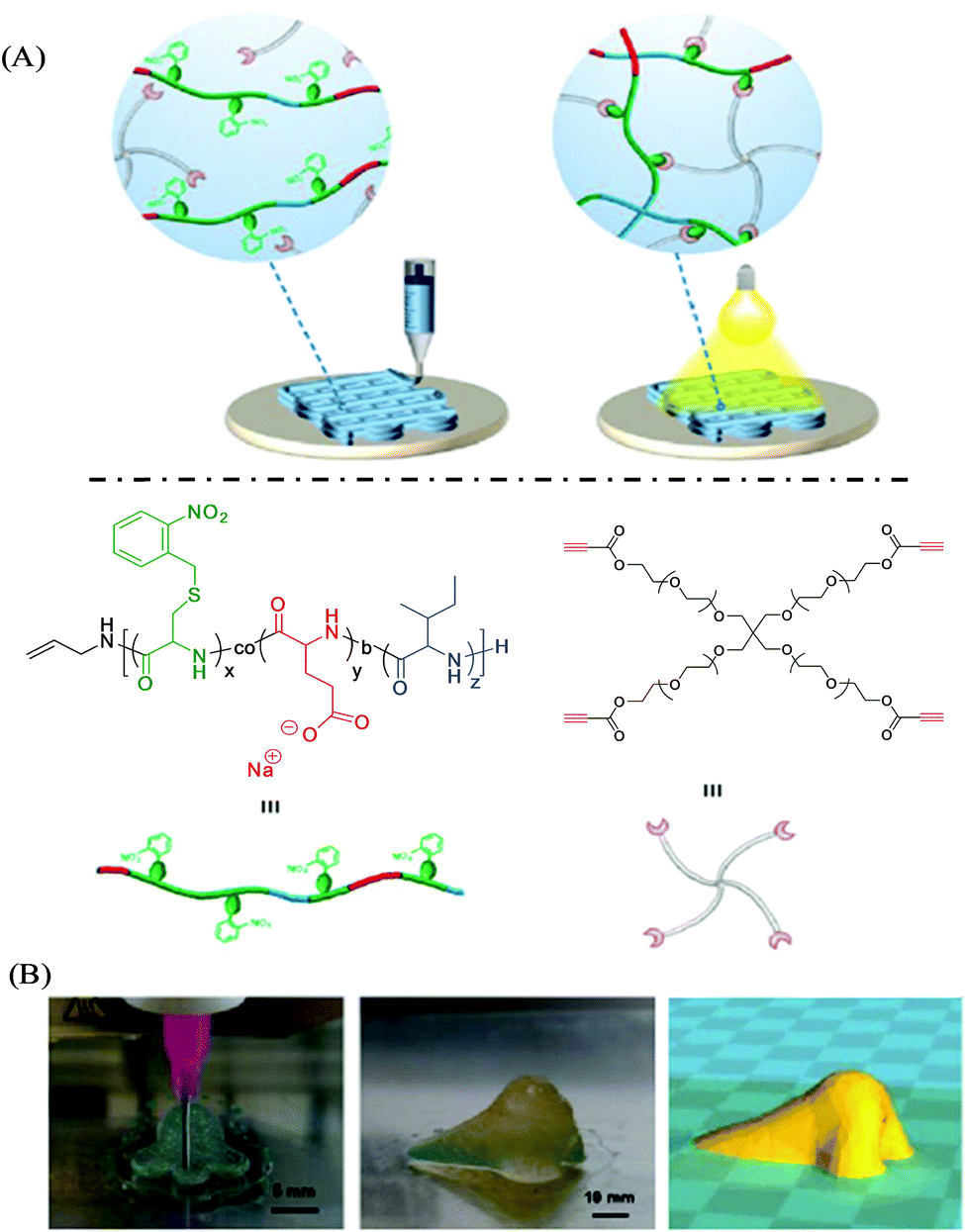 | ||
| Fig. 8 (A) Schematic illustration of photoresponsive copolypeptide hydrogel-based 3D-extrusion printing. Photocleavage of nitrobenzyl protecting groups-generated free thiol residues, which further reacted with the alkyne functional groups of 4-arm PEG-propiolate. (B) The design and development of a human nose shape construct by 3D printing with hydrogel ink. Adapted from ref. 96 with permission from The Royal Society of Chemistry. | ||
In 2011, Hennink and coworkers developed a biodegradable, photopolymerizable, and thermosensitive (as dual stimuli-responsive) ABA triblock copolymer hydrogel for 3D bioprinting-based cartilage tissue engineering applications (Fig. 9).97 A free radical copolymerization method was used to prepare this thermosensitive copolymer (with cloud point 11 °C) comprised of A-blocks based on biodegradable poly(N-(2-hydroxypropyl)methacrylamide lactate) (p(HPMAm-lac)) (Mw ∼ 23.5 kDa) partially modified by methacrylate groups to access photo cross-linking, and B-block derived from the hydrophilic PEG (Mw ∼ 10 kDa), yielding p(HPMA-lac)–PEG–p(HPMA-lac). The as-developed p(HPMA-lac)–PEG–p(HPMA-lac)-based hydrogels, with thermally and chemically cross-linked networks, displayed strain-softening behaviour and illustrated semi-flexible properties similar to various natural polymers including collagen. Moreover, the copolymeric hydrogels (25 wt%) were found to be suitable for the printing of porous 3D constructs with subsequent photopolymerization to exhibit an elastic modulus of 119 kPa (for the 0.6 cm construct) and a degradation time of around 190 days (for the 12-layered construct), and offer mechanical support to the encapsulated cells for the long term until new tissue is formed. Different fluorescent microspheres (fluorescent orange and lemon)-loaded hydrogels displayed the accurate and precise localization of such cell mimicking microspheres encapsulated within the copolymer to their 3D fiber deposition. The cell-laden photopolymerized hydrogel (25 wt%) revealed the homogeneous distribution of chondrocytes over the entire hydrogel with excellent viability of 94% after 1 day, with no adverse effects found due to the exposure of UV.
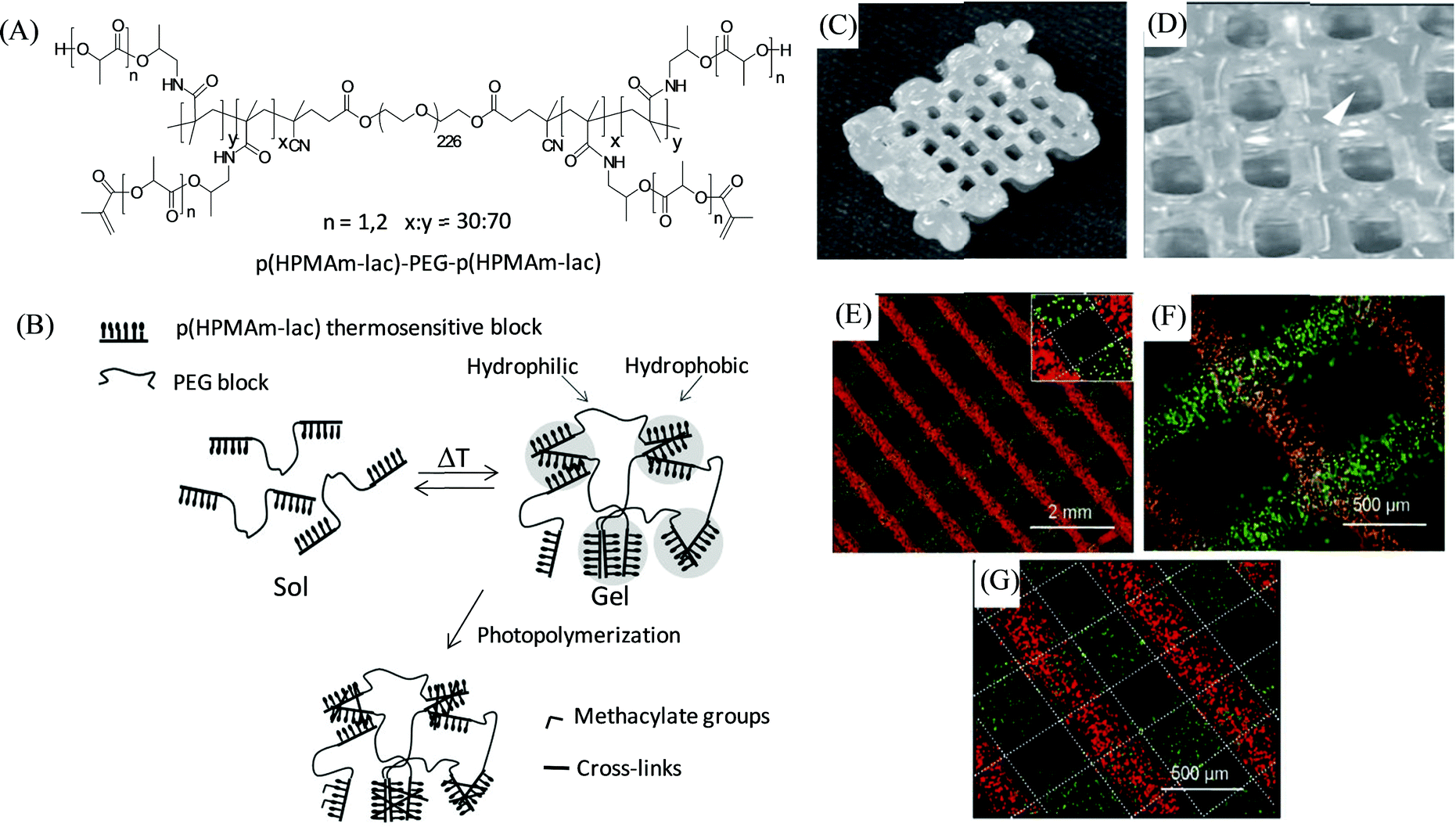 | ||
| Fig. 9 (A) Chemical structure of the thermosensitive triblock copolymer. (B) A cartoon representation of the effect of temperature and photopolymerization on the polymer illustrating hydrogel formation above the cloud point followed by chemical cross-linking within the hydrophobic domain. (C and D) Photographs of the 3D printed construct from 25 wt% copolymer-based hydrogels with dimensions of 1 × 1.5 × 0.6 cm, and strand spacing of 1.5 mm. (E, F, and G) Microscopic images of the 3D printed layers of fluorescent microspheres-loaded hydrogels with 1.5 mm strand spacing of 2- and 3-layer angled constructs, and a 0.8 mm strand spacing of a 2-layer construct, respectively. Adapted with permission from ref. 97. Copyright 2011 John Wiley and Sons. | ||
3.3 Nanoengineered polymers
Nanoengineered polymers are hybrid materials in which a polymeric matrix is dispersed with one or more types of inorganic or organic nanofillers such as silica or metallic nanoparticles, nanoclay, nanorods, carbon nanotubes and nanofibers, graphene, metallic nanowires, quantum dots, etc.98,99 Due to the several advantages imparted by the incorporated nanomaterials (via choosing proper mixing strategies) within the polymer matrix, such fabricated polymer nanocomposite materials exhibit significantly improved rheological performances, functionalities and properties (e.g., mechanical, electrical, optical, thermal, biological and magnetic properties) with respect to the native polymers,100,101 and therefore, make the polymer nanocomposite a suitable candidate to enable additive manufacturing based highly-customized complex and functional 3D constructs.102,103 Meng et al. documented a poly(vinyl alcohol) (PVA)-based nanocomposite hydrogel as artificial cartilage constructed by extrusion-based 3D printing (Fig. 10).104 Incorporation of graphene oxide (GO)-hydroxyapatite (HAp) in the nanocomposite PVA/GO-HAp (25 wt%) highly improved the dynamic viscosity of the materials and exhibited good shear-thinning properties in the extrusion shear rate range with a reduced Barus effect in order to obtain the stable printed constructs with high printing accuracy. The 3D printed PVA/GO-HAp hydrogels displayed biomimetic gradient porous structures with excellent bio-mechanical and bio-frictional behaviors attributed to their promising potential for the exact repair of articular cartilage. Shah and coworkers demonstrated the extrusion-based 3D printing of polymer nanocomposite inks comprised of a biocompatible copolymer polylactide-co-glycolide (PLG) (85:15) with high content of graphene (60 vol% of solid) to construct a mechanically robust and flexible scaffold (with strands ranging from 100 to 1000 μm in diameter) with shape-fidelity and enhanced electrical conductivities >800 S m−1 for electronic applications.105 The printed 3D scaffold further revealed their potential biomedical application towards nerve tissue engineering and regeneration by exhibiting their significant cellular response (attachment, proliferation, and neurogenic differentiation) to human mesenchymal stem cells (hMSCs) and viability to multiple, distinct cell types, including hMSCs.
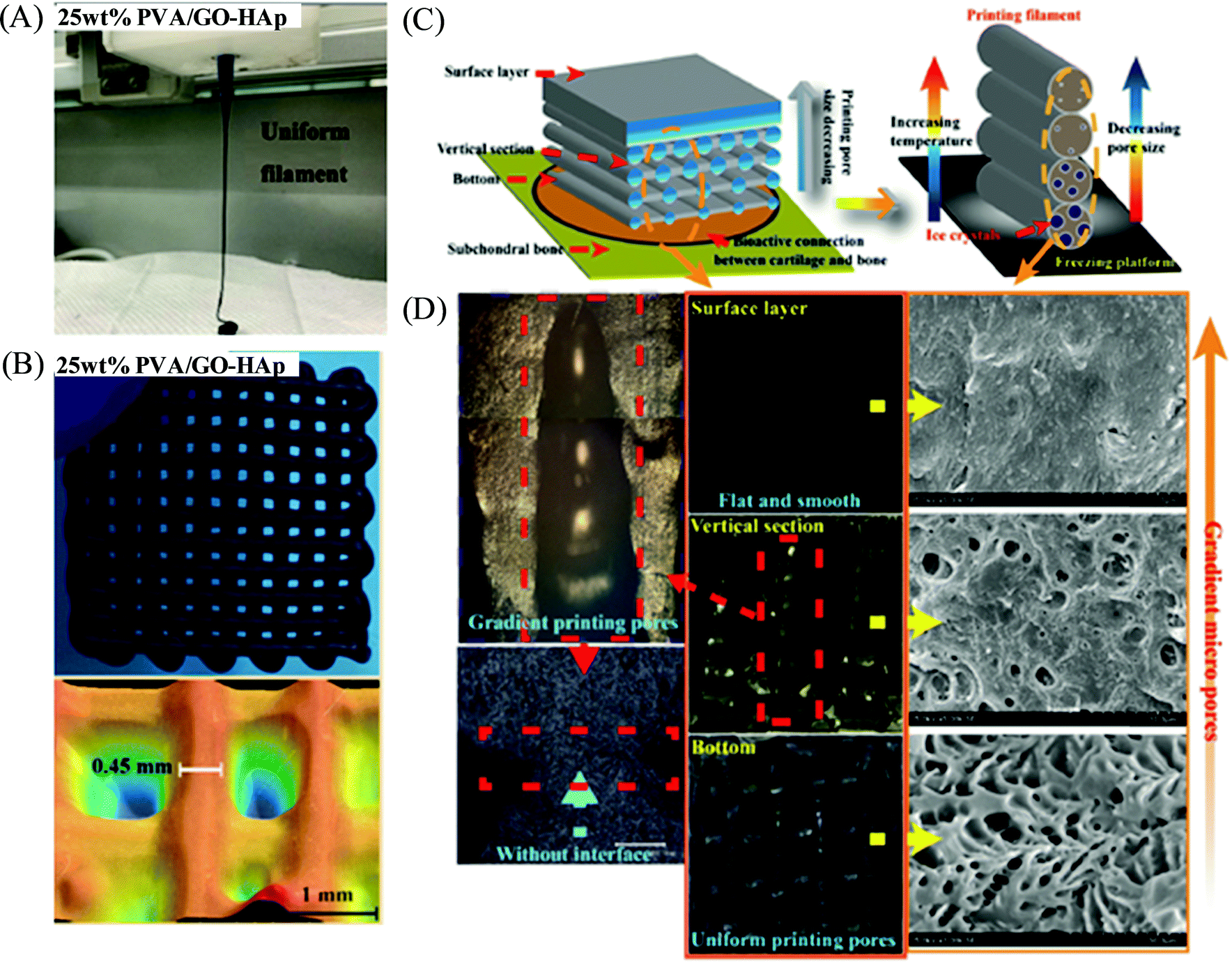 | ||
| Fig. 10 (A) Digital photograph of a 3D printing filament of the PVA/GO-HAp nanocomposite solution with 25 wt% PVA concentration. (B) Digital photograph and structural morphology of the 3D printed sample for the above nanocomposite-based hydrogel having a reticular porous structure. (C) Schematic design of the PVA/GO-HAp nanocomposite-based biomimetic gradient porous structure as artificial cartilage. (D) Digital photograph and SEM images of the 3D printed constructs of PVA/GO-HAp (25 wt%) nanocomposite-based artificial cartilage. Adapted from ref. 104 with permission from The Royal Society of Chemistry. | ||
Polymer/clay nanocomposite ink materials based on PEG precursor colloidal solutions incorporated with biocompatible and bioactive disk-shaped 2D LAPONITE® (XLG) nanoclay were investigated by the Gaharwar group for 3D bioprinting (Fig. 11).106 Such hydrogels based on diacrylated PEG-LAPONITE® demonstrated shear-thinning and self-healing properties due to rapid internal phase rearrangement, thus enabling the printing of a range of complex 3D structures upon UV-based photo-crosslinking. The rheological characteristic of the nanocomposite hydrogels appeared to be independent of the addition of PEG and dominated by the behaviour of the LAPONITE® network, whereas their self-recovery time was found to be controlled by the ratio of PEG:LAPONITE®. Further, the PEG-(4%)LAPONITE®-based bioinks exhibited high cell viability of the encapsulated murine preosteoblasts immediately post-injection, and the viability was further maintained across different volumetric flow rates of 500, 1000, and 200 μL min−1 as indicated by the live/dead assay.
 | ||
| Fig. 11 (A) Digital photograph of the PEG-LAPONITE® colloidal solution-based hydrogel and their printed structure. (B) Schematic of the formation of the internal structure for PEG, LAPONITE®, and PEG-LAPONITE® nanocomposite solutions at the initial time and after 18 hours. (C and D) 3D printed circular and crosshatch structure, respectively, of PEG-(4%) LAPONITE® with preosteoblasts and cell tracker images. Adapted with permission from ref. 106. Copyright 2018 American Chemical Society. | ||
Another example of nanoclay-based polymeric nanocomposite bioink for 3D printing was demonstrated by Zhai et al., derived from supramolecular polymer poly(N-acryloyl glycinamide) (PNAGA).107 The polymer/clay PNAGA-LAPONITE® XLG-based hybrid hydrogel ink provided the fabrication of a 3D high strength composite scaffold with swelling stability (via post-extrusion UV light irradiation) imparted by the dual amide hydrogen bonding interactions of polymer combined with physical cross-linking of nanoclay-polymer chain interactions. Such PNAGA(20%)-LAPONITE® bioscaffolds were able to promote the osteogenic differentiation of primary rat osteoblast (ROB) cells via the release of encapsulated Mg2+ and Si4+ ions from the hydrogel as suggested by the in vitro studies. Importantly, improved cellular adhesion, proliferation, and differentiation bioactivities were observed for ROB seeded within the PNAGA-LAPONITE® composite scaffolds due to the incorporation of clay into such hydrogel systems. The in vivo experiments demonstrated the efficient formation of new bone upon the implantation of the PNAGA(20%)-clay scaffold into tibia defects of rats for 8 weeks.
4. Conclusions and future perspectives
The field of 3D printing-based additive manufacturing has been burgeoning over the past few years and a range of affordable 3D printers are accessible for laboratories to date. A lot of efforts have been devoted to this field, essentially towards (i) the engineering and optimization of the printing techniques for enabling the creation of complex and highly organised structures with controlled processing and higher resolution, and (ii) developing the ink materials with suitable rheological properties and functionality. The particular challenge is the development of (bio)ink materials for the 3D printing of biologically relevant scaffolds/tissue replacement, which allows the cells to adhere, differentiate and proliferate, and preserves the bioactivity of their other embedded compounds such as growth factors, signalling peptides, drugs, etc. Currently, researchers are focusing on the design of (bio)ink materials mainly based on natural polymers or a combination of natural and synthetic polymers; however, this approach is associated with several challenges, and therefore, the field of 3D (bio)printing is still in its infancy. The introduction of synthetic polymers as a single (or major) network precursor of ink, and the involvement of the functionalization and cross-linking chemistry in the ink formulation can lead to the development of a diverse range of new-generation (bio)ink materials with tuneable mechanical, chemical and biological properties, and can therefore emerge as a key player for advancing the field of 3D printing to the next frontier to create synthetic and in-human 3D printed tissues. It is critically important to have a fundamental understanding of engineering the 3D printing technique with controlled processing parameters, evaluating new and advanced polymers-based ink/bioink materials, and designing the biomimetic hard and soft tissue scaffolds with controlled structural integrity and functioning to fabricate the targeted tissue/organ; this will drive further innovation in the field of tissue engineering and regenerative medicines.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The author acknowledge the financial support by Science and Engineering Research Board (SERB), New Delhi, India (Project no.: SRG/2020/000610).References
- R. L. Truby and J. A. Lewis, Nature, 2016, 540, 371–378 CrossRef CAS PubMed.
- (a) S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mulhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed; (b) D. Gräfe, S. L. Walden, J. Blinco, M. Wegener, E. Blasco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2020, 59, 6330–6340 CrossRef PubMed.
- (a) S. H. Park, C. S. Jung and B. H. Min, Tissue Eng. Regener. Med., 2016, 13, 622–635 CrossRef CAS PubMed; (b) A. Shapira and T. Dvir, Adv. Sci., 2021, 8, 2003751 CrossRef CAS PubMed.
- (a) A. Lee, A. R. Hudson, D. J. Shiwarski, J. W. Tashman, T. J. Hinton, S. Yerneni, J. M. Bliley, P. G. Campbell and A. W. Feinberg, Science, 2019, 365, 482–487 CrossRef CAS PubMed; (b) M. S. Mannoor, Z. Jiang, T. James, Y. L. Kong, K. A. Malatesta, W. O. Soboyejo, N. Verma, D. H. Gracias and M. C. McAlpine, Nano Lett., 2013, 13, 2634–2639 CrossRef CAS PubMed.
- M. Nadgorny and A. Ameli, ACS Appl. Mater. Interfaces, 2018, 10, 17489–17507 CrossRef CAS PubMed.
- S. Naficy, R. Gately, R. Gorkin, H. Xin and G. M. Spinks, Macromol. Mater. Eng., 2017, 302, 1600212 CrossRef.
- H. Gudapati, M. Dey and I. Ozbolat, Biomaterials, 2016, 102, 20–42 CrossRef CAS PubMed.
- D. B. Kolesky, R. L. Truby, A. S. Gladman, T. A. Busbee, K. A. Homan and J. A. Lewis, Adv. Mater., 2014, 26, 3124–3130 CrossRef CAS PubMed.
- B. Holmes, K. Bulusu, M. Plesniak and L. G. Zhang, Nanotechnology, 2016, 27, 064001 CrossRef PubMed.
- R. J. Mondschein, A. Kanitkar, C. B. Williams, S. S. Verbridge and T. E. Long, Biomaterials, 2017, 140, 170–188 CrossRef CAS PubMed.
- C. M. B. Ho, A. Mishra, K. Hu, J. An, Y.-J. Kim and Y.-J. Yoon, ACS Biomater. Sci. Eng., 2017, 3, 2198–2214 CrossRef CAS PubMed.
- T. Jungst, W. Smolan, K. Schacht, T. Scheibel and J. Groll, Chem. Rev., 2016, 116, 1496–1539 CrossRef CAS PubMed.
- J. M. Lee and W. Y. Yeong, Adv. Healthcare Mater., 2016, 5, 2856–2865 CrossRef CAS PubMed.
- H. Abe, K. Hayashi and M. Sato, Data book on mechanical properties of living cells tissues, and organs, Springer, 1996 Search PubMed.
- S. S. Athukorala, T. S. Tran, R. Balu, V. K. Truong, J. Chapman, N. K. Dutta and N. R. Choudhury, Polymers, 2021, 13, 474 CrossRef CAS PubMed.
- L. Li, Q. Lin, M. Tang, A. J. E. Duncan and C. Ke, Chem. – Eur. J., 2019, 25, 1–15 CrossRef.
- L. Valot, J. Martinez, A. Mehdi and G. Subra, Chem. Soc. Rev., 2019, 48, 4049–4086 RSC.
- F. You, X. Wu, N. Zhu, M. Lei, B. F. Eames and X. Chen, ACS Biomater. Sci. Eng., 2016, 2, 1200–1210 CrossRef CAS PubMed.
- J. Y. Park, G. Gao, J. Jang and D.-W. Cho, J. Mater. Chem. B, 2016, 4, 7521–7539 RSC.
- V. Mourino and A. R. Boccaccini, J. R. Soc., Interface, 2010, 7, 209–227 CrossRef CAS PubMed.
- I. T. Ozbolat, W. Peng and V. Ozbolat, Drug Discovery Today, 2016, 21, 1257–1271 CrossRef CAS.
- Y. He, F. F. Yang, H. M. Zhao, Q. Gao, B. Xia and J. Z. Fu, Sci. Rep., 2016, 6, 29977 CrossRef CAS PubMed.
- A. Schwab, R. Levato, M. D’Este, S. Piluso, D. Eglin and J. Malda, Chem. Rev., 2020, 120, 11028–11055 CrossRef CAS PubMed.
- M. Bongio, J. J. van den Beucken, M. R. Nejadnik, S. C. Leeuwenburgh, L. A. Kinard, F. K. Kasper, A. G. Mikos and J. A. Jansen, Eur. Cell. Mater., 2011, 22, 359 CrossRef CAS PubMed.
- (a) K. Y. Lee, M. C. Peters, K. W. Anderson and D. J. Mooney, Nature, 2000, 408, 998 CrossRef CAS PubMed; (b) R. Censi, P. Di Martino, T. Vermonden and W. E. Hennink, J. Controlled Release, 2012, 161, 680 CrossRef CAS PubMed.
- K. L. Spiller, S. A. Maher and A. M. Lowman, Tissue Eng., Part B, 2011, 17, 281 CrossRef CAS PubMed.
- H. Gudapati, M. Dey and I. Ozbolat, Biomaterials, 2016, 102, 20–42 CrossRef CAS PubMed.
- I. Freshney, Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications, John Wiley & Sons, 2015 Search PubMed.
- K. Nair, M. Gandhi, S. Khalil, K. C. Yan, M. Marcolongo, K. Barbee and W. Sun, Biotechnol. J., 2009, 4, 1168–1177 CrossRef CAS PubMed.
- J. Liu, L. Sun, W. Xu, Q. Wang, S. Yu and J. Sun, Carbohydr. Polym., 2019, 207, 297–316 CrossRef CAS PubMed.
- C. Mota, S. Camarero-Espinosa, M. B. Baker, P. Wieringa and L. Moroni, Chem. Rev., 2020, 120, 10547–10607 CrossRef CAS PubMed.
- G. Ratheesh, J. R. Venugopal, A. Chinappan, H. Ezhilarasu, A. Sadiq and S. Ramakrishna, ACS Biomater. Sci. Eng., 2017, 3, 1175–1194 CrossRef CAS PubMed.
- M. Guvendiren, J. Molde, R. M. Soares and J. Kohn, ACS Biomater. Sci. Eng., 2016, 2, 1679–1693 CrossRef CAS PubMed.
- S. R. Govindarajan, Y. Xu, J. P. Swanson, T. Jain, Y. Lu, J.-W. Choi and A. Joy, Macromolecules, 2016, 49, 2429–2437 CrossRef CAS.
- D. A. Gopakumar, D. Pasquini, M. A. Henrique, L. C. de Morais, Y. Grohens and S. Thomas, ACS Sustainable Chem. Eng., 2017, 5, 2026–2033 CrossRef CAS.
- L.-Y. Zhou, J. Fu and Y. He, Adv. Funct. Mater., 2020, 30, 2000187 CrossRef CAS.
- B. Narupai and A. Nelson, ACS Macro Lett., 2020, 9, 627–638 CrossRef CAS.
- R. R. Jose, M. J. Rodriguez, T. A. Dixon, F. Omenetto and D. L. Kaplan, ACS Biomater. Sci. Eng., 2016, 2, 1662–1678 CrossRef CAS PubMed.
- C.-C. Lin and K. S. Anseth, Pharm. Res., 2009, 26, 631–643 CrossRef CAS PubMed.
- C. Englert, J. C. Brendel, T. C. Majdanski, T. Yildirim, S. Schubert, M. Gottschaldt, N. Windhab and U. S. Schubert, Prog. Polym. Sci., 2018, 87, 107–164 CrossRef CAS.
- L. Tan, X. Yu, P. Wan and K. Yang, J. Mater. Sci. Technol., 2013, 29, 503–513 CrossRef CAS.
- M. K. Gupta, F. Meng, B. N. Johnson, Y. L. Kong, L. Tian, Y.-W. Yeh, N. Masters, S. Singamaneni and M. C. McAlpine, Nano Lett., 2015, 15, 5321–5329 CrossRef CAS PubMed.
- P. Camacho, H. Busari, K. Seims, P. Schwarzenberg, H. L. Dailey and L. W. Chow, Biomater. Sci., 2019, 7, 4237–4247 RSC.
- A. Rashad, S. Mohamed-Ahmed, M. Ojansivu, K. Berstad, M. A. Yassin, T. Kivijarvi, E. B. Heggset, K. Syverud and K. Mustafa, Biomacromolecules, 2018, 19, 4307–4319 CrossRef CAS PubMed.
- W. J. Peng, D. Unutmaz and I. T. Ozbolat, Trends Biotechnol., 2016, 34, 722–732 CrossRef CAS PubMed.
- M. Askari, M. A. Naniz, M. Kouhi, A. Saberi, A. Zolfagharian and M. Bodaghi, Biomater. Sci., 2021, 9, 535–573 RSC.
- J. Malda, J. Visser, F. P. Melchels, T. Jungst, W. E. Hennink, W. J. Dhert, J. Groll and D. W. Hutmacher, Adv. Mater., 2013, 25, 5011–5028 CrossRef CAS PubMed.
- Z. L. Yue, X. Liu, P. T. Coates and G. G. Wallace, Curr. Opin. Organ Transplant., 2016, 21, 467–475 CrossRef CAS PubMed.
- A. C. Fonseca, F. P. W. Melchels, M. J. S. Ferreira, S. R. Moxon, G. Potjewyd, T. R. Dargaville, S. J. Kimber and M. Domingos, Chem. Rev., 2020, 120, 11093–11139 CrossRef CAS PubMed.
- X. Cui, J. Li, Y. Hartanto, M. Durham, J. Tang, H. Zhang, G. Hooper, K. Lim and T. Woodfield, Adv. Healthcare Mater., 2020, 1901648 CrossRef CAS PubMed.
- Z. Jiang, B. Diggle, M. L. Tan, J. Viktorova, C. W. Bennett and L. A. Connal, Adv. Sci., 2020, 7, 2001379 CrossRef CAS PubMed.
- L. Ouyang, C. B. Highley, W. Sun and J. A. Burdick, Adv. Mater., 2017, 1604983 CrossRef PubMed.
- A. McCormack, C. B. Highley, N. R. Leslie and F. P. W. Melchels, Trends Biotechnol., 2020, 38, 584–593 CrossRef CAS PubMed.
- J. J. Senior, M. E. Cooke, L. M. Grover and A. M. Smith, Adv. Funct. Mater., 2019, 29, 1904845 CrossRef CAS.
- E. Mirdamadi, J. W. Tashman, D. J. Shiwarski, R. N. Palchesko and A. W. Feinberg, ACS Biomater. Sci. Eng., 2020, 6, 6453–6459 CrossRef CAS PubMed.
- E. Davoodi, E. Sarikhani, H. Montazerian, S. Ahadian, M. Costantini, W. Swieszkowski, S. M. Willerth, K. Walus, M. Mofidfar, E. Toyserkani, A. Khademhosseini and N. Ashammakhi, Adv. Mater. Technol., 2020, 5, 1901044 CrossRef CAS PubMed.
- C. Colosi, S. R. Shin, V. Manoharan, S. Massa, M. Costantini, A. Barbetta, M. R. Dokmeci, M. Dentini and A. Khademhosseini, Adv. Mater., 2016, 28, 677–684 CrossRef CAS PubMed.
- (a) S. J. Buwalda, T. Vermonden and W. E. Hennink, Biomacromolecules, 2017, 18, 316 CrossRef CAS PubMed; (b) R. Dong, Y. Pang, Y. Su and X. Zhu, Biomater. Sci., 2015, 3, 937 RSC.
- (a) A. M. Jonker, D. W. P. M. Lowik and J. C. M. van Hest, Chem. Mater., 2012, 24, 759 CrossRef CAS; (b) S. Kumar and W. H. Binder, Soft Matter, 2020, 16, 6964–6968 RSC.
- J. E. Kim, S. H. Kim and Y. Jung, Tissue Eng. Regener. Med., 2016, 13, 636–646 CrossRef CAS PubMed.
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219 RSC.
- J. M. Unagolla and A. C. Jayasuriya, Appl. Mater. Today, 2020, 18, 100479 CrossRef PubMed.
- R. Censi, W. Schuurman, J. Malda, G. di Dato, P. E. Burgisser, W. J. A. Dhert, C. F. van Nostrum, P. di Martino, T. Vermonden and W. E. Hennink, Adv. Funct. Mater., 2011, 21, 1833 CrossRef CAS.
- (a) N. E. Fedorovich, J. R. De Wijn, A. J. Verbout, J. Alblas and W. J. Dhert, Tissue Eng., Part A, 2008, 14, 127 CAS; (b) S. Hong, D. Sycks, H. F. Chan, S. Lin, G. P. Lopez, F. Guilak, K. W. Leong and X. Zhao, Adv. Mater., 2015, 15, 4035–4040 CrossRef PubMed.
- N. E. Fedorovich, I. Swennen, J. Girones, L. Moroni, C. A. van Blitterswijk, E. Schacht, J. Alblas and W. J. Dhert, Biomacromolecules, 2009, 10, 1689 CrossRef CAS PubMed.
- (a) C. M. Smith, A. L. Stone, R. L. Parkhill, R. L. Stewart, M. W. Simpkins, A. M. Kachurin, W. L. Warren and S. K. Williams, Tissue Eng., 2004, 10, 1566 CrossRef CAS PubMed; (b) A. C. Daly, G. M. Cunniffe, B. N. Sathy, O. Jeon, E. Alsberg and D. J. Kelly, Adv. Healthcare Mater., 2016, 5, 2353–2362 CrossRef CAS PubMed.
- H. W. Ooi, C. Mota, A. T. ten Cate, A. Calore, L. Moroni and M. B. Baker, Biomacromolecules, 2018, 19, 3390–3400 CrossRef CAS PubMed.
- A. Skardal, J. Zhang and G. D. Prestwich, Biomaterials, 2010, 31, 6173–6181 CrossRef CAS PubMed.
- S. Joas, G. E. M. Tovar, O. Celik, C. Bonten and A. Southan, Gels, 2018, 4, 69 CrossRef CAS PubMed.
- C. Xu, W. Lee, G. Dai and Y. Hong, ACS Appl. Mater. Interfaces, 2018, 10, 9969–9979 CrossRef CAS PubMed.
- L. Brunsveld, B. Folmer, E. Meijer and R. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- (a) E. Kolomiets and J.-M. Lehn, Chem. Commun., 2005, 1519 RSC; (b) S. Kumar, V. Bheemireddy and P. De, Macromol. Biosci., 2015, 15, 1447–1456 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- (a) J. L. Mann, A. C. Yu, G. Agmon and E. A. Appel, Biomater. Sci., 2018, 6, 10–37 RSC; (b) S. Kumar, R. Acharya, U. Chatterji and P. De, Polym. Chem., 2014, 5, 6039–6050 RSC.
- C. F. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- S.-L. Li, T. Xiao, C. Lin and L. Wang, Chem. Soc. Rev., 2012, 41, 5950–5968 RSC.
- A. M. Pekkanen, R. J. Mondschein, C. B. Williams and T. E. Long, Biomacromolecules, 2017, 18, 2669–2687 CrossRef CAS PubMed.
- C. Echalier, R. Levato, M. A. Mateos-Timoneda, O. Castaño, S. Déjean, X. Garric, C. Pinese, D. Noël, E. Engel, J. Martinez, A. Mehdi and G. Subra, RSC Adv., 2017, 7, 12231–12235 RSC.
- T. Lorson, S. Jaksch, M. M. Lubtow, T. Jungst, J. Groll, T. Luhmann and R. Luxenhofer, Biomacromolecules, 2017, 18, 2161–2171 CrossRef CAS PubMed.
- L. Trachsel, M. Zenobi-Wong and E. M. Benetti, Biomater. Sci., 2021, 9, 2874–2886 RSC.
- Q. Wei, W. Xu, M. Liu, Q. Wu, L. Cheng and Q. Wang, J. Mater. Chem. B, 2016, 4, 6302–6306 RSC.
- Q. Wei, M. Xu, C. Liao, Q. Wu, M. Liu, Y. Zhang, C. Wu, L. Cheng and Q. Wang, Chem. Sci., 2016, 7, 2748–2752 RSC.
- H. Wang, H. Zhu, W. Fu, Y. Zhang, B. Xu, F. Gao, Z. Cao and W. Liu, Macromol. Rapid Commun., 2017, 38, 1600695 CrossRef PubMed.
- S. Salimi, Y. Wu, M. I. E. Barreiros, A. A. Natfji, S. Khaled, R. Wildman, L. R. Hart, F. Greco, E. A. Clark, C. J. Roberts and W. Hayes, Polym. Chem., 2020, 11, 3453–3464 RSC.
- H. Rupp, D. Döhler, P. Hilgeroth, N. Mahmood, M. Beiner and W. H. Binder, Macromol. Rapid Commun., 2019, 40, 1900467 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- (a) Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2017, 2, 16075 CrossRef CAS; (b) S. Kumar, S. Deike and W. H. Binder, Macromol. Rapid Commun., 2018, 39, 1700507 CrossRef PubMed.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- T. M. Reineke, ACS Macro Lett., 2016, 5, 14–18 CrossRef CAS.
- (a) A. J. Boydston, B. Cao, A. Nelson, R. J. Ono, A. Saha, J. J. Schwartz and C. J. Thrasher, J. Mater. Chem. A, 2018, 6, 20621–20645 RSC; (b) H. Rupp and W. H. Binder, Macromol. Rapid Commun., 2020, 42, 2000450 CrossRef PubMed.
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2015, 7, 577–583 CrossRef CAS PubMed.
- M. Zhang, A. Vora, W. Han, R. J. Wojtecki, H. Maune, A. B. A. Le, L. E. Thompson, G. M. McClelland, F. Ribet, A. C. Engler and A. Nelson, Macromolecules, 2015, 48, 6482–6488 CrossRef CAS.
- F. Y. Hsieh, H. H. Lin and S. H. Hsu, Biomaterials, 2015, 71, 48–57 CrossRef CAS PubMed.
- R. D. Murphy, S. Kimmins, A. J. Hibbitts and A. Heise, Polym. Chem., 2019, 10, 4675–4682 RSC.
- R. Censi, W. Schuurman, J. Malda, G. di Dato, P. E. Burgisser, W. J. A. Dhert, C. F. van Nostrum, P. di Martino, T. Vermonden and W. E. Hennink, Adv. Funct. Mater., 2011, 21, 1833–1842 CrossRef CAS.
- S. K. Kumar, B. C. Benicewicz, R. A. Vaia and K. I. Winey, Macromolecules, 2017, 50, 714–731 CrossRef CAS.
- D. A. Gopakumar, A. R. Pai, Y. B. Pottathara, D. Pasquini, L. C. de Morais, M. Luke, N. Kalarikkal, Y. Grohens and S. Thomas, ACS Appl. Mater. Interfaces, 2018, 10, 20032–20043 CrossRef CAS PubMed.
- B. Elder, R. Neupane, E. Tokita, U. Ghosh, S. Hales and Y. L. Kong, Adv. Mater., 2020, 32, 1907142 CrossRef CAS PubMed.
- H. Rupp and W. H. Binder, Adv. Mater. Technol., 2020, 2000509 CrossRef CAS.
- R. D. Farahani and M. Dubé, Adv. Eng. Mater., 2018, 20, 1700539 CrossRef.
- H. Rastin, B. Zhang, A. Mazinani, K. Hassan, J. Bi, T. Tung and D. Losic, Nanoscale, 2020, 12, 16069–16080 RSC.
- Y. Meng, J. Cao, Y. Chen, Y. Yu and L. Ye, J. Mater. Chem. B, 2020, 8, 677–690 RSC.
- A. E. Jakus, E. B. Secor, A. L. Rutz, S. W. Jordan, M. C. Hersam and R. N. Shah, ACS Nano, 2015, 9, 4636–4648 CrossRef CAS PubMed.
- C. W. Peak, J. Stein, K. A. Gold and A. K. Gaharwar, Langmuir, 2018, 34, 917–925 CrossRef CAS PubMed.
- X. Zhai, Y. Ma, C. Hou, F. Gao, Y. Zhang, C. Ruan, H. Pan, W. W. Lu and W. Liu, ACS Biomater. Sci. Eng., 2017, 3, 1109–1118 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |